
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-entropy oxychloride increasing the stability of Li–sulfur batteries†
Markéta Zukalová *a,
Martin Fabiánb,
Olena Porodkob,
Monika Vinarčíkováa,
Barbora Pitňa Láskováa and
Ladislav Kavana
*a,
Martin Fabiánb,
Olena Porodkob,
Monika Vinarčíkováa,
Barbora Pitňa Láskováa and
Ladislav Kavana
aJ. Heyrovský Institute of Physical Chemistry, Czech Acad. Sci., Dolejškova 3, CZ-18223, Prague 8, Czech Republic. E-mail: marketa.zukalova@jh-inst.cas.cz
bInstitute of Geotechnics, Slovak Academy of Sciences, Watsonova 45, 040 01, Košice, Slovak Republic
First published on 7th June 2023
Abstract
A novel lithiated high-entropy oxychloride Li0.5(Zn0.25Mg0.25Co0.25Cu0.25)0.5Fe2O3.5Cl0.5 (LiHEOFeCl) with spinel structure belonging to the cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group is synthesized by a mechanochemical–thermal route. Cyclic voltammetry measurement of the pristine LiHEOFeCl sample confirms its excellent electrochemical stability and the initial charge capacity of 648 mA h g−1. The reduction of LiHEOFeCl starts at ca. 1.5 V vs. Li+/Li, which is outside the electrochemical window of the Li–S batteries (1.7/2.9 V). The addition of the LiHEOFeCl material to the composite of carbon with sulfur results in improved long-term electrochemical cycling stability and increased charge capacity of this cathode material in Li–S batteries. The carbon/LiHEOFeCl/sulfur cathode provides a charge capacity of 530 mA h g−1 after 100 galvanostatic cycles, which represents ca. 33% increase as compared to the charge capacity of the blank carbon/sulfur composite cathode after 100 cycles. This considerable effect of the LiHEOFeCl material is assigned to its excellent structural and electrochemical stability within the potential window of 1.7 V/2.9 V vs. Li+/Li. In this potential region, our LiHEOFeCl has no inherent electrochemical activity. Hence, it acts solely as an electrocatalyst accelerating the redox reactions of polysulfides. This can be beneficial for the performance of Li–S batteries, as evidenced by reference experiments with TiO2 (P90).
m space group is synthesized by a mechanochemical–thermal route. Cyclic voltammetry measurement of the pristine LiHEOFeCl sample confirms its excellent electrochemical stability and the initial charge capacity of 648 mA h g−1. The reduction of LiHEOFeCl starts at ca. 1.5 V vs. Li+/Li, which is outside the electrochemical window of the Li–S batteries (1.7/2.9 V). The addition of the LiHEOFeCl material to the composite of carbon with sulfur results in improved long-term electrochemical cycling stability and increased charge capacity of this cathode material in Li–S batteries. The carbon/LiHEOFeCl/sulfur cathode provides a charge capacity of 530 mA h g−1 after 100 galvanostatic cycles, which represents ca. 33% increase as compared to the charge capacity of the blank carbon/sulfur composite cathode after 100 cycles. This considerable effect of the LiHEOFeCl material is assigned to its excellent structural and electrochemical stability within the potential window of 1.7 V/2.9 V vs. Li+/Li. In this potential region, our LiHEOFeCl has no inherent electrochemical activity. Hence, it acts solely as an electrocatalyst accelerating the redox reactions of polysulfides. This can be beneficial for the performance of Li–S batteries, as evidenced by reference experiments with TiO2 (P90).
1 Introduction
Global climate changes and the rapid increase of fossil fuel prices accelerate the orientation of the world power and automotive industry towards renewable resources of energy. Regarding the fact that green electricity production cannot be synchronized with peak electricity consumption, the availability of balancing and stabilizing large-capacity battery systems represents the key issue of this transformation. Li–sulfur batteries with a theoretical capacity of 1675 mA h g−1 exceeding many times the limits of classical Li-ion batteries offer a cheap and environmentally friendly solution.1 However, despite the decades-lasting effort of the researchers, there remain issues hindering their broad commercialization. The low conductivity of sulfur is compensated by the addition of conductive carbon with a large surface area and appropriate morphology2–10 mediating the reduction of sulfur on the cathode. The main problems are related to the dissolution of lithium polysulfides (PS) in the electrolyte and their parasitic reactions with electrolyte solvents and the Li anode. PS diffusion to the anode compartment of the Li–S battery causes severe redox shuttling between the cathode and Li anode. It results in a low coulombic efficiency for charging/discharging and a fast self-discharge during storage. Hence, effective PS immobilization in the cathode compartment of the battery is essential for the optimum performance and cycling stability of the system. PS can be physically trapped on the surface of carbon or immobilized in the pores of carbonaceous additive. Other alternative methods of PS immobilization include the addition of a barrier layer between the cathode and the separator,11,12 or an appropriate separator modification.13Regarding the fact that PS are bound to the carbon surface by weak van der Waals interactions, the incorporation of oxidic materials providing a strong polar surface for efficient PS trapping further helps to suppress the shuttle effect and subsequently the decay rate per cycle.14,15 TiO2 (ref. 16–18) Magnéli phases,19 and ternary oxides20,21 belong to the most frequently used inorganic additives improving the electrochemical performance of Li–sulfur batteries. High-entropy materials containing five or more elements in near-equiatomic ratios represent a relatively new class of materials with unique electrochemical22 and magnetic23 properties.
High-entropy oxides and their lithiated oxychlorides or oxyfluorides showed promising results in the fields of batteries, supercapacitors, and electrocatalysis.24 Wang et al.25 introduced a new class of high-entropy materials, i.e. rock-salt type lithiated oxyfluorides (Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OFx) as a promising candidate for energy storage applications based on multi-anionic and cationic compounds. Recently, we studied the mechanochemical synthesis of the novel spinel-type high-entropy oxide (aluminate) and its lithiated derivatives-oxychloride and oxyfluoride.26 Although these materials have not yet been optimized from the point of view of their electrochemical properties, they can hardly contribute by their inherent electrochemical activity to the redox processes taking place in Li–sulfur batteries analogously to TiO2,27 because their redox activity occurs at potentials smaller than ca. 1.5 V vs. Li+/Li, which is outside the usual electrochemical window of Li–S batteries.22,26,28 Instead, they can, presumably, serve as efficient PS adsorbents due to the presence of oxygen vacancies in their structure. Our recent studies confirmed the beneficial effect of various inorganic additives on the capacity and cycling stability of the composite cathode in Li–sulfur batteries.27,29,30 This paper aims to broaden the portfolio of promising inorganic additives for the composite cathode in Li–sulfur batteries by lithiated high-entropy oxychloride additive Li0.5(Zn0.25Mg0.25Co0.25Cu0.25)0.5Fe2O3.5Cl0.5 (LiHEOFeCl) and to evaluate its effect on the electrochemical behavior of the system. To the best of our knowledge, there are only few papers reporting the use of high-entropy oxides in Li–sulfur batteries,31,32 but none of them evaluated the inherent electrochemical activity of high-entropy material within the potential window of the Li–sulfur battery. In addition to the adsorption of PS, certain HEOs can also act as electrocatalysts accelerating the redox reactions of PS in Li–S batteries.31,32
2 Experimental
2.1 Sample preparation
The Li0.5(Zn0.25Mg0.25Co0.25Cu0.25)0.5Fe2O3.5Cl0.5 (LiHEOFeCl) powdered samples were prepared by high-energy ball milling of a stoichiometric ratio of precursors: zinc oxide (ZnO, 99.99% purity; Aldrich), copper oxide (CuO, 99.99% purity, Acros Organics), magnesium oxide (MgO, 99.99% purity, Acros Organics), iron oxide (Fe2O3, 99.9% purity; Alfa Aesar) and lithium chloride (LiCl, 98%, Acros Organics). The cobalt oxide CoO precursor was prepared by thermal decomposition of cobalt hydroxide (Co(OH)2, 95% purity, Acros Organics) at 180 °C in vacuum. The precursors were milled at 600 rpm for 390 minutes in a planetary ball mill Pulverisette 7 premium line (Fritsch). A milling chamber (80 cm3 in volume) and balls (10 mm in diameter) made of tungsten carbide (WC) were used. The ball-to-powder weight ratio was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Milling experiments were carried out in Ar atmosphere. After milling, mixtures were calcined at 600 °C in Ar atmosphere for 2 h.
:1. Milling experiments were carried out in Ar atmosphere. After milling, mixtures were calcined at 600 °C in Ar atmosphere for 2 h.
2.2 Electrode preparation
The carbonaceous additive (P_carbon, Penta) was mixed in an agate mortar with sulfur (Aldrich) in a mass ratio of 4:1 (S:P_carbon), mortared, and then treated at 155 °C for 15 hours under Ar atmosphere in a Teflon container. Alternatively, 20 wt% of P_carbon was replaced with the identical mass of LiHEOFeCl, or, alternatively, with P90 (TiO2 from Evonik/Degussa), and the mixture was mortared with sulfur and treated as described above. The product was again mortared, and mixed with a conductive carbon black C65 (Timcal), and a 2% aqueous solution of carboxymethylcellulose (CMC from Sigma). The mass ratio of the P_carbon/sulfur composite to C65 and CMC was 7:2:1 (56 wt% of sulfur in the composite cathode). The mixture was then diluted with deionized water to a consistency of viscous paste and coated by doctor-blading on Al foil. After drying in air at ambient temperature and subsequently at 50 °C in a vacuum overnight, the coated Al foil was cut into disc electrodes of 15 mm in diameter. The areal sulfur loading was adjusted to 1–2 mg cm−2. The electrodes were stored in a glove box with Ar atmosphere.
The electrodes for cyclic voltammetry of pristine LiHEOFeCl were prepared analogously, except for the addition of sulfur. LiHEOFeCl material was mixed with C65 and CMC and treated as described above. The mass ratio of the components was 7:2:1.
2.3 Coin cell assembly
Coin cells were assembled with a cathode described above, a Li-foil anode (14 mm in diameter), a glass microfiber separator (Whatman), and 20 μl of electrolyte solution. The electrolyte/sulfur ratio was 6–12 μl mg−1, which is slightly lower as compared to ref. 32. This separator exhibited an improved performance and higher cycling stability in coin cells as compared to the commonly used polypropylene Celgard separator. The electrolyte used for Li–sulfur cells consisted of 1.0 M lithium bis-(trifluoromethanesulfonyl) imide (LiTFSI, Aldrich) dissolved in a mixture of 1,3-dioxolane and 1,2-dimethoxyethane (1:1 by volume) with 1.0 wt% LiNO3 (Aldrich) as an electrolyte additive. LiTFSI was dried in a vacuum at 130 °C overnight, LiNO3 at 50 °C in a vacuum overnight, and the mixture of organic solvents was dried over a molecular sieve 4 Å (Aldrich). The electrolyte used for cyclic voltammetry of pristine LiHEOFeCl consisted of 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1:1 by volume). Both electrolyte solutions contained 8–12 ppm H2O as determined by Karl Fischer coulometric titration (Mettler Toledo). Electrolytes and solvents were of standard quality (p. a. or electrochemical grade) purchased from Aldrich or Merck.
2.4 Separator modification
A glass microfiber filter (Whatman) was impregnated with P90 (TiO2 from Evonik/Degussa) from ethanolic suspension by dip-coating. The disc filters were dried at ambient temperature in air and then in a vacuum at 55 °C overnight. The details are described elsewhere.332.5 Methods
X-ray powder diffraction (XRPD) patterns were collected using a D8 Advance diffractometer (Bruker) operating with Cu Kα radiation in the Bragg–Brentano configuration. The generator was set up at 40 kV and 40 mA and data were recorded in the range of 20 to 70° 2θ. The ICSD database34 was applied for the determination of phases. XRPD data were quantitatively analyzed by the Le Bail method in the space group Fdm using the FullProf program35 with the application of regular pseudo-Voigt function parameters. Microstructure and morphology of LiHEOFeCl powder were investigated by combined field emission (scanning) transmission microscope (S)TEM (JEOL JEM-2100F), coupled with the energy-dispersive X-ray spectroscopy (EDS) detector (Oxford Instruments, UK). Before the investigations, the sample was crushed in a mortar, dispersed in ethanol, and fixed on a copper-supported carbon grid. ImageJ software36 was used to evaluate the particle size distribution from the (S)TEM micrographs. The atomic absorption spectra were measured using Varian 240RS/240Z, Australia. The powdered sample was dissolved in aqua regia, filtered, and diluted. Adsorption measurements of nitrogen were carried out with the ASAP 2020 apparatus (Micromeritics) at 77 K. All samples were degassed before analysis at 250 °C in vacuum. The surface area was determined by the BET (Brunauer, Emmett, Teller) equation. Electrochemical measurements were carried out with Autolab 302N apparatus (Metrohm) controlled by Nova and Nova Battery SW in 2032 coin-type test cells with Li-metal anode and glass microfiber separator (Whatman) (as received or modified with TiO2). Galvanostatic chronopotentiometry at 0.1C was measured in the 2032 coin-type test cells by Neware Battery Testing System controlled by BTS 7.6 SW. All Li–sulfur battery electrochemical tests were carried out in the potential window from 1.7 V to 2.9 V vs. Li+/Li at the scan rate of 0.1 mV s−1. Cyclic voltammetry of the LiHEOFeCl material was measured in the potential window of 0.65 V/3.0 V vs. Li+/Li with a scan rate of 50, 20, 10, 5, 2, 1, 0.5, 0.2, 0.1 mV s−1. All potentials in this study are referred to the Li+/Li electrode. Electrochemical impedance spectra (EIS) were measured on the 2032 cells by the frequency response analyzer (FRA) interfaced to Autolab 302N. The measurement was carried out in the frequency range from 100 kHz to 0.1 Hz (modulation amplitude 10 mV) at a fixed applied potential of 2.4 V vs. Li+/Li, which is near the open-circuit potential of the cell. Before each EIS measurement, the potential was equilibrated at 2.4 V for 1 min.
3 Results and discussion
3.1 Characterization
As shown in Fig. 1, the La Beil refinement of the synthesized LiHEOFeCl sample confirmed the spinel structure belonging to the cubic Fdm space group. The lattice constant was determined to be 8.3690(2) Å. Partly broad reflections of the particular peaks reveal the nanostructured character of the sample. The average crystallite size, d, estimated by the Scherrer equation37 from the (220), (311), (400), and (440) reflections was found to be ∼65 nm. The value for average crystallite size was higher for the observations performed by (S)TEM electron microscopy, i.e. ∼150 nm (Fig. 2). This is obvious bearing in mind the tendency of mechanochemically prepared particles to agglomerate. Also, as it is distinctive for mechanochemically prepared powders,38 the crystallites are characterized by disordered structure on their surface/interface regions. As is also shown in Fig. 2, the EDS mapping confirms a homogeneous distribution of the particular elements within the prepared sample. The concentration of metals in the LiHEOFeCl sample was determined by atomic absorption spectrometry. The elemental composition of selected metal ions was found to be (in at%): Fe (47.15), Zn (2.62), Mg (2.38), Co (2.63), and Cu (2.35). The corresponding atomic ratios equal: Zn/Fe (0.056), Mg/Fe (0.050), Co/Fe (0.056), Cu/Fe (0.050) which agrees reasonably well with the expected ratio (0.0625) from the stoichiometric formula. The BET surface area of LiHEOFeCl determined by nitrogen adsorption measurements (Fig. S1†) was 4 m2 g−1.
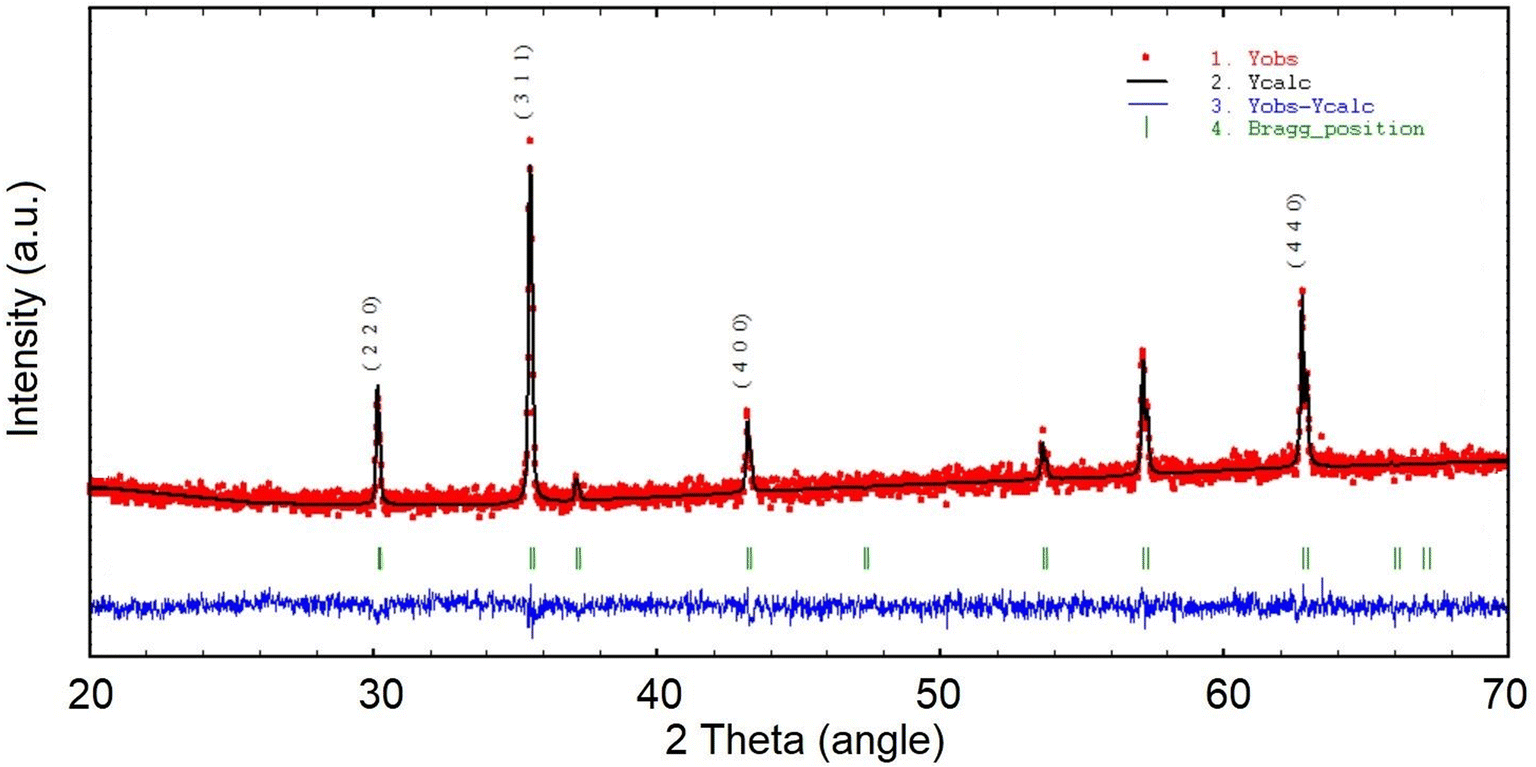 | ||
| Fig. 1 The refined XRDP pattern of the LiHEOFeCl sample prepared by the mechanochemical–thermal route. The Miller indices denote the respective crystal planes. The abbreviations Yobs., Ycalc., Yobs.–Ycalc. stand for the measured data, the calculated fit, and the difference between measured data and calculated fit, respectively. | ||
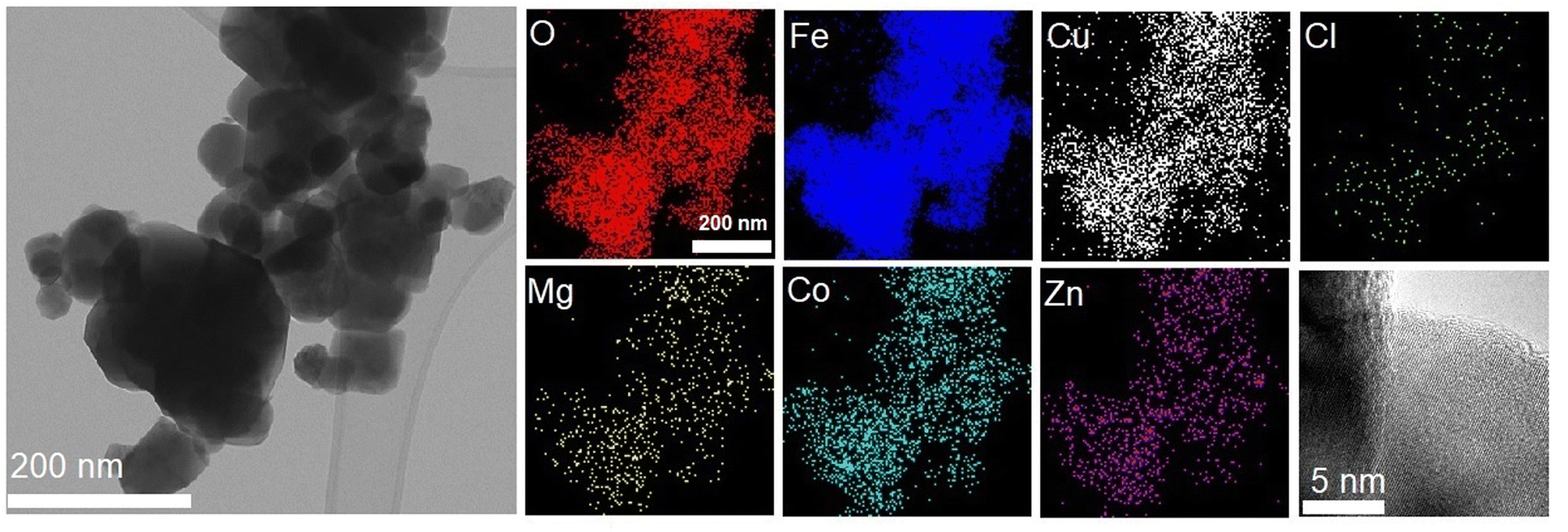 | ||
| Fig. 2 (From left to right), (S)TEM, EDS, and HR-TEM analyses of the investigated LiHEOFeCl. | ||
Carbonaceous additive (P_carbon) used as the conductive component in the sulfur composite cathode, exhibited a BET surface area of 968 m2 g−1 and an external surface area of 345 m2 g−1. Detailed morphology analysis of this material is discussed in ref. 39. The P_carbon/sulfur composite cathode serves as the reference because all its components are commercially available, and the P_carbon exhibits the best compatibility with inorganic additives as far as long-term electrochemical performance is concerned.39
3.2 Electrochemical measurements
The inherent electrochemical behavior of LiHEOFeCl was initially evaluated by cyclic voltammetry and electrochemical impedance spectroscopy. The electrochemical impedance analysis was carried out on the fresh coin cell and then again after the completed set of cyclic voltammetry measurements with different scan rates (see Methods). For the sake of clarity, we show just the last cyclic voltammogram of each completed series measured with the slowest scan rate of 0.1 mV s−1. Inorganic additives in Li–sulfur batteries can serve as the PS adsorbent only,40 or, in addition, can exhibit an inherent electrochemical activity in the particular potential window (1.7 V/2.9 V) as well.27,39The cyclic voltammograms of LiHEOFeCl with the scan rate of 0.1 mV s−1 are shown in Fig. 3a, and the electrochemical impedance spectra before and after each set of cyclic voltammetry measurements are presented in Fig. 3b. The voltammogram of the pristine cathode is exceptional, exhibiting a strong cathodic current at potentials smaller than ca. 0.9 V, which disappears in the follow-up scans. This indicates some parasitic reaction(s) in the first cycle, as reported by others. More specifically, a huge irreversible peak at ca. 0.5 V in the first negative (lithiation) scan has been observed for analogous HEO compounds28 and was ascribed to the electrolyte decomposition, solid electrolyte interface (SEI) formation together with some conversion reactions on the electrode.28 This agrees perfectly with the EIS results evidencing the high value of the charge-transfer resistance (RCT deduced from the high-frequency semicircle on the Nyquist plot; Fig. 3b) in the freshly fabricated coin cell. The RCT is significantly decreased after the first and second cycles, during which the irreversible processes (e.g. the SEI formation, see above) set up. In the next two cycles, the RCT is stabilized with a small tendency to subsequent enhancement, which could be ascribed to the slow degradation of the cathode within progressive cycling. Indeed, the best reversible charge capacity was observed in the second CV cycle. The following scans exhibit one cathodic and one anodic peak (with shoulders at the cathodic and anodic sides, respectively) which is reminiscent of the voltammetric response of some other HEOs possessing the rock salt structure.28 These peaks are assigned to lithiation/delithiation of the LiHEOFeCl structure.
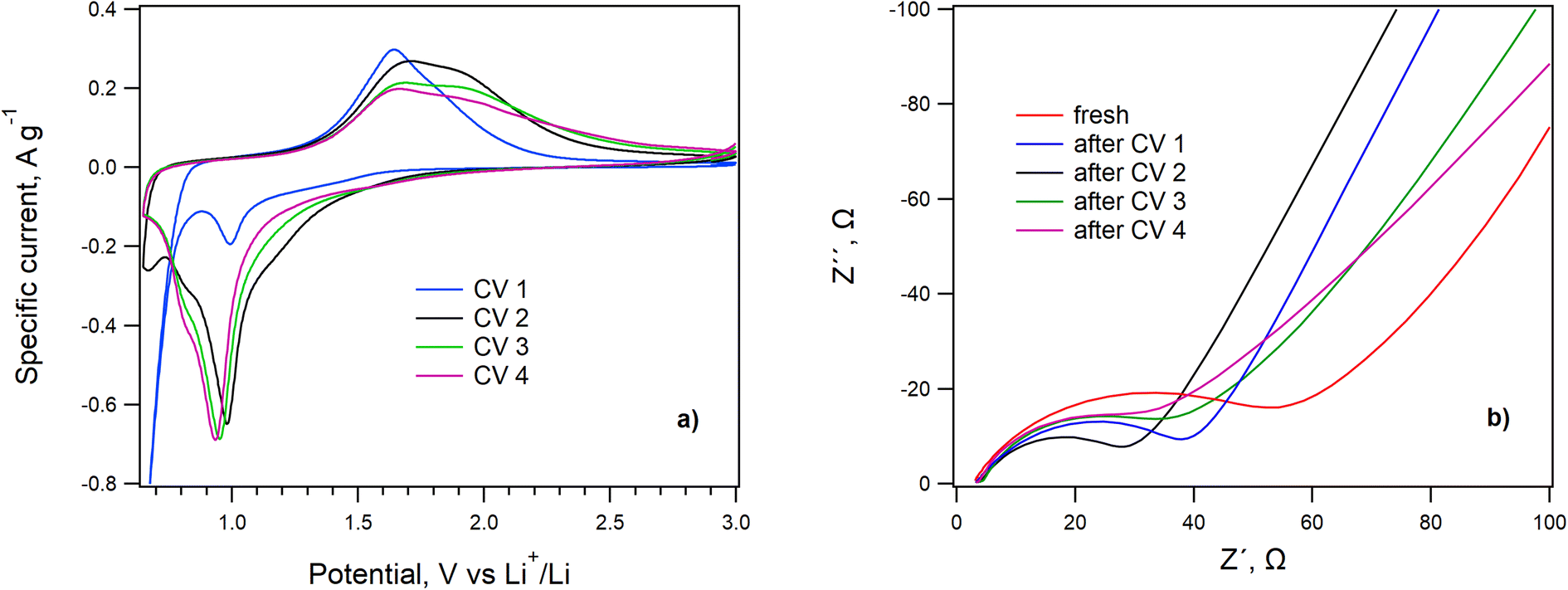 | ||
| Fig. 3 (a) The consecutive cyclic voltammograms of LiHEOFeCl, scan rate 0.1 mV s−1, (b) electrochemical impedance spectra measured on the fresh coin cell and after each set of cyclic voltammetry measurements. Currents in the chart (a) are normalized to the mass of LiHEOFeCl on the electrode. | ||
The system exhibited stable and reversible behavior from the second set of cyclic voltammetry measurements. The charge capacity calculated from the anodic branch of the 2nd cyclic voltammogram is 648 mA h g−1 with a coulombic efficiency of 96%. The slight decrease of the charge capacity in the subsequent voltammetric scans (620 mA h g−1) agrees with EIS data proving the lowest RCT for the spectrum recorded after the 1st set of voltammetric measurements, see above.
In accordance with the EIS results, the capacity drop between consecutive measurements slows down with the number of cycles, which evidences good redox stability of the LiHEOFeCl spinel structure. Nevertheless, lithiation of LiHEOFeCl starts at the potential of ∼1.5 V vs. Li+/Li, which is outside the potential window of the Li–sulfur battery. Hence, the inherent electrochemical activity of LiHEOFeCl cannot contribute to the overall electrochemical performance of the Li–sulfur system, contrary to, for instance, TiO2.27 Consequently, the LiHEOFeCl additive can serve as a pure PS adsorbent. Due to its good electrochemical stability, which manifests itself even outside the usual electrochemical window of the Li–S battery (1.7 to 2.9 V), LiHEOFeCl is a promising candidate for this application.
The electrochemical behavior of the P_carbon_LiHEOFeCl_sulfur composite cathode was evaluated by cyclic voltammetry, galvanostatic chronopotentiometry, and electrochemical impedance spectroscopy. Fig. 4 shows the cyclic voltammogram of the P_carbon_LiHEOFeCl_sulfur composite cathode in a Li–sulfur cell measured with the scan rate of 0.1 mV s−1. The cyclic voltammogram exhibits two reduction peaks at the potentials of 2.28 V and 2.04 V, corresponding to the reduction of sulfur to the low-order PS and the liquid–solid two-phase reduction from the dissolved low-order PS to the insoluble Li2S2 or Li2S, respectively. Due to the sluggish kinetics of the solid–liquid two-phase oxidation from the insoluble Li2S or Li2S2 to low-order PS, the corresponding peak in the oxidation branch of the cyclic voltammogram is overlapped with that corresponding to the low-order PS oxidation to sulfur. Hence, we observe just one oxidation peak at 2.38 V. The potentials of the reduction and oxidation current peak maxima together with the voltammetric charge capacity are listed in Table 1 and compared with those of blank P_carbon_sulfur composite cathode from ref. 39. As expected, the P_carbon_LiHEOFeCl_sulfur composite cathode exhibited a slight shift of the 2nd reduction peak to lower potentials. This is the effect of the slightly larger polarization of this material, presumably due to its lower conductivity, as compared to the pure P_carbon_sulfur composite cathode. For the same reason, the doublet of oxidation peaks (which is clearly distinguished in the voltammogram of pure P_carbon_sulfur composite cathode39) is unresolved in the voltammogram of P_carbon_LiHEOFeCl_sulfur cathode, as discussed above. The values of voltammetric charge capacities of both P_carbon_sulfur and P_carbon_LiHEOFeCl_sulfur cathodes exhibit no pronounced difference.
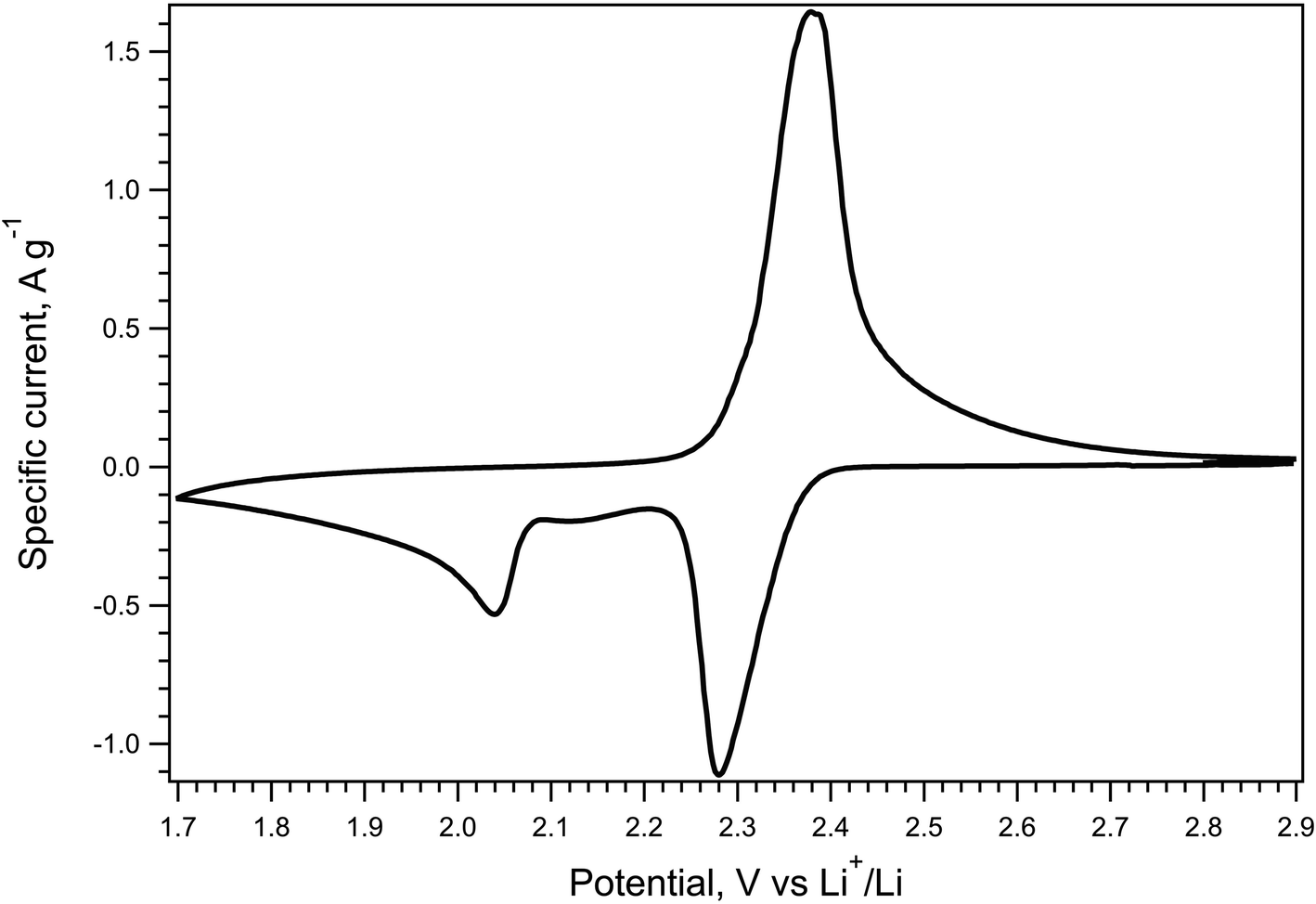 | ||
| Fig. 4 Cyclic voltammogram of the P_carbon_LiHEOFeCl_sulfur composite cathode in Li–sulfur battery. Scan rate 0.1 mV s−1. Current is normalized to the mass of sulfur in the cathode. | ||
| Sample | Capacity mA h g−1 | UR1, V | UR2, V | UO1, V | UO2, V |
|---|---|---|---|---|---|
| P_carbon ref. 39 | 663 | 2.31 | 2.04 | 2.35 | 2.40 |
| P_carbon_LiHEOFeCl | 629 | 2.28 | 2.04 | 2.38 | |
The influence of the LiHEOFeCl additive on the long-term electrochemical performance of the corresponding sulfur composite cathode was studied by galvanostatic chronopotentiometry at the charging/discharging rate of 0.1C. Fig. 5 shows the galvanostatic curve of the P_carbon_LiHEOFeCl_sulfur cathode during 100 galvanostatic cycles with a rate of 0.1C, and it is compared with that of the P_carbon_sulfur cathode without inorganic additives. To avoid casual experimental fluctuations of the capacities measured during the 100 charge/discharge cycles (Fig. 5) we have further processed these data by averaging the capacities found for each ten subsequent discharge cycles. (The discharge capacities were chosen for the analysis because they are assumed to be less influenced by parasitic effects like electrolyte breakdown). The found average capacities over 10 cycles (Cav) are plotted against the cycles' decade (Cd) in Fig. S2.† These values are not only smoothened as compared to the raw data (Fig. 5) but also avoid easy fitting to an exponential function:
| Cav = a + b × exp(c × Cd) | (1) |
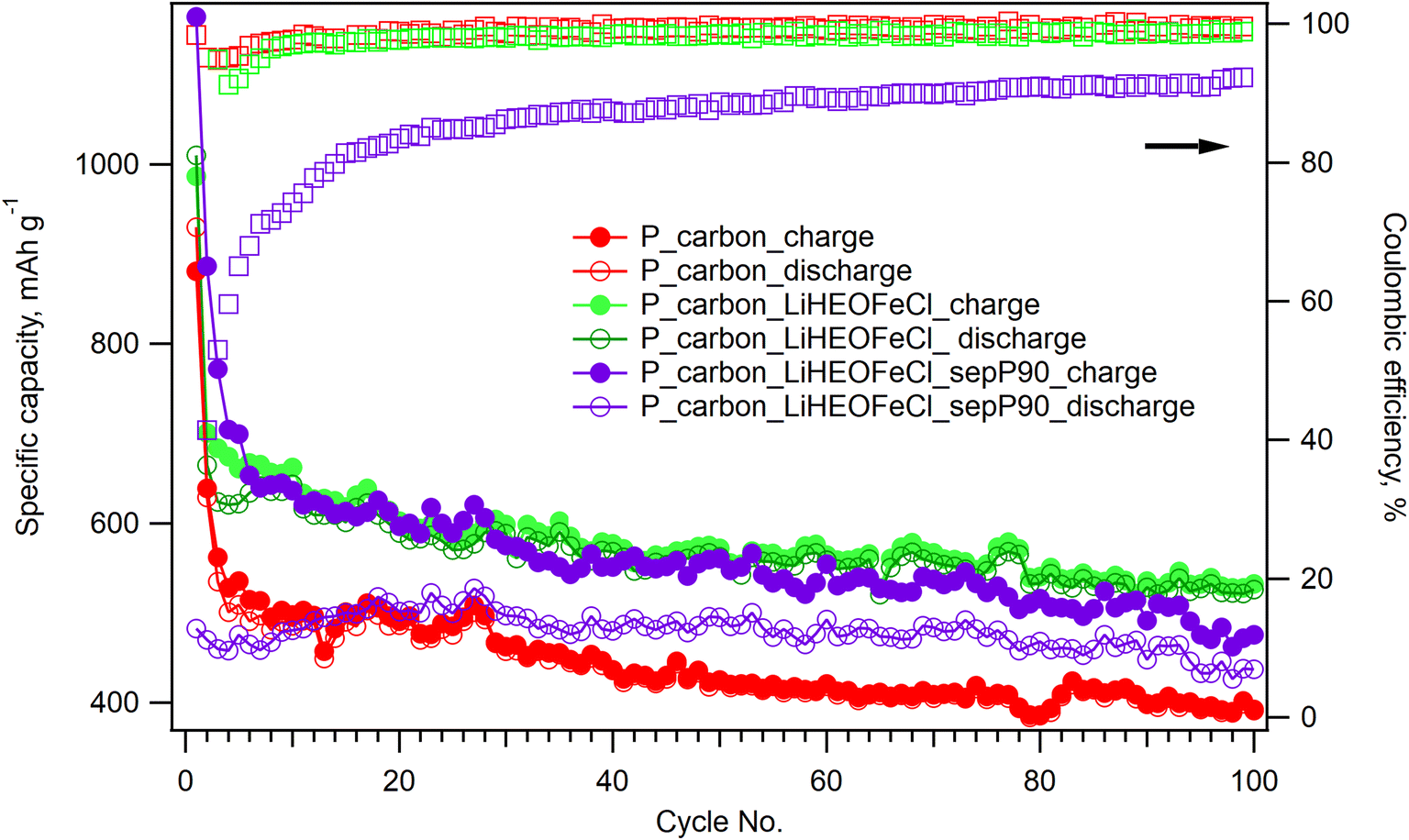 | ||
| Fig. 5 Galvanostatic chronopotentiometry at 0.1C rate measured on the Li–sulfur battery with a sulfur composite cathode containing P_carbon (red circles), P_carbon_LiHEOFeCl (green circles) and P_carbon_LiHEOFeCl with a P90-modified separator (violet circles). Full circles correspond to charge, and empty circles to discharge. The coulombic efficiencies are depicted by empty squares in colors identical to the particular galvanostatic curves. | ||
In addition, the long-term electrochemical performance of the P_carbon_LiHEOFeCl_sulfur cathode was also evaluated in the Li–sulfur battery containing the glass microfiber separator impregnated with TiO2. This modified separator was recently found to be beneficial for effective PS immobilization resulting in ca. 10–20% capacity increase.33
The stabilizing effect of the LiHEOFeCl additive on the long-term performance of the corresponding sulfur composite cathode is obvious. Although the initial values of the charge capacities are comparable for both LiHEOFeCl-containing and LiHEOFeCl-free composite cathodes, the latter exhibits a much faster capacity drop in subsequent cycles. Although this capacity drop between particular cycles of the LiHEOFeCl-free composite cathode declines with progressive cycling, the corresponding charge capacities are significantly lower than those of the P_carbon_LiHEOFeCl_sulfur cathode. The charge capacity increase ascribed to the LiHEOFeCl additive is superior as compared to that of other additives (nano TiO2, Li4Ti5O12 or TiNxOy),30 despite their much larger specific surface area. Obviously, the structural stability of the multicomponent LiHEOFeCl material resulting from its high entropy of mixing41 plays a significant role. This stabilizing effect of the LiHEOFeCl additive effect is interesting and novel. Our previous works studying the effect of more common inorganic additives (TiO2) on the performance of the sulfur/carbon composite cathode evidenced just their contribution to the charge capacity increase.29,30,39 The long-term stability of the corresponding sulfur composite cathode was in all previous studies controlled exclusively by the carbonaceous additive. However, in contrary to LiHEOFeCl, which is electrochemically inactive in the Li–sulfur battery potential window (1.7 V/2.9 V), see above, the previously studied inorganic additive (TiO2) exhibited an inherent electrochemical activity within the operating potential window of the Li–sulfur system.27 Structural changes accompanying this inherent electrochemical activity (such as reversible transformation from tetragonal to orthorhombic phase42) probably limited its possible role as a stabilizer hindering PS diffusion to the anode compartment. Hence, the behavior of the particular inorganic additive for the Li–sulfur cell depends not only on its PS adsorption ability and conductivity, but it seems, that its electrochemical inactivity within the potential window of 1.7 V/2.9 V represents an advantage for the long-term stability of the system. To prove this hypothesis, we present a reference experiment for the P_carbon_P90 sulfur composite. The 100 cycles of galvanostatic chronopotentiometry measured on the coin cell containing P_carbon_P90 sulfur composite (Fig. S3†) show, that although P90 additive in the composite cathode slightly increases the specific charge capacity of the system as compared to the pristine P_carbon_sulfur cathode, no beneficial effect on the cycling stability is observed. This finding illustrates the importance of the detailed study of the inherent electrochemical behavior of the high-entropy material prior to its incorporation in the sulfur composite cathode. The P_carbon_LiHEOFeCl_sulfur cathode provided a charge capacity of 530 mA h g−1 after 100 galvanostatic cycles, which represents ca. 33% increase as compared to the charge capacity of the P_carbon_sulfur composite cathode after 100 cycles (398 mA h g−1). The coulombic efficiency for both systems after the initial formatting cycles reached 98–99%. Concerning the specific surface area of the LiHEOFeCl additive (4 m2 g−1), this improvement cannot be ascribed to the enhanced PS adsorption. Obviously, LiHEOFeCl acts as an electrocatalyst accelerating the redox reactions of PS. This electrocatalytic behavior of high-entropy additives in Li–sulfur batteries has been already reported by others.32 Despite the two orders of magnitude higher specific surface area of P90 additive (105 m2 g−1)27 available for the PS adsorption, no beneficial effect on the cycling stability of the corresponding Li–sulfur battery was observed (Fig. S3†).
Fig. 6 shows the electrochemical impedance spectra of the 2032-coin cells with our two generic sulfur-containing cathode materials, i.e. pure P_carbon (top charts) and P_carbon mixed with LiHEOFeCl. The corresponding Nyquist plots for the freshly assembled and electrochemically treated cell with the P_carbon_S cathode are essentially similar to those reported in our previous works.39,43 Electrochemical cycling causes a decrease of serial resistance, which is evidenced by a downshift of the high-frequency cut-off in the spectrum. This decreased serial resistance is kept up during the whole cyclic test. On the other hand, the charge transfer resistance, deduced from the high-frequency semicircle(s), exhibits complex variations at cycling: it initially drops below the value for a fresh cell, which matches the earlier observations,39,43 but subsequently increases again when the cycling progresses (25–100 cycles) pointing at significant degradation of the cell. The behavior of the cells with the P_carbon_LiHEOFeCl_sulfur cathode is qualitatively different: the series resistance is roughly intact by electrochemical treatment, evidencing improved electrical properties of the cathode already in its pristine form.
 | ||
| Fig. 6 Electrochemical impedance spectra measured at 2.4 V vs. Li+/Li on freshly assembled 2032-coin cells with Li-anode (red lines); and those exposed sequentially to: three consecutive CV scans as in Fig. 4 (blue lines), followed by several galvanostatic cycles at 0.1C: 15 cycles (black) – 25 cycles (green) – 100 cycles (magenta). The top charts are for a cell with the P_carbon_sulfur composite cathode, bottom charts are for P_carbon_LiHEOFeCl_sulfur composite cathode. Charts on the right side are zoomed in to present the high-frequency part in more detail. | ||
Furthermore, the cycling-induced growth of charge-transfer resistance is much smaller compared to that of the reference cathode from pure P_carbon_S. This confirms the enhanced stability of the cathode by the LiHEOFeCl component. Hence the data from EIS and galvanostatic cycling (Fig. 5) are quite consentient and illustrate the beneficial effect of LiHEOFeCl for cell stability. By using the Zview (Scribner) software, the high-frequency part (from 105 to 103 Hz) of our impedance spectra (Fig. 6) was fitted to a Randles-type equivalent circuit composed of a serial resistance Rs and the charge-transfer resistance (RCT), which is parallel to the constant phase element (CPE). This evaluation routine provided the following values for a fresh P_carbon_S: Rs = 4.3 Ω and RCT = 17.9 Ω. The corresponding values for P_carbon_LiHEOFeCl_S were: Rs = 3.9 Ω and RCT = 8.9 Ω. The small decrease of Rs accounts mainly for the electrolyte and contact resistances, which are not assumed to be influenced by the presence of HEO in the cathode composite. On the other hand, the decrease of RCT caused by HEO is significant, by ca. 50% of the control value for P_carbon_S. This clearly demonstrates the beneficial role of our HEO in the cathode material of the Li–sulfur battery.
The effect of the TiO2-modified separator on the long-term electrochemical performance of the Li–sulfur cell containing the P_carbon_LiHEOFeCl_sulfur cathode was insignificant or rather slightly negative (Fig. 5). This agrees with our previous findings as well. The TiO2-modified separator improves the performance of the sulfur/carbon composite cathode without any inorganic additive,33 or, with identical TiO2 (P90) additive both in the cathode and in the separator.27 However, its effect in the Li–sulfur system containing different inorganic component in the cathode is negligible,39 or it is observed solely in the initial charging/discharging cycles.27,39 This behavior can be explained by the competitive PS adsorption on both inorganic components in the separator (TiO2) and the cathode (LiHEOFeCl). Hence, the stabilizing effect of the LiHEOFeCl additive is less pronounced in this particular case.
4 Conclusions
A novel lithiated high-entropy oxychloride Li0.5(Zn0.25Mg0.25Co0.25Cu0.25)0.5Fe2O3.5Cl0.5 (LiHEOFeCl) was synthesized by the mechanochemical–thermal route. The X-ray powder diffraction analysis confirmed its spinel structure belonging to the cubic Fdm space group. (S)TEM electron microscopy proved the presence of agglomerated nanocrystals. Homogeneous distribution of the particular elements within the prepared sample is evidenced by the EDS analysis. Cyclic voltammetry measured on the pristine LiHEOFeCl sample confirmed its excellent electrochemical stability and the initial charge capacity of 648 mA h g−1. The reduction of LiHEOFeCl starts at ca. 1.5 V vs. Li+/Li, which is outside the electrochemical window of the Li–S battery (1.7/2.9 V).
The addition of the LiHEOFeCl material to the P_carbon_sulfur composite cathode in the Li–sulfur battery resulted in improved long-term electrochemical cycling stability and increased charge capacity of the system. The P_carbon_LiHEOFeCl_sulfur cathode provided a charge capacity of 530 mA h g−1 after 100 galvanostatic cycles, which represents ca. 33% increase as compared to the charge capacity of the blank P_carbon_sulfur composite cathode after 100 cycles (398 mA h g−1). This considerable effect of the LiHEOFeCl material is assigned to its excellent structural and electrochemical stability within the potential window of 1.7 V/2.9 V vs. Li+/Li. In this potential region, our LiHEOFeCl exhibits no inherent electrochemical activity and acts solely as an electrocatalyst accelerating the redox reactions of PS. This can be beneficial for the Li–S battery performance, as evidenced by reference experiments with TiO2 (P90).
Author contributions
Conceptualization, M. Z., L. K., and M. F.; methodology, M. Z., M. F., and L. K.; investigation, M. Z., M. F., O. P., L. K., B. P. L., and M. V.; data curation, M. Z., M. F., M. V., O. P., B. P. L., and L. K.; writing—original draft preparation, M. Z., L. K., and M. F.; writing—review and editing, M. Z., L. K., M. F., B. P. L., M. V.; project administration, M. Z., M. F.; funding acquisition, M. Z., M. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr V. Girman and Dr M. Lisnichuk (both Faculty of Natural Sciences, UPJŠ in Košice, Slovakia) for (S)TEM, HR-TEM, and EDS analyses. This research was funded by the Grant Agency of the Czech Republic (contract No. 20-03564S). M. F. and O. P. acknowledge the support by the Slovak Research and Development Agency APVV (contract No. 19-0526).References
- G. He, X. L. Ji and L. Nazar, Energy Environ. Sci., 2011, 4, 2878–2883 RSC
.
- S. Li, B. Jin, X. Zhai, H. Li and Q. Jiang, ChemistrySelect, 2018, 3, 2245–2260 CrossRef CAS
- H. Chen, P. Xia, W. Lei, Y. Pan, Y. Zou and Z. Ma, J. Porous Mater., 2019, 26, 1325–1333 CrossRef CAS
- L. Hencz, H. Chen, H. Y. Ling, Y. Wang, C. Lai, H. Zhao and S. Zhang, Nano-Micro Lett., 2019, 11, 17 CrossRef CAS PubMed
- J. Kim, Y. Kang, S. W. Song and J. Suk, Electrochim. Acta, 2019, 299, 27–33 CrossRef CAS
- J. Wang, Y. Liu, M. Cheng, H. Zhao, J. Wang, Z. Zhao, X. Duan, C. Wang and J. Wang, Electrochim. Acta, 2019, 318, 161–168 CrossRef CAS
- T. Ando, Y. Sato, T. Matsuyama, A. Sakuda, M. Tatsumisago and A. Hayashi, J. Ceram. Soc. Jpn., 2020, 128, 233–237 CrossRef CAS
- S. Li, Z. H. Lin, G. J. He and J. G. Huang, Colloids Surf., A, 2020, 602, 125129 CrossRef CAS
- Y. T. Wang, B. Liu, W. Zhang, C. H. Shao, D. Y. Lan, X. F. Qu, R. F. Chen, W. Q. Zhang, W. M. Zhao, J. Liu, Y. H. Zhang and Z. C. Shi, SN Appl. Sci., 2020, 2, 1276 CrossRef CAS
- Y. Ding and Z. A. Qiao, Adv. Mater., 2022, 34, e2206025 CrossRef PubMed
- Y. P. Huang, X. G. Sun, J. Wang, X. Li, W. Chen, C. C. Wei, H. Hu and G. D. Liang, J. Alloys Compd., 2019, 776, 187–193 CrossRef CAS
- H. Ahn, Y. Kim, J. Bae, Y. K. Kim and W. B. Kim, Chem. Eng. J., 2020, 401, 126042 CrossRef CAS
- T. Yim, S. H. Han, N. H. Park, M.-S. Park, J. H. Lee, J. Shin, J. W. Choi, Y. Jung, Y. N. Jo, J.-S. Yu and K. J. Kim, Adv. Funct. Mater., 2016, 26, 7817–7823 CrossRef CAS
- J. Wu, S. Li, P. Yang, H. Zhang, C. Du, J. Xu and K. Song, J. Alloys Compd., 2019, 783, 279–285 CrossRef CAS
- H. Shao, W. Wang, H. Zhang, A. Wang, X. Chen and Y. Huang, J. Power Sources, 2018, 378, 537–545 CrossRef CAS
- R. Dharmasena, A. K. Thapa, R. K. Hona, J. Jasinski, M. K. Sunkara and G. U. Sumanasekera, RSC Adv., 2018, 8, 11622–11632 RSC
- G. D. Park, J. Lee, Y. Piao and Y. C. Kang, Chem. Eng. J., 2018, 335, 600–611 CrossRef CAS
- M. Wang, S. Tan, S. Kan, Y. Wu, S. Sang, K. Liu and H. Liu, J. Energy Chem., 2020, 49, 316–322 CrossRef
- U. Zubair, J. Amici, C. Francia, D. McNulty, S. Bodoardo and C. O'Dwyer, ChemSusChem, 2018, 11, 1838–1848 CrossRef CAS PubMed
- T. Zeng, P. Ji, B. Shang, Q. Peng, X. Hu and G. Li, Ionics, 2018, 24, 2973–2982 CrossRef CAS
- L. Gao, Y. Cao, J. Wang, H. Ren, J. Wang and J. Huang, Ceram. Int., 2020, 46, 18224–18233 CrossRef CAS
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kubel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed
- P. B. Meisenheimer, L. D. Williams, S. H. Sung, J. Gim, P. Shafer, G. N. Kotsonis, J. P. Maria, M. Trassin, R. Hovden, E. Kioupakis and J. T. Heron, Phys. Rev. Mater., 2019, 3, 104420 CrossRef CAS
- J. W. Sturman, E. A. Baranova and Y. Abu-Lebdeh, Front. Energy Res., 2022, 10, 862551 CrossRef
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC
- O. Porodko, M. Fabián, H. Kolev, M. Lisnichuk, M. Zukalová, M. Vinarčíková, V. Girman, K. L. Da Silva and V. Šepelák, Z. Phys. Chem., 2022, 236, 713–726 CrossRef CAS
- M. Zlámalová, B. Pitňa Lásková, M. Vinarčíková, M. Zukalová and L. Kavan, J. Solid State Electrochem., 2022, 26, 639–647 CrossRef
- T. X. Nguyen, J. Patra, J.-K. Chang and J.-M. Ting, J. Mater. Chem. A, 2020, 8, 18963–18973 RSC
- M. Zukalová, M. Vinarčíkova, M. Bousa and L. Kavan, Nanomaterials, 2021, 11, 541 CrossRef PubMed
- M. Zukalová, M. Vinarčíková, B. Pitňa Lásková and L. Kavan, J. Phys.: Conf. Ser., 2022, 2382, 012001 CrossRef
- Y. Zheng, Y. Yi, M. Fan, H. Liu, X. Li, R. Zhang, M. Li and Z.-A. Qiao, Energy Storage Mater., 2019, 23, 678–683 CrossRef
- L. Tian, Z. Zhang, S. Liu, G. Li and X. Gao, Energy Environ. Mater., 2021, 5, 645–654 CrossRef
- M. Zukalová, M. Vinarčíková, B. Pitňa Lásková and L. Kavan, ECS Trans., 2021, 105, 183–189 CrossRef
- Inorganic Crystal Structure Database (ICSD), Leibniz Institute for Information Infrastructure, FIZ Karlsruhe, Karlsruhe, 2020 Search PubMed
- J. Rodriguez-Carvajal, Fullprof Program, ILL Grenoble, France, 1993 Search PubMed
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS
- M. Fabián, P. Bottke, V. Girman, A. Düvel, K. L. Da Silva, M. Wilkening, H. Hahn, P. Heitjans and V. Šepelák, RSC Adv., 2015, 5, 54321–54328 RSC
- M. Zukalová, M. Vinarčíková, B. Pitňa Lásková and L. Kavan, Mater. Chem. Phys., 2023, 296, 127246 CrossRef
- X. W. Geng, R. W. Yi, X. F. Lin, C. G. Liu, Y. Sun, Y. C. Zhao, Y. Q. Li, I. Mitrovic, R. Liu, L. Yang and C. Z. Zhao, J. Alloys Compd., 2021, 851, 156793 CrossRef CAS
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, e1806236 CrossRef PubMed
- B. Pitna Laskova, L. Kavan, M. Zukalova, K. Mocek and O. Frank, Monatsh. Chem., 2016, 147, 951–959 CrossRef CAS
- L. Kavan, M. Vinarčíková, M. Zlámalová and M. Zukalová, ECS Trans., 2021, 105, 169–176 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01496g |
| This journal is © The Royal Society of Chemistry 2023 |