
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile formation of barium titanium oxyhydride on a titanium hydride surface as an ammonia synthesis catalyst†
Yoshihiro Goto *a,
Masashi Kikugawaa,
Keisuke Kobayashib,
Yuichi Manakab,
Tetsuya Nanbab,
Hideyuki Matsumotobc,
Mitsuru Matsumotoa,
Masakazu Aokia and
Haruo Imagawaa
*a,
Masashi Kikugawaa,
Keisuke Kobayashib,
Yuichi Manakab,
Tetsuya Nanbab,
Hideyuki Matsumotobc,
Mitsuru Matsumotoa,
Masakazu Aokia and
Haruo Imagawaa
aToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute 480-1192, Aichi, Japan. E-mail: yoshihiro-goto@mosk.tytlabs.co.jp
bRenewable Energy Research Center, National Institute of Advanced Industrial Science and Technology, 2-2-9 Machiikedai, Koriyama 963-0298, Fukushima, Japan
cDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152–8552, Japan
First published on 22nd May 2023
Abstract
Oxyhydrides are promising compounds as supports for ammonia synthesis catalysts because they suppress hydrogen poisoning on the catalyst surface and enhance the ammonia synthesis activity. Herein, we developed a facile method for preparing BaTiO2.5H0.5, a perovskite oxyhydride, on a TiH2 surface via the conventional wet impregnation method using TiH2 and Ba hydroxide. Scanning electron microscopy and high-angle annular dark-field scanning transmission electron microscopy observations revealed that BaTiO2.5H0.5 crystallized as nanoparticles of ca. 100–200 nm on the TiH2 surface. The Ru-loaded catalyst Ru/BaTiO2.5H0.5-TiH2 exhibited 2.46 times higher ammonia synthesis activity (3.05 mmol-NH3 g−1 h−1 at 400 °C) than the benchmark Ru catalyst Ru–Cs/MgO (1.24 mmol-NH3 g−1 h−1 at 400 °C) because of the suppression of hydrogen poisoning. The analysis of reaction orders showed that the effect of suppressing hydrogen poisoning on Ru/BaTiO2.5H0.5-TiH2 was equivalent to that of the reported Ru/BaTiO2.5H0.5 catalyst, thus supporting the formation of BaTiO2.5H0.5 perovskite oxyhydride. This study demonstrated that the selection of appropriate raw materials facilitates the formation of BaTiO2.5H0.5 oxyhydride nanoparticles on the TiH2 surface using the conventional synthesis method.
Introduction
Ammonia (NH3), an essential raw material in the production of agricultural fertilizers and synthetic chemicals, has recently attracted attention owing to its applicability as a hydrogen carrier or fuel.1,2 Ammonia is predominantly produced via the Haber–Bosch (HB) process, which accounts for 1–2% of the global energy demand and 2.5% of global CO2 emissions.3 Most of the CO2 emissions are responsible for hydrogen production processes using steam reforming (CH4 + H2O → CO + 3H2) and water gas shift reactions (CO + H2O → CO2 + H2). Replacing these processes with water electrolysis (2H2O → 2H2 + O2) using renewable energy can significantly reduce this CO2 emission.4 However, renewable electricity sources of an intermittent nature are not compatible with ammonia synthesis via the conventional HB process5 because the process is operated on large-scale and steady-state operations. On this basis, ammonia synthesis catalysts need severe reaction conditions (at 450–600 °C and 15–40 MPa).5 Therefore, ammonia synthesis catalysts that work under mild conditions should be developed for ammonia synthesis using renewable electricity sources.The rate-determining step in the synthesis of ammonia (3H2 + N2 → 2NH3) is the dissociation of the N2 triple bond (945 KJ mol−1), which is the strongest bond among those in diatomic molecules.6,7 Supported ruthenium (Ru) catalysts are the most promising candidates for ammonia synthesis under mild conditions because optimum N2 adsorption energy facilitates N2 dissociation on the Ru surface.8 Strongly basic supports (such as CeO2, La0.5Pr0.5O1.75, Ba/Ce0.5La0.5O1.75, CeO2-PrOx, and Ce0.5La0.5−xTixO1.75+0.5x)9–14 further promote N2 dissociation because the basic compounds enhance the electron transfer from the Ru metal to the antibonding orbital of N2.15 However, hydrogen atoms generated by H2 dissociation are often adsorbed on the active sites of the Ru surface, thereby preventing N2 dissociation on the Ru surface.16 In recent years, oxyhydrides such as BaTiO3−xHx, BaCeO3−xHyNz, LaH3−2xOx, GdHO, and SmHO have been reported as supports that suppress hydrogen poisoning and enhance the ammonia synthesis activity of Ru catalysts.17–20 Suppression of hydrogen poisoning is presumed to originate from the diffusivity of hydride (H−), which allows hydrogen spillover from the Ru metal to the surface of the oxyhydride supports.21
Transition-metal (TM) oxyhydride synthesis is generally complicated because the differences in chemical properties (such as reactivity, volatility, and ionic radius) among anions prevent different anions in an identical compound from becoming stable.22 For this reason, solid-state topochemical reactions and/or high-pressure reactions have been used to synthesize TM oxyhydrides.22 BaTiO3−xHx, which is a promising TM perovskite oxyhydride support for ammonia synthesis catalysts, cannot be prepared by simply reducing BaTiO3 with hydrogen. However, this oxyhydride is accessible via a solid-state topochemical reaction involving BaTiO3 and CaH2 because the reaction can provide a metastable phase by exchanging the oxide (O2−) in BaTiO3 with hydride (H−) in CaH2 while maintaining the basic framework structure of BaTiO3.23 However, materials preparation using the topochemical reaction is not suitable for practical use because the reaction involves a multi-step process: (1) mixing BaTiO3 and the moisture-sensitive CaH2 in an inert atmosphere, (2) calcining the mixture for a week under vacuum, and (3) washing in an inert atmosphere to extract product BaTiO3−xHx by removing the residual CaH2 and by-product CaO. The practical utility of BaTiO3−xHx may be limited by low producibility owing to the multi-step process. Thus, developing more facile, efficient ways to prepare BaTiO3−xHx is necessary to accelerate the application of BaTiO3−xHx. Moreover, the development can lead to the discovery of novel oxyhydrides.
Recently, Uchimura et al.24 have reported the direct synthesis of BaTiO3−xHx by a mechanochemical method using BaH2, BaO, and TiO2 and confirmed its performance as a hydrogen-permeable electrode. However, handling in an inert atmosphere is still required because BaH2 and BaO are sensitive to moisture. Herein, we demonstrated the synthesis of BaTiO3−xHx (x = 0.5), a perovskite oxyhydride, on a TiH2 surface via conventional wet impregnation method using TiH2 and Ba(OH)2·8H2O, which are stable in moisture and air. The obtained BaTiO2.5H0.5 was crystallized as fine particles of 100–200 nm size that covered the TiH2 particle surface. The Ru-loaded catalyst, Ru/BaTiO2.5H0.5-TiH2, showed 2.46 times higher ammonia synthesis activity than Ru–Cs/MgO as benchmark Ru catalyst. Moreover, Ru/BaTiO2.5H0.5-TiH2 suppressed hydrogen poisoning, thereby proving the formation of BaTiO2.5H0.5 perovskite oxyhydride.
Methods
BaTiO2.5H0.5-TiH2 was synthesized via the wet impregnation method using TiH2 powder and Ba hydroxide solution. TiH2 (98%, −325 mesh, Sigma-Aldrich) was impregnated with a solution containing the desired amount of Ba(OH)2·8H2O (98.0%, FUJIFILM Wako Chemicals) dissolved in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v) H2O/ethanol at 230 °C in the air, after which it was heated at 350 °C for 3 h in a 10% H2/N2 atmosphere. The Ba addition amounts were varied from 0–15 wt% on a Ba(OH)2 basis. The obtained compounds are hereafter referred to as Ba(α)-TiH2 (α: wt% of Ba(OH)2). Reference compounds Ba(α)-TiO2 (where α = wt% of Ba(OH)2) were prepared using the same protocols as for Ba(α)-TiH2, except TiO2 (99.9%, Rutile, Sigma-Aldrich) was used instead of TiH2. Ba(10)-TiH2 synthesized using Ba(CH3COO)2 (99.0%, FUJIFILM Wako Chemicals) or Ba(NO3)2 (99.0%, FUJIFILM Wako Chemicals) instead of Ba(OH)2·8H2O were also prepared to investigate the effect of Ba sources. Cs/MgO as a benchmark compound was prepared by impregnation using MgO (99%, Sigma-Aldrich) and Cs2CO3 (99.9%, Sigma-Aldrich)/ethanol solution, followed by thermal treatment at 350 °C for 3 h under 10% H2/N2. Ru-loaded catalysts, such as Ru/Ba(α)-TiH2, Ru/Ba(α)-TiO2, and Ru–Cs/MgO, were prepared by impregnation using Ru3(CO)12 (99%, Sigma-Aldrich)/tetrahydrofuran solution. The suspension was stirred for 5 h, and the solvent was subsequently evaporated at 27 °C. The obtained compound was dried at 80 °C for 16 h in the air. The Cs and Ru loading amounts were 1 wt% on a metal basis.
:2 (v/v) H2O/ethanol at 230 °C in the air, after which it was heated at 350 °C for 3 h in a 10% H2/N2 atmosphere. The Ba addition amounts were varied from 0–15 wt% on a Ba(OH)2 basis. The obtained compounds are hereafter referred to as Ba(α)-TiH2 (α: wt% of Ba(OH)2). Reference compounds Ba(α)-TiO2 (where α = wt% of Ba(OH)2) were prepared using the same protocols as for Ba(α)-TiH2, except TiO2 (99.9%, Rutile, Sigma-Aldrich) was used instead of TiH2. Ba(10)-TiH2 synthesized using Ba(CH3COO)2 (99.0%, FUJIFILM Wako Chemicals) or Ba(NO3)2 (99.0%, FUJIFILM Wako Chemicals) instead of Ba(OH)2·8H2O were also prepared to investigate the effect of Ba sources. Cs/MgO as a benchmark compound was prepared by impregnation using MgO (99%, Sigma-Aldrich) and Cs2CO3 (99.9%, Sigma-Aldrich)/ethanol solution, followed by thermal treatment at 350 °C for 3 h under 10% H2/N2. Ru-loaded catalysts, such as Ru/Ba(α)-TiH2, Ru/Ba(α)-TiO2, and Ru–Cs/MgO, were prepared by impregnation using Ru3(CO)12 (99%, Sigma-Aldrich)/tetrahydrofuran solution. The suspension was stirred for 5 h, and the solvent was subsequently evaporated at 27 °C. The obtained compound was dried at 80 °C for 16 h in the air. The Cs and Ru loading amounts were 1 wt% on a metal basis.
X-ray diffraction (XRD) patterns and were collected at room temperature using SmartLab (Rigaku) with Cu Kα radiation (λ = 1.54056 Å). The obtained XRD patterns were analyzed using the JANA2006 software.25 The neutron diffraction (ND) pattern was collected at room temperature using the NOVA time-of-flight (TOF) neutron diffractometer at the J-PARC facility in Japan. The obtained ND pattern was analyzed using Z-Rietveld software.26 Fourier-transform infrared (FT-IR) spectra were recorded using an iS50 spectrometer (Thermo Fisher) equipped with a diffuse reflectance optics accessory. Samples were pretreated at 200 °C for 30 min in flowing He and then examined at 50 °C. Scanning electron microscopy (SEM) images were obtained using a JSM-7400F (JEOL) and a SU3500 (Hitachi) operated at 1.5 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping images were obtained using a Tecnai Osiris (FEI) operated at 200 kV. X-ray photoelectron spectroscopy (XPS) was conducted on a Quantera SXM instrument (ULVAC PHI) using Al Kα radiation (1486.6 eV). CO amounts adsorbed by the catalyst were estimated using a BEL-METAL-3 (MicrotracBEL). CO pulse injections (1.99% CO/He) to the samples were conducted at 50 °C after the pre-treatment at 400 °C for 2 h in 100% H2.
The ammonia synthesis activities under ambient pressure were estimated using a fixed-bed reactor connected to mass flow controllers. A sample (0.2 g) was suspended on a bed of quartz wool in a quartz tube and preheated at 400 °C for 2 h in 75% H2/N2 (H2/N2 = 3) at a flow rate of 80 mL min−1. The activity measures were conducted at 300–400 °C in 75% H2/N2 (H2/N2 = 3) at a flow rate of 80 mL min−1. The ammonia concentrations in the outlet of the quartz tube were monitored by FT-IR spectroscopy and converted into ammonia synthesis rates. Reaction orders of ammonia synthesis with respect to H2, N2, and NH3 were estimated using the method of Aika et al.27
Results and discussion
The XRD patterns of Ba(α)-TiH2 (α = 0, 1, 3, 5, 10, and 15) predominantly showed the cubic phase of the fluorite structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with the lattice parameter of a = 4.4489–4.4519 Å (Fig. 1). The lattice parameter matches that of the reported TiH2 (a = 4.4512(1) Å),28 showing that the cubic phase is TiH2. The XRD patterns of samples with α = 0 and 1 only showed the single phase of TiH2, whereas the XRD patterns of α ≥ 3 samples contained an additional cubic phase of perovskite structure (Pmm; Fig. S1 in the ESI†). The lattice volume was estimated to be V = 64.926–65.073 Å3 (Table 1), which approaches that reported for cubic BaTiO2.38H0.62 perovskite oxyhydride (Pmm, V = 65.140 Å3)23 rather than tetragonal BaTiO3 perovskite oxide (P4mm, V = 64.281 Å3),29 which indicates that the additional cubic phase is BaTiO3−xHx perovskite oxyhydride. The H content x in BaTiO3−xHx (α = 3, 5, 10, and 15) were determined to be x = 0.47–0.57 based on Vegard's law (Table 1; hereafter, the obtained perovskite oxyhydride is referred to as BaTiO2.5H0.5). The formation of oxygen-deficient BaTiO3−δ is unlikely because the lattice volume of BaTiO2.5H0.5 (V = 64.926–65.073 Å3) is larger than that reported for cubic BaTiO3−δ perovskite oxide (δ = 0.25, Pmm, V = 64.337 Å3) where the lattice volume does not change through the formation of oxygen defects.30 The formation of the BaTiO3−δ(OH)δ oxyhydroxide through OH− incorporation is also denied because no peaks associated with OH bonds (observed at 3400 cm−1 in BaTiO3 system)31 were observed by FT-IR spectroscopy (Fig. S2†). Moreover, the XRD patterns of the α = 10 and 15 samples showed that BaCO3 can potentially form through the reaction of unreacted Ba(OH)2 with atmospheric CO2 absorbed during impregnation. Thus, the addition of Ba into TiH2 forms BaTiO2.5H0.5 perovskite oxyhydride and BaCO3.
m) with the lattice parameter of a = 4.4489–4.4519 Å (Fig. 1). The lattice parameter matches that of the reported TiH2 (a = 4.4512(1) Å),28 showing that the cubic phase is TiH2. The XRD patterns of samples with α = 0 and 1 only showed the single phase of TiH2, whereas the XRD patterns of α ≥ 3 samples contained an additional cubic phase of perovskite structure (Pmm; Fig. S1 in the ESI†). The lattice volume was estimated to be V = 64.926–65.073 Å3 (Table 1), which approaches that reported for cubic BaTiO2.38H0.62 perovskite oxyhydride (Pmm, V = 65.140 Å3)23 rather than tetragonal BaTiO3 perovskite oxide (P4mm, V = 64.281 Å3),29 which indicates that the additional cubic phase is BaTiO3−xHx perovskite oxyhydride. The H content x in BaTiO3−xHx (α = 3, 5, 10, and 15) were determined to be x = 0.47–0.57 based on Vegard's law (Table 1; hereafter, the obtained perovskite oxyhydride is referred to as BaTiO2.5H0.5). The formation of oxygen-deficient BaTiO3−δ is unlikely because the lattice volume of BaTiO2.5H0.5 (V = 64.926–65.073 Å3) is larger than that reported for cubic BaTiO3−δ perovskite oxide (δ = 0.25, Pmm, V = 64.337 Å3) where the lattice volume does not change through the formation of oxygen defects.30 The formation of the BaTiO3−δ(OH)δ oxyhydroxide through OH− incorporation is also denied because no peaks associated with OH bonds (observed at 3400 cm−1 in BaTiO3 system)31 were observed by FT-IR spectroscopy (Fig. S2†). Moreover, the XRD patterns of the α = 10 and 15 samples showed that BaCO3 can potentially form through the reaction of unreacted Ba(OH)2 with atmospheric CO2 absorbed during impregnation. Thus, the addition of Ba into TiH2 forms BaTiO2.5H0.5 perovskite oxyhydride and BaCO3.
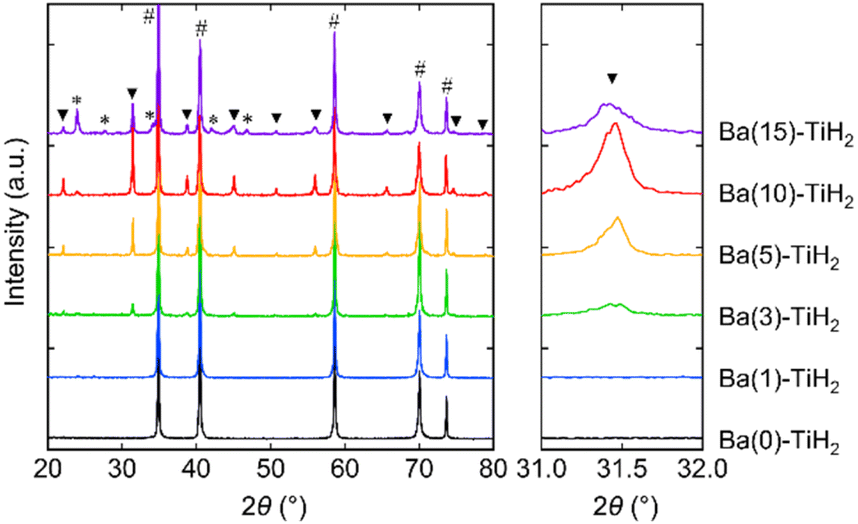 | ||
| Fig. 1 XRD patterns of Ba(α)-TiH2 (α = 0, 1, 3, 5, 10, and 15). Hashtags (#), triangles (▼), and asterisks (*) indicate peaks arising from TiH2, BaTiO2.5H0.5, and BaCO3, respectively. | ||
| Sample | Lattice volume of BaTiO3−xHxa (Å3) | H content xb |
|---|---|---|
| a Calculated from the lattice parameters based on the cubic perovskite structure (Pmm, Z = 1).b Determined from Vegard's law using the lattice volumes of BaTiO3 (V = 64.281 Å3)29 and BaTiO2.38H0.62 (V = 65.140 Å3).23 |
||
| Ba(0)-TiH2 | — | — |
| Ba(1)-TiH2 | — | — |
| Ba(3)-TiH2 | 65.002(2) | 0.52 |
| Ba(5)-TiH2 | 64.926(2) | 0.47 |
| Ba(10)-TiH2 | 65.009(2) | 0.56 |
| Ba(15)-TiH2 | 65.073(3) | 0.57 |
The ND pattern of Ba(10)-TiH2 was collected to investigate the presence of the hydride in BaTiO2.5H0.5 (Fig. S3 and Table S1†). Rietveld refinement was performed by assuming that the secondary phase is a BaTiO3−xHx cubic perovskite with a Pmm structural model, where Ba, Ti, and O/H atoms are placed at the Wyckoff position of 1a (0, 0, 0), 1b (0.5, 0.5, 0.5), and 3c (0, 0.5, 0.5), respectively. Cubic TiH2 (Fmm) was added as a primary phase. Refinement converged to O and H occupancies of g(O) = 0.886(2) and g(H) = 0.114(2), which yields the BaTiO2.658(6)H0.342(6) composition and supports the notion that perovskite phase contains hydride. We note here that the difference in the neutron scattering lengths of oxygen and hydrogen (O: 5.803 fm, H: −3.741 fm)32 gives the composition BaTiO2.438(6) when the refinement is performed by assuming that the secondary phase is oxygen-deficient BaTiO3−δ. However, because the formation of oxygen-deficient BaTiO3−δ is ruled out by considering the lattice volumes estimated by the XRD analysis, the ND results support the formation of an oxyhydride.
The degrees of BaTiO2.5H0.5 and BaCO3 formation were determined by Rietveld refinement of the XRD patterns of Ba(α)-TiH2. We note here that components (less than about 1%) that are not detected in the XRD analysis are not considered. As shown in Fig. 2, the mass fraction of BaTiO2.5H0.5 is 0% at 0 ≤ α ≤ 1 and higher at 3 ≤ α ≤ 10 (1.6% to 8.7%). However, a lower fraction was observed at α = 15 (4.5%). The addition of Ba to TiH2 facilitates the formation of BaTiO2.5H0.5, while excess Ba inhibits its formation. Thus, the optimum amount of Ba(OH)2 needed to form BaTiO2.5H0.5 corresponds to α = 10. The mass fraction of the formed BaCO3 was 0% at 0 ≤ α ≤ 5 but higher at 10 ≤ α ≤ 15 (0.7 to 4.3%), which implies that unreacted Ba(OH)2 remains at α ≥ 10. The samples also exhibited colors that depend on the amounts of BaTiO2.5H0.5 and BaCO3 amounts (Fig. S4†). Ba(0)-TiH2 and Ba(5)-TiH2 are gray, which is typical of TiH2, while Ba(10)-TiH2 is dark blue, which is typical of BaTiO2.5H0.5,23 which also supports the notion that the BaTiO2.5H0.5 perovskite oxyhydride had formed. Ba(15)-TiH2 is brown, which is possibly due to mixed colors associated with TiH2, BaTiO2.5H0.5, and BaCO3.
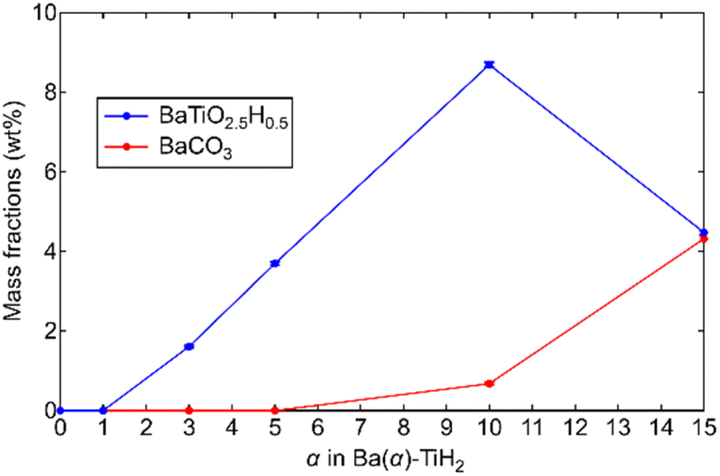 | ||
| Fig. 2 Mass fractions of BaTiO2.5H0.5 and BaCO3 in Ba(α)-TiH2 determined by Rietveld refinements of the XRD patterns of Ba(α)-TiH2. Cubic TiH2 (Fmm), cubic BaTiO2.5H0.5 (Pmm), and orthorhombic BaCO3 (Pnma) phases were applied during the analysis. | ||
The SEM image of Ba(0)-TiH2 revealed that the particle size of TiH2 ranged from several to tens of μm (Fig. S5 and S6†). Particle size was not affected by the amount of added Ba, which agreed with the identical specific surface area of Ba(α)-TiH2 (0 ≤ α ≤ 15; 1.65–1.91 m2 g−1). The particle surface of Ba(0)-TiH2 and Ba(1)-TiH2 were relatively smooth; by comparison, nanoparticles of ca. 100–200 nm were dispersed on the TiH2 surface of Ba(3)-TiH2 and Ba(5)-TiH2 (Fig. 3). The nanoparticles were expected to be BaTiO2.5H0.5 because BaTiO2.5H0.5 in addition to TiH2 was observed in the XRD patterns of Ba(3)-TiH2 and Ba(5)-TiH2 (Fig. 1). The EDX mappings in the HAADF-STEM image of Ru/Ba(5)-TiH2 show that Ba and O elements were localized at the nanoparticles (Fig. 4), thus supporting the identification of the nanoparticles as BaTiO2.5H0.5. The Ba/Ti atomic ratio estimated from the XPS analysis was higher than that of the feed ratio in the preparation, also supporting that the nanoparticles are BaTiO2.5H0.5. The SEM image of Ba(10)-TiH2 showed that the BaTiO2.5H0.5 nanoparticles fully covered the TiH2 particle. Moreover, the SEM image of Ba(15)-TiH2 showed needle-shaped particles on the BaTiO2.5H0.5 nanoparticles. The crystals were identified as BaCO3 according to the XRD pattern because BaCO3 generally formed needle-shape crystals.33 Therefore, BaTiO2.5H0.5 was formed as 100–200 nm nanoparticles at α ≥ 3, and BaCO3 was formed as needle shape particles at α ≥ 10 (Fig. 5).
 | ||
| Fig. 3 SEM images of Ba(α)-TiH2 (α = 0, 5, 10, and 15) at 10000× magnification. | ||
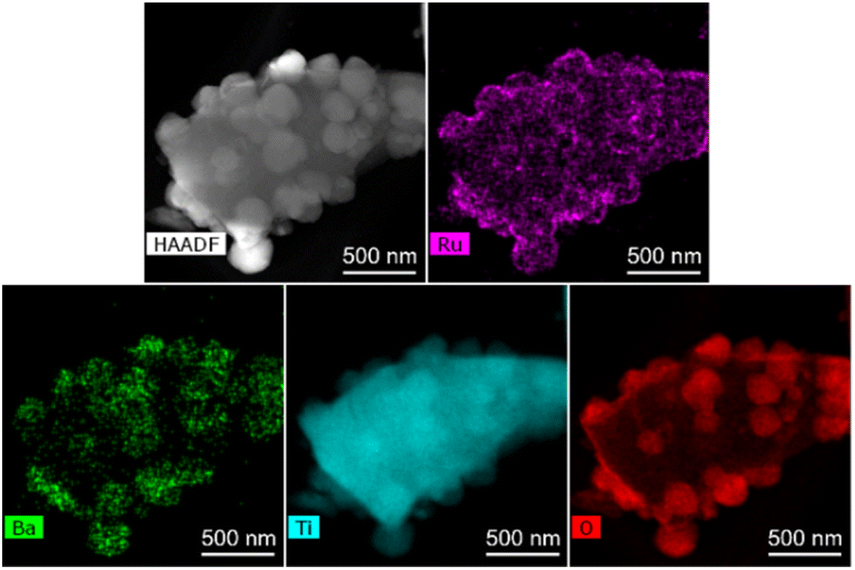 | ||
| Fig. 4 STEM-HAADF and EDX mapping images of Ru/Ba(5)-TiH2. | ||
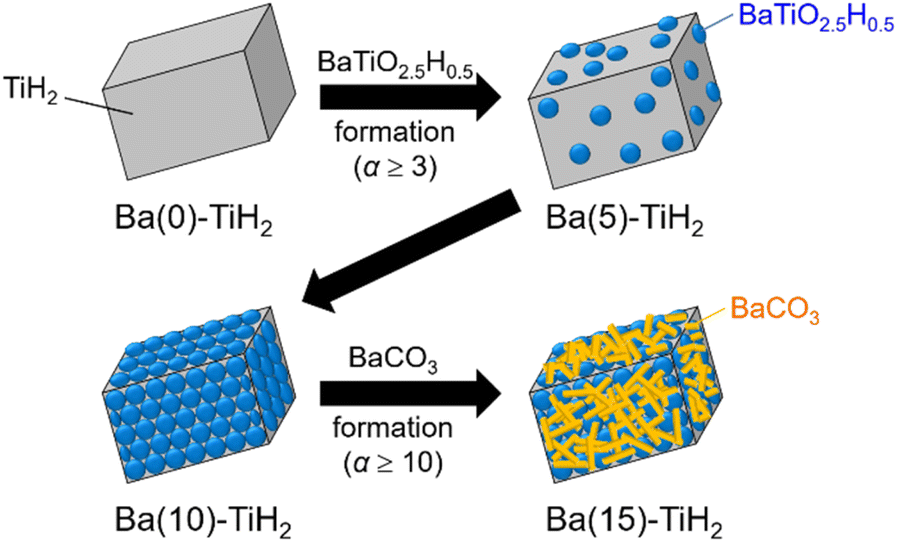 | ||
| Fig. 5 Images of the morphological change of Ba(α)-TiH2. | ||
The XRD patterns of Ba(α)-TiO2 (α = 0, 5, 10, 15) synthesized as comparison are shown in Fig. S8.† All samples showed predominantly rutile TiO2. Ba(0)-TiO2 was the single phase of TiO2, whereas Ba(α)-TiO2 (α = 5, 10, and 15) contained additional Ba2TiO4 and BaCO3. The cubic phase of BaTiO2.5H0.5 formed in Ba(α)-TiH2 was not observed with the addition of any amount of Ba(OH)2. Therefore, TiH2 is essential for BaTiO2.5H0.5 formation.
How is TiH2 involved in the formation of BaTiO2.5H0.5? TiH2 consumed to form BaTiO2.5H0.5 is calculated to be 2.1% for Ba(10)-TiH2 which contains the highest amount of BaTiO2.5H0.5 (8.7%; Fig. 2), suggesting that only the surface part of the TiH2 particles contributes to the formation. This is supported by the SEM and TEM observations (Fig. 3 and 4). Because metal hydrides are generally unstable in an oxidizing atmosphere, the surface of TiH2 particles is known to be covered by an oxide film of TiO2.34 TiH2 (Ti2+) partially oxidized to TiO2 (Ti4+) possibly contributes to the formation of BaTiO2.5H0.5 (Ti3.5+) via the reaction Ba(OH)2 + 0.5xTiH2 + (1 − 0.5x)TiO2 → BaTiO3−xHx + H2O. Moreover, the effect of Ba reagents on BaTiO2.5H0.5 formation was investigated. The XRD patterns of Ba(10)-TiH2 synthesized using Ba(CH3COO)2 or Ba(NO3)2 instead of Ba(OH)2·8H2O are shown in Fig. S9.† The XRD pattern of Ba(10)-TiH2 synthesized via Ba(CH3COO)2 showed only TiH2 and Ba(CH3COO)2 and no formation of BaTiO2.5H0.5. While Ba(10)-TiH2 synthesized via Ba(NO3)2 contained BaTiO2.5H0.5, its mass fraction (1.5%) was only 0.17-times that of the Ba(10)-TiH2 synthesized via Ba(OH)2·8H2O (8.7%). These observations suggest that the hydroxide ion (OH−) promotes the formation of BaTiO2.5H0.5.
The ammonia synthesis activities of Ru/Ba(α)-TiH2 (0 ≤ α ≤ 15) catalysts were examined at 300–400 °C under ambient pressure (Fig. 6a). Ammonia synthesis rates of all catalysts increased at elevated temperature. Ru/Ba(0)-TiH2 exhibited ammonia synthesis activity at ≥375 °C. The ammonia synthesis rate of Ru/Ba(0)-TiH2 (0.05 mmol g−1 h−1 at 400 °C) was 0.04 times that of Ru/Cs–MgO (1.24 mmol g−1 h−1 at 400 °C), which was often called as benchmark Ru catalysts.35,36 The activity of Ru/Ba(1)-TiH2 (0.20 mmol g−1 h−1 at 400 °C) was higher than that of Ru/Ba(0)-TiH2 but remained lower than that of Ru/Cs–MgO. However, by contrast, the activities of Ru/Ba(3)-TiH2, Ru/Ba(5)-TiH2, and Ru/Ba(10)-TiH2 (1.73, 2.08, and 3.05 mmol g−1 h−1 at 400 °C, respectively) were higher than that of Ru–Cs/MgO by a factor of 1.40, 1.68, and 2.46, respectively. Activity increased with the increase in Ba addition amount in 0 ≤ α ≤ 10, whereas the activity decreased at α = 15 (0.32 mmol g−1 h−1 at 400 °C). Therefore, the most active catalyst among Ru/Ba(α)-TiH2 (0 ≤ α ≤ 15) was Ru/Ba(10)-TiH2. The activity of Ru/Ba(10)-TiH2 was higher than that of Ru/Ba(10)-TiO2 (1.09 mmol g−1 h−1 at 400 °C), thereby suggesting that BaTiO2.5H0.5 formation was responsible for the high activity of Ru/Ba(10)-TiH2.
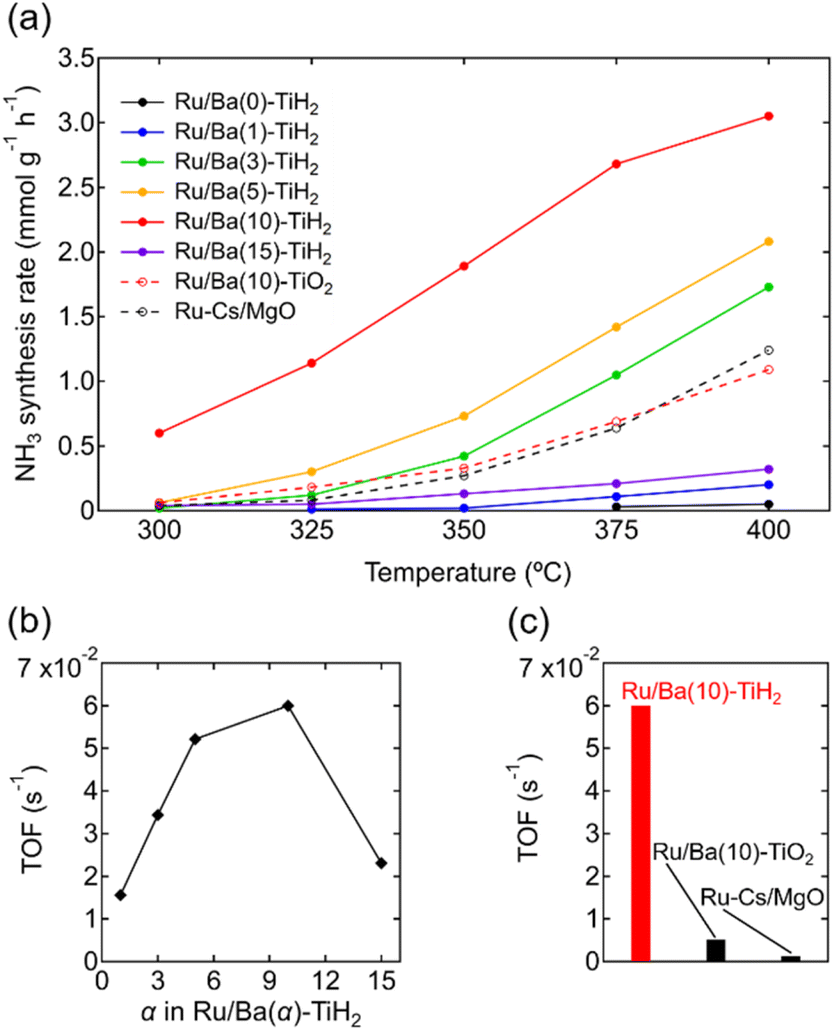 | ||
| Fig. 6 (a) Temperature dependence of the NH3 synthesis rates for Ru/Ba(α)-TiH2 (α = 0, 1, 3, 5, 10, and 15), Ru/Ba(10)-TiO2, and Ru–Cs/MgO (reaction conditions: catalyst, 0.2 g; reaction gas, H2/N2 = 3 at a flow rate of 80 mL min−1; pressure = ambient pressure). (b) TOF of Ru/Ba(α)-TiH2 (α = 0, 1, 3, 5, 10, and 15) at 350 °C as functions of Ba(OH)2 added amount α. (c) TOF of Ru/Ba(10)-TiH2, Ru/Ba(10)-TiO2, and Ru–Cs/MgO at 350 °C. | ||
The TOF for the ammonia synthesis reaction at 350 °C was estimated to obtain a deeper insight into the correlation between ammonia synthesis activity and catalyst composition. As shown in Fig. 6b, the TOF of Ru/Ba(α)-TiH2 increased with increasing amount of Ba addition at 1 ≤ α ≤ 10 (1.56 × 10−2 s−1 to 6.00 × 10−2 s−1) but decreased at α = 15 (2.31 × 10−2 s−1). This trend was consistent with the trend of the mass fraction of BaTiO2.5H0.5 (Fig. 2); these observations support that BaTiO2.5H0.5 formation contributes to the increase in ammonia synthesis activity. This was supported by the fact that the TOF of Ru/Ba(10)-TiH2 (6.00 × 10−2 s−1) was 12 and 46 times larger than those of Ru/Ba(10)-TiO2 (0.51 × 10−2 s−1) and Ru–Cs/MgO (0.13 × 10−2 s−1), respectively (Fig. 6c). Interestingly, the TOF of Ru/Ba(15)-TiH2 (2.31 × 10−2 s−1) was lower than that of Ru/Ba(5)-TiH2 (5.21 × 10−2 s−1) despite the higher mass fraction of BaTiO2.5H0.5 for Ru/Ba(15)-TiH2 (4.48%) than that for Ru/Ba(5)-TiH2 (3.70%). The formation of BaCO3, which partially covered the BaTiO2.5H0.5 particles (Fig. 3), was expected to inhibit the ammonia synthesis reaction of Ru/Ba(15)-TiH2 because BaCO3 was stable in the reaction temperature.37
Finally, reaction orders with respect to H2, N2 and NH3 were investigated using the method of Aika et al.27 (Fig. S10 and Table S3†). The H2 order of Ru/Ba(10)-TiH2 (0.15) was higher than that of Ru–Cs/MgO (−0.59), reflecting that compared to Ru–Cs/MgO, Ru/Ba(10)-TiH2 had less hydrogen poisoning, which prevented N2 dissociation on Ru (Table 2).16 The low hydrogen poisoning effect allowed Ru/Ba(10)-TiH2 to exhibit higher ammonia synthesis activity than Ru–Cs/MgO. The N2 orders of Ru/Ba(10)-TiH2 (0.79) and Ru–Cs/MgO (0.89) were virtually coincident, reflecting that the rate-determining step for both catalysts was the unimolecular cleavage reaction of N2 for both catalysts. Moreover, the orders of Ru/Ba(10)-TiH2 (H2 order: 0.15, N2 order: 0.79) agreed well with the reported orders of Ru/BaTiO2.5H0.5 (H2 order: 0.2, N2 order: 0.7).17 Because Ru/BaTiO3, the reference catalyst for Ru/BaTiO2.5H0.5, exhibits stronger hydrogen poisoning due to the absence of hydride in the support perovskite (H2 order: −0.89, N2 order: 1.2),17 the agreement of the H2 and N2 orders between Ru/Ba(10)-TiH2 and Ru/BaTiO2.5H0.5 supports the formation of BaTiO2.5H0.5 in Ru/Ba(10)-TiH2. The NH3 order of Ru/Ba(10)-TiH2 (−0.36) was higher than that reported for Ru/BaTiO2.5H0.5 (−0.64), indicating that the ammonia decomposition reaction on Ru/Ba(10)-TiH2 was relatively inhibited. The inhibition may be attributed to the reaction pressure of this study (ambient pressure) being lower than that of reported Ru/BaTiO2.5H0.5 (5 MPa) because the ammonia decomposition reaction generally proceeded at a higher pressure.38
| Catalyst | Order | ||
|---|---|---|---|
| H2 | N2 | NH3 | |
| a Estimated from results of kinetic analysis shown in Fig. S8. | |||
| Ru/Ba(10)-TiH2 | 0.15 | 0.79 | −0.36 |
| Ru–Cs/MgO | −0.59 | 0.89 | 0.11 |
Conclusions
The selection of appropriate raw materials allowed for BaTiO2.5H0.5 perovskite oxyhydride nanoparticles to be formed on the TiH2 surface through the conventional wet impregnation method. BaTiO2.5H0.5 crystallized as ca. 100–200 nm sized nanoparticles on the surface of TiH2. Hydroxide in Ba(OH)2 involved BaTiO2.5H0.5 formation. Ru-loaded catalysts inhibited hydrogen poisoning and showed higher ammonia synthesis activity compared to that of the Ru–Cs/MgO benchmark catalyst. We believe that further investigation of oxyhydride prepared via the wet impregnation method accelerates the use of oxyhydride as ammonia synthesis catalysts.Author contributions
Yoshihiro Goto: conceptualization, investigation, writing – original draft, Masashi Kikugawa: investigation, Keisuke Kobayashi: data curation, Yuichi Manaka: validation, Tetsuya Nanba: conceptualization, project administration, Hideyuki Matsumoto: formal analysis Mitsuru Matsumoto: validation, Masakazu Aoki: supervision, project administration, Haruo Imagawa: supervision, writing-review &editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Mr Akira Takatsuki (National Institute of Advanced Industrial Science and Technology, Ibaraki, Japan) for his contribution in collecting the SEM and STEM-HAADF images.Notes and references
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636 CrossRef CAS.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304 RSC.
- P. H. Pfromm, J. Renewable Sustainable Energy, 2017, 9, 034702 CrossRef.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331 RSC.
- M. Ravi and J. W. Makepeace, Chem. Sci., 2022, 13, 890 RSC.
- S. Gambarotta and J. Scott, Angew. Chem., Int. Ed., 2004, 43, 5298 CrossRef CAS PubMed.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555 CrossRef CAS PubMed.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404 CrossRef CAS PubMed.
- Y. Niwa and K. Aika, Chem. Lett., 1996, 3, 3 CrossRef.
- Y. Ogura, K. Tsujimaru, K. Sato, S. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2018, 6, 17258 CrossRef CAS.
- Y. Ogura, K. Sato, S. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230 RSC.
- K. Sato, S. Miyahara, Y. Ogura, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2020, 8, 2726 CrossRef CAS.
- M. Kikugawa, Y. Goto, K. Kobayashi, T. Nanba, H. Matsumoto and H. Imagawa, J. Catal., 2022, 413, 934 CrossRef CAS.
- Y. Goto, M. Kikugawa, K. Kobayashi, T. Nanba, H. Matsumoto and H. Imagawa, Chem. Commun., 2022, 58, 3210 RSC.
- K. Aika, A. Ohya, A. Ozaki, Y. Inoue and I. Yasumori, J. Catal., 1985, 92, 305 CrossRef CAS.
- Y. Kadowaki and K. Aika, J. Catal., 1996, 161, 178 CrossRef CAS.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Loyer, K. Mitsuhara, K. Yamanaka, Y. Tamenori, C. Tassel, T. Yamamoto, T. Tanaka and H. Kageyama, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- M. Kitano, J. Kujirai, K. Ogasawara, S. Matsuishi, T. Tada, H. Abe, Y. Niwa and H. Hosono, J. Am. Chem. Soc., 2019, 141, 20344 CrossRef CAS PubMed.
- K. Ooya, J. Li, K. Fukui, S. Iimura, T. Nakao, K. Ogasawara, M. Sasase, H. Abe, Y. Niwa, M. Kitano and H. Hosono, Adv. Energy Mater., 2021, 11, 2003723 CrossRef CAS.
- H. Yamashita, T. Broux, Y. Kobayashi, F. Takeiri, H. Ubukata, T. Zhu, M. A. Hayward, K. Fujii, M. Yashima, K. Shitara, A. Kuwabara, T. Murakami and H. Kageyama, J. Am. Chem. Soc., 2018, 140, 11170 CrossRef CAS PubMed.
- K. Wang, Z. Wu and D. Jiang, Phys. Chem. Chem. Phys., 2022, 24, 1496 RSC.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- Y. Kobayashi, O. J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami, A. Kitada, M. Ohkura, S. Hosokawa, Z. Li, K. Hayashi, Y. Kusano, J. E. Kim, N. Tsuji, A. Fujiwara, Y. Matsushita, K. Yoshimura, K. Takegoshi, M. Inoue, M. Takano and H. Kageyama, Nat. Mater., 2012, 11, 507 CrossRef CAS PubMed.
- T. Uchimura, F. Takeiri, K. Okamoto, T. Saito, T. Kamiyama and G. Kobayashi, J. Mater. Chem. A, 2021, 9, 20371 RSC.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. Cryst. Mater., 2004, 229, 345 CrossRef.
- R. Oishi, M. Yonemura, Y. Nishimaki, S. Torii, A. Hoshikawa, T. Ishigaki, T. Morishima, K. Mori and T. Kamiyama, Nucl. Instrum. Methods Phys. Res. A: Accel. Spectrom. Detect. Assoc. Equip., 2009, 600, 94 CrossRef CAS.
- K. Aika, M. Kumasaka, T. Oma, O. Kato, H. Matsuda, N. Watanabe, K. Yamazaki, A. Ozaki and T. Onishi, Appl. Catal., 1986, 28, 57 CrossRef CAS.
- P. E. Kalita, S. V. Sinogeikin, K. E. Lipinska-Kalita, T. Hartmann, X. Ke, C. Chen and A. L. Cornelius, J. Appl. Phys., 2010, 108, 043511 CrossRef.
- R. H. Buttner and E. N. Maslen, Acta Crystallogr., 1992, B48, 764 CAS.
- I.-K. Jeong, S. Lee, S. Jeong, C. J. Won, N. Hur and A. Llobet, Phys. Rev. B, 2011, 84, 064125 CrossRef.
- P. R. Arya, P. Jha and A. K. Ganguli, J. Mater. Chem., 2003, 13, 415 RSC.
- V. F. Sears, Neutron News, 1992, 2, 26 CrossRef.
- B. Sreedhar, Ch. Satya Vani, D. Keerthi Devi, V. Sreeram and M. V. Basaveswara Rao, Am. J. Mater. Sci., 2012, 2, 105 CrossRef.
- K. B. Park, J. H. Choi, T. W. Na, J. W. Kang, K. S. Park and H. K. Park, Metals, 2019, 9, 1154 CrossRef CAS.
- K. Aika, H. Hori and A. Ozaki, J. Catal., 1972, 27, 424 CrossRef CAS.
- F. Rosowski, A. Hornung, O. Hinrichsen, D. Herein, M. Muhler and G. Ertl, Appl. Catal., A, 1997, 151, 443 CrossRef CAS.
- X. Zhang, Q. Ye, B. Xu and D. He, Catal. Lett., 2007, 117, 140 CrossRef CAS.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2013, 4, 3124 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01539d |
| This journal is © The Royal Society of Chemistry 2023 |