
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCuI-mediated synthesis of 1-aryl-5,6,7-trimethoxybenzimidazoles as potent antitubulin agents†
Cong-Min Peng‡
a,
Shih-Wei Wang‡ bcd,
Yi-Lin Hwanga,
Wen-Chun Sunbe,
Li-Pin Chiufg,
Yi-Ting Liua,
Yu-Wei Lai*hi and
Hsueh-Yun Lee*ajk
bcd,
Yi-Lin Hwanga,
Wen-Chun Sunbe,
Li-Pin Chiufg,
Yi-Ting Liua,
Yu-Wei Lai*hi and
Hsueh-Yun Lee*ajk
aSchool of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, Taiwan. E-mail: hyl@tmu.edu.tw
bInstitute of Biomedical Sciences, MacKay Medical College, New Taipei City, Taiwan
cDepartment of Medicine, MacKay Medical College, New Taipei City, Taiwan
dSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung, Taiwan
eDivision of Colon and Rectal Surgery, Department of Surgery, MacKay Memorial Hospital, Taipei, Taiwan
fDivision of General Surgery, Taipei City Hospital Chushing Branch, Taipei, Taiwan
gGeneral Education Center, University of Taipei, Taipei, Taiwan
hDivision of Urology, Taipei City Hospital Renai Branch, Taipei, Taiwan
iDepartment of Urology, School of Medicine and Shu-Tien Urological Science Research Center, National Yang Ming Chiao Tung University, Taipei, Taiwan
jPhD Program in Drug Discovery and Development Industry, College of Pharmacy, Taipei Medical University, Taipei, Taiwan
kTMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan
First published on 28th April 2023
Abstract
In situ CuI-mediated cyclization methodology helped yield benzimidazoles with different substitution manner, such as 1,2-diarylbenzimidazoles (4 and 5) and 1-arylbenzimidazoles (6–15). The result of structure–activity relationship (SAR) study confirmed the significance of the 5,6,7-trimethoxybenzimidazole moiety, and the representative derivatives (8–10) exhibited marked antiproliferative activity against A549, HCT-116, and PC-3 cells; in addition, they are able to inhibit the polymerization of tubulin. Among them, compound 10 inhibited the growth of A549, HCT-116, and PC-3 cells with a mean IC50 value of 0.07 μM, and its IC50 value of tubulin polymerization is 0.26 μM.
Introduction
Cell proliferation is a crucial part of cancer progression, and mitosis is what fuels the spread of cancer cells.1 Microtubules are polymers of tubulin and play an essential role in cell mitosis. It has been found that disrupting the function of microtubules, either by inhibiting or stabilizing the polymerization of tubulins can slow down the progress of mitosis.2 Natural products such as vincristine, paclitaxel, and colchicine have been recognized as anti-mitotic agents and are used clinically. Combretastatin A4 (CA-4, 1) was isolated from the bark of the South African tree Combretum caffrum in 1989 and was evaluated as a potent anti-mitotic agent.3 It binds to the colchicine binding site and inhibits tubulin polymerization.4 Structurally, CA-4 possesses a Z-olefinic bridge connecting the 3,4,5-trimethoxyphenyl (A ring) and the 3′-hydroxy-4′-methoxyphenyl (B-ring). It has been shown that the 3,4,5-trimethoxyphenyl and the Z-olefinic bridge as well as the 4′-methoxy group of the B ring are all exceptionally important for binding at the colchicine site and contribute to the potency of the compound. The 3′-hydroxy group of the B ring; however, could be replaced by other substituents without significantly decreasing its potency. The promising anti-mitotic activity of the CA-4 has attracted numerous efforts on the development of potent antitubulin agents (Fig. 1).5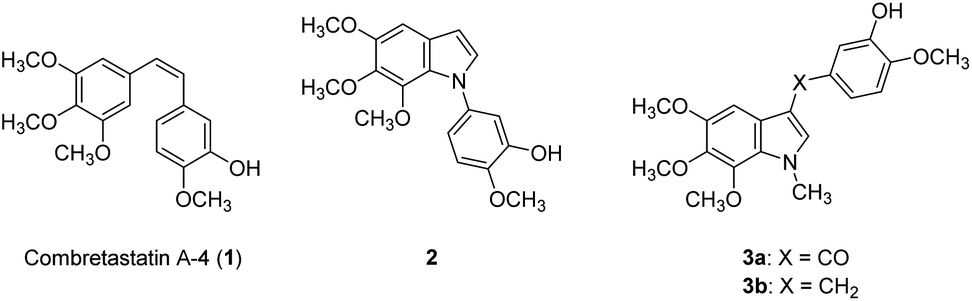 | ||
| Fig. 1 Combretastatin A4 (1, CA-4) and 5,6,7-trimethoxyindoles (2 and 3). | ||
There have been several attempts to modify the A ring, particularly replacing the three methoxy groups on the phenyl ring, but the results were mostly underwhelming.6–9 Nonetheless, the A ring still has the potential to be modified by substituting the phenyl ring into heterocyclic rings. For instance, our previous study found 5,6,7-trimethoxyindole as a surrogate of 3,4,5-trimethoxyphenyl moiety of CA-4 and the resulting compound 2 showed comparable antiproliferative activity to that of CA-4.10 In 2013, Zhao et al. developed C3-substituted N-methyl-5,6,7-trimethoxyindoles (3) showing potent antimitotic and vascular disrupting activity.11 Benzimidazoles are found in a wide range of biologically active compounds, including antimicrobials, antivirals, antiprotozoals, antiproliferative agents, and anti-inflammatory agents;12,13 in addition, it is considered as bioisostere of indole in the field of medicinal chemistry.14 Given the above examples, this study attempted to apply a bioisosterism strategy toward compound 2; as a result, a series of 1,2-diarylbenzimidazoles (4 and 5) and 1-arylbenzimidazoles (6–15, Fig. 2) were synthesized and investigated for biological activities.
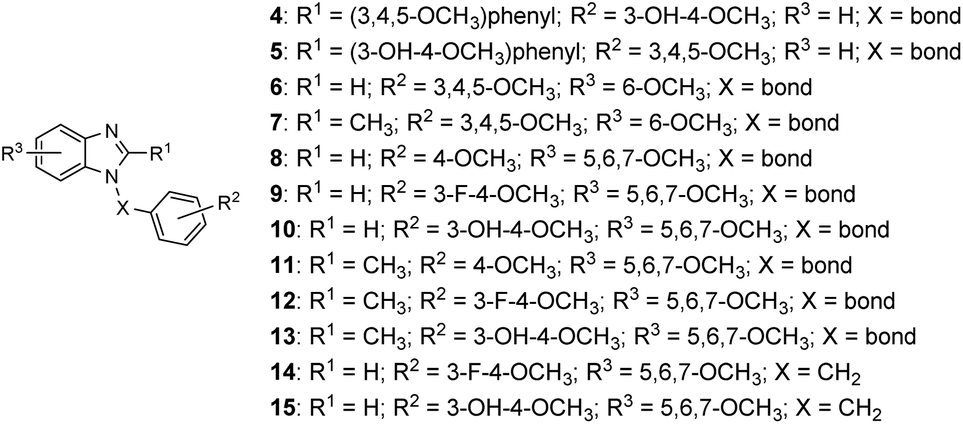 | ||
| Fig. 2 Structures of 1,2-diarylbenzimidazoles (4 and 5) and 1-arylbenzimidazoles (6–15). | ||
The trial using copper-mediated Ullmann arylation of benzimidazole failed the preparation of anticipated compounds. In addition, the synthesis of 5,6,7-trimethoxybenzimidazole is not feasible; therefore, an alternative methodology is required. Yu et al. reported a copper catalyzed approach to obtain 1,2-disubstituted benzimidazoles from imidoyl chlorides and o-haloanilines.15 Liubchak and co-workers published a similar methodology to generate annulated benzimidazoles through CuI-mediated cyclization of amidines which were prepared from lactam and o-bromoaniline.16 Inspired by the cited studies, we applied the reported synthetic methods to the preparation of designed benzimidazole derivatives.
Results and discussion
Chemistry
The synthesis of compounds 4 and 5 is shown in Scheme 1. Compounds 16 and 17 were reacted with triflic anhydride followed by the treatment of o-bromoaniline, and the resulting amidine intermediates 18 and 19 underwent copper catalyzed cyclization by reacting with CuI in the presences of 1,10-phenanthroline to afford 1,2-diarylbenzimidazoles 20 and 21. Removal of benzyl (Bn) protecting group yielded the anticipated compounds 4 and 5.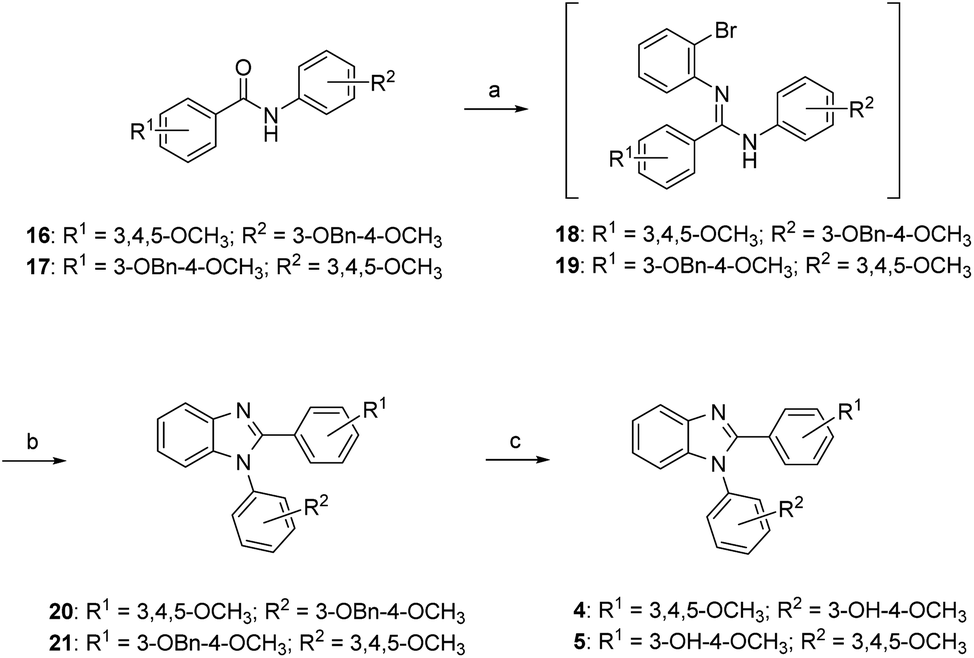 | ||
| Scheme 1 Synthetic approaches to compounds 4 and 5: (a) (i) Tf2O, 2,6-lutidine, DCM, rt; (ii) o-bromoaniline, rt; (b) CuI, 1,10-phenanthroline, ACN, reflux; (c) 10% Pd/C, H2, MeOH, rt. | ||
Compounds 6 and 7 bearing a 3,4,5-trimethoxyphenyl moiety at N1 position were synthesized according to the synthetic route shown in Scheme 2. The reaction of 3,4,5-trimethoxyaniline (22) with formic acid and 2-bromo-4-methoxyaniline in the presence of POCl3 gave a unstable amidine intermediate (23) which was subjected to CuI-mediated cyclization to afford compound 6. On the other hand, the acetamide (25) obtained from the reaction of 3,4,5-trimethoxyaniline (22) with acetic anhydride underwent similar synthetic route toward 6 to generate 2-methylbenzimidazole 7.
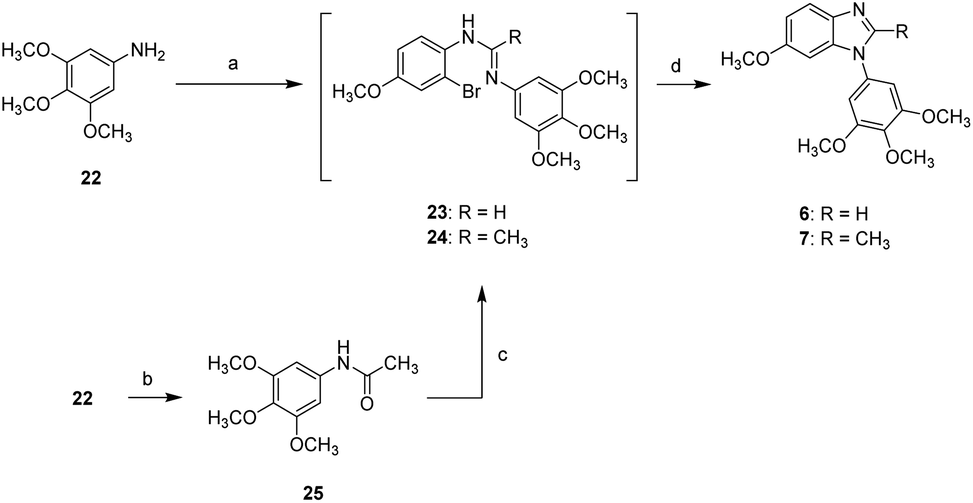 | ||
| Scheme 2 Synthetic approaches to compounds 6 and 7: (a) (i) formic acid, acetic anhydride, DCM, rt; (ii) 2-bromo-4-methoxyaniline, POCl3, toluene, 0 °C to 90 °C; (b) acetic anhydride, DCM, 0 °C to rt; (c) 2-bromo-4-methoxyaniline, POCl3, toluene, 0 °C to 90 °C; (d) CuI, 1,10-phenanthroline, K2CO3, ACN, reflux. | ||
Schemes 3–5 show the synthesis of N1-substituted 5,6,7-trimethoxybenzimidazoles (8–15). Compounds 26 and 27 were converted into the corresponding formamides followed by reaction with 2-bromo-3,4,5-trimethoxyaniline in the presence of POCl3, yielding the amidine intermediates (28 and 30). Following the similar approach, compound 31 generated from 22 through amidation and bromination was converted into the amidine intermediate 29. All amidine intermediates (28–30) subsequently underwent CuI-mediated cyclization to afford compounds 8, 9, and 32. The removal of Bn protecting group of 32 was carried out by hydrogenation to yield compound 10 (Scheme 3).
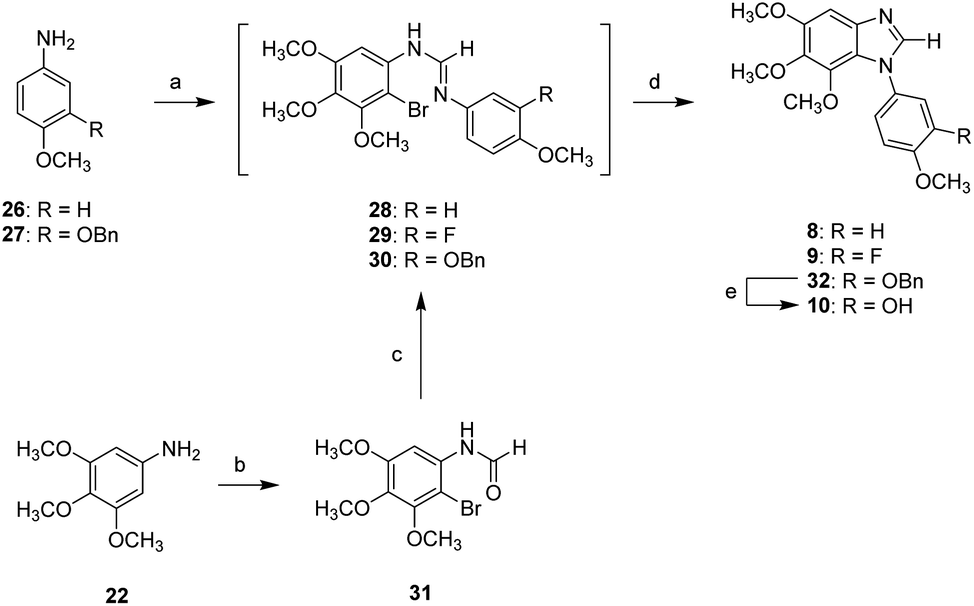 | ||
| Scheme 3 Synthetic approaches to compounds 8–10: (a) (i) formic acid, acetic anhydride, DCM, rt; (ii) 2-bromo-3,4,5-trimethoxyaniline, POCl3, toluene, 0 °C to 90 °C; (b) (i) formic acid, acetic anhydride, DCM, rt; (ii) NBS, DCM, 0 °C; (c) 3-fluoro-4-methoxyaniline, POCl3, toluene, 0 °C to 50 °C; (d) CuI, 1,10-phenanthroline, K2CO3, ACN, reflux; (e) 5% Pd/C, H2, MeOH, rt. | ||
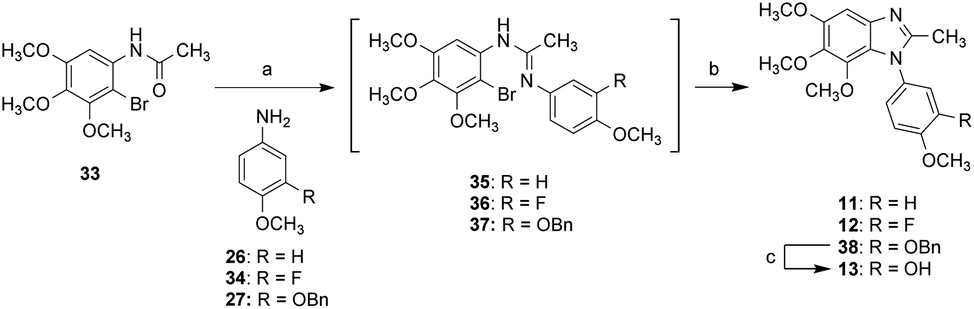 | ||
| Scheme 4 Synthetic approaches to compounds 11–13: (a) for 35 and 37: POCl3, 26 or 27, toluene, 0 °C to 90 °C; for 36: triflic anhydride, 34, 2,6-lutidine, DCM, 0 °C to 50 °C; (b) CuI, 1,10-phenanthroline, K2CO3, ACN, reflux; (c) 10% Pd/C, H2, MeOH, rt. | ||
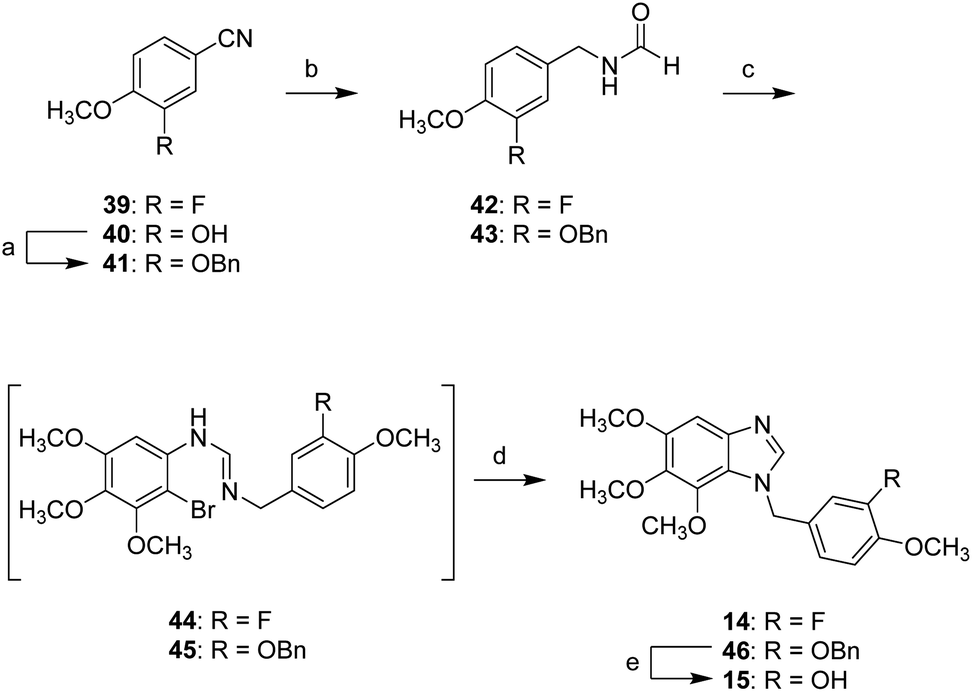 | ||
| Scheme 5 Synthetic approaches to compounds 14 and 15: (a) benzyl bromide, K2CO3, ACN, reflux; (b) (i): LAH, dry THF, 0 °C to rt; (ii): formic acid, acetic anhydride, DCM, rt; (c) (i) 2-bromo-3,4,5-trimethoxyaniline, POCl3, toluene, 0 °C to 90 °C; (d) CuI, 1,10-phenanthroline, K2CO3, ACN, reflux; (e) BCl3 in hexane, DCM, 0 °C. | ||
Scheme 4 shows the synthesis of compounds 11–13. The reaction of compound 33 with POCl3 or triflic anhydride was followed by addition of 26, 27, and 34 to yield the amidine intermediates 35–37. Without further purification, the resulting intermediates (35–37) underwent CuI-mediated cyclization to afford 2-methyl-5,6,7-benzimidazoles 11, 12, and 38. Compound 38 underwent deprotection of Bn group to afford compound 13.
Scheme 5 shows the synthesis of compounds 14 and 15 possessing a methylene linkage. The hydroxyl group of 40 was masked by a Bn group. Then, two benzonitriles (39 and 41) were reduced by LAH, and the resulting products were subsequently reacted with formic acid in the presence of acetic anhydride to afford the corresponding formamides 42 and 43. Conversion of the resulting formamides (42 and 43) to amidine intermediates (44 and 45) followed the similar route to 7 generated compounds 14 and 46. The removal of benzyl group of 46 was carried out by using BCl3 to yield compound 15.
Biological evaluation
All synthesized compounds (4–15) were tested for antiproliferative activity against A549, HCT-116, and PC-3 cells (Table 1). The result from 4 and 5 indicated that replacement of the Z-geometry C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of CA-4 with 1,2-diarylbenzimidazole moiety led to loss of cellular activity. Compounds 6 and 7 bearing 3,4,5-trimethoxyphenyl moiety at N1 position showed no effect on tested cell lines, whereas compounds 8, 9, and 10 exhibited marked cellular activity. This comparison revealed that 1-aryl-5,6,7-trimethoxybenzimidazole moiety is favored; however, there are some limitations as follows. The C2-methyl group led to dramatic loss of cellular activity, from the comparison of 8, 9, and 10 with 11, 12, and 13. In addition, the insertion of a methylene group between N-aryl group and benzimidazole (13 and 14) resulted in marked decrease of activity. Compounds 8, 9, and 10 showed potent antiproliferative activity. Compound 8 possessing a 4-methoxyphenyl group showed antiproliferative activity against A549, HCT-116, and PC-3 cells with GI50 values of 0.4, 0.24, and 0.8 μM, respectively. Addition of an F atom at 3′-position, which results in compound 9, made a slight increase of cellular activity against A549 and HCT-116 cells, as well as three times of cytotoxicity for PC-3 cell than compound 8. Replacement of F atom with –OH (10) contributed approximately 3-fold enhancement of cytotoxic activity. Compound 10 exhibited potent activity against A549, HCT-116, and PC-3 cells with GI50 values of 0.08, 0.05, and 0.09 μM, respectively. In addition, the comparison of 8, 9, and 10 showed that the trend of influence of C3′-substitution is –OH > –F > –H.
C bond of CA-4 with 1,2-diarylbenzimidazole moiety led to loss of cellular activity. Compounds 6 and 7 bearing 3,4,5-trimethoxyphenyl moiety at N1 position showed no effect on tested cell lines, whereas compounds 8, 9, and 10 exhibited marked cellular activity. This comparison revealed that 1-aryl-5,6,7-trimethoxybenzimidazole moiety is favored; however, there are some limitations as follows. The C2-methyl group led to dramatic loss of cellular activity, from the comparison of 8, 9, and 10 with 11, 12, and 13. In addition, the insertion of a methylene group between N-aryl group and benzimidazole (13 and 14) resulted in marked decrease of activity. Compounds 8, 9, and 10 showed potent antiproliferative activity. Compound 8 possessing a 4-methoxyphenyl group showed antiproliferative activity against A549, HCT-116, and PC-3 cells with GI50 values of 0.4, 0.24, and 0.8 μM, respectively. Addition of an F atom at 3′-position, which results in compound 9, made a slight increase of cellular activity against A549 and HCT-116 cells, as well as three times of cytotoxicity for PC-3 cell than compound 8. Replacement of F atom with –OH (10) contributed approximately 3-fold enhancement of cytotoxic activity. Compound 10 exhibited potent activity against A549, HCT-116, and PC-3 cells with GI50 values of 0.08, 0.05, and 0.09 μM, respectively. In addition, the comparison of 8, 9, and 10 showed that the trend of influence of C3′-substitution is –OH > –F > –H.
| Compound | Cell type (GI50 ± SDa, μM) | ||
|---|---|---|---|
| A549 | HCT116 | PC-3 | |
| a SD: standard deviation. GI50: 50% growth inhibition.b Data from ref. 17. | |||
| 4 | >10 | >10 | >10 |
| 5 | 7.30 ± 0.10 | 6.30 ± 0.10 | 7.75 ± 0.05 |
| 6 | >10 | >10 | >10 |
| 7 | >10 | >10 | >10 |
| 8 | 0.40 ± 0.09 | 0.24 ± 0.01 | 0.80 ± 0.03 |
| 9 | 0.27 ± 0.02 | 0.19 ± 0.01 | 0.29 ± 0.02 |
| 10 | 0.08 ± 0.01 | 0.05 ± 0.02 | 0.09 ± 0.01 |
| 11 | >10 | >10 | >10 |
| 12 | >10 | >10 | >10 |
| 13 | >10 | >10 | >10 |
| 14 | 8.60 ± 0.70 | 7.80 ± 0.50 | 8.25 ± 0.15 |
| 15 | >10 | >10 | >10 |
| 1b | 0.02 | 0.079 | 0.01 |
Compounds 8, 9, and 10 were tested for their effects on tubulin polymerization (Fig. 3). Despite their displaying weaker tubulin polymerization inhibition than CA-4, compounds 8, 9, and 10 are able to inhibit polymerization of tubulin in a manner of dose-dependent. The influence of substitution on tubulin polymerization is consistent with that on cellular activity shown in Table 1. The addition of 3′-F resulted in 2.6-fold increase of activity, as compared with compound 8. The placement of 3′-F (9) with the 3′-OH (10) led to a 3.2-fold enhancement of tubulin polymerization inhibition. Among them, 10 showed marked tubulin polymerization inhibition in a dose-dependent manner, with an IC50 value of 0.26 μM.
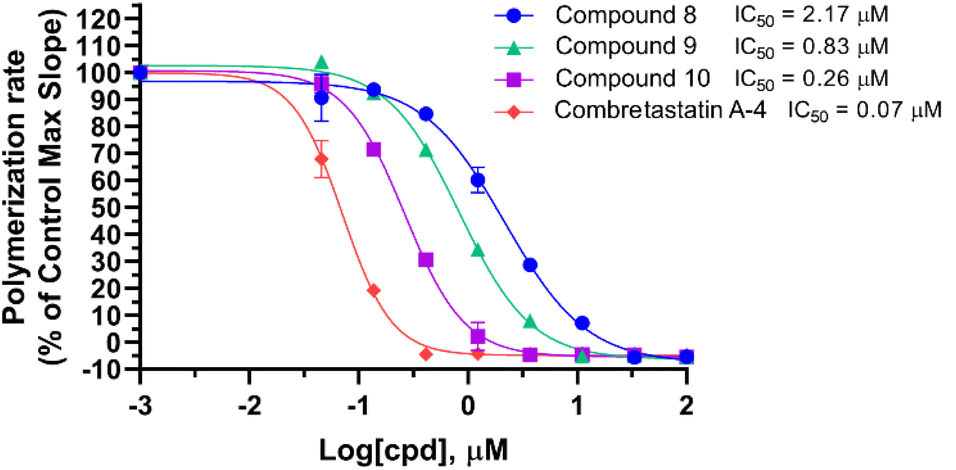 | ||
| Fig. 3 The effect of compounds 8–10 on in vitro tubulin polymerization. | ||
Fig. 4 demonstrates the overlap of compound 10 and CA-4, which was conducted using the Glide module of Schrödinger Maestro. The trimethoxybenzimidazole overlapped with 3,4,5-trimethoxyphenyl moiety of CA-4. In addition, the Z-geometry was retained by benzimidazole ring that had 3′-OH-4′-methoxyphenyl motif directed toward the same orientation as that of CA-4. This result of docking study explains the contribution of benzimidazole to maintain the similar steric alignment.
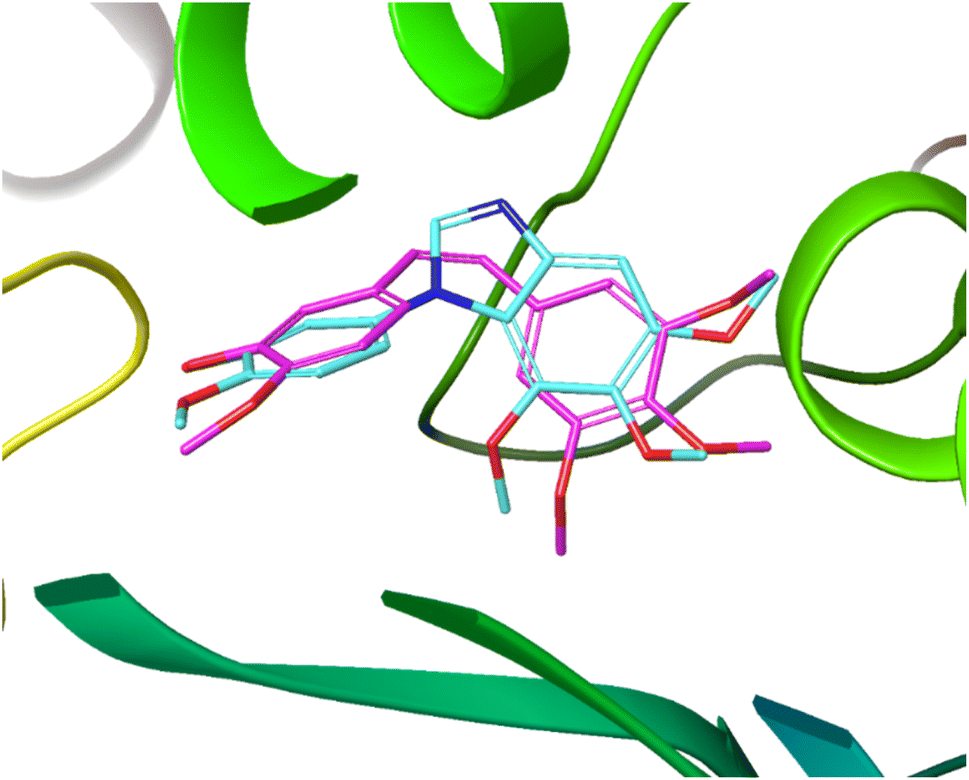 | ||
| Fig. 4 Overlap of 10 (aqua) and CA-4 (pink) at the binding site of tubulin (PDB ID: 5LYJ). | ||
Conclusions
In this study, CuI-mediated cyclization via amidine intermediate fulfilled the synthesis of 1,2-diarylbenzimidazoles (4 and 5) and 1-arylbenzimidazoles (6–15), which helped explore the influence of pattern of substitution (1-aryl and 1,2-diaryl) on biological activity. The result showed that 1-arylbenzimidazole scaffold, especially 1-aryl-5,6,7-trimethoxybenzimidazole (8–10), contributed potent cellular activity against tested cancer cells and were able to inhibit tubulin polymerization. Among them, compound 10 showed the most potent cytotoxicity against A549, HCT-116, and PC-3 cells with GI50 values of 0.08, 0.05, and 0.09 μM, respectively. The IC50 value of 10 toward tubulin polymerization is 0.26 μM. Together with our previous study, it is concluded that 5,6,7-trimethoxyindole and 5,6,7-trimethoxybenzimidazole effectively modified A-ring part of CA-4. Recent studies on antitubulin agents have gradually shifted from single pharmacological function to dual activity.18 Therefore, 5,6,7-trimethoxyindole and 5,6,7-trimethoxybenzimidazole could be templates for advanced development such as multifunctional tubulin agents in the future.Experimental
Chemistry
Nuclear magnetic resonance spectra were obtained with a Bruker DRX-300 spectrometer operating at 300 MHz and with Agilent DD2 600 MHz NMR Spectrometer operating at 600 MHz, with chemical shifts reported in parts per million (ppm, δ) downfield from TMS, an internal standard. High-resolution mass spectra (HRMS) were measured with a JEOL (JMS-700) electron impact (EI) mass spectrometer. Purity of the final compounds was determined using a Hitachi 2000 series HPLC system using C-18 column (Agilent ZORBAX Eclipse XDB-C18 5 μm. 4.6 mm × 150 mm) and was found to be ≥95% in all cases. Flash column chromatography was carried out using silica gel (Merck Kieselgel 60, no. 9385, 230–400 mesh ASTM). All reactions were done under an atmosphere of dry nitrogen.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA = 1:2; Rf = 0.25) to afford crude formamide (2.53 g). To a mixture of resulting formamide (1.14 g, 5.42 mmol) in toluene (10 mL) was added POCl3 (0.60 mL, 2.48 mmol) under ice bath. After stirring at room temperature for 30 minutes, a mixture of 2-bromo-4-methoxyaniline (1.64 g, 8.13 mmol) in toluene (10 mL) was added and stirred at 90 °C for 6 h. The reaction mixture was diluted with H2O, neutralized with 5 N NaOH(aq), extracted with EA, and purified by flash column chromatography on silica gel (Hex:EA = 2:1; Rf = 0.28) to afford crude formamidine 23 (0.55 g). A mixture of the resulting product 23 (0.40 g, 1.00 mmol), CuI (0.01 g, 0.50 mmol), 1,10-phenanthroline (0.09 g, 0.50 mmol), and K2CO3 (0.14 g, 1.00 mmol) in ACN (10 mL) was stirred at reflux for 6 h. The reaction mixture was purified by flash column chromatography on silica gel (Hex:EA = 1:2; Rf = 0.29) and the residue was washed with ether, DCM, and MeOH to afford compound 6 as a pink solid (0.05 g, 3%). Mp = 166.1–166.8 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.38 (s, 1H), 7.64 (d, J = 9.0 Hz, 1H), 7.12 (d, J = 2.4 Hz, 1H), 6.96 (s, 2H), 6.92 (dd, J = 2.6, 9.0 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 156.6, 153.6, 142.5, 137.9, 136.6, 133.8, 131.7, 120.3, 111.8, 101.5, 94.0, 60.1, 56.2, 55.5. HRMS (ESI) for C17H19N2O4 [M + H]+ calculated 315.1345, found 315.1344.:EA = 1:1; Rf = 0.33) to afford crude amidine 24 (0.65 g). The crude product (0.41 g, 1.0 mmol) was dissolved in ACN (10 mL), and then CuI (0.10 g, 0.5 mmol), 1,10-phenanthroline (0.09 g, 0.57 mmol), and K2CO3 (0.14 g, 1.0 mmol) were added. The resulting mixture was heated to reflux for 6 h, and then purified by flash column chromatography on silica gel (Hex:EA = 1:1; Rf = 0.25) and the residue was washed with MeOH to afford compound 7 as a white solid (0.06 g, 13%). Mp = 158.7–159.6 °C; 1H NMR (300 MHz, CDCl3) δ 7.47–7.50 (d, J = 8.7 Hz, 1H), 6.80–6.84 (m, 3H), 6.67 (d, J = 2.4 Hz, 1H), 3.82 (s, 6H), 3.76 (s, 3H), 3.72 (s, 3H), 2.41 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.8, 153.5, 150.4, 137.3, 136.6, 136.5, 131.2, 118.8, 110.8, 104.6, 93.7, 60.1, 56.2, 55.5, 14.1. HRMS (ESI) for C18H21N2O4 [M + H]+ calculated 329.1501, found 329.1500.:EA = 1:2; Rf = 0.25) to afford crude formamide (2.53 g). The resulting product was dissolved in DCM (20 mL), and then NBS (1.1 eq.) was added under ice bath. After stirring at 0 °C for 30 minutes, the reaction mixture was purified by flash column chromatography on silica gel (Hex:EA = 1:2; Rf = 0.28) to afford compound 31 as a pale yellow solid (1.44 g, 72%). 1H NMR (300 MHz, CDCl3) δ 8.47 (d, J = 1.8 Hz, 1H), 7.97 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.86 (s, 3H). 13C NMR (75 MHz, CD3OD) δ 162.1, 154.1, 152.2, 141.5, 132.7, 104.6, 102.1, 61.5, 56.6.:EA = 4:1; Rf = 0.45) to afford compound 41 as a white solid (2.97 g, 93%). 1H NMR (300 MHz, DMSO-d6) δ 7.32–7.49 (m, 7H), 7.14 (d, J = 8.4 Hz, 1H), 5.13 (s, 2H), 3.84 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 153.0, 147.8, 136.3, 128.4, 128.0, 127.9, 126.6, 119.2, 115.8, 112.3, 102.5, 70.1, 55.8.:EA = 1:2; Rf = 0.14) to afford compound 42 as a white solid (1.50 g, 55%). 1H NMR (300 MHz, DMSO-d6) δ 8.46 (br, 1H), 8.11 (d, J = 1.8 Hz, 1H), 7.01–7.14 (m, 3H), 4.23 (d, J = 6.0 Hz, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 161.0, 151.3 (d, J = 242.1 Hz), 146.0 (d, J = 10.4 Hz), 132.0 (d, J = 5.9 Hz), 123.4 (d, J = 3.5 Hz), 114.9 (d, J = 17.8 Hz), 113.7 (d, J = 1.8 Hz), 55.9, 39.8 (d, J = 1.4 Hz).
:EA = 1:2; Rf = 0.25) to afford crude formamide (2.53 g). To a mixture of resulting formamide (1.14 g, 5.42 mmol) in toluene (10 mL) was added POCl3 (0.60 mL, 2.48 mmol) under ice bath. After stirring at room temperature for 30 minutes, a mixture of 2-bromo-4-methoxyaniline (1.64 g, 8.13 mmol) in toluene (10 mL) was added and stirred at 90 °C for 6 h. The reaction mixture was diluted with H2O, neutralized with 5 N NaOH(aq), extracted with EA, and purified by flash column chromatography on silica gel (Hex:EA = 2:1; Rf = 0.28) to afford crude formamidine 23 (0.55 g). A mixture of the resulting product 23 (0.40 g, 1.00 mmol), CuI (0.01 g, 0.50 mmol), 1,10-phenanthroline (0.09 g, 0.50 mmol), and K2CO3 (0.14 g, 1.00 mmol) in ACN (10 mL) was stirred at reflux for 6 h. The reaction mixture was purified by flash column chromatography on silica gel (Hex:EA = 1:2; Rf = 0.29) and the residue was washed with ether, DCM, and MeOH to afford compound 6 as a pink solid (0.05 g, 3%). Mp = 166.1–166.8 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.38 (s, 1H), 7.64 (d, J = 9.0 Hz, 1H), 7.12 (d, J = 2.4 Hz, 1H), 6.96 (s, 2H), 6.92 (dd, J = 2.6, 9.0 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 156.6, 153.6, 142.5, 137.9, 136.6, 133.8, 131.7, 120.3, 111.8, 101.5, 94.0, 60.1, 56.2, 55.5. HRMS (ESI) for C17H19N2O4 [M + H]+ calculated 315.1345, found 315.1344.:EA = 1:1; Rf = 0.33) to afford crude amidine 24 (0.65 g). The crude product (0.41 g, 1.0 mmol) was dissolved in ACN (10 mL), and then CuI (0.10 g, 0.5 mmol), 1,10-phenanthroline (0.09 g, 0.57 mmol), and K2CO3 (0.14 g, 1.0 mmol) were added. The resulting mixture was heated to reflux for 6 h, and then purified by flash column chromatography on silica gel (Hex:EA = 1:1; Rf = 0.25) and the residue was washed with MeOH to afford compound 7 as a white solid (0.06 g, 13%). Mp = 158.7–159.6 °C; 1H NMR (300 MHz, CDCl3) δ 7.47–7.50 (d, J = 8.7 Hz, 1H), 6.80–6.84 (m, 3H), 6.67 (d, J = 2.4 Hz, 1H), 3.82 (s, 6H), 3.76 (s, 3H), 3.72 (s, 3H), 2.41 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.8, 153.5, 150.4, 137.3, 136.6, 136.5, 131.2, 118.8, 110.8, 104.6, 93.7, 60.1, 56.2, 55.5, 14.1. HRMS (ESI) for C18H21N2O4 [M + H]+ calculated 329.1501, found 329.1500.:EA = 1:2; Rf = 0.25) to afford crude formamide (2.53 g). The resulting product was dissolved in DCM (20 mL), and then NBS (1.1 eq.) was added under ice bath. After stirring at 0 °C for 30 minutes, the reaction mixture was purified by flash column chromatography on silica gel (Hex:EA = 1:2; Rf = 0.28) to afford compound 31 as a pale yellow solid (1.44 g, 72%). 1H NMR (300 MHz, CDCl3) δ 8.47 (d, J = 1.8 Hz, 1H), 7.97 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.86 (s, 3H). 13C NMR (75 MHz, CD3OD) δ 162.1, 154.1, 152.2, 141.5, 132.7, 104.6, 102.1, 61.5, 56.6.:EA = 4:1; Rf = 0.45) to afford compound 41 as a white solid (2.97 g, 93%). 1H NMR (300 MHz, DMSO-d6) δ 7.32–7.49 (m, 7H), 7.14 (d, J = 8.4 Hz, 1H), 5.13 (s, 2H), 3.84 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 153.0, 147.8, 136.3, 128.4, 128.0, 127.9, 126.6, 119.2, 115.8, 112.3, 102.5, 70.1, 55.8.:EA = 1:2; Rf = 0.14) to afford compound 42 as a white solid (1.50 g, 55%). 1H NMR (300 MHz, DMSO-d6) δ 8.46 (br, 1H), 8.11 (d, J = 1.8 Hz, 1H), 7.01–7.14 (m, 3H), 4.23 (d, J = 6.0 Hz, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 161.0, 151.3 (d, J = 242.1 Hz), 146.0 (d, J = 10.4 Hz), 132.0 (d, J = 5.9 Hz), 123.4 (d, J = 3.5 Hz), 114.9 (d, J = 17.8 Hz), 113.7 (d, J = 1.8 Hz), 55.9, 39.8 (d, J = 1.4 Hz).Biology
The absorbance data acquired for this compound screen was analyzed as follows:
The maximal velocity (maximum slope, called Vmax) of tubulin polymerization was determined using Spectramax Software. The mean of duplicate maximum slope determinations at each concentration were then normalized to reactions that contained only DMSO (1% final concentration, i.e. buffer control), and were converted to % activity using the following formula: % polymerization rate = 100 × [Vmax(cmpd)/Vmax(DMSO)].
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ministry of Science and Technology, Taiwan (grant no. MOST111-2320-B-038-046). Taipei City Hospital (TCH-10601-62-024; TPCH-108-19).References
- M. A. Feitelson, A. Arzumanyan, R. J. Kulathinal, S. W. Blain, R. F. Holcombe, J. Mahajna, M. Marino, M. L. Martinez-Chantar, R. Nawroth, I. Sanchez-Garcia, D. Sharma, N. K. Saxena, N. Singh, P. J. Vlachostergios, S. Guo, K. Honoki, H. Fujii, A. G. Georgakilas, A. Bilsland, A. Amedei, E. Niccolai, A. Amin, S. S. Ashraf, C. S. Boosani, G. Guha, M. R. Ciriolo, K. Aquilano, S. Chen, S. I. Mohammed, A. S. Azmi, D. Bhakta, D. Halicka, W. N. Keith and S. Nowsheen, Semin. Cancer Biol., 2015, 35(Suppl), S25–S54 CrossRef PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- G. R. Pettit, S. B. Singh, E. Hamel, C. M. Lin, D. S. Alberts and D. Garcia-Kendall, Experientia, 1989, 45, 209–211 CrossRef CAS PubMed.
- G. M. Tozer, C. Kanthou, C. S. Parkins and S. A. Hill, Int. J. Exp. Pathol., 2002, 83, 21–38 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- K. Gaukroger, J. A. Hadfield, N. J. Lawrence, S. Nolan and A. T. McGown, Org. Biomol. Chem., 2003, 1, 3033–3037 RSC.
- A. B. Maya, C. Pérez-Melero, C. Mateo, D. Alonso, J. L. Fernández, C. Gajate, F. Mollinedo, R. Peláez, E. Caballero and M. Medarde, J. Med. Chem., 2005, 48, 556–568 CrossRef CAS PubMed.
- G. R. Pettit, M. D. Minardi, H. J. Rosenberg, E. Hamel, M. C. Bibby, S. W. Martin, M. K. Jung, R. K. Pettit, T. J. Cuthbertson and J. C. Chapuis, J. Nat. Prod., 2005, 68, 1450–1458 CrossRef CAS PubMed.
- G. R. Pettit, C. R. Anderson, D. L. Herald, M. K. Jung, D. J. Lee, E. Hamel and R. K. Pettit, J. Med. Chem., 2003, 46, 525–531 CrossRef CAS PubMed.
- H. Y. Lee, J. Y. Chang, L. Y. Chang, W. Y. Lai, M. J. Lai, K. H. Shih, C. C. Kuo, C. Y. Chang and J. P. Liou, Org. Biomol. Chem., 2011, 9, 3154 RSC.
- D. G. Zhao, J. Chen, Y. R. Du, Y. Y. Ma, Y. X. Chen, K. Gao and B. R. Hu, J. Med. Chem., 2013, 56, 1467–1477 CrossRef CAS PubMed.
- R. S. Keri, A. Hiremathad, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 19–65 CrossRef PubMed.
- S. Tahlan, S. Kumar, S. Kakkar and B. Narasimhan, BMC Chem., 2019, 13, 66 CrossRef PubMed.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- H. Yu, M. S. Zhang and L. R. Cui, Chin. Chem. Lett., 2012, 23, 573–575 CrossRef CAS.
- K. Liubchak, K. Nazarenko and A. Tolmachev, Tetrahedron, 2012, 68, 2993–3000 CrossRef CAS.
- A. Defant and I. Mancini, Molecules, 2019, 24, 2224 CrossRef CAS PubMed.
- W. Shuai, G. Wang, Y. Zhang, F. Bu, S. Zhang, D. D. Miller, W. Li, L. Ouyang and Y. Wang, J. Med. Chem., 2021, 64, 7963–7990 CrossRef CAS PubMed.
- C. L. Yu, M. S. Weng, W. C. Chen, K. T. Chien, C. W. Chi, C. H. Chung, C. W. Huang, P. C. Wang, C. C. Chen, A. C. Tsai, S. C. Liu and S. W. Wang, Int. J. Mol. Sci., 2021, 22, 2930 CrossRef CAS PubMed.
- S. W. Wang, M. H. Lin, F. C. Hsu, M. C. Chen, J. P Liou, Y. T. Liu, S. S. Chen and H. Y. Lee, Bioorg. Med. Chem., 2020, 28, 115250 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC purity data, 1H NMR spectra and 13C NMR spectra of compounds 4–15. See DOI: https://doi.org/10.1039/d3ra01927f |
| ‡ Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |