DOI:
10.1039/D3RA01971C
(Paper)
RSC Adv., 2023,
13, 13337-13353
Exploring the synthetic potential of a g-C3N4·SO3H ionic liquid catalyst for one-pot synthesis of 1,1-dihomoarylmethane scaffolds via Knoevenagel–Michael reaction†
Received
25th March 2023
, Accepted 25th April 2023
First published on 2nd May 2023
Abstract
A highly promising approach for the synthesis of functionalized 1,1-dihomoarylmethane scaffolds (bis-dimedones, bis-cyclohexanediones, bis-pyrazoles, and bis-coumarins) using g-C3N4·SO3H ionic liquid via Knoevenagel–Michael reaction has been developed and the synthesized derivatives were well characterized using spectral studies. The method involved the reaction of C–H activated acids with a range of aromatic aldehydes, in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio catalyzed by a g-C3N4·SO3H ionic liquid catalyst. The use of g-C3N4·SO3H as a catalyst has several benefits, such as low cost, easy preparation, and high stability. It was synthesized from urea powder and chloro-sulfonic acid and was thoroughly characterized using FT-IR, XRD, SEM, and HRTEM. The present work unveils a promising and environmentally friendly method for synthesizing 1,1-dihomoarylmethane scaffolds with high yield, selectivity, and efficiency, using mild reaction conditions, no need for chromatographic separation, and short reaction times. The approach adheres to green chemistry principles and offers a viable alternative to the previously reported methods.
:1 ratio catalyzed by a g-C3N4·SO3H ionic liquid catalyst. The use of g-C3N4·SO3H as a catalyst has several benefits, such as low cost, easy preparation, and high stability. It was synthesized from urea powder and chloro-sulfonic acid and was thoroughly characterized using FT-IR, XRD, SEM, and HRTEM. The present work unveils a promising and environmentally friendly method for synthesizing 1,1-dihomoarylmethane scaffolds with high yield, selectivity, and efficiency, using mild reaction conditions, no need for chromatographic separation, and short reaction times. The approach adheres to green chemistry principles and offers a viable alternative to the previously reported methods.
Introduction
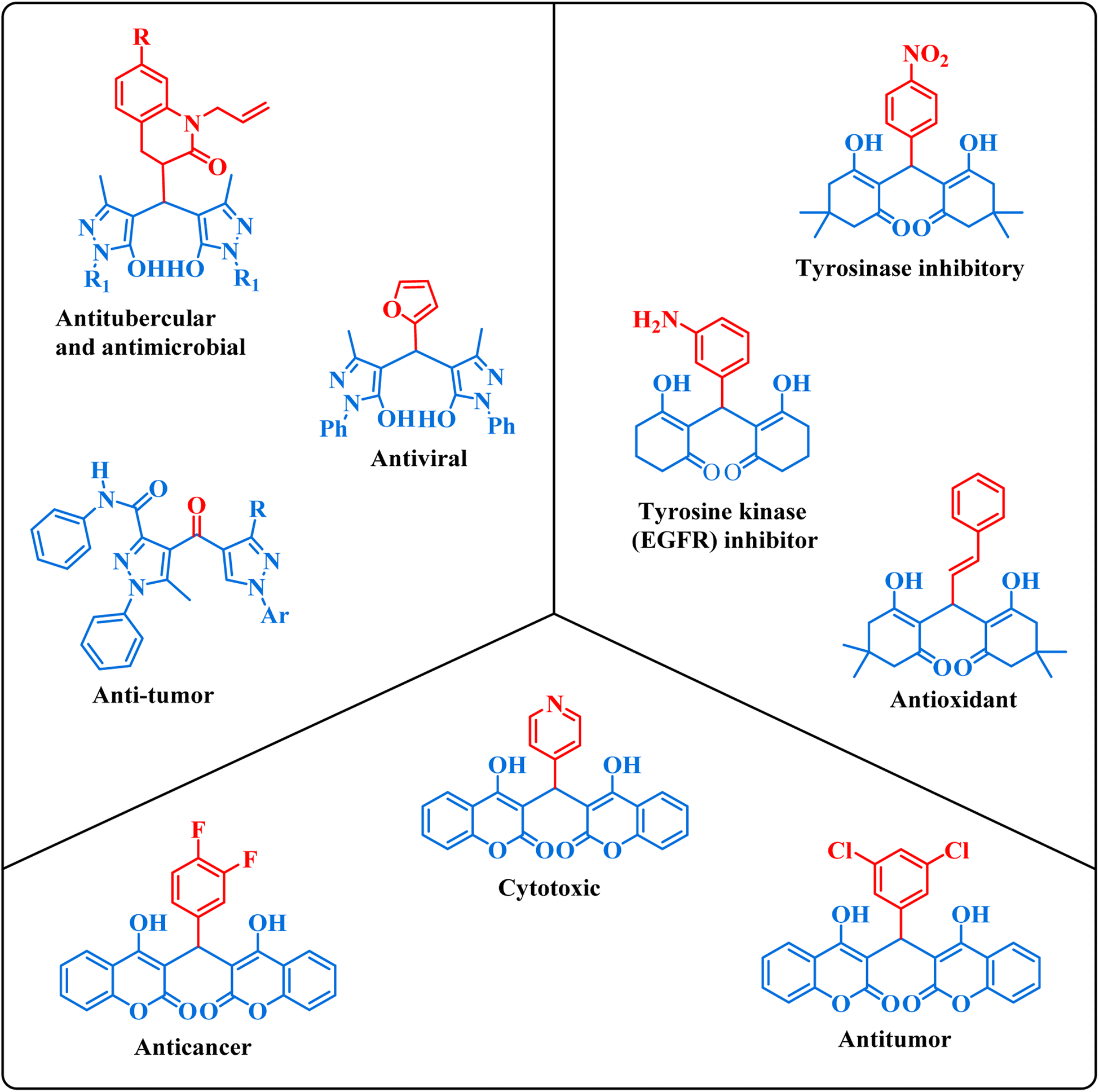
1,1-Dihomoarylmethane scaffolds are well-versed with interesting physical and chemical properties, including high lipophilicity, good bioavailability, and the ability to form stable complexes with metal ions.1 These properties make these scaffolds promising candidates for drug development, as they serve as a basis for the synthesis of bioactive molecules with potential therapeutic applications.1–3 These scaffolds have been explored for a variety of pharmacological activities namely anti-inflammatory, anticancer, and antimicrobial effects, and have shown promising properties in several studies.4–8 The compounds displayed in Fig. 1 represent a subset of bioactive drugs that incorporate 1,1-dihomoarylmethane scaffolds.9–14
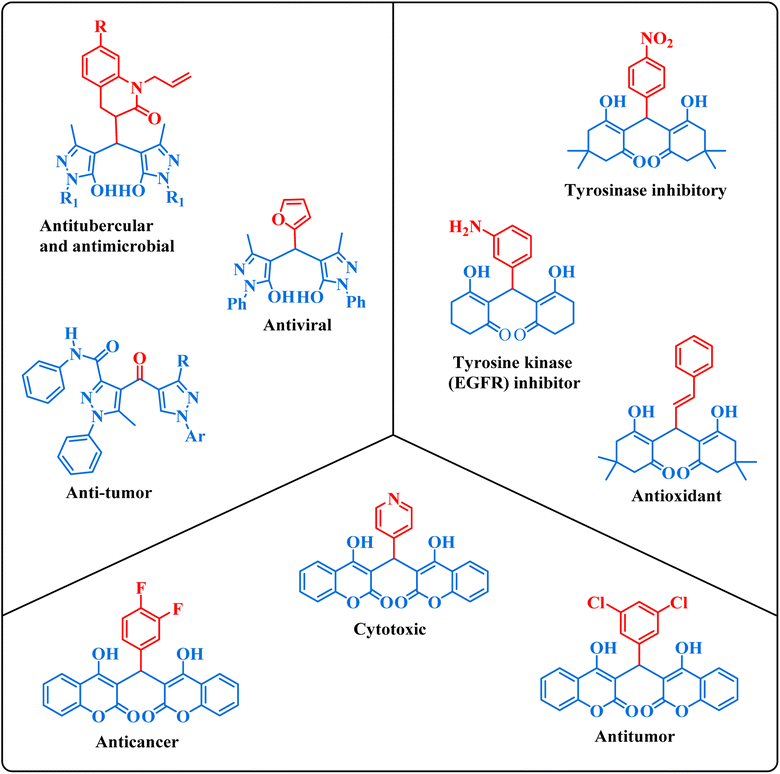 |
| | Fig. 1 Some bioactive drugs possessing 1,1-dihomoarylmethane scaffolds. | |
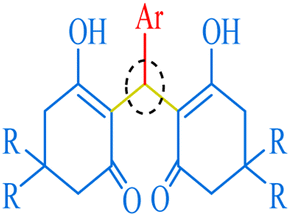
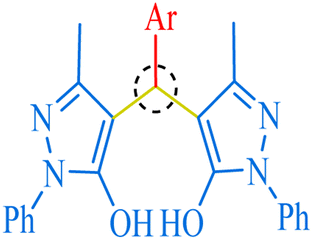
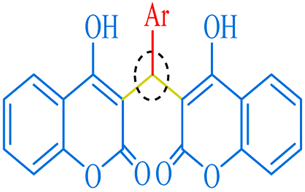
Among 1,1-dihomoarylmethane scaffolds, bis-dimedones, bis-cyclohexanediones, bis-pyrazoles, and bis-coumarins have attracted the attention of the scientific community due to their potential applications in different fields.15–17 Dimedone and 1,3-cyclohexanedione are both cyclic diketones having various applications in organic synthesis, analytical chemistry, materials science, metal detection, and medicinal chemistry.18–25 1-Phenyl-3-methyl-5-pyrazolone (edaravone) is used as a precursor for synthesizing various pyrazolone derivatives with several biological activities viz anti-inflammatory, antimicrobial, and anticancer agents.26,27 It has a wide range of applications in analytical chemistry, pharmaceuticals, dyes, pigments, colorants, chelating agents, and corrosion inhibition.15,28,29 4-Hydroxycoumarin, also known as 4-coumarinol, is a precursor to the synthesis of several coumarin derivatives.30 These derivatives have various biological activities, such as anticoagulant, antifungal, antibacterial, and anticancer effects.31–34 It is commonly used for the synthesis of warfarin, a widely used oral anticoagulant drug.35 It has been shown to promote plant growth and increase resistance to various environmental stresses, such as drought, salinity, and extreme temperatures.36,37 Moreover, it possesses a plethora of applications in medicine, agriculture, and materials science.38
Owing to the utmost significance of 1,1-dihomoarylmethane scaffolds, continuous endeavours have been undertaken by the scientific community to develop practical, eco-benign, and highly efficient approaches for their synthesis. Although most of the reported protocols have several merits, still they have some demerits viz. high reaction time, loading of high catalyst amount, the need for chromatographic separation, by-product formation, etc. These demerits ignite our research group to develop highly sustainable methods for its synthesis. Thus, in concern for developing highly efficient procedures, our research group has developed g-C3N4·SO3H catalyst mediated synthesis of 1,1-dihomoarylmethane scaffolds.
Graphitic carbon nitride (g-C3N4), a polymeric material is composed of carbon and nitrogen atoms arranged in a two-dimensional layered structure.39–41 It is a stable and non-toxic material that is environmentally friendly and has attracted significant attention due to its unique electronic, optical, photocatalytic, and chemical properties.42,43 Its structure can be tuned by changing the preparation conditions and the starting materials used.44 The addition of sulfonic acid groups to the surface of g-C3N4 gives g-C3N4 sulfonic acid (g-C3N4·SO3H). g-C3N4·SO3H has a high surface area and unique electronic properties, making it a promising material for various applications.43,45,46 It has also been used as a heterogeneous acid catalyst for esterification, aldol condensation, and acylation reactions.47–49 Herein we have developed a highly efficient, green, and high-yielding multicomponent reaction for one-pot synthesis of 1,1-dihomoarylmethane scaffolds (bis-dimedones, bis-cyclohexanediones, bis-pyrazoles, and bis-coumarins) using substituted aryl aldehydes and C–H activated compounds, such as dimedone, 1,3-cyclohexanedione, 1-phenyl-3-methyl-5-pyrazolone, and 4-hydroxycoumarin in aqueous ethanol using g-C3N4·SO3H as an ionic liquid catalyst. The reactions were highly efficient, completing in short reaction times and producing high yields, with easy workup procedures.
Result and discussion
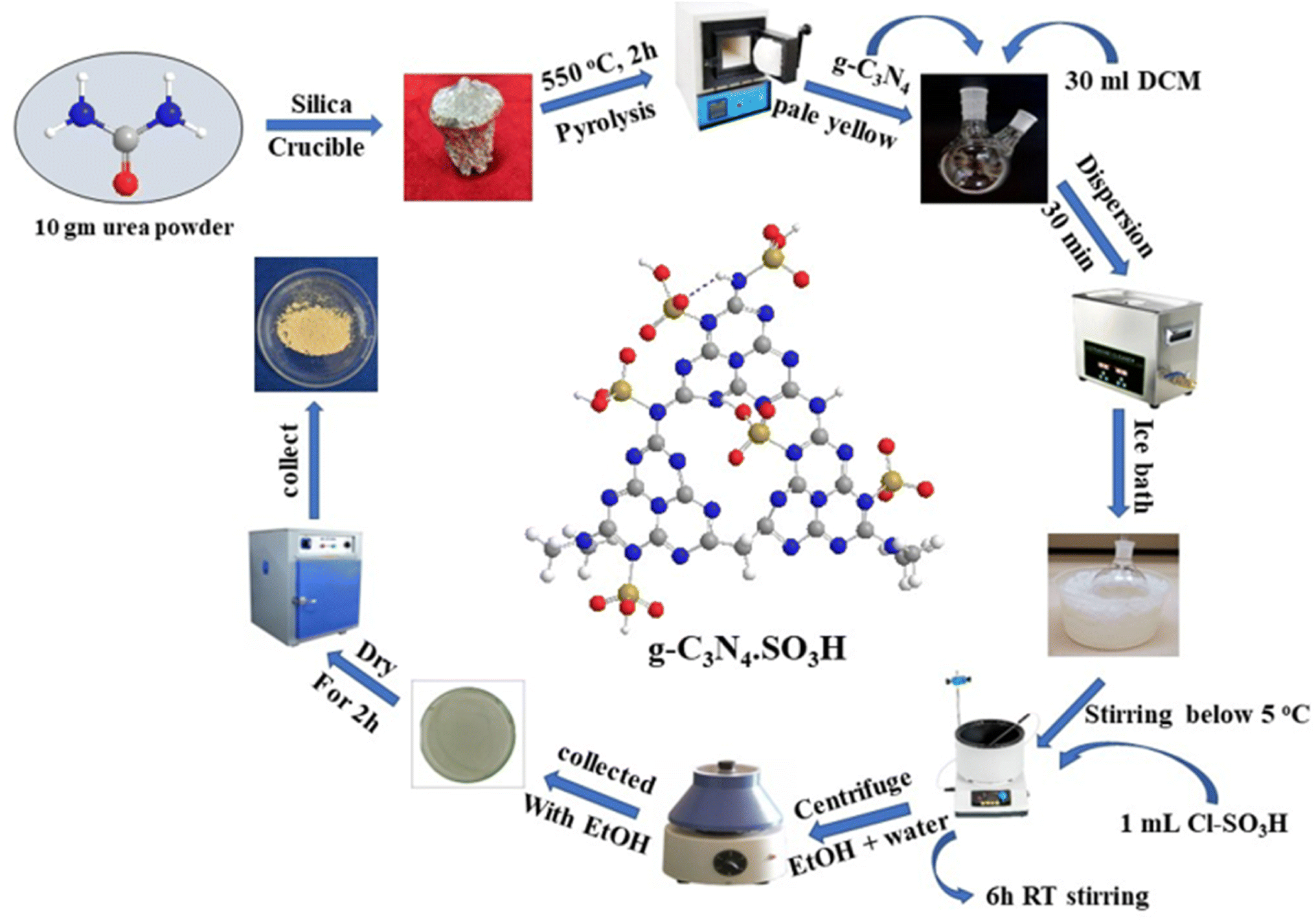
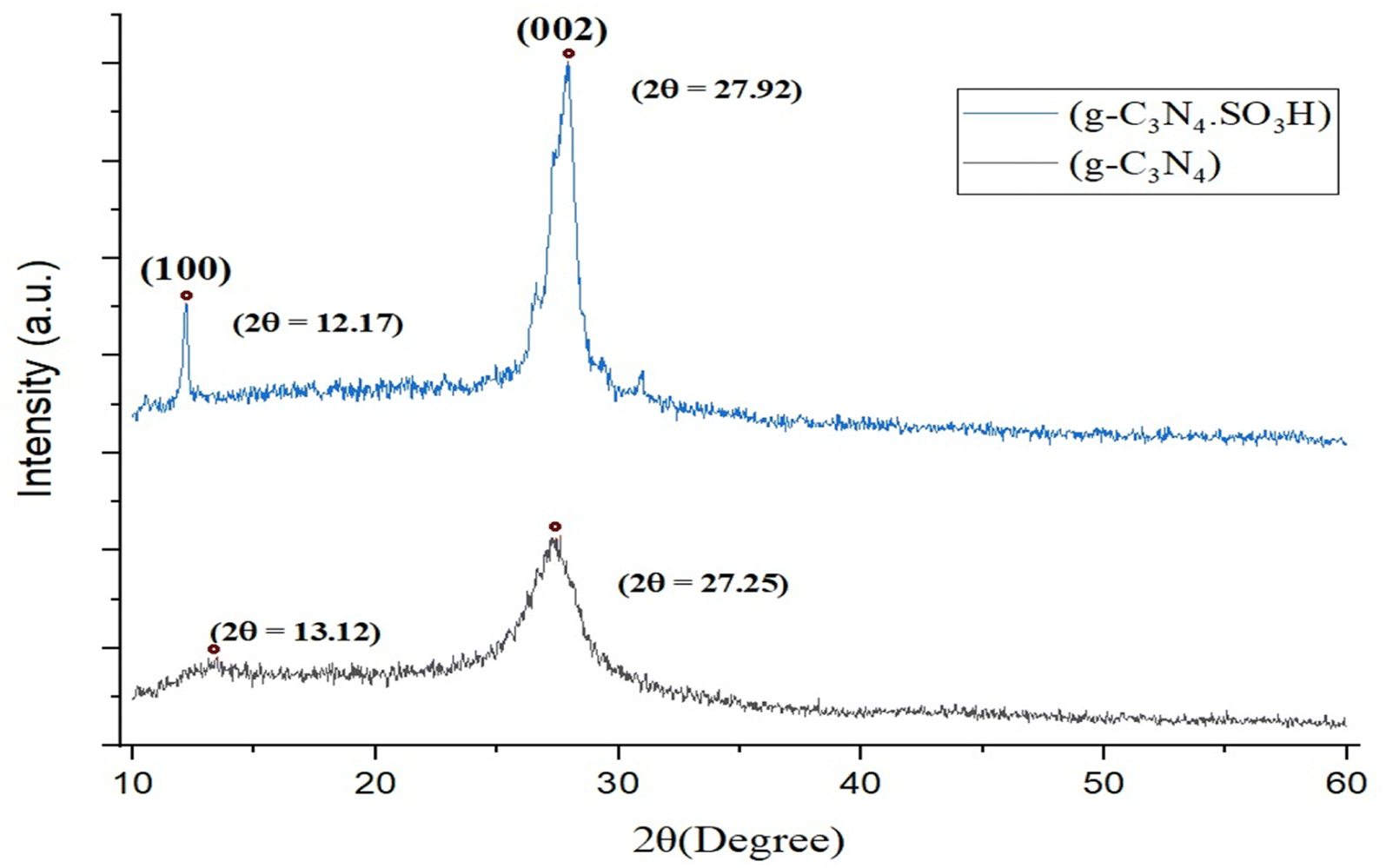
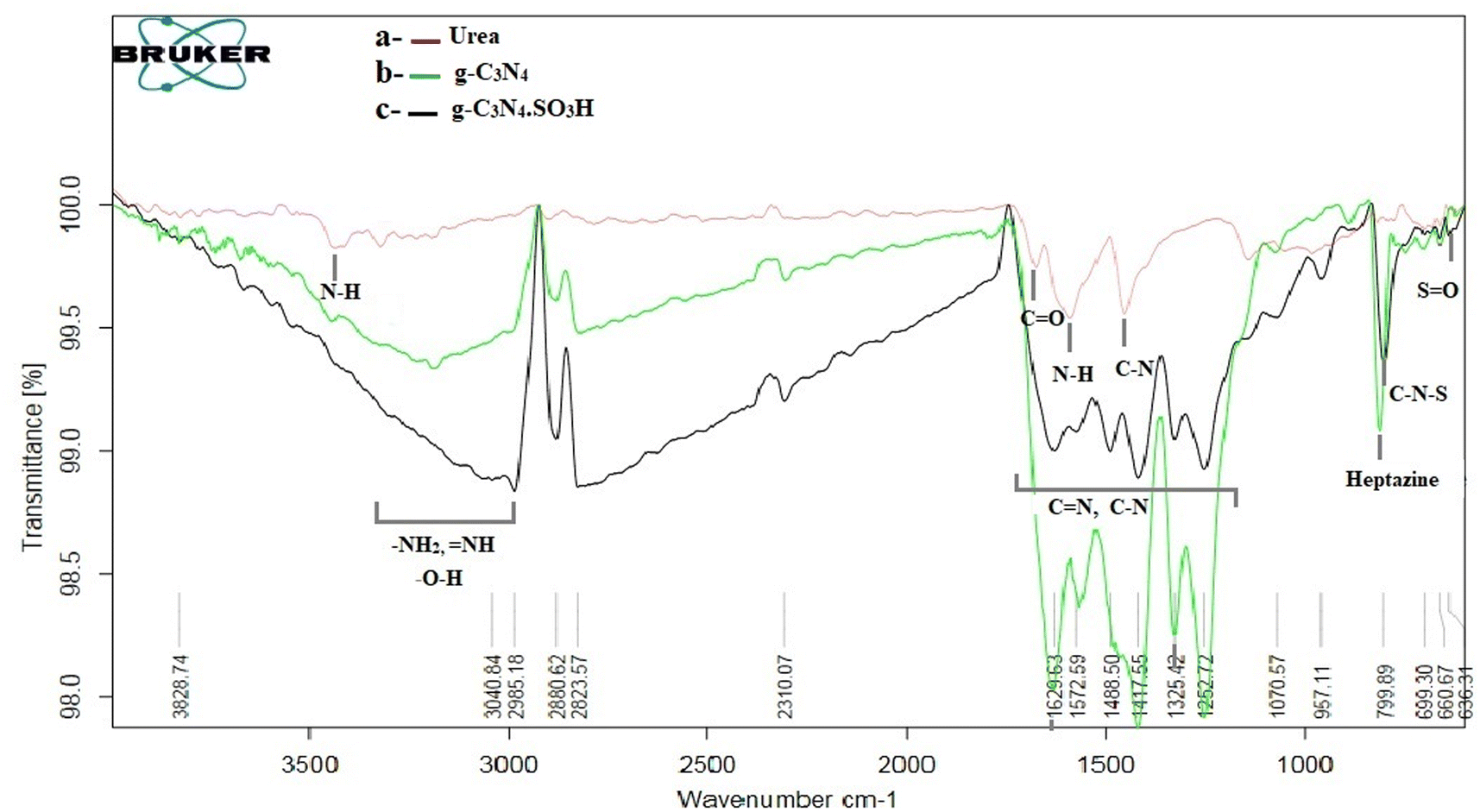
The g-C3N4·SO3H was synthesized using urea and chlorosulfonic acid using a series of steps (Fig. 2) and well characterized by XRD, FT-IR, SEM and HRTEM studies and the results were in full agreement with previously reported literature.43,50,51 The XRD spectrum of g-C3N4 and g-C3N4·SO3H was analysed, as shown in Fig. 3. In Fig. 3, the characteristic peak at 2θ = 13.12° with a plane (100) and the peak with a plane (002) at 2θ = 27.25° were observed for g-C3N4, which matches the JCPDS card 87-1526. The XRD spectral peaks for g-C3N4·SO3H in Fig. 3 was found to be almost identical, 2θ = 12.17° with a (100) plane, and 2θ = 27.92° with a (002) plane, indicating that the functionalization did not result in any significant destruction of the carbon nitride web.43,50 The g-C3N4 and g-C3N4·SO3H was synthesized from urea and a comparative study of the FT-IR spectrum of urea, g-C3N4, and g-C3N4·SO3H was performed over wavenumber 4000–600 cm−1 region. For pure urea in Fig. 4a, the N–H stretching frequency appeared at 3450 cm−1 and 1625 cm−1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency showed its peak at 1670 cm−1. The stretching frequency appeared at 1442 cm−1 that attributed to C–N.51 Fig. 4b and c exhibited a broad peak in 3000 to 3400 cm−1 region that belonged to –NH2, NH stretching frequency of heptazine web and O–H stretching vibration of SO3H. The absorption peak was seen at the 1150–1750 cm−1 region attributed to CN and C–N stretching vibration while the peak at 811 cm−1 was related to the heptazine network. The assured formation of g-C3N4·SO3H was revealed in Fig. 4c by 799 cm−1 and 640 cm−1 of C–N–S and SO stretching frequency respectively.50 The morphology of the synthesized catalyst was studied by SEM in Fig. 5A–C and it was observed that the structure was porous and agglomerated while the sheets were not clearly visible.43,50 The HRTEM analysis in Fig. 5D–F displayed that g-C3N4–SO3H material exhibited a sheet-like structure that appeared to be wrinkled, which could be attributed to the attraction or repulsion of static charges between the layers of g-C3N4–SO3H.50
O stretching frequency showed its peak at 1670 cm−1. The stretching frequency appeared at 1442 cm−1 that attributed to C–N.51 Fig. 4b and c exhibited a broad peak in 3000 to 3400 cm−1 region that belonged to –NH2, NH stretching frequency of heptazine web and O–H stretching vibration of SO3H. The absorption peak was seen at the 1150–1750 cm−1 region attributed to CN and C–N stretching vibration while the peak at 811 cm−1 was related to the heptazine network. The assured formation of g-C3N4·SO3H was revealed in Fig. 4c by 799 cm−1 and 640 cm−1 of C–N–S and SO stretching frequency respectively.50 The morphology of the synthesized catalyst was studied by SEM in Fig. 5A–C and it was observed that the structure was porous and agglomerated while the sheets were not clearly visible.43,50 The HRTEM analysis in Fig. 5D–F displayed that g-C3N4–SO3H material exhibited a sheet-like structure that appeared to be wrinkled, which could be attributed to the attraction or repulsion of static charges between the layers of g-C3N4–SO3H.50
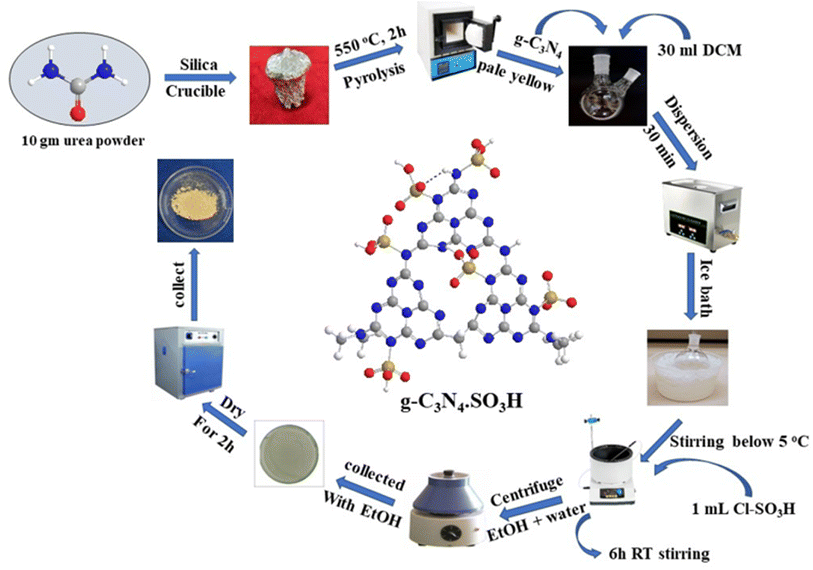 |
| | Fig. 2 The pictorial representation of the synthetic procedure of g-C3N4·SO3H catalyst. | |
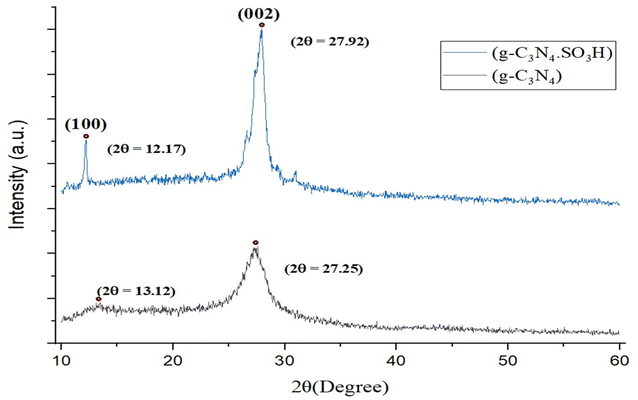 |
| | Fig. 3 XRD spectrum of synthesized g-C3N4 and g-C3N4·SO3H. | |
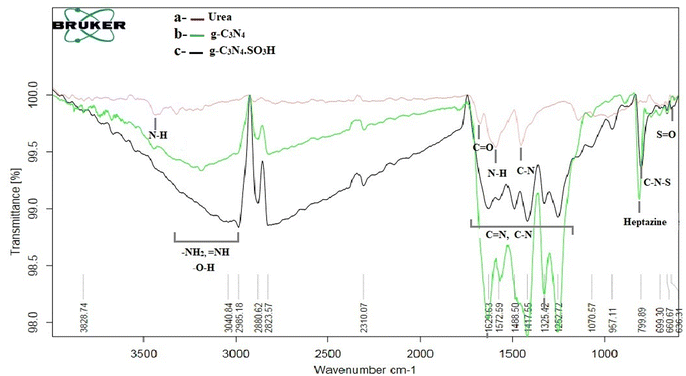 |
| | Fig. 4 FT-IR spectrum of (a) urea and synthesized (b) g-C3N4, (c) g-C3N4·SO3H. | |
 |
| | Fig. 5 SEM (A–C) and HRTEM (D–F) images of synthesized g-C3N4·SO3H with different magnifications. | |
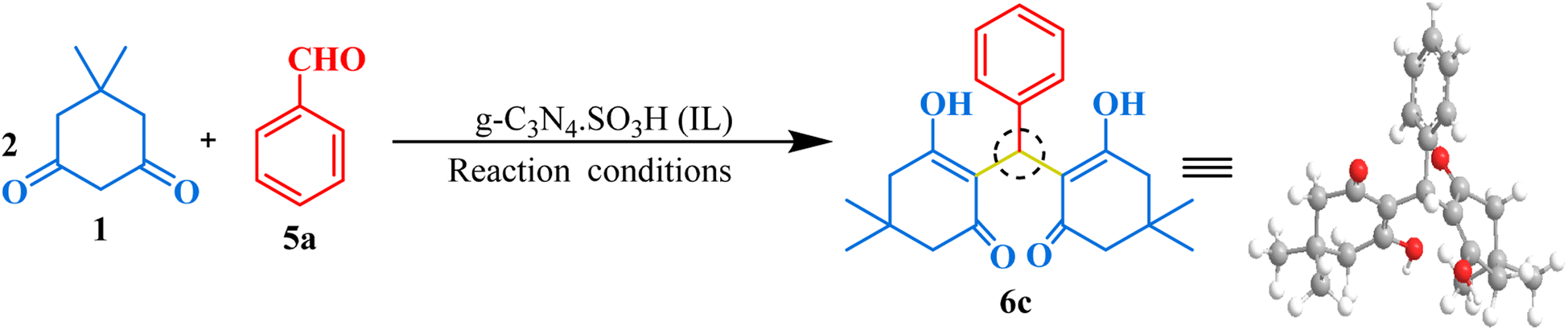
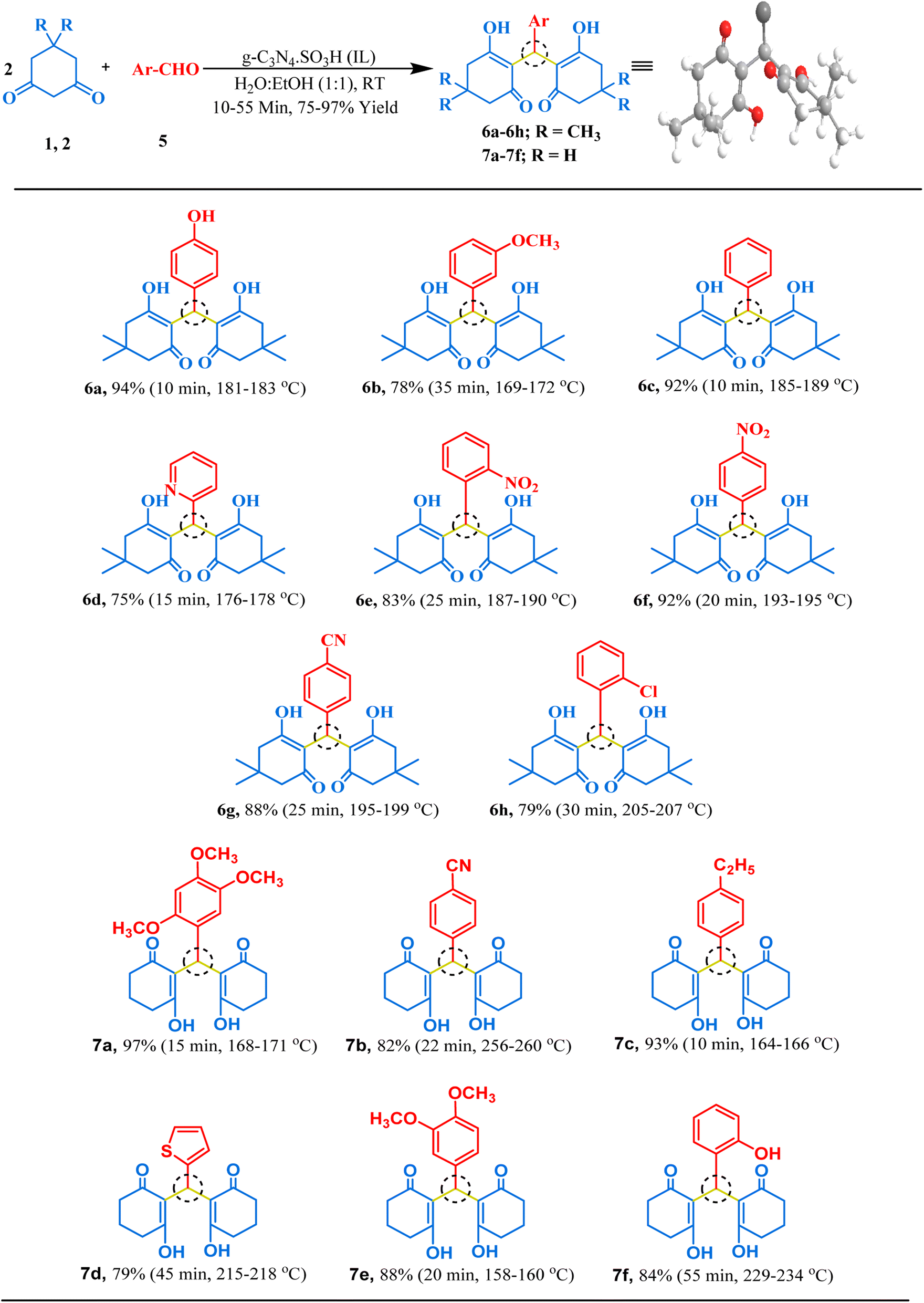
The synthesized g-C3N4·SO3H catalyst was further studied for its catalytic activity. Its effectiveness was evaluated through several trial reactions, including the synthesis of bis-dimedones, bis-cyclohexanediones, bis-pyrazoles, and bis-coumarins. To optimize the reaction conditions for bis-dimedone synthesis, the impact of the amount of catalyst, solvent, and temperature was investigated. Two equivalents of 5,5-dimethyl-1,3-cyclohexanedione and one equivalent of benzaldehyde were used as a standard for the initial studies. In the absence of the catalyst and solvent, the reactants remained unreacted. However, increasing the amount of catalyst to 15 mg in solvent-free conditions resulted in a sequential increase in yield and reduced reaction time (Table 1; entries 4–6). Further increasing the amount of catalyst did not significantly impact the product yield (Table 1; entry 7). Next, various solvents were tested, and the mixture of ethanol and water was found to be the best suitable for the reaction (Table 1; entry 9). However, changing the temperature did not yield satisfactory results (Table 1; entries 11, 12). Further, the model reaction was attempted in the presence of blue light using the g-C3N4·SO3H as a photocatalyst, we found that the results were not significantly improved (Table 1; entry 13). To assess the efficacy of the present catalyst, g-C3N4 was also employed as a catalyst in the model reaction at optimum reaction conditions but the synthesis of the product took longer time than expected and the yield was not optimal (Table 1; entry 3). The most effective reaction condition was found to be 15 mg of catalyst in an ethanol–water (1:1) solvent system at room temperature (RT), yielding a 92% yield in just 10 minutes of reaction time (Table 1; entry 9). Using these optimal reaction conditions, the substrate scope was expanded first with various aromatic aldehydes, and later with cyclohexanedione, a derivative of 5,5-dimethyl-1,3-cyclohexanedione, resulting in excellent yields (Fig. 6). Overall, these findings demonstrated the effectiveness of g-C3N4·SO3H as a catalyst and provided insights into the optimal reaction conditions for bis-dimedone and bis-cyclohexanedione synthesis.
Table 1 Reaction optimization for the synthesis of bis-dimedone scaffolds
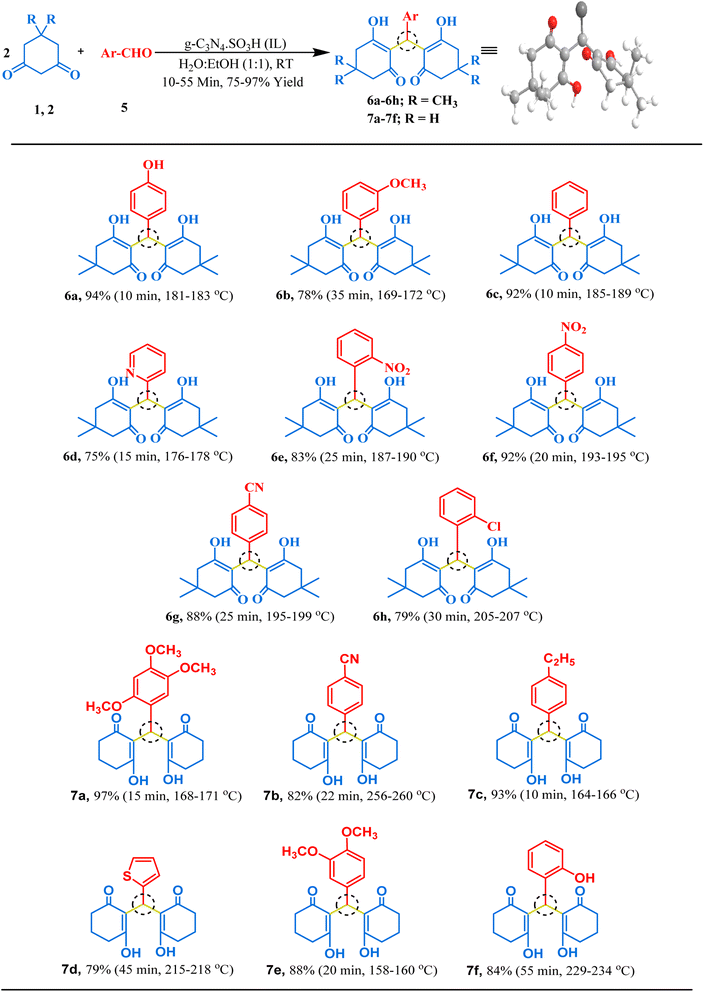 |
| | Fig. 6 The inventory of synthesized bis-dimedone and bis-cyclohexanedione scaffolds. | |
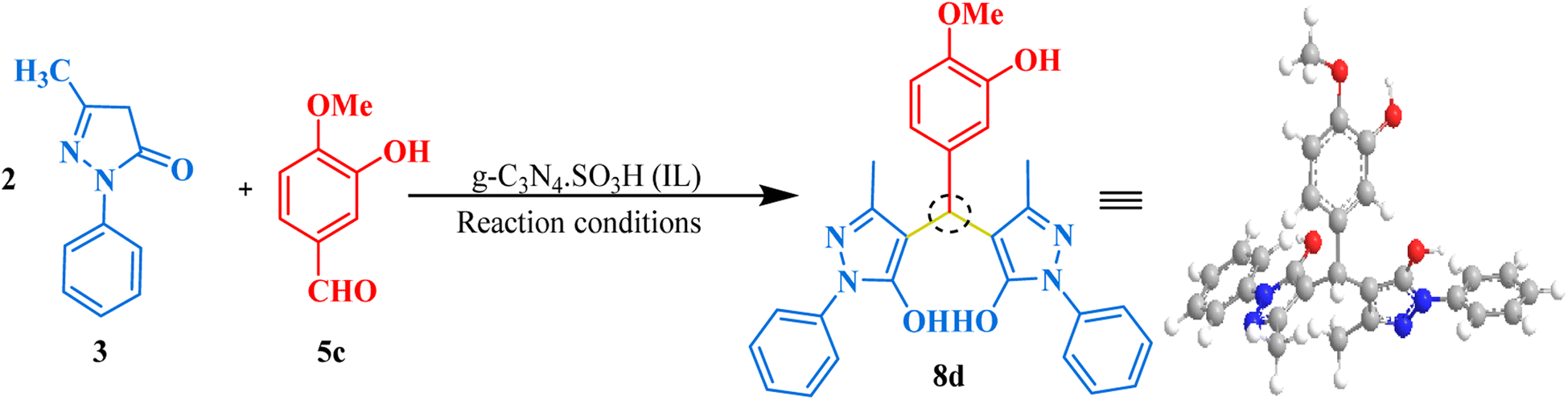
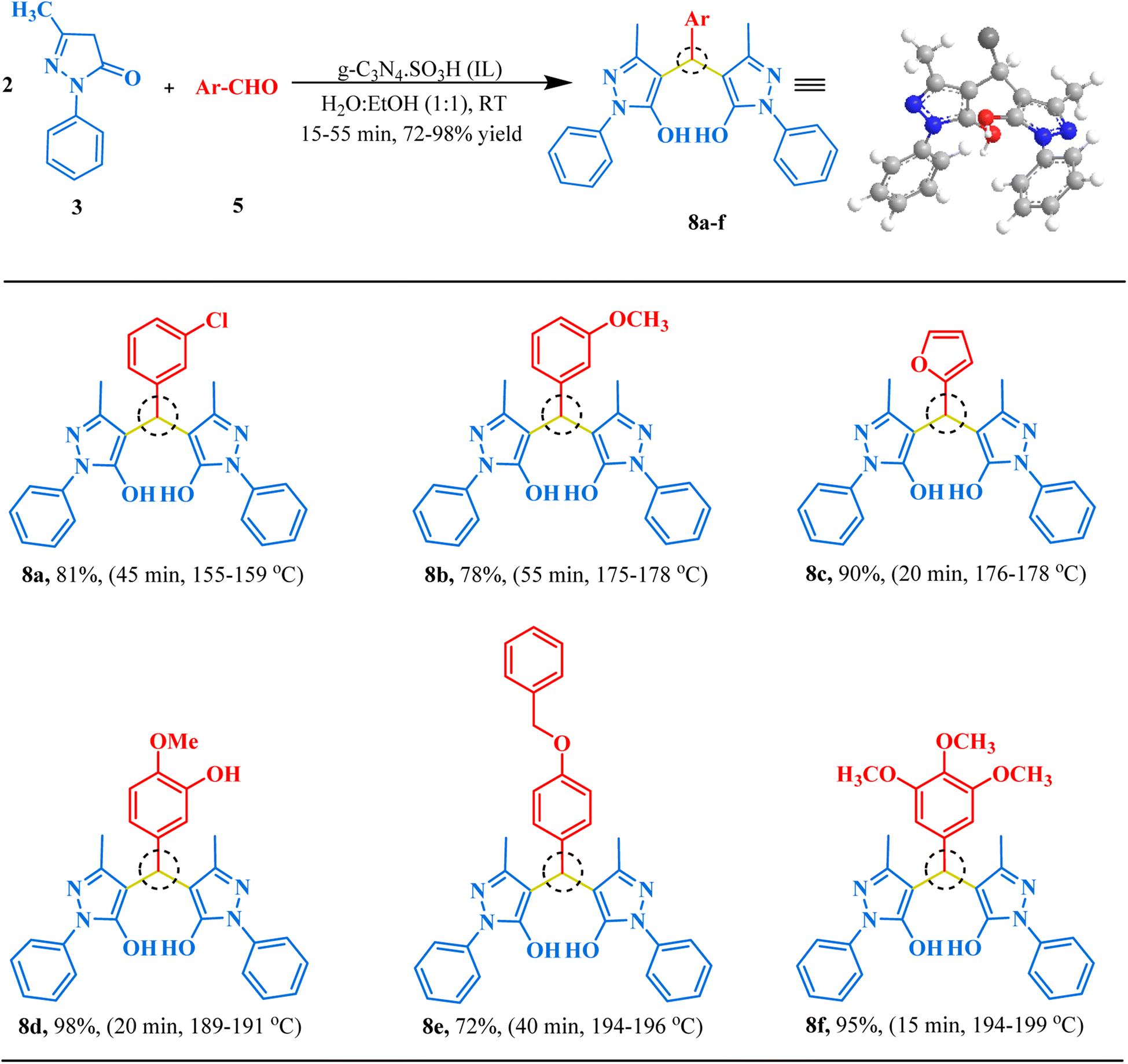
After successfully synthesizing bis-dimedones and bis-1,3-cyclohexanediones, we sought to expand the scope for the synthesis of bis-pyrazoles using 1-phenyl-3-methyl-5-pyrazolone and isovanillin as a model reaction. We found that the optimum conditions for these substrates were the same as for bis-dimedone, 15 mg of catalyst loading, at RT in an ethanol–water (1:1) solvent system. Using these conditions, we were able to synthesize bis-pyrazoles (Table 2) with high yields. We also explored the substrate scope by testing different aromatic aldehydes under optimized conditions. By doing so, we were able to obtain high to excellent yields for the corresponding bis-pyrazole derivatives (Fig. 7).
Table 2 Reaction optimization for the synthesis of bis-pyrazole scaffolds
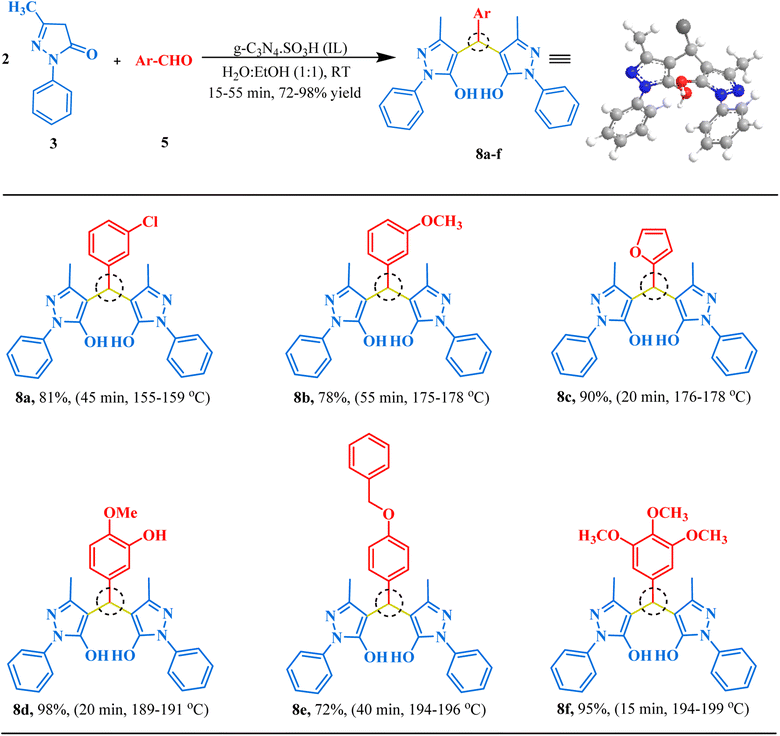 |
| | Fig. 7 The inventory of synthesized bis-pyrazole scaffolds. | |
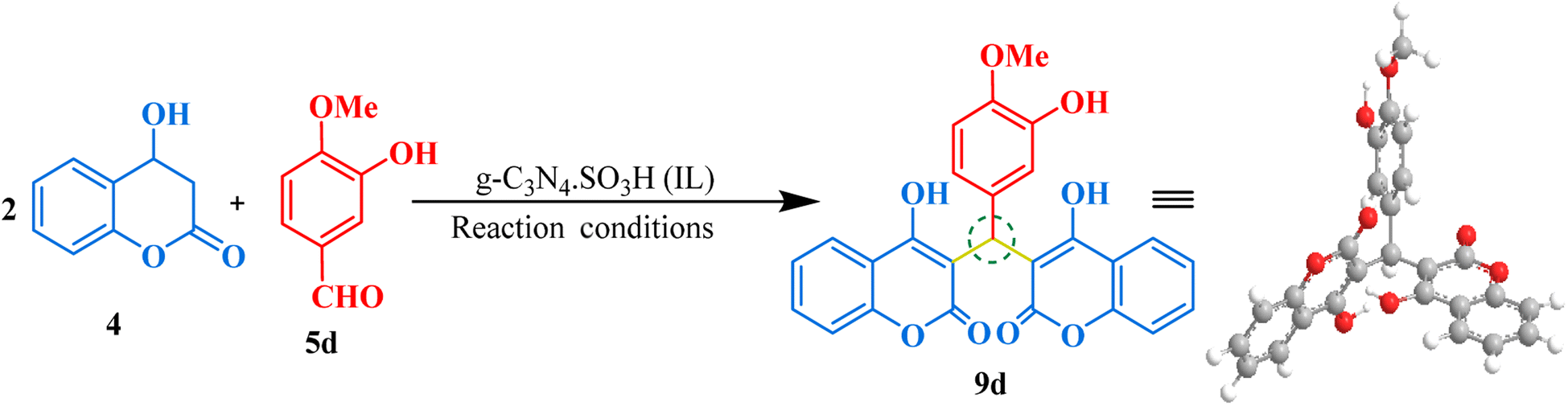
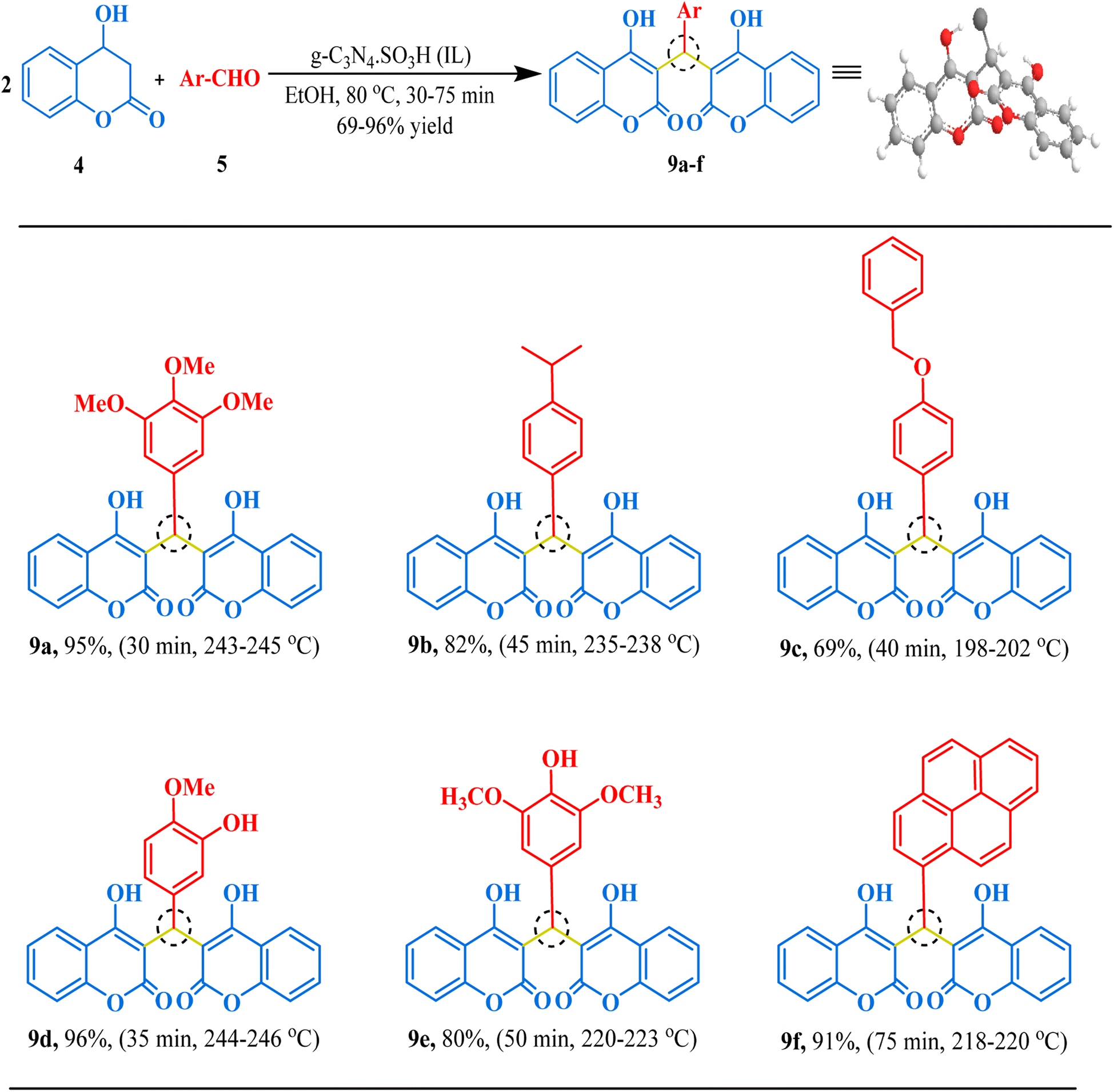
Our next synthesis target was bis-coumarin, for which we selected 4-hydroxycoumarin (2 mmol) and isovanillin (1 mmol) as standard substrates to determine the optimum reaction conditions. We carried out a series of trials to investigate the effect of varying the presence and absence of g-C3N4, and g-C3N4·SO3H, catalyst at different temperature, solvents, and reaction times (Table 3). To our delight, we found that using 20 mg of g-C3N4·SO3H in ethanol solvent at 80 °C for 35 min resulted in the desired bis-coumarin with a 96% yield (Table 3; Entry 15). We confirmed the product's identity by analyzing and characterizing it using TLC and spectral studies (1H-NMR and 13C-NMR). Interestingly, we observed that adding more catalyst did not significantly improve the yield or decrease the reaction time. We summarized our findings in Table 3. We then explored the substrate scope using the optimal conditions by testing different aryl aldehydes with 4-hydroxycoumarin and produced the corresponding biscoumarin derivatives (Fig. 8).
Table 3 Reaction optimization for the synthesis of bis-coumarin scaffolds
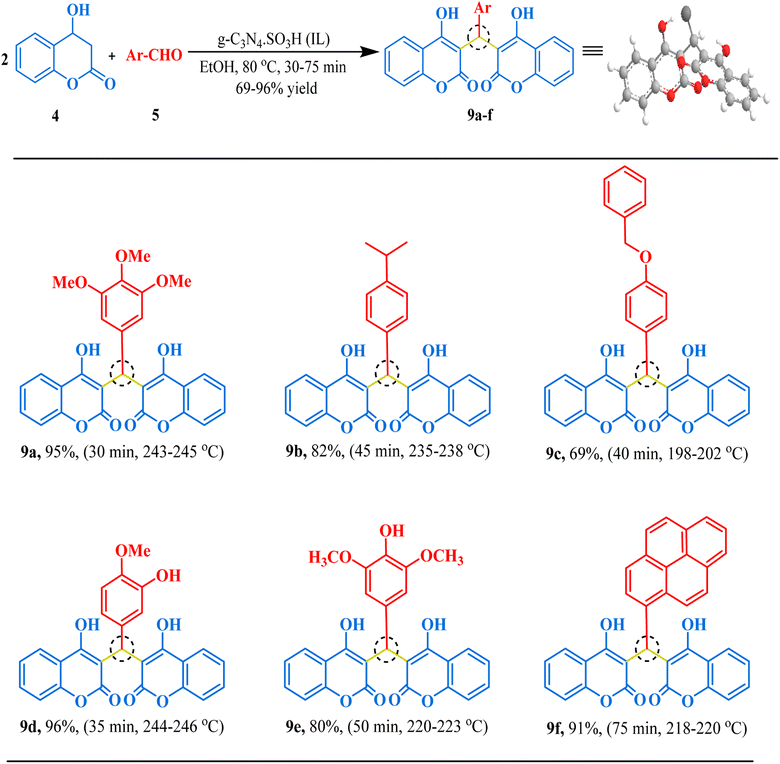 |
| | Fig. 8 The inventory of synthesized bis-coumarin scaffolds. | |
The synthesized products were isolated via washing the obtained crude with hexane in the workup process, followed by recrystallization with ethanol. All the products were characterized depending on their analytical and spectral data including 1H-NMR and 13C-NMR spectrum (in ESI†). The methine bridge exhibit singlet near δ 4.84–6.02 ppm for bis-dimedone, δ 4.62–5.16 ppm for bis-cyclohexanedione, δ 4.68–4.72 ppm for bis-pyrazole, and δ 6.01–6.99 ppm for bis-coumarin, which confirmed the formation of the product.
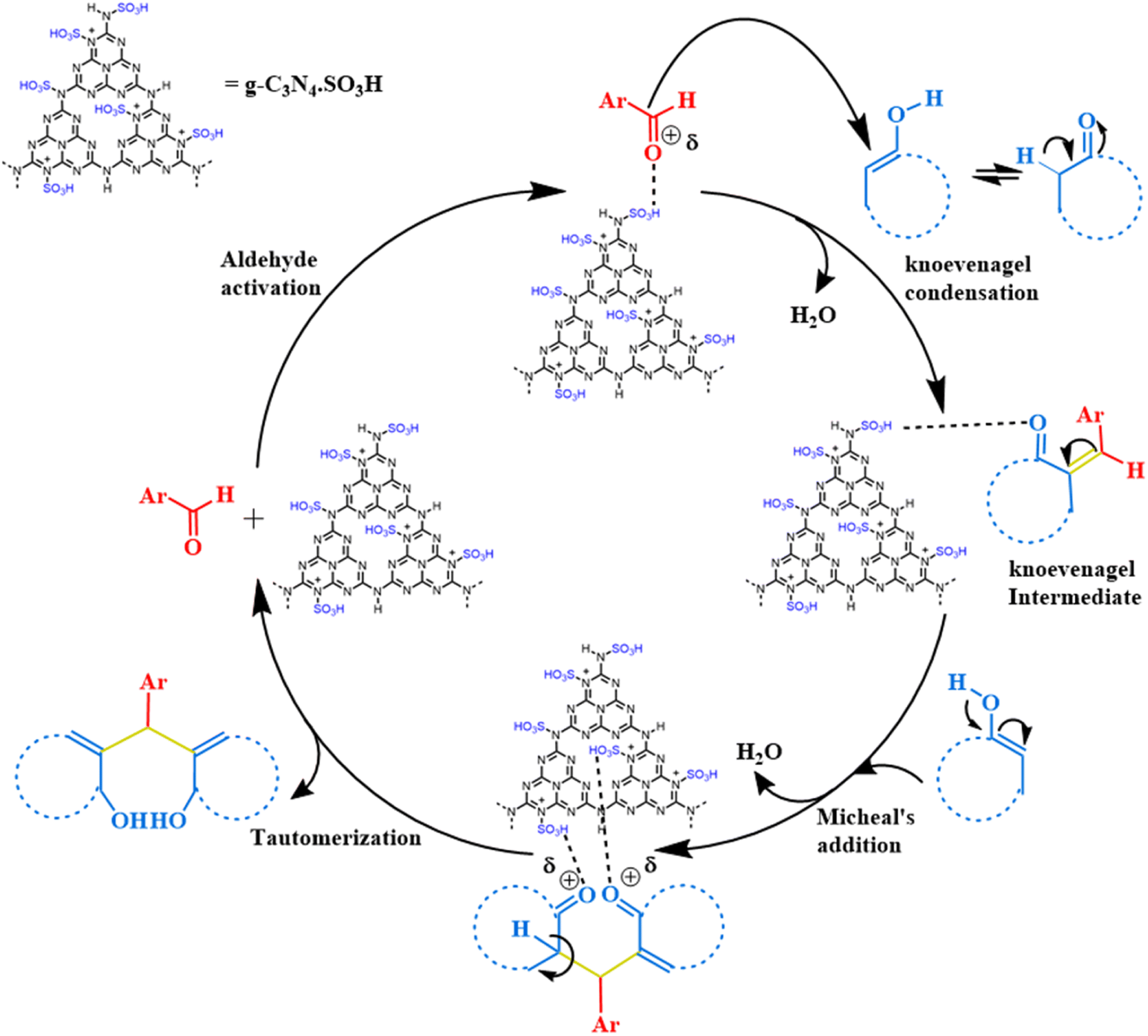
We proposed a plausible mechanism for the synthesis of 1,1-dihomoarylmethane scaffolds from aromatic aldehydes and C–H-activated compounds catalyzed by g-C3N4·SO3H ionic liquid catalyst (Scheme 1). Initially, we suggest that the g-C3N4·SO3H catalyst activated the aldehyde to initiate the reaction. The activated aldehyde then underwent Knoevenagel condensation with the C–H activated compound, resulting in the removal of a water molecule to form an intermediate. Next, the intermediate underwent Michael's addition reaction with another molecule of the C–H-activated compound. Following this, keto–enol tautomerization occurred to form the desired product.
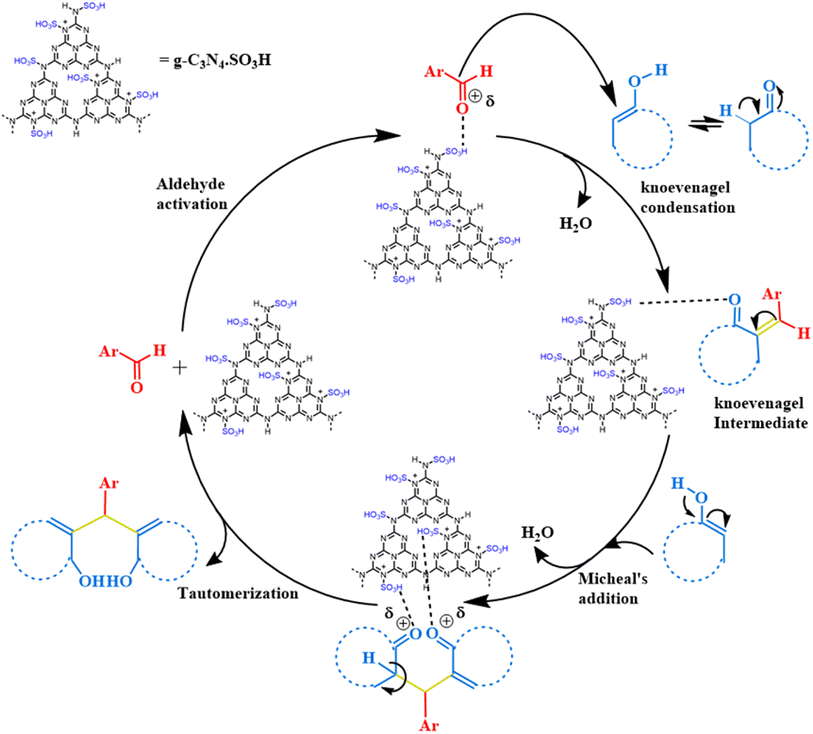 |
| | Scheme 1 The speculated mechanism for the synthesis of 1,1-dihomoarylmethane scaffolds. | |
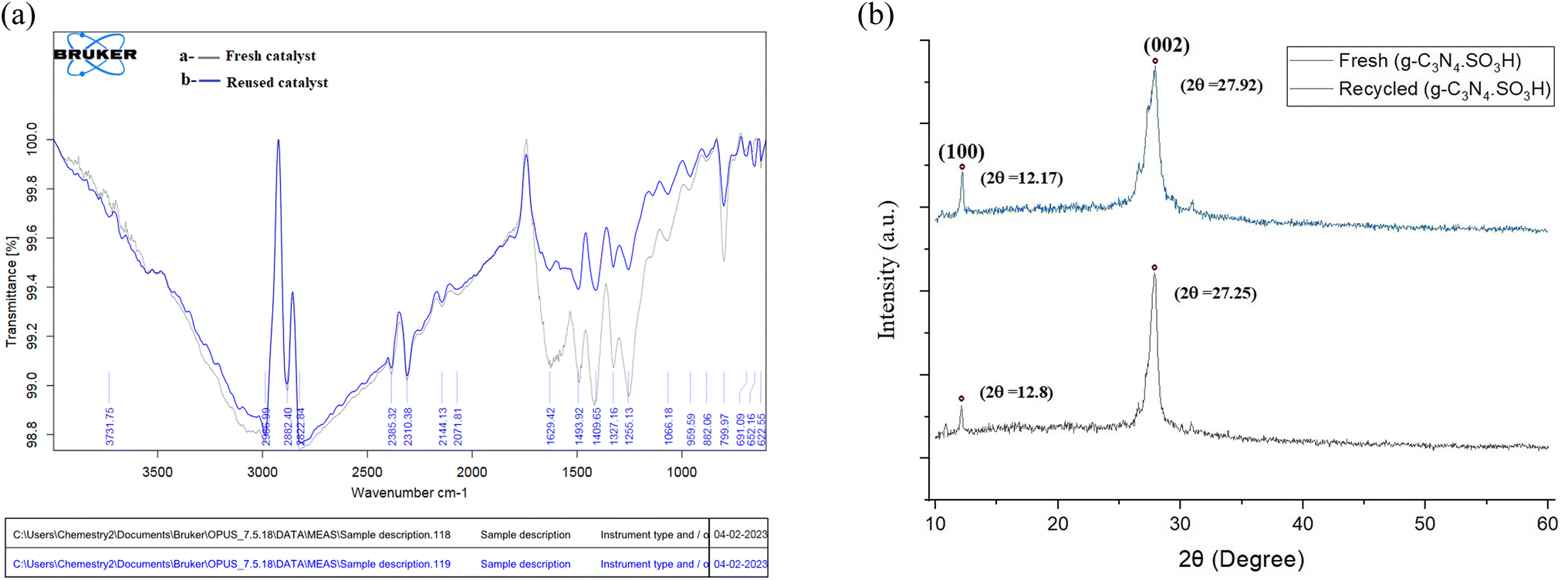
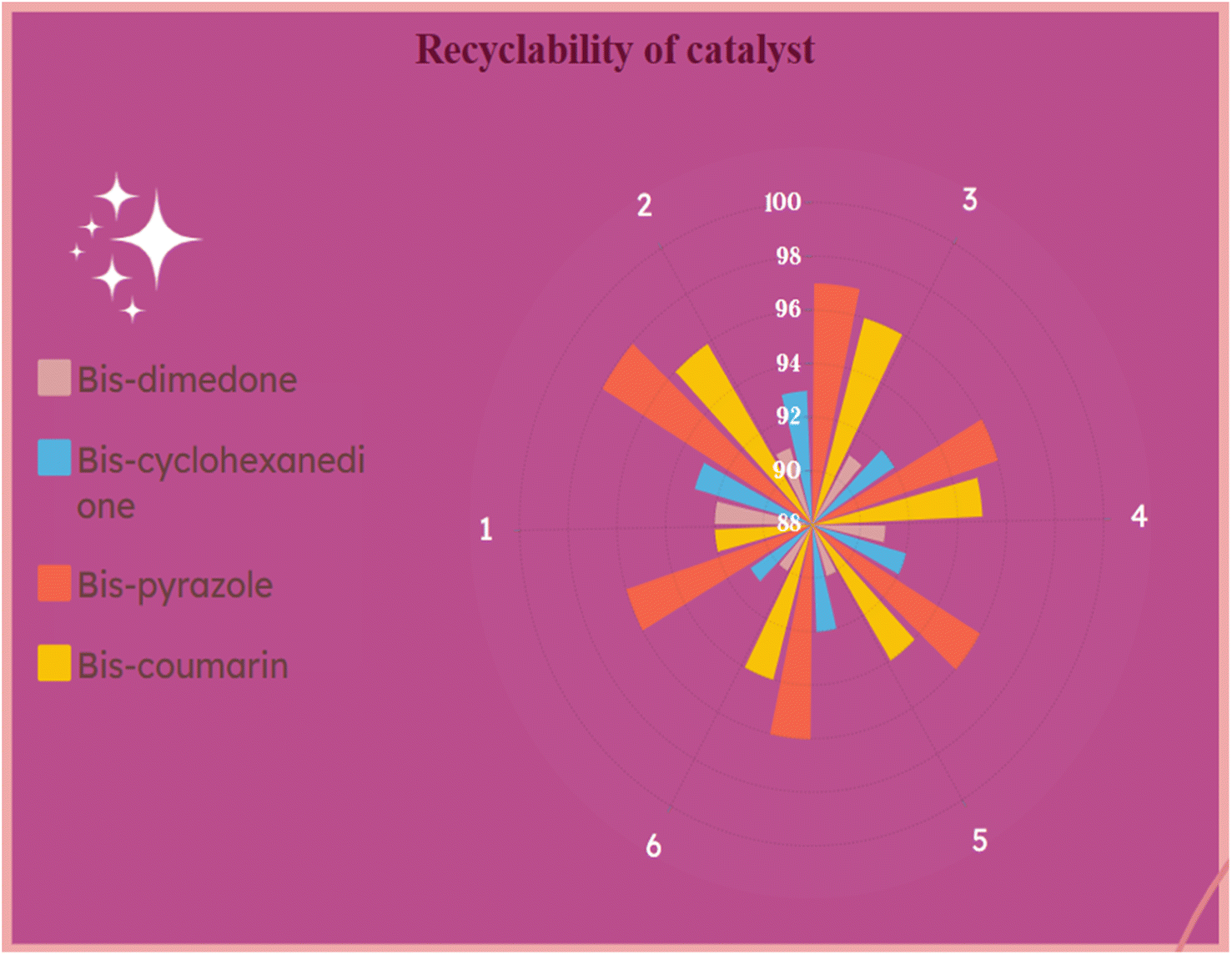
After the optimization of all the reactions, we probed the reusability of the catalyst in the model reactions for several runs. The results are summarized in Fig. 9 and 10. The synthesized g-C3N4·SO3H can be reused and recycled six times without any specific loss in the activity and further verified by FT-IR and XRD studies (Fig. 9A and B). The effectiveness of the protocol was also studied by comparing the present work with previous works (Table 4).
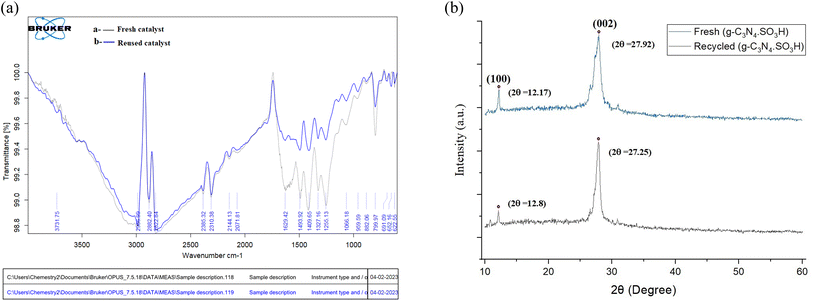 |
| | Fig. 9 The comparative spectral studies of (A) FT-IR and (B) XRD of synthesized fresh and recycled catalyst. | |
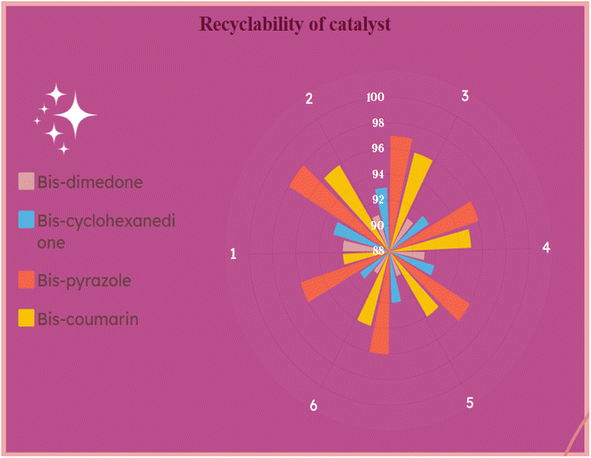 |
| | Fig. 10 Recyclability investigations of g-C3N4·SO3H catalyst (1, 2, 3, 4, 5, 6 represents cycle number and 88–100 indicates yield %). | |
Table 4 The comparative exploration for the synthesis of 1,1-dihomoarylmethane scaffolds with the present method
| Entry |
Product structure |
Catalyst; catalyst loading |
Conditions |
Time (min) |
Yield (%) |
Reusability |
References |
| A |
 |
1. Meglumine; 10 mol% |
H2O + EtOH (1:1)/RT |
6–30 |
64–95 |
4 |
1 |
| 2. Catalyst free |
H2O/RT |
30–240 |
64–98 |
— |
55 |
| 3. CsF; 10 mol% |
EtOH/RT |
10–35 |
84–96 |
— |
56 |
| 4. EDDA; 54 mg |
Reflux |
240–360 |
70–97 |
— |
57 |
| 5. Natural phosphate; 500 mg |
H2O/RT |
120–150 |
90–100 |
— |
58 |
| 6. Taurine; 35 mg |
H2O/reflux |
10–45 |
85–97 |
6 |
59 |
| 7. GO/ZnO; 10 mg |
H2O/reflux |
10–30 |
60–99 |
5 |
11 |
| 8. g-C3N4·SO3H; 15 mg |
H2O + EtOH (1:1)/RT |
10–35 |
78–94 |
6 |
This work |
| B |
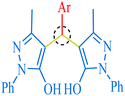 |
1. Xanthan sulfuric acid; 80 mg |
EtOH/reflux |
15–30 |
76–95 |
4 |
60 |
| 2. GO@PyH–CH3SO3; 20 mg |
70 °C |
8–30 |
90–99 |
5 |
27 |
| 3. [Dsim]AlCl4; 1 mol% |
50 °C |
30–60 |
72–91 |
4 |
61 |
| 4. [N2222] [Pro]; 12 mg |
EtOH/reflux |
50–65 |
77–86 |
5 |
62 |
| 5. Phosphomolybdic acid; 10 mol% |
EtOH/RT |
210–270 |
91–96 |
4 |
63 |
| 6. Ph3CCl; 5 mol% |
60 °C |
5–22 |
74–97 |
— |
64 |
| 7. Na+-MMT-[pmim]HSO4; 50 mg |
100 °C |
10–70 |
84–93 |
8 |
65 |
| 8. g-C3N4·SO3H; 15 mg |
H2O + EtOH (1:1)/RT |
15–55 |
72–98 |
6 |
This work |
| C |
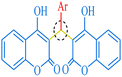 |
1. CSA; 20 mol% |
H2O + EtOH (1:1)/RT |
120–180 |
73–94 |
— |
66 |
| 2. SiO2Cl; 75 mg |
CH2Cl2/40 °C |
120–360 |
68–95 |
— |
67 |
| 3. Mohr's salt hexahydrate; 5 mol% |
H2O/reflux |
25–40 |
85–98 |
— |
68 |
| 4. Fe3O4@GO@Zn–Ni–Fe–LDH; 30 mg |
H2O/reflux |
3–40 |
85–95 |
5 |
69 |
| 5. SiO2–OSO3H NPs |
EtOH/80 °C |
20 |
86–96 |
3 |
70 |
| 6. Fe3O4@SiO2@VB1–Ni MNPs; 10 mg |
110 °C |
30–50 |
65–98 |
5 |
71 |
| 7. Fe3O4@sulfosalicylic acid; 50 mg |
MW/180 W |
10–17 |
73–95 |
6 |
72 |
| 8. g-C3N4·SO3H; 20 mg |
EtOH/80 °C |
30–75 |
69–96 |
6 |
This work |
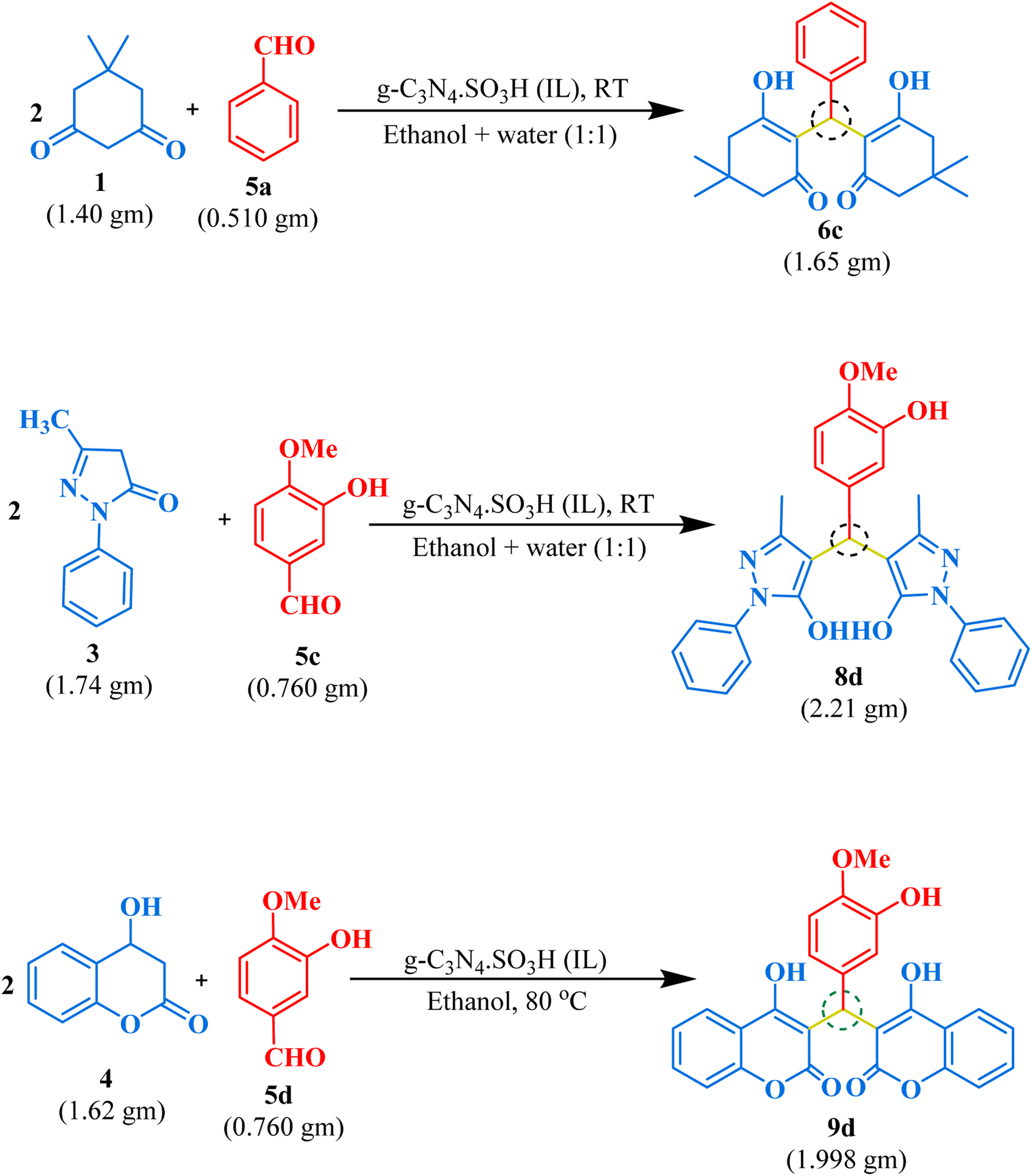
Gram-scale synthesis
We demonstrated the effectiveness of our methodology for industrial applications by performing gram-scale synthesis. Firstly, isovanillin (0.760 gm) and 4-hydroxy coumarin (1.621 gm) with g-C3N4·SO3H (20 mg) were reacted in ethanol solvent at 80 °C for 45 minutes to obtain bis-coumarin with 87.17% yield. Then, we conducted reactions between benzaldehyde (0.51 gm) and dimedone (1.40 gm) for bis-dimedone and isovanillin (0.760 gm) and pyrazolone (1.74 gm) for bis-pyrazolone synthesis in a mixture of ethanol and water (1:1) at room temperature with 15 mg of the catalyst for 20 and 25 minutes, respectively, yielding 89.57% and 91.66% of the products. We confirmed the completion of the reaction by TLC analysis. The catalyst was recovered using chloroform, and the crude product was dried and purified using ethanol to obtain the pure form of the product in high yields (Scheme 2).
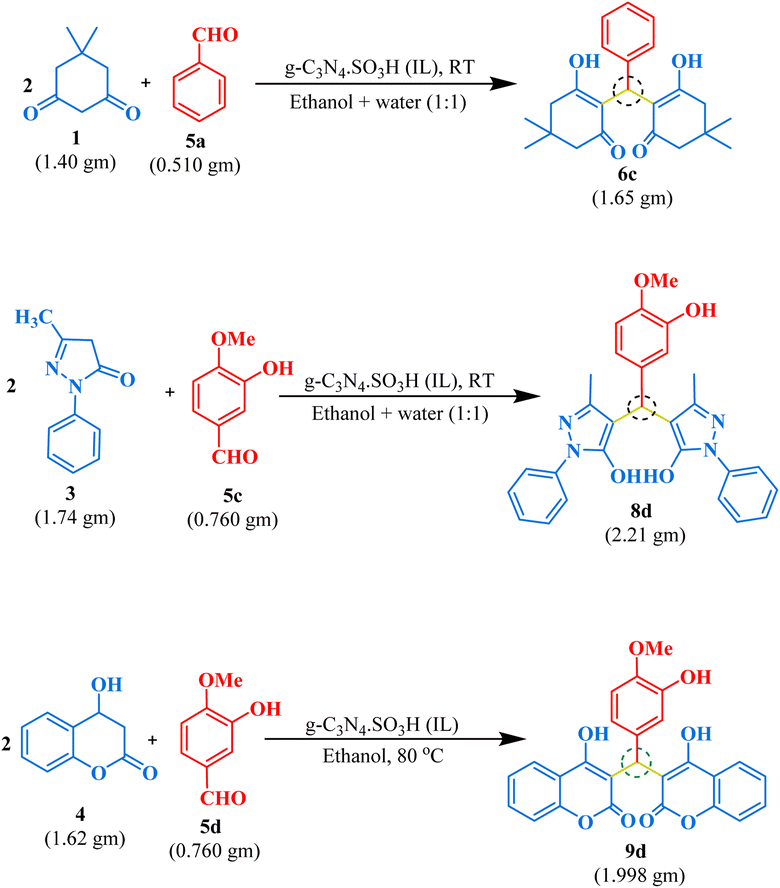 |
| | Scheme 2 Gram-scale synthesis of 1,1-dihomoarylmethane scaffolds. | |
Green chemistry matrix
In recent years, there has been a growing emphasis on developing environmentally friendly and sustainable methods for synthesizing organic compounds. One such approach is green chemistry, which provides a framework for assessing the eco-friendliness of chemical reactions. A promising example of this is the use of g-C3N4·SO3H as a catalyst for synthesizing 1,1-dihomoarylmethane scaffolds. This method has been shown to have a low E-factor (0.14, 0.05, and 0.07), high atom economy (95.34, 96.40, and 96.21%), high reaction mass efficiency (87.46, 94.79, and 92.67%), high process mass intensity (1.16, 1.06, and 1.08), and high eco-score (79.865, 83.165, and 80.155) for bis-dimedone, bis-pyrazolone, and bis-coumarin, respectively. These impressive results demonstrated that the use of g-C3N4·SO3H catalyst for synthesizing 1,1-dihomoarylmethane scaffolds is a sustainable and eco-friendly approach, with minimal impact on the environment. This is a significant step forward in the quest to develop greener and more sustainable methods for synthesizing organic compounds.52–54 [The calculated data is given in the ESI file].
Conclusion
In conclusion, the research team successfully developed a sustainable and highly efficient synthetic route to produce 1,1-dihomoarylmethane scaffolds using g-C3N4·SO3H ionic liquid catalyst with various aromatic aldehydes and C–H-activated compounds. The g-C3N4·SO3H ionic liquid catalyst has several advantages like high catalytic activity, reusability up to six cycles, low toxicity, simple method, and versatility making it a useful tool for chemists and researchers working in a variety of fields. The present findings offer a promising approach for the synthesis of 1,1-dihomoarylmethane scaffolds in high yields, reaction completion in short reaction times, under mild reaction conditions, and with minimal environmental impact. The outcomes reveal that this approach adheres with the principles of green chemistry. This method has been shown to have a low E-factor (0.14, 0.05, and 0.07), high atom economy (95.34, 96.40, and 96.21%), high reaction mass efficiency (87.46, 94.79, and 92.67%), high process mass intensity (1.16, 1.06, and 1.08), and high eco-score (79.865, 83.165, and 80.155) for bis-dimedone, bis-pyrazolone, and bis-coumarin, respectively. Moreover, the present method offers a viable alternative to previously reported methods and holds promise for the efficient synthesis of a wide range of 1,1-dihomoarylmethane scaffolds with potential applications in the pharmaceutical and material industries.
Experimental
General procedure for the preparation of g-C3N4·SO3H (IL) catalyst
To prepare g-C3N4 (graphitic carbon nitride), 10 g of grounded urea was placed in a silica crucible and covered with a lid. The crucible was wrapped in an aluminum foil and heated in a muffle furnace at 550 °C for 2 h. After switching off the furnace, the crucible was carefully removed after 1 h to obtain yellowish-white crude g-C3N4 (0.531 mg). The crude material was sonicated for 30 minutes in 30 ml of dichloromethane (DCM) for dispersion. To synthesize g-C3N4·SO3H, 1 ml of chlorosulfonic acid was added dropwise using a micropipette while stirring the reaction mixture in an ice bath to maintain the temperature below 5 °C. The reaction mixture was stirred for 6 h at RT and then distilled to dryness. The resulting ionic liquid was washed several times with ethanol: water (1:1) by decantation and centrifugation. It was then washed twice with distilled water. The final product (pale yellow, 0.431 mg) was collected with ethanol and dried in a hot air oven for 2 h50 (Fig. 2).
General procedure for the synthesis of 2,2′-(arylmethylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one)/2,2′-((aryl)methylene)bis(3-hydroxy cyclohex-2-en-1-one)/4,4′-((3-chlorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (6a–h, 7a–f, 8a–f)
A mixture of dimedone/1,3-cyclohexanedione/1-phenyl-3-methyl-5-pyrazolone (2 mmol), aryl aldehydes (1 mmol), and g-C3N4·SO3H (15 mg) in 8 ml of ethanol: water (1:1) solvent at RT was stirred in a round bottomed (RB) flask (25 ml) for a certain time. Reaction completion was determined by TLC and then the solvent was evaporated using a rotary evaporator. The catalyst was recovered simply by filtration aid in chloroform solvent which was further washed with ethanol and then dried in a hot air oven at 60 °C for about 3 h, and the pure desired product was collected after crystallization. The after-crystallization purity of the collected product was characterized by 1H-NMR and 13C-NMR spectral analysis.
General procedure for the synthesis of 3,3′-((aryl)methylene)bis(4-hydroxy-2H-chromen-2-one) (8a–f)
In a 25 ml RB flask, a catalytic amount of g-C3N4·SO3H (20 mg) was added to a stirring mixture of aryl aldehydes (1 mmol), and 4-hydroxycoumarin (2 mmol) in ethanol (5 ml) solvent at 80 °C and was refluxed for an appropriate time. After confirming the reaction completion by TLC, the solvent was removed by a rotary evaporator. After that, the catalyst was recovered using the same process (as above mentioned) from the crude. The expected product was then dried and recrystallized using ethanol.
Characterization of synthesized compounds
2,2′-((4-Hydroxyphenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6a). Light yellow solid, yield 94%, m.p. 181–183 °C,73 1H NMR (400 MHz, CDCl3): δ 11.88 (s, 1H, OH), 6.91 (dd, J = 9.0, 1.2 Hz, 2H, Ar–H), 6.68 (d, J = 8.9 Hz, 2H, Ar–H), 5.46 (s, 1H, CH), 2.47–2.27 (m, 8H, 4CH2), 1.25–1.20 (m, 6H, 2CH3), 1.08 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 190.84, 189.70, 153.83, 129.66, 128.02, 115.88, 115.30, 47.09, 46.48, 32.09, 31.50, 31.49, 29.80, 29.76, 27.45; ESI-MS (m/z): 384.19 [M+].
2,2′-((3-Methoxyphenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6b). Off-white crystalline solid, yield: 78%, m.p. 194–196 °C,74 1H NMR (400 MHz, CDCl3): δ 11.95 (s, 1H, OH), 7.19–7.15 (t, J = 8.0 Hz, 1H, Ar–H), 6.72–6.63 (m, 3H, Ar–H), 5.50 (d, J = 1.0 Hz, IH, CH), 3.72 (s, 3H, OCH3), 2.47–2.28 (m, 8H, 4CH2), 1.23 (d, J = 6.0 Hz, 6H, 2CH3), 1.09 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 190.54, 189.53, 159.61, 139.91, 129.19, 119.30, 115.65, 113.04, 111.14, 55.13, 47.11, 46.46, 32.83, 31.46, 29.81, 27.39; ESI-MS (m/z): 398.21 [M+].
2,2′-(Phenylmethylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6c). White solid, yield: 91%, m.p. 185–189 °C,73 1H NMR (400 MHz, CDCl3): δ 11.91 (s, 1H, OH), 7.28–7.24 (m, 2H, Ar–H), 7.18–7.14 (m, 1H, Ar–H), 7.10–7.08 (m, 2H, Ar–H), 5.53 (s, 1H, CH), 2.38 (dq, J = 26.3, 17.7 Hz, 8H, 4CH2), 1.22 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 190.63, 189.54, 138.11, 128.32, 126.86, 125.95, 115.66, 47.11, 46.50, 32.81, 31.50, 29.80, 27.46; ESI-MS (m/z): 368.20 [M+].
2,2′-(Pyridin-2-ylmethylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6d). Brownish black solid, yield: 75%, m.p. 146–149 °C,75 1H NMR (400 MHz, CDCl3): δ 8.37–8.35 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H, Ar–H), 7.59 (dt, J = 7.8, 1.1 Hz, 1H, Ar–H), 7.54 (td, J = 7.6, 1.8 Hz, 1H, Ar–H), 7.00–6.96 (ddd, J = 7.3, 4.9, 1.3 Hz, 1H, Ar–H), 4.84 (s, 1H, CH), 2.53–2.41 (m, 4H, 2CH2), 2.24–2.11 (dt, J = 16.2, 8.6 Hz, 4H, 2CH2), 1.08 (s, 6H, 2CH3), 0.98 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 197.13, 163.48, 161.79, 149.00, 135.82, 125.06, 121.50, 114.39, 50.82, 40.91, 34.53, 32.42, 32.41, 32.39, 29.42, 27.22; ESI-MS (m/z): 369.19 [M+].
2,2′-((2-Nitrophenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6e). Yellow crystalline solid, yield: 83%, m.p. 187–190 °C,73 1H NMR (400 MHz, CDCl3): δ 11.59 (s, 1H, OH), 7.54–7.52 (dd, J = 7.9, 1.4 Hz, 1H, Ar–H), 7.48–7.44 (td, J = 7.7, 1.5 Hz, 1H, Ar–H), 7.33–7.29 (m, 1H, Ar–H), 7.24–7.21 (dt, J = 7.9, 1.2 Hz, 1H, Ar–H), 6.02 (s, 1H, CH), 2.50–2.18 (ddd, J = 56.8, 33.0, 17.1 Hz, 8H, 4CH2), 1.14 (s, 6H, 2CH3), 1.00 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 149.76, 132.17, 131.47, 129.67, 127.27, 124.43, 114.72, 47.03, 46.91, 46.34, 46.21, 31.99, 30.11, 28.77, 28.63, 28.25, 28.16; ESI-MS (m/z): 413.18 [M+].
2,2′-((4-Nitrophenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6f). Dark yellow crystals, yield: 91%, m.p. 193–195 °C,33 1H NMR (400 MHz, CDCl3): δ 11.80 (s, 1H, OH), 8.13–8.11 (m, 2H, Ar–H), 7.24–7.22 (dd, J = 9.0, 1.2 Hz, 2H, Ar–H), 5.53 (s, 1H, CH), 2.50–2.30 (dq, J = 27.6, 17.7 Hz, 8H, 4CH2), 1.22 (s, 6H, 2CH3), 1.10 (s, 6H, 2CH3); ESI-MS (m/z): 413.18 [M+].
4-(Bis(2-hydroxy-4,4-dimethyl-6-oxocyclohex-1-en-1-yl)methyl)benzonitrile (6g). Light yellow solid, yield: 87%,m.p. 195–199 °C,33 1H NMR (400 MHz, CDCl3): δ 11.79 (s, 1H, OH), 7.56–7.54 (d, J = 8.8 Hz, 2H, Ar–H), 7.17–7.19 (dd, J = 8.8, 1.3 Hz, 2H, Ar–H), 5.50 (s, 1H, CH), 2.49–2.28 (ddd, J = 33.7, 28.0, 17.9 Hz, 8H, 4CH2), 1.21 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 191.09, 189.67, 144.40, 132.19, 127.69, 119.05, 114.91, 109.78, 47.03, 46.46, 33.30, 31.55, 31.52, 31.52, 29.69, 27.51; ESI-MS (m/z): 393.19 [M+].
2,2′-((2-Chlorophenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (6h). Off-white crystalline solid, yield: 78%, m.p. 205–207 °C,73 1H NMR (400 MHz, CDCl3): δ 11.88 (s, 1H, OH), 7.38–7.27 (m, 2H, Ar–H), 7.22–7.11 (m, 2H, Ar–H), 5.60 (s, 1H, CH), 2.45–2.03 (ddd, J = 120.2, 61.5, 52.6 Hz, 8H, 4CH2), 1.25–0.93 (m, 12H, 4CH3). 13C NMR (101 MHz, CDCl3): δ 136.61, 133.60, 130.44, 129.46, 127.78, 126.55, 115.78, 115.77, 115.76, 47.11, 46.46, 32.15, 31.59, 29.13; ESI-MS (m/z): 402.16 [M+].
2,2′-((2,4,5-Trimethoxyphenyl)methylene)bis(3-hydroxycyclohex-2-en-1-one) (7a). Light yellow crystals, yield: 97%, m.p. 168–171 °C, 1H NMR (400 MHz, CDCl3): δ 7.10–7.08 (d, J = 8.8 Hz, 1H, Ar–H), 6.56–6.54 (d, J = 8.8 Hz, 1H, Ar–H), 4.76 (s, 1H, CH), 3.86 (s, 3H, OCH3), 3.77 (s, 6H, 2OCH3), 2.59–2.53 (m, 4H, 2CH2), 2.33–2.28 (m, 4H, 2CH2), 2.02–1.91 (ddd, J = 19.0, 11.5, 7.0 Hz, 4H, 2CH2). 13C NMR (101 MHz, CDCl3): δ 197.01, 164.32, 152.74, 152.63, 141.91, 128.91, 126.19, 115.91, 106.43, 60.53, 60.50, 55.81, 37.10, 29.42, 27.33, 20.42; ESI-MS (m/z): 402.17 [M+].
4-(Bis(2-hydroxy-6-oxocyclohex-1-en-1-yl)methyl)benzonitrile (7b). Milky white solid, yield: 82%, m.p. 256–260 °C, 1H NMR (400 MHz, CDCl3): δ 7.51–7.39 (dd, J = 38.8, 8.7 Hz, 4H, Ar–H), 4.80 (s, 1H, CH), 2.65–2.62 (m, 4H, 2CH2), 2.35–2.31 (m, 3H, CH2), 2.04–1.96 (m, 3H, CH2), 1.24 (s, 1H, CH2), 0.86–0.82 (m, 1H, CH2). 13C NMR (101 MHz, CDCl3): δ 196.63, 164.64, 149.75, 132.09, 129.42, 119.20, 115.89, 110.21, 36.90, 32.39, 27.21, 20.30; ESI-MS (m/z): 337.13 [M+].
2,2′-((4-Ethylphenyl)methylene)bis(3-hydroxycyclohex-2-en-1-one) (7c). Cream solid, yield: 93%, m.p. 164–166 °C, 1H NMR (400 MHz, CDCl3): δ 7.20–7.18 (d, J = 8.1 Hz, 2H, Ar–H), 7.04–7.02 (d, J = 8.2 Hz, 2H, Ar–H), 4.77 (s, 1H, CH), 2.57–2.51 (m, 4H, 2CH2), 2.36–2.30 (m, 4H, 2CH2), 2.02–1.97 (ddd, J = 7.0, 6.4, 3.2 Hz, 4H, 2CH2), 1.61 (s, 2H, CH2), 1.18–1.14 (t, J = 7.6 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3): δ 196.79, 163.93, 142.19, 141.72, 128.30, 127.71, 117.12, 37.06, 31.22, 28.50, 27.23, 20.37, 15.41; ESI-MS (m/z): 340.17 [M+].
2,2′-(Thiophen-2-ylmethylene)bis(3-hydroxycyclohex-2-en-1-one) (7d). Munsell yellow, yield: 79%, m.p. 215–218 °C, 1H NMR (400 MHz, CDCl3): δ 7.04–6.83 (m, 3H, Ar–H), 5.16 (s, 1H, CH), 2.66–2.35 (m, 6H, 3CH2), 2.05–2.00 (m, 3H, CH2), 1.24–1.22 (d, J = 7.3 Hz, 1H, CH2), 0.86–0.83 (m, 2H, CH2). 13C NMR (101 MHz, CDCl3): δ 196.64, 164.42, 148.47, 126.91, 125.10, 123.66, 116.48, 37.01, 27.24, 26.22, 20.33; ESI-MS (m/z): 318.09 [M+].
2,2′-((3,4-Dimethoxyphenyl)methylene)bis(3-hydroxycyclohex-2-en-1-one) (7e). White solid, yield: 88%, m.p. 158–160 °C,76 1H NMR (400 MHz, CDCl3): δ 6.98 (d, J = 2.1 Hz, 1H, Ar–H), 6.69–6.68 (m, 2H, Ar–H), 4.75 (s, 1H, CH), 3.87 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 2.61 (s, 4H, 2CH2), 2.36–2.30 (dd, J = 14.2, 8.9 Hz, 4H, 2CH2), 2.03–2.00 (m, 4H, 2CH2). 13C NMR (101 MHz, CDCl3): δ 196.88, 163.96, 148.45, 147.56, 137.30, 119.64, 117.03, 112.65, 110.87, 55.84, 37.06, 31.01, 27.24, 20.41; ESI-MS (m/z): 372.16 [M+].
2,2′-((2-Hydroxyphenyl)methylene)bis(3-hydroxycyclohex-2-en-1-one) (7f). Chrome yellow, yield: 83%, m.p. 229–234 °C,55 1H NMR (400 MHz, CDCl3): δ 10.84 (s, 1H, Ar-OH), 7.17–7.12 (m, 1H, Ar–H), 7.02–6.99 (m, 3H, Ar–H), 4.62 (s, 1H, CH), 2.77–2.71 (dt, J = 17.5, 4.5 Hz, 1H, CH2), 2.58–2.50 (m, 3H, CH2), 2.45–2.40 (ddd, J = 11.5, 5.2, 2.6 Hz, 2H, CH2), 2.11 (s, 1H, CH2), 2.06–1.97 (ddd, J = 19.2, 13.9, 8.7 Hz, 3H, CH2), 1.82–1.73 (dd, J = 18.0, 16.3 Hz, 2H, CH2). 13C NMR (101 MHz, CDCl3): δ 201.62, 197.20, 172.96, 171.28, 150.95, 128.15, 127.62, 124.73, 124.69, 119.92, 115.62, 112.37, 37.06, 36.09, 29.81, 28.08, 19.99, 19.70; ESI-MS (m/z): 328.13 [M+].
4,4′-((3-Chlorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8a). Golden yellow solid, yield: 81%, m.p. 155–159 °C,64 1H NMR (400 MHz, CDCl3): δ 7.57–7.55 (d, J = 7.5 Hz, 4H, Ar–H), 7.27 (s, 4H, Ar–H), 7.14–7.10 (m, 6H, Ar–H), 4.71 (s, 1H, CH), 2.06 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 147.96, 146.58, 142.79, 134.30, 129.72, 129.07, 127.44, 126.80, 126.46, 125.57, 124.27, 121.42, 118.44, 31.69, 22.76; ESI-MS (m/z): 470.15[M+].
4,4′-((3-Methoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8b). Bronze brown solid, yield: 78%, m.p. 175–178 °C,64 1H NMR (400 MHz, CDCl3) δ 7.59–7.57 (d, J = 5.5 Hz, 4H, Ar–H), 7.33 (s, 4H, Ar-H), 7.09–7.07 (d, J = 8.1 Hz, 4H, Ar–H), 6.76–6.74 (d, J = 8.3 Hz, 2H, Ar–H), 4.71 (s, 1H, CH), 3.72 (s, 3H, OCH3), 2.06–2.03 (d, J = 9.5 Hz, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 159.05, 155.78, 154.77, 146.64, 132.29, 129.01, 128.27, 126.25, 121.33, 113.78, 60.54, 55.32, 11.75; ESI-MS (m/z): 466.20 [M+].
4,4′-(Furan-2-ylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8c). Charcoal black crystals, yield: 90%, m.p. 176–178 °C,64 1H NMR (400 MHz, CDCl3): δ 7.53–7.51 (d, J = 7.8 Hz, 5H, Ar–H), 7.25–7.22 (d, J = 6.1 Hz, 4H, Ar–H), 7.08–7.05 (t, J = 7.3 Hz, 2H, Ar–H), 6.23 (s, 1H, Ar–H), 6.15 (s, 1H, Ar–H), 4.70 (s, 1H, CH), 2.04 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 153.18, 146.23, 141.39, 136.95, 128.99, 126.33, 121.50, 121.40, 110.59, 106.91, 28.95, 11.57; ESI-MS (m/z): 426.17 [M+].
4,4′-((3-Hydroxy-4-methoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8d). Yellow solid, yield: 98%, m.p. 189–191 °C,26 1H NMR (400 MHz, CDCl3): δ 8.19 (s, 1H, OH), 7.56–7.54 (d, J = 9.0 Hz, 5H, Ar–H), 7.23–7.21 (s, 2H, Ar–H), 7.08 (t, J = 7.8 Hz, 2H, Ar–H), 6.70–6.65 (d, J = 19.3 Hz, 4H, Ar–H), 4.68 (s, 1H, CH), 3.74 (s, 3H, OCH3), 2.12 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO-d6): δ 146.79, 146.65, 146.42, 135.12, 129.49, 129.02, 126.14, 121.27, 121.20, 121.04, 120.84, 118.13, 115.30, 112.62, 112.57, 56.17, 32.81, 12.14; ESI-MS (m/z): 482.20 [M+].
4,4′-((4-(Benzyloxy)phenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8e). Cream solid, yield: 71%, m.p. 194–196 °C,27 1H NMR (400 MHz, CDCl3): δ 7.38–7.35 (d, J = 7.8 Hz, 4H, Ar–H), 7.33 (dd, J = 12.1, 7.2 Hz, 7H, Ar–H), 7.24–7.22 (d, J = 7.8 Hz, 2H, Ar–H), 7.08–7.06 (m, 4H, Ar–H), 6.83–6.81 (d, J = 8.7 Hz, 2H, Ar–H), 4.96 (s, 2H, CH2), 4.68 (s, 1H, CH), 2.03 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3): δ 157.44, 151.31, 146.57, 137.13, 132.83, 129.17, 129.00, 128.66, 128.29, 128.02, 127.58, 126.29, 121.37, 119.03, 114.68, 70.06, 32.94, 11.66; ESI-MS (m/z): 542.23 [M+].
4,4′-((3,4,5-Trimethoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol) (8f). Light yellow crystals, yield: 95%, m.p. 194–199 °C,26 1H NMR (400 MHz, CDCl3): δ 7.63–7.61 (d, J = 9.1 Hz, 4H, Ar–H), 7.32–7.28 (dd, J = 10.5, 4.8 Hz, 4H, Ar–H), 7.15–7.11 (m, 2H, Ar–H), 6.49 (s, 2H, Ar–H), 4.72 (s, 1H, CH), 3.77–3.3.72 (d, J = 20.3 Hz, 9H, 3OCH3), 2.16 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO-d6): δ 153.06, 153.03, 153.02, 139.13, 136.38, 129.70, 129.49, 121.29, 120.90, 105.31, 60.45, 56.30, 34.33, 26.86; ESI-MS (m/z): 526.22 [M+].
3,3′-((3,4,5-Trimethoxyphenyl)methylene)bis(4-hydroxy-2H-chromen-2-one) (9a). White solid, yield: 95%, m.p. 243–245 °C,77 1H NMR (400 MHz, CDCl3) δ 8.08–8.06 (d, J = 6.6 Hz, 1H, Ar–H), 7.65–7.61 (ddd, J = 8.6, 7.3, 1.7 Hz, 2H, Ar–H), 7.42–7.40 (d, J = 9.0 Hz, 4H, Ar–H), 6.40 (d, J = 1.3 Hz, 3H, Ar–H), 6.06 (m, 1H, CH), 3.84 (s, 3H, OCH3), 3.70 (s, 6H, 2OCH3); ESI-MS (m/z): 502.13 [M+].
3,3′-((4-Isopropylphenyl)methylene)bis(4-hydroxy-2H-chromen-2-one) (9b). Off-white crystals, yield: 82%, m.p. 235–238 °C, 1H NMR (400 MHz, CDCl3) δ 11.50 (s, 1H, OH), 8.07–8.05 (dd, J = 27.2, 8.7 Hz, 2H, Ar–H), 8.00–7.98 (ddd, J = 8.5, 7.3, 1.7 Hz, 2H, Ar–H), 7.64–7.60 (m, 4H, Ar–H), 7.16–7.13 (ddd, J = 8.7, 7.7, 3.7 Hz, 4H, Ar–H), 6.06 (s, 1H, CH), 2.92–2.85 (dt, J = 14.3, 7.2 Hz, 1H, CH), 1.24–1.22 (d, J = 7.0 Hz, 6H, CH3). 13C NMR (101 MHz, CDCl3): δ 147.52, 132.90, 132.44, 126.79, 126.48, 124.95, 124.47, 116.72, 35.95, 33.70, 24.07; ESI-MS (m/z): 454.14 [M+].
3,3′-((4-(Benzyloxy)phenyl)methylene)bis(4-hydroxy-2H-chromen-2-one) (9c). Light yellow solid, yield: 69%, m.p. 198–202 °C,33 1H NMR (400 MHz, CDCl3): δ 11.51 (s, 1H, OH), 8.06–8.04 (dd, J = 25.3, 7.6 Hz, 2H, Ar–H), 8.00–7.98 (m, 2H, Ar–H), 7.40 (m, 9H, Ar–H), 7.38–7.36 (m, 2H, Ar–H), 6.93–6.91 (d, J = 8.9 Hz, 2H, Ar–H), 6.04 (s, 1H, CH), 5.03 (s, 2H, CH2); ESI-MS (m/z): 518.14 [M+].
3,3′-((3-Hydroxy-4-methoxyphenyl)methylene)bis(4-hydroxy-2H-chromen-2-one) (9d). Off-white crystals, yield: 96%, m.p. 244–246 °C,33 1H NMR (400 MHz, CDCl3): δ 11.57 (s, 1H, OH), 8.03–7.98 (m, 2H, Ar–H), 7.6–7.59 (m, 2H, Ar–H), 7.40–7.38 (d, J = 8.3 Hz, 4H, Ar–H), 6.78–6.77 (m, 2H, Ar–H), 6.76 (ddd, J = 8.3, 2.3, 1.3 Hz, 1H, Ar–H), 6.01 (d, J = 1.1 Hz, 1H, CH), 3.86 (s, 3H, OCH3); ESI-MS (m/z): 458.10 [M+].
3,3′-((4-Hydroxy-3,5-dimethoxyphenyl)methylene)bis(4-hydroxy-2H-chromen-2-one) (9e). Yellow solid, yield: 79%, m.p. 192–194 °C,78 1H NMR (400 MHz, CDCl3): δ 11.56 (s, 1H, OH), 8.08–8.01 (d, J = 25.7 Hz, 2H, Ar–H), 7.65–7.60 (m, 2H, Ar–H), 7.42–7.25 (d, J = 8.4 Hz, 4H, Ar–H), 6.41–6.40 (d, J = 1.3 Hz, 2H, Ar–H), 6.07 (s, 1H, CH), 3.74 (s, 6H, 2OCH3); ESI-MS (m/z): 488.11 [M+].
3,3′-(Pyren-1-ylmethylene)bis(4-hydroxy-2H-chromen-2-one) (9f). Brownish-yellow solid, yield: 90%, m.p. 218–220 °C, 1H NMR (400 MHz, CDCl3): δ 11.41 (s, 1H, OH), 8.16–8.14 (d, J = 7.6 Hz, 3H, Ar–H), 8.10–8.03 (dd, J = 17.2, 8.4 Hz, 4H, Ar–H), 7.96–7.90 (m, 4H, Ar–H), 7.63–7.60 (t, J = 7.3 Hz, 2H, Ar–H), 7.45–7.33 (dd, J = 37.6, 7.4 Hz, 4H, Ar–H), 6.99 (s, 1H, CH). 13C NMR (101 MHz, CDCl3): δ 133.02, 131.40, 130.91, 130.43, 128.92, 128.60, 127.84, 127.51, 126.05, 125.59, 125.56, 125.48, 125.28, 124.86, 122.26, 117.33, 116.76, 116.68, 35.62; ESI-MS (m/z): 536.13 [M+].
Conflicts of interest
The authors confirmed that this article has no conflict of interest.
Acknowledgements
The authors are thankful to Department of Chemistry, MLSU, Udaipur for providing research facilities, and Department of Physics, MLSU Udaipur, CUG, Gandhinagar and CIL, Punjab for XRD, SEM, and TEM studies. N. Sahiba and P. Teli wish to acknowledge CSIR, India (09/172(0088)2018-EMR-I) (09/172(0099)2019-EMR-I) for senior research fellowship as financially support. S. Agarwal sincerely acknowledges Ministry of Education, Government of India and Ministry of Higher Education, Government of Rajasthan, India for providing NMR facility under RUSA 2.0, Research, and Innovation project.
References
- M. S. Patel, J. N. Parekh, D. D. Chudasama, H. C. Patel, P. Dalwadi, A. Kunjadiya, V. Bhatt and K. R. Ram, ACS Omega, 2022, 7, 30420–30439 CrossRef CAS.
- T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed.
- H. Wu, H. Li and Z. Fang, Green Chem., 2021, 23, 6675–6697 RSC.
- Q.-C. Ren, C. Gao, Z. Xu, L.-S. Feng, M.-L. Liu, X. Wu and F. Zhao, Curr. Top. Med. Chem., 2018, 18, 101–113 CrossRef CAS PubMed.
- F. G. Medina, J. G. Marrero, M. Macías-Alonso, M. C. González, I. Córdova-Guerrero, A. G. Teissier García and S. Osegueda-Robles, Nat. Prod. Rep., 2015, 32, 1472–1507 RSC.
- M. A. Wani, J. A. Farooqi and W. A. Shah, J. Pharm. Appl. Chem., 2016, 2, 53–57 CrossRef.
- C. Chen and L. He, Eur. J. Med. Chem., 2020, 203, 112577 CrossRef CAS PubMed.
- S. A.-G. Abdel-Aziz, T. E.-S. Ali, K. M. El-Mahdy and S. M. Abdel-Karim, Eur. J. Chem., 2011, 2, 25–35 CrossRef CAS.
- A. B. Danne, M. V. Deshpande, J. N. Sangshetti, V. M. Khedkar and B. B. Shingate, ACS Omega, 2021, 6, 24879–24890 CrossRef CAS PubMed.
- W. F. Rodhan, S. S. Kadhium, Z. Z. M. Ali, A. G. Eleiwi, R. F. Abbas, I. R. Mohamed and Z. A. Hussein, J. Phys.: Conf. Ser., 2021, 1853, 012059 CrossRef CAS.
- S. Hasanzadeh Banakar, M. G. Dekamin and A. Yaghoubi, New J. Chem., 2018, 42, 14246–14262 RSC.
- S. Shafiu, E. I. Edache, U. Sani and M. Abatyough, J. Pharmaceut. Med. Res., 2017, 1, 78–80 Search PubMed.
- G. Brahmachari and S. Begam, ChemistrySelect, 2019, 4, 5415–5420 CrossRef CAS.
- Y.-P. Sui, H.-R. Huo, J.-J. Xin, J. Li, X.-J. Li, X.-L. Du, H. Ma, H.-Y. Zhou, H.-D. Zhan, Z.-J. Wang, C. Li, F. Sui and M.-K. Li, Molecules, 2015, 20, 17614–17626 CrossRef CAS PubMed.
- X. Zhang, L. Hu, X. Wang, Y. Zhao and X. Chen, SSRN Electron. J., 4261720, DOI:10.2139/ssrn.4261720.
- S. Parihar, S. Pathan, R. N. Jadeja, A. Patel and V. K. Gupta, Inorg. Chem., 2012, 51, 1152–1161 CrossRef CAS.
- R. Xu, M. Liang, F. Yin, S. Li, Y. Huang, Y. Zhaoa, J. Liu and W. Ma, Inorg. Chem. Commun., 2014, 40, 120–123 CrossRef CAS.
- A. Barakat, A. M. Al-Majid, M. S. Islam, I. Warad, V. H. Masand, S. Yousuf and M. Iqbal Choudhary, Res. Chem. Intermed., 2016, 42, 4041–4053 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, B. Fattahi and N. Nazari, Curr. Org. Chem., 2016, 20, 1676–1735 CrossRef CAS.
- G. F. Woods and I. W. Tucker, J. Am. Chem. Soc., 1948, 70, 2174–2177 CrossRef CAS.
- E. A. Shokova, J. K. Kim and V. V. Kovalev, Russ. J. Org. Chem., 2015, 51, 755–830 CrossRef CAS.
- P. Teli, N. Sahiba, A. Sethiya, J. Soni and S. Agarwal, J. Heterocycl. Chem., 2021, 58, 1393–1407 CrossRef CAS.
- V. M. Nurchi, R. Cappai, G. Crisponi, G. Sanna, G. Alberti, R. Biesuz and S. Gama, Front. Chem., 2020, 8, 597400 CrossRef CAS PubMed.
- L. Bai, W. Sun, M. Huang, L. Li, C. Geng, K. Liu and D. Yan, Crit. Rev. Anal. Chem., 2020, 50, 78–89 CrossRef CAS.
- P. Teli, N. Sahiba, A. Manhas, P. C. Jha, P. Meena and S. Agarwal, ChemistrySelect, 2023, 8, 15 CrossRef.
- N. G. Shabalala, N. Kerru, S. Maddila, W. E. van Zyl and S. B. Jonnalagadda, Chem. Data Collect., 2020, 28, 100467 CrossRef CAS.
- E. Rostami and Z. Kordrostami, Asian J. Nanosci. Mater., 2020, 3, 203 CAS.
- R. Sharma, P. A. Chawla, V. Chawla, R. Verma, N. Nawal and V. Gupta, Mini-Rev. Med. Chem., 2021, 21, 1770–1795 CrossRef CAS PubMed.
- S. Parihar, S. Pathan, R. N. Jadeja, A. Patel and V. K. Gupta, Inorg. Chem., 2012, 51, 1152–1161 CrossRef CAS.
- B. Borah, K. Dhar Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 3101–3126 CrossRef CAS.
- J. Soni, N. Sahiba, A. Sethiya, P. Teli, D. Kr. Agarwal, A. Manhas, P. C. Jha, D. Joshi and S. Agarwal, Polycyclic Aromat. Compd., 2022, 42, 2970–2990 CrossRef CAS.
- Z. Nofal, M. El-Zahar and S. Abd El-Karim, Molecules, 2000, 5, 99–113 CrossRef CAS.
- P. Teli, A. Sethiya and S. Agarwal, Res. Chem. Intermed., 2022, 48, 731–750 CrossRef CAS.
- P. Teli, A. Sethiya and S. Agarwal, ChemistrySelect, 2019, 4, 13772–13787 CrossRef CAS.
- Y. Lin, X. Shen, Q. Yuan and Y. Yan, Nat. Commun., 2013, 4, 2603 CrossRef PubMed.
- R. Z. Batran, M. A. Khedr, N. A. Abdel Latif, A. A. Abd El Aty and A. N. Shehata, J. Mol. Struct., 2019, 1180, 260–271 CrossRef CAS.
- W. Wang, B. Vinocur and A. Altman, Planta, 2003, 218, 1–14 CrossRef CAS PubMed.
- H. A. Garro, G. Petroselli, C. R. Pungitore, C. E. Tonn, and R. E. Balsells, 2015.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- Y. Guan, S. Hu, G. Gu, G. Lu, X. Yuan and J. Bai, Nano, 2020, 15, 2050083 CrossRef CAS.
- Z. Zeng, Y. Chen, X. Zhu and L. Yu, Chin. Chem. Lett., 2023, 34, 107728 CrossRef CAS.
- T. O. Ajiboye, O. A. Oyewo and D. C. Onwudiwe, J. Inorg. Organomet. Polym. Mater., 2021, 31, 1419–1442 CrossRef CAS.
- H. Venkatesvaran, S. Balu, A. Chowdhury, S. W. Chen and T. C. K. Yang, Catalysts, 2022, 142, 104637 Search PubMed.
- S. Zuluaga, L.-H. Liu, N. Shafiq, S. M. Rupich, J.-F. Veyan, Y. J. Chabal and T. Thonhauser, Phys. Chem. Chem. Phys., 2015, 17, 957–962 RSC.
- H. Venkatesvaran, S. Balu, B.-S. Tsai and T. C.-K. Yang, J. Taiwan Inst. Chem. Eng., 2023, 142, 104637 CrossRef CAS.
- H. Li, Q. Zhang, X. Liu, F. Chang, Y. Zhang, W. Xue and S. Yang, Bioresour. Technol., 2013, 144, 21–27 CrossRef CAS PubMed.
- M. L. Testa and V. La Parola, Catalysts, 2021, 11, 1143 CrossRef CAS.
- L. J. Konwar, P. Mäki-Arvela and J. P. Mikkola, Chem. Rev., 2019, 119 Search PubMed.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118 Search PubMed.
- H. Veisi, P. Mohammadi and T. Ozturk, J. Mol. Liq., 2020, 303, 112625 CrossRef CAS.
- M. Manivannan and S. Rajendran, J. Eng. Sci. Technol., 2011, 3, 8048–8060 Search PubMed.
- K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 1, 3 Search PubMed.
- A. Lapkin and D. J. C. Constable, Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, Wiley, Chichester, 2008, pp. 91–96 Search PubMed.
- N. Sahiba, A. Sethiya, J. Soni and S. Agarwal, ChemistrySelect, 2020, 42, 13076–13080 CrossRef.
- J. J. Yu, L. M. Wang, J. Q. Liu, F. Lou Guo, Y. Liu and N. Jiao, Green Chem., 2010, 12, 216–219 RSC.
- K. P. Nandre, V. S. Patil and S. V. Bhosale, Chin. Chem. Lett., 2011, 22, 777–780 CrossRef CAS.
- D. H. Jung, Y. R. Lee, S. H. Kim and W. S. Lyoo, Bull. Korean Chem. Soc., 2009, 30, 1989–1995 CrossRef CAS.
- A. Fallah, M. Tajbakhsh, H. Vahedi and A. Bekhradnia, Res. Chem. Intermed., 2017, 43, 29–43 CrossRef CAS.
- F. Shirini and N. Daneshvar, RSC Adv., 2016, 111, 110190–110205 RSC.
- B. S. Kuarm and B. Rajitha, Synth. Commun., 2012, 42, 2382–2387 CrossRef CAS.
- A. Khazaei, M. A. Zolfigol, A. R. Moosavi-Zare, Z. Asgari, M. Shekouhy, A. Zare and A. Hasaninejad, RSC Adv., 2012, 2, 8010 RSC.
- N. G. Khaligh, T. Mihankhah, H. Gorjian and M. R. Johan, Synth. Commun., 2020, 50, 3276–3286 CrossRef CAS.
- K. R. Phatangare, V. S. Padalkar, V. D. Gupta, V. S. Patil, P. G. Umape and N. Sekar, Synth. Commun., 2012, 42, 1349–1358 CrossRef CAS.
- A. Zare, M. Merajoddin, A. R. Moosavi-Zare and M. Zarei, Chin. J. Catal., 2014, 35, 85–89 CrossRef CAS.
- F. Shirini, M. Seddighi, M. Mazloumi, M. Makhsous and M. Abedini, J. Mol. Liq., 2015, 208, 291–297 CrossRef CAS.
- G. Kaur, D. Singh, A. Singh and B. Banerjee, Synth. Commun., 2021, 51, 1045–1057 CrossRef CAS.
- R. Karimian, F. Piri, A. A. Safari and S. J. Davarpanah, J. Nanostruct. Chem., 2013, 3, 1–6 Search PubMed.
- S. Khodabakhshi and M. Baghernejad, J. Chin. Chem. Soc., 2013, 60, 495–498 CrossRef CAS.
- B. Zeynizadeh and M. Gilanizadeh, New J. Chem., 2019, 43, 18794–18804 RSC.
- B. Sadeghi and T. Ziya, J. Chem., 2013, 179013 Search PubMed.
- N. Azizi, F. Abbasi and M. Abdoli-Senejani, ChemistrySelect, 2018, 3, 3797–3802 CrossRef CAS.
- Z. Zare-Akbari, S. Dastmalchi, L. Edjlali, L. Dinparast and M. Es’haghi, Appl. Organomet. Chem., 2020, 34, e5649 CrossRef CAS.
- B. M. Sapkal, P. K. Labhane and J. R. Satam, Res. Chem. Intermed., 2017, 43, 4967–4979 CrossRef CAS.
- M. S. Esmaeili, Z. Varzi, R. Taheri-Ledari and A. Maleki, Res. Chem. Intermed., 2021, 47, 973–996 CrossRef CAS.
- M. Sigalov, B. Shainyan, P. Krief, I. Ushakov, N. Chipanina and L. Oznobikhina, J. Mol. Struct., 2011, 1006, 234–246 CrossRef CAS.
- F. E. King and D. G. I. Felton, J. Chem. Soc. (Resumed), 1948, 1371–1372 RSC.
- G. Brahmachari and B. Banerjee, RSC Adv., 2015, 49, 39263–39269 RSC.
- V. D. Kancheva, P. V. Boranova, J. T. Nechev and I. I. Manolov, Biochimie, 2010, 92, 1138–1146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information is available for readers in supplementary file which includes 1H and 13C NMR of synthesized compounds. See DOI: https://doi.org/10.1039/d3ra01971c |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
: