
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew cyclic glycolipids from Silene succulenta promote in vitro MCF-7 breast carcinoma cell apoptosis by cell cycle arrest and in silico mitotic Mps1/TTK inhibition†
Sarah A. Badawy a,
Ahmed R. Hassan*a,
Rawah H. Elkousyb,
Salwa A. Abu El wafab and
Abd-El salam I. Mohammadc
a,
Ahmed R. Hassan*a,
Rawah H. Elkousyb,
Salwa A. Abu El wafab and
Abd-El salam I. Mohammadc
aMedicinal and Aromatic Plants Department, Desert Research Center, El-Matariya 11753, Cairo, Egypt. E-mail: ahmedhassan@drc.gov.eg
bDepartment of Pharmacognosy, Faculty of Pharmacy (for Girls), Al-Azhar University, Nasr City 11651, Cairo, Egypt
cDepartment of Pharmacognosy, Faculty of Pharmacy (for Boys), Al-Azhar University, Nasr City 13129, Cairo, Egypt
First published on 20th June 2023
Abstract
In vitro anticancer screening of Silene succulenta Forssk. aerial parts (Caryophyllaceae) showed that the n-hexane fraction was a highly effective fraction against breast carcinoma cell lines (MCF-7) with IC50 = 15.5 μg mL−1. The bioactive-guided approach led to the isolation of two new cyclic glucolipids from the n-hexane fraction, identified as a 1,2′-cyclic ester of 11-oxy-(6′-O-acetyl-β-D-glucopyranosyl) behenic acid (1) as a C-11 epimeric mixture and 11(R)-oxy-(β-D-glucopyranosyl)-1,2′-cyclic ester of behenic acid (2). An in vitro cytotoxicity study showed the potential suppression of MCF-7 cells with IC50 values of 11.7 ± 0.04 and 6.6 ± 0.01 μg mL−1 for compounds 1 and 2, respectively, compared to doxorubicin (IC50 = 3.83 ± 0.01 μg mL−1). Accordingly, only cell cycle tracking for the most active compound (2) was assessed. The cell cycle investigation showed that compound 2 altered the cell cycle at G0/G1, S, and G2/M phases in MCF-7 treated cells. In addition, its powerful apoptotic ability resulted in a significant increase in the early and late stages of apoptosis. Moreover, molecular docking analysis, which was performed against the anticancer mitotic (or spindle assembly) checkpoint target Mps1 kinase, showed that the two new cyclic glycolipids (1 and 2) possess high binding affinity of −7.7 and − 7.6 kcal mol−1, respectively, compared to its ATP ligand. Overall, this report emphasizes that natural cyclic glycolipids can be used as potential antitumour breast cancer agents.
1. Introduction
Breast cancer (BC) spreads worldwide and is the main reason for cancer-related deaths in women aged from 20 to 59.1 Numerous studies have shown how the development of BC depends on hormones and how effective hormone antagonists are.2,3 Although estrogen receptor (ER)-positive cancers are still best managed by hormone therapy, this form of treatment efficacy is severely constrained by developed endocrine resistance.4–6 Additionally, in triple-negative breast cancer (TNBC), where the ER and human epidermal growth factor receptor-2 (HER2) targets are inactive, it loses its efficacy.7 In addition, the long-term use of chemotherapy medications primarily reduces the effectiveness of the medication because of the development of chemoresistance and undesirable side effects.8 Therefore, new methods of treatment must be discovered. Mitotic kinases, such as CDKs, PLKs, and Auroras, which are overexpressed in propagated malignant cells, are important for cell division and are believed to be appealing targets for stopping cancer cell growth.9 In the clinic, some inhibitors of mitotic kinase are being studied. Herein, we were interested in evaluating our new compounds as Mps1/TTK inhibitors. Mps1 kinase, a serine/threonine and tyrosine protein kinase with dual selectivity, is required for the correct chromosomal attachment to the mitotic spindle as it is a pivotal part of SAC (spindle assembly checkpoint). For the promotion of faithful chromosome segregation, it regulates the interaction of microtubules and kinetochores in prometaphase until all chromosomes are aligned properly.10–12 Chromosome missegregation caused by Mps1 inhibition has been linked to cell death. Along with its function in mitosis, Mps1 also plays a role in centrosome duplication, checkpoint response of DNA damage, meiosis, cell differentiation, and chromosomal mutability.9 Mps1 is expressed considerably more strongly in solid tumors than in normal cells. It is considered to be one of the leading 25 genes overexpressed in malignancies.13–15 Excluding the testis and placenta, it is expressed at modest levels in normal organs but is extremely elevated in breast cancer cells.16Natural compounds may be utilized in combined treatment therapies because of their potential as anti-cancer and structural resemblance to synthetic drugs.17 The Caryophyllaceae family, which comprises more than 2600 species in 80 genera, is widely distributed in temperate climate zones. Most species of this family, more than 750 species, belong to the genus Silene.18–20 Silene (Caryophyllaceae) is a blooming plant that is mainly spread out in Africa, North America and Eurasia.18 For both pharmacological and ethnomedicinal studies, Silene is one of the most extensively investigated genera in the Caryophellaceae family. Species of Silene exhibit various pharmacological activities, such as anti-inflammatory,21 antimicrobial,22 antiviral,23 hepatoprotection,24 and antioxidant.25 Ethnopharmacological study on the Silene genus shows that its plants have anticancer properties, including Silene firma Siebold & Zucc. Rohrb. root extract, Silene viridiflora L. methanol extract, and Silene fortunei Vis. root extract.26 In addition, the plant under investigation, Silene succulenta Forssk., which grows on the seashores of various Mediterranean islands and in North Africa, was indexed as a medicinal plant, and its strong cytotoxic effect on RAW cells was indicated.27 Our previous study on the total hydro-methanolic extract and its fractions of S. succulenta aerial parts against different cancer cell lines revealed that the highest activity was against breast carcinoma cell lines.28
Moreover, the previously reported phytochemical studies on Silene genus plants led to the isolation of many phytoconstituents from different chemical classes comprising benzenoids,29 cyclic fatty acyl glycosides,30 flavonoids,31 phytoecdysteroids,32,33 terpenoids,34 terpenoidal saponins,35 and sterols.36 For glycolipid structures isolated from Caryophyllaceae plants, a series of gallicasides A–H from Silene gallica was reported,30 and a series of glomerasides A–N from Cerastium glomeratum was discovered.37 These uncommon cyclic glycolipid structures inspired us to investigate phytochemically the Caryophyllaceous plant (S. succulenta) in more detail in addition to continuing our phytochemical and biological studies on the interesting Egyptian desert plants growing on the Northwestern coast of Egypt38 as well as the search for anticancer candidates from the promising wild plants.39–41
According to these previous studies, the real intention was to explore the cytotoxic activity of S. succulenta aerial parts against MCF-7. Moreover, the isolation and identification of some constituents responsible for this activity were done via bioassay guided fractionation using various chromatographic and spectroscopic techniques. Furthermore, an assessment of the potential cytotoxicity mechanism for these isolated compounds was considered.
2. Results and discussion
2.1. Chemistry
| No. | 1a | 2 |
|---|---|---|
| 2 | 2.39 (m, 2H) | 2.40 (t, J = 7.3 Hz, 2H) |
| 3 | 1.66 (m, 1H) | 1.60 (m, 1H) |
| 1.80 (m, 1H) | 1.80 (m, 1H) | |
| 4–21 | 1.60–1.22 (m, 34H) | 1.59–1.21 (m, 34H) |
| 11 | 3.57 (m, 1H) | 3.56 (m, 1H) |
| 22 | 0.87 (t, J = 6.8 Hz, 3H) | 0.87 (t, J = 6.7 Hz, 3H) |
| 1′ | 4.47 (d, J = 7.9 Hz, 1H) | 4.48 (d, J = 7.9 Hz, 1H) |
| 2′ | 4.72 (dd, J = 9.4, 7.8 Hz, 1H) | 4.72 (t, J = 8.5 Hz, 1H) |
| 3′ | 3.57 (m, 1H) | 3.58 (brt, J = 9.2 Hz, 1H) |
| 4′ | 3.46 (m, 1H) | 3.64 (brt, J = 9.2 Hz, 1H) |
| 5′ | 3.44 (m, 1H) | 3.32 (m, 1H) |
| 6′ | 4.39 (dd, J = 12.0, 4.3 Hz, 1H) | 3.85 (m, 2H, overlapped) |
| 4.32 (dd, J = 12.0, 2.2 Hz, 1H) | ||
| Ac (CH3) | 2.10 (s, 3H) | — |
| No. | 1a | 2 |
|---|---|---|
| 1 | 174.73 | 174.36 |
| 2 | 32.32 | 32.16 |
| 3 | 22.70 | 22.51 |
| 4–19 | 35.04 | 34.88 |
| 34.45 | 34.21 | |
| 32.41 | 29.97 | |
| 29.89 | 29.90 | |
| 29.85 | 29.85 | |
| 29.81 | 29.82 | |
| 29.77 | 29.80 | |
| 29.50 | 29.78 | |
| 27.25 | 29.50 | |
| 27.16 | 27.13 | |
| 26.61 | 26.22 | |
| 26.22 | 26.15 | |
| 25.60 | 25.68 | |
| 25.03 | 25.11 | |
| 24.24 | 24.15 | |
| 20 | 32.06 | 32.06 |
| 21 | 22.83 | 22.83 |
| 11 | 81.43 | 81.15 |
| 22 | 14.26 | 14.25 |
| 1′ | 100.87 | 100.85 |
| 2′ | 74.90 | 74.44 |
| 3′ | 76.28 | 75.89 |
| 4′ | 71.15 | 70.75 |
| 5′ | 73.34 | 75.11 |
| 6′ | 63.47 | 62.15 |
| Ac(CH3) | 20.98 | — |
| CH3CO | 171.74 | — |
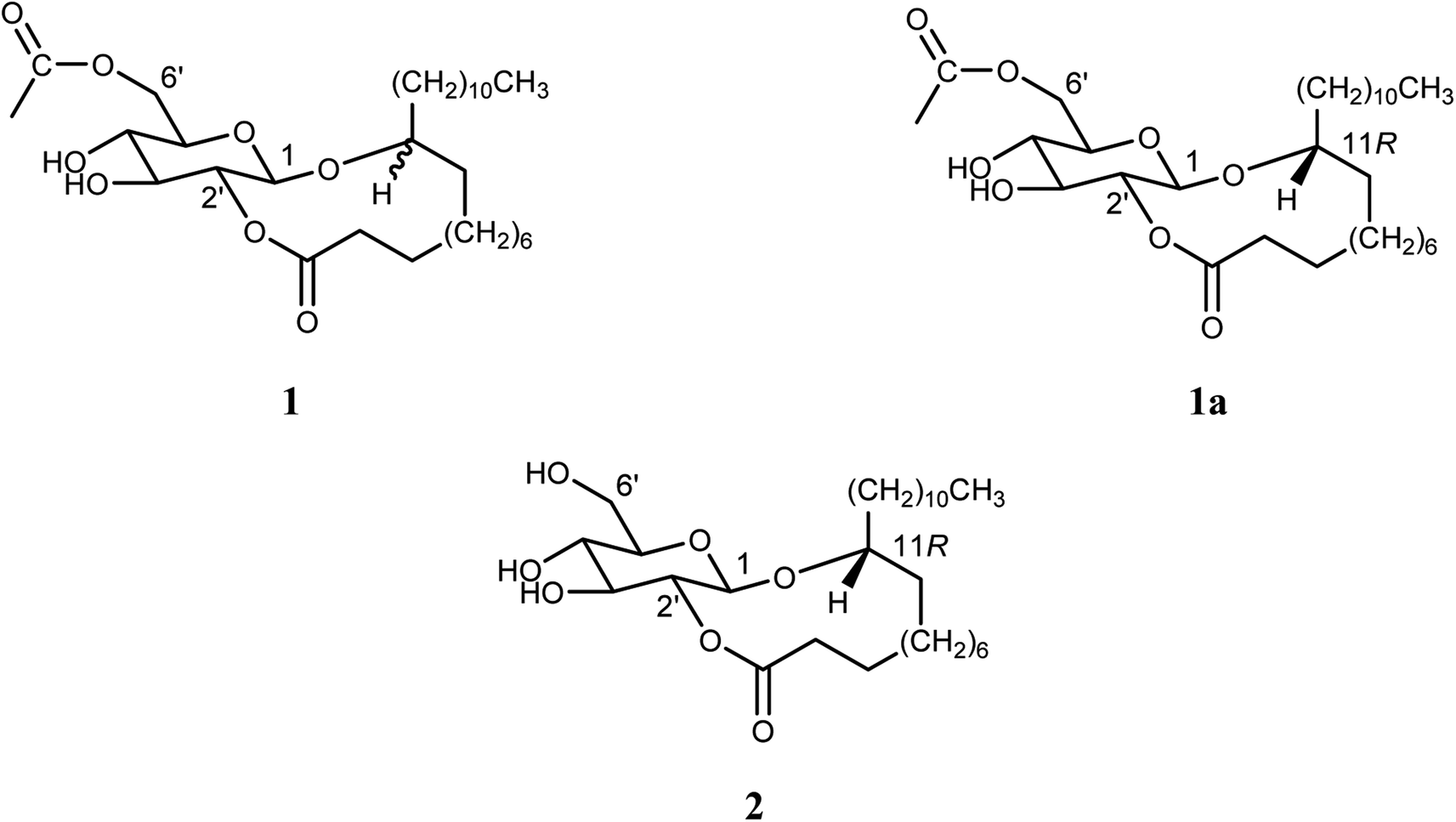 | ||
| Fig. 1 Compounds 1, 1a and 2. | ||
 | ||
| Fig. 2 Structure of hydroxy fatty acid methyl ester 6. | ||
Compound 2 (colorless oil) has a molecular ion peak m/z 501.3855 [M + H]+ that corresponds to C28H52O7. 1H NMR data (Table 1 and ESI Fig. S7†) have shown seven protons signals that represented a β-glucopyranosyl moiety; δH 4.48 (H-1′), 4.72 (H-2′), 3.58 (H-3′), 3.64 (H-4′), 3.32 (H-5′), 3.85 (H2-6′, overlapped). In addition, particular signals of the fatty acyl group were detected: terminal –CH3 protons that appeared triplet at δH 0.87, a chain of methylene protons from δH 1.21 to 1.59 and the attached CH2– protons to the CO– group at δH 2.40. Moreover, an oxymethine proton shifted downfield at δH 3.56 (m, H-11). The signals of 13C NMR (Table 2 and ESI Fig. S8†) confirmed the linearity of the mono-oxygenated fatty acyl moiety: δC 14.25 for terminal –CH3, and δC 81.15 and δH 3.56 for oxymethine. The degree of unsaturation in compound 2 indicates an additional site of cyclization. HMBC spectrum (ESI Fig. S10†) assured the two positions of cyclization; anomeric H-1′ at δH 4.48 correlated with the oxymethine carbon at δC 81.15 and H-2′ at δH 4.72 correlated with C-1 of the fatty acyl moiety at δC 174.36 through glycosidic bonds. According to the chemical formula, the oxygenated fatty acyl molecule has a carbon number of C22. The exact oxymethine position was C-11 of the docosanoyl (behenyl) moiety, which was determined using the same methods as compound 1. Therefore, methyl 11(R)-hydroxydocosanoate [methyl 11(R)-hydroxybehenate] was established. Based on previous findings, compound 2 was approved as 11(R)-oxy-(β-D-glucopyranosyl)-1,2′-cyclic ester of behenic acid.
2.2. Biological evaluation
| Sample | IC50 | |
|---|---|---|
| (μg mL−1) | (μM) | |
| Total hydro-methanolic extract | 18 ± 0.02 | — |
| n-Hexane fraction | 15.5 ± 0.01 | — |
| Ethyl acetate (EtOAc) fraction | 33 ± 0.007 | — |
| n-Butanol (n-BuOH) fraction | 33 ± 0.005 | — |
| 1 | 11.7 ± 0.04 | 21.53 |
| 2 | 6.6 ± 0.01 | 13.16 |
| Doxorubicin | 3.83 ± 0.01 | 7.04 |
As evidenced by the results, the n-hexane fraction and compounds 1 and 2 exhibited potential cytotoxic activity against the MCF-7 cell line with IC50 = 15.5 ± 0.01, 11.7 ± 0.04 and 6.6 ± 0.01 μg mL−1 comparable to doxorubicin (IC50 = 3.83 ± 0.01 μg mL−1).
Consequently, compound 2 was chosen for an additional study to clarify the mechanism behind its potent cytotoxic effect against MCF-7.
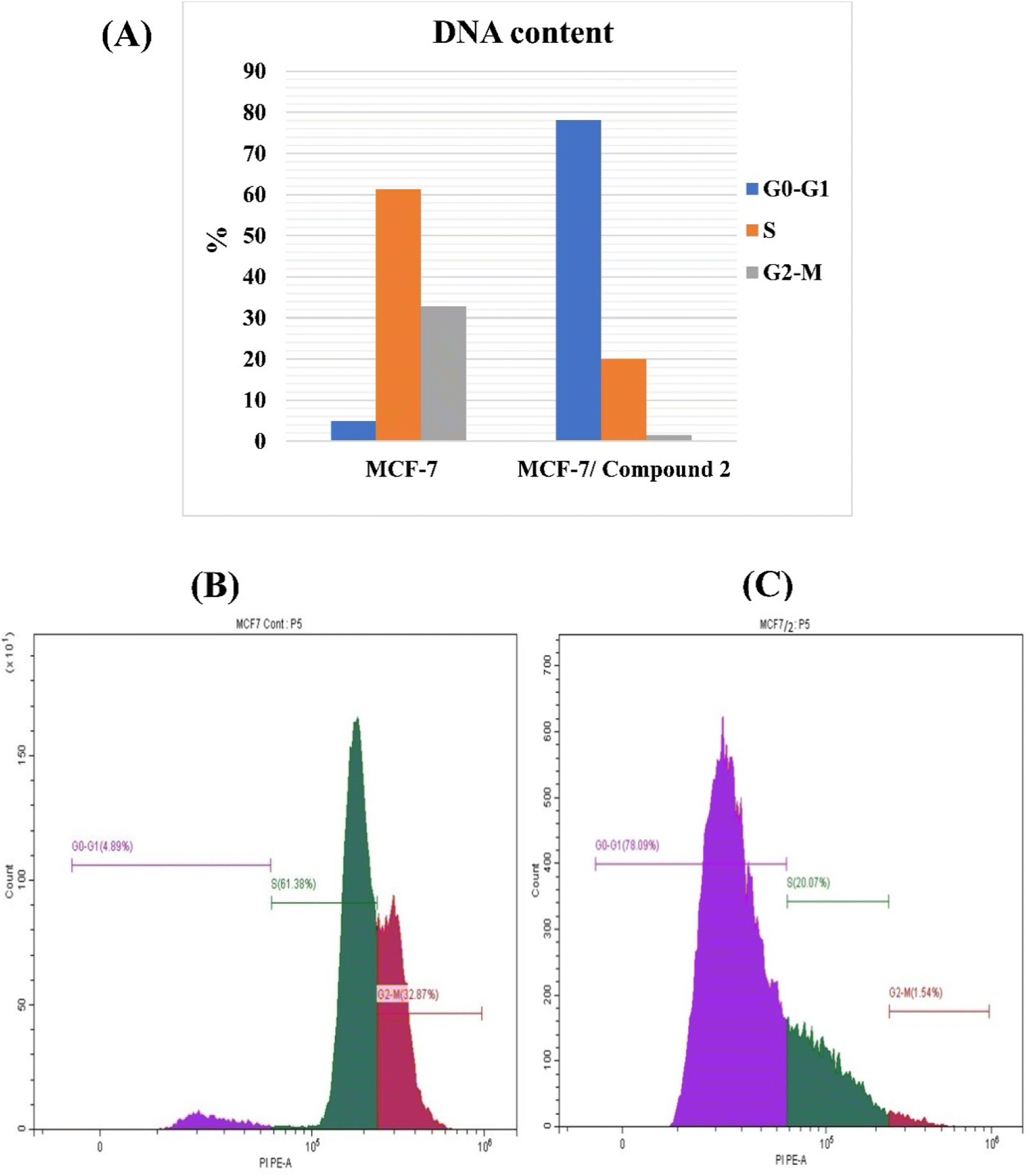 | ||
| Fig. 3 (A) Bar chart of DNA content distribution for both MCF-7 treated cells with 2 and controlled cells in G0/G1, S and G2/M phases. Distribution of DNA content in a cell cycle experiment using flow cytometry for PI-stained untreated (B) and treated MCF-7 cells (C). | ||
| Compound no. | DNA content | ||
|---|---|---|---|
| % G0–G1 | % S | % G2/M | |
| Control (MCF-7) | 4.89 ± 0.001 | 61.38 ± 0.81 | 32.87 ± 0.74 |
| MCF-7 treated with 2 | 78.09 ± 0.47 | 20.07 ± 0.47 | 1.54 ± 0.02 |
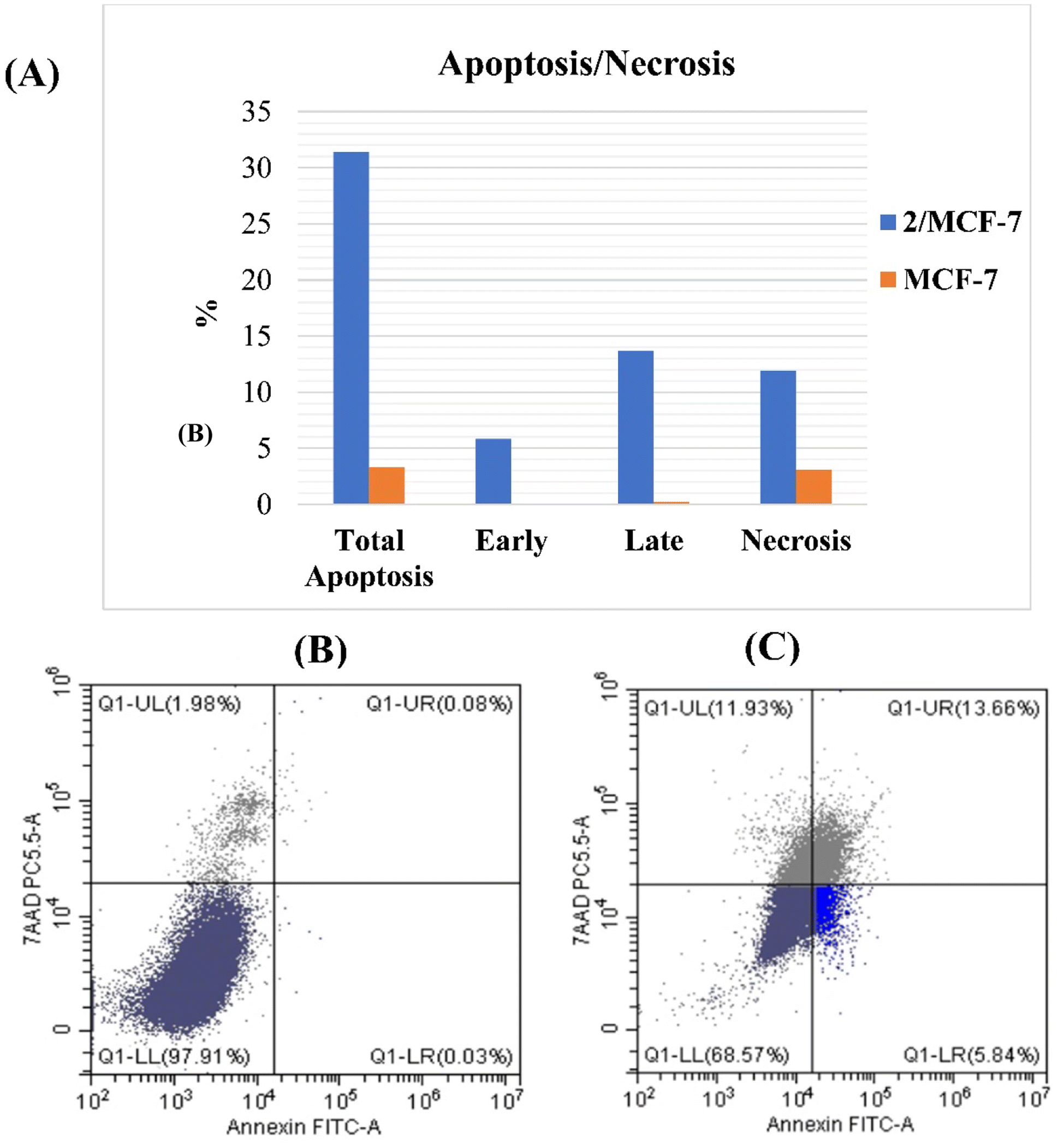 | ||
| Fig. 4 (A) Bar chart of percentages of apoptotic control cells and treated MCF-7 cells with compound 2 by annexin V-FITC/7-AAD flow cytometry analysis. Percentage of cell distribution between early apoptosis, late apoptosis, and necrotic stage of control MCF-7 cells (B) and treated MCF-7 cells with compound 2 (C). | ||
2.3. Molecular docking study
To clarify the interactions and binding affinities of compounds 1 and 2 inside the Mps1/TTK protein kinase active site, a molecular modeling analysis was conducted by applying AutoDock Vina. ATP re-docking revealed an identical co-crystallized ligand with RMSD equals 1.3 Å. As illustrated previously,46 the region of Mps1, which acts as the hinge, Glu603–Gly605, is the critical binding domain for the ATP adenine base. In addition, few preserved hydrogen bonds are formed between Gly605 (N1 and NH) and Glu603 (N6 and carbonyl oxygen). Additionally, in the C-terminal domain, van der Waals forces between the Ile586, Leu654, Val539, and Met602 residues with the adenine base of ATP are formed. The docked compounds 1 and 2 showed high binding affinity, which achieved docking energy scores of −7.7 and −7.6 kcal mol−1, respectively (Table 5). Docking of compound 1 in Mps1 at the ATP-binding site reveals a three-point hinge binding interaction between Gly605 (NH and OH as H-donors and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as H-bond acceptor) and the acetylated glucopyranosyl moiety (6′-acetyl CO and 4′-OH group as H-acceptor, and 4′-OH as H-donor) (Fig. 5). For compound 2, Gly605 (CO as H-acceptor) interacts with the glucopyranosyl part (3′-OH as H-donor) by one H-bond. In addition, various interactions are displayed in Fig. 5. These vital interactions direct the remaining cyclic fatty acid chain toward the ribose-binding pocket while fixing the compound in the receptor active site. Another intriguing aspect of these structures is the organized conformation of the activation loop, which is made up of the residues Met671 and Pro673, and which, along with the P-loop, forms an antiparallel β-sheet to obtain a distinct and Mps1-specific hydrophobic pocket in the ribose region.47 To conclude, the docking study indicated the contribution of Gly605 amino acid residue, which has a crucial role in Mps1/TTK kinase activity inhibition. Therefore, the cytotoxicity of these compounds is related to Mps1 kinase inhibition.
O as H-bond acceptor) and the acetylated glucopyranosyl moiety (6′-acetyl CO and 4′-OH group as H-acceptor, and 4′-OH as H-donor) (Fig. 5). For compound 2, Gly605 (CO as H-acceptor) interacts with the glucopyranosyl part (3′-OH as H-donor) by one H-bond. In addition, various interactions are displayed in Fig. 5. These vital interactions direct the remaining cyclic fatty acid chain toward the ribose-binding pocket while fixing the compound in the receptor active site. Another intriguing aspect of these structures is the organized conformation of the activation loop, which is made up of the residues Met671 and Pro673, and which, along with the P-loop, forms an antiparallel β-sheet to obtain a distinct and Mps1-specific hydrophobic pocket in the ribose region.47 To conclude, the docking study indicated the contribution of Gly605 amino acid residue, which has a crucial role in Mps1/TTK kinase activity inhibition. Therefore, the cytotoxicity of these compounds is related to Mps1 kinase inhibition.
| Compound no. | E-Score (kcal mol−1) | Amino acid residue | Distance [Å] | Functional group |
|---|---|---|---|---|
| 1 | −7.7 | Gly605 | 2.29 | CO (H-acceptor) |
| 2.32 | OH (H-acceptor) | |||
| 2.59 | OH (H-donor) | |||
| Cys604 | 2.49 | CO (H-acceptor) | ||
| Ile607 | 1.79 | CO (H-acceptor) | ||
| 2 | −7.6 | Gly605 | 2.20 | OH (H-donor) |
| Ile607 | 1.74 | CO (H-acceptor) |
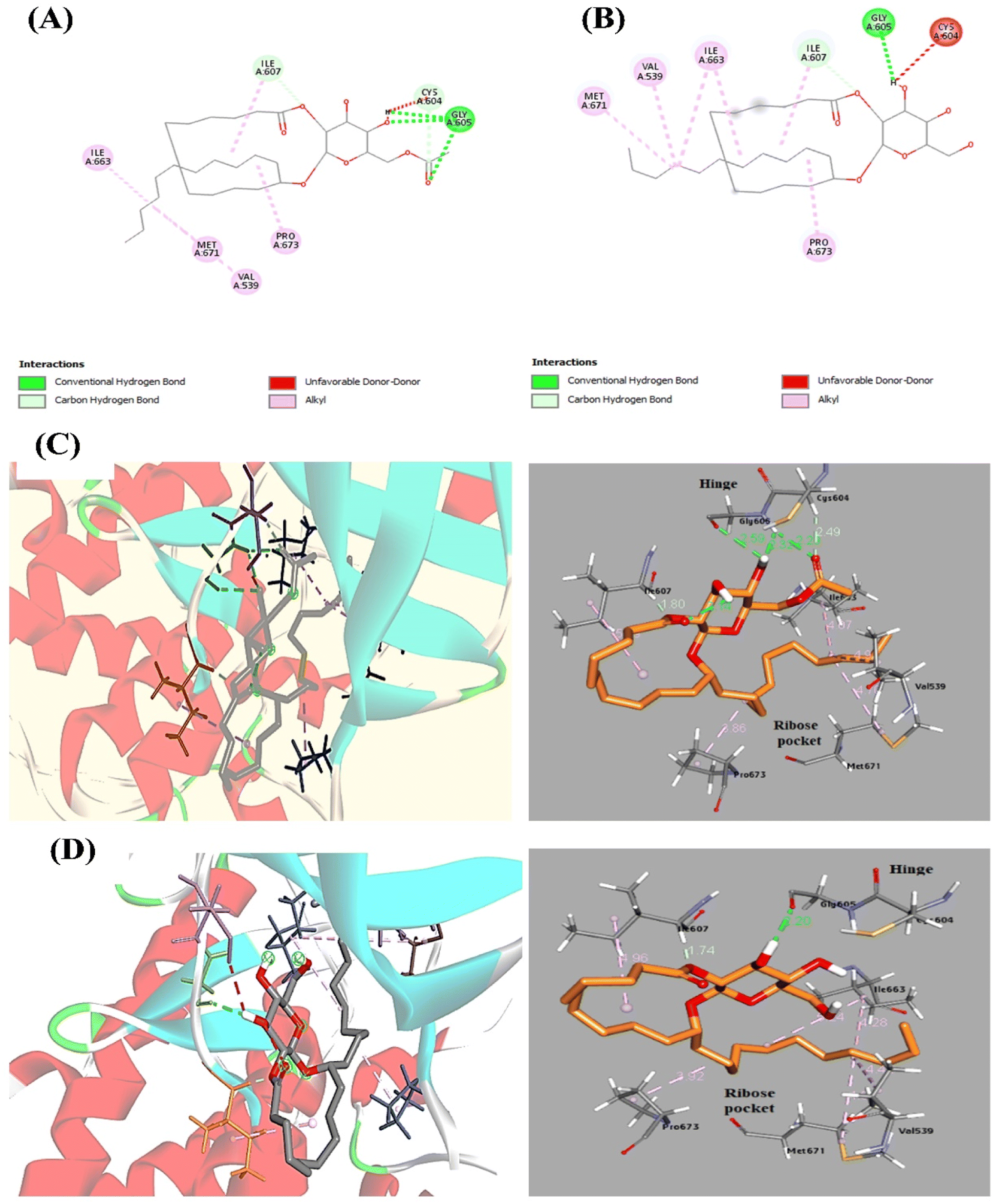 | ||
| Fig. 5 2D interactions of 1 (A) and 2 (B) with Mps1 amino acid residues showing the H-bond interactions with Gly605 residue inside the active site of the Mps1/TTK protein kinase receptor. 3D-conformational change in Mps1 upon binding with compounds 1 (C) and 2 (D) and interacted bond distances with amino acid residues. | ||
The inhibition of Mps1 has been proven to result in chromosomal missegregation, which leads to apoptosis. Mps1 has recently been investigated as a new target for small molecule cancer medications. Numerous new-structured Mps1 inhibitors have been created. To date, five Mps1 inhibitors have been used in clinical studies.46,48 Therefore, there is a lot of interest in creating Mps1 inhibitors with significant cytotoxic activity. Additionally, interest in plant-based cancer treatments has grown because of the enormous prospect of medicinal herbs as an origin for drug discovery.
Herein, we concentrate on the isolation of distinctive compounds, cyclic glycolipids, which are responsible for the cytotoxic activity of S. succulenta and also characteristic compounds of the Caryophyllaceae family.37 We choose this plant that belongs to the genus Silene of the Caryophyllaceae family to be under study. S. succulenta has attracted great interest owing to its lack of phytochemical and biological studies. Therefore, S. succulenta aerial part total extract and its fractions were screened for their cytotoxic activity against the breast carcinoma cell line (MCF-7). The n-hexane fraction showed the highest MCF-7 cytotoxic activity (IC50 value of 15.5 μg mL−1). Therefore, the n-hexane fraction was subjected to the isolation and purification of its major compounds.
The succulent hairy plant, S. succulenta, n-hexane fraction provides two new cyclic glycolipids (compounds 1 and 2). This is the first evidence provided by this study for the existence of 1,2′ cyclic glycosyloxy-docosanoate in plants, especially for the Caryophyllaceae family. The structures of these glycolipids differed significantly from those of S. gallica's gallicasides, which were 1,2′-cyclic esters of (β-D-glucopyranosyloxy) octadecanoic acid,30 and Stellaria dichotoma was found to contain 1,2′-cyclic esters of 5-(β-D-xylopyranosyloxy)fatty acids.49 1,6′ Cyclic glycosyloxy-docosanoic acid esters were also reported.37 Based on these previous findings, it was hypothesized that cyclic glycolipids are particular to some plants of the Caryophyllaceae family, which have a special ability to attach a hydroxyl moiety at different places in fatty acids for the creation of a glycosidic linkage.37 The chemical nature of the cyclic glucolipids 1 and 2 and the lack of their biological studies led us to the evaluation of their cytotoxic activity against the MCF-7 cell line compared to doxorubicin as a reference drug. A comparison between IC50 of doxorubicin (3.83 mg mL−1) and the newly isolated compound 2 (6.6 mg mL−1) showed that compound 2 has a higher anti-breast cancer activity and could be used as a complementary medicine for breast cancer patients.
To investigate the anti-proliferative mechanism, the most potent cytotoxic compound 2 was chosen to be the best representative for evaluating cell cycle and apoptosis. An analysis of cell cycle phases revealed a decrease in cells at S and G2/M phases whereas an increase at G0/G1 phase. In addition, the most active compound's powerful apoptotic ability resulted in a 195- and 171-fold increase in the early and late stages of apoptosis, respectively. The G0/G1 phase is a resting stage in the cell cycle, where cells are not dividing. If the G0/G1 phase increases after treatment, this could mean that the cells have been arrested in the cell cycle and are not progressing. This can be desirable in cancer therapy, as it can prevent cancer cells from dividing and multiplying. An increase in the G0/G1 phase can also indicate that cancer cells are undergoing apoptosis, or programmed cell death, which is a desirable effect of some chemotherapy drugs.7,8
The expected mechanism at the G2/M phase was evaluated using molecular docking, which uses the active side of the key enzyme Mps1/TTK protein kinase. Owing to the critical role of Mps1 in the spindle assembly checkpoint (SAC), the metaphase checkpoint, once there is any perturbation at the M phase of the cell cycle, it is responsible for arresting cell division.48,50 Although the presence of free hydroxyl groups and/or attached acetyl groups on the glucopyranoside moiety of the two studied components was shown to play a significant role in the Mps1 active binding site, docking studies suggested a competitive type of enzyme inhibition for compounds 1 and 2. However, the structure–activity relationship (SAR) between the two glycolipids can be inspected from the in vitro assay. This suggests that the acetylation of the hydroxyl group present at position-6′ of glucose moiety reduces glycolipid cytotoxicity against MCF-7. To corroborate our findings, additional in silico and in vitro research on this chemical class is advised.
3. Experimental
3.1. Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (100:0 → 30:70). The obtained sub-fractions were mixed according to their TLC patterns to yield the five main sub-fractions (H-I to H-V). Collective sub-fraction H-II (0.2 g, eluted with n-hexane:EtOAc, 7:3) and sub-fraction H-III (0.3 g, eluted with n-hexane–EtOAc, 1:1) were further purified using preparative NP TLC with toluene–EtOAc–formic acid, 10:4:1 and repurified by sephadex LH-20 CC, eluted with CH2Cl2–MeOH, 1:1 to afford 1 (13 mg) and 2 (16 mg), respectively. In the same way, the sub-fractions of EtOAc residue (7.58 g) were exposed to the CC of silica gel using n-hexane:EtOAc (100:0 → 30:70) and gathered to give the four main sub-fractions (E-I to E-IV). Collective sub-fraction E-III (0.4 g, eluted with n-hexane–EtOAc, 3:7) was purified using preparative NP TLC with DCM–MeOH, 9.8:0.2 and repurified by sephadex LH-20 CC using 100% MeOH to afford 3 (12 mg). Moreover, n-BuOH residue (27.77 g) was chromatographed on polyamide CC eluted by saturated n-BuOH with water to afford five collective sub-fractions (Bu-I to Bu-V). By crystallization followed by repurification using sephadex LH-20 CC (100% MeOH) of collective sub-fraction Bu-II, compound 4 (20 mg) was obtained. Furthermore, collective sub-fraction Bu-IV was re-chromatographed in a sequence of chromatographic techniques, including silica gel CC eluted with (EtOAc–MeOH–H2O, 70:5:4), followed by reversed-phase C18-silica gel CC using MeOH–H2O (1:1) and then purified on sephadex gel filtration (100% MeOH) to obtain compound 5 (6 mg).
:EtOAc (100:0 → 30:70). The obtained sub-fractions were mixed according to their TLC patterns to yield the five main sub-fractions (H-I to H-V). Collective sub-fraction H-II (0.2 g, eluted with n-hexane:EtOAc, 7:3) and sub-fraction H-III (0.3 g, eluted with n-hexane–EtOAc, 1:1) were further purified using preparative NP TLC with toluene–EtOAc–formic acid, 10:4:1 and repurified by sephadex LH-20 CC, eluted with CH2Cl2–MeOH, 1:1 to afford 1 (13 mg) and 2 (16 mg), respectively. In the same way, the sub-fractions of EtOAc residue (7.58 g) were exposed to the CC of silica gel using n-hexane:EtOAc (100:0 → 30:70) and gathered to give the four main sub-fractions (E-I to E-IV). Collective sub-fraction E-III (0.4 g, eluted with n-hexane–EtOAc, 3:7) was purified using preparative NP TLC with DCM–MeOH, 9.8:0.2 and repurified by sephadex LH-20 CC using 100% MeOH to afford 3 (12 mg). Moreover, n-BuOH residue (27.77 g) was chromatographed on polyamide CC eluted by saturated n-BuOH with water to afford five collective sub-fractions (Bu-I to Bu-V). By crystallization followed by repurification using sephadex LH-20 CC (100% MeOH) of collective sub-fraction Bu-II, compound 4 (20 mg) was obtained. Furthermore, collective sub-fraction Bu-IV was re-chromatographed in a sequence of chromatographic techniques, including silica gel CC eluted with (EtOAc–MeOH–H2O, 70:5:4), followed by reversed-phase C18-silica gel CC using MeOH–H2O (1:1) and then purified on sephadex gel filtration (100% MeOH) to obtain compound 5 (6 mg).
1,2′-Cyclic ester of 11-oxy-(6′-O-acetyl-β-D-glucopyranosyl) behenic acid (1). Colorless oil (13 mg); ESI-MS m/z 543.3905 [M + H]+ (calcd for C30H55O8+, 543.3897), spectroscopic identification of the major compound [11(R)-oxy-(6′-O-acetyl-β-D-glucopyranosyl) 1,2′-cyclic behanic acid ester] (1a); 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) are shown in Tables 1 and 2. For further analysis, a small portion of compound 1 was hydrolyzed completely to produce the corresponding hydroxy fatty acid and glucose.30 As glucose (water-soluble fraction) was identified by comparing it with an authentic glucose sample using TLC, the hydrolyzed hydroxy fatty acid portion was subjected to methylation, followed by silylation to obtain its corresponding TMS ether for GC-MS detection. NMR data of the minor epimer [see the ESI of compound 1 (ESI Fig. S2–S5)†].
1,2′-Cyclic ester of 11(R)-oxy-(β-D-glucopyranosyl) behenic acid (2). Colorless oil (16 mg); ESI-MS m/z: 501.3855 [M + H]+ (equivalent to C28H53O7+, 501.3791); 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) are depicted in Tables 1 and 2. Further investigation was carried out in the same manner as described for 1.
3.2. Evaluation of cytotoxic activity
3.3. Molecular docking
The docking was established using AutoDock vina modeling software to interpret binding affinities and bond interactions between compounds 1, 2 and the active site of Mps1/TTK protein kinase. Protein Data Bank (https://www.rcsb.org/structure/PDB ID 7CJA)54 provided us with the crystal structure of Mps1/TTK protein kinase. The 3D structures of the new compounds 1 and 2 were drawn by Chem3D Ultra 8.0, and the ligand 3D structure (ATP) was obtained from PubChem. Chimera 1.16 allows the application of energy minimization and protonation before docking to achieve the best results. Docking of the ligand was followed by compounds 1 and 2 inside the Mps1/TTK protein kinase active site relative to RMSD. Discovery Studio 2021 was used for the result processing. Selection of the conformer, which achieved good binding with the amino acid residues of the enzyme active pocket, and illustration of its hydrogen bonding interactions and their length were provided.3.4. Statistical analysis
The results of the cytotoxic activity and cell cycle experiments are shown as mean ± standard deviation of the mean for n = 3. By graphing the % inhibition versus concentration, a dose–response curve was created to obtain IC50.4. Conclusions
The results provided evidence that S. succulenta may be effective in the treatment of breast cancer (MCF-7). Additionally, our findings suggest that new glycolipids 1 and 2 are thought to be interesting cytotoxic alternatives for the development of newer, more efficient anti-breast cancer drugs. In vivo studies are recommended to confirm our conclusion. Furthermore, to the best of our knowledge, the three known flavonoids (3, 4, and 5) were reported for the first time in this plant.Author contributions
ARH, SAA and AIM designed the study. SAB and ARH performed the laboratory experiments. SAB, ARH, SAA and AIM analyzed the data. SAB wrote the original draft. ARH, RHE, SAA and AIM revised the manuscript and prepared the final version. The final manuscript was read and approved by all authors.Conflicts of interest
The authors state that they have no conflicts of interest.Acknowledgements
The authors would like to acknowledge Dr Omran Ghaly (Doctor Researcher of plant taxonomy, Desert Research Center, Egypt) for identifying the plant and we are also grateful to NMR unit of Faculty of Pharmacy, Cairo University, Egypt for NMR results. Further, National Cancer Institute in Egypt is gratefully acknowledged. This research did not receive any specific support from funding agencies in the public, commercial, or not-for-profit sectors.References
- R. Kumilau, F. Hayati, J. E. S. Liew, S. Z. Sharif and N. A. Sahid Nik Lah, Short term recurrence and survival rate of breast cancer patients post-surgical treatment; north Borneo experience, Ann. Med. Surg., 2022, 81, 104560, DOI:10.1016/j.amsu.2022.104560.
- R. Addo, M. Haas and S. Goodall, The Cost-effectiveness of adjuvant tamoxifen treatment of hormone receptor–positive early breast cancer among premenopausal and perimenopausal Ghanaian women, Value Health Reg. Issues, 2021, 25, 196–205, DOI:10.1016/j.vhri.2021.05.005.
- L. Jung, A. Miske, A. Indorf, K. Nelson, V. K. Gadi and K. Banda, A retrospective analysis of metronomic cyclophosphamide, methotrexate, and fluorouracil (CMF) versus docetaxel and cyclophosphamide (TC) as adjuvant treatment in early stage, hormone receptor positive, HER2 negative breast cancer, Clin. Breast Cancer, 2022, 22, e310–e318, DOI:10.1016/j.clbc.2021.09.007.
- C. K. Osborne and R. Schiff, Mechanisms of endocrine resistance in breast cancer, Annu. Rev. Med., 2011, 62, 233–247, DOI:10.1146/annurev-med-070909-182917.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease, Nat. Rev. Clin. Oncol., 2016, 13, 674–690, DOI:10.1038/nrclinonc.2016.66.
- M. C. Papadimitriou, A. Pazaiti, K. Iliakopoulos, M. Markouli, V. Michalaki and C. A. Papadimitriou, Resistance to CDK4/6 inhibition: Mechanisms and strategies to overcome a therapeutic problem in the treatment of hormone receptor-positive metastatic breast cancer, Biochim. Biophys. Acta, Mol. Cell Res., 2022, 1869, 119346, DOI:10.1016/j.bbamcr.2022.119346.
- K. Thu, I. Soria-Bretones, T. Mak and D. Cescon, Targeting the cell cycle in breast cancer: towards the next phase, Cell Cycle, 2018, 17, 1871–1885, DOI:10.1080/15384101.2018.1502567.
- R. Fatehi, M. Rashedinia, A. R. Akbarizadeh, M. zamani and N. Firouzabadi, Metformin enhances anti-cancer properties of resveratrol in MCF-7 breast cancer cells via induction of apoptosis, autophagy and alteration in cell cycle distribution, Biochem. Biophys. Res. Commun., 2023, 644, 130–139, DOI:10.1016/j.bbrc.2022.12.069.
- X. Liu and M. Winey, The Mps1 family of protein kinases, Annu. Rev. Biochem., 2012, 81, 561–585, DOI:10.1146/annurev-biochem-061611-090435.
- M. G. Bursavich, D. Dastrup, M. Shenderovich, K. M. Yager, D. M. Cimbora, B. Williams and D. V. Kumar, Novel Mps1 kinase inhibitors: From purine to pyrrolopyrimidine and quinazoline leads, Bioorg. Med. Chem. Lett., 2013, 23, 6829–6833, DOI:10.1016/j.bmcl.2013.10.008.
- Y. Hiruma, A. Koch, N. Hazraty, F. Tsakou, R. H. Medema, R. P. Joosten and A. Perrakis, Understanding inhibitor resistance in Mps1 kinase through novel biophysical assays and structures, J. Biol. Chem., 2017, 292, 14496–14504, DOI:10.1074/jbc.M117.783555.
- X. Li, W. Wei, L. Tao, J. Zeng, Y. Zhu, T. Yang, Q. Wang, M. Tang, Z. Liu and L. Yu, Design, synthesis and biological evaluation of a new class of 7H-pyrrolo[2,3-d]pyrimidine derivatives as Mps1 inhibitors for the treatment of breast cancer, Eur. J. Med. Chem., 2022, 114887, DOI:10.1016/j.ejmech.2022.114887.
- S. L. Carter, A. C. Eklund, I. S. Kohane, L. N. Harris and Z. Szallasi, A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers, Nat. Genet., 2006, 38, 1043–1048, DOI:10.1038/ng1861.
- R. Brough, J. R. Frankum, D. Sims, A. Mackay, A. M. Mendes-Pereira, I. Bajrami, S. Costa-Cabral, R. Rafiq, A. S. Ahmad, M. A. Cerone, R. Natrajan, R. Sharpe, K.-K. Shiu, D. Wetterskog, K. J. Dedes, M. B. Lambros, T. Rawjee, S. Linardopoulos, J. S. Reis-Filho, N. C. Turner, C. J. Lord and A. Ashworth, Functional viability profiles of breast cancer, Cancer Discovery, 2011, 1, 260–273, DOI:10.1158/2159-8290.CD-11-0107.
- D. J. Gordon, B. Resio and D. Pellman, Causes and consequences of aneuploidy in cancer, Nat. Rev. Genet., 2012, 13, 189–203, DOI:10.1038/nrg3123.
- Y. Xie, A. Wang, J. Lin, L. Wu, H. Zhang, X. Yang, X. Wan, R. Miao, X. Sang and H. Zhao, Mps1/TTK: a novel target and biomarker for cancer, J. Drug Targeting, 2017, 25, 112–118, DOI:10.1080/1061186X.2016.1258568.
- C. L. Soave, T. Guerin, J. Liu and Q. P. Dou, Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing, Cancer Metastasis Rev., 2017, 36, 717–736, DOI:10.1007/s10555-017-9705-x.
- N. Mamadalieva, R. Lafont and M. Wink, Diversity of secondary metabolites in the genus Silene L. (Caryophyllaceae)—structures, distribution, and biological properties, Diversity, 2014, 6, 415–499, DOI:10.3390/d6030415.
- F. Jafari, S. Zarre, A. Gholipour, F. Eggens, R. K. Rabeler and B. Oxelman, A new taxonomic backbone for the infrageneric classification of the species-rich genus Silene (Caryophyllaceae), Taxon, 2020, 69, 337–368, DOI:10.1002/tax.12230.
- N. A. Moiloa, M. Mesbah, S. Nylinder, J. Manning, F. Forest, H. J. de Boer, C. D. Bacon and B. Oxelman, Biogeographic origins of southern African Silene (Caryophyllaceae), Mol. Phylogenet. Evol., 2021, 162, 107199, DOI:10.1016/j.ympev.2021.107199.
- S. Boukhira, L. E. Mansouri, M. Bouarfa, A. Ouhammou, S. Achour, M. Khadhr and D. Bousta, Phytochemical screening, anti-Inflammatory and analgesic activities of formulation cream of Silene vulgaris, Res. J. Med. Plant, 2016, 10, 150–158, DOI:10.3923/rjmp.2016.150.158.
- G. Zengin, M. F. Mahomoodally, A. Aktumsek, R. Ceylan, S. Uysal, A. Mocan, M. A. Yilmaz, C. M. N. Picot-Allain, A. Ćirić, J. Glamočlija and M. Soković, Functional constituents of six wild edible Silene species: A focus on their phytochemical profiles and bioactive properties, Food Biosci., 2018, 23, 75–82, DOI:10.1016/j.fbio.2018.03.010.
- I. Orhan, D. Deliorman-Orhan and B. Özçelik, Antiviral activity and cytotoxicity of the lipophilic extracts of various edible plants and their fatty acids, Food Chem., 115, 701–705, DOI:10.1016/j.foodchem.2009.01.024.
- H. S. Yusufoglu, G. A. Soliman, A. I. Foudah, M. S. Abdelkader, A. Alam and M. A. Salkini, Anti-inflammatory and hepatoprotective potentials of the aerial parts of Silene villosa Caryophyllaceae methanol extract in rats, Trop. J. Pharm. Res., 2018, 17, 117, DOI:10.4314/tjpr.v17i1.17.
- R. Karamian and F. Ghasemlou, Screening of total phenol and flavonoid content, antioxidant and antibacterial activities of the methanolic extracts of three Silene species from Iran, Int. J. Agric. Crop Sci., 2013, 5(3), 305–312 Search PubMed.
- S. Chandra and D. S. Rawat, Medicinal plants of the family Caryophyllaceae: a review of ethno-medicinal uses and pharmacological properties, Integr. Med. Res., 2015, 4, 123–131, DOI:10.1016/j.imr.2015.06.004.
- M. Ghonime, M. Emara, R. Shawky, H. Soliman, R. El-Domany and A. Abdelaziz, Immunomodulation of RAW 264.7 murine macrophage functions and antioxidant activities of 11 plant extracts, Immunol. Invest., 2015, 44, 237–252, DOI:10.3109/08820139.2014.988720.
- S. Mahmoud, A. Hassan, S. Abu El Wafa and A. E.-S. Mohamed, UPLC-MS/MS profiling and antitumor activity of Silene succulenta Forssk. growing in Egypt, Azhar Int. J. Pharm. Med. Sci., 2021, 1(2), 58–62, DOI:10.21608/aijpms.2021.57206.1039.
- S. Dötterl, L. M. Wolfe and A. Jürgens, Qualitative and quantitative analyses of flower scent in Silene latifolia, Phytochemistry, 2005, 66, 203–213, DOI:10.1016/j.phytochem.2004.12.002.
- T. Asai and Y. Fujimoto, Cyclic fatty acyl glycosides in the glandular trichome exudate of Silene gallica, Phytochemistry, 2010, 71, 1410–1417, DOI:10.1016/j.phytochem.2010.05.008.
- S. Bechkri, A. A. Magid, A. Khalfallah, L. Voutquenne-Nazabadioko, A. Kabouche, C. Sayagh, D. Harakat and Z. Kabouche, Antioxidant activity-guided isolation of flavonoids from Silene gallica aerial parts, Phytochem. Lett., 2022, 50, 61–66, DOI:10.1016/j.phytol.2022.05.002.
- Y. Meng, P. Whiting, L. Zibareva, G. Bertho, J.-P. Girault, R. Lafont and L. Dinan, Identification and quantitative analysis of the phytoecdysteroids in Silene species (Caryophyllaceae) by high-performance liquid chromatography: Novel ecdysteroids from S. pseudotites, J. Chromatogr. A, 2001, 935, 309–319, DOI:10.1016/S0021-9673(01)00893-7.
- E. Claude, R. Lafont, R. S. Plumb and I. D. Wilson, High performance Reversed-Phase Thin-Layer Chromatography-Desorption electrospray ionisation - time of flight high resolution mass spectrometric detection and imaging (HPTLC/DESI/ToFMS) of phytoecdysteroids, J. Chromatogr. B, 2022, 1200, 123265, DOI:10.1016/j.jchromb.2022.123265.
- B. Azadi and Y. Sohrabi, Chemical composition of Silene morganae Freyn volatile oil, Nat. Prod. Res., 2015, 29, 791–794, DOI:10.1080/14786419.2014.980251.
- N. Takahashi, W. Li and K. Koike, Oleanane-type triterpenoid saponins from Silene armeria, Phytochemistry, 2016, 129, 77–85, DOI:10.1016/j.phytochem.2016.07.011.
- H. Kılınç, M. Masullo, A. Bottone, T. Karayıldırım, Ö. Alankuş and S. Piacente, Chemical constituents of Silene montbretiana, Nat. Prod. Res., 2019, 33, 335–339, DOI:10.1080/14786419.2018.1451998.
- T. Asai, Y. Nakamura, Y. Hirayama, K. Ohyama and Y. Fujimoto, Cyclic glycolipids from glandular trichome exudates of Cerastium glomeratum, Phytochemistry, 2012, 82, 149–157, DOI:10.1016/j.phytochem.2012.07.001.
- A. R. Hassan, I. M. Sanad, A. E. Allam, M. E. Abouelela, A. M. Sayed, S. S. Emam, El-S. M. Kousy and K. Shimizu, Chemical constituents from Limonium tubiflorum and their in silico evaluation as potential antiviral agents against SARS-CoV-2, RSC Adv., 2021, 11(51), 32346–32357, 10.1039/d1ra05927k.
- H. M. El-Tantawy, A. R. Hassan and H. E. Taha, Antioxidant potential and LC/MS metabolic profile of anticancer fractions from Echium angustifolium Mill. aerial parts, J. Appl. Pharm. Sci., 2021, 11(12), 200–208, DOI:10.7324/JAPS.2021.1101220.
- H. M. El-Tantawy, A. R. Hassan and H. E. Taha, Anticancer mechanism of the non-polar extract from Echium angustifolium Mill. aerial parts in relation to its chemical content. Egypt, J. Chem., 2022, 65(10), 17–26, DOI:10.21608/ejchem.2022.130795.5757.
- A. R. Hassan, Chemical profile and cytotoxic activity of a polyphenolic-rich fraction from Euphorbia dendroides aerial parts, S. Afr. J. Bot., 2022, 147, 332–339, DOI:10.1016/j.sajb.2022.01.035.
- H. G. T. Castañeda, A. J. Colmenares Dulcey and J. H. Isaza Martínez, Flavonoid glycosides from Siparuna gigantotepala leaves and their antioxidant activity, Chem. Pharm. Bull., 2016, 64, 502–506, DOI:10.1248/cpb.c15-00788.
- C. Xie, N. C. Veitch, P. J. Houghton and M. S. J. Simmonds, Flavone C-Glycosides from Viola yedoensis MAKINO, Chem. Pharm. Bull., 2003, 51(10), 1204–1207, DOI:10.1248/cpb.51.1204.
- Z. Ye, J.-R. Dai, C.-G. Zhang, Y. Lu, L.-L. Wu, A. G. W. Gong, H. Xu, K. W. K. Tsim and Z.-T. Wang, Chemical differentiation of Dendrobium officinale and Dendrobium devonianum by using HPLC fingerprints, HPLC-ESI-MS, and HPTLC analyses, J. Evidence-Based Complementary Altern. Med., 2017, 1–9, DOI:10.1155/2017/8647212.
- C. Zhou, Y. Luo, Z. Lei and G. Wei, UHPLC-ESI-MS analysis of purified flavonoids fraction from stem of Dendrobium denneaum Paxt. and its preliminary study in inducing apoptosis of HepG2 cells, J. Evidence-Based Complementary Altern. Med., 2018, 1–10, DOI:10.1155/2018/8936307.
- S. Wang, M. Zhang, D. Liang, W. Sun, C. Zhang, M. Jiang, J. Liu, J. Li, C. Li, X. Yang and X. Zhou, Molecular design and anticancer activities of small-molecule monopolar spindle 1 inhibitors: A Medicinal chemistry perspective, Eur. J. Med. Chem., 2019, 175, 247–268, DOI:10.1016/j.ejmech.2019.04.047.
- K. Kusakabe, N. Ide, Y. Daigo, T. Itoh, T. Yamamoto, E. Kojima, Y. Mitsuoka, G. Tadano, S. Tagashira, K. Higashino, Y. Okano, Y. Sato, M. Inoue, M. Iguchi, T. Kanazawa, Y. Ishioka, K. Dohi, Y. Kido, S. Sakamoto, S. Ando, M. Maeda, M. Higaki, H. Yoshizawa, H. Murai and Y. Nakamura, A unique hinge binder of extremely selective aminopyridine-based Mps1 (TTK) kinase inhibitors with cellular activity, Bioorg. Med. Chem., 2015, 23, 2247–2260, DOI:10.1016/j.bmc.2015.02.042.
- Y. Sugimoto, D. B. Sawant, H. A. Fisk, L. Mao, C. Li, S. Chettiar, P.-K. Li, M. V. Darby and R. W. Brueggemeier, Novel pyrrolopyrimidines as Mps1/TTK kinase inhibitors for breast cancer, Bioorg. Med. Chem., 2017, 25, 2156–2166, DOI:10.1016/j.bmc.2017.02.030.
- T. V. Ganenko, A. A. Semenov, A. L. Vereschagin, I. A. Ushakov and Yu. A. Chuvashov, Cyclic glycolipids of Stellaria dichotoma L. above-ground part, Rastit. Resur., 2001, 37, 43–48 CAS.
- M. Malumbres and M. Barbacid, Cell cycle, CDKs and cancer: a changing paradigm, Nat. Rev. Cancer, 2009, 9, 153–166, DOI:10.1038/nrc2602.
- S. M. El-Kousy, S. S. Emam, A. R. Hassan and I. M. Sanad, Metabolites profiling of Limonium tubiflorum (Delile) Kuntze var tubiflorum via UPLC-qTOF-MS technique in relation to its cytotoxic activity, Jordan J. Biol. Sci., 2021, 14(4), 664–669, DOI:10.54319/jjbs/140405.
- L. M. Al-Harbi, E. A. Al-Harbi, R. M. Okasha, R. A. El-Eisawy, M. A. A. El-Nassag, H. M. Mohamed, A. M. Fouda, A. A. Elhenawy, A. Mora, A. M. El-Agrody and H. K. A. El-Mawgoud, Discovery of benzochromene derivatives first example with dual cytotoxic activity against the resistant cancer cell MCF-7/ADR and inhibitory effect of the P -glycoprotein expression levels, J. Enzyme Inhib. Med. Chem., 2023, 38, 2155814, DOI:10.1080/14756366.2022.2155814.
- S. Gajjar, V. Bora and B. M. Patel, Repositioning of simvastatin for diabetic colon cancer: role of CDK4 inhibition and apoptosis, Mol. Cell. Biochem., 2023, 1–13, DOI:10.1007/s11010-023-04663-w.
- Y. Lee, H. Kim, H. Kim, H. Y. Cho, J.-G. Jee, K.-A. Seo, J. B. Son, E. Ko, H. G. Choi, N. D. Kim and I. Kim, X-ray crystal structure-guided design and optimization of 7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile scaffold as a potent and orally active Monopolar Spindle 1 inhibitor, J. Med. Chem., 2021, 64, 6985–6995, DOI:10.1021/acs.jmedchem.1c00542.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01793a |
| This journal is © The Royal Society of Chemistry 2023 |