
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of fusion peptide release for the spike protein of SARS-CoV-2
Jie Yu†
,
Zhi-Wei Zhang†,
Han-Yu Yang†,
Chong-Jin Liu† and
Wen-Cai Lu† *
*
College of Physics, Qingdao University, Qingdao 266071, Shandong, P. R. China. E-mail: wencailu@qdu.edu.cn
First published on 6th June 2023
Abstract
The spike protein of SARS-CoV-2 can recognize the ACE2 membrane protein on the host cell and plays a key role in the membrane fusion process between the virus envelope and the host cell membrane. However, to date, the mechanism for the spike protein recognizing host cells and initiating membrane fusion remains unknown. In this study, based on the general assumption that all three S1/S2 junctions of the spike protein are cleaved, structures with different forms of S1 subunit stripping and S2′ site cleavage were constructed. Then, the minimum requirement for the release of the fusion peptide was studied by all-atom structure-based MD simulations. The results from simulations showed that stripping an S1 subunit from the A-, B- or C-chain of the spike protein and cleaving the specific S2′ site on the B-chain (C-chain or A-chain) may result in the release of the fusion peptide, suggesting that the requirement for the release of FP may be more relaxed than previously expected.
1. Introduction
SARS-CoV-2, an enveloped positive single-stranded RNA virus, belongs to severe acute respiratory syndrome-related coronavirus species and the β coronavirus family. There are 24–40 randomly arranged spike proteins distributed outside the spherical phospholipid coating of SARS-CoV-2. The spike protein is a highly glycosylated homotrimeric protein, and its monomer has two subunits, i.e., S1 and S2.1 S1 is composed of an N-terminal domain (NTD) and a receptor-binding domain (RBD),1 and the receptor binding motif (RBM) on the RBD binds with angiotensin converting enzyme type 2 (ACE2) on the host cell.2 S2 consists of fusion peptide (FP), heptad repeats 1 and 2 (HR1 and HR2), transmembrane domain (TM), and cytoplasmic tail (CP).1 The spike protein has been vividly described as a “metastable spring-loaded fusion machine”, which controls the membrane fusion of the virus and target cell.3 In addition, the spike protein is covered with a shielding layer of glycan, which makes it almost impossible to be recognized by the immune system.3One of the functions of the spike protein is to mediate membrane fusion. The process of the membrane fusion is related to the cleavage at the S1/S2 junction and the S2′ site at arginine 815 and the release of FPs.4 The S1/S2 site is also known as the furin cleavage site, and S1/S2 cleavage is essential for spike protein-mediated cell–cell fusion and entry into human lung cells.5 One study highlighted that the S1/S2 site is cleaved during virus assembly.6 The S1 and S2 subunits are linked in the form of non-covalent bond interaction after the cleavage of the S1/S2 junction.7 On the post-fusion spike protein, the S1 subunit did not exist, and the incomplete S2 subunit was cleaved at the S2′ sites.8 A study on the cell entry mechanisms of SARS-CoV-2 suggested that it relies on a second strategy, i.e., host protease activation.4 Furin, transmembrane serine protease 2 (TMPRSS2), and cathepsin L (CatL) are considered to play a key role in the host protease activation.4
In an earlier study, Dodero-Rojas et al.9 conducted molecular dynamics (MD) simulations with the all-atom structure-based model (SMOG)9 and found that the spike glycoprotein with all three S1 subunits stripped off and all three S2′ sites cleaved would release its FP, and the sugar chains on the S2 subunit hindered the release of FP.10 To the best of our knowledge, there are no other simulation studies on the mechanism of FP release of the spike protein reported to date, except for the above-mentioned study.10 In this work, we focused on the minimum requirement for FP release.
On the one hand, it was reported that the interaction between ACE2 and the RBD region on the S1 subunit can force the RBD region to change to the UP conformation, reducing the contact between S1 and S2.11 This process also leads to the refolding of the S1 subunit, disrupting the interaction between the residues related to ASP614 and near the S2′ sites.11 The final effect of ACE2 may remove the S1 subunit from the spike protein. However, simulation of ACE2 disrupting the S1 and S1 interaction, and finally removing the S1 subunit is difficult and quite time-consuming. Thus, for simplicity, we mimicked the effect of ACE2 on the spike protein by directly removing the S1 subunit.
On the other hand, conformational changes of the spike protein may happen during the viral binding and interaction with the cell. In this study, advance induced conformational changes were not considered, instead the native spike protein was used. It can be expected that the fusion peptide will be more easily released from the spike protein with induced conformational changes due to the viral binding with the cell. Therefore, the real condition for the release of the fusion peptide can be expected to be reduced compared to that obtained using the native spike protein.
We built a series of artificially modified structures for the spike protein and performed MD simulations. The system of the spike protein is large, and thus we applied the SMOG model in the MD simulation study of the release of FP of the spike protein. The SMOG model is an all-atom structure-based model, in which the potential energy function is defined based on the knowledge of stable conformations of systems.12 The MD simulations with the SMOG model were successfully applied in describing the protein folding dynamics,13,14 and the all-atom variants were later used to simulate rearrangements of large biomolecular structures, such as ribosomes15,16 and viral capsids.17,18
To characterize the release of the FP, θFP was used to describe the angle of FP relative to the long trimer interface helix of the spike protein, i.e., residue 816 Cα pointing to residue 855 Cα with respect to residue 990 Cα pointing to residue 1035 Cα. The calculated root-mean-square-deviation (RMSD) for the fusion peptide was used to estimate the stability of FP. The dot product of the two vectors were calculated, and then the θFP values were obtained.
The simulation results showed that when an S1 subunit of the A, B or C chain is stripped off and the S2′ site of the B, C or A chain is cleaved, the FP on the chain with the S2′ site cleavage can be released, suggesting the minimum requirement for the release of FP from the spike protein is more relaxed than previously expected, i.e., stripping off all the S1 subunits and cleaving all the S2′ sites.10
We also performed simulations of the modified spike protein structures A_S2′/B/C-S1, A-S1/BSS2′/C, and A/B-S1/CS2′ with the classical D614G mutation. The simulation results showed that for the three structures with the mutation, the fusion peptide was released more frequently for the repeated ten times of simulations (Table 1).
| A/B/C | A-S1 | B-S1 | C-S1 | A-S1/B-S1 | B-S1/C-S1 | C-S1/A-S1 | A-S1/B-S1/C-S1 | |
|---|---|---|---|---|---|---|---|---|
| A_S2′ | 0 | 0 | 0 | 5 | 0 | 5 | 4 | 4 |
| B_S2′ | 0 | 9 | 0 | 0 | 10 | 1 | 8 | 9 |
| C_S2′ | 0 | 0 | 0 | 0 | 4 | 8 | 0 | 9 |
| A_S2′/B_S2′ | 0 | 9 | 0 | 10 | 9 | 9 | 10 | 10 |
| B_S2′/C_S2′ | 0 | 10 | 0 | 0 | 10 | 1 | 10 | 10 |
| C_S2′/A_S2′ | 0 | 0 | 1 | 1 | 9 | 10 | 10 | 10 |
| A_S2′/B_S2′/C_S2′ | 0 | 9 | 4 | 6 | 10 | 10 | 10 | 10 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| D614G | ||||||||
| A_S2′ | 7 | |||||||
| B_S2′ | 9 | |||||||
| C_S2′ | 0 | |||||||
2. Structural modelling and simulation method
2.1 Structural modelling
The PDB file of the SARS-CoV-2 fully glycosylated full-length spike protein1 was downloaded from the SARS-CoV-2 section at the CHARMM-GUI website and was used as the initial structural template. We used the names A, B and C in the PDB file to mark the three chains of the spike protein. The representations of -S1 and _S2′ indicate the stripping of an S1 subunit and the cleavage at the S2′ site, respectively. Given that the sugar chains were suggested to be like a damper and did not affect the result of conformational changes,10 all the sugar chains were removed from the spike protein (Fig. 1).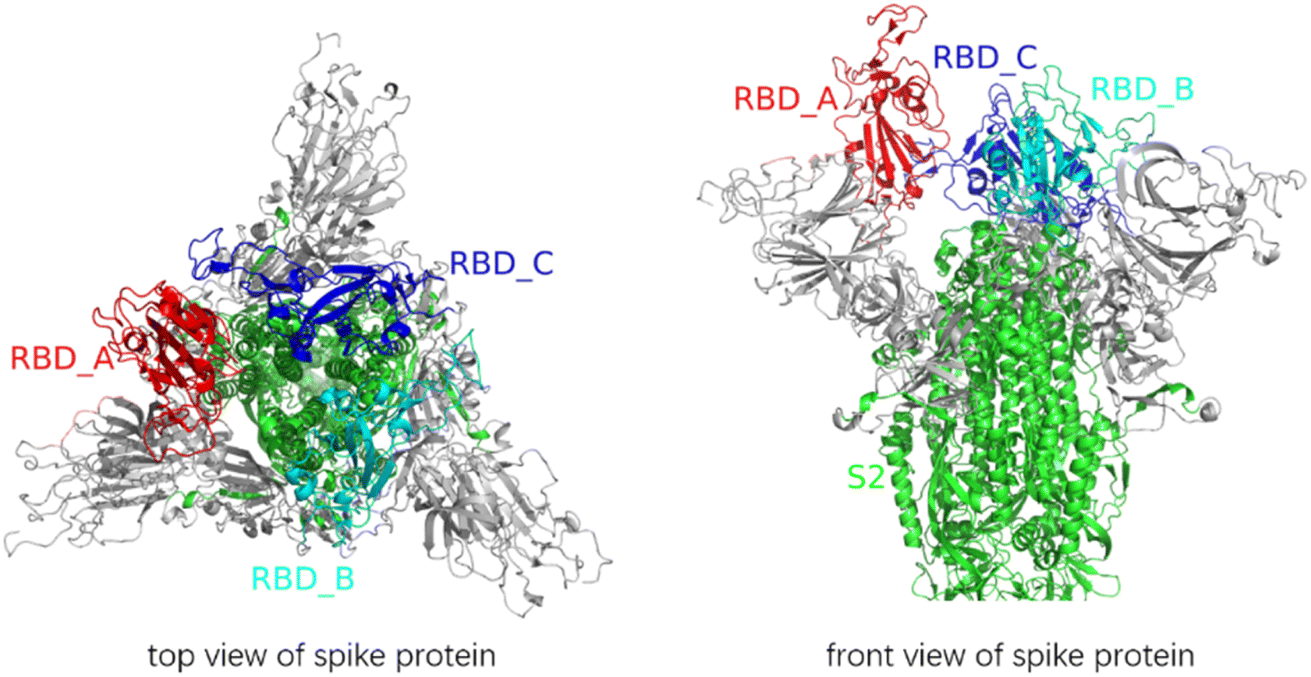 | ||
| Fig. 1 Vertical and front views of the structure of the spike protein. All glycans were removed. The RBD of A-chain kept on an “up” conformation, and the RBDs of B-chain and C-chain were at the “down” conformation. | ||
Among the structural modifications of the spike protein, all the S1/S2 sites were cleaved as a common requirement. Then, the artificially modified structures of the spike protein were constructed, in which one, two or three S2′ sites were cleaved, as follows: A_S2′/B/C, A/B_S2′/C, A/B/C_S2′; A_S2′/B_S2′/C, A/B_S2′/C_S2′, A_S2′/B/C_S2′; and A_S2′/B_S2′/C_S2′. Meanwhile, the modifications of stripping off one, two or three S1 subunits compared to that without S1 subunit removal were constructed, as follows: A/B/C; A-S1/B/C, A/B-S1/C, A/B/C-S1; A-S1/B-S1/C, A/B-S1/C-S1, A-S1/B/C-S1; and A-S1/B-S1/C-S1. Consequently, we constructed 7 × 8 = 56 types of modifications for the spike protein, which were used as the inputs of MD simulations with the SMOG model.
2.2 MD simulation with the SMOG model
The SMOG method, an all-atom structure-based model, was employed in the MD simulations of the spike protein. We used the SMOG2 force field file (AA_glycans_Dodero21.v1) in the force field repository at the SMOG2 official website (https://smog-server.org). The values of bond lengths and bond angles were provided by the Amber03 force field, and the strengths of non-planar dihedron and contact were consistent with earlier implementations of the structure-based model. The SMOG model was described in detail in the literature.9The all-atom structure-based MD simulations were performed with Gromacs (v2021.4).19 The 1 ns normal MD simulations of the modified structures, A_S2′/B/C, A/B_S2′/C, A/B/C_S2′; A_S2′/B_S2′/C, A/B_S2′/C_S2′, A_S2′/B/C_S2′; and A_S2′/B_S2′/C_S2′, with 7 types of S2′ site cleavages under the condition that all three S1/S2 sites were cleaved, were performed for pre-equilibrium. We retained the outputs of the gro files and used the trjconv module of Gromacs to output the protein coordinates in the PDB format.
SMOG 2 topology files were written in reduced units. The length unit was nano meters, and the mass of each bead was set to 1, corresponding to a mass unit of about 12 amu.20 Gromacs.mdp expects the temperature to be in Kelvin, where a value of Boltzmann's constant kB (0.00831451 kJ mol−1 K−1) is used internally.20 Thus, in a Gromacs.mdp file, a temperature of 120.3 K corresponds to a reduced temperature of 1. The time scale was estimated to be increased by a factor of 1000–10000.21
Based on the above-mentioned 7 structures with various types of S2′ site cleavages, we modified them further according to the 8 cases of stripping off S1 subunits, as described in the Structural modelling section. Then, the 56 designed structures were simulated with the SMOG (2.4.4) model using Gromacs. The parameters for MD simulations were the default values in the MDP file of Gromacs 5 provided by SMOG-2.4.4. In the MD simulations, the step size was 0.002, temperature was controlled at around 130 with the Langevin thermostat method, and 5000000 steps were run. Each of the SMOG MD simulations were repeated 10 times.
2.3 Structural metrics
To characterize the release of FP, θFP was used to describe the angle of FP relative to the long trimer interface helix of the spike protein, i.e., residue 816 Cα pointing to residue 855 Cα with respect to residue 990 Cα pointing to residue 1035 Cα. The calculated root-mean-square-deviation (RMSD) for the fusion peptide was used to estimate the stability of FP. The dot product of the two vectors was calculated, and then the θFP values were obtained.3. Results and discussion
Fig. 2 shows the RMSD curves for the A_S2′/B/C, A/B_S2′/C, A_S2′/B/C_S2′, A_S2′/B_S2′/C, A/B_S2′/C_S2′, A_S2′/B/C_S2′, and A_S2′/B_S2′/C_S2′ structures with various S2′ cleavages. All 7 structures tended towards equilibrium after 1 ns MD simulations with the charmm36-jul2021 force field. After these 7 structures with various S2′ cleavages were relaxed, the S1 subunits were removed and the SMOG MD simulations for 5000000 steps with a time step of 0.002 were performed and FP was found to be released for some modified spike proteins.
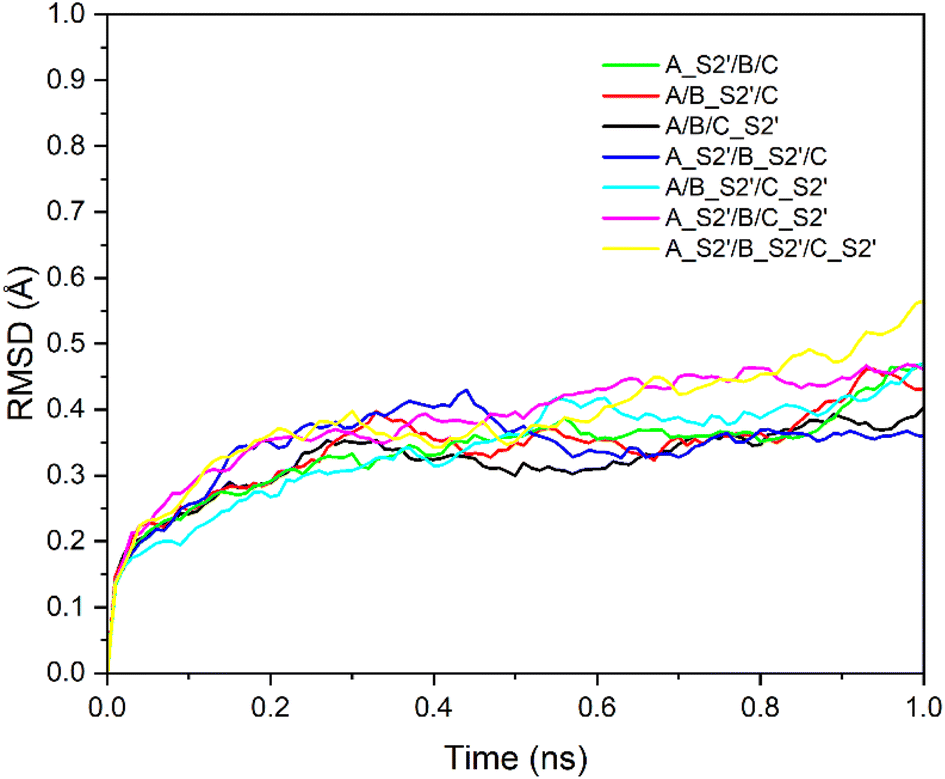 | ||
| Fig. 2 RMSD of seven structures: A_S2′/B/C, A/B_S2′/C, A_S2′/B/C_S2′, A_S2′/B_S2′/C, A/B_S2′/C_S2′, A_S2′/B/C_S2′ and A_S2′/B_S2′/C_S2′ with time. All seven systems tend towards equilibrium. | ||
We constructed 56 variously modified structures of the spike protein. Various cleavages of the S2′ sites and various stripping of S1 subunits were considered under the common condition that all three S1/S2 sites were cleaved. Each MD simulation with the SMOG model for the 56 modified structures of the spike protein was repeated 10 times. The MD simulation results for the modified structures with A_S2′, A_S2′/B_S2′ and A_S2′/B_S2′/C_S2′ are displayed in Fig. 3–11.
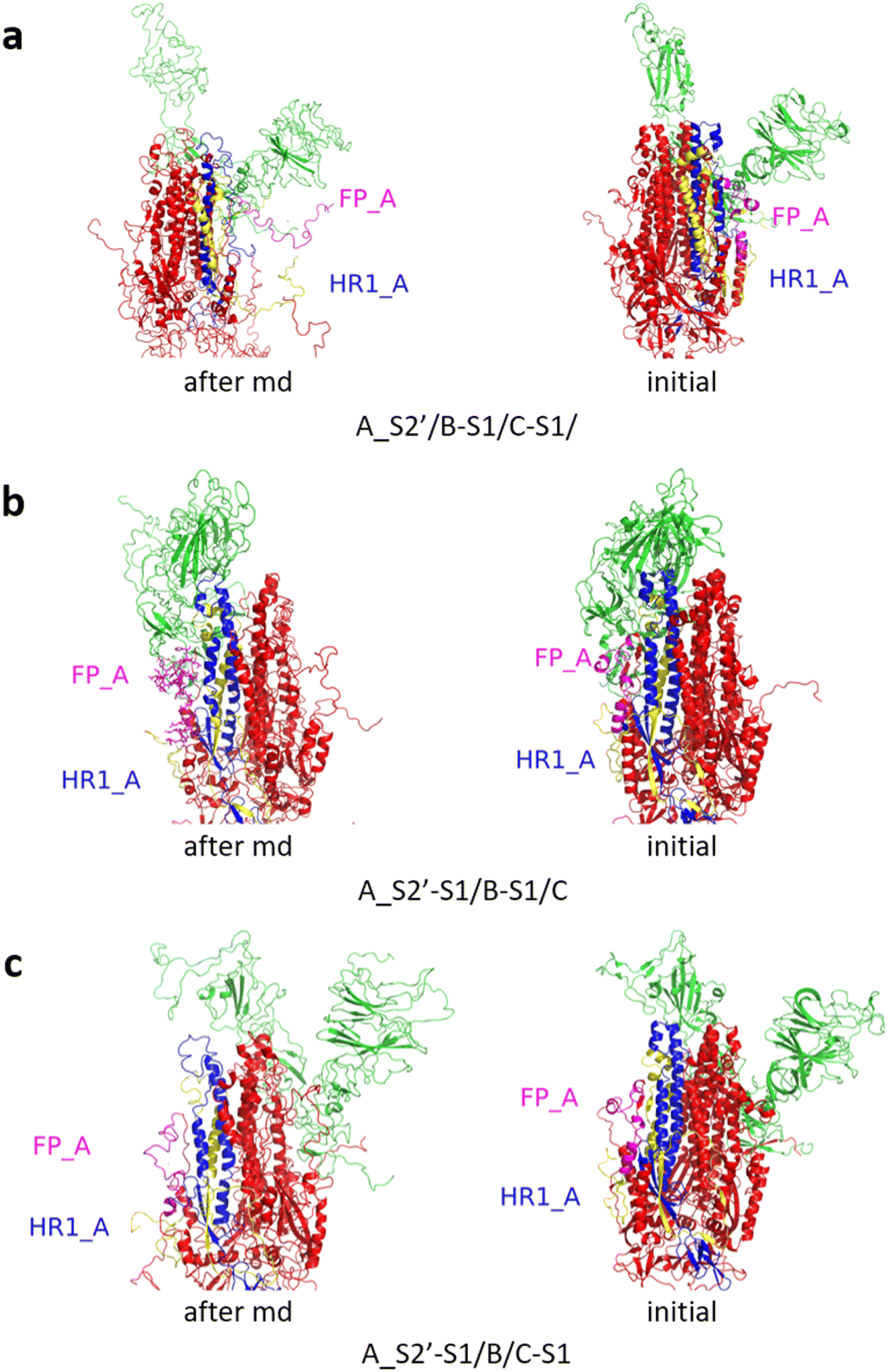
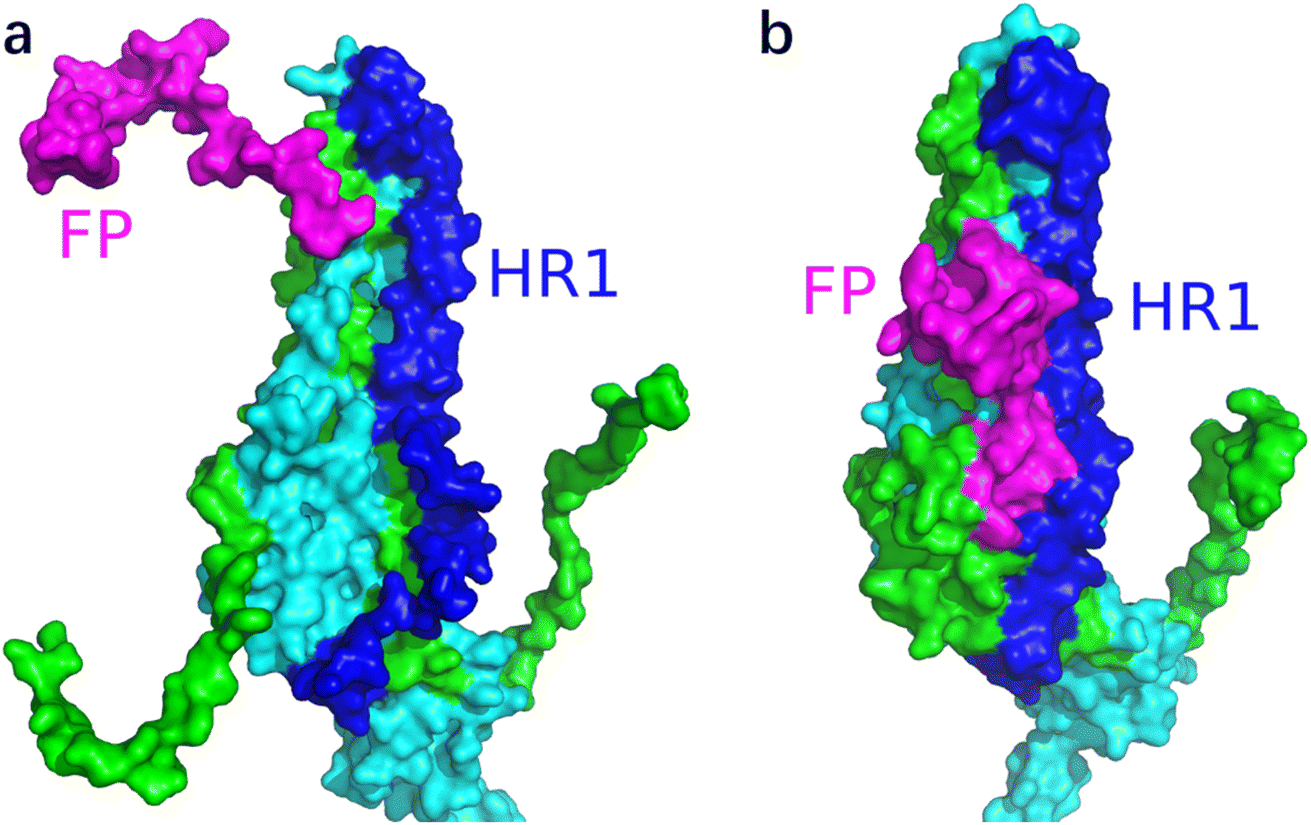
Fig. 3 shows the MD simulation results when all the S1 subunits were stripped or retained after the S2′ site on A-chain was cleaved. A_S2′-S1/B-S1/C-S1 showed obvious conformational changes for FP and HR1, while A_S2′/B/C did not show obvious outward motions of FP and HR1. Fig. 4 shows the results in the case that one S1 subunit of (a) C-chain, (b) B-chain, or (c) A-chain was stripped after cleaving the S2′ site of A-chain. A_S2′/B/C-S1 showed obvious outward motions of FP and unwinding of HR1, while A_S2′/B-S1/C and A_S2′-S1/B/C did not show obvious conformational changes for FP and HR1. Fig. 5 shows the results in the case that two S1 subunits of (a) B-chain and C-chain, (b) A-chain and B-chain, and (c) A-chain and C-chain were stripped after the S2′ site of A-chain was cleaved. A_S2′/B-S1/C-S1 showed obvious outward motions of FP and unwinding of HR1, whereas A_S2′-S1/B-S1/C and A_S2′-S1/B/C-S1 did not have obvious conformational changes for FP and HR1. Fig. 6 shows the results when (a) all the S1 subunits were stripped and (b) all S1 subunits were retained after cleaving two S2′ sites on A-chain and B-chain. A_S2′-S1/B_S2′-S1/C-S1 showed obvious outward motions of FP and HR1, and A_S2′/B_S2′/C did not have obvious conformational changes for FP and HR1. Fig. 7 shows the results when one S1 subunit of (a) A-chain, (b) C-chain or (c) B-chain was stripped after cleaving two S2′ sites on A-chain and B-chain. A_S2′-S1/B_S2′/C and A_S2′/B_S2′/C-S1 exhibited obvious outward motions of FP and unwinding of HR1, while A_S2′/B_S2′-S1/C did not have obvious conformational changes for FP and HR1. Fig. 8 shows the results when the S1 subunits of (a) A-chain and B-chain, (b) B-chain and C-chain, and (c) A-chain and C-chain were stripped after cleaving two S2′ sites on A-chain and B-chain. A_S2′-S1/B_S2′-S1/C, A_S2′/B_S2′-S1/C-S1 and A_S2′-S1/B_S2′/C-S1 showed obvious outward motions of FP and HR1. Fig. 9 shows the results when all the S1 subunits were stripped or retained after cleaving three S2′ sites on A-chain, B-chain and C-chain. A_S2′-S1/B_S2′-S1/C_S2′-S1 showed obvious outward motions of FP and HR1, while A_S2′/B_S2′/C_S2′ did not have obvious conformational changes for FP and HR1. Fig. 10 shows the results in the case that one S1 subunit of A-, B- or C-chain was stripped after cleaving all the three S2′ sites, where all the A_S2′-S1/B_S2′/C_S2′, A_S2′/B_S2′-S1/C_S2′ and A_S2′/B_S2′/C_S2′-S1 structures showed obvious outward motions of FP and HR1. Fig. 11 shows the results when two S1 subunits on (a) A-chain and B-chain, (b) B-chain and C-chain, and (c) A-chain and C-chain were stripped after cleaving all three S2′ sites, where all A_S2′-S1/B_S2′-S1/C_S2′, A_S2′/B_S2′-S1/C_S2′-S1 and A_S2′-S1/B_S2′/C_S2′-S1 showed obvious outward motions of FP and HR1.
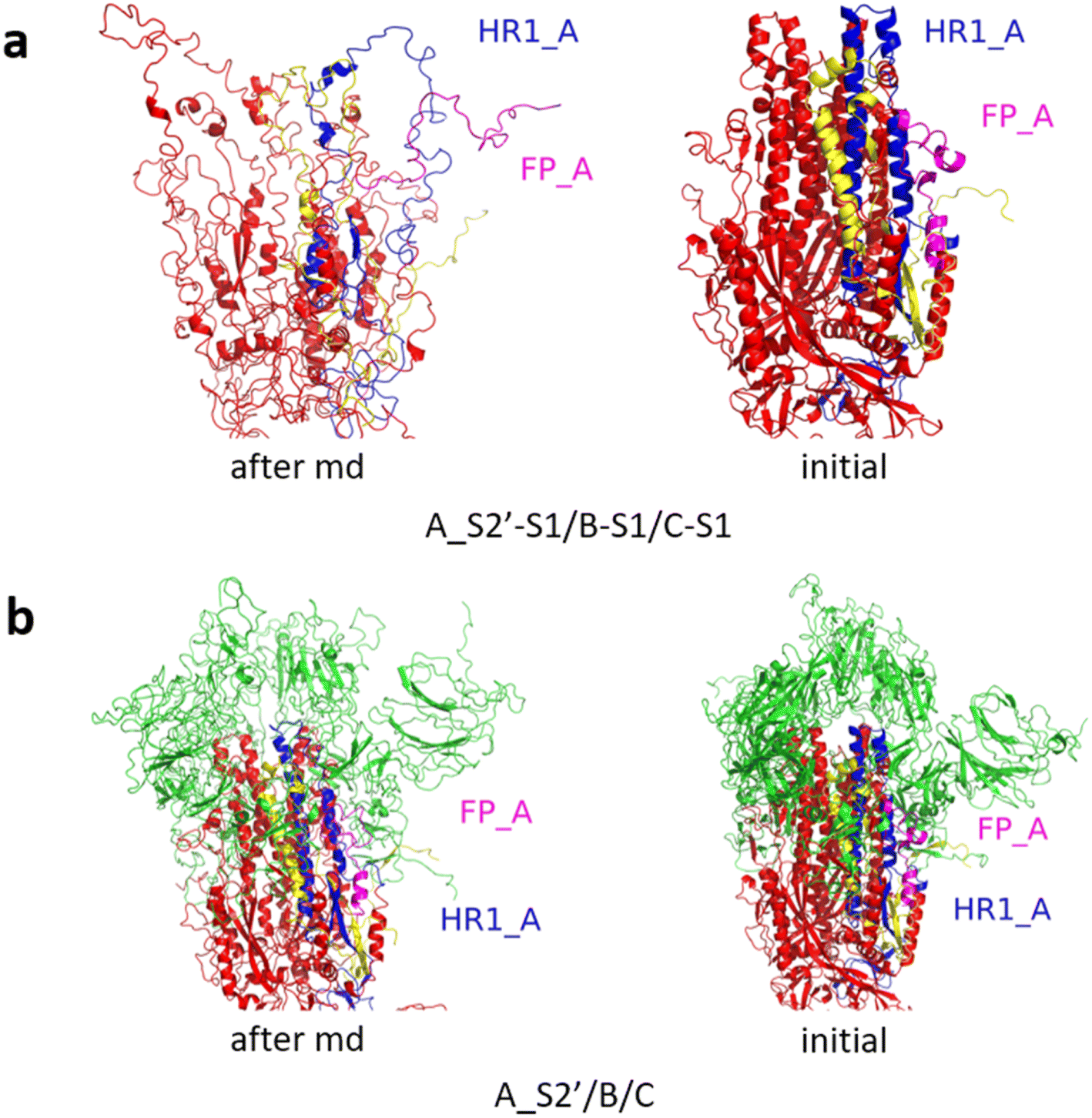 | ||
| Fig. 3 MD simulation results of the modified spike proteins. (a) A_S2′-S1/B-S1/C-S1 showed obvious conformational changes for FP and HR1. (b) A_S2′/B/C did not show obvious outward motions of FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for the other residues. The MD simulations were performed using the SMOG model.9 | ||
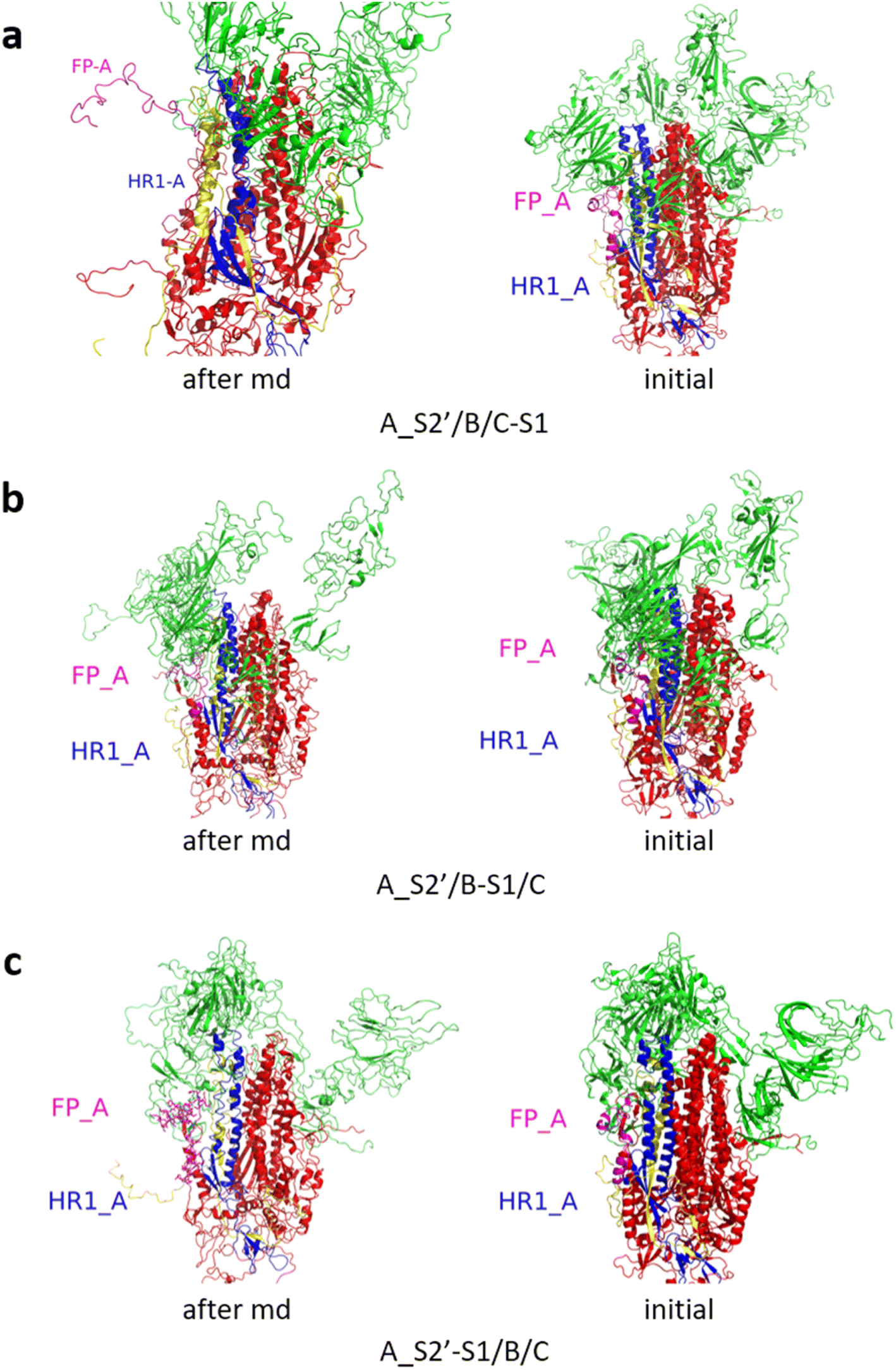 | ||
| Fig. 4 (a) A_S2′/B/C-S1 showed obvious outward motions of FP, and (b) A_S2′/B-S1/C and (c) A_S2′-S1/B/C did not show obvious conformational changes for FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
 | ||
| Fig. 5 (a) A_S2′/B-S1/C-S1 showed obvious outward motions of FP and HR1 was unwinding, and (b) A_S2′-S1/B-S1/C and (c) A_S2′-S1/B/C-S1 did not show obvious conformational changes for FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
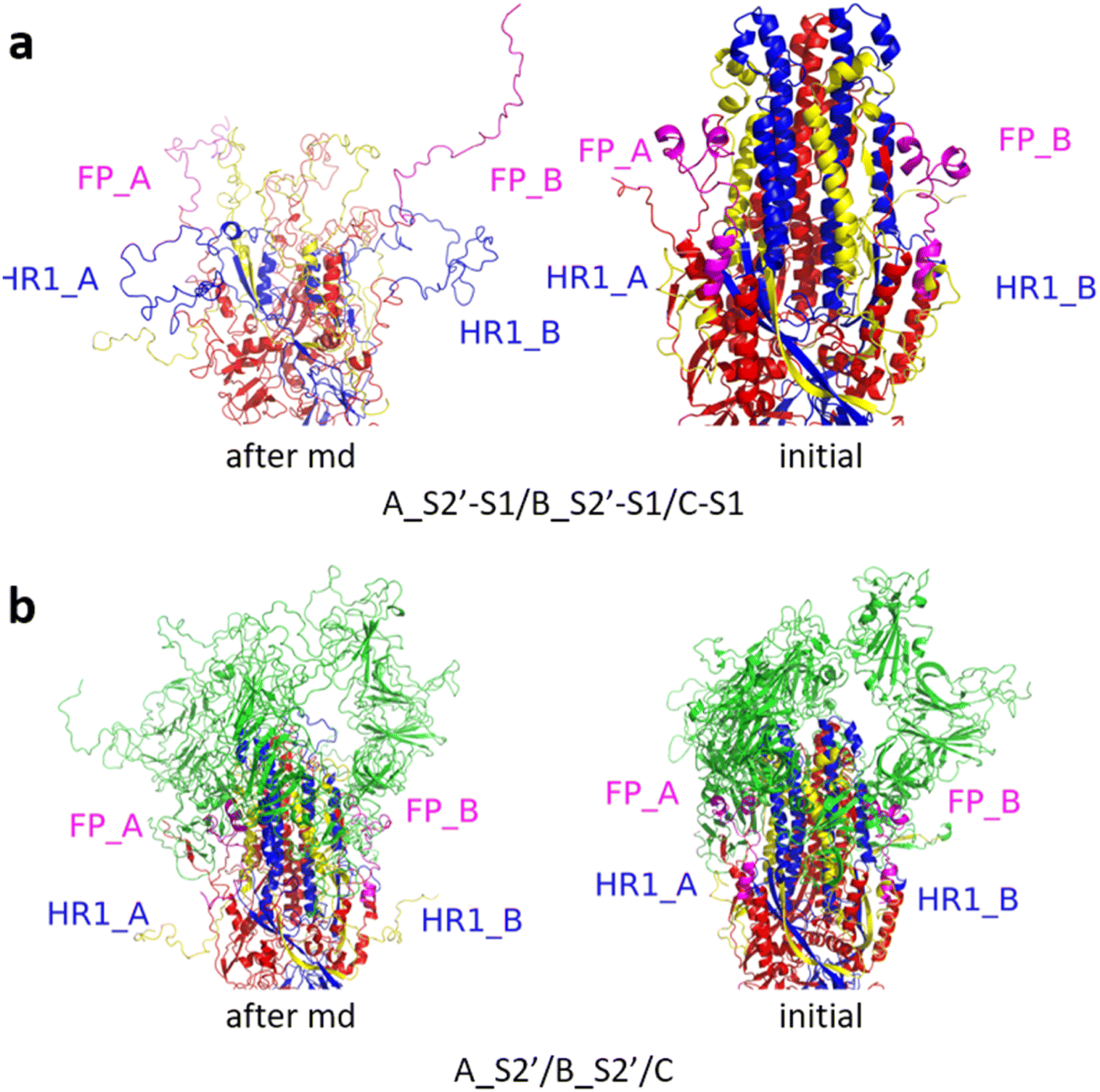 | ||
| Fig. 6 (a) A_S2′-S1/B_S2′-S1/C-S1 showed obvious outward motions of FP and HR1, and (b) A_S2′/B_S2′/C did not show obvious conformational changes for FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
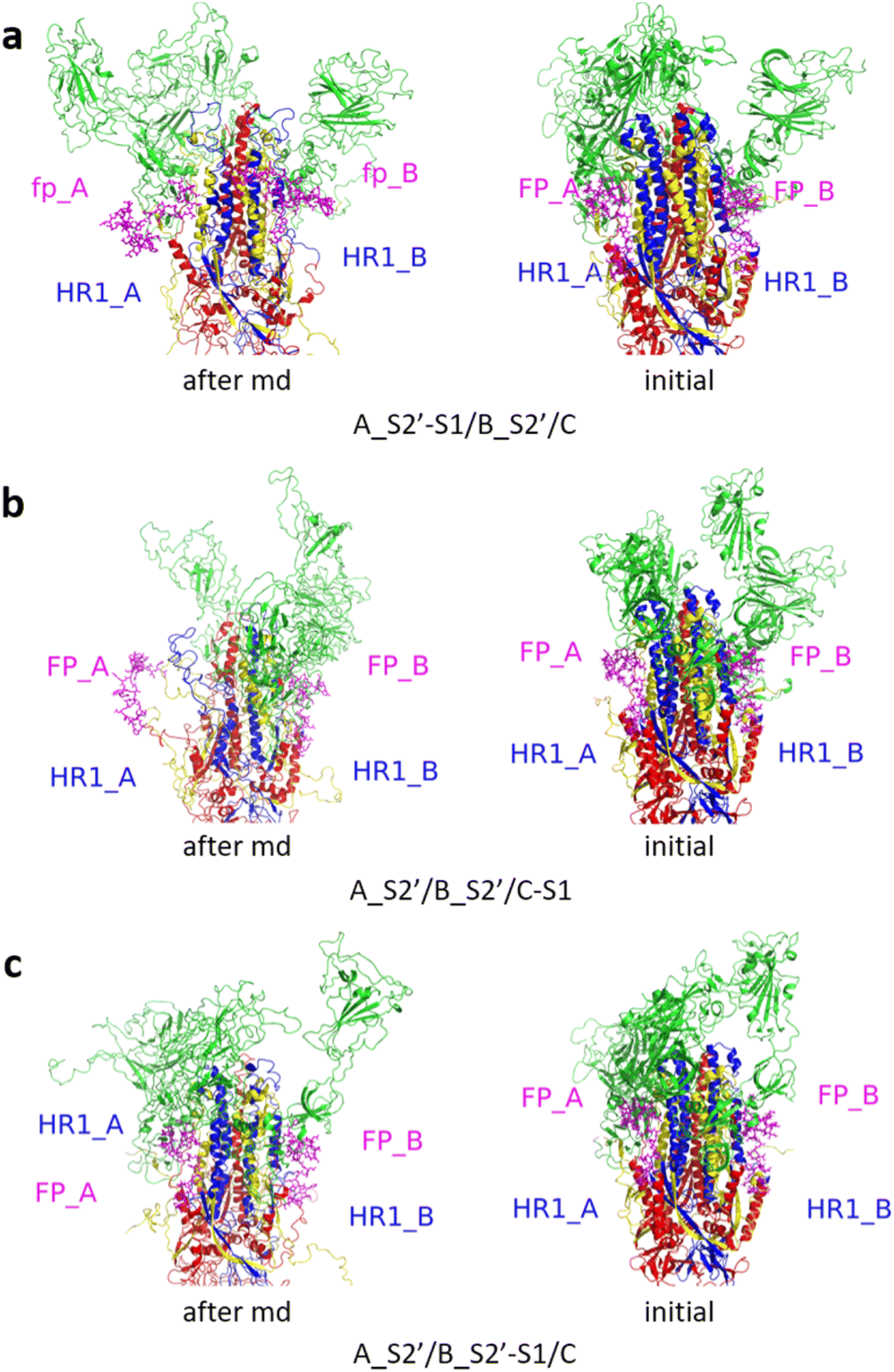 | ||
| Fig. 7 (a) A_S2′-S1/B_S2′/C and (b) A_S2′/B_S2′/C-S1 showed obvious outward motions of FP and HR1 was unwinding, and (c) A_S2′/B_S2′-S1/C did not show obvious conformational changes for FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
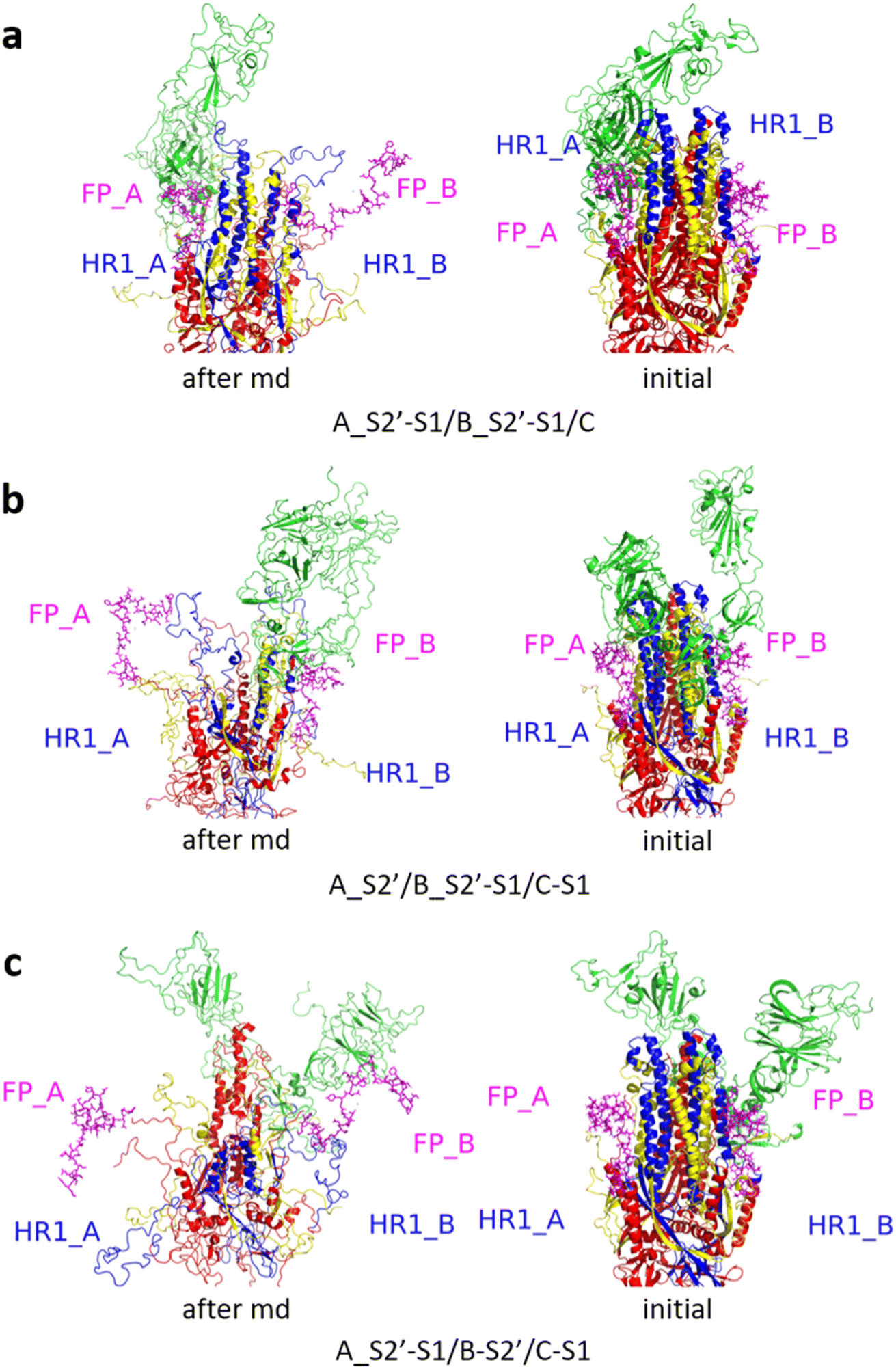 | ||
| Fig. 8 (a) A_S2′-S1/B_S2′-S1/C, (b) A_S2′/B_S2′-S1/C-S1 and (c) A_S2′-S1/B_S2′/C-S1 showed obvious outward motions of FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
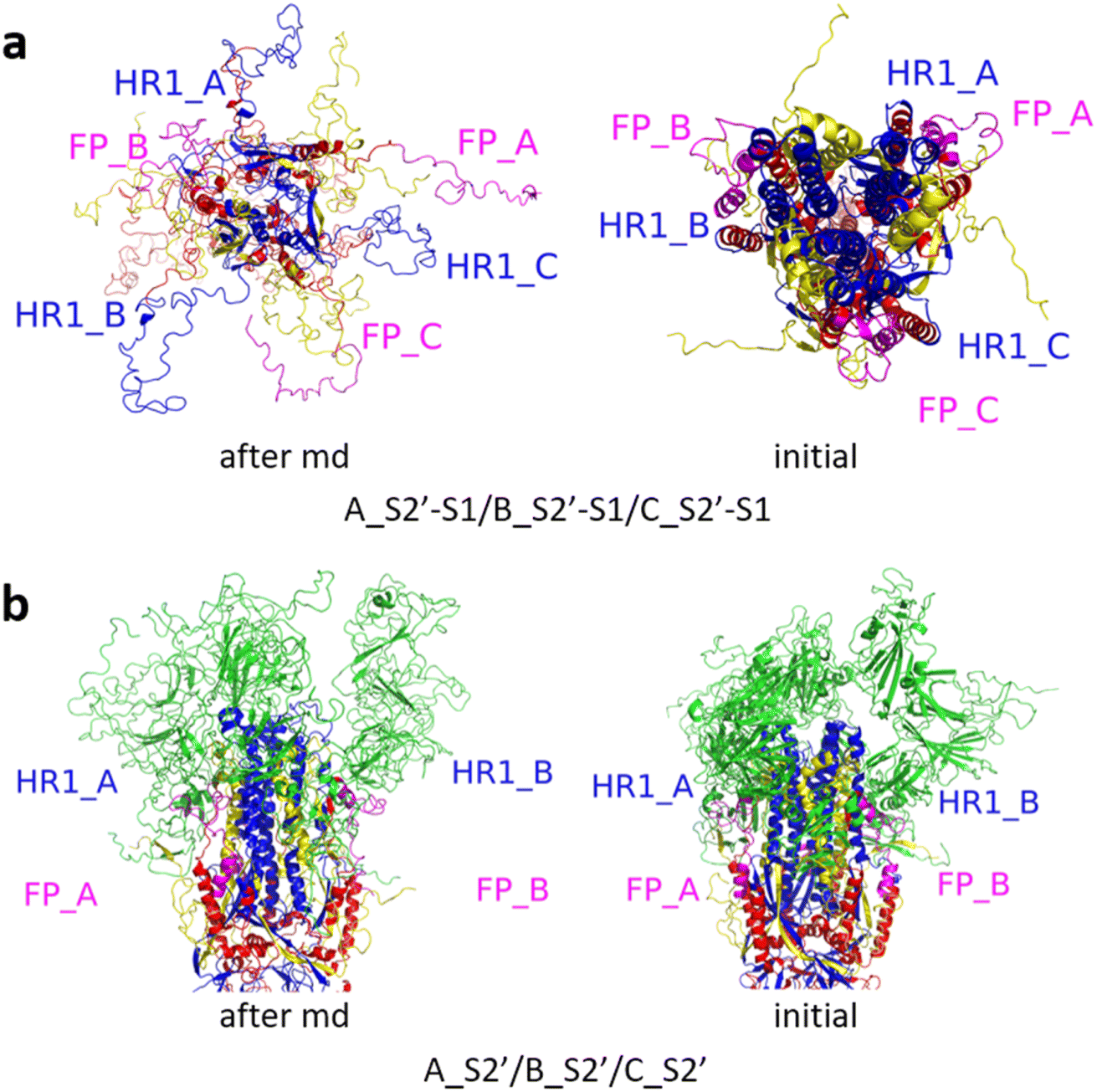 | ||
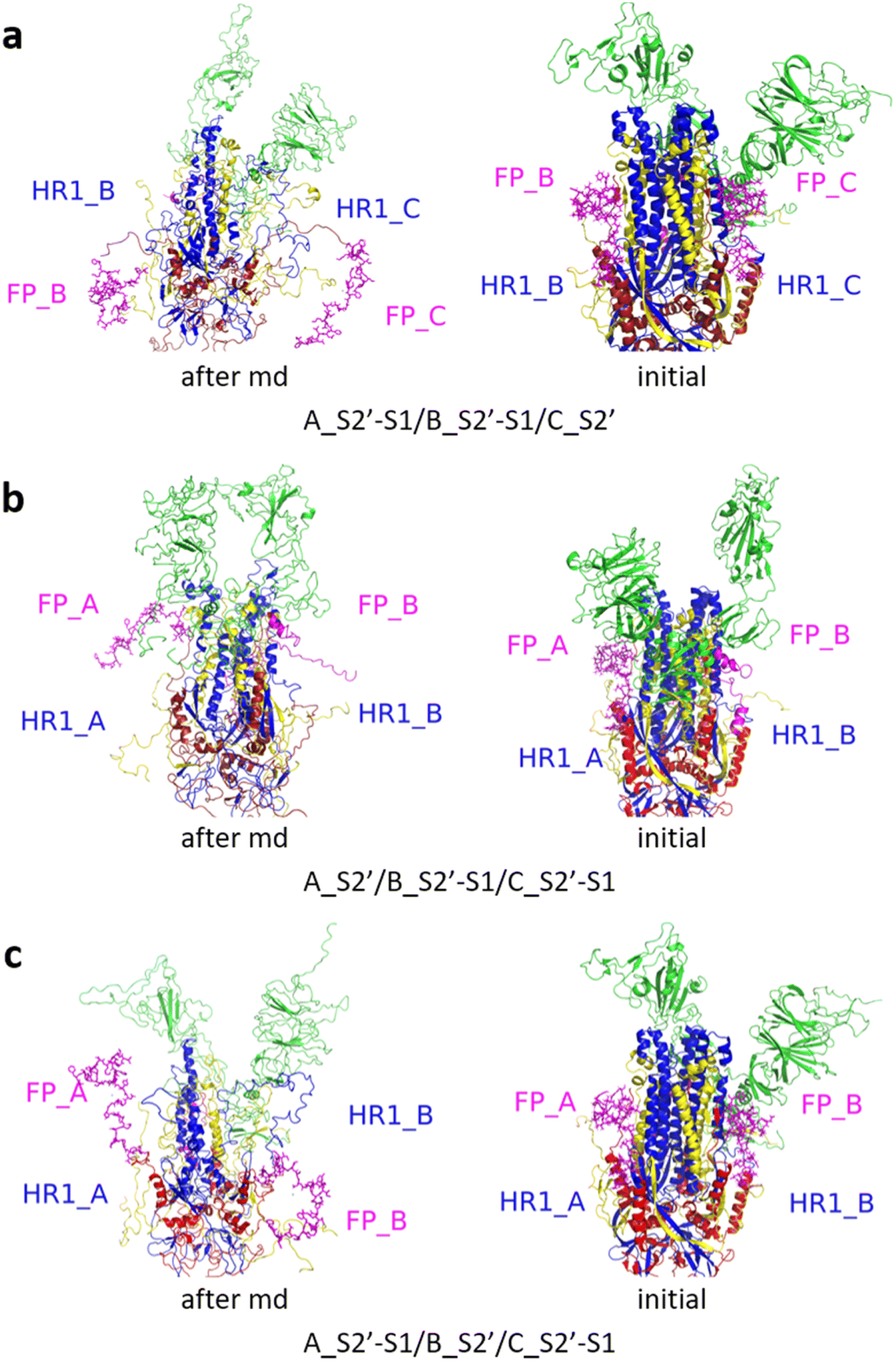
| Fig. 9 (a) A_S2′-S1/B_S2′-S1/C_S2′-S1 showed obvious outward motions of FP and HR1, and (b) A_S2′/B_S2′/C_S2′ did not show obvious conformational changes for FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
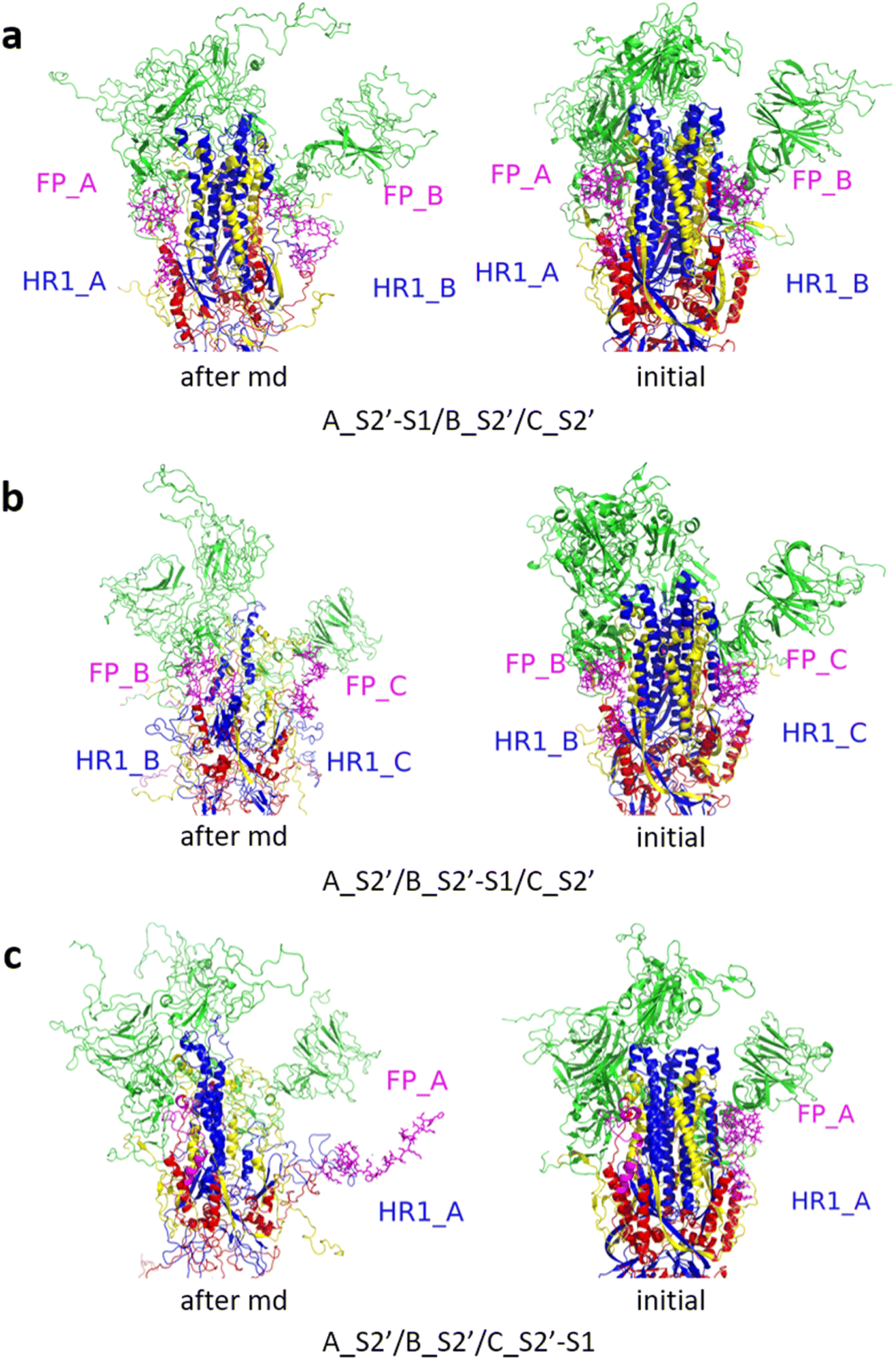 | ||
| Fig. 10 (a) A_S2′-S1/B_S2′/C_S2′ showed obvious outward motions of FP and HR1 was unwinding, and (b) A_S2′/B_S2′-S1/C_S2′, and (c) A_S2′/B_S2′/C_S2′-S1 showed obvious outward motions of FP and HR1. Green represented the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
 | ||
| Fig. 11 (a) A_S2′-S1/B_S2′-S1/C_S2′, (b) A_S2′/B_S2′-S1/C_S2′-S1, and (c) A_S2′-S1/B_S2′/C_S2′-S1 showed obvious outward motions of FP and HR1. Green represents the S1 subunit, blue for HR1, magenta for FP, yellow for residues 685–815 connecting S2 and S2′ cleavage sites, and red for other residues. The MD simulations were performed using the SMOG model.9 | ||
Fig. 12 shows the S1 subunit of chain C and the S2 subunit of chain A, given that the latter was covered by the former. As shown in Fig. 12, before removing the S1 subunit, the FP on the S2 subunit was bound in a groove under S1. The S1 key resisting the FP release was mainly the ASP614 residue.11 When S1 was removed, the key ASP614 residue of S1 was removed together, and thus the head of the S2 subunit was exposed. This provided the possibility for FP release, which was also dependent on the S2′ site cleavage. Fig. 13 shows that after the MD simulation, the FP obviously stretched out.
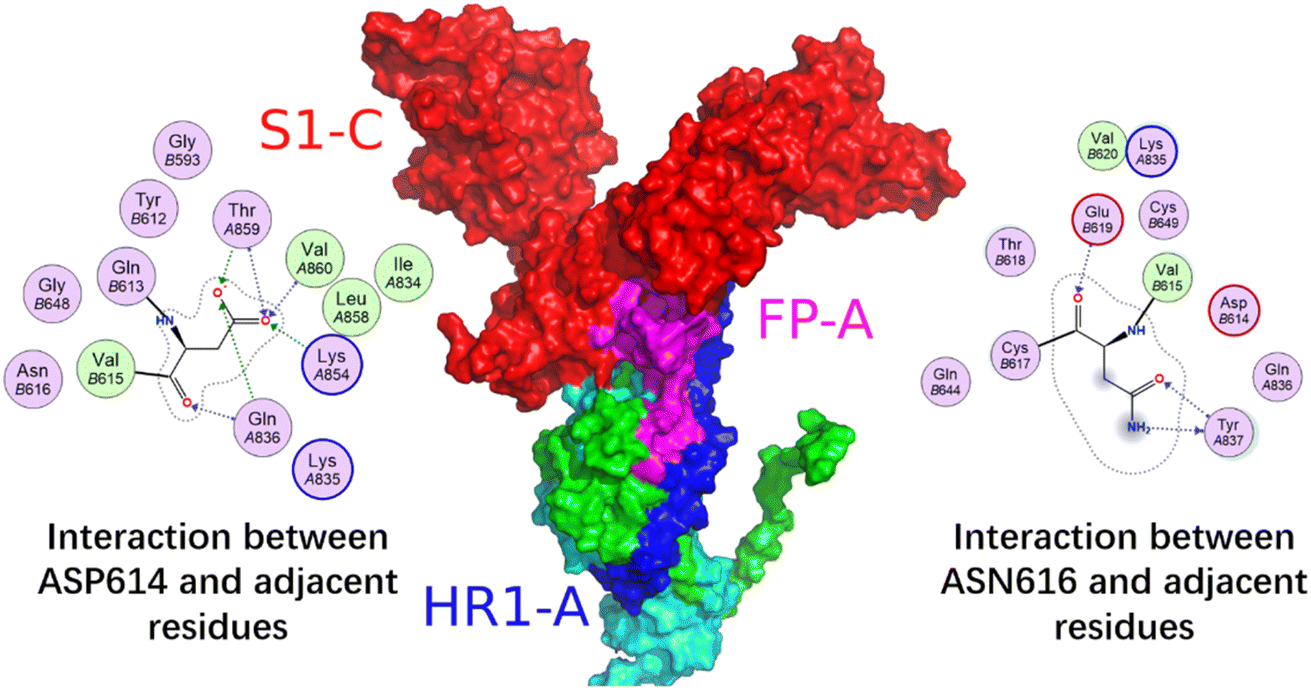 | ||
| Fig. 12 Interaction between the S1 subunit of chain C and the S2 subunit of chain A. Red is for S1 (residues 1–685), blue for HR1 (residues 912–984), magenta for FP (residues 816–855), green for residues 685–815 connecting S2 and S2′ cleavage sites, and cyan for other residues. The 2D graph show the interactions between ASP614 and ASN616 of S1 subunits and adjacent residues from left to right, respectively. | ||
 | ||
| Fig. 13 Conformational changes of the S2 subunit of chain A for the A_S2′/B/C-S1 structure (a) after and (b) before the MD simulation for 500000 steps with a time step of 0.002 using the SMOG model. | ||
Fig. 12 shows the local conformational changes, in which only the S1 subunit of the C chain and S2 subunit of the A chain remained. Using the ligand interaction module of MOE, it was found that the LYS835 residue of FP interacted with the ASN616 residue on the A-chain by hydrogen bonding, the GLN836 and LYS854 residues of FP formed hydrogen bond interactions with the ASP614 residue of the A-chain S1 subunit, and the ARG983 residue of HR1 interacted via π–H with the HIS519 residue of the A-chain RBD region.
It was obvious that the fusion peptide was masked by the S1 subunit before it was removed, and after the S1 subunit was stripped, the C-terminal of the fusion peptide was exposed. At the end of the MD, the N-terminal of the fusion peptide moved away from its original position and the angle with the S2 subunit backbone was close to 90°.
To characterize the release of FP, the angle of FP relative to the long trimer interface helix of the spike protein (θFP) was defined, i.e., the angle between the vector of the 816 Cα residue pointing to the 855 Cα residue and the vector of the 990 Cα residue pointing to the 1035 Cα residue. The dot product of the two vectors was calculated, and then the θFP value was obtained. For FP release in the structure A_S2′/B/C-S1, there was a cliff descent for θFP at about 6 ns (SMOG time) simulations compared to the initial structure, showing that FP was released (Fig. 14).
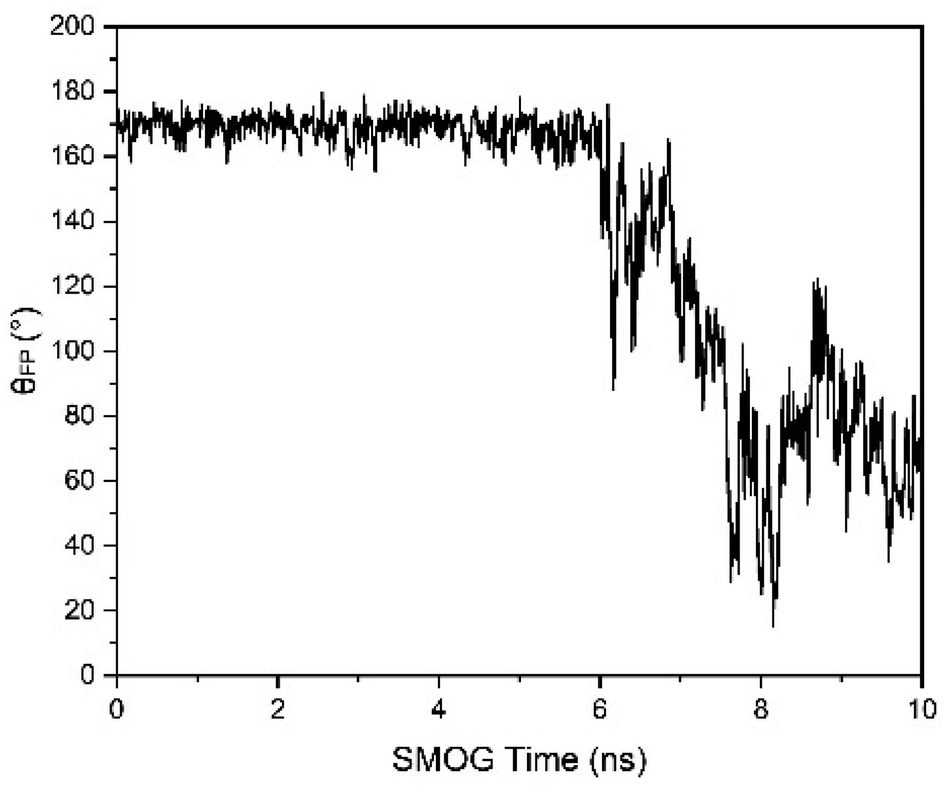 | ||
| Fig. 14 θFP changes with time for the A_S2′/B/C-S1 structure. At approximately 6 ns of SMOG time. The SMOG time should be multiplied by a factor larger than 1000 to correspond to the actual simulation time. There was a cliff descent for θFP at about 6 ns of SMOG time compared to the initial structure, indicating the release of FP. | ||
It was interesting to find that FP could be released when only one S1 subunit was stripped off and one S2′ site was cleaved under the general condition of cleaving all three S1/S2 junctions. The FP release process for the spike protein was similar to that observed in the video file provided in a previous study,10 where all three S1 subunits were stripped off and all three S2′ sites were cleaved. In this study, the simulation results showed that the requirement for the release of FP may be more relaxed than previously expected. The times for the release of the FP for 10 repeated MD simulations are summarized in Table 1.
When all the S1 subunits of A-chain, B-chain and C-chain were reserved, the release of FP could not occur even if all three S2′ sites and all three S1/S2 sites were cleaved. The FP of the spike protein could be released in the following cases: (1) when one S2′ site on A-chain (B-chain or C-chain) was cleaved, the minimum requirement for the release of FP was stripping off the S1 subunit on C-chain (A-chain or B-chain); (2) when two S2′ sites were cleaved, one specific S1 subunit needed to be stripped off for the release of FP; and (3) when all S2′ sites were cleaved, removal of any one S1 could lead to the release of FP. The case (1) indicated that the FP could be released for the S2′ site cleavage of A-chain and the S1 subunit stripping of C-chain, or the S2′ site cleavage of B-chain and the S1 subunit stripping of A-chain, or the S2′ site cleavage of C-chain and the S1 subunit stripping of B-chain, which had similar release tendencies. For example, the stripping of the S1 subunit of A-chain may be considered to control the release of FP from B-chain.
As given in Table 1, the MD simulations of A-S1/B-S1/B_S2′ and A-S1/B-S1/C_S2′ showed the possible releases of the FP on B-chain and C-chain, B-S1/C-S1/C_S2′ and A_S2′/B-S1/C-S1 showed the possible release of the FP on C-chain and A-chain, and A-S1/B_S2′/C-S1 and A-S1/A_S2′/C-S1 showed the possible release of the FP on B-chain and A-chain. For the A-S1/B-S1/C-S1 structure, cleavage of any S2′ could induce the release of FP.
The MD simulation results of the 56 artificially modified structures of the spike protein suggested that the minimum requirement for the release of FP from the spike protein may be more relaxed than the harsh condition where all three S1/S2 and three S2′ sites were cleaved and all three S1 subunits were stripped off.10 In this study, when only one S1 subunit was stripped off and one specific S2′ site was cleaved under the common condition of three S1/S2 cleavages, the release of FP could occur according to the MD simulations with the SMOG model. Of course, when two or three S2′ sites were cleaved and one S1 subunit was removed, FP could also be released.
The simulation results in this study suggest that the minimum requirement for releasing the FP from the spike protein may be more relaxed than previously expected. It will be easier to strip off one S1 unit and cleave one S2′ site compared to stripping off all three S1 subunits and cleaving all three S2′ sites. The simulation results may provide a new way to think about the mechanism of FP release, and the FPs can be expected to be released one by one in an asymmetric and non-simultaneous process.
The modification treatments for the spike protein in this study were reasonable to explore the mechanism of the FP release from the spike protein. In the previous study,10 the condition for the release of FP was assumed to be all three S1 subunits were stripped off and all the three S2′ sites were cleaved. Here, we found that the minimum requirement for releasing the FP may be more relaxed than expected and an asynchronous FP release could occur based on the results of the MD simulations with the all-atom structure-based SMOG model.
Moreover, the spike protein mutations may have an effect on the release of FPs. It has been shown that the mutations in individual residues of the spike protein have a significant impact on the pathological function of the spike protein.22 As an example, it was reported that the D614G mutation enhanced the density and infectivity of the virus spike protein, suggesting that this mutation may be associated with fewer S1 subunit shedding.11,23,24 It was pointed out that Asp614 of the S1 subunit forms a salt bridge with Lys854 of the S2 subunit, and Asp614 is often replaced by glycine, the mutation of which will remove key salt bridges.11
Early studies have shown that the D614G mutation may affect the detachment of the S1 subunit, but it has not been mentioned whether it will affect the release of fusion peptides, except that this mutation will cause the disappearance of the salt bridge between ASP614 and LYS854. We supplemented the md of the spike protein with D614G mutation. We selected the three structures(A_S2′/B/C-S1, A-S1/B_S2′/C and A/B-S1/C_S2′) with the lowest possible requirements for releasing fusion peptides as the control group to observe the effect of D614G mutation on the release of fusion peptides. Ten repetitive and independent MD results demonstrated that the fusion peptide release frequency of the mutant protein does not differ significantly from that of the wild-type structure across three different structures. For the (A_S2′/B/C-S1) structure, the fusion peptide was released in seven results, for the (A-S1/B-S2′/C) structure, the fusion peptide was released in ten results, and for the (A/B-S1/C-S2′) structure, no fusion peptide was released. This indicates that the D614G mutation had a small impact on the fusion peptide.
4. Conclusion
In this study, MD simulations with the all-atom structure-based SMOG model were performed for 56 modified structures of the spike protein to study the minimum requirements for the release of FP. Each MD simulation was repeated 10 times. The MD simulation results showed that when one S1 subunit of A-chain (B-chain or C-chain) was stripped off and one specific S2′ site of B-chain (C-chain or A-chain) was cleaved, the release of FP could occur, suggesting a more relaxed requirement for the release of FP than expected previously. This asynchronous release of the FPs may facilitate a flexible mechanism for spike protein targeting on the host cells.Conflicts of interest
No conflict of interest exits in the submission of this manuscript.References
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Cell entry mechanisms of SARS-CoV-2, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(21), 11727–11734 CrossRef CAS PubMed.
- C. Xu, Y. Wang, C. Liu, C. Zhang, W. Han, X. Hong and Y. Cong, et al., Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM, Sci. Adv., 2021, 7(1), eabe5575 CrossRef CAS PubMed.
- L. Casalino, Z. Gaieb, J. A. Goldsmith, C. K. Hjorth, A. C. Dommer, A. M. Harbison and R. E. Amaro, et al., Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein, ACS Cent. Sci., 2020, 6(10), 1722–1734 CrossRef CAS PubMed.
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Cell entry mechanisms of SARS-CoV-2, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(21), 11727–11734 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber and S. Pöhlmann, A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells, Mol. Cell, 2020, 78(4), 779–784 CrossRef CAS PubMed.
- T. P. Peacock, D. H. Goldhill, J. Zhou, L. Baillon, R. Frise, O. C. Swann and W. S. Barclay, et al., The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets, Nat. Microbiol., 2021, 6(7), 899–909 CrossRef CAS PubMed.
- M. A. Tortorici, A. C. Walls, Y. Lang, C. Wang, Z. Li, D. Koerhuis and D. Veesler, et al., Structural basis for human coronavirus attachment to sialic acid receptors, Nat. Struct. Mol. Biol., 2019, 26(6), 481–489 CrossRef PubMed.
- L. Tai, G. Zhu, M. Yang, L. Cao, X. Xing, G. Yin and Y. Zhu, et al., Nanometer-resolution in situ structure of the SARS-CoV-2 postfusion spike protein, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(48), e2112703118 CrossRef CAS PubMed.
- P. C. Whitford, J. K. Noel, S. Gosavi, A. Schug, K. Y. Sanbonmatsu and J. N. Onuchic, An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields, Proteins: Struct., Funct., Bioinf., 2009, 75(2), 430–441 CrossRef CAS PubMed.
- E. Dodero-Rojas, J. N. Onuchic and P. C. Whitford, Sterically confined rearrangements of SARS-CoV-2 Spike protein control cell invasion, Elife, 2021, 10, e70362 CrossRef CAS PubMed.
- D. J. Benton, A. G. Wrobel, P. Xu, C. Roustan, S. R. Martin, P. B. Rosenthal and S. J. Gamblin, et al., Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion, Nature, 2020, 588(7837), 327–330 CrossRef CAS PubMed.
- C. Clementi, H. Nymeyer and J. N. Onuchic, Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins, J. Mol. Biol., 2000, 298(5), 937–953 CrossRef CAS PubMed.
- S. Gosavi, L. L. Chavez, P. A. Jennings and J. N. Onuchic, Topological frustration and the folding of interleukin-1β, J. Mol. Biol., 2006, 357(3), 986–996 CrossRef CAS PubMed.
- J. E. Shea, J. N. Onuchic and C. L. Brooks III, Exploring the origins of topological frustration: design of a minimally frustrated model of fragment B of protein A, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(22), 12512–12517 CrossRef CAS PubMed.
- K. Nguyen and P. C. Whitford, Steric interactions lead to collective tilting motion in the ribosome during mRNA–tRNA translocation, Nat. Commun., 2016, 7(1), 10586 CrossRef CAS PubMed.
- M. Levi, K. Walak, A. Wang, U. Mohanty and P. C. Whitford, A steric gate controls P/E hybrid-state formation of tRNA on the ribosome, Nat. Commun., 2020, 11(1), 5706 CrossRef CAS PubMed.
- J. K. Noel, M. Levi, M. Raghunathan, H. Lammert, R. L. Hayes, J. N. Onuchic and P. C. Whitford, SMOG 2: a versatile software package for generating structure-based models, PLoS Comput. Biol., 2016, 12(3), e1004794 CrossRef PubMed.
- P. C. Whitford, W. Jiang and P. Serwer, Simulations of phage T7 capsid expansion reveal the role of molecular sterics on dynamics, Viruses, 2020, 12(11), 1273 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- M. Levi, P. Bandarkar, H. Yang, A. Wang, U. Mohanty, J. K. Noel and P. C. Whitford, Using SMOG 2 to simulate complex biomolecular assemblies, Biomolecular Simulations: Methods and Protocols, 2019, pp. 129–151 Search PubMed.
- M. Kouza, M. S. Li, E. P. O'Brien, C. K. Hu and D. Thirumalai, Effect of finite size on cooperativity and rates of protein folding, J. Phys. Chem. A, 2006, 110(2), 671–676 CrossRef CAS PubMed.
- F. Tian, B. Tong, L. Sun, S. Shi, B. Zheng, Z. Wang and P. Zheng, et al., N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2, Elife, 2021, 10, e69091 CrossRef CAS PubMed.
- L. Zhang, C. B. Jackson, H. Mou, A. Ojha, H. Peng, B. D. Quinlan and H. Choe, et al., SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity, Nat. Commun., 2020, 11(1), 6013 CrossRef CAS PubMed.
- Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus.
Footnote |
| † Jie Yu, Zhi-Wei Zhang, Han-Yu Yang and Chong-Jin Liu are postgraduate students engaged in research in computational materials. Wen-Cai Lu is a professor engaged in research on theoretical and computational materials. |
| This journal is © The Royal Society of Chemistry 2023 |