
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew pyrazolopyridine and pyrazolothiazole-based compounds as anti-proliferative agents targeting c-Met kinase inhibition: design, synthesis, biological evaluation, and computational studies†
Zahra M. Alamshanya,
Eman M. Algamdia,
Ismail M. M. Othmanb,
Manal M. Anwar *c and
Eman S. Nossier*de
*c and
Eman S. Nossier*de
aDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O. Box 42805, Jeddah 21551, Saudi Arabia
bDepartment of Chemistry, Faculty of Science, Al-Azhar University, Assiut 71524, Egypt
cDepartment of Therapeutic Chemistry, National Research Centre, Dokki, Cairo 12622, Egypt. E-mail: manal.hasan52@live.com
dDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo 11754, Egypt. E-mail: dr.emannossier@gmail.com
eThe National Committee of Drugs, Academy of Scientific Research and Technology, Cairo, 11516, Egypt
First published on 25th April 2023
Abstract
c-Met tyrosine kinase plays a key role in the oncogenic process. Inhibition of the c-Met has emerged as an attractive target for human cancer treatment. This work deals with the design and synthesis of a new set of derivatives bearing pyrazolo[3,4-b]pyridine, pyrazolo[3,4-b]thieno[3,2-e]pyridine, and pyrazolo[3,4-d]thiazole-5-thione scaffolds, 5a,b, 8a–f, and 10a,b, respectively, utilizing 3-methyl-1-tosyl-1H-pyrazol-5(4H)-one (1) as a key starting material. All the new compounds were evaluated as antiproliferative agents against HepG-2, MCF-7, and HCT-116 human cancer cell lines utilizing 5-fluorouracil and erlotinib as two standard drugs. Compounds 5a,b and 10a,b represented the most promising cytotoxic activity of IC50 values ranging from 3.42 ± 1.31 to 17.16 ± 0.37 μM. Both 5a and 5b showed the most cytotoxicity and selectivity toward HepG-2, with IC50 values of 3.42 ± 1.31 μM and 3.56 ± 1.5 μM, respectively. The enzyme assay demonstrated that 5a and 5b had inhibition potency on c-Met with IC50 values in nanomolar range of 4.27 ± 0.31 and 7.95 ± 0.17 nM, respectively in comparison with the reference drug cabozantinib (IC50; 5.38 ± 0.35 nM). The impact of 5a on the cell cycle and apoptosis induction potential in HepG-2 and on the apoptotic parameters; Bax, Bcl-2, p53, and caspase-3 was also investigated. Finally, the molecular docking simulation of the most promising derivatives 5a and 5b was screened against c-Met to investigate the binding patterns of both compounds in the active site of the c-Met enzyme. In silico ADME studies were also performed for 5a and 5b to predict their physicochemical and pharmacokinetic characteristics.
1. Introduction
C-Mesenchymal epithelial transition factor (c-Met) is a tyrosine kinase receptor encoded by the proto-oncogene MET. It is a high-affinity receptor for hepatocyte growth factor (HGF).1 When HGF binds to c-Met, it causes c-Met to dimerize, resulting in the phosphorylation of numerous tyrosine residues, which activates a number of intricate signalling pathways involved in cell growth, differentiation, migration, apoptosis, induction of cell polarity, scattering, angiogenesis and invasion.2–4 Accordingly, dysregulation of c-Met/HGF signalling pathways that occurred due to abnormal c-Met expression or their stimulation via mutation, amplification, translocation, or HGF stimulation were confirmed to be related to the development and/or progression of a wide range of human cancers.5,6 Recently, the pharmacological inhibition of c-Met activity is considered as one of the most important strategic path ways in cancer therapy.7–9Many biological studies have exhibited that there are two groups of c-Met kinase inhibitors based on their structural features and their binding modes with the active site: group I inhibitors and group II inhibitors.10–12 The inhibitors of type I are ATP analogues and compete with ATP to bind to the extremely conservative hinge region of c-Met kinase. On the other hand, type II inhibitors interact with the same region as type I inhibitors and interact with the allosteric site located beyond the entrance of the active site of c-Met via hydrogen and hydrophobic bindings. According to recent reports, some mutations close to the c-Met active site may result in type I inhibitor resistance. Consequently, type II inhibitors may be more successful than type I inhibitors against mutations close to the c-Met active site due to their multi-binding nature.2,13–15 Fig. 1 exhibits different compounds possessing different scaffolds as type II c-Met inhibitors, where some of them have been launched or are in clinical trials.16–22
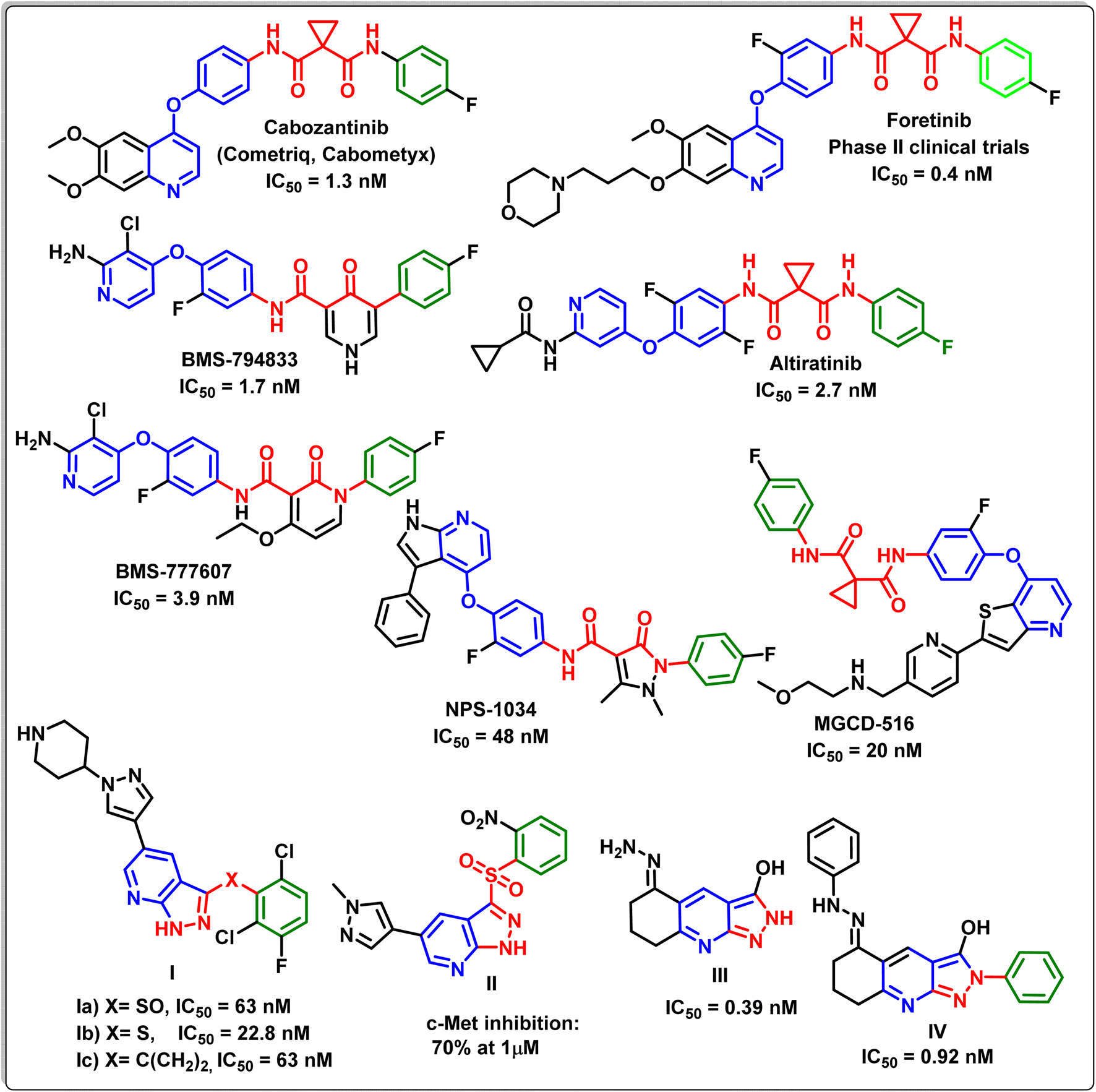 | ||
| Fig. 1 Different examples of type II c-Met inhibitors bearing different scaffolds. | ||
Recent studies showed that the structures of type II c-Met inhibitors can be divided into three units (A–C).13 Unit A of c-Met inhibitors is usually a phenyl or p-substituted phenyl moiety, which generates hydrophobic interactions with the hydrophobic pocket of the c-Met protein. Unit B is a linker bridge that could be a linear chain or a heterocyclic ring and should possess hydrogen-bond donors or acceptors. On the other hand, unit C exhibits a high degree of structural diversity, such as pyridine or its analogues attached to a different substituted phenyl moiety, resulting in the creation of hydrogen bonds and π–π stacking interactions with the backbone of the c-Met enzyme23–25 (Fig. 2).
 | ||
| Fig. 2 The design strategy and the molecular structures of the new target derivatives. | ||
Considered one of the most promising privileged scaffolds is pyrazolo[3,4-b]pyridine. Many small compounds with pyrazolo[3,4-b]pyridine bases have demonstrated a clinical value in a number of medical research disciplines. It is interesting to note that pyrazolo[3,4-b]pyridine derivatives have been linked to a wide range of pharmacological actions.26
Of particular interest, the anticancer activity of pyrazolo[3,4-b]pyridines is one of the most significant pharmacological actions. It has been reported that the pyrazolo[3,4-b]pyridine derivatives can exert their antitumor impact via various mechanisms such as cell growth inhibition,27 apoptosis induction,28 angiogenesis inhibition29 as well as by targeting diverse tyrosine, serine/threonine and lipid kinases including for example adenosine 5′-monophosphate-activated protein kinase (AMPK),30 B-Raf kinase,31,32 epidermal growth factor receptor kinase (EGFR),32 checkpoint kinase 1 (CHK1),33 c-Met kinase,34 fibroblast growth factor receptor (FGFR),35 PIM-1 kinase,36 glycogen synthase kinase-3 (GSK-3),37 and cyclin-dependent kinases (CDKs).38
Liu et al.,39 have designed and synthesized new pyrazolo[3,4-b]pyridine derivatives Ia–c, with a sulfoxide, sulfur, and methylene as a linker. The enzyme assay and cell assay in vitro exhibited the strong c-Met kinase inhibitory activity of the latter derivatives with IC50 values in the nano range. Also Ma et al.,40 have synthesized the metabolically stable pyrazolo[3,4-b]pyridine derivative II, which inhibited c-Met at concentration of 1 μM. Furthermore, it has been reported that both compounds III and IV exhibited potent c-Met inhibitory activity of IC50 values of 0.39 and 0.92 nM, respectively.41
Cancer continues to be a major public health concern on a global scale.42,43 Over 10 million people die from cancer each year, and the number of cases has increased dramatically worldwide. Although surgery, chemotherapy, and radiation (RT) are currently the primary methods for treating cancer, there have been many instances in which these therapies have had serious negative effects on healthy tissues or have failed to prevent tumor spread or recurrence.44,45 Accordingly, with the aim of exploring new effective type II c-Met inhibitors with minimal side effects and based on the structure–activity relationships (SARs) and structural characteristic pharmacophores mentioned earlier, 1-tosyl-pyrazolo[3,4-b]pyridine fragments were introduced into block C aiming to create hydrogen bonds and π–π stacking interaction with c-Met backbone and different substituents were tested on block A to evaluate their influences on the c-Met inhibitory activity. The linker bridge which is a part of the heterocyclic pyridine ring (NH-CS-C-CN) in block B was retained to form vital hydrogen bonds with the receptor residues.
Several studies have found that the thiazole heterocycle has intriguing anti-cancer activity against various human cancer cells via diverse mechanisms such as apoptosis, DNA intercalation, and enzyme inhibition, including epidermal growth factor receptors and B-RAF kinase.47–50 Thus, bioisostere replacement of the pyrazolo[3,4-b]pyridine scaffold with the pyrazolo[3,4-d]thiazole scaffold was also studied to see how it affected anticancer and c-Met inhibitory activity.
All the target compounds were evaluated against three cancer cell lines; the liver cancer cell line (HepG-2), breast cancer cell line (MCF-7), and colorectal cancer cell line (HCT), in addition to their cytotoxic activity against the normal WI-38 cell line. The most promising candidates were further assessed as c-Met kinase inhibitors. Extra investigations were also carried out to investigate the mechanistic pathways of the antiproliferative activity of the most potent compounds, such as cell cycle analysis, apoptosis, and the impacts of these derivatives on the oncogenic markers; Bax, Bcl-2, p53, and caspase-3. Moreover, molecular docking research was carried out on the most potent c-Met inhibitors to highlight and justify their modes of action. In silico ADMET prediction was also performed for the potent compounds to investigate their drug-likeness characteristics.
2. Experimental
2.1. Chemistry
The instrumental devices used for measuring the melting points, spectral data (IR, Mass, 1H NMR and 13C NMR) and elemental analyses are provided in details in ESI.†2.1.1.1. 3-Methyl-4-phenyl-6-thioxo-1-tosyl-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile (5a). Yield: 88%; yellow crystals; mp 222–224 °C; FT-IR (KBr) (νmax/cm−1): 3306 (NH), 3058 (CH-aromatic), 2979 (CH-aliphatic), 2215 (CN); 1H NMR (300 MHz, DMSO-d6) δ = 2.24 (s, 3H, CH3), 2.36 (s, 3H, CH3), 7.32–7.86 (m, 9H, Ar-H), 11.76 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δ: 14.1 (CH3), 22.4 (CH3), 101.9, 106.7, 115.6, 127.1, 128.1, 128.2, 128.7, 131.8, 134.8, 137.9, 140.4, 140.7, 151.9, 162.9 (Ar-C), 179.6 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S); EI-MS m/z (%): 420 [M+] (41.16); anal. calcd for C21H16N4O2S2 (420.51): C, 59.98; H, 3.84; N, 13.32; S, 15.25%; found: C, 59.76; H, 3.65; N, 13.53; S, 15.47%.
S); EI-MS m/z (%): 420 [M+] (41.16); anal. calcd for C21H16N4O2S2 (420.51): C, 59.98; H, 3.84; N, 13.32; S, 15.25%; found: C, 59.76; H, 3.65; N, 13.53; S, 15.47%.
2.1.1.2. 4-(4-Chlorophenyl)-3-methyl-6-thioxo-1-tosyl-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-5- carbonitrile (5b). Yield: 85%; yellow crystals; mp 217–219 °C; FT-IR (KBr) (νmax/cm−1): 3340 (NH), 3054 (CH-arom.), 2943 (CH-aliph.), 2208 (CN); 1H NMR (300 MHz, DMSO-d6) δ = 2.33 (s, 3H, CH3), 2.62 (s, 3H, CH3), 6.76–6.87 (2d, J = 7.5 Hz, 2H, Ar-H), 7.05–7.08 (2d, J = 7.5 Hz, 2H, Ar-H), 7.41–7.43 (2d, J = 7.5 Hz, 2H, Ar-H), 7.81–7.84 (2d, J = 7.5 Hz, 2H, Ar-H), 11.58 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δ: 12.1 (CH3), 21.8 (CH3), 100.2, 108.2, 115.1, 126.1, 127.7, 128.3, 130.4, 133.5, 134.1, 136.0, 138.1, 140.3, 149.5, 161.3 (Ar-C), 178.5 (C
S); EI-MS m/z (%): 4454 [M+] (30.21); anal. calcd for C21H15Cl N4O2S2 (454.95): C, 55.44; H, 3.32; Cl, 7.79; N, 12.31; S, 14.10%; found: C, 55.32; H, 3.54; Cl, 7.58; N, 12.53; S, 14.31%.
2.1.2.1. 5-Amino-3-methyl-4-phenyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridine-6-carboxamide (8a). Yield: 67%; yellow crystals; mp 272–274 °C; FT-IR (KBr) (νmax/cm−1): 3422, 3339, 3315, 3148 (2NH2), 3081 (CH-aromatic), 2921 (CH-aliphatic), 1680 (C
O); 1H NMR (300 MHz, DMSO-d6) δ: 2.02 (s, 3H, CH3), 2.26 (s, 3H, CH3), 5.92 (s, 2H, NH2, D2O exchangeable), 6.83–7.84 (m, 11H, Ar-H + NH2); 13C NMR (100 MHz, DMSO-d6) δ: 13.5 (CH3), 21.6 (CH3), 110.1, 122.3, 124.0, 126.0, 126.9, 127.7, 129.4, 130.5, 130.9, 134.5, 134.8, 138.1, 138.7, 155.7, 159.2 (aromatic-C), 164.6 (CO); EI-MS m/z (%): 477 [M+] (30.51); anal. calcd for C23H19N5O3S2 (477.56): C, 57.85; H, 4.01; N, 14.66; S, 13.43%; found: C, 57.63; H, 4.22; N, 14.87; S, 13.65%.
2.1.2.2. 1-(5-Amino-3-methyl-4-phenyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridin-6-yl)ethan-1-one (8b). Yield: 70%; yellow crystals; mp 287–289 °C; FT-IR (KBr) (νmax/cm−1): 3421, 3279 (NH2), 3030 (CH-aromatic), 2952 (CH-aliphatic), 1692 (C
O); 1H NMR (300 MHz, DMSO-d6) δ: 2.06 (s, 3H, CH3), 2.22 (s, 3H, CH3), 2.63 (s, 3H, CH3), 6.38 (s, 2H, NH2, D2O exchangeable), 7.20–7.71 (m, 9H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 15.8 (CH3), 22.1 (CH3), 25.5 (CH3), 111.3, 120.1, 124.1, 124.3, 128.6, 128.7, 129.5, 133.8, 133.9, 135.0, 135.7, 139.7, 142.3, 148.2, 157.7, 157.9 (aromatic-C), 182.4 (CO); EI-MS m/z (%): 476 [M+] (35.31); anal. calcd for C24H20N4O3S2 (476.57): C, 60.49; H, 4.23; N, 11.76; S, 13.46%; found: C, 60.71; H, 4.45; N, 11.54; S, 13.67%.
2.1.2.3. 5-Amino-4-(4-chlorophenyl)-3-methyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridine-6-carbonitrile (8c). Yield: 71%; yellow crystals; mp 276–278 °C; FT-IR (KBr) (νmax/cm−1): 3405, 3319 (NH2), 3057 (CH-aromatic), 2965 (CH-aliphatic), 2220 (CN); 1H NMR (300 MHz, DMSO-d6) δ: 2.12 (s, 3H, CH3), 2.35 (s, 3H, CH3), 6.45 (s, 2H, NH2, D2O exchangeable), 7.14–7.17 (2d, J = 7.5 Hz, 2H, Ar-H), 7.21–7.24 (2d, J = 7.5 Hz, 2H, Ar-H), 7.33–7.35 (2d, J = 7.5 Hz, 2H, Ar-H), 7.53–7.55 (2d, J = 7.5 Hz, 2H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 11.6 (CH3), 25.0 (CH3), 102.3, 107.9, 113.6, 121.0, 126.9, 127.9, 128.7, 130.4, 130.8, 133.4, 133.9, 136.5, 138.1, 142.1, 149.4, 156.1, 159.5 (aromatic-C); EI-MS m/z (%): 493 [M+] (30.31); anal. calcd for C23H16Cl N5O2S2 (493.99): C, 55.92; H, 3.26; Cl, 7.18; N, 14.1 8; S, 12.98%; found: C, 55.70; H, 3.74; Cl, 7.39; N, 14.38; S, 12.76%.
2.1.2.4. Ethyl 5-amino-4-(4-chlorophenyl)-3-methyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridine-6-carboxylate (8d). Yield: 72%; yellow crystals; mp 267–269 °C; FT-IR (KBr) (νmax/cm−1): 3346, 2294 (NH2), 3068 (CH-aromatic), 2976 (CH-aliphatic), 1720 (C
O); 1H NMR (300 MHz, DMSO-d6) δ: 1.28 (t, J = 7.5 Hz, 3H, OCH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 2.04 (s, 3H, CH3), 2.47 (s, 3H, CH3), 4.27 (q, J = 7.5 Hz, 2H, OC2CH3), 6.24 (s, 2H, NH2, D2O exchangeable), 6.92–6.94 (2d, J = 7.5 Hz, 2H, Ar-H), 7.32–7.35 (2d, J = 7.5 Hz, 2H, Ar-H), 7.50–7.53 (2d, J = 7.5 Hz, 2H, Ar-H), 7.54–7.57 (2d, J = 7.5 Hz, 2H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 12.2 (OCH2
3), 2.04 (s, 3H, CH3), 2.47 (s, 3H, CH3), 4.27 (q, J = 7.5 Hz, 2H, OC2CH3), 6.24 (s, 2H, NH2, D2O exchangeable), 6.92–6.94 (2d, J = 7.5 Hz, 2H, Ar-H), 7.32–7.35 (2d, J = 7.5 Hz, 2H, Ar-H), 7.50–7.53 (2d, J = 7.5 Hz, 2H, Ar-H), 7.54–7.57 (2d, J = 7.5 Hz, 2H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 12.2 (OCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 12.6 (CH3), 22.7 (CH3), 60.2 (OH2CH3), 110.3, 119.1, 123.7, 126.8, 127.9, 129.4, 130.6, 130.7, 134.3, 134.8, 136.5, 138.8, 146.2, 153.1 (aromatic-C), 160.7 (CO); EI-MS m/z (%): 541 [M+] (45.25); anal. calcd for C25H21ClN4O4S2 (541.04): C, 55.50; H, 3.91; Cl, 6.55; N, 10.36; S, 11.85%; found: C, 55.71; H, 3.69; Cl, 6.77; N, 10.58; S, 11.64%.
H3), 12.6 (CH3), 22.7 (CH3), 60.2 (OH2CH3), 110.3, 119.1, 123.7, 126.8, 127.9, 129.4, 130.6, 130.7, 134.3, 134.8, 136.5, 138.8, 146.2, 153.1 (aromatic-C), 160.7 (CO); EI-MS m/z (%): 541 [M+] (45.25); anal. calcd for C25H21ClN4O4S2 (541.04): C, 55.50; H, 3.91; Cl, 6.55; N, 10.36; S, 11.85%; found: C, 55.71; H, 3.69; Cl, 6.77; N, 10.58; S, 11.64%.
2.1.2.5. 1-(5-Amino-4-(4-chlorophenyl)-3-methyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridin-6-yl)ethan-1-one (8e). Yield: 65%; yellow crystals; mp 281–283 °C; FT-IR (KBr) (νmax/cm−1): 3423, 3371 (NH2), 3028 (CH-aromatic), 2981 (CH-aliphatic), 1685 (C
O); 1H NMR (300 MHz, DMSO-d6) δ: 2.19 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.78 (s, 3H, CH3), 6.25 (s, 2H, NH2, D2O exchangeable), 7.02–7.04 (2d, J = 7.5 Hz, 2H, Ar-H), 7.19–7.21 (2d, J = 7.5 Hz, 2H, Ar-H), 7.42–7.44 (2d, J = 7.5 Hz, 2H, Ar-H), 7.65–7.68 (2d, J = 7.5 Hz, 2H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 16.7 (CH3), 22.1 (CH3), 27.1 (CH3), 111.0, 120.9, 124.1, 127.7, 127.9, 133.3, 133.6, 135.9, 136.8, 137.01, 137.04, 140.7, 144.7, 156.0, 158.8 (aromatic-C), 181.8 (CO); EI-MS m/z (%): 511 [M+] (35.11); anal. calcd for C24H19ClN4O3S2 (511.02): C, 56.41; H, 3.75; Cl, 6.94; N, 10.96; S, 12.55%; found: C, 56.63; H, 3.95; Cl, 6.73; N, 10.75; S, 12.78%.
2.1.2.6. 5-Amino-4-(4-chlorophenyl)-3-methyl-1-tosyl-1H-pyrazolo[3,4-b]thieno[3,2-e]pyridine-6-carboxylic acid (8f). Yield: 62%; yellow crystals; mp 264–266 °C; FT-IR (KBr) (νmax/cm−1): 3455 (OH), 3406, 3390 (NH2), 3057 (CH-aromatic), 2928 (CH-aliphatic), 1688 (C
O); 1H NMR (300 MHz, DMSO-d6) δ: 2.03 (s, 3H, CH3), 2.37 (s, 3H, CH3), 6.29 (s, 2H, NH2, D2O exchangeable), 6.85–6.87 (2d, J = 7.2 Hz, 2H, Ar-H), 7.04–7.06 (2d, J = 7.2 Hz, 2H, Ar-H), 7.72–7.74 (2d, J = 7.2 Hz, 2H, Ar-H), 7.83–7.85 (2d, J = 7.2 Hz, 2H, Ar-H), 11.78 (s, 1H, OH, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δ: 12.0 (CH3), 20.1 (CH3), 110.4, 118.3, 123.3, 127.6, 27.7, 128.5, 130.1, 131.2, 131.9, 135.7, 139.75, 139.76, 142.3, 156.2, 158.6 (aromatic-C), 165.5 (CO); EI-MS m/z (%): 512 [M+] (40.25); anal. calcd for C23H17ClN4O4S2 (512.99): C, 53.85; H, 3.34; Cl, 6.91; N, 10.92; S, 12.50%; found: C, 53.64; H, 3.56; Cl, 6.70; N, 10.69; S, 12.73%.
2.1.3.1. 3,6-Dimethyl-1-tosyl-1,6-dihydro-5H-pyrazolo[3,4-d]thiazole-5-thione (10a). Yield: 59%; brown crystals; mp 291–293 °C; FT-IR (KBr) (νmax/cm−1): 3037 (CH-aromatic), 2972 (CH-aliphatic), 1276 (C
S); 1H NMR (300 MHz, DMSO-d6) δ: 2.18 (s, 3H, CH3), 2.37 (s, 3H, CH3), 2.99 (s, 3H, CH3), 7.23–7.25 (2d, J = 7.2 Hz, 2H, Ar-H), 7.79–7.81 (2d, J = 7.2 Hz, 2H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 17.8 (CH3), 21.6 (CH3), 31.5 (CH3), 102.2, 127.1, 130.0, 134.5, 136.3, 137.8, 143.4, 154.6 (aromatic-C), 182.1 (CS); EI-MS m/z (%): 339 [M+] (30.29); anal. calcd for C13H13N3O2S3 (339.46): C, 46.00; H, 3.86; N, 12.38; S, 28.34%; found: C, 46.21; H, 3.64; N, 12.59; S, 28.57%.
2.1.3.2. 3-Methyl-6-phenyl-1-tosyl-1H-pyrazolo[3,4-d]thiazole-5(6H)-thione (10b). Yield: 61%; brown crystals; mp 295–297 °C; FT-IR (KBr) (νmax/cm−1): 3045 (CH-aromatic), 2964 (CH-aliphatic), 1273 (C
S); 1H NMR (300 MHz, DMSO-d6) δ: 2.01 (s, 3H, CH3), 2.82 (s, 3H, CH3), 7.05–7.89 (m, 9H, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ: 10.3 (CH3), 23.7 (CH3), 101.5, 127.3, 127.7, 128.01, 128.04, 130.8, 132.1, 132.4, 138.2, 140.2, 155.0 (aromatic-C), 180.1 (CS); anal. calcd for C18H15N3O2S3 (401.53): C, 53.84; H, 3.77; N, 10.47; S, 23.96%; found: C, 53.63; H, 3.56; N, 10.68; S, 23.73%.
2.2. Biological assays
2.3. Computational studies
3. Results and discussion
3.1. Chemistry
The new target compounds were synthesized according to the synthetic pathways outlined in Schemes 1–3. Their structures were confirmed based on their elemental analyses and spectral data (ESI†). According to Scheme 1, the starting compound 3-methyl-1-tosyl-1H-pyrazol-5(4H)-one (1) was condensed with different arylidine cyanothioacetamide derivatives, namely, 2-cyano-3-phenylprop-2-enethioamide and 3-(4-chlorophenyl)-2-cyanoprop-2-enethioamide 2a and 2b, respectively, in absolute ethanol containing a catalytic amount of piperidine, as a basic medium to afford the corresponding thioxopyrazolo[3,4-b]pyridines 5a,b (Scheme 1). IR spectra of compounds 5a,b were characterized by the appearance of absorption peaks in the ranges 3340–3306 and 2215–2208 cm−1 related to NH and CN groups, respectively. Also, 1H NMR spectra of 5a,b showed singlet signals at δ = 2.24–2.70 ppm attributed to the CH3 moieties, the appearance of different multiplet signals at δ = 6.70–7.86 ppm related to the aromatic protons, and a D2O exchangeable singlet signal at δ = 11.58–11.76 ppm assignable to the NH group. In addition, 13C NMR spectra of 5a,b exhibited the two methyl groups as two singlets at δ = 12.1–22.4 ppm, the aromatic carbons as singlet signals in the region δ = 100.2–163.0 ppm, whereas CS group appeared as a singlet signal at 178.5–179.6 ppm.
 | ||
| Scheme 1 Synthetic approach of thioxopyrazolo[3,4-b]pyridine derivatives 5a,b. | ||
In addition, Scheme 2 depicts the cyclo-condensation of the thioxopyrazolo[3,4-b]pyridines 5a,b with various alkyl halides, such as 2-chloroacetamide, 1-chloropropan-2-one, 2-chloroacetonitrile, ethyl 2-chloroacetate, and 2-chloroacetic, in absolute ethanol containing sodium ethoxide, leading to the formation of the three corresponding cyclic pyrazolo[3,4-b]thieno[3,2-e]pyridine compounds 8a–f. The proposed mechanistic route for the formation of the latter derivatives 8a–f involves the nucleophilic attack of the thiol group (–SH) of compounds 5a,b on the used halogen compounds, followed by the elimination of the HCl molecule to yield the non-isolated intermediates 7a–f, which were then cyclized to yield the final products 8a–f. IR spectra of the target derivatives 8a–f displayed various absorption stretching bands in the region 3405–3352 cm−1 corresponding to the NH2 groups and at 2221 cm−1 due to CN group of the compound 8c. Furthermore, 1H NMR spectra of 8a–f revealed the protons of the parent groups (CH3, aromatic, and NH2) in their expected regions in addition to a singlet signal at δ 2.78 ppm due to 6-CH3 of compounds 8b and 8e. The characteristic triplet–quartet signals of OCH2–CH3 of compound 8d appeared at δ 1.20, and 4.13 ppm. 13C NMR spectra of the derivatives 8a–f illustrated signals at δ 11.6–25.0 ppm due to CH3 functionalities and at δ 110.0–148.2 ppm due to the aromatic carbons. Also, the compounds 8a,b and 8d–f revealed singlet signals in the range δ 160.7–181.8 ppm referring to CO groups. Compound 8d exhibited an additional singlet at δ 60.2 ppm assignable to the methylene carbon of OH2CH3 group (Scheme 2).
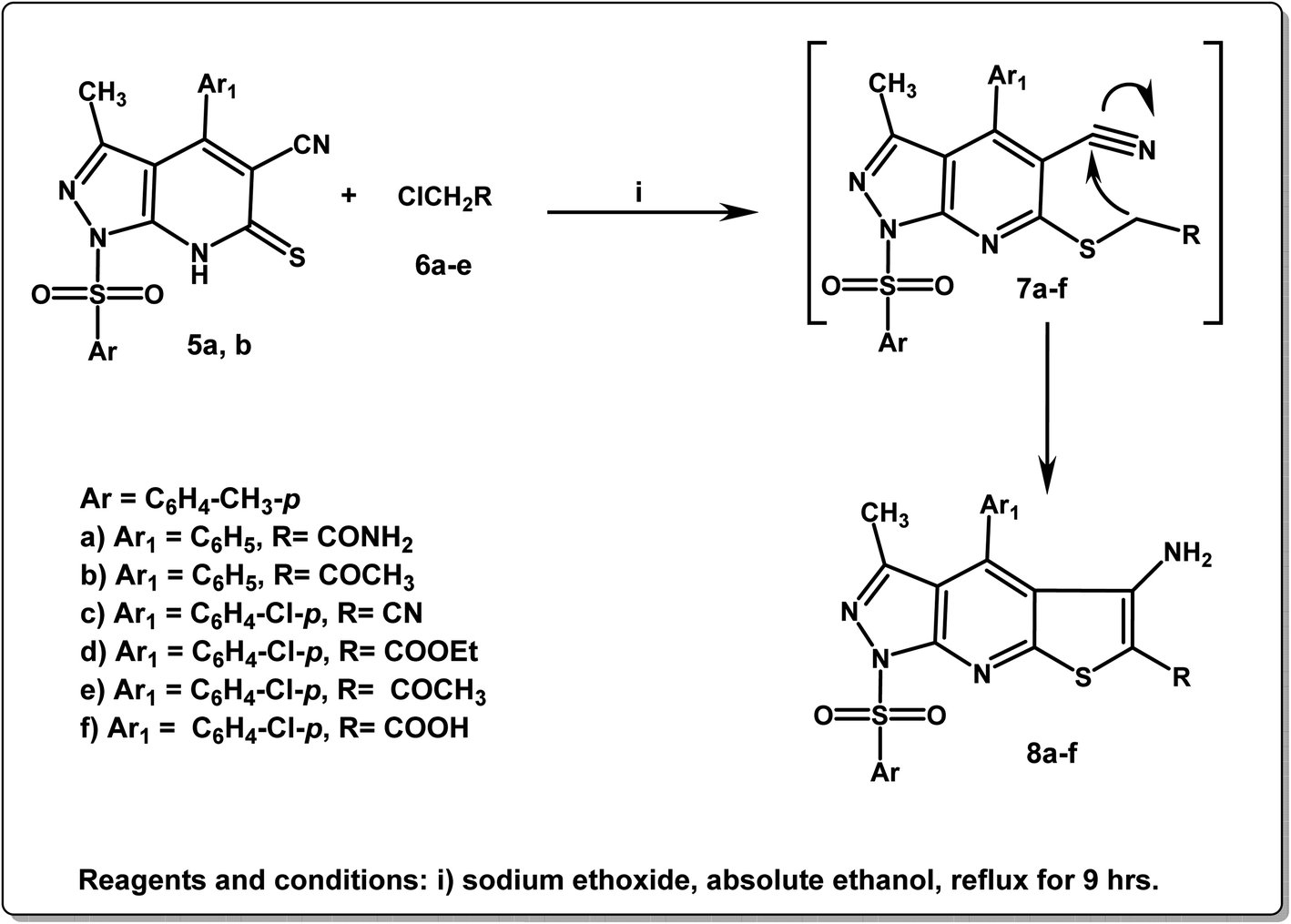 | ||
| Scheme 2 Synthetic approach of pyrazolo[3,4-b]thieno[3,2-e]pyridine derivatives 8a–f. | ||
Utilizing Hantzsch reaction for thiazole synthesis led to the formation of the corresponding pyrazolo[3,4-d]thiazole-5-thione derivatives 10a,b. The reaction was carried out via the treatment of 3-methyl-1-tosyl-1H-pyrazol-5(4H)-one (1) with elemental sulfur and methyl/phenylisothiocyanate in dimethylformamide/ethanol solution containing a few drops of triethylamine.
1H NMR spectra of 10a,b revealed the protons of the parent groups (2CH3 and aromatic protons) in their expected regions in addition to a singlet signal at δ 2.99 ppm due to N–CH3 of compound 10a. Moreover, 13C NMR spectra of the derivatives 10a,b illustrated singlet signals in the regions δ 10.3–31.5 ppm due to CH3 moieties, δ 101.0–155.0 ppm referring to the aromatic carbons and δ 180.0.1–182.2 ppm referring to CS groups (Scheme 3).
 | ||
| Scheme 3 Synthetic approach of pyrazolo[3,4-d]thiazole-5-thione derivatives 10a,b. | ||
The mass spectra of all new compounds represented molecular ion peaks which were in agreement with molecular formulae.
3.2. Biological activity
| Tested compounds | IC50 valuesa (μM) against tumor cell lines | |||
|---|---|---|---|---|
| HepG-2 | MCF-7 | HCT-116 | WI-38 | |
| a IC50 values = mean ± SD of three independent determinations. | ||||
| 5a | 3.42 ± 1.31 (SI = 23.52) | 5.25 ± 1.04 (SI = 15.32) | 6.27 ± 1.15 (SI = 12.83) | 80.46 ± 0.33 |
| 5b | 3.56 ± 1.5 (SI = 23.90) | 6.82 ± 1.1 (SI = 12.47) | 9.21 ± 0.02 (SI = 9.23) | 85.10 ± 0.32 |
| 8a | 30.58 ± 0.01 | 43.02 ± 0.24 | 47.38 ± 0.11 | |
| 8b | 52.08 ± 0.71 | 60.23 ± 0.17 | 67.84 ± 0.02 | |
| 8c | 25.37 ± 0.5 | 36.25 ± 0.13 | 41.77 ± 0.4 | |
| 8d | 23.12 ± 0.08 | 32.14 ± 0.19 | 38.11 ± 0.3 | |
| 8e | 12.31 ± 0.1 | 14.18 ± 0.33 | 18.21 ± 0.62 | |
| 8f | 15.44 ± 0.3 | 19.27 ± 0.45 | 18.86 ± 0.12 | |
| 10a | 10.23 ± 0.15 | 13.65 ± 0.7 | 17.16 ± 0.37 | |
| 10b | 6.75 ± 0.62 (SI = 12.65) | 8.13 ± 0.4 (SI = 10.50) | 9.36 ± 0.45 (SI = 9.12) | 85.41 ± 0.51 |
| 5-Fluorouracil | 2.87 ± 0.15 (SI = 27.77) | 3.58 ± 0.45 (SI = 22.26) | 5.26 ± 0.31 (SI = 15.15) | 79.70 ± 0.64 |
| Erlotinib | 8.19 ± 0.4 (SI = 2.94) | 4.16 ± 0.2 (SI = 5.79) | 7.41 ± 1.12 (SI = 3.25) | 69.99 ± 1.08 |
As shown in Table 1, the derivatives 5a,b with a common structural feature of thioxopyrazolo[3,4-b]pyridine nucleus exhibited the most growth inhibitory activity against the three examined cell lines with IC50 values ranging from 3.42 ± 1.31 to 9.21 ± 0.02 μM. In comparison to erlotinib, the compound 5a and its chlorinated analogue 5b were approximately 2.4-fold more potent against the HepG-2 cancer cell line (IC50 values: 3.42 ± 1.3, 3.56 ± 1.5, respectively; IC50 erlotinib, 8.19 ± 0.40 μM). Both compounds were approximately equipotent or slightly less potent against the other two cancer cell lines (MCF-7 and HCT) representing IC50 values ranging from 4.16 ± 0.2 to 9.21 ± 0.02 μM and IC50 erlotinib, 4.16 ± 0.2 and 7.41 ± 1.12 μM, respectively. Meanwhile, a slight decrease in the target activity was detected by both 5a and 5b in comparison with 5-FU, which represented IC50s ranging from 2.87 ± 0.15 to 5.26 ± 0.31 μM. It is evident that the HepG-2 cancer cell line was more sensitive to both compounds 5a and 5b than the MCF-7 and HCT-116 cell lines. Moreover, the promising sensitivity of the HepG-2 cancer cell line was maintained by the compound bearing the thioxopyrazolo[3,4-d]thiazole structural feature 10b in comparison with erlotinib with IC50 value of 6.75 ± 0.62 μM, but 2.3-fold less when compared with 5-FU. Moreover, a detectable decrease in the activity of 10b was also reported against the other two cancer cell lines (MCF-7 and HCT) in comparison to both reference drugs, 5-FU and erlotinib, with IC50 values of 8.13 ± 0.4 and 9.36 ± 0.45 μM, respectively. A detectable reduction in the target potency was detected by the methyl analogue 10a against the three cancer cell lines (IC50 values: 10.23 ± 0.15, 13.65 ± 0.7, and 17.16 ± 0.37, respectively), in comparison with both standard drugs. This indicated that the substitution of the thiazolidine ring with a phenyl ring is more favorable for the anticancer activity via increasing the lipophilicity of the compound. In addition, the fusion of a thiophene ring to the parent thioxopyrazolo[3,4-b]pyridine nucleus resulting in the thioxopyrazolo[3,4-b]thieno[3,2-e]pyridine derivatives 8a–f was unfavorable for the anti-proliferative activity producing a significant drop against the three cancer cell lines with IC50 values ranging from 12.31 ± 0.11 to 67.84 ± 0.02 μM. This result could be explained by the increase in the size of the molecule leads to its improper orientation and inadequate fitting of the new molecule in the active site of the target protein.
Consequently, the selectivity profile of the three most promising compounds, 5a, 5b, and 10b, towards cancer cells rather than normal cells was valuable to be examined. Accordingly, the cytotoxic activity of the latter analogues was screened in human lung fibroblasts (WI-38) normal cells utilizing MTT assay. Fortunately, the evaluated compounds were more selective for the tested cancer cells than for the normal cell line WI-38, producing IC50 values, 80.46 ± 0.33, 85.10 ± 0.32, and 85.41 ± 0.51 μM, in comparison with 5-FU and erlotinib of IC50 values of 79.70 ± 0.64 and 69.99 ± 1.08 μM. Based on the previous results, the structural thioxopyrazolo[3,4-b]pyridine and thioxopyrazolo[3,4-d]thiazole features as compounds 5a, 5b, and 10b, respectively are promising cytotoxic scaffolds with significant selectivity indices against normal cells (Table 1) (Fig. 3). On the other hand, the tricyclic ring system such as the thioxopyrazolo[3,4-b]thieno[3,2-e]pyridine scaffold of compounds 8a–f is not preferred for the target anticancer activity.
 | ||
| Fig. 3 Cell cycle analysis of 5a in HepG-2 cell line. | ||
| Compound no. | c-Met |
|---|---|
| IC50 (mean ± SEM) (nM) | |
| a IC50: compound concentration required to inhibit the enzyme activity by 50%, SEM: standard error mean; each value is the mean of three values. | |
| 5a | 4.27 ± 0.31 |
| 5b | 7.95 ± 0.17 |
| Cabozantinib | 5.38 ± 0.52 |
It could be noted that both compounds showed the most favorable antiproliferation effects and selectivity in the cell assays and also showed promising performance in the enzymatic assay. According to these findings, the pyrazolo[3,4-b]pyridine fragment is a privileged scaffold for c-Met inhibitory activity and antiproliferative effect.
The dot plot flow cytometry data of the stained cells is represented in Fig. 4. HepG-2 cancer cells were treated with the IC50 concentration of 5a (3.42 μM) and incubated for 24 h. The obtained data showed a significant increase in the percent of positive apoptotic cells in the early and late stages (lower and upper right quadrants of the cytogram) from 0.39 and 0.14% to 24.63 and 16.28%, respectively, which comprises about 63 and 116 folds with respect to the untreated cells (Fig. 4, Table 3). Moreover, there is an increase in the percentage of the necrotic cells from 1.08% (untreated cells) to 6.24% (5a-treated cells). This result exhibited that the total percentages of the early and late apoptotic cells are greater than the percentage of the necrotic cells, confirming that the apoptosis process is one of the main mechanistic pathways of cell death caused by 5a (Table 3).
 | ||
| Fig. 4 The cell cycle analysis and apoptosis induction study of the control untreated and 5a-treated HepG-2 cell line utilizing Annexin V/PI. | ||
| Apoptosis | Necrosis | |||
|---|---|---|---|---|
| Total | Early | Late | ||
| 5a/HepG-2 | 47.15 | 24.63 | 16.28 | 6.24 |
| Cont./HepG-2 | 1.61 | 0.39 | 0.14 | 1.08 |
The mitochondrial apoptotic process is synchronised by the Bcl-2 protein family.
Two of these proteins, Bcl-2 and Bax, fine-tune this predetermined process, with Bcl-2 acting as an anti-apoptotic and Bax as an inducer (pro-apoptotic). The ability of the cell to undergo apoptosis is determined by the precise balance between these two distinct proteins.65,66 The tumor suppressor gene, p53, is also an essential component of the cellular emergency response mechanism that results in cell death or the arrest of cell growth.
Cancer can be stopped in its tracks by p53 activation resulting in apoptosis, or a G1 cell cycle halt.
The retention of genomic integrity and p53 inactivation in malignancies may be the cause of accelerated cell proliferation and resistance to genotoxic anticancer therapies. An essential part of the cellular emergency response system that causes cell growth arrest or apoptosis is p53.67
Furthermore, the Cysteinyl Aspartate-Specific Proteinase family, which includes Caspases-3, is essential for starting programmed cell death (apoptosis). All apoptotic processes, such as cell shrinkage, chromatin condensation, and DNA fragmentation, depend on caspase-3.68,69
Accordingly, it was of interest to study the effects of compound 5a on the hallmarks levels; Bax, Bcl-2, p53, and caspase-3 in HepG-2 cell line using ELISA process.67–69 HepG-2 cells were treated with the IC50 concentration of compound 5a (3.42 μM) for 24 h utilizing erlotinib as a reference drug. The obtained data were expressed in terms of pg mL−1 for Bax and p53 and as ng mL−1 for both Bcl-2 and caspase-3 levels in the 5a-treated HepG-2 cells. It has been investigated that compound 5a boosted the level of the pro-apoptotic protein Bax by ∼5.4 folds and reduced the antiapoptotic protein Bcl-2 level by ∼3.9 folds in the treated HepG-2 cells in comparison with the untreated cells. Due to its importance, the ratio of the two latter proteins Bax/Bcl-2 was calculated for compound 5a to be 21 times higher than that in the untreated cells. This result confirmed that the compound 5a has moved the cells toward apoptosis. Moreover, the results showed that compound 5a upregulated the levels of p53 and caspase-3 in the tested HepG-2 cells by ∼6.9 and 23.8 folds, respectively compared to the control (Table 4). The reported data in Table 4 represents that the activities of the evaluated compound 5a was approximately similar to that obtained by the standard drug erlotinib (Table 4).
| Compound 5a | Bax (pg mL−1) | Bcl-2 (ng mL−1) | Bax/Bcl-2 | p53 (pg mL−1) | Casp-3 (ng mL−1) |
|---|---|---|---|---|---|
| 5a/HepG-2 | 211.40 ± 0.25 | 2.09 ± 0.15 | 101.14 | 825.22 ± 0.40 | 42.60 ± 0.10 |
| Cont./HepG-2 | 38.80 ± 0.10 | 8.16 ± 0.24 | 4.75 | 119.52 ± 0.02 | 1.79 ± 0.15 |
| Erlotinib/HepG-2 | 200.20 ± 0.25 | 1.69 ± 0.10 | 118.84 | 890.30 ± 0.01 | 58.41 ± 0.12 |
4. Computational studies
4.1. Molecular modeling studies
Utilizing the Molecular Operating Environment software (MOE-Dock) version 2014.0901,70,71 molecular docking was carried out to clarify the variance in the in vitro inhibitory activities of the evaluated compounds 5a and 5b against c-Met kinase. The native co-crystallized ligand, N-{3-fluoro-4-[(7-methoxyquinolin-4-yl)oxy]phenyl}-1-[(2R)-2-hydroxypropyl]-5-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide, was re-docked in the active binding site of c-Met (PDB code: 3U6I)72 to validate the docking process. This resulted in energy score of −9.72 kcal mol−1 at root mean square deviation (RMSD) value equal to 0.79 Å, indicating that the docking process was confirmed. Then, our targets 5a and 5b were docked and the results were depicted in Fig. 5 and 6.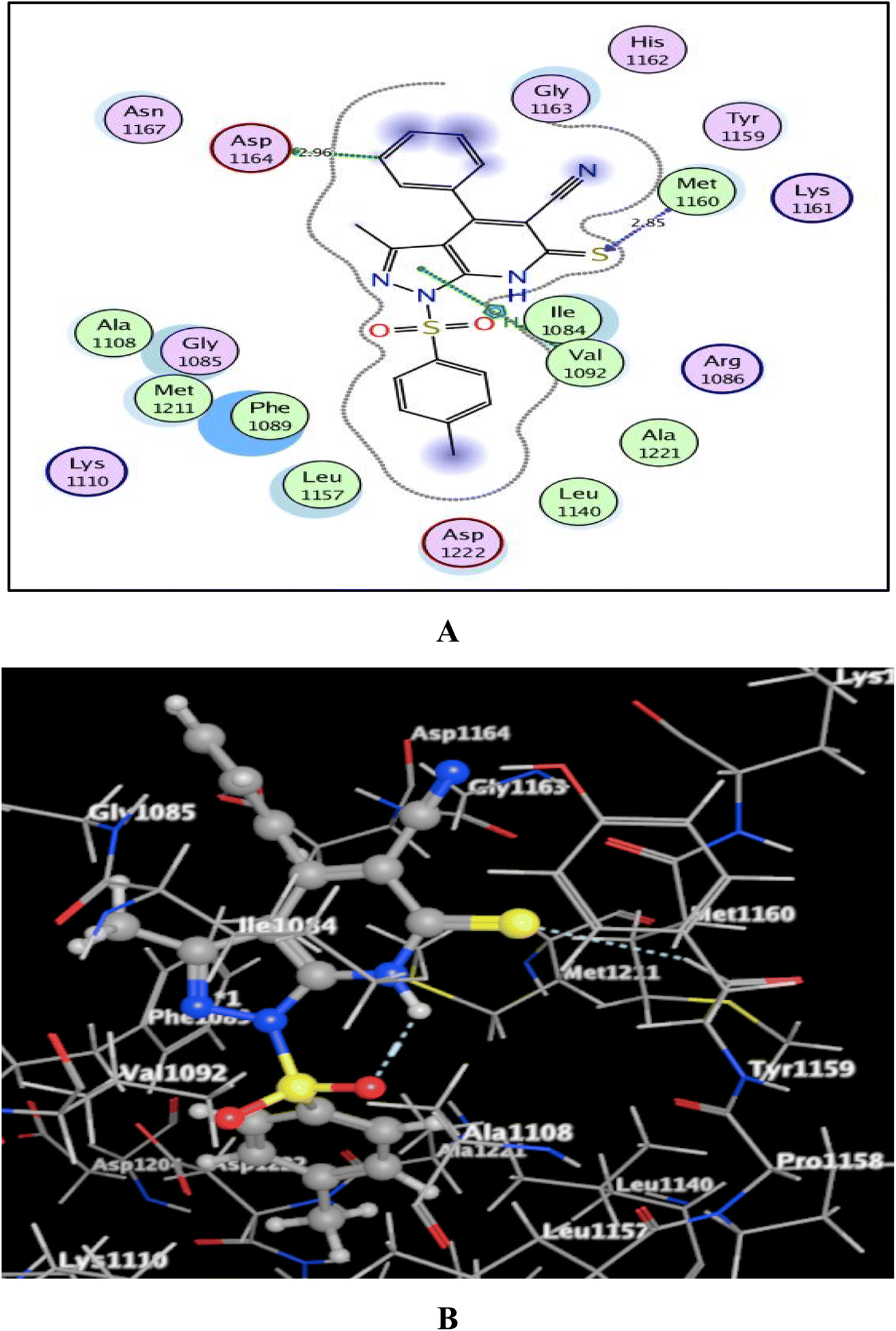 | ||
| Fig. 5 (A) & (B) patterns illustrating the 2D and 3D binding interactions of the promising pyrazolo[3,4-b]pyridine target 5a into active pocket of c-Met kinase (PDB code: 3U6I). | ||
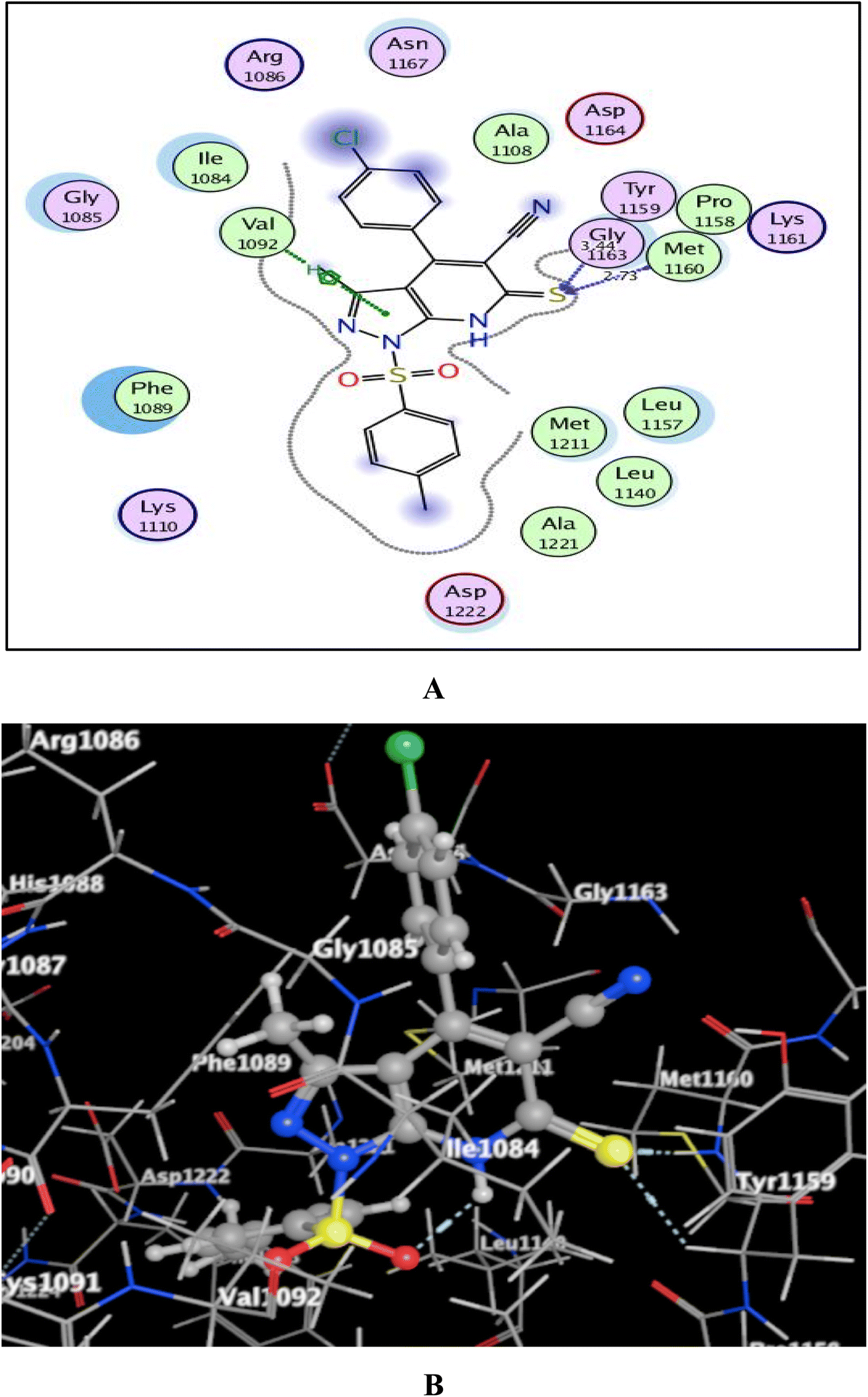 | ||
| Fig. 6 (A) & (B) patterns illustrating the 2D and 3D binding interactions of the promising pyrazolo[3,4-b]pyridine target 5b into active pocket of c-Met kinase (PDB code: 3U6I). | ||
As shown in Fig. 5 and 6, the screened promising targets 5a and 5b were fixed well within the active pocket of c-Met with favorable energy scores of −10.22 and −9.83 kcal mol−1, respectively. It was observed in both derivatives that the sulfonyl-4-methylbenzene fragment at p-1 of the pyrazolo[3,4-b]pyridine core is located facing and surrounded by the essential amino acids Lys1110, Leu1140, Leu1157, Met1211, Ala1221, and Asp1222 through hydrophobic interactions. On the other hand, the 6-thioxopyrazolo[3,4-b]pyridine-5-carbonitrile scaffold afforded arene-H interactions between the centroid of pyrazole and the amino acid Val1092. Moreover, the sulfur atom formed H-bond acceptor with the key amino acid Met1160. The phenyl ring at p-4 of pyrazolo[3,4-b]pyridine in 5a illustrated an H-bond donor between C-3 and the sidechain of Asp1164 (distance: 2.96 Å). While substitution of the phenyl ring with Cl atom in the para position devoted 5b from the previous hydrogen binding, and that could be the reason for its lower inhibitory activity than 5a.
4.2. In silico ADME prediction study
Studying absorption, distribution, metabolism, and excretion (ADME) for the targeted substances might provide important data regarding the ideal pharmaceutical choice. This predicted investigation was assisted through the utilization of SwissADME, a free online application.73–75 The Veber rule (TPSA ≤ 140 Å2 and number of rotatable bonds ≤ 10) and Lipinski's rule (molecule with number of hydrogen bond acceptors ≤ 10, number of hydrogen bond donors ≤ 5 MW ≤ 500 and MLogP ≤ 4.15) govern the optimal medication for oral administration. The 6-thioxopyrazolo[3,4-b]pyridine-5-carbonitrile target 5a was found to be in compliance with the prior rules with no violations, while the derivative 5b exhibited MW > 500 and one violation (Table 5).| Compd. | MWa | MLogPb | TPSAc (Å2) | nHBDd | nHBAe | nRBf | Violationsg |
|---|---|---|---|---|---|---|---|
| a Molecular weight.b Calculated lipophilicity (MLogPo/w).c Topological polar surface area.d Number of hydrogen bond donor.e Number of hydrogen bond acceptor.f Number of rotatable bond.g Violations from Lipinski and Veber rules. | |||||||
| 5a | 420.51 | 3.17 | 132.01 | 1 | 4 | 3 | 0 |
| 5b | 518.54 | 1.66 | 137.57 | 0 | 8 | 6 | 1 |
Considering the bioavailability radar chart (Fig. 7), it was found that the pyrazolo[3,4-b]pyridine-5-carbonitrile derivatives 5a and 5b are located in the ideal range (pink zone) concerning to five key factors LIPO (lipophilicity), SIZE, POLAR (polarity), INSOLU (solubility) and FLEX (flexibility), but were distanced from the optimal region of INSATU (saturation).
 | ||
| Fig. 7 The bioavailability radar chart of pyrazolo[3,4-b]pyridine-5-carbonitrile derivatives 5a and 5b. The pink region illustrated the optimal range for each of the oral bioavailability parameters and the predicted ones for the screened compounds were displayed as red lines. | ||
The pharmacokinetic properties of the promising pyrazolo[3,4-b]pyridine-5-carbonitriles 5a and 5b were represented and analyzed in Fig. 8 and Table 6 (ESI†). All the screened compounds are predicted to be neither brain-penetrated nor gastrointestinally absorbed (outside the egg). Therefore, they are not predicted to be administered orally and could be used parenterally. Also, they are only employed to treat peripheral illnesses and are not thought to disrupt the central nervous system. These derivatives also got a satisfactory bioavailability grade of 0.55 and are not supposed to trigger a PAIN warning. Additionally, the red dots in Fig. 8 indicate that the two derivatives are not substrates for p-glycoprotein (P-gp, a drug efflux transporter), revealing a low chance of their efflux out of the cell with their highest activity.
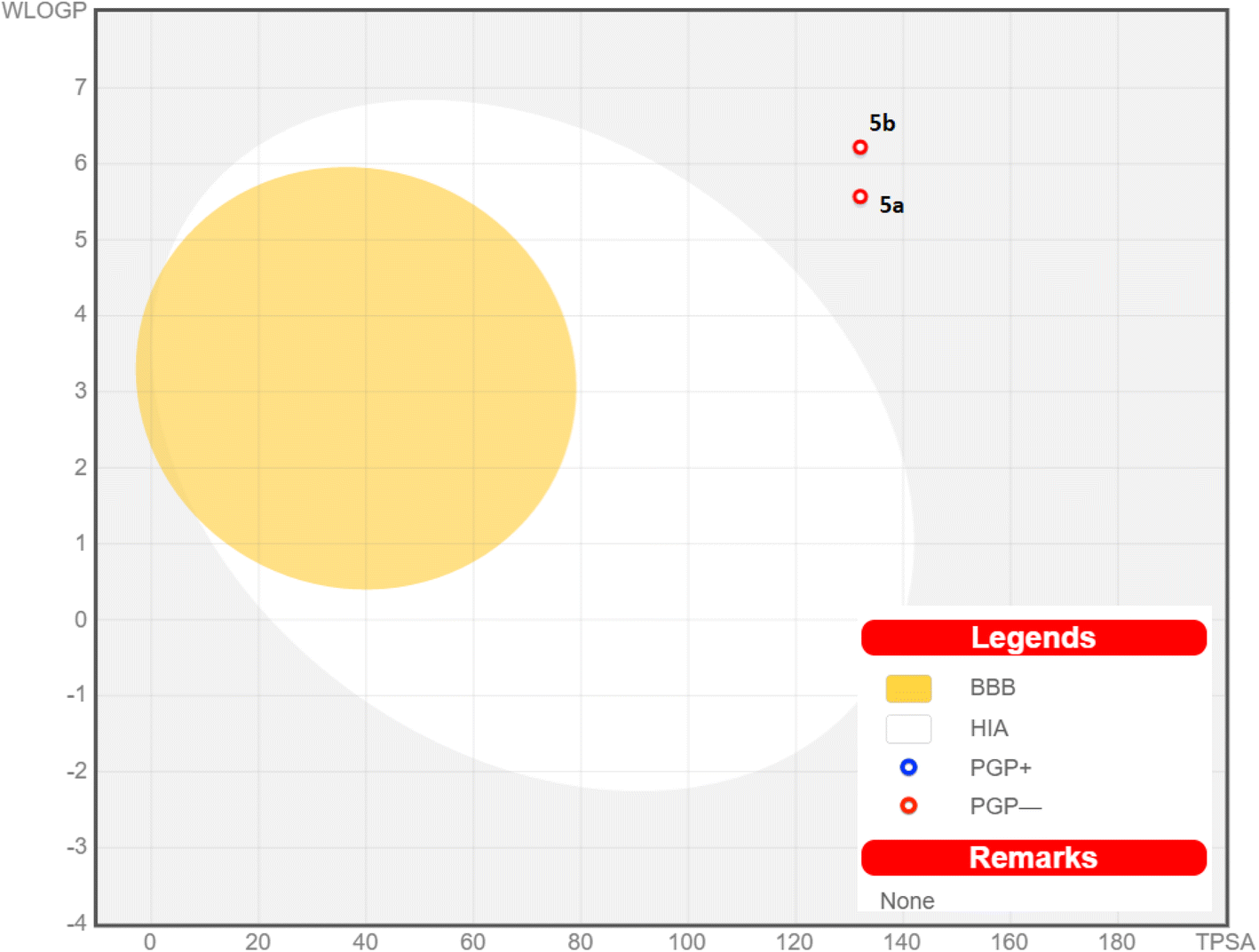 | ||
| Fig. 8 Chart of boiled-egg revealing the BBB penetration capability and gastrointestinal absorption of pyrazolo[3,4-b]pyridine-5-carbonitrile derivatives 5a and 5b; PGP+: substrate of p-glycoprotein; PGP−: non-substrate of p-glycoprotein. | ||
5. Conclusion
In summary, a new set of derivatives bearing pyrazolo[3,4-b]pyridine, pyrazolo[3,4-b]thieno[3,2-e]pyridine, and pyrazolo[3,4-d]thiazole-5-thione scaffolds 5a,b, 8a–f, and 10a,b, respectively, were designed, synthesized, and evaluated for their biological impacts as antiproliferative agents targeting c-Met inhibition. The screening of anticancer activity resulted in the identification of the two most promising compounds, 5a and 5b, with IC50 values ranging from 3.42 ± 1.31 to 9.21 ± 0.02 μM against HepG-2, MCF-7, and HCT-116 human cancer cell lines. c-Met assay represented the promising inhibitory activities of both compounds, with IC50 values of 4.27 ± 0.31 and 7.95 ± 0.17 nM, respectively. Meanwhile, cell cycle and apoptosis assays in HepG-2 cells by flow cytometry indicated that compound 5a arrested the cell cycle at the G1/S phase, inducing apoptosis of the cells. In addition, 5a up-regulated the levels of Bax/Bcl-2, p53, and caspase-3 in the tested HepG-2 cells by 21, 6.9, and 23.8 folds, respectively, compared to the untreated HepG-2 cells. Molecular docking study showed that both compounds 5a and 5b were fixed well within the active pocket of c-Met with favorable energy scores of −10.22 and −9.83 kcal mol−1, respectively. In addition, the ADME prediction study showed that 5a and 5b fulfilled the criteria of the drug likeness approach. Accordingly, the pyrazolo[3,4-b]pyridine scaffold is a promising template in the field of drug discovery and optimization.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The research work was funded by institutional fund projects under grant no. IFPIP-1237-247-1443. Therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.References
- Y. Dong, H. Hu, Y. Sun, M. Qin, P. Gong, Y. Hou and Y. Zhao, Design, synthesis and biological evaluation of novel c-Met/HDAC dual inhibitors, Bioorg. Med. Chem. Lett., 2020, 30(23), 127610 CrossRef CAS.
- Z. Wang, J. Shi, X. Zhu, W. Zhao, Y. Gong, X. Hao, Y. Hou, Y. Liu, S. Ding, J. Liu and Y. Chen, Design, synthesis and biological evaluation of novel 4-phenoxypyridine based 3-oxo-3, 4-dihydroquinoxaline-2-carboxamide derivatives as potential c-Met kinase inhibitors, Bioorg. Chem., 2020, 105, 104371 CrossRef CAS PubMed.
- M. Mortazavi, E. Raufi, T. Damghani, M. Khoshneviszadeh, N. Edraki, M. Eskandari, E. Giovannetti, G. J. Peters, S. Pirhadi and O. Firuzi, Discovery of anticancer agents with c-Met inhibitory potential by virtual and experimental screening of a chemical library, Eur. J. Pharmacol., 2023, 938, 175395 CrossRef CAS PubMed.
- C. H. Gow, M. S. Hsieh, Y. L. Chen, Y. N. Liu, S. G. Wu and J. Y. Shih, Survival outcomes and prognostic factors of lung cancer patients with the MET exon 14 skipping mutation: A single-center real-world study, Front. Oncol., 2023, 13, 925 Search PubMed.
- J. A. Bikker, N. Brooijmans, A. Wissner and T. S. Mansour, Kinase domain mutations in cancer: implications for small molecule drug design strategies, J. Med. Chem., 2009, 52, 1493–1509 CrossRef CAS PubMed.
- R. Robert Jr, Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update, Pharmacol. Res., 2020, 152, 104609 CrossRef PubMed.
- P. Su, M. Zhang and X. Knag, Targeting c-Met in the treatment of urologic neoplasms: Current status and challenges, Front. Oncol., 2023, 13, 1071030 CrossRef.
- W. M. Rashed, C-MET as a potential target therapy toward personalized therapy in some pediatric tumors: An overview, Crit. Rev. Oncol./Hematol., 2018, 131, 7–15 CrossRef PubMed.
- S. Pilotto, L. Carbognin, N. Karachaliou, P. C. Ma, R. Rosell, G. Tortora and E. Bria, Tracking MET de-addiction in lung cancer: A road towards the oncogenic target, Cancer Treat. Rev., 2017, 60, 1–11 CrossRef CAS PubMed.
- J. Hu, L. Chen, Z. Lu, H. Yao, Y. Hu, L. Feng, Y. Pang, J. Q. Wu, Z. Yu and W. H. Chen, Design, Synthesis and Antitumor Activity of Novel Selenium-Containing Tepotinib Derivatives as Dual Inhibitors of c-Met and TrxR, Molecules, 2023, 28(3), 1304 CrossRef CAS.
- H. Morgan and K. John, The conformational plasticity of protein kinases, Cell, 2002, 109, 275–282 CrossRef PubMed.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, The protein kinase complement of the human genome, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- S. Berthou, D. M. Aebersold, L. S. Schmidt, D. Stroka, C. Heigl, B. Streit, D. Stalder, G. Gruber, C. Liang, A. R. Howlett, D. Candinas, R. H. Greiner, K. E. Lipson and Y. Zimmer, The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants, Oncogene, 2004, 23, 5387–5393 CrossRef CAS PubMed.
- M. H. Norman, L. Liu, M. Lee, N. Xi, I. Fellows, N. D. D'Angelo, C. Dominguez, K. Rex, S. F. Bellon, T. S. Kim and I. Dussault, Structure-based design of novel class II c- Met inhibitors: 1. Identification of pyrazolone-based derivatives, J. Med. Chem., 2012, 55, 1858–1867 CrossRef CAS PubMed.
- N. Liu, Y. Wang, G. Huang, C. Ji, W. Fan, H. Li, Y. Cheng and H. Tian, Design, synthesis and biological evaluation of 1H-pyrrolo[2,3-b]pyridine and 1H-pyrazolo[3,4-b]pyridine derivatives as c-Met inhibitors, Bioorg. Chem., 2016, 65, 146–158 CrossRef CAS PubMed.
- F. M. Yakes, J. Chen, J. Tan, K. Yamaguchi, Y. Shi, P. Yu, F. Qian, F. Chu, F. Bentzien, B. Cancilla, J. Orf, A. You, A. D. Laird, S. Engst, L. Lee, J. Lesch, Y. C. Chou and A. H. Joly, Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth, Mol. Cancer Ther., 2011, 10, 2298–2308 CrossRef CAS PubMed.
- F. Qian, S. Engst, K. Yamaguchi, P. Yu, K. A. Won, L. Mock, T. Lou, J. Tan, C. Li, D. Tam, J. Lougheed, F. M. Yakes, F. Bentzien, W. Xu, T. Zaks, R. Wooster, J. Greshock and A. H. Joly, Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases, Cancer Res., 2009, 69, 8009–8016 CrossRef CAS.
- Z. Wang, J. Shi, X. Zhu, W. Zhao, Y. Gong, X. Hao, Y. Hou, Y. Liu, S. Ding, J. Liu and Y. Chen, Design, synthesis and biological evaluation of novel 4-phenoxypyridine based 3-oxo-3, 4-dihydroquinoxaline-2-carboxamide derivatives as potential c-Met kinase inhibitors, Bioorg. Chem., 2020, 105, 104371 CrossRef CAS.
- B. D. Smith, M. D. Kaufman, C. B. Leary, B. A. Turner, S. C. Wise, Y. M. Ahn, R. J. Booth, T. M. Caldwell, C. L. Ensinger, M. M. Hood, W. P. Lu, T. W. Patt, W. C. Patt, T. J. Rutkoski, T. Samarakoon, H. Telikepalli, L. Vogeti, S. Vogeti, K. M. Yates, L. Chun, L. J. Stewart, M. Clare and D. L. Flynn, Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2, Mol. Cancer Ther., 2015, 14, 2023 CrossRef CAS PubMed.
- Y. Dai and D. W. Siemann, BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro, Mol. Cancer Ther., 2010, 9, 1554–1561 CrossRef CAS PubMed.
- J. K. Rho, Y. J. Choi, S. Y. Kim, T. W. Kim, E. K. Choi, S. J. Yoon, B. M. Park, E. Park, J. H. Bae, C. M. Choi and J. C. Lee, MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation, Cancer Res., 2014, 74, 253–262 CrossRef CAS PubMed.
- Q. D. Tang, L. X. Wang, Y. L. Duan, W. H. Wang, S. M. Huang, J. Zhi, S. Jia, W. F. Zhu, P. Wang, R. Luo and P. W. Zheng, Discovery of novel 7-azaindole derivatives bearing dihydropyridazine moiety as c-Met kinase inhibitors, Eur. J. Med. Chem., 2017, 133, 97–106 CrossRef CAS PubMed.
- Q. D. Tang, L. X. Wang, Y. Y. Tu, W. F. Zhu, R. Luo, Q. D. Tu, P. Wang, C. J. Wu, P. Gong and P. W. Zheng, Discovery of novel pyrrolo[2,3-b]pyridine derivatives bearing 1,2,3-triazole moiety as c-Met kinase inhibitors, Bioorg. Med. Chem. Lett., 2016, 26, 1680–1684 CrossRef CAS PubMed.
- J. Liu, Y. L. Gong, J. T. Shi, X. C. Hao, Y. Wang, Y. P. Zhou, Y. L. Hou, Y. J. Liu, S. Ding and Y. Chen, Design, synthesis and biological evaluation of novel N-[4-(2-fluorophenoxy)pyridin-2-yl]cyclopropanecarboxamide derivatives as potential c-Met kinase inhibitors, Eur. J. Med. Chem., 2020, 194, 112244 CrossRef CAS PubMed.
- G. Luo, Y. Ma, X. Liang, G. Xie, Y. Luo, D. Zha, S. Wang, L. Yu, X. Zheng, W. Wu and C. Zhang, Design, synthesis and antitumor evaluation of novel 5-methylpyrazolo [1,5-a] pyrimidine derivatives as potential c-Met inhibitors, Bioorg. Chem., 2020, 104, 104356 CrossRef CAS PubMed.
- R. F. Barghash, W. M. Eldehna, M. Kovalova, V. Vojáčková, V. Kryštof and H. A. Abdel-Aziz, One-pot three-component synthesis of novel pyrazolo [3,4-b] pyridines as potent antileukemic agents, Eur. J. Med. Chem., 2022, 227, 113 CrossRef.
- I. H. Eissa, A. M. El-Naggar and M. A. El-Hashash, Design, synthesis, molecular modeling and biological evaluation of novel 1H-pyrazolo[3,4-b]pyridine derivatives as potential anticancer agents, Bioorg. Chem., 2016, 67, 43–56 CrossRef CAS PubMed.
- A. Papastathopoulos, N. Lougiakis, I. K. Kostakis, P. Marakos, N. Pouli, H. Pratsinis and D. Kletsas, New bioactive 5-arylcarboximidamido pyrazolo[3,4-c]pyridines: synthesis, cytotoxic activity, mechanistic investigation and structure activity relationships, Eur. J. Med. Chem., 2021, 218, 113387 CrossRef CAS PubMed.
- M. Michailidou, V. Giannouli, V. Kotsikoris, O. Papadodima, G. Kontogianni, I. K. Kostakis, N. Lougiakis, A. Chatziioannou, F. N. Kolisis, P. Marakos, N. Pouli and H. Loutrari, Novel pyrazolopyridine derivatives as potential angiogenesis inhibitors: synthesis, biological evaluation and transcriptome-based mechanistic analysis, Eur. J. Med. Chem., 2016, 121, 143e15 CrossRef.
- B. Zheng, Y. Peng, W. Wu, J. Ma, Y. Zhang, Y. Guo, S. Sun, Z. Chen, Q. Li and G. Hu, Synthesis and structure–activity relationships of pyrazolo[3,4-b]pyridine derivatives as adenosine 5′-monophosphate-activated protein kinase activators, Arch. Pharm. Chem. Life Sci., 2019, 352, 1900066 CrossRef PubMed.
- Y. Li, H. Cheng, Z. Zhang, X. Zhuang, J. Luo, H. Long, Y. Zhou, Y. Xu, R. Taghipouran, D. Li, A. Patterson, J. Smaill, Z. Tu, D. Wu, X. Ren and K. Ding, N-(3-Ethynyl-2,4-difluorophenyl)sulfonamide derivatives as selective raf inhibitors, ACS Med. Chem. Lett., 2015, 6(5), 543–547 CrossRef CAS PubMed.
- H. Cheng, Y. Chang, L. Zhang, J. Luo, Z. Tu, X. Lu, Q. Zhang, J. Lu, X. Ren and K. Ding, Identification and optimization of new dual inhibitors of B-raf and epidermal growth factor receptor kinases for overcoming resistance against vemurafenib, J. Med. Chem., 2014, 57, 2692–2703 CrossRef CAS PubMed.
- A.-S. Huart, B. Saxty, A. Merritt, M. Nekulova, S. Lewis, Y. Huang, B. Vojtesek, C. Kettleborough and T. R. Hupp, A Casein kinase 1/Checkpoint kinase 1 pyrazolo-pyridine protein kinase inhibitor as novel activator of the p53 pathway, Bioorg. Med. Chem. Lett., 2013, 23, 5578–5585 CrossRef CAS PubMed.
- N. Liu, Y. Wang, G. Huang, C. Ji, W. Fan, H. Li, Y. Cheng and H. Tian, Design, synthesis and biological evaluation of 1H-pyrrolo[2,3-b]pyridine and 1H-pyrazolo[3,4-b]pyridine derivatives as c-Met inhibitors, Bioorg. Chem., 2016, 65, 146–158 CrossRef CAS.
- B. Zhao, Y. Li, P. Xu, Y. Dai, C. Luo, Y. Sun, J. Ai, M. Geng and W. Duan, Discovery of substituted 1H-pyrazolo[3,4-b]pyridine derivatives as potent and selective FGFR kinase inhibitors, ACS Med. Chem. Lett., 2016, 7, 629–634 CrossRef CAS PubMed.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- J. Witherington, V. Bordas, D. Haigh, D. M. B. Hickey, R. J. Ife, A. D. Rawlings, B. P. Slingsby, D. G. Smith and R. W. Ward, 5-Aryl-pyrazolo[3,4-b]pyridazines: potent inhibitors of glycogen synthase kinase-3 (GSK-3), Bioorg. Med. Chem. Lett., 2003, 13, 1581–1584 CrossRef CAS PubMed.
- G. S. Hassan, H. H. Georgey, E. Z. Mohammed, R. F. George, W. R. Mahmoud and F. A. Omar, Mechanistic selectivity investigation and 2D-QSAR study of some new antiproliferative pyrazoles and pyrazolopyridines as potential CDK2 Inhibitors, Eur. J. Med. Chem., 2021, 218, 113389 CrossRef CAS.
- N. Liu, Y. Wang, G. Huang, C. Ji, W. Fan, H. Li, Y. Cheng and H. Tian, Design, synthesis and biological evaluation of 1H-pyrrolo[2,3-b]pyridine and 1H-pyrazolo[3,4-b] pyridine derivatives as c-Met inhibitors, Bioorg. Chem., 2016, 65, 146–158 CrossRef CAS PubMed.
- Y. Ma, G. Sun, D. Chen, X. Peng, Y. L. Chen, Y. Su, Y. Ji, J. Liang, X. Wang, L. Chen and J. Ding, Design and optimization of a series of 1-sulfonylpyrazolo [4,3-b] pyridines as selective c-Met inhibitors, J. Med. Chem., 2015, 58(5), 2513–2529 CrossRef CAS PubMed.
- R. M. Mohareb, K. M. Hilmy and Y. A. Elshehawy, Discovery of new thiophene, pyrazole, isoxazole derivatives as antitumor, c-Met, tyrosine kinase and Pim-1 kinase inhibitors, Bull. Chem. Soc. Ethiop., 2018, 32(2), 285–308 CrossRef CAS.
- J. Li, W. Lu, Y. Yang, R. Xiang, Y. Ling, C. Yu and Y. Zhou, Hybrid Nanomaterials for Cancer Immunotherapy, Adv. Sci. Lett., 2022, 5, 2204932 Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209 CrossRef.
- (a) W. Song, S. N. Musetti and L. Huang, Nanomaterials for cancer immunotherapy, Biomaterials, 2017, 148, 16 CrossRef CAS PubMed; (b) H. Phuengkham, L. Ren, I. W. Shin and Y. T. Lim, Nanoengineered immune niches for reprogramming the immunosuppressive tumor microenvironment and enhancing cancer immunotherapy, Adv. Mater., 2019, 31, 1803322 CrossRef PubMed.
- S. Yan, X. Jiao, H. Zou and K. Li, Prognostic significance of c-Met in breast cancer: a meta-analysis of 6010 cases, Diagn. Pathol., 2015, 10, 62 CrossRef PubMed.
- J. S. Pyo, G. Kang, W. J. Cho and S. B. Choi, Clinicopathological significance and concordance analysis of c-MET immunohistochemistry in non-small cell lung cancers: A meta-analysis, Pathol., Res. Pract., 2016, 212, 710–716 CrossRef CAS PubMed.
- S. Yu, Y. Yu, N. Zhao, J. Cui, W. Li and T. Liu, C-Met as a prognostic marker in gastric cancer: a systematic review and meta-analysis, PLoS One, 2013, 8, e79137 CrossRef CAS PubMed.
- J. H. Kim, B. J. Kim and H. S. Kim, Clinicopathological impacts of high c-Met expression in head and neck squamous cell carcinoma: a meta-analysis and review, Oncotarget, 2017, 8, 113120–113128 CrossRef PubMed.
- J. Peng, S. Qi, P. Wang, W. Li, C. Liu and F. Li, Diagnosis and prognostic significance of c-Met in cervical cancer: a meta-analysis, Dis. Markers, 2016, 2016, 6594016 CrossRef PubMed.
- J. H. Kim, H. S. Kim, B. J. Kim, J. Lee and H. J. Jang, Prognostic value of c-Met overexpression in pancreatic adenocarcinoma: a meta-analysis, Oncotarget, 2017, 8, 73098–73104 CrossRef PubMed.
- J. H. Kim, H. S. Kim, B. J. Kim, H. J. Jang and J. Lee, Prognostic value of c-Met overexpression in hepatocellular carcinoma: a meta-analysis and review, Oncotarget, 2017, 8, 351–357 Search PubMed.
- J. H. Kim, B. J. Kim and H. S. Kim, Clinicopathological impacts of high c-Met expression in renal cell carcinoma: a meta-analysis and review, Oncotarget, 2017, 8, 75478–75487 CrossRef PubMed.
- C. A. Larsen and R. H. Dashwood, Suppression of Met activation in human colon cancer cells treated with (−)-epigallocatechin-3-gallate: Minor role of hydrogen peroxide, Biochem. Biophys. Res. Commun., 2009, 389(3), 527–530 CrossRef CAS PubMed.
- A. A. El-Sayed, E. S. Nossier, A. A. Almehizia and A. E. G. E. Amr, Design, synthesis, anti-cancer evaluation and molecular docking study of novel 2,4-dichlorophe-noxymethyl-based derivatives linked to nitrogenous heterocyclic ring systems as potential CDK-2 inhibitors, J. Mol. Struct., 2022, 1247, 131285 CrossRef CAS.
- E. S. Nossier, S. M. El-hallouty and E. R. Zaki, Synthesis, anticancer evaluation and molecular modeling of some substituted thiazolidinonyl and thiazolyl pyrazole derivatives, Int. J. Pharm. Pharm. Sci., 2015, 7, 353–359 CAS.
- I. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, E. S. Nossier, M. M. Anwar and H. A. Radwan, Chemical synthesis and molecular docking study of new thiazole, thiophene, and thieno[2,3-d]pyrimidine derivatives as potential antiproliferative and antimicrobial agents, J. Mol. Struct., 2022, 15, 133926 CrossRef.
- Y. Sun, Y. Liu, X. Ma and H. Hu, The influence of cell cycle regulation on chemotherapy, Int. J. Mol. Sci., 2021, 22(13), 6923 CrossRef CAS PubMed.
- I. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. Gad-Elkareem, S. S. Abd El-Karim and E. S. Nossier, Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitors, RSC Adv., 2022, 12(1), 561–577 RSC.
- E. A. Abd El-Meguid, G. O. Moustafa, H. M. Awad, E. R. Zaki and E. S. Nossier, Novel benzothiazole hybrids targeting EGFR: Design, synthesis, biological evaluation and molecular docking studies, J. Mol. Struct., 2021, 1240, 130595 CrossRef.
- Z. M. Alamshany, N. Y. Tashkandi, I. M. Othman, M. M. Anwar and E. S. Nossier, New thiophene, thienopyridine and thiazoline-based derivatives: Design, synthesis and biological evaluation as antiproliferative agents and multitargeting kinase inhibitors, Bioorg. Chem., 2022, 127, 105964 CrossRef CAS PubMed.
- K. M. Amin, Y. M. Syam, M. M. Anwar, H. I. Ali, T. M. Abdel-Ghani and A. M. Serry, Synthesis and molecular docking study of new benzofuran and furo[3,2-g]chromone-based cytotoxic agents against breast cancer and p38α MAP kinase inhibitors, Bioorg. Chem., 2018, 76, 487–500 CrossRef CAS PubMed.
- W. A. El-Sayed, F. M. Alminderej, M. M. Mounier, E. S. Nossier, S. M. Saleh and A. F. Kassem, Novel 1,2,3-triazole-coumarin hybrid glycosides and their tetrazolyl analogues: design, anticancer evaluation and molecular docking targeting EGFR, VEGFR-2 and CDK-2, Molecules, 2022, 27(7), 2047 CrossRef CAS PubMed.
- W. A. El-Sayed, F. M. Alminderej, M. M. Mounier, E. S. Nossier, S. M. Saleh and A. F. Kassem, New 1,2,3-Triazole-Coumarin-Glycoside Hybrids and Their 1,2,4-triazolyl thioglycoside analogs targeting mitochondria apoptotic pathway: Synthesis, anticancer activity and docking simulation, Molecules, 2022, 27(17), 5688 CrossRef CAS PubMed.
- Y. M. Syam, M. M. Anwar, S. S. Abd El-Karim, K. M. Elokely and S. H. Abdelwahed, New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies, Molecules, 2022, 27(15), 4924 CrossRef CAS PubMed.
- E. A. Abd El-Meguid, E. M. M. El-Deen, G. O. Moustafa, H. M. Awad and E. S. Nossier, Synthesis, anticancer evaluation and molecular docking of new benzothiazole scaffolds targeting FGFR-1, Bioorg. Chem., 2022, 119, 105504 CrossRef CAS PubMed.
- A. E. G. E. Amr, E. A. Elsayed, M. A. Al-Omar, H. O. Eldin Badr, E. S. Nossier and M. M. Abdallah, Design, synthesis, anticancer evaluation and molecular modeling of novel estrogen derivatives, Molecules, 2019, 24(3), 416 CrossRef PubMed.
- Z. He, W. Y. Ma, T. Hashimoto, A. M. Bode, C. S. Yang and Z. Dong, Induction of apoptosis by caffeine is mediated by the p53, Bax, and caspase 3 pathways, Cancer Res., 2003, 63(15), 4396–4401 CAS.
- G. O. Moustafa, A. Shalaby, A. M. Naglah, M. M. Mounier, H. El-Sayed, M. M. Anwar and E. S. Nossier, Synthesis, characterization, in vitro anticancer potentiality, and antimicrobial activities of novel peptide–glycyrrhetinic-acid-based derivatives, Molecules, 2021, 26(15), 4573 CrossRef CAS PubMed.
- A. S. Hassan, G. O. Moustafa, H. M. Awad, E. S. Nossier and M. F. Mady, Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids, ACS Omega, 2021, 6, 12361–12374 CrossRef CAS PubMed.
- H. H. Elganzory, F. M. Alminderej, M. N. El-Bayaa, H. M. Awad, E. S. Nossier and W. A. El-Sayed, Design, synthesis, anticancer activity and molecular docking of new 1,2,3-triazole-based glycosides bearing 1,3,4-thiadiazolyl, indolyl and arylacetamide scaffolds, Molecules, 2022, 27(20), 6960 CrossRef CAS PubMed.
- A. E. G. E. Amr, R. E. Abdel Mageid, M. El-Naggar, A. M. Naglah, E. S. Nossier and E. A. Elsayed, Chiral Pyridine-3,5-bis-(L-phenylalaninyl-L-leucinyl) Schiff Base Peptides as Potential Anticancer Agents: Design, synthesis, and molecular docking studies targeting lactate dehydrogenase-A, Molecules, 2020, 25(5), 1096 CrossRef PubMed.
- M. H. Norman, L. Liu, M. Lee, N. Xi, I. Fellows, N. D. D'Angelo, C. Dominguez, K. Rex, S. F. Bellon, T. S. Kim and I. Dussault, Structure-based design of novel class II c-Met inhibitors: 1. Identification of pyrazolone-based derivatives, J. Med. Chem., 2012, 55(5), 1858–1867 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7(1), 1–13 CrossRef PubMed.
- E. M. Mohi El-Deen, E. S. Nossier and E. A. Karam, New quinazolin-4(3H)-one derivatives incorporating hydrazone and pyrazole scaffolds as antimicrobial agents targeting DNA gyraze enzyme, Sci. Pharm., 2022, 90(3), 52 CrossRef CAS.
- I. M. Othman, M. A. Gad-Elkareem, A. E. G. E. Amr, M. A. Al-Omar, E. S. Nossier and E. A. Elsayed, Novel heterocyclic hybrids of pyrazole targeting dihydrofolate reductase: design, biological evaluation and in silico studies, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1491–1502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01931d |
| This journal is © The Royal Society of Chemistry 2023 |