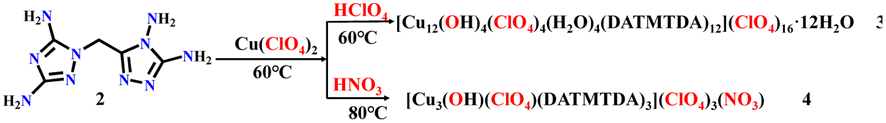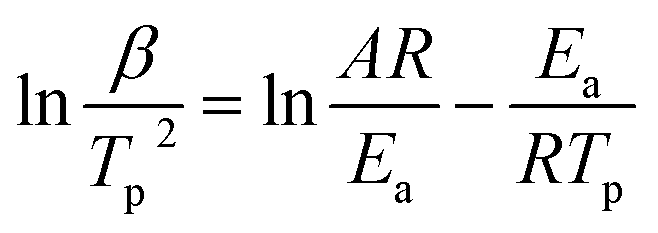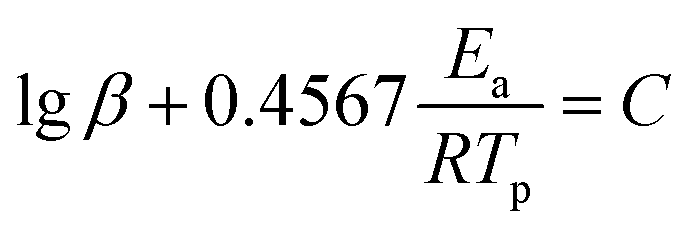DOI:
10.1039/D3RA01739G
(Paper)
RSC Adv., 2023,
13, 22549-22558
Cupric coordination compounds with multiple anions: a promising strategy for the regulation of energetic materials†
Received
16th March 2023
, Accepted 10th July 2023
First published on 25th July 2023
Abstract
To seek new high energetic materials, N-methylene-C-bridged nitrogen-rich heterocycle 1-((4,5-diamino-4H-1,2,4-triazol-3-yl)methyl)-1H-1,2,4-triazol-3,5-diamine (DATMTDA) (2) was first synthesized, and two copper coordination compounds ([Cu12(OH)4(ClO4)4(H2O)4(DATMTDA)12](ClO4)16·12H2O (3) and [Cu3(OH)(ClO4)(DATMTDA)3](ClO4)3(NO3) (4)) based on 2 were formed by introducing different anions. These compounds were characterized by elemental analysis, IR spectroscopy and single-crystal X-ray diffraction analysis. The crystal structures of compounds 3 and 4 are similar and crystallize in monoclinic systems with the P21/c space group, while the central copper atoms show different coordination behaviors. However, the structure of compounds 3 and 4 is analogous to a three dimensional structure owing to the O atom of OH−, forming coordinate bonds with three copper cations. The NBO charge of 2 was calculated using density functional theory to understand its coordination modes. The Hirshfeld surface calculation reveals that 3 and 4 have strong intermolecular interactions. The thermal decomposition processes, non-isothermal kinetics, and enthalpies of formation and sensitivities of these compounds were investigated. By introducing one NO3− of compound 4 to replace one ClO4− in compound 3, compound 4 shows lower density and lower decomposition peak temperature but lower sensitivity and a higher formation enthalpy than compound 3. The complex 4 possesses an outstanding catalytic effect for the decomposition of AP than that of complex 3. The results illustrate the possibility of introducing various anions into energetic coordination compounds for the regulation of energetic materials.
Introduction
Energetic materials with high-energy density have been widely used in civil and military fields.1–3 However, high-energy density is usually followed by high sensitivity, which is usually accompanied and contradictory.4 Therefore, the development of novel energetic materials with suitable security and excellent performance has become an attractive topic in the field of energetic materials. Several strategies have been applied to develop new energetic compounds, such as constructing nitrogen-rich energetic cocrystals, designing, and synthesizing novel nitrogen-rich energetic compounds, forming nitrogen-rich energetic salts and energetic coordination complexes (ECCs).5–9 Moreover, owing to ECCs usually consisting of three moieties (metal cations, energy-storage ligands, and anions), ECCs can easily adjust their performance by changing the three moieties.5 Therefore, many energetic coordination complexes have been prepared with excellent properties that can be used as catalysts to promote ammonium perchlorate decomposition, as primary explosives, or as promoters for ionic liquid hypergolic ignition.10–12 Moreover, they exhibit excellent performance or application by introducing different kinds of ligands, metal cations, and anions, increasing the variety of metal cations as well as the variety of anions.13–17 For example, N3− (azide ions) gives the complex higher mechanical sensitivity and strong detonation ability, while DCA− (dicyandiamide ions) gives the complex better thermal stability, lower sensitivity, and higher combustion heat.18,19 Another example is the preparation of Cu(N3)2(L) (L = 1-methyltetrazole (MTZ), 1-ethyltetrazole (ETZ), and 1-propyltetrazole (PTZ)), which regulates the stability and mechanical sensitivity of copper azide by changing the ligand.20 By changing the central metal ion, mechanical sensitivity, thermal decomposition temperature, and combustion heat of the energetic compounds can all be changed.21
Apart from ECCs, the design and synthesis of novel nitrogen-rich energetic compounds are also efficient strategies for balancing security with detonation performance. Owing to the high nitrogen content, high thermal stability, insensitivity and hypotoxicity, nitrogen-rich heterocycle compounds, such as triazole, tetrazole, tetrazine, bitriazole, bitetrazole and their derivatives, have attracted extensive attention. Compared with monoazole compounds, bicycloazole compounds have more modifiable sites, which can be modified by introducing energetic groups (–NO2, –N3, –C(NO2)3, –NHNO2, and –OH) according to the required performance.22–25 Moreover, the introduction of bridging blocks, such as imino,26 azo,27 alkyl,28,29 carbonyl,30,31 nitromethyl,32 and hydrazine groups,33 can combine two or more azolyl components effectively to form bicycloazole compounds, and the performance of these bridging bicycloazole compounds can also be modified via changing bridging blocks. Furthermore, methylene-bridged bicycloazole compounds usually show excellent thermostable and low sensitivity.29 Finally, triazole energetic compounds usually tend to exhibit positive heat of formation (HOF), insensitivity towards mechanical stimuli, high thermal stability and acceptable detonation performance.34–37 At present, there are many studies on methylene bridged bis(triazole), but mainly on C-methylene-C bridged compounds, such as 3,3′-bis (5-amino-1,2,4-triazolyl)methane, 3,3′-bis(5-nitroamino-1,2,4-triazolyl)methane, 3,3′-bis(4,5-diamino-1,2,4-triazol)methane, and 3,3′-bis(5-nitro-1,2,4-triazolyl)methane, while there are few studies on N-methylene-C bridged bis(triazole).29,38–40 Owing to the bond energy of N–C bonds being higher than that of C–C bonds, N-methylene-C-bridged compounds exhibit higher energy than C-methylene-C-bridged compounds because of N–C bonds at multiple bridging positions.41
Ammonium perchlorate (AP) is a widely used solid oxidant, especially in solid propellants.42 Its thermal decomposition characteristics directly affect the combustion performance of propellants, but the high decomposition temperature and slow decomposition of AP limit the combustion rate of propellants. Usually, the addition of catalysts can reduce the thermal decomposition temperature and increase the decomposition rate of the AP. Therefore, it is meaningful to find an efficient catalyst to promote the decomposition of AP.43 In recent years, owing to their superior energy and catalytic performance, high-energy metal complexes have been used as catalysts for AP and have shown good catalytic effects.10,14
Based on the above considerations, a novel N-methylene-C-bridged nitrogen-rich heterocycle 1-((4,5-diamino-4H-1,2,4-triazol-3-yl)methyl)-1H-1,2,4-triazol-3,5-diamine (DATMTDA) was first synthesized, and two copper energetic compounds with different energetic anions based on DATMTDA were prepared using a simple process. The crystal structures of the newly prepared compounds were characterized by X-ray analysis. Moreover, the thermal behavior, and sensitivity of these compounds were investigated. Finally, the catalytic effect of the two copper complexes on AP was tested using DSC.
Experimental section
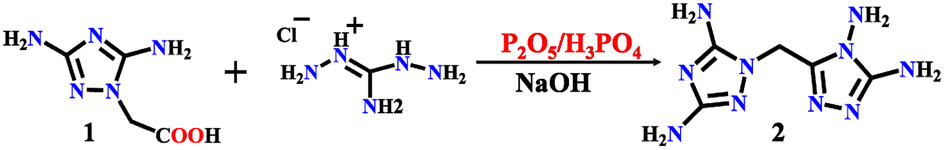
All the chemical reagents used in this study were obtained from commercial channels and used without further purification. 1-Carboxymethyl-3,5-diamino-1,2,4-triazole (1) was prepared according to ref. 44, and compound 2–4 was prepared as shown in Schemes 1 and 2.
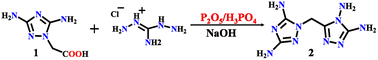 |
| | Scheme 1 Syntheses of compound 2. | |
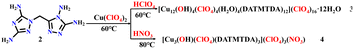 |
| | Scheme 2 Syntheses of compounds 3 and 4. | |
Synthesis of 1-((4,5-diamino-4H-1,2,4-triazol-3-yl)methyl)-1H-1,2,4-triazol-3,5-diamine (2)
Compound 2 was synthesized in three steps. Diaminoguanidine hydrochloride (25.12 g, 20 mmol) mixed with compound 1 (31.4 g, 20 mmol) was added into a mixed liquid of phosphoric acid (120 g) and phosphorus pentoxide (40 g) with strong stirring at 50 °C for 30 min and then heated to 120 °C for 12 h. Along with cooling to room temperature. A sodium hydroxide aqueous solution (10 mol L−1, 350 mL) was added. The reaction mixture was heated to 140 °C with stirring for 1 h. When the reaction solution was cooled to room temperature, a white precipitate of DATMTDA (2) was obtained by filtration, washed with water and methanol, and dried. Product 28.2 g, yield: 67.1%. The colorless block crystal was separated from the water. Tdec. 308.0 °C. IR (KBr): 3409, 3305, 3177, 1651, 1593, 1494, 1417, 1225, 1093, 983, 775, and 720 (cm−1). Elemental analysis: C5H10N10 (MW 210.23 g mol−1). Calculation for C 28.54%, H 4.76%, N 66.59%; found for C 28.56%, H 4.77%, N 66.56%.
Synthesis of compound 3
2 (0.21 g, 1 mmol) was added into 10 mL of methanol at 60 °C with stirring. Cu(ClO4)2·6H2O (0.37 g, 1.0 mmol) in 10 mL of methanol was then added dropwise into the reaction solution. After 10 min, the 69–72% HClO4 (0.144 g, 1 mmol) was added dropwise to the solution, and the solution was maintained at 60 °C for 45 min. Dark green block crystal was separated from the reaction solution with the following yield: 0.356 g, 75.87%. Tdec. 240.5 °C. IR (KBr): 3354, 1654, 1623, 1424, 1096, 1059, 842, 774, 661, and 629 (cm−1). Elemental analysis: C60H156Cl20Cu12N120O100 (MW 5630.52 g mol−1). Calculation for C 12.79%, H 2.77%, Cl 12.61%, Cu 13.64%, N 29.84%, O 28.42%; found for C 12.81%, H 2.78%, Cl 12.64%, Cu 13.62%, N 29.82%, O 28.40%.
Synthesis of compound 4
2 (0.21 g, 1 mmol) was added into 10 mL of water at 60 °C with stirring. Cu(ClO4)2·6H2O (0.37 g, 1.0 mmol) in 10 mL of water was then added dropwise into the reaction solution. After 10 min, 65% HNO3 (0.063 g, 1 mmol) was added dropwise to the solution. Then, the solution was heated to 80 °C and remained at 80 °C for 45 min. Dark green block crystal was separated from the reaction solution with the following yield: 0.336 g, 77.65%. Tdec. 218.4 °C. IR (KBr): 3364, 1654, 1622, 1541, 1452, 1096, 842, 725, 678, and 628 (cm−1). Elemental analysis: C15H31Cl4Cu3N31O20 (MW 1298.13 g mol−1). Calculation for C 13.87%, H 2.39%, Cl 10.94%, Cu 14.79%, N 33.43%, O 24.65%; found for C 13.86%, H 2.40%, Cl 10.96%, Cu 14.81%, N 33.41%, O 24.63%.
Characterization
Crystals suitable for X-ray diffraction measurements were obtained, as described in the experimental section. Data were collected using a Bruker SMART CCD diffractometer. All the crystals were irradiated with graphite monochromatic Mo Kα radiation (λ = 0.71073 Å) at 298 K or 296 K in multi-scan mode. The structures of compound 2 were solved using SHELXS-97 and refined using full matrix least-square procedures on F2 with SHELXL-97. The structures of compounds 3 and 4 were solved using SHELXS-2014 and refined by applying full matrix least-squares procedures on F2 using SHELXL-2014.
Elemental analyses were carried out using a Flash EA 1112 analyzer. FT-IR spectra were obtained using a Thermos Nicolet iS20 spectrometer. The FT-IR spectra of compound 2–4 are shown in Fig. S1–S3.† The thermal decomposition behaviors of these compounds were investigated by differential scanning calorimetry (DSC) using CDR-4 (Shanghai Precision & Scientific Instrument Co., Ltd.). The TG-DTG experiment was carried out using a TGA/DSC 3+ apparatus (METTLER TOLEDO). The experimentally determined constant-volume energies of combustion were tested using a Parr-6200 bomb calorimeter (static jacket) with a type 6510 water handling system. Impact and friction sensitivities were determined using a BFH-10 BAM fall hammer and an FSKM-10 BAM friction apparatus, respectively.
Results and discussion
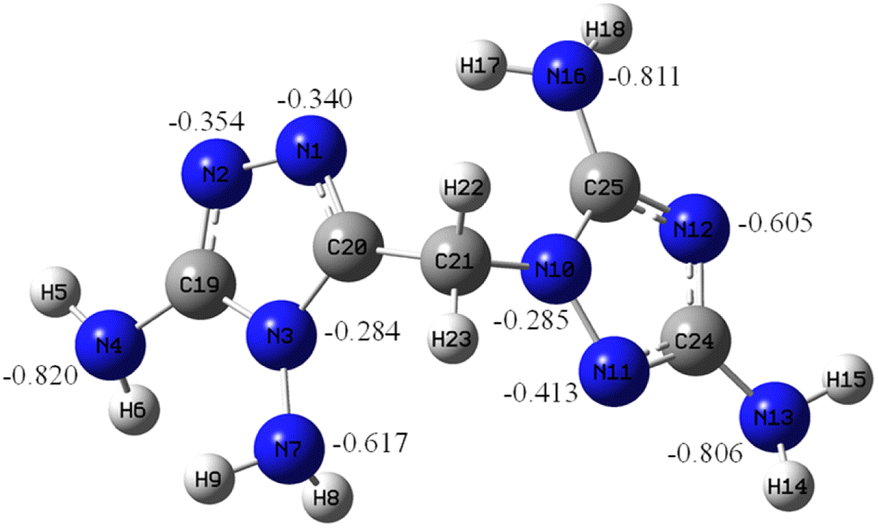
Natural bond orbital (NBO) of DATMTDA
To obtain a better understanding of DATMTDA (2), the molecular orbital of compound 2 was analyzed using the B3LYP method with 6-311++G** basis sets. The optimized structures were based on the true local energy minima on the potential energy surface. The NBO charges of the N atoms in DATMTDA are shown in Fig. 1. As shown in Fig. 1, the N atoms (N4, N7, N13 and N16) of NH2 have more negative charges than N atoms located in the triazole heterocycle, and the N atoms of C–NH2 showed more negative charges than N–NH2. The negative charges of N atoms located in the triazole heterocycle increase in the following order: N1 < N2 < N11 < N12, which means that the electrostatic attraction of the N12 atom with metal ions is strongest than others, followed by N11, N2 and N1. The tendency of metal ions to form coordinate bonds with N atoms exhibits the following order: N12, N11, N2 and N1. As shown in compounds 3 and 4, the coordinate bonds of metal and N11, N2 and N1 atoms were formed, but this may be because of the positive charge of the hydrogen atoms in the two NH2 groups. The N12 atom cannot form a coordination bond with the metal.
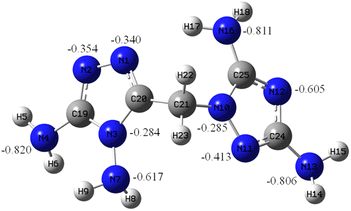 |
| | Fig. 1 NBO charge of DATMTDA. | |
X-ray crystallography
Crystals of compound 2–4 suitable for X-ray diffraction measurements were obtained by different routes. Compound 2 was obtained by slow evaporation from the recrystallized aqueous solution, while compounds 3 and 4 were obtained by slow evaporation from the reaction solution. The crystallographic data and experimental details of the structural analysis are summarized in Table 1. The bond lengths, bond angles, hydrogen bonds and torsion angles of these studding compounds are presented in Table S1–S12.† The CCDC numbers of these compounds are 2221146, 2221150, and 2221154.
Table 1 X-ray crystallographic data for compound 2–4
| |
2 |
3 |
4 |
| Empirical formula |
C5H10N10 |
C60H156Cl20Cu12N120O100 |
C15H31Cl4Cu3N31O20 |
| Formula weight/g mol−1 |
210.23 |
5630.52 |
1298.13 |
| Temperature/K |
298(2) |
296(2) |
296(2) |
| Crystal system |
Orthorhombic |
Monoclinic |
Monoclinic |
| Space group |
Pbca |
P21/c |
P21/c |
| a/Å |
7.7917(7) |
22.1106(10) |
22.0613(13) |
| b/Å |
10.5981(9) |
14.1198(7) |
13.9583(8) |
| c/Å |
20.3073(18) |
14.6843(6) |
14.5135(8) |
| α/° |
90 |
90 |
90 |
| β/° |
90 |
94.7250(10) |
95.2030(10) |
| γ/° |
90 |
90 |
90 |
| Volume/Å3 |
1676.9(3) |
4568.8(4) |
4450.8(4) |
| Z |
8 |
1 |
4 |
| ρ/g cm−3 |
1.665 |
2.046 |
1.937 |
| μ/mm−1 |
0.124 |
1.797 |
1.771 |
| F(000) |
880 |
2844 |
2612 |
| Size/mm3 |
0.28 × 0.25 × 0.12 |
0.200 × 0.200 × 0.200 |
0.200 × 0.200 × 0.200 |
| λ (Mo Kα)/Å |
0.71073 |
0.71073 |
0.71073 |
| 2θ range/° |
3.30: 25.01 |
2.004: 25.028 |
2.028: 25.052 |
| Index ranges |
−9 ≥ h ≥ 7 |
−26 ≥ h ≥ 26 |
−26 ≥ h ≥ 23 |
| −12 ≥ k ≥ 12 |
−16 ≥ k ≥ 16 |
−16 ≥ k ≥ 15 |
| −24 ≥ l ≥ 21 |
−17 ≥ l ≥ 17 |
−17 ≥ l ≥ 17 |
| Reflect. coll. |
7797 |
102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
66258 |
| Independ. refl. |
1478 |
8041 |
7873 |
| Rint |
0.1006 |
0.0741 |
0.124 |
| S |
1.075 |
1.033 |
1.028 |
| R1; wR2[I ≥ 2σ(I)] |
0.0652; 0.1575 |
0.0430; 0.0893 |
0.0740; 0.1665 |
| R1; wR2[all data] |
0.0814; 0.1675 |
0.0680; 0.1018 |
0.1327; 0.2038 |
| Resd. dens./e Å−3 |
0.466/−0.397 |
0.709/−0.733 |
1.081/−0.770 |
| CCDC |
2221146 |
2221150 |
2221154 |
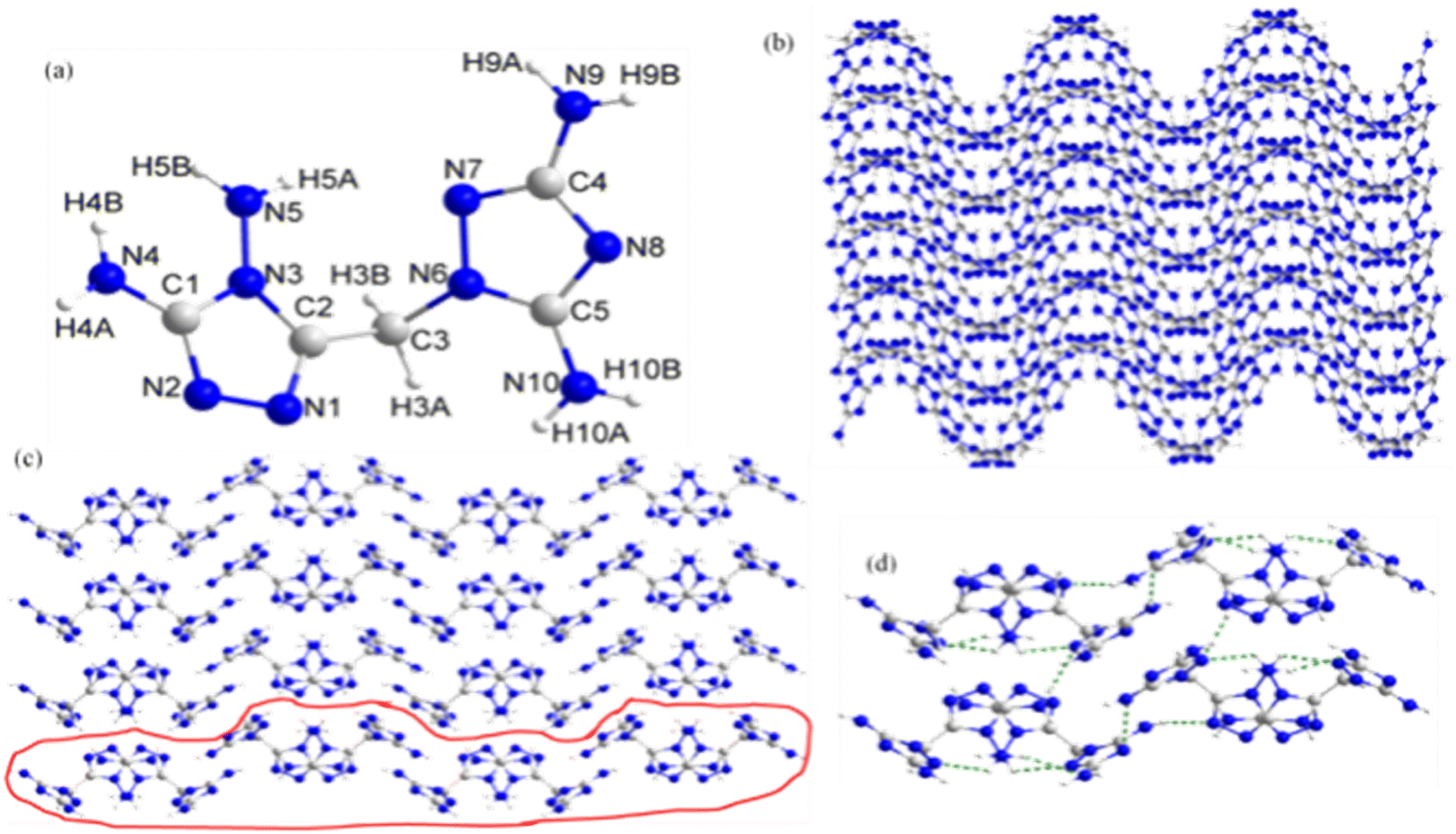
Crystal structure of DATMTDA (2). The molecular structure of compound 2 belongs to the monoclinic system and Pbca space group with a density of 1.665 g cm−3 and a volume of 1676.9 Å3 at 298 K, and there were eight formula units per unit cell. The molecular unit of 2 has one 3,5-diamino-1,2,4-triazole and one 3,4-diamino-1,2,4-triazole ring bridged by methylene, as shown in Fig. 2(a). The lengths of C1–N2, C1–N3, C1–N4, C2–N1, C2–N3, C3–N6, C4–N7, C4–N8, C4–N9, C5–N10, N1–N2, N3–N5 and N6–N7 bonds are 1.304 Å, 1.349 Å, 1.330 Å, 1.290 Å, 1.348 Å, 1.446 Å, 1.307 Å, 1.346 Å, 1.348 Å, 1.316 Å, 1.328 Å, 1.339 Å, 1.395 Å, 1.385 Å and 1.383 Å, respectively. These bond lengths are shorter than C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1.47 Å) or NN (1.48 Å) but longer than C–N (1.22 Å) or N–N (1.2 Å). Thus, conjugate effects were found in the atoms of the heterocyclic rings. Moreover, the torsion angle of N3–C2–C3–N6 was 74.3°, suggesting that the two triazole rings in the structure were non-planar. As shown in Fig. 2(b)–(d), viewed along the c-axis and a-axis, compound 2 exhibits wavelike stacking and layer-by-layer stacking, respectively, and the layers are connected through hydrogen bonds.
N (1.47 Å) or NN (1.48 Å) but longer than C–N (1.22 Å) or N–N (1.2 Å). Thus, conjugate effects were found in the atoms of the heterocyclic rings. Moreover, the torsion angle of N3–C2–C3–N6 was 74.3°, suggesting that the two triazole rings in the structure were non-planar. As shown in Fig. 2(b)–(d), viewed along the c-axis and a-axis, compound 2 exhibits wavelike stacking and layer-by-layer stacking, respectively, and the layers are connected through hydrogen bonds.
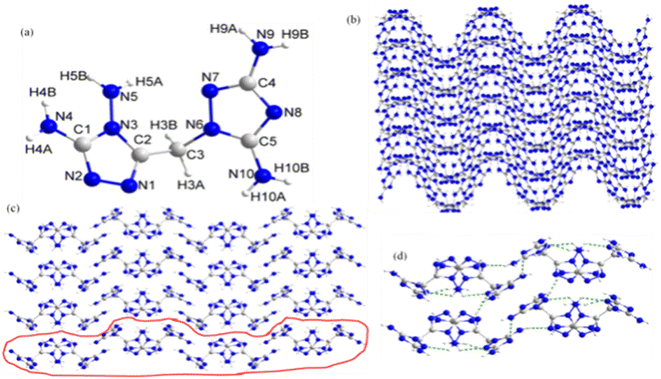 |
| | Fig. 2 (a) The molecular structure of 2. (b) The packing diagram of 2 viewed along the c-axis. (c) The packing diagram of 2 viewed along the a-axis. (d) Hydrogen bonding networks in 2 viewed along the a-axis. | |
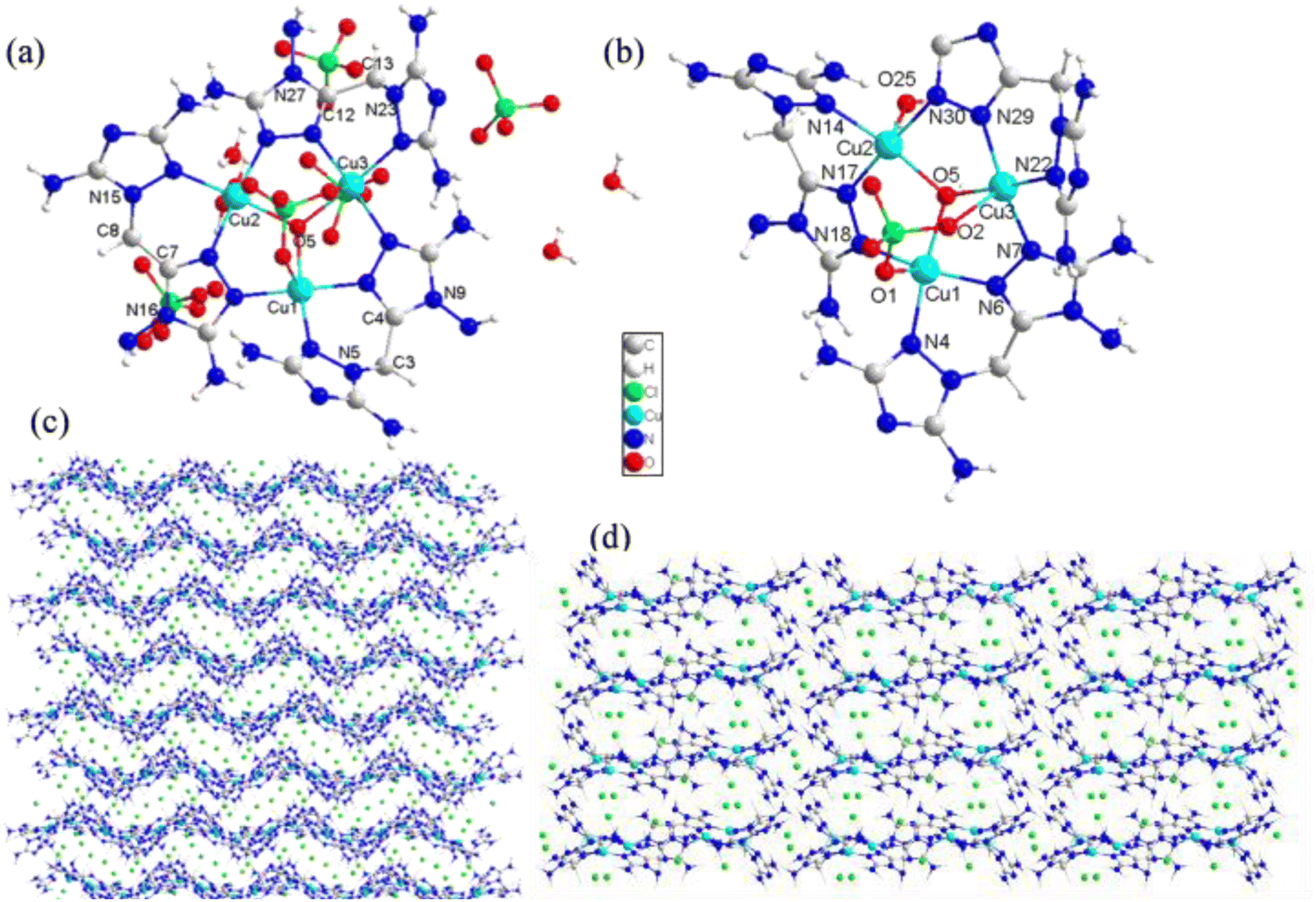
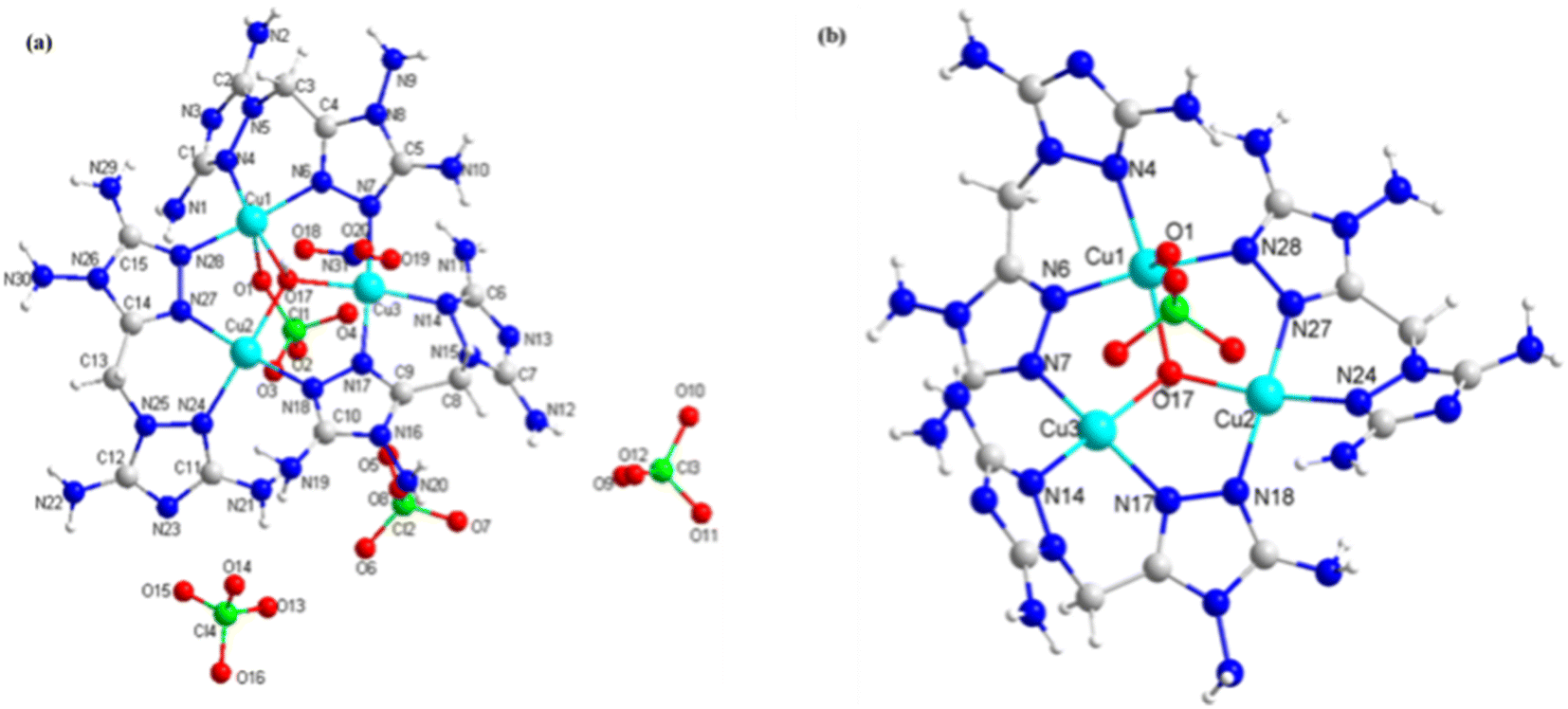
Compounds 3 and 4 were copper energetic compounds of 2, differing by the kind of anion in the crystal. They all crystallized in the monoclinic P21/c space group. The calculated densities of the two compounds at 296 K were different: the density of 3 was 2.046 g cm−3, while that of 4 was 1.937 g cm−3. For compound 3, every unit cell included one formula unit, with the molecular unit consisting of twelve copper atoms, two kind anions (four OH− and twenty ClO4−), twelve DATMTDA ligands, four coordination water molecules and twelve crystal water molecules. All three central copper atoms were penta-coordinated. Cu1 and Cu3 were penta-coordinated with two DATMTDA ligands, one ClO4− molecule and one OH−. Cu2 was penta-coordinated with two DATMTDA ligands, one water molecule and one OH−, as shown in Fig. 3(b). All metal atoms and ligands exhibit wavelike stacking viewed along the a-axis, which can also be called layer-by-layer stacking (Fig. 3(c)). Anions are distributed in the middle of the interlayer gap and viewed along the b-axis; the metal atoms and ligands also exhibit layer-by-layer stacking (Fig. 3(d)). Moreover, three copper atoms and O5 in the structure are non-planar (Cu2–O5–Cu1 = 115.56(13)°, Cu2–O5–Cu3 = 115.08(13)°, and Cu1–O5–Cu3 = 113.82 (13)°) but formed a trigonal pyramid. Two triazole rings of the DATMTDA ligand (N5–C3–C4–N9 = 152.2(4)°, N16–C7–C8–N15 = 151.5(4)°, and N27–C12–C13–N23 = 148.2 (4)°) in the structure were also non-planar but closer to coplanar than compound 2. For compound 4, there were 4 formula units per unit cell, with the molecular unit consisting of three copper atoms, three kind anions (one OH−, four ClO4− and one NO3−), and three DATMTDA ligands (Fig. 4(a)). Compound 4 shows two kinds of central copper atoms with diverse coordination modes (Fig. 4(b)). Cu1 was penta-coordinated with two DATMTDA ligands, one ClO4− molecule and one OH−. Cu2 and Cu3 were tetra-coordinated with two DATMTDA ligands and one OH−. The packing diagram of compound 4 is the same as that of compound 3 (Fig. 3(c) and (d)). All metal atoms and ligands also exhibit a wavelike stacking viewed along the a-axis and b-axis (Fig. S4(a) and (b)†). The three copper atoms and O17 in the structure are also non-planar (Cu2–O17–Cu1 = 114.7(3)°, Cu2–O17–Cu3 = 115.3(3)°, and Cu1–O17–Cu3 = 113.8(2)°) and formed a trigonal pyramid. Moreover, two triazole rings of the DATMTDA ligand (N5–C3–C4–N8 = 149.4(8)°, N15–C8–C9–N16 = 146.1(8)°, and N25–C13–C14–N26 = 148.7(7)°) in the structure were non-planar as compound 3 but closer to coplanar than compound 2.
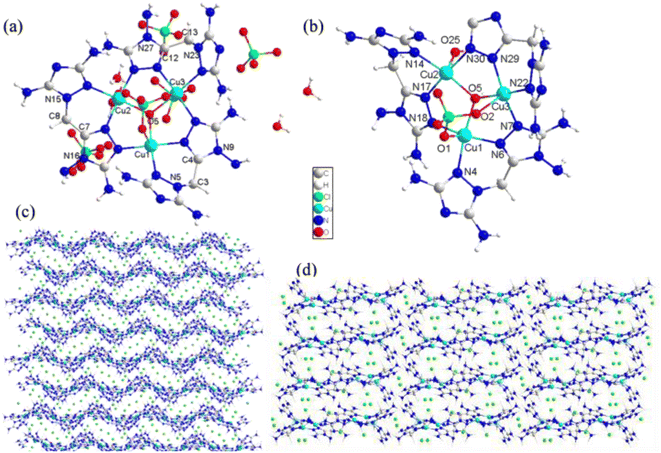 |
| | Fig. 3 (a) Molecular structure of 3, (b) coordination environment of copper cations, and (c) the packing diagram of 3 viewed along the a-axis, and (d) the packing diagram of 3 viewed along the b-axis. | |
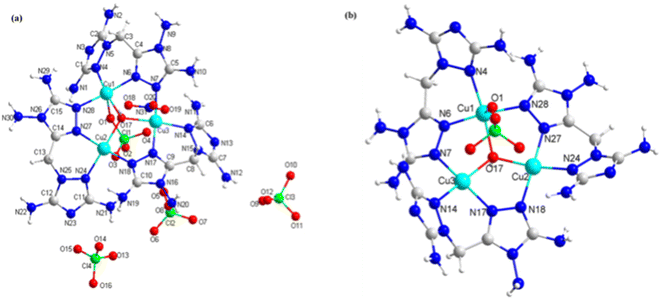 |
| | Fig. 4 (a) The molecular structure of 4 and (b) coordination environment of copper cations. | |
Thermal decomposition behaviour
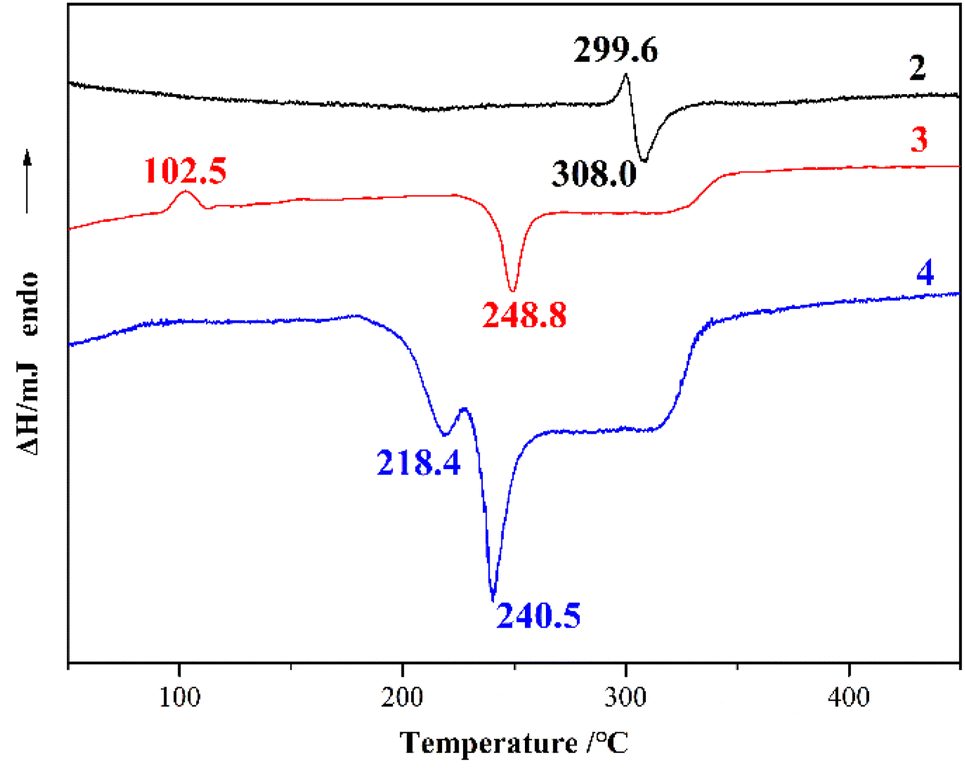
The thermal decomposition processes of DATMTDA (2) and compound 3–4 were investigated by applying differential scanning calorimetry (DSC) with a heating rate of 10 °C min−1 in air atmosphere, as well as thermogravimetric analysis (TGA) at a heating rate of 10 °C min−1 in dry nitrogen atmosphere with a flow rate of 20 mL min−1. The DSC curves are shown in Fig. 5, and the TGA curves are shown in Fig. S5–S7.† Compound 2 shows an exothermic signal (308.0 °C) directly after the endothermic signal (299.6 °C), and no other signals were observed. Compound 3 shows an exothermic signal (248.8 °C) and an endothermic signal (102.5 °C). According to the TGA curve, the endothermic peak is the loss of crystal water. Compound 4 shows two exothermic signals (218.4 °C and 240.5 °C). The results indicated that these compounds exhibit acceptable thermal stabilities, and compound 2 shows the best thermal stability. The decomposition temperature of compound 3 is higher than that of compound 4. The same trend was observed in the TGA curves (Fig. S5–S7†). This phenomenon may be because compound 3 contains water molecules, whereas compound 4 does not, and these water molecules can provide a large number of hydrogen bonds.
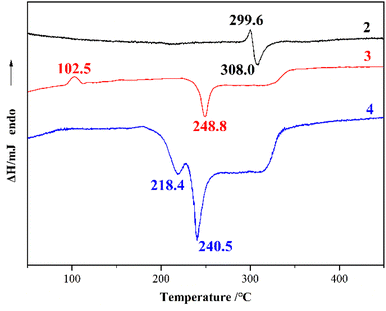 |
| | Fig. 5 DSC curves of compound 2–4. | |
Non-isothermal kinetic analysis
To better analyze the thermal decomposition behavior, according to the non-isothermal kinetic DSC method, the apparent activation energies (Ea) and the pre-exponential factor (A) based on the first exothermal decomposition peak temperatures of compounds under different heating rates (5, 10, 15, 20 °C min−1) were calculated by applying Kissinger's method45 and Ozawa–Doyle's method,46 and the equations for Kissinger's method (eqn (1)) and Ozawa–Doyle's method (eqn (2)) are listed below. The corresponding temperature of the first exothermic decomposition peak is shown in Table S13,† and the calculated kinetic parameters are listed in Table 2. Moreover, the Arrhenius equations based on Kissinger's method can be expressed as eqn (3)–(5) for compound 2–4. The apparent activation energies (kJ mol−1) calculated using Kissinger's method for compound 2–4 are 209.0 kJ mol−1, 240.0 kJ mol−1 and 120.8 kJ mol−1, respectively.| |
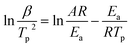 | (1) |
| |
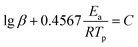 | (2) |
where Tp is the peak temperature (K) of the first reaction process, A is the pre-exponential factor (s−1), Ea is the apparent activation energy (kJ mol−1), R is the gas constant (J mol−1 K−1), and β is the heating rate (K min−1) and C is a constant.
Table 2 Non-isothermal reaction kinetic parameters and thermodynamic parameters of compound 2–4
| Compound |
Kissinger's method |
Ozawa's method |
Tp0 (K) |
Tb (K) |
ΔS‡ (J K−1 mol−1) |
ΔH‡ (kJ mol−1) |
ΔG‡ (kJ mol−1) |
| EK (kJ mol−1) |
lgAK |
EO (kJ mol−1) |
| 2 |
209.0 |
16.85 |
208.0 |
561.65 |
574.79 |
79.20 |
204.33 |
163.67 |
| 3 |
240.0 |
22.18 |
236.5 |
517.35 |
526.97 |
175.11 |
235.70 |
145.10 |
| 4 |
120.8 |
10.78 |
122.7 |
475.45 |
492.12 |
−42.42 |
116.85 |
137.05 |
DATMTDA (2):
| | |
lnk = 38.8 − 209 × 103/(RT)
| (3) |
[Cu12(OH)4(ClO4)4(H2O)4(DATMTDA)12](ClO4)16·12H2O (3):
| | |
lnk = 51.1 − 240 × 103/(RT)
| (4) |
[Cu3(OH)(ClO4)(DATMTDA)3](ClO4)3(NO3) (4):
| | |
lnk = 24.8 − 120.8 × 103/(RT)
| (5) |
To evaluate the thermal safety of compound 2–4, the thermodynamic parameters (Tp0, Tb, ΔG‡, ΔS‡ and ΔH‡) of these compounds were obtained by employing Zhang's calculation equations,47 and the equations are listed below (eqn (6)–(10)). The calculated thermodynamic parameters are listed in Table 2. The results reveal that the enthalpy of activation and free energy of activation of compound 2–4 are positive numbers, indicating that the thermal decomposition processes of these compounds are non-spontaneous endothermic processes. Moreover, the Tb of compound 4 is the minimum among these compounds, which illustrates its poor thermal safety.
| | |
Tpi = Tp0 + aβ + bβ2 + cβ3
| (6) |
| | |
Tb = [EK − (EK2 − 4EKRTp0)−0.5] /2R
| (7) |
| | |
A = (kBTp0/h)exp(1 + ΔS‡/R)
| (8) |
Here,
a,
b and
c are coefficients,
kB is the Boltzmann constant (1.381 × 10
−23 J K
−1) and
h is the Planck constant (6.626 × 10
−34 J s).
Tp0 is the value of the peak temperature corresponding to
β → 0,
Tb is the corresponding critical temperature of the thermal explosion, Δ
S‡ is the entropy of activation, Δ
H‡ is the enthalpy of activation, Δ
G‡ is the free energy of activation.
Enthalpy of formation
To evaluate the energetic performance of the compounds, the energy of combustion was measured by oxygen bomb calorimetry, and the formation enthalpies of compound 2–4 were calculated based on the energy of combustion, the combustion reactions (eqn (11)–(13)) and the Hess thermochemical cycle. The measured constant volume combustion energies and the calculated formation enthalpies are listed in Table 3. As shown in Table 3, compound 3 presents the minimum values of formation enthalpies and combustion heats.
Table 3 Physicochemical properties of compound 2–4
| Compound |
ΔcUa (MJ kg−1) |
ΔfHθ b (kJ mol−1) |
OBc (%) |
ρd (g cm−3) |
Te (°C) |
ISf (J) |
FSg (N) |
| Constant-volume combustion energies. Calculated enthalpies of formation. Oxygen balance, OB = 1600 × (o − (b − d)/2 − 2 × a − e)/M, CaHbCldCueNcOo, M: molar weight of compound, g mol−1. The density was obtained from the single-crystal X-ray diffraction. Peak temperatures of the first exothermal decomposition processes. Impact sensitivity. Friction sensitivity. |
| 2 |
−15.35 |
−175.83 |
114.16 |
1.665 |
308.0 |
>40 |
>360 |
| 3 |
−6.64 |
−9632.51 |
−28.42 |
2.046 |
248.8 |
2 |
108 |
| 4 |
−7.56 |
−834.43 |
−32.66 |
1.937 |
218.4 |
2 |
120 |
2:
| | |
C5H10N10(s) + 7.5O2(g) → 5CO2(g) + 5H2O(l) + 5N2(g)
| (11) |
3:
| | |
C60H156Cl20Cu12N120O100(s) + 50O2(g) → 60CO2(g) + 68H2O(l) + 60N2(g) + 12CuO(s) + 20HCl(g)
| (12) |
4:
| | |
C15H31Cl4Cu3N31O20(s) + 13.25O2(g) → 15CO2(g) + 13.5H2O(l) + 15.5N2(g) + 3CuO(s) + 4HCl(g)
| (13) |
Sensitivity
Mechanical sensitivity is an important parameter for evaluating the safety of energetic materials. The sensitivities of the studied compounds toward impact and friction were measured according to the corresponding BAM standard methods. As shown in Table 3, the results showed that compound 2 can be classified as an insensitive energetic material (FS > 360 N, IS > 40 J), but compounds 3 and 4 are sensitive toward impact (2 J) and friction (108–120 N), and 3 has a higher friction sensitivity.
Effects of catalysis on the thermal decomposition of AP
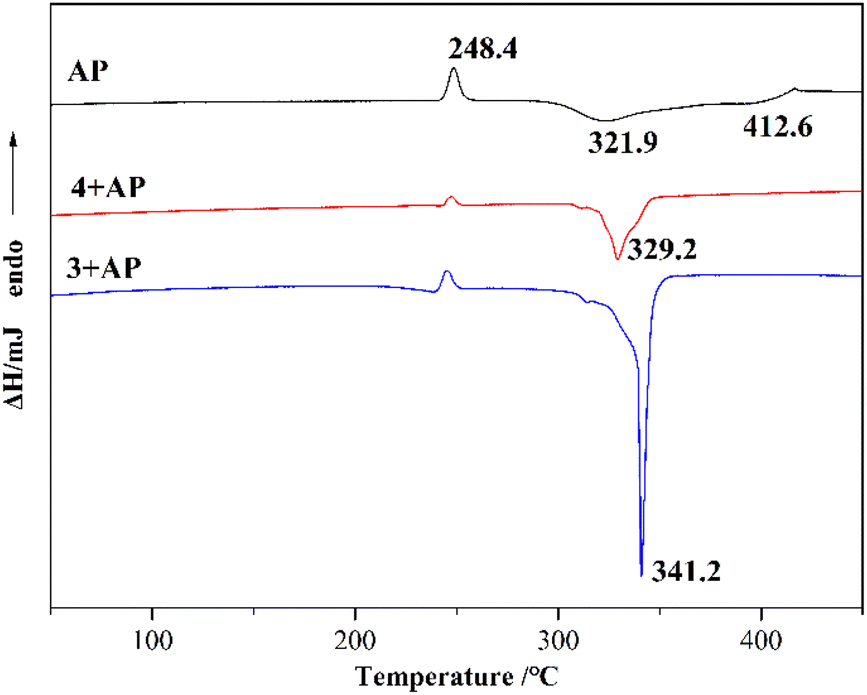
To explore the catalytic effect of two copper complexes on the thermal decomposition of AP, the complexes and AP were mixed at a mass ratio of 1:9 and ground into powder by physical grinding. The mixed samples were studied by differential scanning calorimetry (DSC) at a heating rate of 10 °C min−1 in an air atmosphere. Fig. 6 shows the DSC curves of AP, 3 + AP and 4 + AP. The DSC curve of pure AP has one endothermic peak (phase transition) located at 248.4 °C and two exothermic peaks located at 321.9 °C (low-temperature decomposition, LTD) and 412.6 °C (high-temperature decomposition, HTD), respectively. The phase transition peak of the mixed samples was the same as that of the pure AP. The peak temperatures of the HTD of 3 + AP and 4 + AP decreased to 341.2 °C and 329.2 °C, and the exothermic peaks of 3 + AP were drastically sharpened, indicating the rapid decomposition process of AP. The experimental results reveal that the decomposition of AP is accelerated by compounds 3 and 4. Complex 4 possesses an outstanding catalytic effect for the decomposition of AP (Table 4).
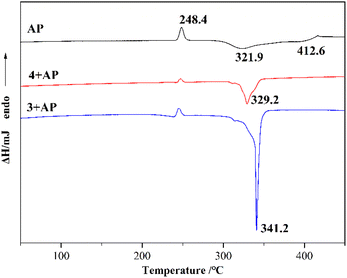 |
| | Fig. 6 DSC curves of AP, 3 + AP and 4 + AP. | |
Table 4 The catalytic effect of compounds 3 and 4 on AP decomposition
| Sample |
Decomposition (DSC) |
| HTD (°C) |
Δ (°C) |
| AP |
412.6 |
— |
| 3 + AP |
341.2 |
−71.4 |
| 4 + AP |
329.2 |
−83.4 |
Hirshfeld surface analysis
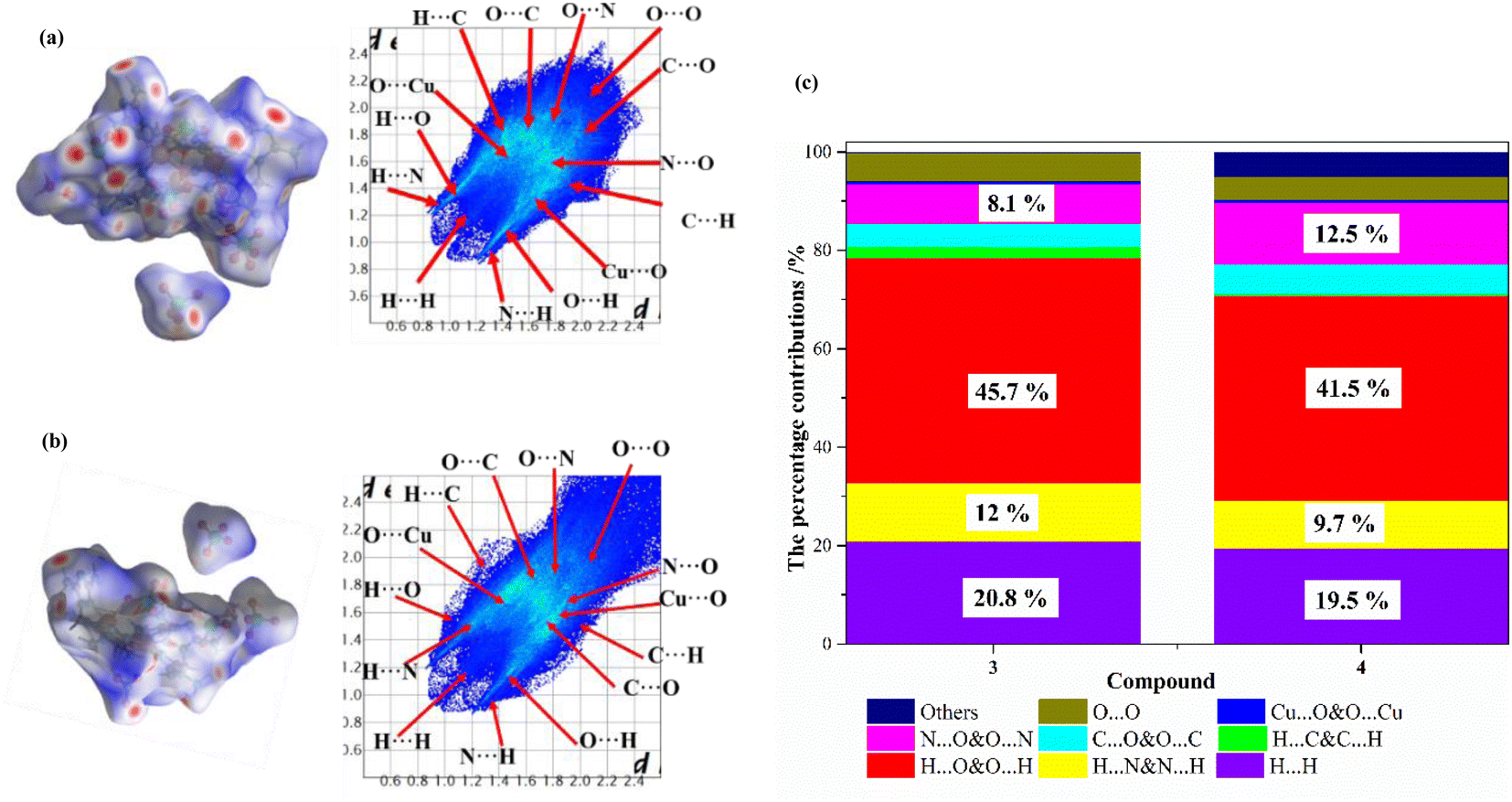
As can be seen from the above analysis, the crystal structures of compounds 3 and 4 are similar, but their physicochemical properties are different. To obtain a better understanding of the relationship between crystal packing and intermolecular interactions, the Hirshfeld surface and 2D finger plots of compounds 3 and 4 are analyzed using the Crystalexplorer program.48 The Hirshfeld surface and 2D fingerprint plots of the compounds are shown in Fig. 7(a) and (b), respectively. As shown in Fig. 7, the color coding on the Hirshfeld surface represents the proximity of close contacts around compound molecules: blue indicates weak interactions, which normally belong to π–π stacking, such as C⋯O, O⋯N and N⋯N interactions; white indicates van der Waals contact; red indicates strong interaction, which mainly denotes intermolecular H⋯N&N⋯H and O⋯H&H⋯O interactions.49,50 Additionally, 2D fingerprints can obtain the percentage contributions of the intermolecular interaction forces in the crystal structure. The percentage contributions for the main type of interaction to the total weak interactions are shown in Fig. 7(c) and are listed in Table S14.† For compound 3, the percentages of H⋯N&N⋯H and H⋯O&O⋯H interactions are 12% and 45.7%, respectively, and those for compound 4 are 9.7% and 41.5%, respectively. These results show that the percentage of hydrogen bonding in compound 3 is higher than that in compound 4. The total percentages of N⋯O&O⋯N and C⋯O&O⋯C interactions for compounds 3 and 4 are 12.8% and 18.4%, respectively. These results show that the percentage of π–π interactions in compound 4 is higher than that in compound 3. Owing to the extensive hydrogen bonding interactions and π–π interactions, compounds 3 and 4 show a high density. The π–π interactions can result in better mechanical stability, but O⋯O contacts can increase the possibility of unexpected explosions stability.50 For compounds 3 and 4, the percentages of O⋯O contacts are 5.7% and 4.6%. According to these results, it is not surprising that compounds 3 and 4 have high sensitivities toward friction.
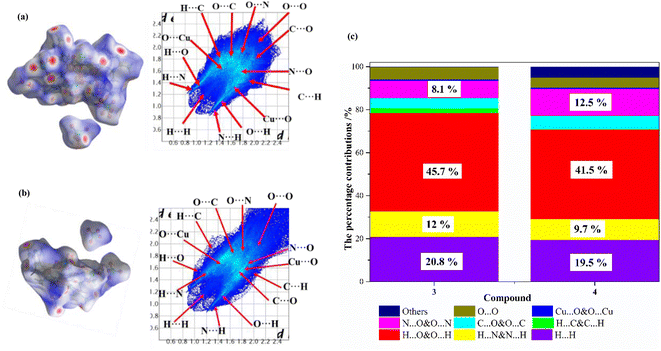 |
| | Fig. 7 Hirshfeld surface calculations of 3 and 4 as well as the associated 2D fingerprint plots in the crystal structures. (a) Hirshfeld surface and 2D fingerprint plots of 3, (b) Hirshfeld surface and 2D fingerprint plots of 4, and (c) the percentage contributions of individual atomic contacts to the Hirshfeld surface. | |
Conclusions
In summary, a novel thermally stable and mechanically insensitive energetic compound 1-((4,5-diamino-4H-1,2,4-triazol-3-yl)methyl)-1H-1,2,4-triazol-3,5-diamine (2) and its copper energetic compounds were synthesized and thoroughly characterized. The crystal structures of 3 and 4 were confirmed using single-crystal X-ray diffraction, showing that the two compounds are a mixed-ligand and mixed-anion copper coordination polymer. Significantly, owing to the extensive and intensive hydrogen bonding and the π–π stacking effect, the two compounds exhibited excellent densities (2.046 g cm−3 (3) and 1.937 g cm−3 (4)). These two compounds exhibited acceptable thermal stability (Tdec. > 218.4 °C). Moreover, the apparent activation energy was calculated by applying the Kissinger–Ozawa method, and the formation enthalpy of these compounds was calculated. Compound 4 shows a higher formation enthalpy than compound 3. Moreover, compound 3 shows higher sensitivities toward impact and friction than 4. Complexes 3 and 4 exhibit excellent catalytic performance for AP decomposition, and complex 4 shows a better catalytic effect than complex 3. The results indicate that by introducing two or more different types of anions, the catalytic performance of energetic compounds can be improved, and better energetic catalysts can be obtained.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge financial support from the State Key Laboratory of Explosion Science and Technology (No. YBKT22-03) and the Young Talent Program of Xingtai (No. 2020ZZ042).
Notes and references
- C. Qi, S. H. Li, Y. C. Li, Y. Wang, X. X. Zhao and S. P. Pang, Chemistry, 2012, 18, 16562–16570 CrossRef CAS PubMed.
- X. Yang, Y. Wang, W. Zhang, Y. Yin, Z. Li and T. Zhang, CrystEngComm, 2020, 22, 4573–4579 RSC.
- X. Zhao, C. Qi, L. Zhang, Y. Wang, S. Li, F. Zhao and S. Pang, Molecules, 2014, 19, 896–910 CrossRef PubMed.
- V. Thottempudi, F. Forohor, D. A. Parrish and J. M. Shreeve, Angew. Chem., Int. Ed. Engl., 2012, 51, 9881–9885 CrossRef CAS PubMed.
- H. Li, Y. Wang, Z. Wei, L. Xia, Z. Li and T. Zhang, New J. Chem., 2022, 46, 4462–4469 RSC.
- N. Şen, J. Mol. Struct., 2019, 1179, 453–461 CrossRef.
- J. Ren, W. Zhang, T. Zhang, Z. Li, Q. Zeng and T. Zhang, J. Mol. Struct., 2021, 1223, 128955–128962 CrossRef CAS.
- Y. Liu, C. He, Y. Tang, G. H. Imler, D. A. Parrish and J. M. Shreeve, Dalton Trans., 2019, 48, 3237–3242 RSC.
- C. He, G. H. Imler, D. A. Parrish and J. n. M. Shreeve, J. Mater. Chem. A, 2018, 6, 16833–16837 RSC.
- H. Li, W. Zhang, Z. Wei, L. Xia, M. Long, Z. Li and T. Zhang, Eur. J. Inorg. Chem., 2022, 2022, e202200008 CAS.
- Z. Li, Y. Zhong, L. Liang, Y. Feng, J. Zhang, T. Zhang and Y. Zhang, Chem. Eng. J., 2021, 423, 130187 CrossRef CAS.
- J. W. Fronabarger, M. D. Williams, W. B. Sanborn, J. G. Bragg, D. A. Parrish and M. Bichay, Propellants, Explos., Pyrotech., 2011, 36, 541–550 CrossRef CAS.
- W. S. Dong, W. L. Cao, Q. U. Tariq, X. W. Wu, Y. Hu, C. Zhang and J. G. Zhang, Dalton Trans., 2022, 51, 9894–9904 RSC.
- X. Yang, Y. Wang, Z. Li, J. Du, L. Wang and T. Zhang, New J. Chem., 2019, 43, 14159–14165 RSC.
- L. Liang, Y. Zhong, J. Chen, J. Zhang, T. Zhang and Z. Li, Inorg. Chem., 2022, 61, 14864–14870 CrossRef CAS PubMed.
- H. Li, Y. Wang, Z. Wei, X. Yang, L. Liang, L. Xia, M. Long, Z. Li and T. Zhang, Chem. Eng. J., 2022, 430, 132739 CrossRef CAS.
- B. Westwater, H. J. Lloyd, I. J. Vitorica-Yrezabal, A. Fong, P. McMaster, M. Sloan, B. M. Coaker, C. R. Pulham and P. Portius, Dalton Trans., 2020, 49, 14975–14984 RSC.
- S. Rahali, R. Zarrougui, M. Marzouki and O. Ghodbane, J. Electroanal. Chem., 2020, 871, 114289 CrossRef CAS.
- M. H. H. Wurzenberger, M. S. Gruhne, M. Lommel, N. Szimhardt and J. Stierstorfer, Mater. Adv., 2022, 3, 579–591 RSC.
- M. H. H. Wurzenberger, M. Lommel, M. S. Gruhne, N. Szimhardt and J. Stierstorfer, Angew. Chem., Int. Ed. Engl., 2020, 59, 12367–12370 CrossRef CAS PubMed.
- Y. Feng, S. Chen, M. Deng, T. Zhang and Q. Zhang, Inorg. Chem., 2019, 58, 12228–12233 CrossRef CAS PubMed.
- V. D. Ghule, J. Phys. Chem. C, 2013, 117, 16840–16849 CrossRef CAS.
- T. Zhang, J. Du, Z. Li, X. Lin, L. Wang, L. Yang and T. Zhang, CrystEngComm, 2019, 21, 765–772 RSC.
- T. Liu, X. Qi, K. Wang, J. Zhang, W. Zhang and Q. Zhang, New J. Chem., 2017, 41, 9070–9076 RSC.
- D. Izsák and T. M. Klapötke, Crystals, 2012, 2, 294–305 CrossRef.
- T. M. Klapötke, P. Mayer, J. Stierstorfer and J. J. Weigand, J. Mater. Chem., 2008, 18, 5248–5258 RSC.
- D. Fischer, T. M. Klapötke, J. Stierstorfer and N. Szimhardt, Chem.–Eur. J., 2016, 22, 4966–4970 CrossRef CAS PubMed.
- J. Singh, R. J. Staples and J. n. M. Shreeve, Mater. Adv., 2022, 3, 6062–6068 RSC.
- R. S. Mathpati, A. K. Yadav, V. D. Ghule and S. Dharavath, Energ. Mater. Front., 2022, 3, 90–96 CrossRef CAS.
- Y. Wang, X. Yang, W. Zhang, H. Li, Z. Li, L. Wang and T. Zhang, CrystEngComm, 2019, 21, 6452–6459 RSC.
- Y.-H. Joo, B. Twamley and J. M. Shreeve, Chem.–Eur. J., 2009, 15, 9097–9104 CrossRef CAS PubMed.
- G. Zhao, C. He, H. Gao, G. H. Imler, D. A. Parrish and J. n. M. Shreeve, New J. Chem., 2018, 42, 16162–16166 RSC.
- D. Chand, D. A. Parrish and J. n. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383–15389 RSC.
- M. Wang, Z. Wang, B. Wang, J. Zhang and J. Zhang, J. Mol. Struct., 2021, 1227, 129536 CrossRef CAS.
- Y. Tang, Z. Yin, A. K. Chinnam, R. J. Staples and J. M. Shreeve, Inorg. Chem., 2020, 59, 17766–17774 CrossRef CAS PubMed.
- W. Geng, Y. Jia, Y. Chen, Q. Ma, G. Fan and L. Liao, CrystEngComm, 2020, 22, 3144–3154 RSC.
- H. Xiong, H. Yang, G. Cheng and Z. Zhang, ChemistrySelect, 2019, 4, 8876–8881 CrossRef CAS.
- H. Li, B. Yan, H. Ma, X. Ma, Z. Sun and Y. Ma, Acta Crystallogr., Sect. C: Struct. Chem., 2020, 76, 965–971 CrossRef CAS PubMed.
- S. E. Creegan, M. Zeller, E. F. C. Byrd and D. G. Piercey, Z. Anorg. Allg. Chem., 2022, 648, 1–7 CrossRef.
- Y. Yin, E. Yao, L. Xiao, Z. Wang, Y. Ren, J. Bai, H. Ma and F. Zhao, J. Mol. Struct., 2023, 1281, 134957–134967 CrossRef CAS.
- P. Yin, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 4778–4786 CrossRef CAS PubMed.
- W.-y. Zhao, T.-l. Zhang, L.-n. Zhang, L. Yang and Z.-n. Zhou, J. Ind. Eng. Chem., 2016, 38, 73–81 CrossRef CAS.
- Q.-L. Yan, F.-Q. Zhao, K. K. Kuo, X.-H. Zhang, S. Zeman and L. T. DeLuca, Prog. Energy Combust. Sci., 2016, 57, 75–136 CrossRef.
- T. M. Klapötke, A. Nordheider and J. Stierstorfer, New J. Chem., 2012, 36, 1463–1468 RSC.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- T. Zhang, R. Hu, Y. Xie and F. Li, Thermochim. Acta, 1994, 244, 171–176 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- S. Chen, Y. Liu, Y. Feng, X. Yang and Q. Zhang, Chem. Commun., 2020, 56, 1493–1496 RSC.
- Q. Yu, G. H. Imler, D. A. Parrish and J. M. Shreeve, Org. Lett., 2019, 21, 4684–4688 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yan-na Wangb,
Xiao-ming Yangc,
Lin-na Lianga,
Zhi-min Li
a,
Yan-na Wangb,
Xiao-ming Yangc,
Lin-na Lianga,
Zhi-min Li