
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel entecavir analogues having 4′-cyano-6′′-fluoromethylenecyclopentene skeletons as an aglycone moiety as highly potent and long-acting anti-hepatitis B virus agent†
Hiroki Kumamoto*a,
Nobuyo Higashi-Kuwata b,
Sanae Hayashic,
Debananda Dasf,
Haydar Bulutf,
Ryoh Tokudad,
Shuhei Imotod,
Kengo Onitsukab,
Yuka Hondae,
Yuki Odanakae,
Satoko Shimbara-Matsubayashie,
Kazuhiro Haraguchia,
Yasuhito Tanakac and
Hiroaki Mitsuyabfg
b,
Sanae Hayashic,
Debananda Dasf,
Haydar Bulutf,
Ryoh Tokudad,
Shuhei Imotod,
Kengo Onitsukab,
Yuka Hondae,
Yuki Odanakae,
Satoko Shimbara-Matsubayashie,
Kazuhiro Haraguchia,
Yasuhito Tanakac and
Hiroaki Mitsuyabfg
aDepartment of Pharmaceutical Sciences, Nihon Pharmaceutical University, Saitama, Japan. E-mail: h-kumamoto@nichiyaku.ac.jp
bDepartment of Refractory Viral Infection, National Center for Global Health & Medicine Research Institute, Tokyo, Japan
cDepartment of Gastroenterology and Hepatology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan
dFaculty of Pharmaceutical Sciences, Sojo University, Kumamoto, Japan
eSchool of Pharmacy, Showa University, Tokyo, Japan
fExperimental Retrovirology Section, HIV and AIDS Malignancy Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
gDepartment of Clinical Sciences, Kumamoto University Hospital, Kumamoto, Japan
First published on 31st May 2023
Abstract
Encouraged by our recent findings that 4′-cyano-deoxyguanosine (2), entecavir analogues 4 and 5 are highly potent anti-hepatitis B virus (HBV) agents, we designed and synthesized 6 having a hybridized structure of 4 and 5. The chiral quaternary carbon portion at the 4′-position, which is substituted by cyano- and 5′-hydroxymethyl groups, was stereospecifically constructed by radical-mediated 5-exo-dig mode cyclization of 10. The introduction of the fluorine atom into the 6′′-position was achieved by radical-mediated stannylation of sulfide (E)-11 and subsequent electrophilic fluorination of (E)-12. The desired (E)-6 was obtained after the introduction of the guanine base into (E)-18 under Mitsunobu conditions and following global deprotection. The stereoisomer (Z)-6 was also prepared by the same procedure using (Z)-12. Compound (E)-6 showed highly potent anti-HBV activity (EC50 = 1.2 nM) with favorable cytotoxicity (CC50 = 93 μM).
1. Introduction
The infection of hepatitis B virus (HBV)1,2 causes a serious hepatopathy, which develops chronic hepatitis B (CHB), cirrhosis, and hepatocellular carcinoma (HCC).3 HBV possesses a double-strand circular DNA as a genome, which is regenerated from pre-genomic RNA catalyzed by its own reverse transcriptase (RT) for efficient replication.4,5 Lately, the anti-HBV chemotherapy by typically employing nucleoside analogue RT inhibitors (NRTIs),6,7 such as entecavir (1, Fig. 1),8–10 has improved the morbidity and mortality of CHB, cirrhosis, and HCC. Despite this effective treatment, the longtime continuous use of NRTIs is necessary, which in turn leads to the emergence of drug-resistant HBV variants, which pose serious challenges.11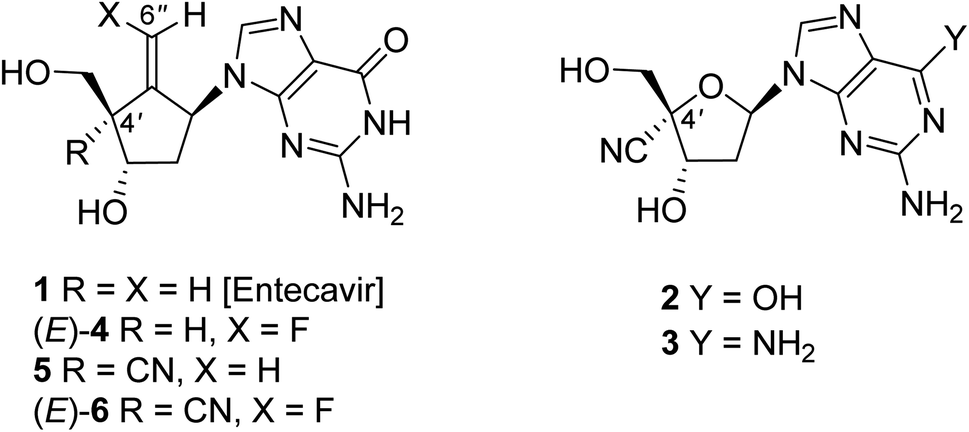 | ||
| Fig. 1 Structures of compounds 1–6. | ||
We have previously reported that purine nucleosides having a cyano group at the 4′-position on the deoxyribosyl moiety, such as 2 and 3, exert potent anti-HBV activity compared to 1 not only against wild type but also against entecavir-resistant HBV.12 Molecular models of HBV-reverse transcriptase (RT) with corresponding triphosphate of 2 and 3 indicate that, 2 or 3-triphosphate is well accommodated in HBV-RT due to an excellent interaction between hydrophobic pockets of HBV-RT and 4′-cyano group.12 Unfortunately, these purine nucleosides proved to be overly cytotoxic. One of our recent studies towards the development of nucleoside anti-HBV agents has revealed that the entecavir analogue (4) possessing a fluorine atom at the olefin terminal is more potent against wild-type HBV than the parent compound 1 and less cytotoxic albeit the actual roes of the fluorine atom located at the olefin terminal have been unclear.13 Recently, we found that compound 5, which is a 4′-cyano-derivative of entecavir (1), possesses potent activity against an entecavir-resistant HBV variant without causing significant cytotoxicity.14 Based on these findings, we designed, synthesized, and identified a novel nucleoside analogue (E)-6 that possesses a hybridized structure between (E)-4 and 5,15 which exerts a long-acting and highly potent anti-HBV activity without significant cytotoxicity. In this study, we wish to report the details of the synthesis of (E)-6 and its stereoisomer (Z)-6 and their biological and structural features.
2. Results and discussion
2.1. Chemistry
We have established a diastereoselective synthetic route for (E)-4 through a radical-mediated 5-exo-dig mode cyclization of the selenide through the construction of the methylenecyclopentene unit.13 The highly stereochemical outcome at the 4-position might be derived from the steric repulsion between the trityloxymethyl function and the cyclic acetonide. One would anticipate that if this radical technique was applied for the preparation of title molecule 6, then the stereoselectivity for the newly generated quaternary carbon center would decrease, due to the steric repulsion between the cyclic acetonide and the cyano group (or its masked form). Accordingly, we planned the novel synthetic route for 6 as depicted in Scheme 1 as a retrosynthetic analysis.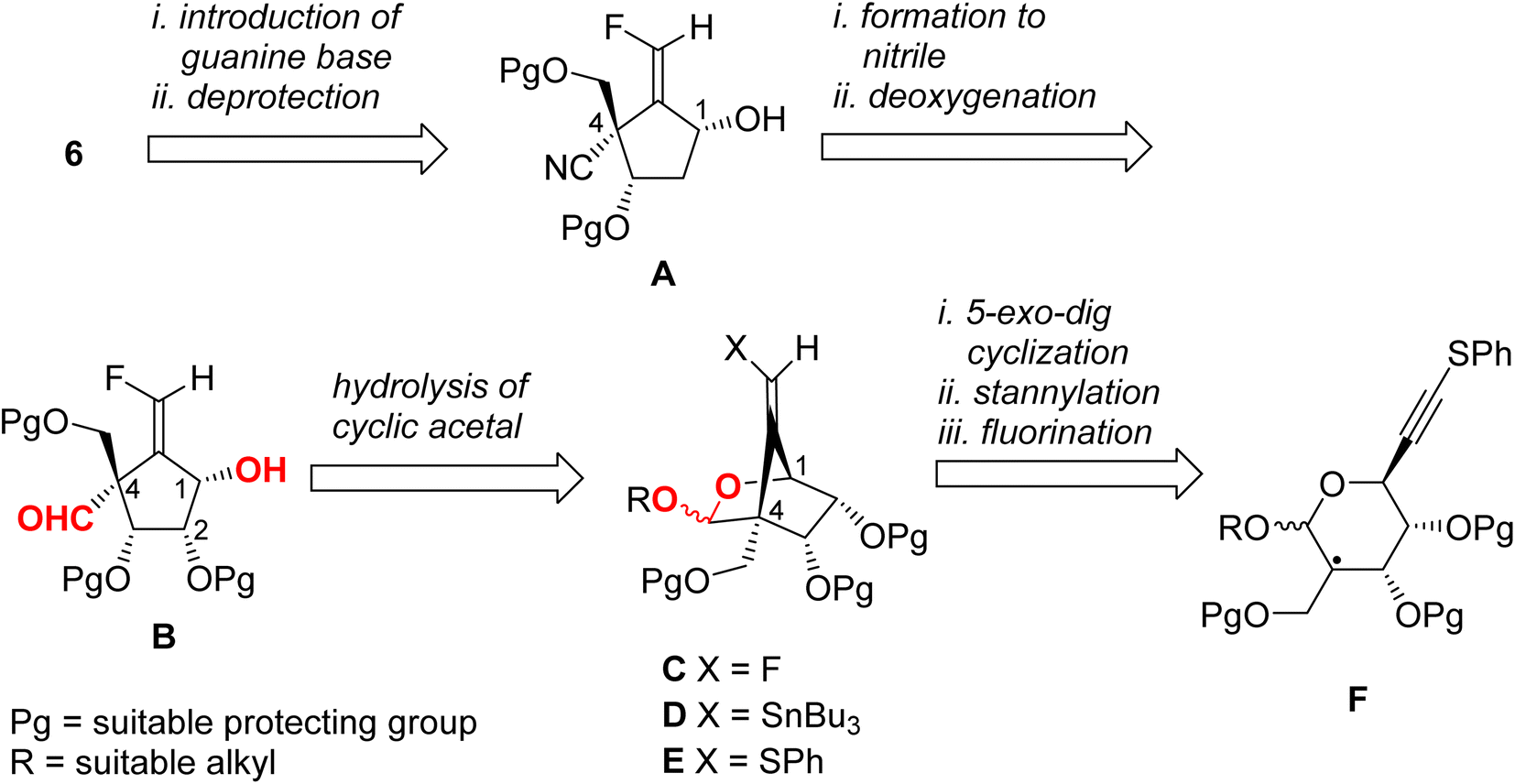 | ||
| Scheme 1 Retrosynthetic analysis for 6. | ||
Target molecule 6 was prepared from alcohol A by the introduction of a guanine base under SN2 manner and deprotection. Cyanide A was prepared from B by deoxygenation of the 2-hydroxyl group and nitrile formation of the 4-formyl group. Aldehyde B was derived from cyclic acetal C by hydrolysis. The relative configuration of the 1-hydroxyl group and the 4-formyl group must be fixed at this stage. Fluoride C was prepared from stannane D by electrophilic fluorination. The stannane D was converted from sulfide E employing the radical-mediated stannylation.13 The bicyclo[2.2.1] system of E may be stereospecifically constructed by a radical-mediated 5-exo-dig cyclization of carbon-centered radical F. Therefore, we started to prepare the corresponding radical precursor for F.
The ketone 7 having a 1-(S) configuration was stereoselectively protected from D-ribose by a reported method (Scheme 2).16 Treatment of 7 with the ylide (Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOMe) prepared from Ph3PCH2OMeCl and BuLi gave the enol ether 8 in 77% yield as a diastereomeric mixture (ca. 7
CHOMe) prepared from Ph3PCH2OMeCl and BuLi gave the enol ether 8 in 77% yield as a diastereomeric mixture (ca. 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, the stereochemistries of the isomers were not determined) after desilylation using Bu4NF. When the enol ether 8 was reacted with PhSeCl in the presence of a stoichiometric amount of pyridine at −80 °C, the required intramolecular seleno-etherification readily proceeded to form seleno methyl glycoside 9 as a mixture of three diastereomers (ca. 20:12:1, the actual stereochemistries of the diastereomers were not determined at this stage). The desired radical precursor 10 was obtained in moderate yield after the introduction of the PhS group at the terminal alkyne moiety of 9 via lithium acetylide (generated by LDA).
:3, the stereochemistries of the isomers were not determined) after desilylation using Bu4NF. When the enol ether 8 was reacted with PhSeCl in the presence of a stoichiometric amount of pyridine at −80 °C, the required intramolecular seleno-etherification readily proceeded to form seleno methyl glycoside 9 as a mixture of three diastereomers (ca. 20:12:1, the actual stereochemistries of the diastereomers were not determined at this stage). The desired radical precursor 10 was obtained in moderate yield after the introduction of the PhS group at the terminal alkyne moiety of 9 via lithium acetylide (generated by LDA).
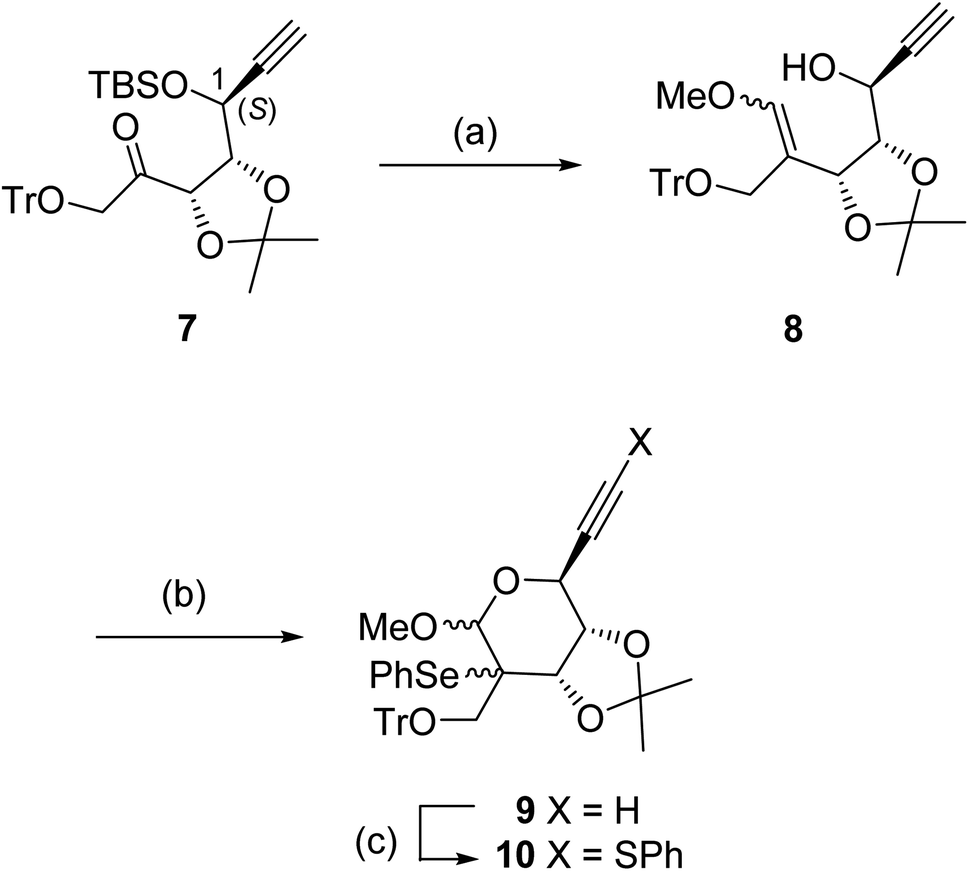 | ||
| Scheme 2 Conditions: (a) (i) Ph3PCH2OmeCl, BuLi, THF, 0 °C, (ii) Bu4NF, THF (77% for two steps); (b) PhSeCl, pyridine, CH2Cl2, −80 °C to RT (84%); (c) PhSO2SPh, LDA, THF, −80 °C (79%). | ||
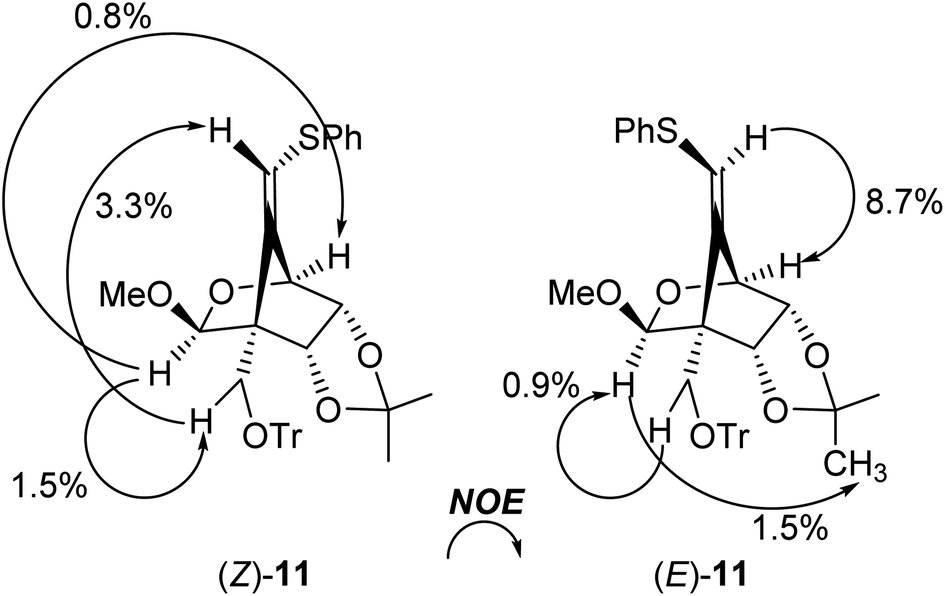
We next examined the radical-mediated cyclization of the precursor 10 (Table 1). When the selenide 10 was reacted with tris(trimethylsilyl)silane [(Me3Si)3SiH] in the presence of a catalytic amount of AIBN as an initiator in hot toluene, the desired 5-exo-dig mode cyclization readily proceeded to afford the desired cyclic products having a methylenecyclopentane skeleton (Z)- and (E)-11 [the ratio of the (E)-/(Z)-configuration of 6′-position was 8.0:1, entry 1]. The stereochemistry of both isomers was confirmed by NOE experiments as illustrated in Table 1 (see the Experimental section). The stereochemistries of the 4-position of both (Z)- and (E)-11 were completely controlled as (S)-configuration, as we expected before. The free rotation of both 3,4- and 4,5-carbon–carbon bonds of the radical intermediate F′ are certainly forbidden, due to the presence of the oxo(methoxy)methano bridge between 1- and 4-position. Therefore, the radical cyclization stereospecifically proceeded to give only (4S)-products, since the stereochemistry of the 1-position possessing a radical acceptor moiety of F′ is (S)-configuration.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reductant | Initiator | Temp. (°C) | Time (h) | Yield of (E)-11/(Z)-11 (%) | Ratio of (E)-11/(Z)-11 |
| a A trace amount of 10 was observed on TLC. | ||||||
| 1 | [(Me3Si)3SiH] | AIBN | 100 | 1.5 | 80/10 | 8.0 |
| 2 | [(Me3Si)3SiH] | Et3B | Rt | 0.5 | 82/7 | 11.7 |
| 3 | Bu3SnH | Et3B | Rt | 0.5 | 71/10 | 7.1 |
| 4 | [(Me3Si)3SiH] | Et3B | −50 | 8 | 90/5 | 18 |
| 5 | [(Me3Si)3SiH] | Et3B | −85 | 23.5a | 89/4a | 22.3 |
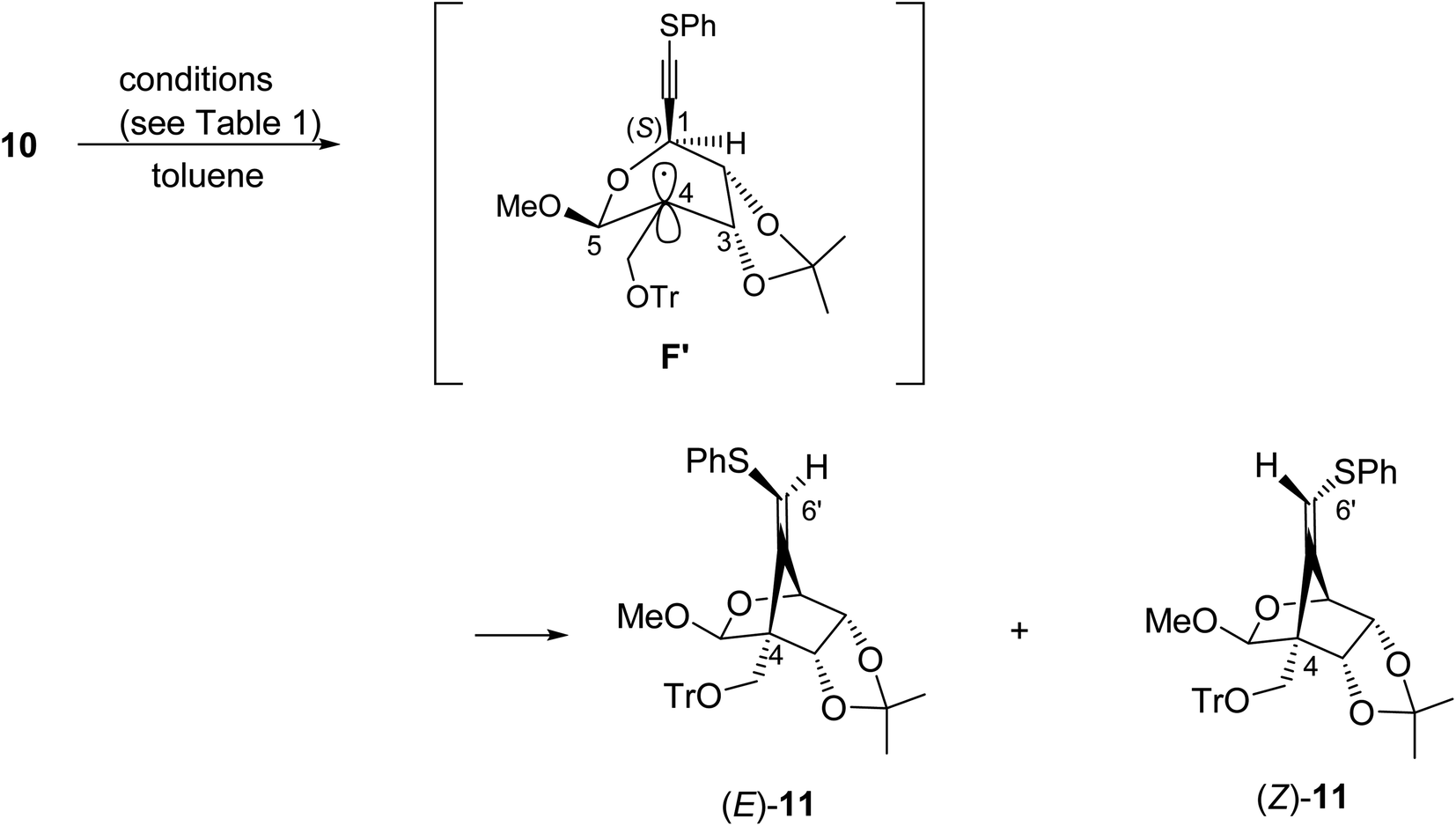
The ratio of the (E)-/(Z)-configuration of the 6′-position was increased to 11.7:1 when the reaction was carried out at room temperature using Et3B as an initiator (entry 2). The use of Bu3SnH instead of (Me3Si)3SiH, on the other hand, lowered the ratio to 7.1:1 (entry 3). Further increase of the stereoselectivity was observed along with the decrease of the reaction temperature (entries 4 and 5). The high (E)-stereoselectivity of the 6′-position might be derived from the steric repulsion between the bulky reductant (Me3Si)3SiH and trithyloxymethyl (TrOCH2–) groups of the generated vinyl radical intermediate G, which may prevent an approach from the same face of the TrOCH2– group (Fig. 2).
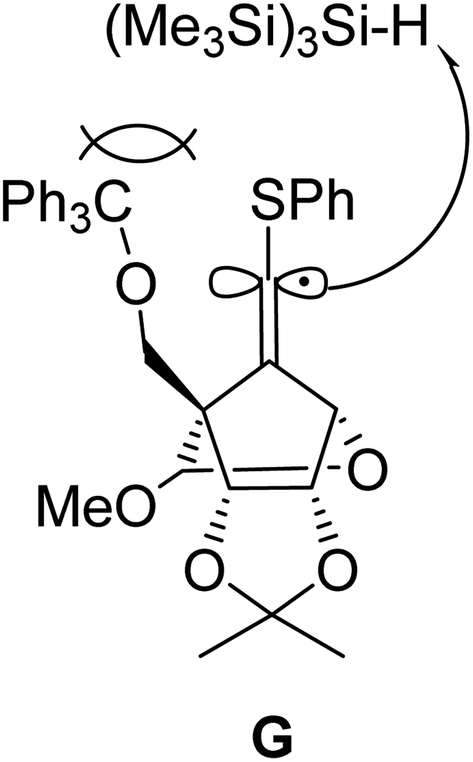 | ||
| Fig. 2 Reduction of radical intermediate G by (Me3Si)3SiH. | ||
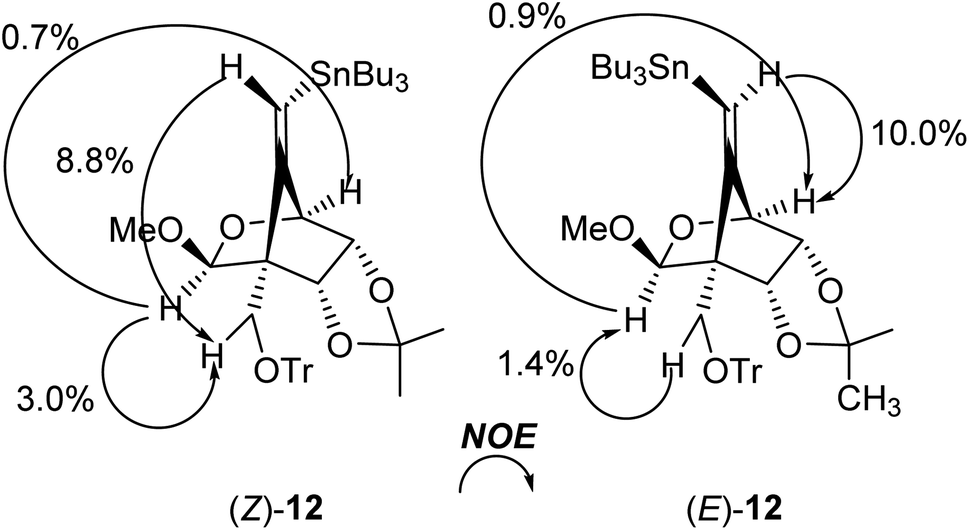
The radical-mediated sulfur extrusive stannylation of 11 was next carried out (Scheme 3). When the sulfide (E)-11 was treated with Bu3SnH and AIBN in the presence of i-Pr2NEt in hot toluene, the vinylstannanes (E)-12 (65%) and (Z)-12 (34%) were obtained, respectively. Stereochemical analysis of 12 was also confirmed by NOE experiments as depicted in Scheme 3 (see Experimental section). In the case of radical-stannylation for (Z)-11, on the other hand, (Z)-12 (83%) was the sole product. Similar results for these stereoselectivities were previously observed.13
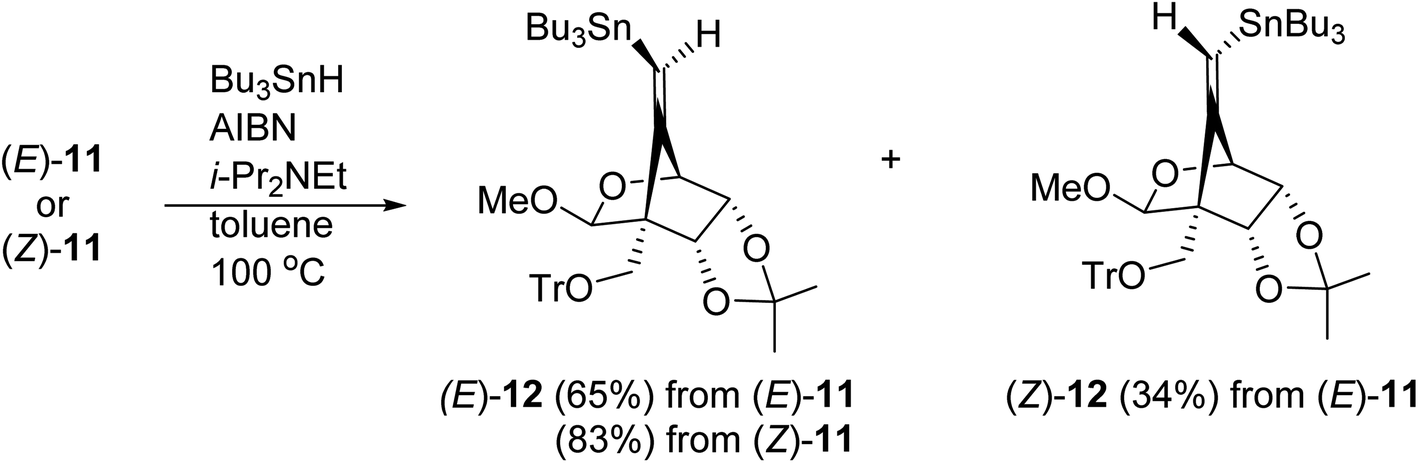 | ||
| Scheme 3 Radical-mediated sulfur-extrusive stannylation of 11. | ||
We also confirmed the possibility of the direct one-pot conversion of sulfide 10 into stannane 12 (Scheme 4). When the sulfide 10 was reacted with Bu3SnH and AIBN in the presence of i-Pr2NEt in heating toluene, the stannanes (E)-12 (49%) and (Z)-12 (33%) were obtained, respectively, and the ratio of (E)-12:(Z)-12 was 1.48:1, Scheme 4a. As illustrated in Scheme 4b on the other hand, the yield of 12 was slightly increased when the residue containing (E)- and (Z)-11, which was prepared in a described manner in Table 1; entry 4, was reacted with Bu3SnH and AIBN. The ratio of (E)-12:(Z)-12 was consistent with the results of Table 1 and Scheme 3. Therefore, the one-pot conversion without purification described in Scheme 4b should be comparable to the sequential manner.
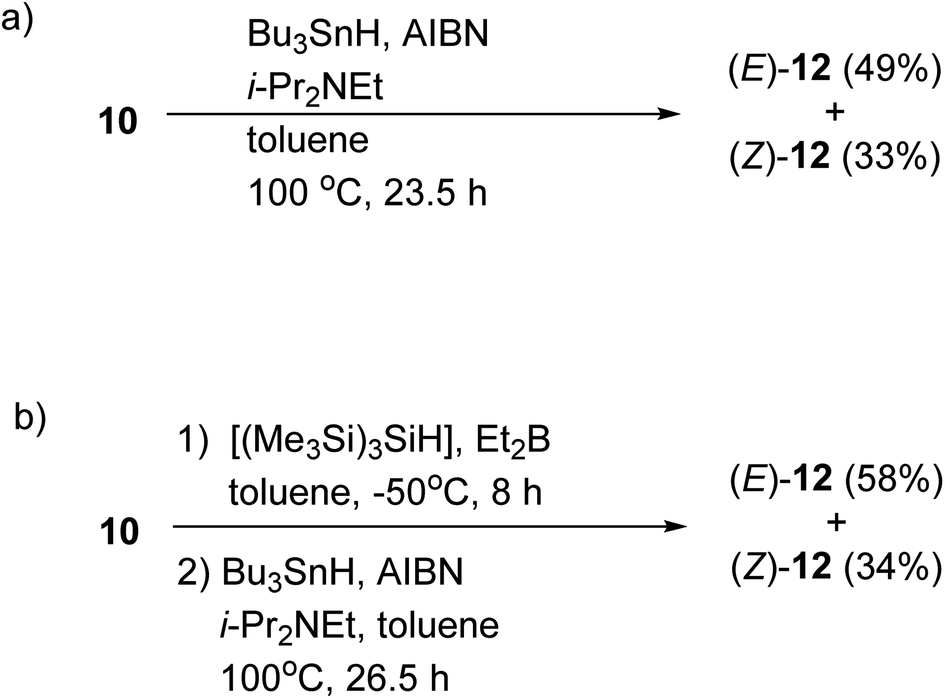 | ||
| Scheme 4 One-pot conversion of sulfide 10 into stannane 12. (a) The reaction was carried out at 100 °C. (b) The reaction was carried out under the condition described in entry 4 (Table 1) before a treatment with Bu3SnH and AIBN. | ||
Electrophilic fluorination of vinylstannanes 12 readily proceeded by treatment with XeF2 and AgOTf in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP).17,18 This gave the corresponding vinyl fluorides (E)-13 and (Z)-13, respectively (Scheme 5). The vinyl fluorides (E)-13 and (Z)-13 were converted to the cyanides (E)-14 and (Z)-14, respectively, by the following four-step sequence: (1) acidic hydrolysis of all of the acid labile protecting groups including the cyclic acetal function; (2) transformation of the generated formyl group into the corresponding oxime; (3) acetylation of all of the hydroxy groups include the oxime hydroxyl group and (4) β-elimination of the oxime acetate by the treatment with heating AcOH in the presence of a catalytic amount of NaOAc. In order to discriminate between the four hydroxyl groups of 14, we selected a cyclic protection technique, which has been frequently employed, especially for the selective protection of 3′ and 5′ hydroxyl groups of nucleosides. After deacetylation of (E)-14 by treatment with NH3/MeOH, the resulting tetraol was reacted with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2) in cold pyridine. Unexpectedly, the sole product was silyl ether (E)-15 in which the primary alcohol remained and the two secondary alcohols were protected instead. A similar result was observed in the case of (Z)-14; this might be due to the steric hindrance around the primary alcohol derived from (E)- and (Z)-14. Protection of these free hydroxyl groups in (E)-15 and (Z)-15 was carried out by the reaction with BOMCl. cis-Diols (E)-16 and (Z)-16 were prepared, respectively, after desilylation in moderate yields. The secondary alcohol of (E)-16 was selectively protected again into (E)-17 using bulky TBSCl. However, in the case of (Z)-16, mono silylether (Z)-17 was obtained as an inseparable mixture with (Z)-17a (ca. 3:2 of diastereomeric mixture).
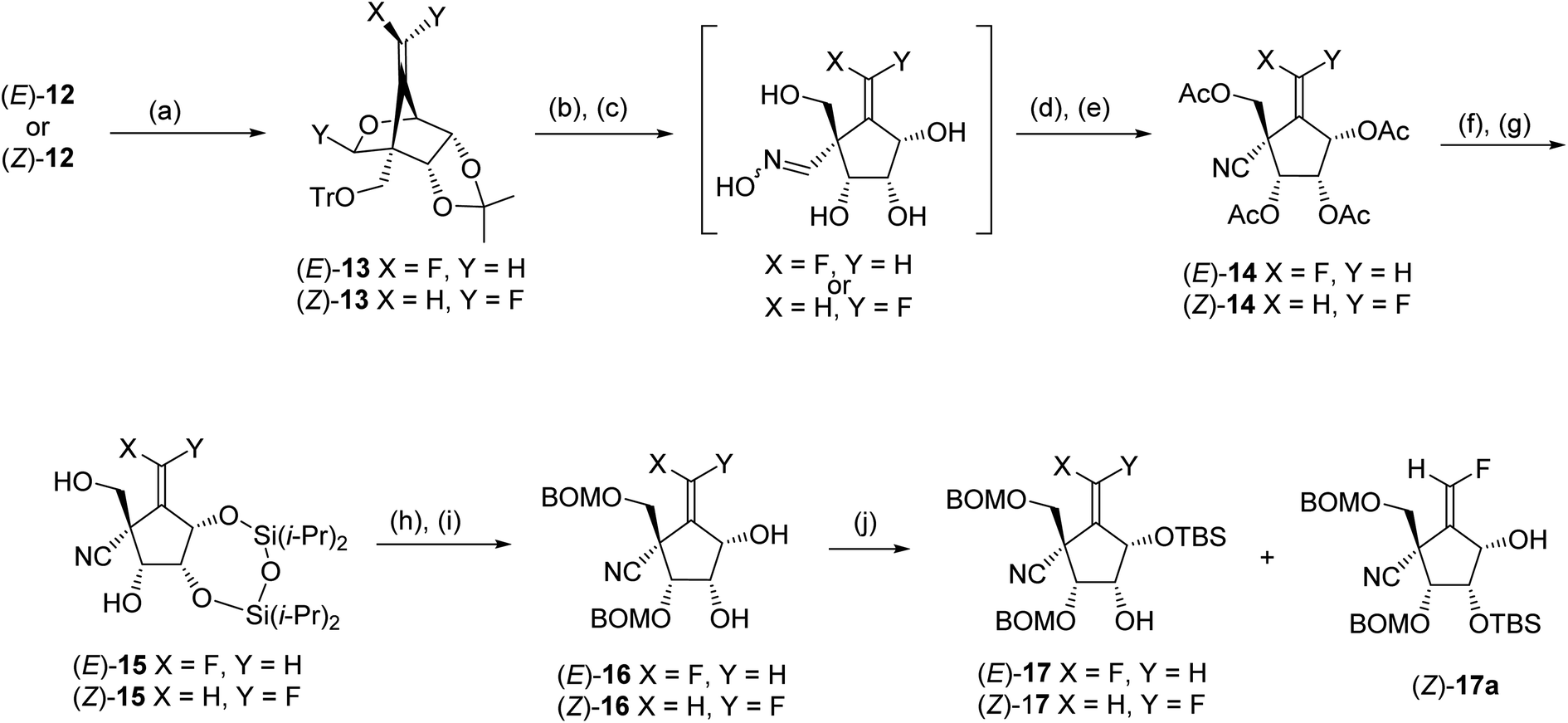 | ||
| Scheme 5 Conditions: (a) XeF2, AgOTf, DTBMP, CH2Cl2; (b) 80% AcOH, THF, 80 °C; (c) H2N–OH·HCl, pyridine; (d) Ac2O, pyridine; (e) NaOAc, AcOH, 80 °C; (f) NH3/MeOH, 4 °C; (g) TIPDSCl2, pyridine, −30 °C; (h) BOMCl, i-Pr2NEt, Bu4NI, toluene, 80 °C; (i) Bu4NF, THF; (j) TBSCl, imidazole, CH2Cl2. | ||
In order to reduce the free 2-hydroxyl group, (E)- and (Z)-17 were treated with triflic anhydride into the corresponding triflate (Scheme 6); the reaction of the triflate with lithium iodide gave iodides, respectively. The desired carbocyclic units (E)- and (Z)-18 were prepared by radical-mediated deiodination using (Me3Si)3SiH and subsequent desilylation by treatment with Bu4NF [the pure (Z)-18 was isolated at this stage]. The introduction of guanine base for (E)- and (Z)-18 was carried out under Mitsunobu19 condition employing the Boc-protected 2-amino-6-chloropurine (19)20,21 and bis(methoxyethyl)azodicarboxylate (DMEAD),22 the carbocyclic nucleosides (E)-20 (53%) and (Z)-20 (92%) were gained, respectively. Finally, the global deprotection of (E)- and (Z)-20 was conducted by treatment with 80% TFA, and the synthesis of title molecules (E)-6 and (Z)-6 was achieved after HPLC purification.
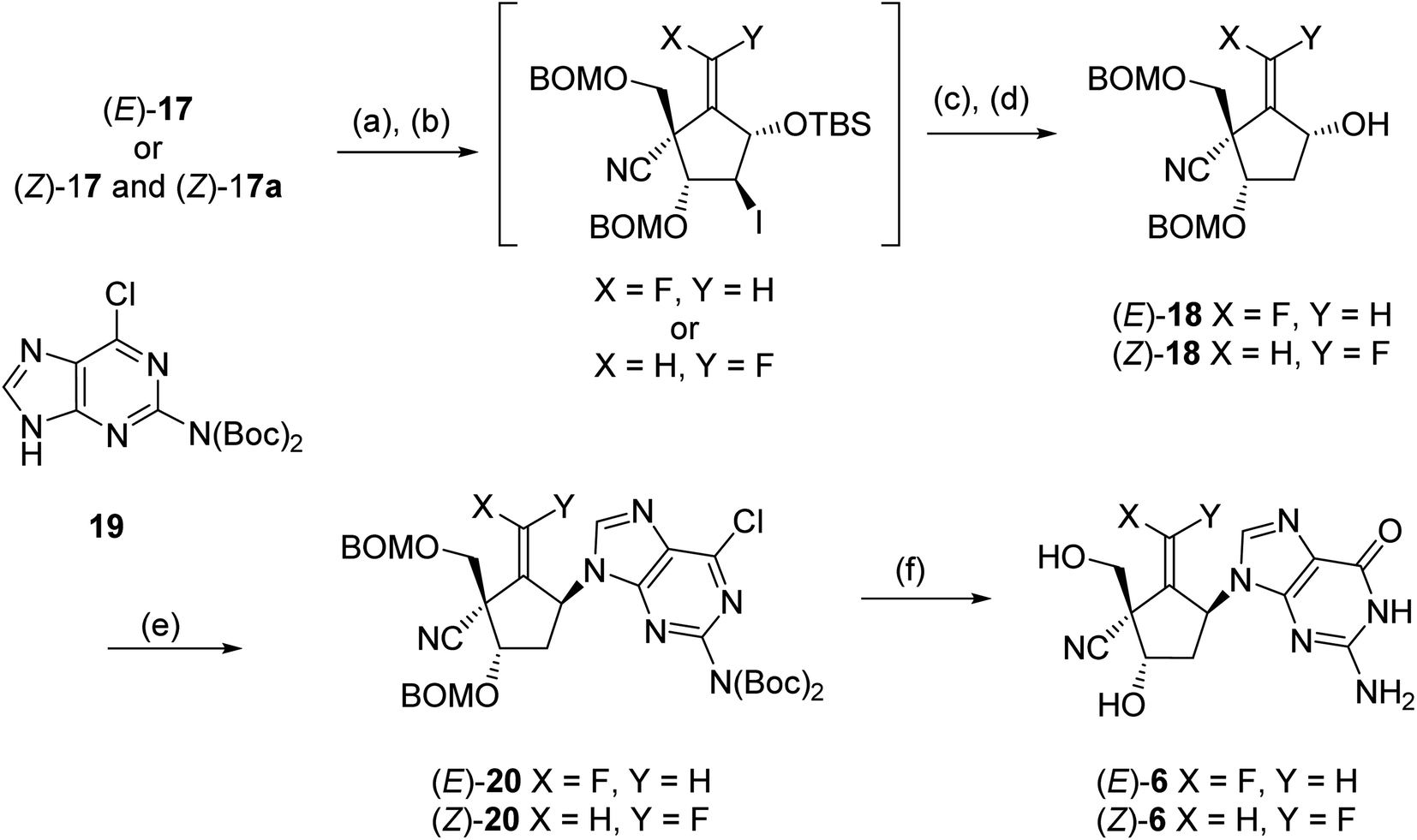 | ||
| Scheme 6 Conditions: (a) Tf2O, pyridine; (b) LiI. DMF; (c) (Me3Si)SiH, Et3B, toluene; (d) Bu4NF, THF; (e) 19, DMEAD, Ph3P, toluene; (f) 80% TFA, HPLC purification. | ||
2.2. Biological evaluation
The anti-HBV activity and cytotoxicity of compounds were determined using the HBV-producing HepG2 2.2.15 human hepatoblastoma cell line.12,14,15 The anti-HBV activity and cytotoxicity of compounds 1–6 are summarized in Table 2. Entecavir served as a control. In the present study, compound (E)-6 showed significant anti-HBV activity (EC50 = 1.2 nM) with favorable cytotoxicity (CC50 = 93 μM). By contrast, compound (Z)-6 displayed ∼500 times lower anti-HBV activity compared to compound (E)-6 and showed no cytotoxicity up to 100 μM. These results indicate that E-positioned fluorine in the cyclopentyl moiety contributes to compound (E)-6's potent anti-HBV activity.| Compound | EC50 (nM) against HBV | CC50 (μM) in HepG2.2.15 |
|---|---|---|
| 1 | 2.3 ± 0.6 | 63 |
| 2 | 0.3 ± 0.1 | 3.0 ± 0.2 |
| 3 | 0.5 ± 0.2 | 3.6 ± 0.2 |
| (E)-4 | 0.9 ± 0.3 | 83 ± 14 |
| 5 | 3.5 ± 23 | >100 |
| (E)-6 | 1.2 ± 0.7 | 93 |
| (Z)-6 | 585 ± 358 | >100 |
2.3. Structural models of the interactions of (E)-6-TP with HBVWT-RT
Using sequence alignment and homology between HBV-RT and HIV-RT, we built a structure of an HBVWT-RT primer/template (p/t) complex.12,14 Coordinates of the complex of ETV with HBVWT-RT (p/t) were used to generate a model of the interactions of (E)-6-triphosphate (TP) with HBVWT-RT (p/t) (Fig. 3). The aminopurine group of (E)-6-TP formed hydrogen bond interactions with Met-171. The magnesium ion coordinated with the triphosphate group, and has polar interactions with Asp83, Val84, and Asp205. The triphosphate moiety has hydrogen bond interactions with the side chain atoms of Ser85. The 4′-cyano substituent had a hydrogen bond interaction with the backbone NH of Phe-88. The fluorine of (E)-6-TP forms polar interactions with Met-204. This favorable interaction is not possible for (Z)-6-TP and may account for the difference in activities between these stereoisomers. (E)-6-TP also has many favorable van der Waals interactions with HBVWT-RT (p/t) as shown in Fig. 3.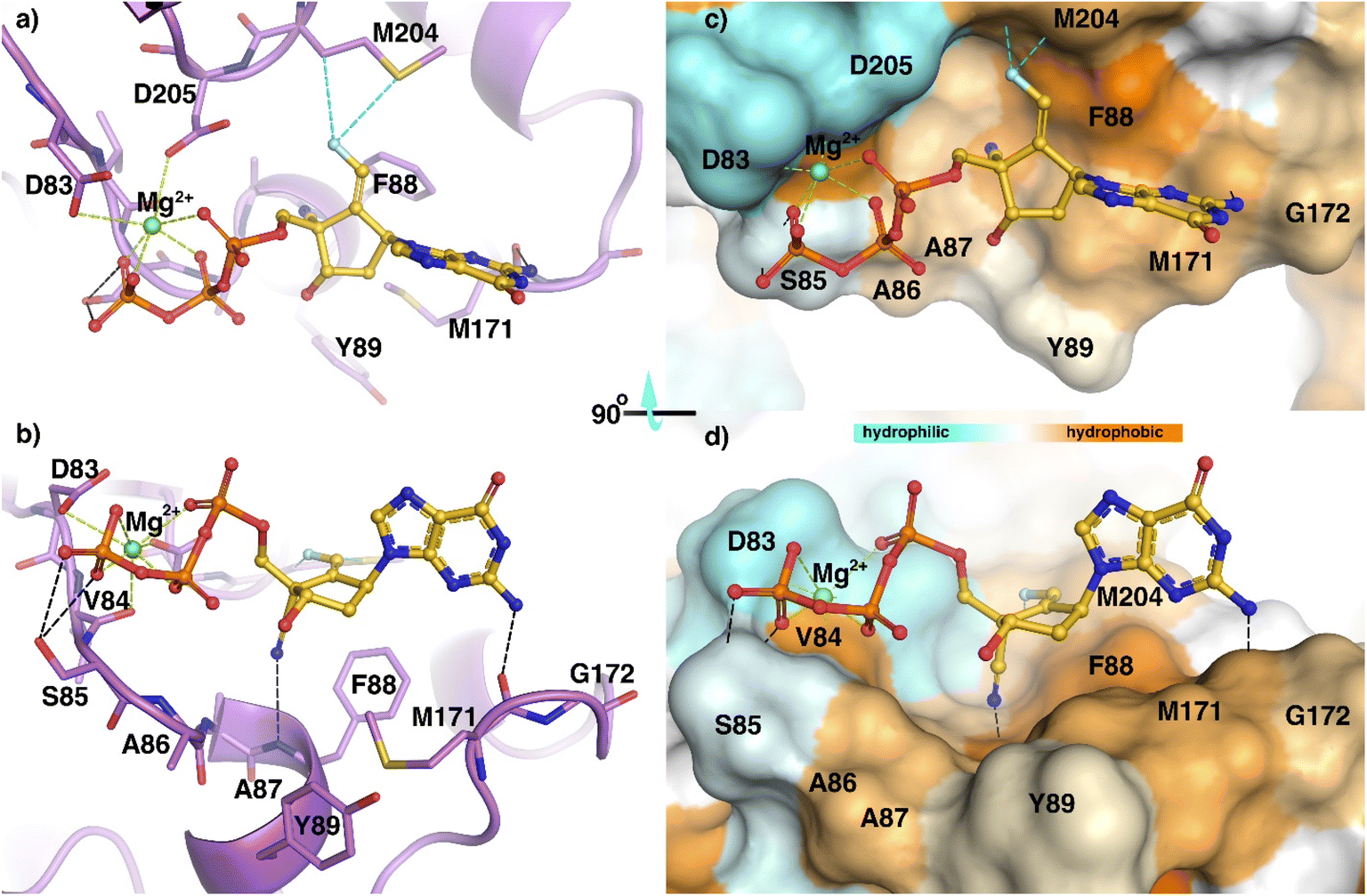 | ||
| Fig. 3 Model of structural interactions of (E)-6-TP inside the active site of HBVWT-RT in stick mode (a and b) and as a surface representation of HBVWT-RT with the hydrophobicity scale (c and d). | ||
The figure shows a ribbon representation of HBVWT-RT (purple) with active site residues in stick mode (a and b). The (E)-6-TP forms multiple interactions inside the active site. Hydrogen bonds are shown as black dashed lines, while fluorine contacts are shown as cyan dashed lines. While the aminopurine forms a hydrogen bond with the backbone carbonyl oxygen of M-171, the cyano group forms an additional hydrogen bond with the backbone amino group of F88. The side chain atoms of Ser85 form bifurcate hydrogen bond interactions with the triphosphate group. Magnesium ion (Mg2+) at the active-site periphery is shown in green and metal coordinations (under 3 Å) are highlighted in green color. The magnesium ion coordinated between the triphosphate group and the side chain carboxyl groups of D-83, D-205, and the main chain carbonyl oxygen of V-84. Fluorine atoms engage in interactions such as hydrogen bonds with the side chain of M-204. Surface representation of HBVWT-RT with the hydrophobicity scale (c and d). The center part of (E)-6-TP sandwiched between F-88 and Y-89 and aminopurine moiety was further stretched along the hydrophobic residue M171. On the opposite side, the cyan color indicates negatively-charged residues around the phosphate group coordinated with Mg2+.
3. Conclusion
Our previous findings that nucleosides 2, 3, (E)-4, and 5 are strong candidates for anti-HBV agents encouraged us to carry out this study. Synthesis of the entecavir analogues 6 possessing a cyano group and fluorine atom at 4′- and 6′′-position was accomplished based on radical-mediated stereospecific 5-exo dig mode cyclization of 10, sulfur-extrusive stannylation of 11 and subsequent electrophilic fluorination into 13, and construction of the cyano group employing the formyl group generated by hydrolysis of 13, which was located at the chiral quaternary carbon, as representative steps. Both diastereomers (E)- and (Z)-6 were synthesized after deoxygenation, the introduction of guanine base, and global deprotection. Of the two diastereomers 6 synthesized in this study, (E)-6 showed higher anti-HBV activity than (Z)-6 and parent compound 1 with lower cytotoxicity than that of 1. Molecular models suggested that both the cyano group and fluorine atoms of (E)-6 have strong interactions with HBVWT-RT and must be partly responsible for its high anti-HBV activity.4. Experimental section
4.1. Chemistry
(S)-1-((4S,5R)-5-(1-Methoxy-3-(trityloxy)prop-1-en-2-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-ol (8). To an anhydrous THF (200 mL) suspension of Ph3PCH2OMeCl (47.4 g, 138.4 mmol), BuLi (2.66 mol L−1 in hexane, 49.5 mL, 131.6 mmol) was added dropwise at −80 °C under a positive pressure of dry Ar. The resulting suspension was stirred for 2 h at rt. To this was dropwise added a THF (150 mL) solution of 7 (15.8 g, 27.7 mmol) at 0 °C. The resulting mixture was further stirred at rt for 22 h. The resulting dark brown mixture was partitioned between aqueous saturated NH4Cl and AcOEt. After evaporation of all the volatiles in the organic layer, the residue was dissolved in THF (100 mL). To this was added Bu4NF (1.0 mol L−1 in THF, 30.5 mL, 30.5 mmol), then stirred at rt for 5 h. The resulting mixture was partitioned between aqueous saturated NH4Cl and AcOEt. After evaporation of all the volatiles of the organic layer, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 5/1). This gave 8 (10.34 g, 77%, ca. 7
:3 of diastereomeric mixture) as a foam. [α]26D −26.5 (c 0.68, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.31 (s, 2.1H), 1.325 (s, 0.9H), 1.330 (s, 0.9H), 1.34 (s, 2.1H), 2.31 (d, J = 2.3 Hz, 0.3H), 2.43 (d, J = 2.3 Hz, 0.7H), 3.24 (d, J = 5.8 Hz, 0.3H), 3.53 (d, J = 5.2 Hz, 0.7H), 3.68 (s, 0.9H), 3.71 (d, J = 10.0 Hz, 0.7H), 3.73 (s, 2.1H), 3.94 (d, J = 10.0 Hz, 0.7H), 4.01 (dd, J = 8.0 and 6.3 Hz, 0.7H), 4.17–4.18 (m, 0.7H), 4.24 (dd, J = 7.5 and 6.9 Hz, 0.3H), 4.51 (d, J = 5.2 Hz, 0.7H), 5.10 (dd, J = 5.2 and 2.3 Hz, 0.3H), 6.21 (d, J = 1.1 Hz, 0.3H), 6.46 (s, 0.7H), 7.11–7.25 (m, 3H), 7.29–7.32 (m, 6H), 7.44–7.49 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 24.9, 25.2, 26.7, 27.3, 58.7, 60.4, 60.5, 61.5, 61.9, 62.5, 73.2, 73.4, 73.5, 79.9, 80.1, 82.8, 83.2, 87.7, 88.0, 107.1, 107.8, 108.5, 110.7, 127.1, 127.9, 128.8, 143.4, 143.8, 148.5, 150.8. ESI-MS (m/z): 507 [M+ + Na]. HRMS (ESI+): calcd for C31H32O5Na 507.2142, found 507.2155 [M+ + Na].
(3aS,4S,7aS)-4-Ethynyl-6-methoxy-2,2-dimethyl-7-(phenylselanyl)-7-((trityloxy)methyl)tetrahydro-4H-[1,3]dioxolo[4,5-c]pyran (9). To a CH2Cl2 (200 mL) solution of 8 (14.6 g, 30.12 mmol) and pyridine (24.4 mL, 301.2 mmol) was added PhSeCl (6.07 g, 31.6 mmol) in one portion at −80 °C. The resulting mixture was stirred at the same temperature for 5 h, and then stirred further for 20 h at rt. The mixture was partitioned between aqueous saturated NaHCO3 and CH2Cl2. After evaporation of all the volatiles in the organic layer, the residue was purified by column chromatography on silica gel (toluene). This gave 9 (16.24 g, 84%, ca. 20
:12:1 of inseparable diastereomeric mixture) as a foam. [α]26D +38.1 (c 1.08, CHCl3); 1H NMR (CDCl3, 500 MHz) of the two major diastereomers δ 1.19 (s, 1.89H), 1.22 (s, 1.11H), 1.48 (s, 1.11H), 1.51 (s, 1.89H), 2.53 (d, J = 1.7 Hz, 0.37H), 2.54 (s, J = 2.3 Hz, 0.63H), 3.33 (d, J = 9.8 Hz, 0.37H), 3.34 (d, J = 10.3 Hz, 0.63H), 3.47 (s, 1.11H), 3.50 (s, 1.89H), 3.54–3.58 (m, 0.93H), 3.80 (d, J = 10.3 Hz, 1H), 4.01 (d, J = 5.2 Hz, 0.63H), 4.19–4.21 (m, 1H), 4.40 (d, J = 4.0 Hz, 0.37H), 7.11–7.33 (m, 12H), 7.40–7.46 (m, 6H), 7.59–7.60 (m, 2H). 13C NMR (CDCl3, 125 MHz) of the two major diastereomers δ 25.55, 25.58, 28.3, 28.4, 51.8, 52.7, 57.26, 57.34, 62.8, 66.3, 67.0, 67.1, 73.7, 74.0, 74.4, 74.8, 75.9, 77.1, 77.2, 78.4, 80.7, 80.8, 87.0, 87.5, 102.2, 104.0, 108.9, 109.7, 125.4, 125.5, 126.88, 126.92, 127.5, 127.6, 127.8, 127.9, 128.3, 128.56, 128.61, 128.7, 128.8, 128.9, 129.0, 129.2, 138.4, 138.7, 143.6, 143.7. ESI-MS (m/z): 663 [M+ + Na]. HRMS (ESI+): calcd for C37H36O5NaSe 663.1624, found 663.1635 [M+ + Na].
(3aS,4S,7aS)-6-Methoxy-2,2-dimethyl-7-(phenylselanyl)-4-((phenylthio)ethynyl)-7-((trityloxy)methyl)tetrahydro-4H-[1,3]dioxolo[4,5-c]pyran (10). To an anhydrous THF (60 mL) solution of 9 (4.05 g, 5.87 mmol) was dropwise added LDA (1.5 mol L−1 in THF, 7.83 mL, 11.75 mmol) at −80 °C under positive pressure of dry Ar. After 10 min stirring of the mixture, this was stirred for 10 min at rt. To this was added a THF (25 mL) solution of PhSO2SPh (2.94 g, 11.75 mmol) at −80 °C, then the mixture was stirred further for 30 min at the same temperature. After the addition of further LDA (3.92 mL, 5.87 mmol), the mixture was partitioned between aqueous saturated NH4Cl and AcOEt. After evaporation of all the volatiles in the organic layer, the residue was purified by column chromatography on silica gel (toluene). This gave 10 (3.46 g, 79%, ca. 100
:33:7 of inseparable diastereomeric mixture) as a foam. [α]26D +18.8 (c 0.49, CHCl3); 1H NMR (CDCl3, 500 MHz) of the two major diastereomers δ 1.21 (s, 2.25H), 1.23 (s, 0.75H), 1.48 (s, 0.75H), 1.52 (s, 2.25H), 3.35 (d, J = 9.5 Hz, 0.25H), 3.39 (d, J = 10.9 Hz, 0.75H), 3.48 (s, 0.75H), 3.50 (dd, J = 9.5 and 4.6 Hz, 0.75H), 3.51 (s, 2.25H), 3.58 (d, J = 9.5 Hz, 0.25H), 3.80 (d, J = 10.9 Hz, 0.75H), 4.06 (d, J = 4.6 Hz, 0.75H), 4.20 (dd, J = 9.2 and 4.6 Hz, 0.25H), 4.39 (d, J = 9.5 Hz, 0.75H), 4.41 (d, J = 9.2 Hz, 0.25H), 4.44 (d, J = 4.6 Hz, 0.25H), 4.74 (s, 0.75H), 4.80 (s, 0.25H), 7.15–7.22 (m, 3H), 7.23–7.25 (m, 8H), 7.29–7.36 (m, 3H), 7.39–7.41 (m, 6H), 7.43–7.45 (m, 3H), 7.62–7.64 (m, 2H). 13C NMR (CDCl3, 125 MHz) of the two major diastereomers δ 25.6, 26.1, 28.3, 28.4, 52.0, 52.9, 57.3, 57.4, 62.6, 66.2, 68.17, 68.24, 73.2, 73.5, 74.3, 74.8, 77.2, 78.5, 87.0, 87.5, 95.5, 95.6, 102.1, 104.0, 108.9, 109.6, 126.57, 126.62, 126.7, 126.9, 127.0, 127.1, 127.5, 127.8, 128.3, 128.6, 128.7, 128.8, 128.9, 129.0, 129.2, 129.4, 138.4, 138.7. ESI-MS (m/z): 749 [M+ + H], HRMS (ESI+): calcd for C43H41O5SSe 749.1840, found 749.1875 [M+ + H].
Entry 1. A toluene (7 mL) solution of 10 (500 mg, 0.67 mmol), (Me3Si)3SiH (413 μL, 1.34 mmol) and AIBN (21 mg, 0.13 mmol) was heated at 100 °C for 1.5 h. After evaporation of all the volatile components, the residue was purified by column chromatography on the silica gel. This gave (Z)-11 (hexane/AcOEt = 8
:1, 41 mg, 10% as a foam) and (E)-11 (hexane/AcOEt = 6:1, 317 mg, 80% as a foam), respectively.
Entry 2. To a toluene (7 mL) solution of 10 (500 mg, 0.67 mmol) and (Me3Si)3SiH (413 μL, 1.34 mmol) was added Et3B (1.0 mol L−1 in THF, 13.4 mL, 1.34 mmol) in one portion at RT. The resulting mixture was stirred further for 30 min at the same temperature. After evaporation of all of the volatile components, the residue was purified by column chromatography on silica gel. This gave (Z)-11 (hexane/AcOEt = 8
:1, 26 mg, 7%) and (E)-11 (hexane/AcOEt = 6:1, 327 mg, 82%) respectively.
Entry 3. To a toluene (7 mL) solution of 10 (500 mg, 0.67 mmol) and Bu3SnH (360 μL, 1.34 mmol) was added Et3B (1.0 mol L−1 in THF, 13.4 mL, 1.34 mmol) in one portion at rt. The resulting mixture was stirred further for 30 min at the same temperature. After evaporation of the volatile components, the residue was purified by column chromatography on silica gel. This gave (Z)-11 (hexane/AcOEt = 8
:1, 41 mg, 10%) and (E)-11 (hexane/AcOEt = 6:1, 282 mg, 71%).
Entry 4. To a toluene (7 mL) solution of 10 (500 mg, 0.67 mmol) and (Me3Si)3SiH (413 μL, 1.34 mmol) was added Et3B (1.0 mol L−1 in THF, 13.4 mL, 1.34 mmol) in one portion at −50 °C. The resulting mixture was stirred for a further 8 h at the same temperature. The mixture was treated in the same manner as described in Entry 2. This gave (Z)-11 (20 mg, 5%) and (E)-11 (358 mg, 90%) respectively.
Entry 5. To a toluene (7 mL) solution of 10 (500 mg, 0.67 mmol) and (Me3Si)3SiH (413 μL, 1.34 mmol) was added Et3B (1.0 mol L−1 in THF, 13.4 mL, 1.34 mmol) in one portion at −80 °C. The resulting mixture was stirred further for 23.5 h at the same temperature. The mixture was treated in the same manner as described in Entry 2. This gave (Z)-11 (14 mg, 4%) and (E)-11 (353 mg, 89%) respectively.
Physical data for (Z)-11. [α]27D +90.3 (c 1.26, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.41 (s, 3H), 1.63 (s, 3H), 3.318 (d, J = 9.6 Hz, 1H), 3.321 (s, 3H), 3.64 (d, J = 9.6 Hz, 1H), 4.36 (dd, J = 8.0 and 2.0 Hz, 1H), 4.69 (d, J = 2.0 Hz, 1H), 4.76 (d, J = 8.0 Hz, 1H), 5.07 (s, 1H), 5.86 (s, 1H), 7.23–7.29 (m, 14H), 7.44–7.46 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 25.6, 25.7, 55.3, 57.4, 58.6, 74.9, 78.0, 78.3, 76.6, 86.6, 101.8, 114.6, 115.4, 126.5, 127.1, 127.5, 127.8, 128.8, 129.0, 135.3, 143.5, 143.7. ESI-MS (m/z): 615 [M+ + Na]. HRMS (ESI+): calcd for C37H36O5NaS 615.2176, found 615.2190 [M+ + Na].
Physical data for (E)-11. [α]26D +70.0 (c 0.66, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.40 (s, 3H), 1.70 (s, 3H), 3.42 (s, 3H), 3.64 (d, J = 9.2 Hz, 1H), 3.81 (d, J = 9.2 Hz, 1H), 4.31 (dd, J = 10.3 and 2.3 Hz, 1H), 4.32 (s, 1H), 4.58 (d, J = 10.3 Hz, 1H), 5.31 (s, 1H), 6.25 (s, 1H), 7.19–7.25 (m, 1H), 7.28–7.31 (m, 5H), 7.52–7.53 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 25.5, 25.8, 55.7, 58.1, 61.0, 78.6, 79.0, 79.6, 86.5, 102.5, 115.3, 118.4, 126.9, 127.4, 127.6, 129.0, 129.6, 135.2, 139.5, 163.9. ESI-MS (m/z): 593 [M+ + H], HRMS (ESI+): calcd for C37H37O5S 593.2381, found 593.2388 [M+ + H].
 :1, 10.3 g, 34% as an oil) and (E)-12 (hexane/AcOEt = 15:1, 19.8 g, 65% as an oil).
:1, 10.3 g, 34% as an oil) and (E)-12 (hexane/AcOEt = 15:1, 19.8 g, 65% as an oil).
Physical data for (Z)-12. [α]27D + 60.8 (c 1.69, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 0.82–0.94 (m, 15H), 1.23–1.30 (m, 6H), 1.40 (s, 3H), 1.41–1.45 (m, 6H), 1.63 (s, 3H), 3.24 (d, J = 9.2 Hz, 1H), 3.33 (s, 3H), 3.52 (d, J = 9.2 Hz, 1H), 4.09 (d, J = 2.0 Hz, 1H), 4.26 (dd, J = 8.0 and 2.0 Hz, 1H), 4.29 (d, J = 8.0 Hz, 1H), 5.14–5.15 (m, 1H), 5.45–5.56 (m, 1H), 7.22–7.25 (m, 3H), 7.28–7.31 (m, 6H), 7.48–7.49 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 10.4, 13.6, 25.5, 25.8, 27.2, 28.9, 54.9, 57.3, 59.5, 78.2, 78.6, 78.9, 86.3, 101.8, 114.7, 119.1, 126.9, 127.7, 128.9, 144.0, 156.3, 78.7, 80.8, 81.1, 86.1, 102.8, 114.6, 119.6, 126.8, 127.7, 128.8, 144.2, 156.2. ESI-MS (m/z): 797 [M+ + Na]. HRMS (ESI+): calcd for C43H58O5NaSn 797.3208, found 797.3218 [M+ + Na].
Physical data for (E)-12. [α]26D +56.6 (c 0.71, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 0.53–0.62 (m, 6H), 0.78–0.81 (m, 9H), 1.09–1.16 (m, 6H), 1.24–1.28 (m, 6H), 1.40 (s, 3H), 1.68 (s, 3H), 3.29 (d, J = 8.1 Hz, 1H), 3.38 (s, 3H), 3.63 (d, J = 8.1 Hz, 1H), 4.13 (d, J = 1.7 Hz, 1H), 4.27 (dd, J = 8.1 and 1.7 Hz, 1H), 4.36 (d, J = 8.1 Hz, 1H), 5.34 (s, 1H), 5.66–5.74 (m, 1H), 7.21–7.24 (m, 3H), 7.28–7.31 (m, 6H), 7.53–7.54 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 11.0, 13.7, 25.4, 25.8, 27.1, 28.9, 55.7, 57.1, 60.6, 78.7, 80.7, 81.1, 86.1, 102.8, 114.6, 119.6, 126.8, 127.7, 128.8, 144.2, 156.2. ESI-MS (m/z): 797 [M+ + Na]. HRMS (ESI+): calcd for C43H58O5NaSn 797.3208, found 797.3214 [M+ + Na].
 :1, 185 mg, 34%) and (E)-12 (hexane/AcOEt = 15:1, 301 mg, 58%).
:1, 185 mg, 34%) and (E)-12 (hexane/AcOEt = 15:1, 301 mg, 58%).
(3aS,4S,6S,7S,7aR,E)-8-(Fluoromethylene)-6-methoxy-2,2-dimethyl-7-((trityloxy)methyl)tetrahydro-4H-4,7-methano[1,3]dioxolo[4,5-c]pyran [(E)-13]. To a mixture of (E)-12 (19.8 g, 25.6 mmol), 2,6-di-tert-butyl-4-methylpyridine (11.8 g, 57.6 mmol) and XeF2 (4.56 g, 26.9 mmol) in anhydrous CH2Cl2 (200 mL) was added AgOTf (1.64 g, 6.4 mmol). The resulting mixture was stirred for 100 min at room temperature. The mixture was partitioned between CH2Cl2 and aqueous-saturated NaHCO3. After evaporation of the volatiles in the organic layer, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 3
:1). This gave (E)-13 (11.26 g, 88%) as a solid. Mp 228–231 °C; [α]26D +91.2 (c 1.05, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.39 (s, 3H), 1.66 (s, 3H), 3.40 (s, 3H), 3.51 (d, J = 9.5 Hz, 1H), 3.75 (d, J = 9.5 Hz, 1H), 4.29 (dd, J = 8.0, and 2.0 Hz, 1H), 4.39 (t, JH,F = 2.0 Hz and J = 2.0 Hz, 1H), 4.54 (d, J = 8.0 Hz, 1H), 5.25 (s, 1H), 6.60 (d, JH,F = 82.5 Hz, 1H), 7.23–7.26 (m, 3H), 7.29–7.32 (m, 6H), 7.50–7.51 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 25.5, 25.7, 55.7, 57.5, 60.3 (d, JC,F = 2.4 Hz), 74.8 (d, JC,F = 10.8 Hz), 79.7, 79.9 (d, JC,F = 3.5 Hz), 86.4, 102.1, 115.9, 122.4 (d, JC,F = 7.2 Hz), 126.9, 127.6, 128.9, 141.9 (d, JC,F = 262.3 Hz), 143.9. 19F NMR (CDCl3, 470 MHz) δ −136.2 (d, JH,F = 73.3 Hz). ESI-MS (m/z): 525 [M+ + Na]. HRMS (ESI+): calcd for C31H31FNaO5 525.2048, found 525.2048 [M+ + Na].
(3aS,4S,6S,7S,7aR,Z)-8-(Fluoromethylene)-6-methoxy-2,2-dimethyl-7-((trityloxy)methyl)tetrahydro-4H-4,7-methano[1,3]dioxolo[4,5-c]pyran [(Z)-13]. Compound (Z)-17 (10.8 g, 14.0 mmol) was treated with the same procedure described for (E)-13. This gave (Z)-13 (3.27 g, 47%) as a foam. [α]26D +77.5 (c 0.23, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.40 (s, 3H), 1.61 (s, 3H), 3.24 (d, J = 9.8 Hz, 1H), 3.32 (s, 3H), 3.58 (d, J = 9.8 Hz, 1H), 4.39 (dd, J = 8.0 and 2.0 Hz, 1H), 4.77 (d, J = 8.0 Hz, 1H), 4.83 (d, J = 2.0 Hz, 1H), 5.03 (d, JH,F = 4.0 Hz, 1H), 6.17 (d, JH,F = 83.8 Hz, 1H), 7.24–7.26 (m, 3H), 7.30–7.33 (m, 6H), 7.44–7.47 (m, 6H). 13C NMR (CDCl3, 125 MHz) δ 25.5, 25.7, 55.3 (d, JC,F = 3.8 Hz), 55.4, 58.3, 72.9, 77.3, 78.3 (d, JC,F = 3.6 Hz), 79.2, 86.6, 101.8, 116.0, 123.7 (d, d, JC,F = 11.9 Hz), 127.1, 127.9, 128.7, 140.3 (d, JC,F = 258.7 Hz), 143.6. 19F NMR (CDCl3, 470 MHz) δ −132.5 (d, JH,F = 73.4 Hz). ESI-MS (m/z): 525 [M+ + Na]. HRMS (ESI+): calcd for C31H31FNaO5 525.2048, found 525.2061 [M+ + Na].
(1S,2R,3R,4S,E)-4-(Acetoxymethyl)-4-cyano-5-(fluoromethylene)cyclopentane-1,2,3-triyl triacetate [(E)-14]. To a THF (20 mL) solution of (E)-13 (11.0 g, 22.0 mmol) was added 80% AcOH (100 mL). The resulting suspension was stirred at 80 °C for 5 h. After evaporation of all of the volatiles of the mixture, the residue was dissolved in pyridine (100 mL). To this was added HO–NH2·HCl (3.1 g, 43.1 mmol) at 0 °C, then, the mixture was stirred for 30 h at room temperature. After monitoring the disappearance of an intermediate aldehyde and generation of oxime by TLC (CHCl3/MeOH = 3
:1), to this was added Ac2O (31.1 mL, 329.3 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 40 h. After the addition of MeOH (50 mL), the mixture was partitioned between aqueous saturated NaHCO3 and AcOEt. Column chromatography on silica gel (hexane/AcOEt = 1:1) of the organic layer gave crude pentaacetate (7.02 g) as an oil. To an AcOH (100 mL) solution of the oil was added NaOAc (100 mg). The mixture was stirred at 80 °C for 96 h. After evaporation of all the volatile components, the residue was partitioned between aqueous-saturated NaHCO3 and AcOEt. Column chromatography on silica gel (hexane/AcOEt = 1:1) of the organic layer gave (E)-14 (5.01 g, 64% for four steps) as an oil. [α]26D +9.5 (c 0.43, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.119 (s, 3H), 2.122 (s, 3H), 2.16 (s, 3H), 2.18 (s, 3H), 4.41 (d, J = 11.5 Hz, 1H), 4.44 (d, J = 11.5 Hz, 1H), 5.38 (d, J = 3.5 Hz, 1H), 5.46–5.47 (m, 1H), 5.64–5.66 (m, 1H), 6.92 (dd, JH,F = 77.9 Hz and J = 2.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 20.47, 20.51, 20.6, 45.0, 63.1 (d, JC,F = 2.4 Hz), 68.9 (d, JC,F = 8.3 Hz), 70.5, 71.1, 114.8, 116.6 (d, JC,F = 7.2 Hz), 151.6 (d, JC,F = 271.8 Hz), 169.3, 169.6, 169.7, 169.8. 19F NMR (CDCl3, 470 MHz) δ −118.9 (d, JH,F = 73.4 Hz). ESI-MS (m/z): 372 [M+ + H], HRMS (ESI+): calcd for C16H19FNO8 372.1095, found 372.1079 [M+ + H].
(1S,2R,3R,4S,Z)-4-(Acetoxymethyl)-4-cyano-5-(fluoromethylene)cyclopentane-1,2,3-triyl triacetate [(Z)-14]. Compound (Z)-13 (3.3 g, 6.6 mmol) was treated with the same procedure described for (E)-14. This gave (Z)-14 (1.01 g, 42% for four steps) as a solid. Mp 157–158 °C; [α]26D −7.0 (c 0.33, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.12 (s, 3H), 2.14 (s, 3H), 2.16 (s, 3H), 2.18 (s, 3H), 4.16 (d, J = 11.5 Hz, 1H), 4.35 (d, J = 11.5 Hz, 1H), 5.29 (d, J = 4.0 Hz, 1H), 5.45 (dd, J = 5.2 and 4.0 Hz, 1H), 6.02–6.04 (m, 1H), 7.03 (dd, JH,F = 76.8 Hz and J = 2.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 20.38, 20.42, 20.46, 20.53, 45.1 (d, JC,F = 6.0 Hz), 65.1, 67.4, 70.0, 71.6, 116.4, 118.0 (d, JC,F = 10.8 Hz), 152.1 (d, JC,F = 273.5 Hz), 169.3, 169.46, 169.52, 169.6. 19F NMR (470 MHz, CDCl3) δ −118.3 (d, J = 73.4 Hz). MS (ESI+) m/z C16H18FNNaO8, 394 [M+ + Na]. HRMS (ESI+): calcd for C16H18FNNaO8 394.0909, found 394.0900 [M+ + Na].
(5aS,7S,8R,8aR,E)-6-(Fluoromethylene)-8-hydroxy-7-(hydroxymethyl)-2,2,4,4-tetraisopropyltetrahydro-6H-cyclopenta[f][1,3,5,2,4]trioxadisilepine-7-carbonitrile [(E)-15]. To a THF (10 mL) solution of (E)-14 (5.0 g, 13.5 mmol) was added NH3/MeOH (saturated at 0 °C, 70 mL). The resulting solution was kept at 4 °C for 80 h. After evaporation of all the volatile components, the crude residue was azeotropic and dried with toluene. The residue was dissolved in anhydrous pyridine (60 mL). To this was dropwise added 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (4.68 mL, 14.8 mmol) at −50 °C. The mixture was stirred further for 45 h at −30 °C. After the addition of MeOH (100 mL) and evaporation of all volatiles, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 7
:5). This gave (E)-15 (3.07 g, 51%) as a solid. Mp 84–86 °C; [α]26D −6.1 (c 0.88, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.04–1.10 (m, 28H), 2.06 (dd, J = 7.5 and 5.5 Hz, 1H), 3.09 (d, J = 12.4 Hz, 1H), 3.99 (dd, J = 10.9 and 7.5 Hz, 1H), 4.04 (dd, J = 10.9 and 5.5 Hz, 1H), 4.28 (d, J = 12.4 and 3.2 Hz, 1H), 4.42 (dd, J = 6.3 and 3.2 Hz, 1H), 4.67–4.69 (m, 1H), 6.89 (dd, JH,F = 80.8 Hz and J = 2.9 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 12.86, 12.91, 13.0, 14.2, 16.7, 16.9, 17.20, 17.29, 17.35, 17.41, 17.5, 48.3, 63.4, 71.7, 73.7 (d, JC,F = 7.2 Hz), 75.5, 116.4, 121.6 (d, JC,F = 6.0 Hz), 149.3 (d, JC,F = 261.5 Hz). 19F NMR (470 MHz, CDCl3) δ −130.5 (d, J = 88.0 Hz). ESI-MS (m/z): 446 [M+ + H], HRMS (ESI+): calcd for C20H37FNO5Si2 446.2194, found 446.2199 [M+ + H].
(5aS,7S,8R,8aR,Z)-6-(Fluoromethylene)-8-hydroxy-7-(hydroxymethyl)-2,2,4,4-tetraisopropyltetrahydro-6H-cyclopenta[f][1,3,5,2,4]trioxadisilepine-7-carbonitrile [(Z)-15]. Compound (Z)-14 (1.01 g, 2.72 mmol) was treated with the same procedure described for (E)-15. This gave (Z)-15 (877 mg, 72%) as a solid. Mp 100–103 °C; [α]27D +1.8 (c 0.46, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.05–1.12 (m, 28H), 2.24 (dd, J = 8.0 and 5.4 Hz, 1H), 3.21 (d, J = 10.9 Hz, 1H), 3.73 (dd, J = 10.9 and 8.0 Hz, 1H), 3.87 (dd, J = 10.9 and 5.4 Hz, 1H), 4.02 (dd, J = 10.9 and 4.0 Hz, 1H), 4.33 (t, J = 4.0 Hz, 1H), 5.007–5.011 (m, 1H), 7.03 (dd, JH,F = 79.0 Hz and J = 2.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 12.9, 13.2, 13.5, 16.9, 16.95, 17.0, 17.1, 17.25, 17.32, 17.4, 17.5, 49.4 (d, JC,F = 6.0 Hz), 66.4, 72.6, 73.7, 74.9, 118.4, 121.9 (d, JC,F = 8.4 Hz), 151.2 (d, JC,F = 271.1 Hz). 19F NMR (CDCl3, 470 MHz) δ −122.7 (d, JH,F = 73.4 Hz). ESI-MS (m/z): 446 [M+ + H], HRMS (ESI+): calcd for C20H37FNO5Si2 446.2194, found 446.2202 [M+ + H].
(1S,2R,3S,4S,E)-2-((Benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-5-(fluoromethylene)-3,4-dihydroxycyclopentane-1-carbonitrile [(E)-16]. To a mixture of (E)-15 (3.07 g, 6.89 mmol), i-Pr2NEt (14.4 mL, 82.7 mmol) and Bu4NI (15.3 g, 41.3 mmol) in anhydrous toluene (60 mL) was added BOMCl (6.35 mL, 58.1 mmol). The resulting mixture was stirred at 80 °C for 6 days. After the addition of MeOH (10 mL), the mixture was partitioned between aqueous saturated NaHCO3 and AcOEt, and the organic layer was further washed with brine. After the evaporation of all of the volatiles in the organic layer, the residue was used for the next step without further purification. To a THF (100 mL) solution of the residue was added Bu4NF (1.0 mol L−1 in THF, 17.2 mL, 17.2 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 22 h. After the addition of AcOH (900 μL) and evaporation of all the volatiles of the mixture, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 7
:5). This gave (E)-16 (2.81 g, 92% for two steps) as an oil. [α]26D −34.8 (c 0.27, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.58 (d, J = 10.9 Hz, 1H), 2.89 (d, J = 3.5 Hz, 1H), 3.83 (d, J = 9.7 Hz, 1H), 3.92 (d, J = 9.7 Hz, 1H), 4.06–4.10 (m, 1H), 4.25 (d, J = 3.4 Hz, 1H), 4.40–4.43 (m, 1H), 4.56 (d, J = 12.1 Hz, 1H), 4.58 (d, J = 12.1 Hz, 1H), 4.72 (d, J = 12.3 Hz, 1H), 4.74 (t, J = 6.3 Hz, 2H), 4.75 (d, J = 12.3 Hz, 1H), 4.97 (t, J = 7.2 Hz, 2H), 6.90 (dd, JH,F = 79.0 Hz and J = 2.3 Hz, 1H), 7.28–7.37 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ 46.4, 67.6, 70.1, 70.9, 71.0 (d, JC,F = 3.6 Hz), 72.3, 78.2, 94.8, 95.1, 116.8, 122.6 (d, JC,F = 2.4 Hz), 127.9, 127.95, 128.03, 128.2, 128.5, 128.6, 136.9, 137.2, 150.4 (d, JC,F = 266.3 Hz). 19F NMR (CDCl3, 470 MHz) δ −125.7 (d, JH,F = 73.4 Hz). ESI-MS (m/z): 444. [M+ + H], HRMS (ESI+): calcd for C24H27FNO6 444.1822, found 444.1834 [M+ + H].
(1S,2R,3S,4S,Z)-2-((Benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-5-(fluoromethylene)-3,4-dihydroxycyclopentane-1-carbonitrile [(Z)-16]. Compound (Z)-15 (825 mg, 1.85 mmol) was treated with the same procedure described for (E)-16. This gave (Z)-16 (752 mg, 92%) as an oil. [α]27D +9.2 (c 0.27, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.60 (d, J = 9.2 Hz, 1H), 3.13 (d, J = 6.9 Hz, 1H), 3.57 (d, J = 9.8 Hz, 1H), 3.60 (d, J = 9.8 Hz, 1H), 4.05–4.06 (m, 1H), 4.18 (d, J = 2.9 Hz, 1H), 4.62 (t, J = 12.3 Hz, 2H), 4.68–4.71 (m, 1H), 4.75 (t, J = 12.0 Hz, 2H), 4.79 (t, J = 7.7 Hz, 2H), 4.95 (d, J = 7.2 Hz, 1H), 5.02 (d, J = 7.2 Hz, 1H), 7.04 (dd, JH,F = 79.0 Hz and J = 1.7 Hz, 1H), 7.29–7.38 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ 46.9 (d, JC,F = 6.0 Hz), 69.3, 70.3, 70.4, 70.9, 71.6, 79.5, 94.8, 95.3, 118.7, 123.1 (d, JC,F = 10.8 Hz), 127.9, 128.0, 128.1, 128.6, 137.0, 137.2, 152.3 (d, JC,F = 268.7 Hz). 19F NMR (470 MHz, CDCl3) δ −120.4 (d, J = 73.3 Hz). ESI-MS (m/z): 466 [M+ + Na], HRMS (ESI+): calcd for C24H26FNNaO6 466.1636, found 466.1629 [M+ + Na].
(1S,2R,3R,4S,E)-2-((Benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-4-((tert-butyldimethylsilyl)oxy)-5-(fluoromethylene)-3-hydroxycyclopentane-1-carbonitrile [(E)-17]. To a mixture of (E)-16 (204 mg, 0.46 mmol) and imidazole (94 mg, 1.38 mmol) in CH2Cl2 (5 mL) was added TBSCl (167 mg, 1.1 mmol) at 0 °C. The resulting mixture was stirred at rt for 4.5 h. After the addition of MeOH (1 mL), the mixture was partitioned between aqueous saturated NaHCO3 and CH2Cl2. Column chromatography on silica gel (hexane/AcOEt = 3
:1) of the organic layer gave (E)-17 (205 mg, 80%) as an oil. [α]27D −20.0 (c 0.37, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 0.12 (s, 3H), 0.13 (s, 3H), 0.93 (s, 9H), 2.67 (d, J = 2.3 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.94 (d, J = 10.9 Hz, 1H), 4.04–4.05 (m, 1H), 4.20 (d, J = 3.4 Hz, 1H), 4.45–4.47 (m, 1H), 4.56 (s, 2H), 4.73 (d, J = 12.0 Hz, 1H), 4.74 (s, 2H), 4.78 (d, J = 12.0 Hz, 1H), 4.96 (t, J = 7.4 Hz, 2H), 6.68 (dd, JH,F = 79.6 Hz and J = 2.3 Hz, 1H), 7.29–7.37 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ −4.9, −4.8, 18.1, 25.7, 46.0, 67.7, 69.9, 70.4, 71.6 (d, JC,F = 7.2 Hz), 72.6, 77.4, 94.8, 94.9, 116.6, 122.1 (d, JC,F = 4.8 Hz), 127.8, 127.9, 128.0, 128.1, 128.5, 137.2, 137.3, 149.3 (d, JC,F = 263.9 Hz). 19F NMR (CDCl3, 470 MHz) δ −127.6 (d, JH,F = 73.3 Hz). MS (ESI+) m/z C30H40FNNaO6Si, 580 [M+ + Na]. HRMS (ESI+): calcd for C30H40FNNaO6Si 580.2501, found 580.2512 [M+ + Na].
Silylation of Z-21; formation to (1S,2R,3R,4S,Z)-2-((benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-4-((tert-butyldimethylsilyl)oxy)-5-(fluoromethylene)-3-hydroxycyclopentane-1-carbonitrile [(Z)-17] and its regioisomer [(Z)-17a]. Compound (Z)-16 (646 mg, 1.46 mmol) was treated with the same procedure described for (E)-17. This gave an inseparable mixture of (Z)-17 and (Z)-17a (721 mg, ca. 3
:2 of mixture, 89%) as an oil. The residue was used for the next step without further separation. 1H NMR of the mixture of (Z)-17 and (Z)-17a (CDCl3, 500 MHz) δ 0.14 (s 1.2H), 0.16 (s, 3.6H), 0.17 (s, 1.2H), 0.93 (s, 5.4H), 0.95 (s, 3.6H), 2.80 (d, J = 10.5 Hz, 0.4H), 3.02 (d, J = 8.2 Hz, 0.6H), 3.61 (d, J = 9.7 Hz, 0.4H), 3.62 (d, J = 9.8 Hz, 0.6H), 3.67–3.69 (m, 1H), 3.97–4.02 (m, 1.2H), 4.10 (dd, J = 5.2 and 3.5 Hz, 0.4H), 4.16 (d, J = 3.5 Hz, 0.4H), 4.60–4.63 (m, 3H), 4.70–4.78 (m, 4H), 4.88–4.98 (m, 4H), 6.96 (dd, JH,F = 80.2 Hz and J = 2.3 Hz, 0.6H), 7.03 (dd, JH,F = 79.0 Hz and J = 1.7 Hz, 0.4H), 7.30–7.35 (m, 10H). 13C NMR of the mixture of (Z)-22 and (Z)-22a (CDCl3, 125 MHz) δ −5.24, −5.17, −5.15, −4.8, −4.7, 18.2, 18.3, 25.7, 25.8, 46.4 (d, JC,F = 7.2 Hz), 46.7 (d, JC,F = 6.0 Hz), 69.7, 70.0, 70.1, 70.2, 70.3, 70.4, 70.46, 70.53, 71.8, 73.3, 78.3, 78.6, 94.7, 94.81, 94.84, 118.7, 118.8, 122.7 (d, JC,F = 10.8 Hz), 123.6 (d, JC,F = 10.8 Hz), 127.91, 127.93, 128.2, 128.5, 137.1, 137.2, 137.4, 150.8 (d, JC,F = 268.7 Hz), 151.9 (d, JC,F = 268.7 Hz). 19F NMR (CDCl3, 470 MHz) δ −123.1 (d, JH,F = 73.4 Hz) and −121.8 (d, JH,F = 73.3 Hz). MS (ESI+) m/z C30H40FNO6Si, 580 [M+ + Na]. HRMS (ESI+): calcd for C30H40FNNaO6Si 580.2501, found 580.2519 [M+ + Na].
(1S,3R,5S,E)-5-((Benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-2-(fluoromethylene)-3-hydroxycyclopentane-1-carbonitrile [(E)-18]. To a CH2Cl2 (35 mL) solution of (E)-17 (2.5 g, 4.48 mmol) and pyridine (1.45 mL, 17.9 mmol) was dropwise added trifluoromethanesulfonic anhydride (1.51 mL, 8.96 mmol) at −30 °C. The resulting mixture was stirred for 40 min at the same temperature, then this was stirred further for 2 h at RT. The mixture was partitioned between aqueous saturated NaHCO3 and CH2Cl2. After the evaporation of all of the volatiles in the organic layer, the residue was used for the next step without further purification. To a DMF (50 mL) solution of the residue was added LiI (6.0 g, 44.8 mmol) at rt. The resulting solution was stirred at rt for 60 h in the dark. The resulting brown solution was partitioned between brine and AcOEt. After evaporation of all the volatiles of the organic layer gave a crude iodide. To a toluene (40 mL) solution of the iodide and (Me3Si)3SiH (2.75 mL, 8.96 mmol) was added Et3B (1.0 mol L−1 in THF, 4.48 mL, 4.48 mmol) in one portion. The resulting mixture was stirred for 20 min at rt an open air. After evaporation of all volatiles, the residue was roughly purified by column chromatography on silica gel (hexane/AcOEt = 4
:1). This gave a crude silyl ether (ca. 2.3 g) as an oil. This was used for the next step without further purification. To a THF (20 mL) solution of the crude product was dropwise added Bu4NF (1.0 mol L−1 in THF, 5.38 mL, 5.38 mmol) at rt. The resulting mixture was stirred for a further 1.5 h at the same temperature. After the addition of AcOH (300 μL) and evaporation of all the volatile components, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 1:1). This gave (E)-18 (1.18 g, 62% for four steps) as an oil. [α]26D −15.6 (c 1.53, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.01 (ddd, J = 14.9, 5.2 and 4.6 Hz, 1H), 2.08 (ddd, J = 14.9, 5.2 and 2.3 Hz, 1H), 2.74 (d, J = 11.5 Hz, 1H), 3.37 (d, J = 9.8 Hz, 1H), 3.94 (d, J = 9.8 Hz, 1H), 4.53–4.56 (m, 2H), 4.57 (d, J = 12.0 Hz, 1H), 4.65 (d, J = 12.0 Hz, 1H), 4.68 (d, J = 11.5 Hz, 1H), 4.75 (d, J = 11.5 Hz, 1H), 4.77 (d, J = 6.9 Hz, 1H), 4.81 (d, J = 6.9 Hz, 1H), 4.91 (d, J = 7.4 Hz, 1H), 4.95 (d, J = 7.4 Hz, 1H), 7.03 (d, JH,F = 79.6 Hz, 1H), 7.29–7.40 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ 39.2, 50.3, 67.3, 70.2, 71.2, 71.3, 79.3, 93.7, 94.8, 117.8, 124.3, 127.9, 128.0, 128.1, 128.5, 137.2, 137.3, 151.4 (d, JC,F = 271.1 Hz). 19F NMR (CDCl3, 470 MHz) δ −121.3 (d, JH,F = 88.0 Hz). ESI-MS (m/z): 428 [M+ + H], HRMS (ESI+): calcd for C24H27FNO5 428.1873, found 428.1878 [M+ + H].
(1S,3R,5S,Z)-5-((Benzyloxy)methoxy)-1-(((benzyloxy)methoxy)methyl)-2-(fluoromethylene)-3-hydroxycyclopentane-1-carbonitrile [(Z)-17]. A mixture of compounds (Z)-17 and (Z)-17a (636 mg, 1.14 mmol, ca. 3
:2 of the mixture) was treated with the same procedure described for (E)-18. This gave (Z)-18 (227 mg, 47%, for four steps) as an oil. [α]26D +2.0 (c 0.3, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 2.05 (ddd, J = 14.9, 6.3 and 4.0 Hz, 1H), 2.11 (dt, J = 14.9 and 2.3 Hz, 1H), 2.58 (d, J = 8.6 Hz, 1H), 3.47 (d, J = 10.0 Hz, 1H), 3.50 (d, J = 10.0 Hz, 1H), 4.39–4.40 (m, 1H), 4.60 (d, J = 11.5 Hz, 1H), 4.64 (d, J = 11.5 Hz, 1H), 4.68 (d, J = 12.1 Hz, 1H), 4.74 (d, J = 12.1 Hz, 1H), 4.78 (d, J = 9.8 Hz, 1H), 4.80 (d, J = 9.8 Hz, 1H), 4.88–4.91 (m, 1H), 4.90 (d, J = 10.3 Hz, 1H), 4.92, (d, J = 10.3 Hz, 1H), 6.97 (dd, JH,F = 79.6 Hz and J = 1.2 Hz, 1H), 7.29–7.39 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ 38.4, 50.1 (d, JC,F = 6.0 Hz) 0.69.2, 70.0 (d, JC,F = 2.4 Hz), 70.2, 70.3, 79.1, 93.6, 94.8, 119.0, 126.2 (d, JC,F = 6.6 Hz), 127.97, 127.99, 128.1, 128.5, 128.6, 137.1, 137.2, 150.5 (d, JC,F = 266.3 Hz). 19F NMR (470 MHz, CDCl3) δ −121.5 (d, J = 73.3 Hz). 19F NMR (CDCl3, 470 MHz) δ −121.5 (d, JH,F = 73.3 Hz). ESI-MS (m/z): 428 [M+ + H], HRMS (ESI+): calcd for C24H27FNO5 428.1873, found 428.1878 [M+ + H].
Mitsunobu reaction of (E)-18; formation to (E)-20. To a toluene (8 mL) solution of (E)-18 (272 mg, 0.64 mmol), protected 2-amino-6-chloropurine (19) (473 mg, 1.28 mmol) and Ph3P (336 mg, 1.28 mmol), a toluene (4 mL) solution of DMEAD (300 mg, 1.28 mmol) was added dropwise at 0 °C. The resulting mixture was stirred for 1 h at room temperature. The mixture was partitioned between brine and AcOEt. After evaporation of all volatiles of the organic layer, the residue was roughly purified by column chromatography on silica gel (hexane/AcOEt = 1
:1). The obtained crude product was further purified by HPLC (hexane/AcOEt = 1:1, 20 mL min−1, Rt = 7.6 min). This gave (E) −20 (265 mg, 53%) as a foam. [α]26D +33.8 (c 0.3, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.47 (s, 18H), 2.46 (ddd, J = 1.29, 6.9 and 5.2 Hz, 1H), 2.58 (ddd, J = 12.9, 8.0 and 4.6 Hz, 1H), 4.07 (d, J = 9.8 Hz, 1H), 4.10 (d, J = 9.8 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.57–4.59 (m, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.66 (d, J = 12.0 Hz, 1H), 4.77 (d, J = 12.0 Hz, 1H), 4.83 (t, J = 6.9 Hz, 2H), 4.88 (d, J = 7.2 Hz, 1H), 4.92 (d, J = 7.2 Hz, 1H), 5.75–5.76 (m, 1H), 6.67 (dd, JH,F = 77.3 Hz and J = 1.8 Hz, 1H), 7.32–7.36 (m, 10H), 8.20 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 27.9, 36.5, 50.2 (d, JC,F = 7.2 Hz), 67.2, 70.5 (d, JC,F = 9.6 Hz), 84.0, 94.2, 95.0, 116.5, 119.6 (d, JC,F = 6.0 Hz), 127.9, 128.0, 128.1, 128.6, 130.4, 137.1, 137.2, 144.3, 150.6, 151.4 (d, JC,F = 271.2 Hz), 151.8, 152.0, 152.1. 19F NMR (CDCl3, 470 MHz) δ −119.5 (d, JH,F = 73.4 Hz). ESI-MS (m/z): 801 [M+ + Na]. HRMS (ESI+): calcd for C39H44ClFN6NaO8 801.2785, found 801.2799 [M+ + Na].
Mitsunobu reaction of (Z)-18; formation to (Z)-20. Compound (Z)-18 (74 mg, 0.173 mmol) was treated with same procedure described for (E)-20. This gave (Z)-20 (124 mg, 92%) as a foam. [α]26D +54.6 (c 0.44, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.45 (s, 18H), 2.54–2.57 (m, 2H), 3.95 (d, J = 10.0 Hz, 1H), 4.13 (d, J = 10.0 Hz, 1H), 4.64–4.69 (m, 4H), 4.76 (d, J = 12.1 Hz, 1H), 4.83 (s, 2H), 4.90 (d, J = 7.5 Hz, 1H), 4.93 (d, J = 7.5 Hz, 1H), 5.79–5.82 (m, 1H), 7.01 (dd, JH,F = 77.9 Hz and J = 2.3 Hz, 1H), 7.31–7.36 (m, 10H), 8.10 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 27.9, 36.4, 50.3 (d, JC,F = 4.8 Hz), 53.3, 69.5, 70.4 (d, JC,F = 6.0 Hz), 78.0, 83.9, 94.2, 94.9, 118.4, 120.1 (d, JC,F = 9.6 Hz), 127.9, 128.1, 128.5, 128.6, 130.5, 137.1, 137.3, 145.3, 151.5, 151.57, 151.58 (d, JC,F = 271.1 Hz), 151.7. 19F NMR (470 MHz, CDCl3) δ −119.0 (d, J = 73.3 Hz). 19F NMR (CDCl3, 470 MHz) δ −119.0 (d, JH,F = 73.3 Hz). ESI-MS (m/z): 801 [M+ + Na]. HRMS (ESI+): calcd for C39H44ClFN6NaO8 801.2785, found 801.2801 [M+ + Na].
(1S,3S,5S,E)-3-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile (E)-6. To a THF (1 mL) solution of (E)-20 (84 mg, 0.11 mmol) was added 80% TFA (4 mL). The resulting mixture was stirred for 72 h at rt. After evaporation of all volatile components of the mixture, the residue was treated with NH3/MeOH (saturated at 0 °C, 4 mL). This was kept at 4 °C for 14 h, then volatiles were evaporated. The residue was purified by RP-HPLC (30% MeOH, 10 mL min−1, Rt = 8.72 min). This gave (E)-6 (28 mg, 81%) as a solid. Mp > 200 °C decomp.; [α]24D + 55.7 (c 0.11, MeOH); 1H NMR (DMSO-d6, 500 MHz) δ 2.11–2.13 (m, 1H), 2.49–2.50 (m, 1H), 3.77 (dd, J = 11.5 and 6.3 Hz, 1H), 3.82 (dd, J = 11.5 and 5.7 Hz, 1H), 4.42 (br-s, 1H), 5.52–5.56 (m, 1H), 5.87 (t, J = 6.3 Hz, 1H), 6.08 (d, J = 4.6 Hz, 1H), 6.44 (br-s, 2H), 6.75 (dd, JH,F = 79.1 Hz and J = 2.3 Hz, 1H), 7.80 (s, 1H), 10.40 (br-s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 37.5, 51.5 (d, JC,F = 6.0 Hz), 53.6, 61.9, 70.7, 116.7, 118.9, 121.4 (d, JC,F = 4.8 Hz), 135.9, 150.4 (d, JC,F = 262.7 Hz), 150.9, 153.5, 156.8. 19F NMR (DMSO-d6, 470 MHz) δ −126.7 (d, JH,F = 73.3 Hz). ESI-MS (m/z): 321 [M+ + H], 343 [M+ + Na]. HRMS (ESI+): calcd for C13H14FN6O3 321.1106, found 321.1112 [M+ + H].
(1S,3S,5S,Z)-3-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile (Z)-6. Compound (Z)-20 (264 mg, 0.34 mmol) was treated with the same procedure described for (E)-6. This gave (Z)-6 (47 mg, 43%, Rt = 9.92 min) as a solid. Mp > 240 °C decomp.; [α]25D + 31.2 (c 0.12, MeOH); 1H NMR (DMSO-d6, 500 MHz) δ 2.17–2.22 (m, 1H), 2.27–2.32 (m, 1H), 3.69 (dd, J = 11.5 and 5.7 Hz, 1H), 3.93 (dd, J = 11.5 and 5.7 Hz, 1H), 4.38 (d, J = 3.4 Hz, 1H), 5.63–5.67 (m, 1H), 5.83 (t, J = 5.7 Hz, 1H), 6.04 (d, J = 5.2 Hz, 1H), 6.41 (br-s, 1H), 7.18 (dd, JH,F = 79.0 Hz and J = 2.9 Hz, 1H), 7.70 (s, 1H), 10.59 (br-s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 38.6, 51.2, 53.2 (d, JC,F = 4.8 Hz), 64.1, 71.2, 116.4, 120.2, 121.4 (d, JC,F = 8.4 Hz), 136.1, 150.3 (d, JC,F = 263.9 Hz), 150.8, 153.3, 156.7. 19F NMR (DMSO-d6, 470 MHz) δ −124.2 (d, JH,F = 88.0 Hz). ESI-MS (m/z): 321 [M+ + H], 343 [M+ + Na]. HRMS (ESI+): calcd for C13H14FN6O3 321.1106, found 321.1110 [M+ + H].
4.2. Anti-HBV and cytotoxicity assay
Anti-HBV assays were conducted as previously described.14,15 Briefly, HepG2 2.15 cells were seeded in 96-well cell culture plates at a density of 4 × 103 cells in 200 μL per well together with various concentrations of a compound. On days 3 and 7 after plating, the culture medium was removed and the fresh medium and a drug were replenished. On day 14, the cells were harvested for DNA collection and the DNA samples were extracted from the cells using QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions and were re-suspended in 100 μL Tris–EDTA buffer. Total cellular DNA samples were amplified using the Genesig Advanced Kit for quantification of hepatitis B virus genomes (PrimerDesign™Ltd., Southampton, UK) and Applied Biosystems® 7500 Fast Real-time PCR System (Life Technologies Japan Ltd., Tokyo Japan). PCR preparations were incubated at 95 °C for 10 min, followed by 50 cycles at 95 °C for 10 s and 60 °C for 1 min. The data were analyzed using 7500 Fast Software Version 2.0.6 (Life Technologies Japan Ltd., Tokyo Japan), and threshold cycle (CT) values were obtained. To derive the HBV copy numbers from the CT values, a standard curve was generated with 10-fold serial dilutions of an HBV plasmid. The amount of HBV DNA in each assay sample with a test compound was compared to no-compound control samples, and 50% effective concentration (EC50) was determined using the Forecast function of Microsoft Excel software. The cytotoxicities of the compounds in HepG2.2.15 cells were also determined. Cells were plated in a 96-well plate at a density of 2 × 103 cells in 200 μL per well and were continuously exposed to the compound at a certain concentration for 14 days. The number of viable cells in each well was determined using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan), compared to no-compound control samples, and 50% cytotoxicity concentration (CC50) values were determined.4. 3. Methods – molecular modeling
We used the previously published12,14 ternary complexes of HBV-reverse transcriptase (RT) with entecavir triphosphate (ETV-TP) as a template for building a model of the structural interactions of (E)-6 with HBV-RTWT in the presence of primer-templates (pt). The HBV-RT model was built by homology modeling using sequence homology between HBV-RT and HIV-RT.12 (E)-6-TP was generated by suitable modifications to ETV-TP. Minimization of the (E)-6-HBV-RTWT (pt) complex in the OPLS3 force field as implemented in Maestro version 10.7.015 (Schrödinger, LLC, New York) was carried out. Figures of structural interactions were generated using Pymol version 2.5.4.Author contributions
H. K.: conceptualization, investigation, synthetic methodology, and writing-original draft. N. H.-K.: investigation and writing a part of the original draft. S. H.: investigation. D. D.: molecular modeling and writing a part of the original draft. H. B.: molecular modeling and writing a part of the original draft. R. T.: investigation. S. I.: investigation. K. O.: investigation. Y. H.: investigation. Y. O.: formal analysis. S. S.-M.: formal analysis. K. H.: investigation. Y. T.: conceptualization, methodology, and investigation. H. M.: conceptualization, methodology, supervision, and writing-review and editing. All authors have read, commented, and given approval to the final version of the manuscript.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The present work was supported in part by a grant for the Development of Novel Drugs for Treating HBV Infection from the Japan Agency for Medical Research and Development (YT; JP20fk0310101, HM: JP16fk0310501 and JP20fk0310113); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development under grant number JP18fk0410001 (HM); grants from Japan Society for the Promotion of Sciences (HM: 17H04222 and 17K19577), and the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM, DD). The authors are grateful to Kyoko Hattori and Asuka Fujiwara for their technical help. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH, Bethesda, MD (https://hpc.nih.gov).References
- C. L. Lai, V. Ratziu, M. F. Yuen and T. Poynard, Lancet, 2003, 362, 2089–2094 CrossRef CAS PubMed.
- C. W. Shepard, E. P. Simard, L. Finelli, A. E. Fiore and B. P. Bell, Epidemiol. Rev., 2006, 28, 112–125 CrossRef PubMed.
- W. M. Lee, N. Engl. J. Med., 1997, 337, 1733–1745 CrossRef CAS PubMed.
- M. Nassal, Virus Res., 2008, 134, 235–249 Search PubMed.
- J. Fung, C. L. Lai, W. K. Seto and M. F. Yuen, J. Antimicrob. Chemother., 2011, 66, 2715–2725 CrossRef CAS PubMed.
- V. Levy and R. M. Grant, Clin. Infect. Dis., 2006, 43, 904–910 CrossRef CAS PubMed.
- M. Nassal, Future Virol., 2009, 4, 55–70 CrossRef CAS.
- S. F. Innaimo, M. Seifer, G. S. Bisacchi, D. N. Stabdring, R. Zahaler and R. J. Colonno, Antimicrob. Agents Chemother., 1997, 41, 1444–1448 CrossRef CAS PubMed.
- G. Yamanaka, T. Wilspn, S. F. Innaimo, P. Bisacchi Egli, J. K. Rinehart, R. Zahler and R. J. Colonno, Antimicrob. Agents Chemother., 1999, 43, 190–193 CrossRef CAS PubMed.
- D. R. Langley, A. W. Walsh, C. J. Baldick, B. J. Eggers, R. E. Rose, S. M. Levine, A. J. Kapur and R. J. Colonno, J. Virol., 2007, 81, 3992–4001 CrossRef CAS PubMed.
- S. Levine, D. Hernandez, G. Yamanaka, S. Zhang, R. Rose, S. Weinheimer and R. J. Colonno, Antimicrob. Agents Chemother., 2002, 46, 2525–2532 CrossRef CAS PubMed.
- Y. Takamatsu, Y. Tanaka, S. Kohgo, S. Murakami, K. Singh, D. Das, D. Venzon, M. Amano, N. Higashi-Kumara, M. Aoki, N. S. Delino, S. Hasashi, S. Takahashi, Y. Sukenaga, K. Haraguchi, S. G. Salafianos, K. Maeda and H. Mitsuya, Hepatology, 2015, 62, 1024–1036 CrossRef CAS PubMed.
- H. Kumamoto, M. Fukano, T. Nakano, K. Iwagami, C. Takeyama, S. Kohgo, S. Imoto, M. Amano, N. Higashi-Kuwata, M. Aoki, H. Abe, H. Mitsuya, K. Fukuhara and K. Haraguchi, J. Org. Chem., 2016, 81, 2827–2836 CrossRef CAS PubMed , most of references for the sulfur-extrusive stannylation re cited..
- N. Higashi-Kuwata, S. Hayashi, D. Das, S. Kohgo, S. Murakami, S. Hattori, S. Imoto, D. J. Venzon, K. Singh, S. G. Sarafianos, Y. Tanaka and H. Mitsuya, Antimicrob. Agents Chemother., 2019, 63, e02413–e02418 CrossRef PubMed.
- N. Higashi-Kuwata, S. Hayashi, H. Kumamoto, H. Ogata-Aoki, D. Das, D. Venzon, S. Hattori, H. Bulut, M. Hashimoto, M. Otagiri, N. Takamune, N. Kishimoto, D. A. Davis, S. Misumi, M. Kakumi, Y. Tanaka and H. Mitsuya, J. Hepatol., 2021, 74, 1075–1086 CrossRef CAS PubMed.
- R. Samunuri, A. K. Jha and C. Bal, Nucleosides, Nucleotides Nucleic Acids, 2019, 38, 391–399 CrossRef CAS PubMed.
- M. A. Tius and J. K. Kawakami, Synlett, 1993, 207–209 CrossRef CAS.
- M. A. Tius, Tetrahedron, 1995, 51, 6605–6634 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
- S. Day and P. Garner, J. Org. Chem., 2000, 65, 7697–7699 CrossRef PubMed.
- X. Yin, W. Li and S. W. Schneller, Tetrahedron Lett., 2006, 47, 9187–9189 CrossRef CAS.
- T. Sugimura and K. Hagiya, Chem. Lett., 2007, 36, 566–567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01750h |
| This journal is © The Royal Society of Chemistry 2023 |