
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomerization and reaction process of N2O4(H2O)n
Yi Guo a,
Zhiyong Huanga,
Gan Tiana,
Wei Wub,
Jie Linc and
Xinlong Chang*a
a,
Zhiyong Huanga,
Gan Tiana,
Wei Wub,
Jie Linc and
Xinlong Chang*a
aSchool of Missile Engineering, Rocket Force University of Engineering, Xi'an, 710025, China. E-mail: xinlongch@vip.sina.com
bCenter of Engineering Quality Supervision, Logistics Support Department, Beijing, 100142, China
cSchool of Electronic Information and Communication, Huazhong University of Science and Technology, Wuhan, 430074, China
First published on 21st April 2023
Abstract
Liquid propellant N2O4 is prone to absorb H2O to form an N2O4(H2O)n system during long-term storage, ultimately generating HNO3, HNO2, and other substances capable of corroding the storage tank, which will adversely affect the performance of weapons and equipment. In this work, the reaction process of the N2O4(H2O)n system is simulated using density functional theory, and the potential energy surface, the geometric configurations of the molecules, the charge distribution, and the bond parameters of the reaction course at n = 0–3 are analyzed. The results show that the potential energy of the system is lower and the structure is more stable when the H2O in the N2O4(H2O)n system is distributed on the same side. When n = 1 or 2, the reaction profiles are similar, and the systems are partly ionic, although still mainly covalently bonded. When n = 3, the charge on the trans-ONONO2 group and the ON–ONO2 bond length change abruptly to −0.503 a.u. and 2.57 Å, respectively, at which point the system is dominated by ionic bonds. At n = 2, a proton-transfer phenomenon occurs in the reaction course, with partial reverse charge-transfer from NO3− to NO+, making the ON–ONO2 bond less susceptible to cleavage, further verifying that N2O4(H2O)n tends to afford the products directly in one step as H2O accumulates in the system.
1 Introduction
Liquid-fuelled strategic missiles mainly use N2O4 and unsym-dimethylhydrazine (UDMH) as propellants, and during long-term storage, N2O4 as an oxidant will absorb moisture from the surrounding environment to produce an N2O4(H2O)n system, ultimately generating HNO3, HNO2, and other corrosive substances, accelerating corrosion of the receptacle and potentially allowing propellants to leak.1 Traditional liquid-fuelled strategic missiles are constrained by this factor and can only be temporarily refilled with N2O4 before launch to shorten the contact time between N2O4 and the receptacle, but this process leads to long preparation cycles and slow operational response of such missiles, to the detriment of their operational performance.N2O4 is a volatile, reddish-brown transparent liquid at room temperature. It is a highly toxic chemical, which hampers experimental studies. N2O4, as a dimer of NO2, is an important intermediate (IM) in the hydrolysis of NO2 and plays an important role in the formation of acid rain.2 Due to its noxious nature, the use of quantum calculations to probe the evolution of the N2O4(H2O)n system is a trend of current research. Some studies have been carried out on the isomerization and self-ionization of N2O4 and the dimerization of NO2, but the understanding of the chemical mechanism is still incomplete.3–14 In particular, the effect of H2O on the N2O4(H2O)n system needs to be explored more deeply. Pimentel et al.5 found that N2O4 isomerization and NO2 dimerization in the presence of H2O can directly form ONO–NO2. Miller et al.4 simulated the interaction of H2O with ONO–NO2 at room temperature. For the N2O4(H2O)n system, the ionization rate is two orders of magnitude lower when n = 1 and 2 than when n > 2, indicating that, in the latter case, the reaction is transient and there is no IM. Medeiros et al.6 mentioned that local minima for the N2O4(H2O)n system can only be found when n is odd. Putikam et al.9 found a “roaming-like” transition state f formed in ONO–NO2 during the collision with H2O by the stretching of the N–N bond and the rotation of the –NO2 group. Finlayson-Pitts et al.10 proposed a mechanism for the hydrolysis of N2O4:
| 2NO2(g) ↔ N2O4(g) | (1) |
| N2O4(g) ↔ N2O4(surface) | (2) |
| N2O4(surface) → ONONO2(surface) | (3) |
| ONONO2(surface) → NO+NO3−(surface) | (4) |
| NO+NO3−(surface) + H2O → HONO(g) + HNO3(surface) | (5) |
N2O4 first forms sym-N2O4, which isomerizes to ONO–NO2 and then reacts with H2O to form HONO and HNO3. The above hydrolysis mechanism reveals the complex diversity of species in the N2O4(H2O)n system and the high number of isomers of N2O4.3 The precise structures of the resulting HONO and HNO3 after combining with H2O remain uncertain.
In the propellant storage environment, N2O4 is distinct from that in the atmospheric environment and exists mainly in liquid form. The reaction profile of the N2O4(H2O)n system after absorbing H2O from air and forming the N2O4(H2O)n system is unclear. Therefore, in this study, we have investigated the isomerization and reaction profile of the N2O4(H2O)n system based on density functional theory (DFT), focusing on the following issues:
(1) The effects on the N2O4(H2O)n system of n being an odd or even number.
(2) The route by which the N2O4(H2O)n system directly forms NO+ and NO3−.
(3) The variations in the charges on the groups during the reaction course of the N2O4(H2O)n system when n = 0–3.
2 Calculation method
Based on the conclusions of Pimentel et al.,5 the DFT/B3LYP method was used to optimize N2O4 single-molecule isomerization at the 6-311++G (3df, 2p) basis group level, considering all species in the reaction of the N2O4(H2O)n system. The intrinsic reaction coordinates (IRC) were established through standard 6-311++G (3df, 2p) basis group calculations to confirm the connection of each transition state (TS) to the specified intermediate (IM). The resulting reactant, product, and IM frequencies were positive, with one and only one imaginary frequency for the TS. Single-point energies were re-determined at the B2PLYP/Def2-TZVP basis set level.15 To take into account the effect of the solvent medium, the self-consistent reaction field (SCRF) method and the implicit solvation model of the SMD model were used at n = 3, in conjunction with the conclusions of Miller4 and Putikam.9 All calculations were performed using the Gaussian 16 software package.163 Results and discussion
3.1 Isomerization of N2O4 (n = 0)
Experimental studies have shown that N2O4 has three main conformations; in addition to sym-N2O4, there are two asymmetric N2O4 isomers, namely trans-N2O4 and cis-N2O4.17–19 Zhu et al.8 predicted the possible geometric conformations of N2O4 isomers based on ab initio molecular dynamics, and identified the most stable isomers as sym-N2O4 and trans-N2O4. Meanwhile, the spontaneous dissociation of N2O4 to NO2 is less likely in the liquid propellant storage environment. Therefore, the work in this section mainly considers the mutual isomerization between sym-N2O4, cis-N2O4, and trans-N2O4. The potential energy surface (PES) and the geometric configurations and bond parameters of the conformers during the isomerization are shown in Fig. 1. The optimized sym-N2O4 has an N–N bond length of about 1.80 Å and N–O bond lengths of about 1.18 Å, in good agreement with reported values determined experimentally by electron diffraction analysis at 252 K.20 When sym-N2O4 isomerizes to cis-N2O4, the N–N bond length in TS1 is 2.68 Å and the dihedral angle O4–N2–O6–N1 is 177°. The associated potential energy barrier is 166.91 kJ mol−1, which further completes the isomerization process of NO2 proposed by Liu et al.3 In contrast, the “roaming-like” TS structure proposed by Putikam et al.9 is more relaxed, with an N–N bond length of 3.70 Å, a dihedral angle O4–N2–O6–N1 of 76°, and a potential energy barrier of 34.31 kJ mol−1, significantly different from the values calculated here. For the isomerization of sym-N2O4 to trans-N2O4 via TS3, the two –NO2 groups of the reactants and products are coplanar, but the structure of TS3 is vertical, the N–N bond length is only 2.08 Å, and the associated potential energy barrier is about 175.94 kJ mol−1. In contrast, in the TS structure reported in the literature, the N–N bond length reaches 3.57 Å, the two –NO2 groups are almost dissociated, and the electron density between the interacting groups is reduced, so the TS potential energy barrier (53.56 kJ mol−1) is much lower than that calculated here.9 The course of the isomerization of cis-N2O4 to trans-N2O4 is similar to that calculated in the literature.21 | ||
| Fig. 1 Potential energy surface of N2O4 isomerization and the geometries and bond parameters of the conformers (key length unit: Å, key angle unit: °). | ||
3.2 Isomerization and reaction processes of N2O4(H2O) and N2O4(H2O)2 (n = 1, 2)
From Section 3.1, it is clear that the relative energy of trans-N2O4 is lower than that of cis-N2O4, indicating that the former is more stable in the course of the isomerization. Hence, only trans-N2O4 is considered in the following calculations. Fig. 2 shows the PES and the geometric configurations of the molecules with bond parameters for the isomerization and reaction course of N2O4(H2O). When N2O4 absorbs H2O, a complex such as IM1 is produced due to van der Waals forces. At this point, two pathways are available for IM1 for the next reaction step. | ||
| Fig. 2 Potential energy surface of N2O4(H2O) isomerization and reaction process, and the geometries and bond parameters of molecules (key length unit: Å, key angle unit: °). | ||
(1) IM1 may be directly converted to HNO3 + trans-HONO via TS4, as shown in Fig. 2. TS4 is a compact five-membered-ring structure with an associated potential barrier of 138.62 kJ mol−1. This value is similar to that reported by Chou et al.22 Upon further conversion of TS4, H8 in H2O gradually detaches and combines with one of the –NO2 groups to produce trans-HONO. The remaining –OH of H2O combines with the other –NO2 group to produce HNO3.
(2) IM1 may be converted to TS5 to generate trans-ONONO2. In this case, the structure of TS5 is more similar to TS3. In the course of isomerization of sym-N2O4 to trans-N2O4, one of the –NO2 groups rotates about the N–N bond, thus causing a shift of H2O, and the distance between N1 and O7 reaches 4.02 Å. When structurally stable trans-ONONO2 is generated, the distance between N1 and O7 is 2.85 Å. As the reaction continues, trans-ONONO2 produces HNO3 + trans-HONO and HNO3 + cis-HONO through two six-membered-ring transition states, TS6 and TS7, respectively. In TS6, H8 of H2O is transferred to O5 in –NO3, eventually forming HNO3, while the remaining –OH of H2O combines with –NO. The structure of TS7 is similar to that of TS6, except that H2O is rotated by a certain angle. It follows a similar reaction course to finally form HNO3 + cis-HONO, the reaction potential of which is 2.41 kJ mol−1 higher than that of HNO3 + trans-HONO.
In Fig. 2, it can be seen that the energy of trans-HONO is lower than that of cis-HONO, indicating that the former is more stable and the reaction pathway is more likely to occur. Therefore, the reaction course of cis-HONO is not considered in the following calculations. When the content of H2O is further increased, N2O4 combines with more H2O in the vicinity to produce N2O4(H2O)2. The PES and geometric configurations of the molecules with bond parameters for the isomerization and reaction course are shown in Fig. 3.
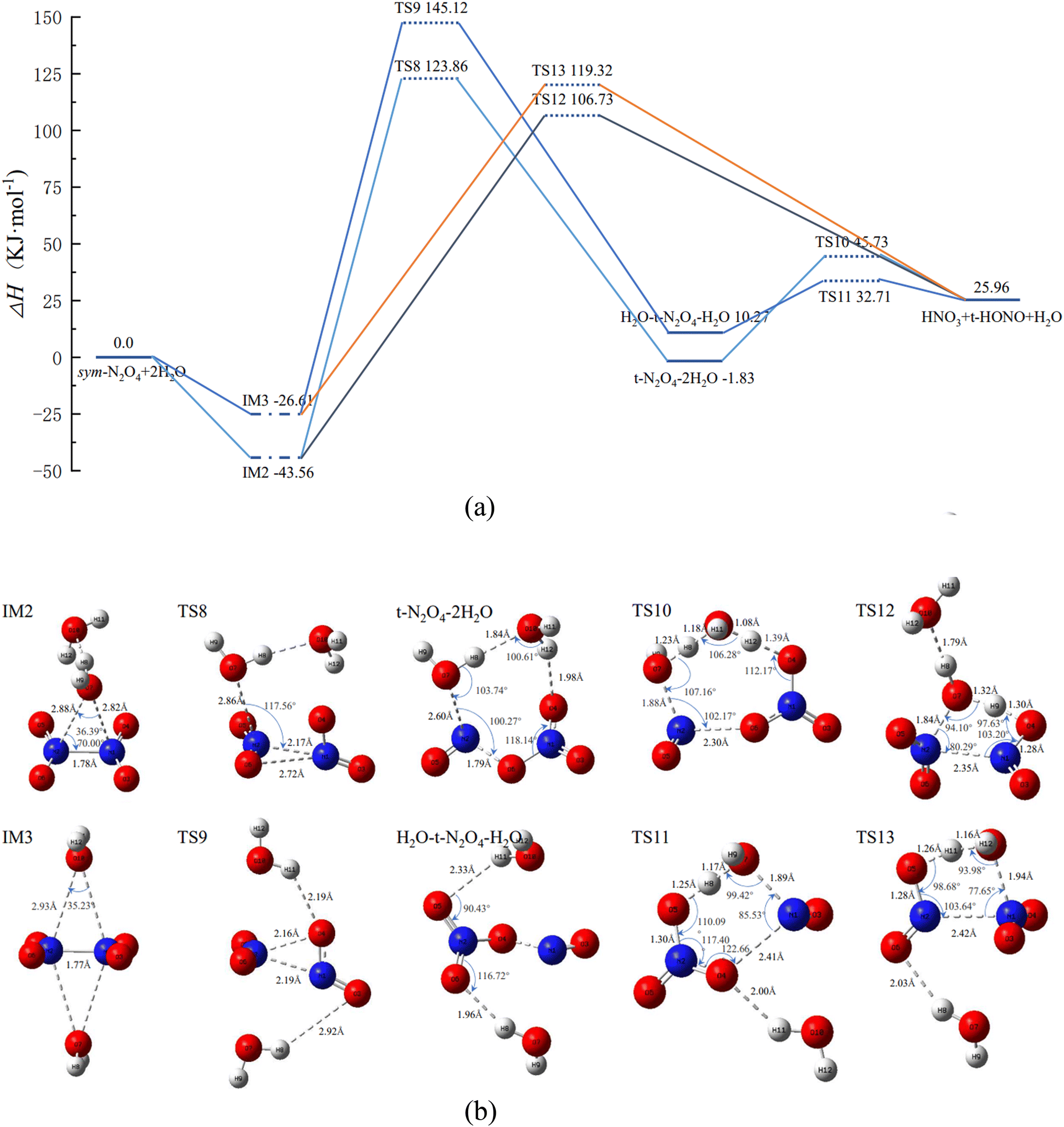 | ||
| Fig. 3 (a) PES of N2O4(H2O)2 isomerization and reaction course. (b) Molecular geometrical configurations and bond parameters for N2O4–(H2O)2 isomerization and reaction course (bond lengths in Å, bond angles in °). | ||
Similar to IM1 in Fig. 2, when N2O4 is combined with 2 H2O, two N2O4(H2O)2 complexes, IM2 and IM3, are produced, as shown in Fig. 3(b), in which the 2 H2O molecules are located on the same or opposite sides of N2O4, respectively. Therefore, its reaction course will follow two main pathways.
② IM2 first passes through TS8 to form trans-N2O4–2H2O. Both H2O molecules are involved in the reaction course, and the product trans-N2O4–2H2O has an eight-membered-ring structure. Upon further reaction of trans-N2O4–2H2O, the final product is formed through TS10. During this process, both H2O molecules engage in proton-transfer phenomena, i.e., O7 and H10 of one H2O molecule combine with NO to form trans-HONO, while H12 of the other H2O molecule combines with NO3− to form HNO3. The remaining H8 and O10 of the two H2O molecules recombine with H11 to form a new H2O molecule.
② IM3 can also go through TS9 to first form H2O–trans-N2O4–H2O, and then O7 of H2O, which is closer to N1, binds to N1 to form the six-membered-ring structure TS11. The latter is lower in energy and more stable than the eight-membered-ring structure of TS10. Of the H2O (O7) molecule involved in the reaction, H8 combines with NO3 to form HNO3, while O7 and H9 combine with NO to form trans-HONO.
3.3 Isomerization and reaction profile of N2O4(H2O)3 (n = 3)
In N2O4(H2O)3, there are two possible distributions of H2O molecules, i.e., all three H2O molecules residing on one side of N2O4, or two H2O molecules on one side and the third on the other side. Our calculations reveal that N2O4–3H2O is lower in energy than 2H2O–N2O4–H2O. Combined with the results in Section 3.2, 2H2O–N2O4–H2O can be studied with reference to the isomerization and reaction profiles of IM2 and IM3, and so this section focuses on the isomerization and reaction profiles of N2O4–3H2O, for which the PES and the geometric configurations and bond parameters of the molecules are shown in Fig. 4. | ||
| Fig. 4 Potential energy surface of N2O4(H2O)3 isomerization and reaction process, and the geometries and bond parameters of molecules (key length unit: Å, key angle unit: °). | ||
(1) IM4 may directly generate HNO3 + trans-HONO + 2H2O via TS16. In IM4, the three H2O molecules are connected sequentially to form a ring structure with one of the –NO2 groups, while O10 of the H2O molecule located above N2O4 forms a stable triangular structure with both N atoms. The two N–O bonds are almost equal in length. Although only one H2O molecule is involved in the reaction, H2O (O10), in TS16 it forms both an eight-membered-ring structure with the remaining two H2O molecules and a five-membered-ring structure with N2O4.
(2) IM4 first forms trans-N2O4–3H2O via TS14, and the three H2O molecules also break from the ring structure into a single chain and then rejoin with O3 to form a ring. The trans-N2O4–3H2O then undergoes proton transfer, with H2O (O7) providing –OH and H2O (O10) providing H to form IM5. IM5 already has the structure of the products, but the molecules are still connected to each other and only form the products after bond-breaking.
N2O4(H2O)n has one and only one stable structure during the isomerization and reaction course when n < 2. When n ≥ 2, the distribution of H2O affects the stable structure of N2O4(H2O)n, resulting in different energies. If H2O is distributed on the same side of N2O4, with fewer interconnected chemical bonds and shorter bond lengths, the energy is lower and the structure is more stable, as in IM2 and IM3 in Fig. 3(b). Irrespective of whether n is odd or even, multiple pathways are available for N2O4(H2O)n to undergo isomerization reactions. To verify the speculation of Miller et al.,4 the potential energy barriers associated with different pathways of N2O4(H2O)n isomerization for different values of n and pertaining to the most stable conformation are plotted in Fig. 5.
 | ||
| Fig. 5 Potential energy barriers associated with different reaction paths for N2O4(H2O)n isomerization reactions. | ||
The energy of 166.01 kJ mol−1 for n = 0, i.e., the direct ionization of N2O4 to form NO + NO3, and combining TS4, TS12, and TS16 as the reaction potential for the direct reaction of N2O4(H2O)n to form the product pathway; the first step of isomerization process of N2O4(H2O)n through multiple steps to generate products as the reaction potential of its multi-step reaction pathway, i.e., N2O4(H2O)n → trans-N2O4–(H2O)n: TS3, TS5, TS8, and TS14. From Fig. 5, it can be seen that the potential energy barrier of N2O4(H2O)n gradually decreases as n is increased and more H2O is involved in the reaction. When n = 0–2, the differences in the potential energy barriers associated with the direct and multi-step reaction paths are small, such that N2O4(H2O)n may follow both reaction paths at the same time. When n = 3, however, the difference in potential energy barriers associated with the two paths reaches 28.4 kJ mol−1, indicating that N2O4(H2O)3 will preferentially pass through TS16 to afford the products directly. Combined with the finding in Fig. 4 that TS15 is lower in energy than the reaction potential of the products when N2O4(H2O)3 undergoes a multi-step reaction path, it is clear that the multi-step reaction path of N2O4–(H2O)3 is difficult to reach. On the basis of thermodynamics, Mulliken charge analysis was performed on the intermediate product trans-ONONO2–(H2O)n, and the results in terms of charge on the trans-ONONO2 group and ON–ONO2 bond length are shown in Table 1.
| n | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| Electric charge/a.u. | 0 | −0.019 | −0.025 | −0.503 |
| Key length/Å | 1.62 | 1.76 | 1.79 | 2.57 |
As can be seen from Table 1, the charge on the trans-ONONO2 group gradually becomes more negative as the H2O content in trans-ONONO2–(H2O)n is increased, indicating increased polarity. Concomitantly, the bond length of ON–ONO2 also increases, and –ON gradually dissociates and from the trans-ONONO2 group. It can be seen that the differences in charge on the trans-ONONO2 group and ON–ONO2 bond length are not large when n = 1 and 2. However, when n = 3, the charge on the trans-ONONO2 group and the ON–ONO2 bond length change abruptly, reaching −0.503 a.u. and 2.57 Å, respectively. These results support the conjecture of Miller et al.;4 when n = 1 or 2, trans-ONONO2-(H2O)n is partly ionically bonded, but still mainly covalently bonded. When n = 3, however, ionic bonding becomes dominant. Meanwhile, combined with the reaction rate results of Miller et al.4 and analyzed by the potential energy surfaces in Fig. 2–4, the reaction rate constant increases by two orders of magnitude when n = 3 compared with n < 3. In Section 3.2.1, trans-N2O4–2H2O was seen to undergo a proton-transfer phenomenon as the reaction proceeds. Luo et al.14 confirmed that the formation of hydrogen bonds and a polar environment are prerequisites for intermolecular proton transfer. Proton-transfer due to trans-ONONO2 dominates in the production of –OH, which is also supported by experimental results,23 leading to a shorter lifetime of trans-ONONO2 with the increase of H2O. All charge-transfer steps result in partial reverse charge-transfer from NO3− to NO+, making the ON–ONO2 bond less susceptible to cleavage, further verifying that when n = 3, the N2O4(H2O)3 system is dominated by ionic bonds between molecules. Due to the short-lived existence of trans-ONONO2 and the difficulty of breaking the ON–ONO2 bond, the reaction can be considered as being completed instantaneously.24 That is to say, IM4 mainly passes through TS16 to directly generate the final products HNO3 + trans-HONO + 2H2O.
4 Conclusions
(1) N2O4(H2O)n system structure reaction course of the local minimum is not related to the parity of the n value. When H2O molecules are distributed on the same side, the potential energy of the system is lower, and the structure is more stable. When n = 1 or 2, the two reaction pathways are similar in energy. The systems are partly ionically bonded, but mainly still covalently bonded.(2) When n = 3, the charge on the trans-ONONO2 group and the ON–ONO2 bond length change abruptly, reaching −0.503 a.u. and 2.57 Å, respectively, and the system is then dominated by ionic bonds.
(3) When n = 2, the reaction course shows the phenomenon of proton-transfer, with partial reverse charge-transfer from NO3− to NO+, making cleavage of the ON–ONO2 bond more difficult. Combining the change of potential energy surface, the charge on the ONONO2 group, and the molecular bond parameters during the course of the reaction, it is again verified that N2O4(H2O)n tends to afford the products directly in one step with increasing H2O content in the system.
Author contributions
Y. Guo: conceptualization, investigation, methodology, writing – original draft. Z. Y. Huang: data curation, formal analysis. G. Tian: project administration, validation. W. Wu: resources, supervision. J. Lin: software, validation. X. L. Chang: funding acquisition, writing – review & editing.Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work.Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 52272446) and Natural Science Foundation of Shaanxi Province (No. 2021JM-250).References
- Y. Guo, X. L. Chang, G. Tian, D. J. Liu and C. Pang, Rare Met. Mater. Eng., 2022, 51, 3459–3465 Search PubMed.
- D. J. Liu, G. Tian, Z. W. Yang, G. F. Jin, W. Zhang, Y. Wang and H. L. Wei, Chin. J. Aeronaut., 2022, 36, 304–315 CrossRef.
- W. G. Liu and A. G. William, J. Am. Chem. Soc., 2012, 134, 12970–12978 CrossRef CAS PubMed.
- Y. Miller, B. J. Finlayson-Pitts and R. B. Gerber, J. Am. Chem. Soc., 2009, 131, 12180–12185 CrossRef CAS PubMed.
- A. S. Pimentel, C. A. L. Francisco and A. B. F. da Silva, J. Phys. Chem. A, 2007, 111, 2913–2920 CrossRef CAS PubMed.
- D. J. Medeiros and A. S. Pimentel, J. Phys. Chem. A, 2011, 115, 6357–6365 CrossRef CAS PubMed.
- I. I. Zakharov, Theor. Exp. Chem., 2012, 48, 233–239 CrossRef CAS.
- R. S. Zhu, K. Y. Lai and M. C. Lin, J. Phys. Chem. A, 2012, 116, 4466–4472 CrossRef CAS PubMed.
- R. Putikam and M. C. Lin, Int. J. Quantum Chem., 2017, 118, 25560–25569 CrossRef.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, Phys. Chem. Chem. Phys., 2003, 5, 223 RSC.
- T. C. Marilia, J. M. Anglada, J. S. Francisco and M. F. Ruiz-Lopez, J. Am. Chem. Soc., 2020, 142, 20937–20941 CrossRef PubMed.
- F. Menezes and G. M. Popowicz, Phys. Chem., 2022, 23, 395 Search PubMed.
- Y. Miller and R. B. Gerber, Phys. Chem. Chem. Phys., 2008, 10, 1091–1093 RSC.
- G. F. Luo and X. B. Chen, J. Phys. Chem. Lett., 2012, 3, 1147–1153 CrossRef CAS PubMed.
- J. S. Zhao, Z. Y. Huang, G. F. Jin, M. N. Gao and H. X. Zhu, Chin. J. Energ. Mater., 2021, 29, 1125–1131 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Givan and A. Loewenschuss, J. Chem. Phys., 1989, 90, 6135 CrossRef CAS.
- A. Givan and A. Loewenschuss, J. Chem. Phys., 1989, 91, 5126 CrossRef CAS.
- A. Givan and A. Loewenschuss, J. Chem. Phys., 1991, 94, 7562 CrossRef CAS.
- B. W. McClelland, G. Gundersen and K. Hedlberg, J. Chem. Phys., 1972, 56, 4541 CrossRef CAS.
- K. Y. Lai, MS thesis, National Chiao Tung University, Taiwan, 2009.
- A. Chou, R. Zhi and F. M. Tao, J. Phys. Chem. A, 1999, 103, 7848 CrossRef CAS.
- T. Kinugawa, S. Enami, A. Yabushita, M. Kawasaki, M. Hoffmann and A. Colussi, Phys. Chem. Chem. Phys., 2011, 13, 5144–5149 RSC.
- A. Yabushita, S. Enami, Y. Sakamoto and M. Kawasaki, J. Phys. Chem. A, 2009, 113, 4844–4848 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |