
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGemini surfactant-like peptide-based nanocages with β-sheet-enhanced stability and encapsulation efficiency of hydrophobic anticancer drugs†
Feng Gao‡
a,
Xinmin Zhao‡b,
Qiankang Sib,
Xingkun Niub,
Shaojie Houc,
Shihao Liuc,
Jun Guo*c,
Liping Wang*c and
Feng Zhang *abc
*abc
aSchool of Life Science, Inner Mongolia Agricultural University, Hohhot 010010, China
bQuantum Biophotonic Lab, Key Laboratory of Optical Technology and Instrument for Medicine, Ministry of Education, School of Optical-Electrical and Computer Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China. E-mail: fzhang@usst.edu.cn
cWenzhou Institute, University of Chinese Academy of Sciences, Wenzhou 325001, China. E-mail: guojun-nbm@wiucas.ac.cn; lpwang-wiucas@ucas.ac.cn
First published on 25th April 2023
Abstract
Peptide-based scaffolds have been widely applied to drug delivery because of their ease and high yields of synthesis, well-defined structure, biocompatibility, diversity, tunability of properties, and molecular recognition abilities. However, the stability of peptide-based nanostructures highly depends on the intermolecular assembling manner, e.g., α-helix based coiled coils, β-sheet. Inspired by the robust protein fibril structures in amyloidosis, herein we constructed a β-sheet-forming gemini surfactant-like peptide to self-assemble into nanocages with the help of molecular dynamics simulation. As expected, the experimental results showed that nanocages can be formed with the inner diameter of up to ∼400 nm, which were robust enough even under both transmission electron microscopy and atomic force microscopy, indicating the significant contribution of β-sheet conformation. The β-nanocages can load hydrophobic anticancer drugs, e.g., paclitaxel with a very high encapsulation efficiency, which holds great potential for clinic drug delivery due to the improved anticancer effect as compared with paclitaxel alone.
1. Introduction
Since human beings started developing and synthesizing drugs on their own, approximately 70% of drug molecules, especially for anticancer drugs have exhibited poor water solubility.1 The water solubility of new drug molecules is a crucial factor affecting their effectiveness, which dramatically hinders their wide applications in the biomedical field.2 Therefore, it is critical to construct a drug delivery system that is safe, effective and meets the required quality standards.3In recent years, a large number of studies have proposed different approaches to improve the solubility and bioavailability of hydrophobic drugs,4 including the use of various novel delivery materials such as hydrogel carriers,5,6 magnetic ionic liquids,7 lipids,8,9 and surfactants.10 However, these materials have gradually exposed their problems in clinical applications. For example, hydrogel carriers have inherently poor mechanical properties and are costly. Lipid carriers are prone to decomposition and exhibiting poor stability during long-term storage, and most of them need to be synthesized in organic solvents, which inevitably introduces risk and challenges in terms of biosafety. In contrast, self-assembling peptides have shown unique advantages in drug delivery due to their excellent biocompatibility, chemical tailorability and structural designability.11–13
Similar to other biomolecules, many papers have reported the use of peptides to construct drug carriers for encapsulating hydrophobic drugs.14 Among these reports, the surfactant-like peptides (SLP) have shown notable efficiency in the solubilization of hydrophobic drugs.15,16 Sometimes, SLP is also called lipid-like peptide, for instance Karavasili et al. designed a peptide with a length similar to that of cellular membrane lipids and verified their permeability for facilitating transport through the intestinal barrier.17 Peng et al. designed a Gemini-type SLPs (GSLPs) composed of mere natural amino acids that can efficiently encapsulate different hydrophobic drugs by designing six alanine amino acid for forming β-sheet conformation.18 However, the pure hydrophobic amino acids e.g., alanine cannot avoid the nonspecific co-assembling with other hydrophobic molecules including drug themselves. To solve this issue, it is interesting to discover novel type of GSLPs containing specific β-sheet forming peptide sequences.
Inspired by the β-sheet-forming proteins in amyloidosis, herein we designed a novel GSLP with the sequence of GNNQQNY-PKKP-GNNQQNY. In this design, we employed an antiparallel β-sheet-forming peptide sequence GNNQQNY (G7) to replace the twin hydrophobic tails (here actually a little hydrophilic) and two proline (P) for flexible turning around and two lysine (K) residue to achieve the positively charged cages in physiological environments. The rational designed GSLPs can effectively self-assemble into β-nanocages and successfully encapsulate hydrophobic anticancer drugs. In contrast to the usual SLPs, the new design in this work successfully replaced the conventional hydrophobic tails with β-sheet-forming hydrophilic peptides, which greatly enhances the solubility of precursors and products.
2. Materials and methods
2.1 Materials
Peptide (GNNQQNY-PKKP-GNNQQNY) is a freeze-dried powder purchased from TG peptide Biotechnology Co., Ltd. (Nanjing, China) with a purity greater than 95%. Paclitaxel (PTX, >99%) was purchased from Aladdin's reagent (Shanghai, China). CCK-8 staining solution and Calcein-AM/PI Double Stain Kit was purchased from Yeasen (Shanghai, China). The A549 cells (human non-small cell lung cancer cells) used in this experiment were purchased from ATCC (Manassas, VA, USA).2.2 Preparation of β-nanocages and encapsulating hydrophobic molecules
The GSLPs were accurately weighed and then dissolved in phosphate buffered saline (PBS, pH = 7.4) to a concentration of 1 mM. The dissolved peptide solution was sonicated for 5 min and shaken on a shaking table overnight to form stable β-nanocages.For drug encapsulation, 1 mL of peptide solution (1 mM, pH = 7.4) was prepared using PBS and sonicated for 5 min, then 1 mL of PTX or pyrenes dissolved in chloroform (0.5 mg mL−1) was added into the peptide solution. And the mixtures were emulsified under ultrasonication for 2 min (5 s on and 3 s off for each cycle) with a sonicating probe (30% power) in ice-water bath. Then the emulsified solution was vacuumed under vigorous stirring in vacuum desiccator for 5 h. The obtained solutions then were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min at 4 °C, and the supernatant was collected.
000 rpm for 30 min at 4 °C, and the supernatant was collected.
2.3 Molecular dynamics simulation
AlphaFold2 was used to predict the peptide structure,19 and VMD was used to display its structure,20 and Packmol21 was used to add 10 peptide structures in a water-filled box (10 × 10 × 10 nm3). The calculation was performed using the Gromacs 2019.3 software package.22 The Gromos 54a7 combined force field parameters included in the program was used for the force field, and the simulation temperature of the system was set to 25 °C. The solvent model was the water, and SPC/E model was used at the same time with the chloride ion neutralization system. The periodic boundary mode was used for the simulation. Firstly, the system was optimized for 10000 steps to promote the relaxation of the structure and eliminate the unreasonable conformation of the structure, so that the system energy was at the lowest state, and then both the Berendsen heat bath method and Parrinello–Rahman pressure bath method were used to balance it for 100 ps. In the calculation process, the simulation step size was 2 fs, and the electrostatic interaction was calculated by PME. The LINCS algorithm was used to constrain the bond length of the molecule, and the PME method was selected to calculate the long-range electrostatic interaction. Finally, molecular dynamics simulations were performed for 100 ns for each system, and the simulated trajectories were analyzed using Gromacs.
2.4 Critical assembling concentration (CAC)
Peptide self-assembled nanocages with different concentrations were prepared by dissolving peptides in pure water. The absorbance at 280 nm of peptide solutions with different concentrations was measured on an ultraviolet-visible-near infrared spectrometer (Agilent, USA), and the measured values were plotted against the logarithm of peptide concentrations. The intersection of two lines was determined as the CAC value.2.5 Atomic force microscopy (AFM)
To study the surface structure of peptide nanocages, AFM (Dimension Icon, Bruker) was used to compare the structure of the pure GSLP assemblies with that after encapsulating hydrophobic drugs. 5 μL GSLP or GSLP-drug complex solution (1 mM) were adsorbed on the freshly cleaved mica surface for 3 min. Then the droplets were sucked away and the mica surface was rinsed gently with ∼3 mL ultrapure water. After vacuum drying, the mica surface was scanned by AFM to observe the structural information of β-nanocages.23,242.6 Dynamic light scattering (DLS)
DLS is used to detect the size distribution of β-nanocages in suspension. 1 mL sample was placed into a low-capacity disposable cuvette and equilibrated at 25 °C for 2 min before measurement. The data was collected on a DLS instrument (Zetasizer Nano ZS ZEN 3600). Each sample was measured three times, and the results were averaged from 20–25 measurements.2.7 Transmission electron microscopy (TEM)
10 μL of sample solution was dropped on the surface of a 400-mesh copper grid and left for 5 min, and then the solution was blotted off with a piece of filter paper. Then 10 μL of staining solution (2% phosphotungstic acid) was dropped on the grid and kept staining for 2 min. The dye solution was blotted off with the filter paper, washed once with deionized water, and dried for 12 h. The nanocages were observed by a TEM (F200S, FEI Talos).2.8 Cytotoxicity assay
A549 cells were seeded in 96-well plates with a density of 5 × 103 cells per mL and cultured for 24 h. To compare the nanocage encapsulated PTX (GSLP-PTX) and PTX alone, the cell medium was replaced with a fresh medium containing the same concentration of PTX and the GSLP-PTX solution (pH = 8.09). After 24 h of culture, CCK-8 staining solution was used to stain the cells, and the cells were cultured in the incubator for another 3 h. The OD value was detected at 450 nm using the microplate reader (Synergy H1, BioTek), and the cell survival rate was calculated by the following equation:| Cell viability (%) = (ODtest − ODblank)/(ODcontrol − ODblank) × 100% |
2.9 Fluorescence microscopy
A549 cells were planted into 48-well plates with a density of 5 × 103 cells per mL and cultured overnight. Cell apoptosis was detected using the Calcein-AM/PI Double Stain Kit (live/dead stain kit). Cells were washed three times with 1× assay buffer (by diluting the 10× assay buffer with 1× PBS) and stained with a staining solution in the dark for 20 min. After that, cells were washed three times with 1× assay buffer and imaged using an inverted fluorescence microscope (ZEISS Axio, Germany). Fluorescence images were acquired by ZEISS Axio Vert.A1 software (ZEISS, Germany).3. Results and discussion
3.1 Peptide design and self-assembling simulation
The peptide sequence we designed possesses surfactant-like properties. To ensure its solubility in water, we used two highly soluble lysine residues as the head of the hydrophilic terminals. For the hydrophobic terminals, we employed two antiparallel β-sheet-forming peptide sequences: G7 (GNNQQNY), derived from yeast protein Sup 35, which can form β-sheet nanofibers and achieve a similar effect to conventional hydrophobic ends. The flexible hinge provided by the proline (P) residues connecting the two terminals of the GSLP peptide sequence mimics the turning structure of natural proteins, enabling the two antiparallel peptide sequences G7 to form stable β-sheet conformations in solution. This combination gives rise to the new GSLP sequence (G7-PKKP-G7) (Fig. 1a).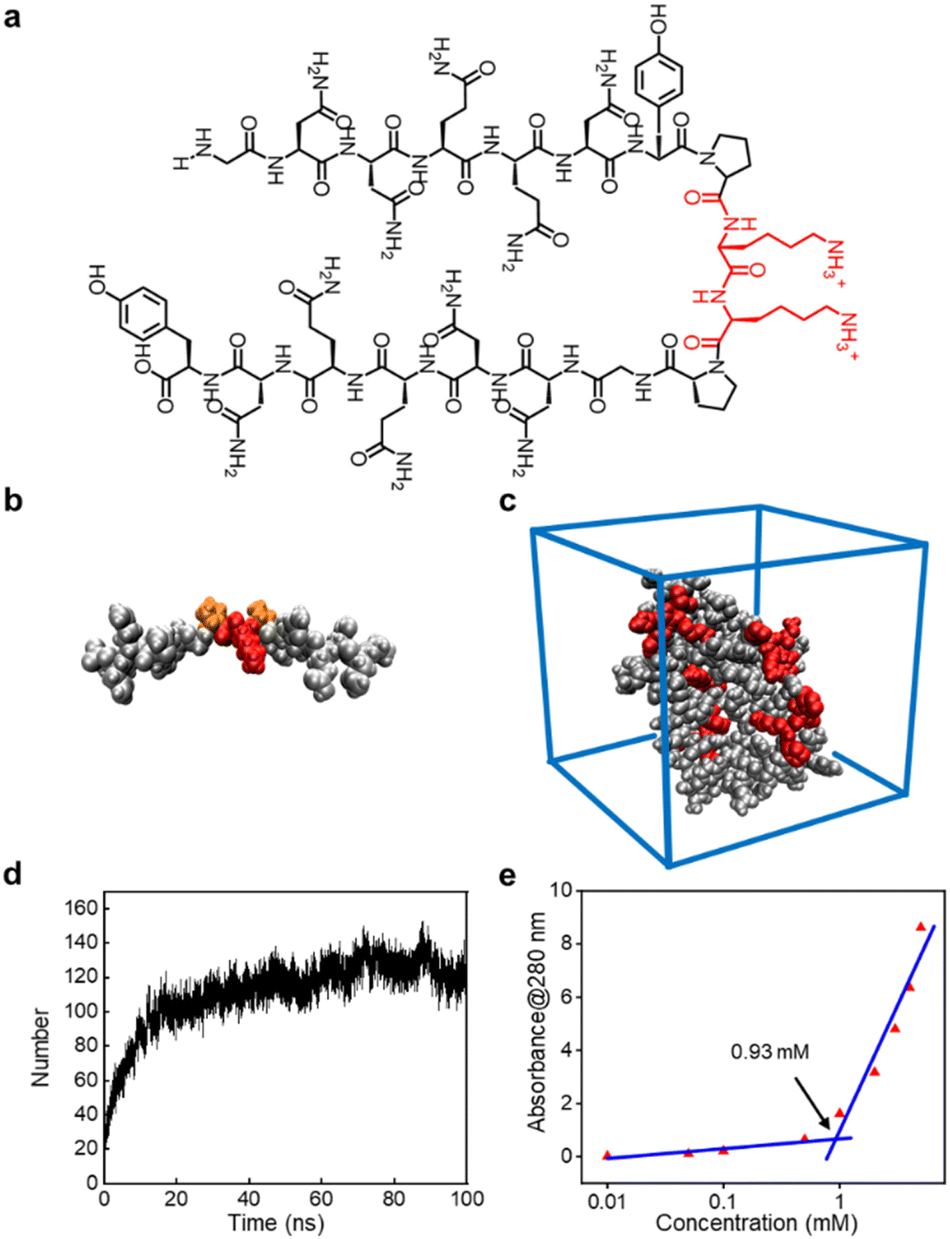 | ||
| Fig. 1 Design and self-assembly of GSLPs. (a) Structural design of the GSLP. Black is the “tail” that can be assembled and folded to form a hydrophobic structure, and red is the “head” of the hydrophilic structure composed of hydrophilic amino acids. (b) The 3D structure of the GSLP model, with the hydrophilic end in red, the hydrophobic tail in black, and the two prolines providing the corner in orange. (c) Molecular dynamics simulation of self-assembly results. (d) The number of hydrogen bonds formed by Gromacs analysis of self-assembly between GSLPs with time. (e) The plot of absorbance of the GSLP at 280 nm versus GSLP concentration. The intersection of the two lines is the CAC of the GSLP, 0.93 mM. | ||
The CD analysis of the peptide solution revealed the presence of β-sheets, which can be attributed to the self-assembly and drug encapsulation. Specifically, the β-sheet content was found to be as high as 49% before encapsulation and increased to 63% after the drug was encapsulated (Fig. S1†). We used AlphaFold2 to predict the secondary structure of the peptide sequence (Fig. 1b) and performed simple molecular dynamics analysis of the predicted structure using Gromacs to visualize the structural state more dynamically. Our analysis verified that the designed peptides have a strong tendency to self-assemble (Fig. 1c). We observed the self-assembly of the peptides through hydrogen bonds between the GSLP molecules by monitoring the number of hydrogen bonds between the GSLPs over time (Fig. 1d).
3.2 GSLPs self-assembling into β-nanocages
The formation of GSLP nanocages with cavity structures depends on their concentrations in solution (Fig. 2a). An easy and fast method to determine the critical assembling concentration (CAC) is by measuring the UV absorbance at a specific wavelength as a function of concentration, which changes with micelle formation. A sudden change in slope above a certain concentration indicates the CAC, as aggregates like micelles and free molecular species contribute to the UV adsorption differently.25 We measured the absorption spectra of GSLP solutions at different concentrations (Fig. S2†) and observed a concentration-dependent maximum peak at 280 nm for all nanostructures (Fig. 1e). The corresponding CAC value of the peptide was around 1 mM, indicating that stable nanocages were more likely to form at this concentration.26 The particle size of the self-assembled GSLPs was 78.36 nm (Fig. 2b), and electron microscopy showed a hollow structure with darker edges and a lighter middle27 (Fig. 2c). AFM images confirmed the hollow “cage” structure on the mica surface with a height difference of about 7.65 nm between the edge and the middle (Fig. 2d and e). TEM and AFM observations suggested that GSLPs self-assemble spontaneously via a concentration-dependent mechanism, where their surfactant nature amplifies their tendency to aggregate during the assembly process, facilitating the formation of stable nanocages. To evaluate the thermal stability of the GSLP nanocages, we increased the temperature of nanocage solution up to 42 °C for 120 minutes, and monitored the cage's size. It turned out that the size of nanocage kept almost same (81.35 nm) as that at 25 °C (Fig. S3†). This result indicates that the cage size did not change significantly compared to that at normal temperature, and suggests that the peptide is stable at the tested temperature range. These properties make GSLP cages an ideal carrier for encapsulated drugs.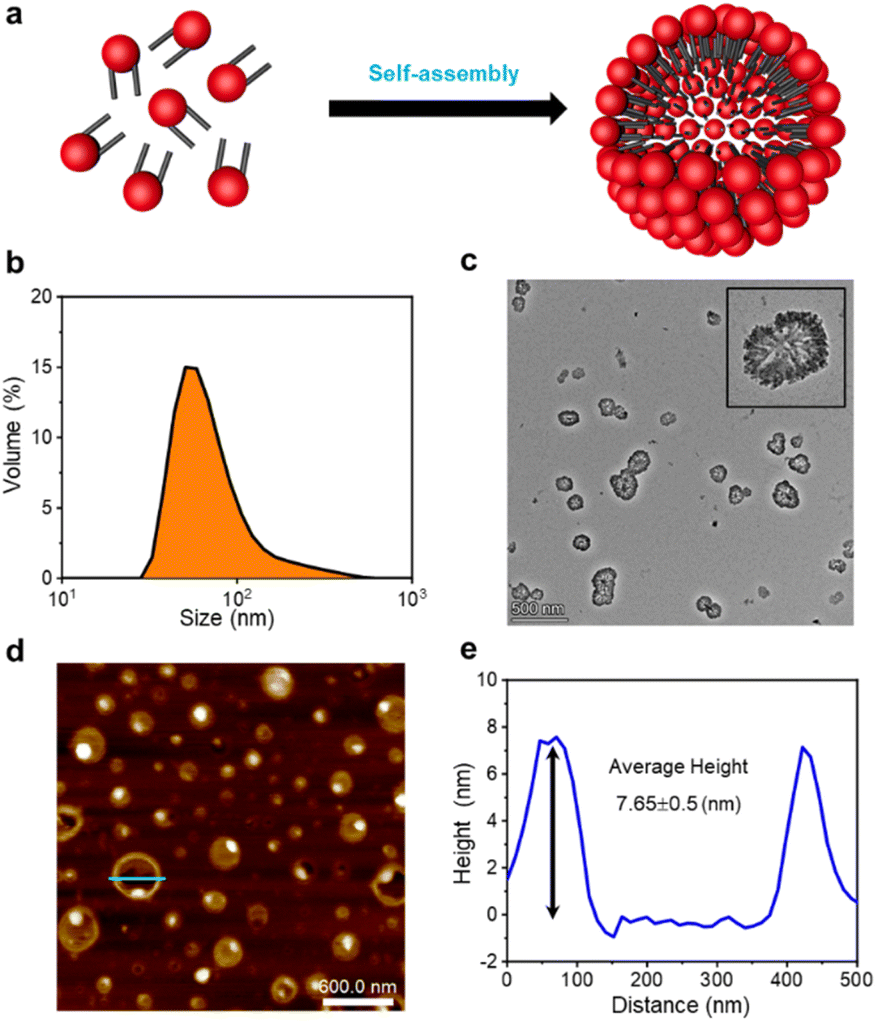 | ||
| Fig. 2 GSLPs self-assembling into β-nanocages. (a) Schematic illustration for GSLP self-assembling into β-nanocages. (b) Hydrodynamic size distribution of GSLP β-nanocages measured by the dynamic light scattering method. TEM (c) and AFM (d) images of self-assembled β-nanocages. The scale bars for TEM and AFM are 500 nm and 600 nm, respectively. The inset figure in (d) is an amplified β-nanocage structure in a vacuum state. (e) Cross-section analysis of the broken hollow nanocage structure as cut by a green line in (e). | ||
3.3 Hydrophobic molecule encapsulation of β-nanocages
After confirming the stable cavity structure of the assembled GSLPs, we investigated their potential for encapsulating hydrophobic molecules (Fig. 3a). To assess the embedding ability of drugs, we analysed the fluorescence spectra, particle size distribution, and morphology of the GSLP nanocages before and after encapsulation of hydrophobic drugs, using pyrene as a model molecule. Pyrene is widely studied due to its strong hydrophobic and fluorescent properties.28 Given that peptides can self-assemble into nanocages with a hydrophobic inner space, which could attract hydrophobic molecules such as pyrene, leading to the increase of nanocage sizes. To test this, we measured the hydrodaynic size of GSLP nanocage alone, which was 78.36 nm, and after co-assembling with pyrene, resulting in a larger size of 117.3 nm (Fig. 3c). The entry of pyrene into the cavity could increase the cage size of the entire structure, supporting our hypothesis. In addition, upon excitation with UV light of 280 nm, a light blue fluorescence is visible in the solution, and the fluorescence spectrum (Fig. S4†) also showed an emission peak at 470 nm, indicating the presence of nanocages containing pyrene. TEM and AFM images also confirmed that the GSLPs self-assembled into a spherical, well-encapsulated nanostructure after encapsulating the pyrene molecule (Fig. 3d and e).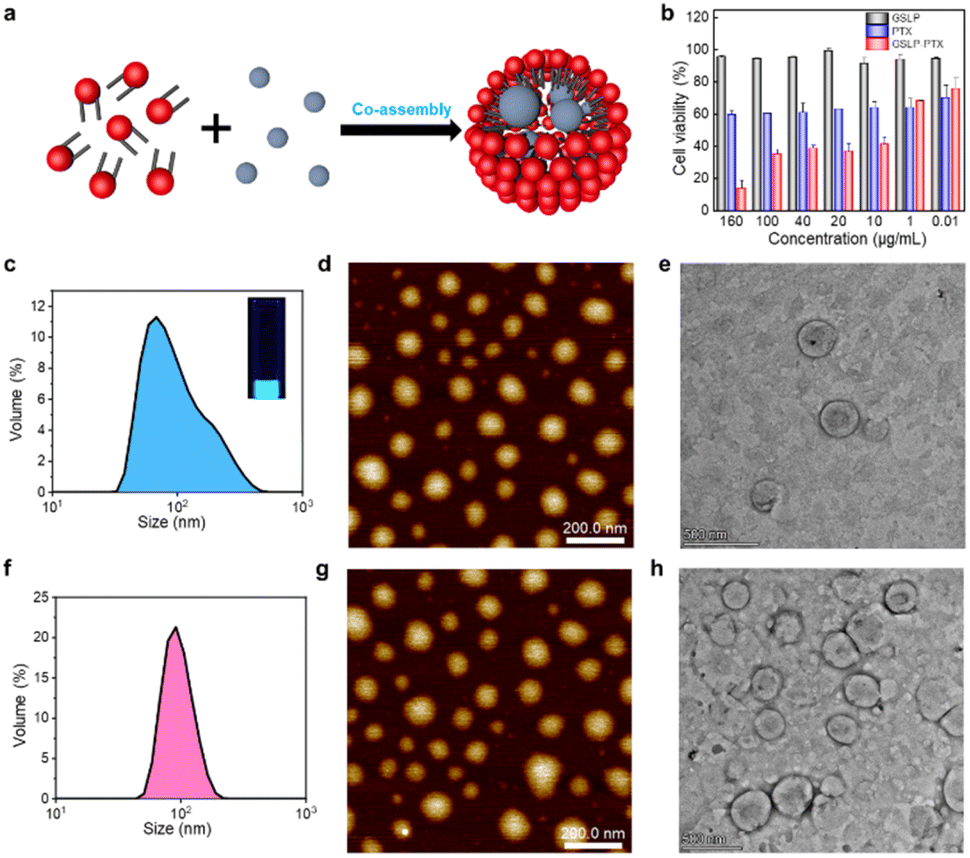 | ||
| Fig. 3 Hydrophobic molecule encapsulation and cytotoxicity of β-nanocages. (a) Schematic diagram of self-assembled β-nanocages encapsulating hydrophobic molecules. Red balls represent hydrophilic heads, black rods are hydrophobic β-tails, and the gray blue balls represent hydrophobic molecules. (b) Histogram comparison of the cytotoxicity to A549 cells. GSLP-PTX represents the β-caged PTX nanostructures. (c and f) Hydrodynamic diameter measurement of β-nanocages after encapsulating hydrophobic molecules such as pyrene (c) and anticancer drug PTX (f). The photograph inset in (c) was taken under 280 nm ultraviolet illumination. AFM (e and h) and TEM (d and g) images of β-nanocages encapsulating hydrophobic molecules such as pyrene (d and e) and anticancer drug PTX (g and h). Scale bars for AFM and TEM images are 200 nm and 500 nm, respectively. | ||
3.4 Cytotoxicity of anticancer drug-encapsulated β-nanocages
Paclitaxel (PTX) is a potent chemotherapeutic drug that has been used to treat a variety of cancers.29 However, its poor water solubility limits its application in the medical field. To improve its water solubility, many methods have been developed. In this study, we aimed to explore the potential of GSLP nanocages for delivering hydrophobic antitumor drugs by encapsulating PTX in the GSLP nanocages, following our successful encapsulation of the signature hydrophobic drug pyrene in GSLP nano microspheres. Like the encapsulated pyrene, we believe that PTX can also be encapsulated by GSLP nanocages. Because the absorption peak of PTX is covered by the absorption peak of the peptides' (280 nm), thus cannot be detected like pyrene. Therefore, we have to use other means to prove the successful encapsulation of PTX by GSLP nanocages. Firstly, we prepared GSLP-PTX nanostructures with an excess of peptide, and no precipitation of PTX was observed. Secondly, we conducted cytotoxicity tests, and found that GSLP alone was not concentration-dependent toxic, while the GSLP-PTX nanostructures exhibited concentration-dependent toxicity. Finally, we observed a change in the particle size of GSLP-PTX nanostructures, which increased to 102.3 nm (Fig. 3f), indicating the entry of PTX molecules into the nanocage cavity. Based on these findings, we concluded that PTX should be successfully encapsulated by GSLP nanocages. AFM images showed that the GSLP nanocages tightly encapsulated paclitaxel to form sphere nanostructures (Fig. 3g). TEM images clearly showed that the vast majority of nanocages successfully wrapped the anticancer drug, with an encapsulation rate of about 93%, while only a few particles were empty (Fig. 3h). These findings demonstrate the potential of GSLP nanocages as an effective carrier for the delivery of hydrophobic drugs such as paclitaxel.We assessed the inhibitory effect of PTX-encapsulated GSLP nanocages and PTX alone on A549 lung cancer cells. Our findings revealed that at concentrations of GSLP-PTX nanostructures ranging from 10–160 μg mL−1, the cell viability was significantly lower than that treated with PTX alone. At the highest concentration of 160 μg mL−1, the cell survival rate of PTX alone was approximately 60%, while that of PTX-encapsulated GSLP nanostructures dropped to 14% (Fig. 3b). The low water solubility of PTX limits its efficacy in cancer treatment, but encapsulation in GSLP nanocages improves its solubility and concentration-dependent cytotoxicity. Furthermore, GSLPs are biocompatible and do not exhibit cytotoxicity, facilitating drug uptake by cells. The GSLP nanocage did not hinder its drug release inside the cell, and the cytotoxic effect of the GSLP drug was confirmed by inverted fluorescence microscopy imaging (green for live cells, red for dead cells) (Fig. 4). These findings suggest that GSLPs are non-toxic carriers that can effectively deliver hydrophobic drugs to tumor cells.
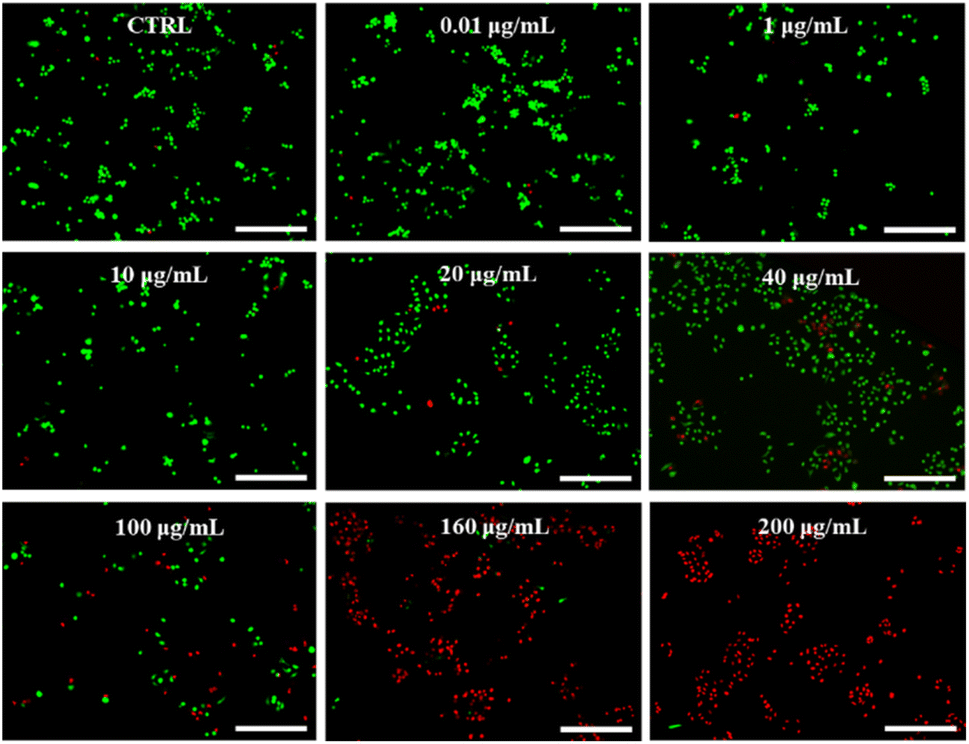 | ||
| Fig. 4 The dose-dependent anticancer capability of PTX-encapsulated β-nanocages. A549 cells treated with different concentrations of β-cage PTX. The CTRL was 0 μg mL−1, as shown in figures. All scale bars were 50 μm. | ||
4. Conclusions
In summary, inspired by the β-sheet-forming proteins in amyloidosis, here we provide a novel strategy to use specific β-sheet-forming hydrophilic GSLP sequence G7 to replace the conventional hydrophilic ones in the design of GSLPs. With this replacement, the whole GSLPs show much better water solubility than those completely using hydrophobic amino acid resides e.g., alanine (A), and the assembled nanostructure, i.e., nanocages can be merely well-controlled by the critical assembling concentration, reaching much higher efficiency for both drug loading and encapsulation, since the inner diameter of GSLP nanocages can be greater than ∼400 nm. On the other hand, the positively charged lysine (K) residues and β-turn component proline (P) together formed a cellular membrane-accessible surface for the β-nanocage, which also holds great potential for antimicrobial applications. By demonstrating the encapsulation of a model hydrophobic anticancer drug paclitaxel, we found that this strategy can be used for rational design the effective and suitable drug delivery platform and will find wider clinical applications, such as incorporation of tumor-targeting peptide RGD sequences and as antimicrobial agents.12,30Author contributions
Conceptualization, Feng Zhang and Liping Wang and Jun Guo; methodology, Feng Gao; investigation, Feng Gao, Qiankang Si, Xingkun Niu and Shihao Liu; data curation, Feng Gao; writing-original draft preparation, Feng Gao and Jun Guo; writing-review and editing, Feng Zhang and Liping Wang; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Key Research and Development Program of China (No. 2021YFA1200402), the National Nature Science Foundation of China (No. 32271298, T2241002), and Wenzhou Institute of the University of Chinese Academy of Sciences (WIUCASQD2021003) for support.Notes and references
- L. Z. Benet, F. Broccatelli and T. I. Oprea, AAPS J., 2011, 13, 519–547 CrossRef CAS PubMed.
- C. Fink, D. Sun, K. Wagner, M. Schneider, H. Bauer, H. Dolgos, K. Mader and S. A. Peters, Clin. Pharmacol. Ther., 2020, 107, 650–661 CrossRef CAS PubMed.
- K. Rajpoot, M. Tekade, N. Sreeharsha, M. C. Sharma and R. K. Tekade, in The Future of Pharmaceutical Product Development and Research, ed. R. K. Tekade, Academic Press, 2020, pp. 109–144 Search PubMed.
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, ISRN Pharm., 2012, 2012, 195727 Search PubMed.
- E. Larraneta, S. Stewart, M. Ervine, R. Al-Kasasbeh and R. F. Donnelly, J. Funct. Biomater., 2018, 9, 13–33 CrossRef PubMed.
- R. Narayanaswamy and V. P. Torchilin, Molecules, 2019, 24, 603 CrossRef PubMed.
- A. Kulshrestha, P. S. Gehlot and A. Kumar, J. Mater. Chem. B, 2020, 8, 3050–3057 RSC.
- D. Irby, C. Du and F. Li, Mol. Pharm., 2017, 14, 1325–1338 CrossRef CAS PubMed.
- H. Shrestha, R. Bala and S. Arora, J. Pharm., 2014, 2014, 801820 Search PubMed.
- Y. Chen, C. Tang, J. Zhang, M. Gong, B. Su and F. Qiu, Int. J. Nanomed., 2015, 10, 847–858 Search PubMed.
- U. Anand, A. Bandyopadhyay, N. K. Jha, J. M. Pérez de la Lastra and A. Dey, BioFactors, 2022, 49, 251–269 CrossRef PubMed.
- C. H. Chen, T. H. Weng, K. Y. Huang, H. J. Kao, K. W. Liao and S. L. Weng, Int. J. Med. Sci., 2022, 19, 2008–2021 CrossRef PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- A. C. Lee, J. L. Harris, K. K. Khanna and J. H. Hong, Int. J. Mol. Sci., 2019, 20, 2383 CrossRef PubMed.
- S. Tayyab, S. E. Sam, M. Z. Kabir, N. F. W. Ridzwan and S. B. Mohamad, Spectrochim. Acta, Part A, 2019, 214, 199–206 CrossRef CAS PubMed.
- F. Bugli, F. Massaro, F. Buonocore, P. R. Saraceni, S. Borocci, F. Ceccacci, C. Bombelli, M. Di Vito, R. Marchitiello, M. Mariotti, R. Torelli, M. Sanguinetti and F. Porcelli, Int. J. Mol. Sci., 2022, 23, 2164 CrossRef CAS PubMed.
- C. Karavasili, M. Spanakis, D. Papagiannopoulou, I. S. Vizirianakis, D. G. Fatouros and S. Koutsopoulos, J. Pharm. Sci., 2015, 104, 2304–2311 CrossRef CAS PubMed.
- F. Peng, Y. Chen, J. Liu, Z. Xing, J. Fan, W. Zhang and F. Qiu, J. Colloid Interface Sci., 2021, 591, 314–325 CrossRef CAS PubMed.
- P. Cramer, Nat. Struct. Mol. Biol., 2021, 28, 704–705 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- Y. Cai, B. Liang, S. Chen, Q. Zhu, T. Tu, K. Wu, Q. Cao, L. Fang, X. Liang and X. Ye, Biosens. Bioelectron., 2020, 165, 112408 CrossRef CAS PubMed.
- Y.-X. Bao, M. Yuan, Q. Du, Y.-B. Li, J.-Y. Gao, A. J. Khan and F. Zhang, Nucl. Sci. Tech., 2019, 30, 117 CrossRef.
- T. Lutzenburg, N. Burdina, M. S. Scholz and I. Neundorf, Pharmaceutics, 2021, 13, 2075 CrossRef PubMed.
- S. Shimizu and N. Matubayasi, Phys. Chem. Chem. Phys., 2021, 23, 8705–8716 RSC.
- V. Lapeyre, N. Renaudie, J. F. Dechezelles, H. Saadaoui, S. Ravaine and V. Ravaine, Langmuir, 2009, 25, 4659–4667 CrossRef CAS PubMed.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- F. Y. Alqahtani, F. S. Aleanizy, E. El Tahir, H. M. Alkahtani and B. T. AlQuadeib, Profiles Drug Subst., Excipients, Relat. Methodol., 2019, 44, 205–238 Search PubMed.
- C. Zhang, Y. Gao, N. Yang, T. You, H. Chen and P. Yin, Microchim. Acta, 2018, 185, 90 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01950k |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2023 |