
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid organic–inorganic nanoparticles with associated functionality for catalytic transformation of biomass substrates†
Alicia Coloma,
Alexandra Velty * and
Urbano Díaz*
* and
Urbano Díaz*
Instituto de Tecnología Química, Universitat Politècnica de València, Consejo Superior de Investigaciones Científicas, 46022 Valencia, Spain. E-mail: avelty@itq.upv.es; udiaz@itq.upv.es
First published on 30th March 2023
Abstract
We present the one-pot synthesis of functionalized organosilica nanoparticles to generate multi-functional hybrid catalysts. Octadecyl, alkyl-thiol and alkyl-amino moieties were used separately and in different combinations, to generate different hybrid spherical nanoparticles with tunable acidic, basic and amphiphilic properties, covalently incorporating up to three organic functional elements onto the surface of the nanoparticles. Several parameters were optimised such as the concentration of the base employed during the hydrolysis and condensation synthesis process that showed a strong influence on the particle size. The physico-chemical properties of the hybrid materials were fully characterized by XRD, elemental and thermogravimetric analysis, electron microscopy, nitrogen adsorption isotherms and 13C and 29Si NMR spectroscopy. Finally, the potential uses of the prepared materials as amphiphilic catalysts, with acidic or basic properties for the conversion of biomass molecules into platform chemicals were evaluated.
Introduction
Climate change is one of the main challenges that humanity faces today. Greenhouse gases are responsible for global warming and carbon dioxide (CO2) is the most important among them, with human activities altering the carbon cycle by adding more CO2 to the atmosphere. Decarbonization has become a global imperative and a priority to save the planet. Then, the use of biomass as a renewable feedstock for the production of fuels and chemicals has received much attention by the scientific community to ensure the sustainable development of human society.1,2 The design and production of chemicals and fuels while reducing or eliminating the use and generation of hazardous substances is the goal of green chemistry. Therein, catalysis plays a key role, and in particular heterogeneous catalysis because of the environmentally friendly nature of its recyclable nature attributed to simple separation and recovery, as well as its applicability to flow reactors. Owing to high surface area, tunable composition (multiple active species), adjustable particle morphology and porous architecture, heterogeneous catalysts offer versatile physicochemical and sorption properties for the design and development of efficient and green catalytic processes.Anchoring organocatalysts on inorganic surfaces, it was a successful approach to heterogenize active and selective functions to obtain specific multi-functional organic–inorganic materials. These hybrid solids contain several supported well-defined active species regularly distributed and separated at controlled molecular distances, without loss of activity or deactivation.3,4 Among the different supporting surfaces, amorphous silica works as excellent support for the synthesis of functional catalysts due to the large number of hydroxyl groups located on its external surface.5 Furthermore, they have high thermal stability and recyclability.6,7 In addition, silica alkoxides (organosilanes) can be incorporated on the silica surface by reaction with the mentioned silanol groups through hydrolysis (acid or basic) processes. In this way, organic functional groups can be introduced into a solid matrix, as well as modify the hydrophobic/hydrophilic properties of the silica used as support.8,9 Within siliceous materials, the formation of compact silica nanoparticles has gained great interest in recent years due to their high stability, low toxicity and fast and simple synthesis. Moreover, they can be used as individual elements in which a variety of well-dispersed active sites can be placed, being also possible stabilize the control of the nanoparticle size.10 The functionalization of these mentioned silica nanoparticles would allow the design of hybrid organic–inorganic materials with unique properties for their application in specific fields.5,11 These materials have been already used as sensors,12 insulators,13 in nanomedicine14,15 or in catalysis.16 In addition, multi-functional hybrid catalysts are of great interest for carrying out cascade reactions.
In the present work, we describe the one-pot preparation of different mono-, bi- and tri-functionalized silica nanoparticles to generate multi-functional catalysts. For this purpose, octadecyl, alkyl thiol and alkyl amine groups, from monomeric organosilane precursors, were used separately and in different combinations. Several parameters were optimised such as the concentration of the base employed during the synthesis process that showed a strong influence on the particle size. The physico-chemical properties of the hybrid materials were fully characterized by XRD, elemental and thermogravimetric analysis, electron microscopy, nitrogen adsorption isotherms and 13C and 29Si NMR spectroscopy. Non-porous organosilica materials were obtained with a spherical morphology and a thermal stability up to 250–300 °C. Spectroscopic studies confirmed the covalent incorporation of the distinct organic moieties on the surface of the silica nanoparticles and their integrity.
Next, the catalytic properties of the prepared materials as amphiphilic catalysts with acidic or basic properties were evaluated in the conversion of biomass molecules into valuable platform chemicals. Over the past five decades, 5-hydroxymethylfurfural (HMF) has emerged as one of the most promising biochemical platforms as a new and sustainable alternative to value chains based on increasingly scarce fossil resources. The most widely described route for the production of HMF is the acid catalysed dehydration of a hexose (fructose). Currently, there are no cost-effective processes to obtain HMF and worldwide production is not sufficient to use HMF as a renewable and readily available platform molecule, due to the low yield and stability of HMF in acidic media. It has been widely reported that the best solvent for the dehydration of fructose to HMF to inhibit self-condensation and rehydration reactions is DMSO.17–19 However, the energetic cost of distillation as well as the loss of yield during distillation force to look for other alternatives, such as the development of biphasic media using Lewis and Brønsted acids and organocatalysts,20 allowing the continuous extraction of HMF from the aqueous phase and from contact with the catalyst to avoid side reactions,21,22 or the use of ionic liquids.23 Nevertheless, the main issue remains the production of HMF with high isolated yield, selectivity and purity. Herein, the benefits of C18 hydrocarbon chains (–C18) over the catalytic performance of mono-functionalized silica nanoparticles with propyl-sulfonic groups (–SO3H) were shown for the fructose dehydration in a biphasic system. On the other hand, the isomerization of glucose to fructose is an important intermediate step for the conversion of cellulosic biomass to renewable fuels and chemical platforms such as HMF and levulinic acid. Lewis acids and Brønsted bases have been commonly employed to catalyse the isomerization of glucose to fructose, whereas the base-catalysed route has received significantly less attention due to its low yield (<10%) and selectivity to fructose.24–27 Tertiary amines have exhibited good selectivity towards fructose, and have been shown to be a viable catalyst for fructose production with 32% yield.28 Accordingly, the catalytic performance of organosilica nanoparticles containing isolated –NH2 and both –NH2 and –C18 moieties was finally explored for the isomerization of glucose to fructose in water.
Experimental section
Synthesis of silica nanoparticles
The synthesis of silica nanoparticles (NPs-SiO2) was carried out following the method previously described by Zhou et al. in 2014 (ref. 16) (Scheme 1). A solution (A) consisting of 316 g ethanol, 56 g H2O Milli-Q® and ammonium hydroxide solution (NH4OH) was prepared. Meanwhile, 18.6 g of tetraethylorthosilicate (TEOS) and 18.6 g of ethanol were mixed (solution B). Solution A was stirred for 10 min with reflux at 40 °C, while solution B was stirred for 5 min at room temperature. Solution B was then added to solution A and left under stirring with reflux at 40 °C for 30 min. The resulting solution was centrifuged in an AVANTI J-25 XP centrifuge at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min and washed successively with water at the same speed and time, until neutral pH. Finally, the solid obtained was dried at 60 °C overnight. The amount of ammonium hydroxide used was varied between 3.5 and 29 g, depending on the desired particle size. Tetraethylorthosilicate (98%) and NH4OH solution (28–30% NH3 in H2O) were purchased from Sigma-Aldrich. Absolute ethanol was supplied by Scharlab. Pure silica nanoparticles were labelled as NPs-SiO2.
000 rpm for 10 min and washed successively with water at the same speed and time, until neutral pH. Finally, the solid obtained was dried at 60 °C overnight. The amount of ammonium hydroxide used was varied between 3.5 and 29 g, depending on the desired particle size. Tetraethylorthosilicate (98%) and NH4OH solution (28–30% NH3 in H2O) were purchased from Sigma-Aldrich. Absolute ethanol was supplied by Scharlab. Pure silica nanoparticles were labelled as NPs-SiO2.
 | ||
| Scheme 1 Stöber method scheme and conditions used for nanoparticles synthesis and functionalization. | ||
Synthesis of functionalized NPs-SiO2
For obtaining functional NPs-SiO2, the synthesis of silica NPs was carried out as previously described,16 with minor modifications (Scheme 1). For the synthesis of mono-functionalized NPs-SiO2, the corresponding organosilane precursor was added 5 min after the pre-hydrolysis of TEOS, at 40 °C in a solution of ethanol–water. Then, the solution was left for another 30 min with reflux at 40 °C and stirring, before centrifugation and washing process as described above. When more than one function was incorporated, various organosilane precursors were added in the following order: trimethoxy(octadecyl)silane (ODTMSi), (3-mercaptopropyl)trimethoxysilane (MPTMSi) and (3-amino-propyl)trimethoxysilane (APTMSi), stirring the solution for 5 min between the addition of each precursor, and another 30 min after the addition of the last one. Then, the same synthesis process described before was carried out to obtain the final product. (3-Mercaptopropyl)trimethoxysilane (95%), (3-amino-propyl)trimethoxysilane (97%) and trimethoxy(octadecyl)silane (technical grade) were supplied by Sigma-Aldrich. Functional silica nanoparticles were named as NPs-SiO2–R, where R corresponds to the organic moieties incorporated (–SH/SO3H, –NH2, –C18).Oxidation of thiol groups
The oxidation procedure of the thiol groups to sulfonic groups was carried out in two stages: a first oxidation with H2O2 and a second acid process with H2SO4. For the oxidation, 1 g of sample was added over 30 mL of H2O2. The solution was left under stirring at room temperature for 4 h. The resulting solid was recovered by centrifugation at 12000 rpm for 10 min and washed with distilled water to neutral pH. Finally, it was dried overnight at 60 °C. The solid obtained was added over a 0.8 M H2SO4 solution (30 mL of H2SO4 solution per gram of sample) and left stirring at room temperature for 2 h. Again, the final solid was obtained as explained before. Hydrogen peroxide solution (30% w/w) and sulphuric acid (ACS grade, 95–98%) were provided by Sigma-Aldrich.
Fructose dehydration. General procedure
The procedure described by Bispo et al.29 was followed. Specifically, 44 wt% fructose solution was prepared by dissolving 4.4 g of fructose in 5.6 g of Milli-Q® water. For each reaction, solid catalyst, 300 mg of fructose solution and 900 mg of a methyl-isobutyl ketone–butanol (MIBK:BuOH) 70:30 solution were weighed into a 2 mL microwave vial reactor. The reaction was carried out in a Biotage® Initiator+ microwave apparatus. After the reaction, the crude was filtered to recover the catalyst. Then, 10 mL H2O and 10 mL of MIBK:BuOH 70:30 solution were added to the reaction medium. Then, aliquot of the reaction was monitored in a LC-2030C Plus high-performance liquid chromatography (HPLC) system. For solvent testing, MIBK:BuOH solution was substituted by alternative solvents: MeOH, 1,4-dioxane, DMSO and acetone. D-(−)-Fructose (≥99%), 4-methyl-2-pentanone HPLC grade (≥99%), 1-butanol HPLC grade (≥99.7%), methanol HPLC grade (99.99%) and anhydrous 1,4-dioxane (99.8%) were purchased from Sigma-Aldrich.
Glucose isomerization. General procedure
Reaction was carried out according to Liu et al.30 procedure. Typically, solid catalyst, 5 g of water and 0.5 g of glucose were added into a 5 mL microwave vial reactor. The reaction was carried out in a Biotage® Initiator+ microwave apparatus with 2 min pre-stirring. After the reaction, the crude was filtered to recover the catalyst. D-(+)-Glucose (≥99.5%) was purchased from Sigma-Aldrich.HPLC analysis of reaction samples
The aqueous phases of the different samples were analysed using an Aminex® HPX-874 300 × 7.8 mm column from Bio-Rad Laboratories. A refractive index detector RID-20A detector from Shimadzu Corporation was used to determine the remaining fructose, while a UV-Vis detector was used to quantify the HMF in the aqueous phase. The organic phase was separated on xBridge® C18 5 μm 4.6 × 150 mm column by WatersTM with UV-Vis detection for the analysis of HMF in this phase. A wavelength of 253 nm was selected for the UV-Vis detector. Samples from single-phase systems were analysed on the Aminex® HPX-874 column, using both detectors. External standard method was used for quantification. For that, a fructose and HMF calibration curve were obtained, independently, using solutions of pure reactants.Characterization techniques
CHNS content was determined with a EuroEA3000 elemental analyser (Eurovector). Thermogravimetric and differential thermal analysis (TGA-DTA) were performed in an airstream with STA 449F3 Jupiter® analysed (NETZSCH). Nitrogen adsorption isotherms were obtained at 77 K with a Micromeritics ASA™ 2420 volumetric adsorption apparatus, with no sample pre-treatment. The Brunauer–Emmett–Teller (BET) method was followed for surface area and pore volume determinations.31 Surface area was calculated with the data in the range from 0.04 to 0.2. The total pore volume was obtained from the amount of N2 adsorbed at a relative pressure of 0.99. Finally, the external surface area and micropore volume were estimated using the t-plot method in the t range from 3.2 to 5. Solid-state MAS-NMR spectra were obtained at room temperature under magic angle spinning (MAS) in AV-400 and AV-III HD-400 spectrometers. The single pulse 29Si spectra were obtained with a 7 mm Bruker BL-7 probe, at 79.5 MHz and using pulses of 3.5 ms, corresponding to a flip angle of 3/4 p radians, and a recycle delay of 240 s for the 13C cross-polarization (CP) spectra. 13C and 29Si NMR spectra were referenced to adamantane and tetramethylsilane, respectively. For liquid samples, ASCEND 400 and UltraShield 300 systems were used. All equipment was provided by Bruker. Transmission electron microscopy (TEM) was performed in a JEOL model JEM-1400Flash with a STEM detector. For the analysis, a small amount of the sample was dispersed in dichloromethane. Then, a drop was placed on a nickel grid and dried under a lamp for 2 h. HR-FESEM analysis were performed with a ZEISS GeminiSEM500 microscope. Specifically, average size of the different silica nanospheres obtained in this study was confirmed after the measurement of 100–130 individual particles from different TEM images (Table S1†).Results and discussion
Nanoparticles synthesis
In general, these silica nanospheres materials are synthesized through sol–gel processes that consist of the hydrolysis of an inorganic salt or an alkoxide in the presence of an acid or a base as a catalyst.6,9 Synthesis in basic media allows the formation of monodisperse spherical particles of a wide range of sizes, unlike acid-catalysed systems. Other chemical synthesis routes such as chemical vapour deposition (CVD) have proven to be more toxic, expensive and energy-consuming.9 Therefore, the Stöber method is the most widely used approach to obtain silica nanoparticles. Both the temperature and time used during this process vary considerably between the procedures followed by different research groups.16,32–34 In this work, the mildest possible conditions were used. Specifically, pure silica nanoparticles were synthesized based on the Stöber method described by Zhou et al. (2014),16 as shown in Scheme 1. Specifically, during the synthesis process, first, hydrolysis of the silica precursors in the presence of water and basic media takes places. Subsequently, the hydrolysed silylated product can condense with itself or with other silicon precursors, resulting polymerized silica in the form of regular-size nanoparticles (Scheme S1†). | ||
| Fig. 1 Electronic microscopy images of silica nanoparticles. (a) SEM [NH4OH] = 1.6 M, (b) SEM [NH4OH] = 0.39 M, (c) TEM [NH4OH] = 1.6 M, (d) TEM [NH4OH] = 0.20 M. | ||
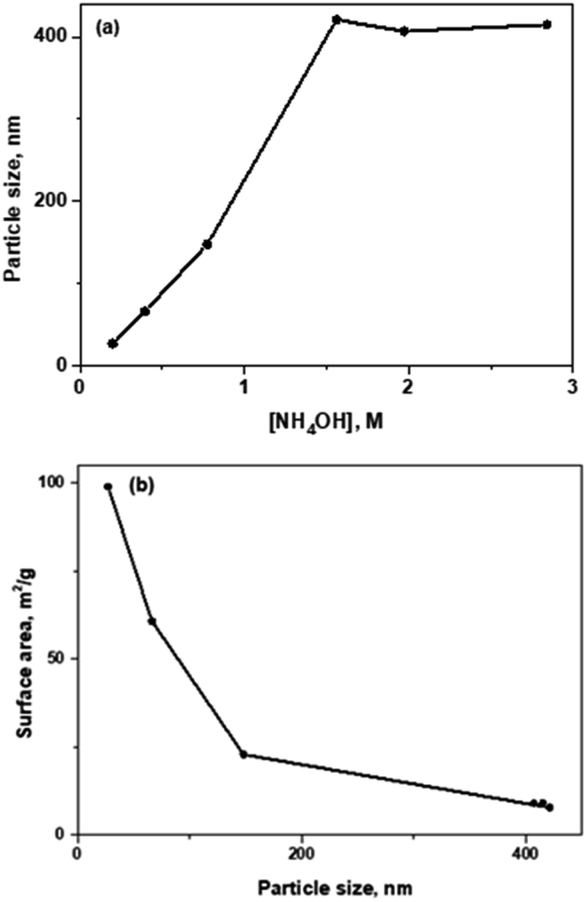 | ||
| Fig. 2 (a) Influence of NH4OH concentration in particle size. (b) Correlation between particle size and surface area. | ||
Propyl-thiol (–SH), propyl-amino (–NH2) groups and C18 hydrocarbon chains (–C18), independently, were directly incorporated on the surface of silica nanoparticles, following the synthesis process previously discussed (Scheme 2). The mono-functional catalyst containing propyl-amino moieties was also synthetized at smaller particle size for further catalytic applications, using a more reduced concentration of NH4OH base catalyst.
 | ||
| Scheme 2 Mono-functional silica nanoparticles synthetized. (a) NPs-SiO2–SO3H (acid sites), (b) NPs-SiO2–NH2 (basic sites), (c) NPs-SiO2–C18 (hydrophobic character). | ||
The chemical composition of the mono-functionalized NPs-SiO2 materials was obtained by CHNS analysis (Table 1). Total organic content was calculated using data both from elemental and thermogravimetric analysis (TGA). Within these materials, the highest organic content was reached when incorporating –C18 moieties, due to the higher molecular weight of the long hydrocarbon chains present in the composition of the hybrid nanoparticles. Moreover, sulphur-containing nanoparticles showed lower sulphur incorporation than expected. A higher incorporation of the organic fragments was observed when small particle size nanoparticles were synthetized. When comparing the amino mono-functionalized nanoparticles (Table 1, entries 3 and 4), the nitrogen percentage was almost the double when lowing the particle size.
:4, SH/NH2 1:1
| Entry | Catalyst | [NH4OH] | % N | % C | % H | % S | % Organic contenta (EA) | % Organic contentb (TGA) | C/Sc | C/Sd | C/Nc | C/Nd | N/Sc | N/Sd |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated without taking into account the % N in materials that did not contain the precursor APTMSi.b Calculated from the loss between (200–600 °C), without considering the adsorbed water molecules.c Theoretical molar ratio.d Experimental molar ratio. | ||||||||||||||
| 1 | NPs-SiO2–SO3H | 1.6 | 0.15 | 1.3 | 1.4 | 0.77 | 3.5 | 4.2 | 3.0 | 4.6 | — | — | — | — |
| 2 | NPs-SiO2–C18 | 1.6 | 0 | 37 | 6.8 | 0 | 44 | 42 | — | — | — | — | — | — |
| 3 | NPs-SiO2–NH2 | 1.6 | 3.7 | 9.4 | 3.0 | 0 | 16 | 13 | — | — | 3.0 | 3.0 | — | — |
| 4 | NPs-SiO2–NH2 | 0.20 | 6.1 | 16 | 4.3 | 0 | 27 | 22 | — | — | 3.0 | 3.1 | — | — |
| 5 | NPs-SiO2–C18–SO3H | 1.6 | 0 | 3.0 | 1.2 | 0.43 | 4.6 | 6.7 | 7.5 | 19 | — | — | — | — |
| 6 | NPSi–SiO2–C18–SO3H | 0.20 | 0 | 14 | 3.4 | 4.0 | 21 | 25 | 7.5 | 8.9 | — | — | — | — |
| 7 | NPs-SiO2–C18–NH2 | 1.6 | 2.9 | 17 | 4.2 | 0 | 24 | 22 | — | — | 7.5 | 7.1 | — | — |
| 8 | NPs-SiO2–C18–NH2 | 0.20 | 3.7 | 22 | 4.9 | 0 | 31 | 26 | — | — | 7.5 | 7.1 | — | — |
| 9 | NPs-SiO2–C18–SO3H–NH2 | 0.20 | 2.1 | 20 | 4.3 | 5.6 | 32 | 33 | 11 | 9.4 | 11 | 11 | 1.0 | 0.88 |
Same tendency was observed when other functional groups were also present (Table 1, entries 7 and 8) and when comparing sulphur content in materials containing sulphonic groups (Table 1, entries 5 and 6) in bi-functional organosilica nanoparticles.
Thermal stability of the obtained materials was considered by thermogravimetrical analysis (TGA). All samples showed an initial loss (I) between 20 °C and 120 °C associated with adsorbed water and remaining NH4OH. A second weight loss (II) was observed from 250–300 °C to 600–700 °C, corresponding to the degradation of the organic moieties present in the silica surface of nanoparticles. Water dehydroxylation was also detected in a third loss (III). This phenomenon corresponds to the water formed at high temperature because of the condensation of silanol groups. Therefore, the materials synthetized were stable up to 250–300 °C, approximately. Fig. 3(a)–(d) shows the thermogravimetrical analyses obtained for mono-functional NPs-SiO2, being observed the more pronounced loss in the organosilica NPs containing –C18 groups due to the higher molecular weight of these surface moieties, such as was also detected by elemental analysis (Table 1).
 | ||
| Fig. 3 Thermogravimetrical analysis of the different materials: mass loss (TGA, black lines) and derivative curves (DTA, red lines) of (a) NPs-SiO2–SH, (b) NPs-SiO2–SO3H, (c) NPSi–SiO2–C18, (d) NPs-SiO2–NH2 and (e) NPs-SiO2–C18–SO3H–NH2. | ||
Subsequently, two organosilane precursors were simultaneously incorporated on the nanoparticle surface in order to introduce two types of functional groups available on the surface of the same material (Scheme S2†). Next, the three organic chains were incorporated simultaneously to maximize the functionalities of the silica nanoparticles. Bi-functional NPs-SiO2 containing –NH2 groups were incorporated in a molar ratio –C18/–NH2 1:4 and –SH/–NH2 1:1, respectively. The synthesis was first carried out with [NH4OH] = 1.6 M in order to be comparable with all materials previously described. Tri-functional nanoparticles were synthesised with molar ratios C18/–NH2 1:4, C18/–SH 1:4 and –SH/–NH2 1:1. Tri-functional and bi-functional materials containing both C18 and NH2 moieties, and C18 together with –SH moieties, were additionally obtained using [NH4OH] = 0.20 M, to generate multi-functionalized materials with higher external area.
The results of the elemental analyses of the above multi-functional materials are summarized in Table 1 (entries 5–9). According to the above results, the effective incorporation of organic fragments was confirmed without notable differences between the values determined by elemental or thermogravimetrical (TGA) analyses. In addition, similar theoretical and experimental C/S, C/N and N/S molar ratios were reached in most cases. Moreover, no difference was seen between oxidized and non-oxidized materials, in the case of nanoparticles containing thiol and derived sulfonic groups. It should be noticed that small nitrogen amount in samples which propyl-amino chain is not present was detected, corresponding to the remaining NH4OH used as base catalyst during the synthesis process. Referred to TGA, no notable differences were observed between all the materials synthesized and the previous ones (Fig. 3e and S4†), being 250–300 °C the stability range temperature of the materials, considering the thermal degradation of the external organic moieties.
Specifically, when adding –SH and –C18 chains simultaneously (Table 1, entry 5), the organic incorporation was lower than expected. On the contrary, when using 0.20 M as NH4OH concentration and thus decreasing the particle size, the organic incorporation achieved was as expected, being close to the experimental and theoretical C/S molar ratios (Table 1, entry 6). Furthermore, an increase from 5% to 21 wt% in the total organic content was observed, proving that better results are obtained by decreasing the particle size down to ∼30 nm. Regarding bi-functional nanoparticles containing –NH2 moieties (Table 1, entries 7–8) and tri-functional nanoparticles (Table 1, entry 9), both theoretical and experimental ratios resembled and similar percentages of organic content were reached, corroborating the presence and preservation of organic moieties such as those present in the starting precursors.
Additionally, all samples were further characterized using electronic microscopy to analyze the morphology of the obtained organosilica nanoparticles. Non-functionalized NPs-SiO2 showed a smooth surface such as it was observed in Fig. 4. All other materials showed an external roughness morphology associated with surface organic functionalization, obtaining spherical particles in most cases. It was observed that a partial loss of spherical morphology occurred when propyl-amino groups were incorporated on the outer surface of the nanoparticles (Fig. 4d). In these cases, mostly small clusters of incomplete spherical particles of heterogeneous size were detected. This result can be attributed to the difference in the polymerization rate of the alkyl-amino silylated precursors. In these cases, a faster polymerization rate and a higher incorporation of the organic moieties happened, being this matter corroborated by thermogravimetrical analysis, as mentioned before (Table 1). A higher rate would favour the generation of incomplete nanospheres with irregular surface and shape. These non-regular spherical particles were also observed when adding functionalization to small-sized particles, not involving propyl-amino chains (Fig. S5†). In this case, the reason could be the higher molecular size (–C18) of the organic moieties in comparison to the size of silica nanoparticles. Moreover, the interaction of the organic fragments from different small nanoparticles could be favoured resulting in a higher agglomeration.
 | ||
| Fig. 4 SEM (a)–(d) images of: (a) NPs-SiO2; (b) NPs-SiO2–SO3H; (c) NPs-SiO2–C18; (d) NPs-SiO2–NH2 [NH4OH] = 1.6 M. | ||
Textural properties obtained from the respective nitrogen isotherms demonstrate the synthesis of non-porous multi-functional materials. It is important to notice that higher external surface when decreasing particle size was observed, for the same composition, as it is shown in Table S2† (entries 3–8). As it can be seen, the highest external surface area was reached in small-sized bi-functional NPs-SiO2 containing C18 and SO3H moieties (Table S2,† entry 6).
The integrity of the incorporated organic fragments was verified by 13C NMR spectroscopy. Spectra of individual monomeric organosilanes were simulated and chemical shifts were assigned to each carbon atom present in the functional groups to establish a comparison with the obtained organosilica nanoparticles. In particular, the chemical shift corresponding to organic thiol groups was observed at 24 ppm, alkyl-sulfonic groups at 54 ppm, alkyl-amino groups at 42 ppm and the most characteristic band of the C18 chains at 30 ppm (Fig. 5a). Multi-functional NPs-SiO2 materials showed chemical shifts corresponding to the organic moieties incorporated in each case, proving the effective incorporation and preservation of the functional groups (Fig. 5b). On the other side, no difference was observed in any of these spectra when varying the nanoparticle size. Thus, the 13C NMR spectra showed, for all materials, the efficient incorporation and preservation of the functional groups such as they were in their initial composition.
 | ||
| Fig. 5 (a) 13C NMR spectra of mono-functional silica nanoparticles: NPs-SiO2–C18 (red line), NPs-SiO2–SH (black line), NPs-SiO2–SO3H (blue line), NPs-SiO2–NH2 (green line). (b) 13C NMR spectra of bi-functional NPs-SiO2–C18–NH2, NPs-SiO2–C18–SO3H (red and black lines) and tri-functional NPs-SiO2–C18–NH2–SO3H (blue line) silica nanoparticles. | ||
Functionalization percentage of silica nanoparticles was confirmed by NMR spectroscopy. 29Si NMR showed up to 71% of Q4-type (Si(OSi)4) and 29% of Q3-type (Si(OH)(OSi)3) silicon atoms for NPs-SiO2. When adding external functionalization, an additional chemical shifts were observed corresponding to T silicon atoms (C–Si(OSi)3 and C–Si(OH)(OSi)2) between −55 and −80 ppm. This signal corroborates the presence of Si–C bonds in the NPs-SiO2, as in the case of the starting organosilanes. This verification confirms the covalent integration of the organic fragments in the silica matrix of the nanoparticles. Moreover, the chemical shift of the T silicon atoms varies when comparing the solid material and the corresponding liquid pure monomeric precursors due to the different chemical environment. Specifically, chemical shift can be found at around −40 ppm in the spectra of the liquid organosiliceous precursor, corresponding to atoms in T1 position (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Si(OEt)3) (Fig. S3†). On the other hand, in solid state, this chemical shift is found at around −60 ppm (Fig. 6). It can also be observed a chemical shift at −100 ppm in the liquid precursor corresponding to a partial hydrolysis of some organosilane monomers. For mono-functional materials containing alkyl-thiol/sulfonic group, only a slight peak was detected corresponding to this T-type chemical shift, showing less incorporation level for this organosilane (Fig. 6a and Table 2, entry 1). In addition, low proportion of silicon atoms Q4-type detected in the catalyst containing alkyl-amino groups (55%) with respect to the others mono-functionalized materials (Table 2, entries 1–3) confirmed the possible difference in the polymerization rate between TEOS and the monomeric organosilanes used during the synthesis process. So, in the NPs-SiO2 containing the alkyl-amino moiety, –NH2 competes with TEOS leading to a worse polymerization and a higher number of external hydroxyl groups. This basic moiety (–NH2) has a strong influence when –C18 and –SO3H are cohabitating in the organosilica NPs because it favours their incorporation (Table 2, entries 7–9).
C–Si(OEt)3) (Fig. S3†). On the other hand, in solid state, this chemical shift is found at around −60 ppm (Fig. 6). It can also be observed a chemical shift at −100 ppm in the liquid precursor corresponding to a partial hydrolysis of some organosilane monomers. For mono-functional materials containing alkyl-thiol/sulfonic group, only a slight peak was detected corresponding to this T-type chemical shift, showing less incorporation level for this organosilane (Fig. 6a and Table 2, entry 1). In addition, low proportion of silicon atoms Q4-type detected in the catalyst containing alkyl-amino groups (55%) with respect to the others mono-functionalized materials (Table 2, entries 1–3) confirmed the possible difference in the polymerization rate between TEOS and the monomeric organosilanes used during the synthesis process. So, in the NPs-SiO2 containing the alkyl-amino moiety, –NH2 competes with TEOS leading to a worse polymerization and a higher number of external hydroxyl groups. This basic moiety (–NH2) has a strong influence when –C18 and –SO3H are cohabitating in the organosilica NPs because it favours their incorporation (Table 2, entries 7–9).
 | ||
| Fig. 6 (a) 29Si BD NMR spectra of mono-functional silica nanoparticles. (b) 29Si BD NMR spectra of bi-functional (red and black lines) and tri-functional (blue line) silica nanoparticles. [NH4OH] = 1.6 M, except NPs-SiO2–C18–SO3H–NH2 prepared with [NH4OH] = 0.20 M. | ||
:4, SH/NH2 1:1
| Entry | Catalyst | [NH4OH] | % Q3 | % Q4 | % T |
|---|---|---|---|---|---|
| a Chemical sight detectable from NMR 29Si CP spectra but not quantifiable from NMR 29Si BD spectra. | |||||
| 1 | NPs-SiO2–SO3H | 1.6 | 29 | 71 | a |
| 2 | NPs-SiO2–C18 | 1.6 | 16 | 81 | 3 |
| 3 | NPs-SiO2–NH2 | 1.6 | 38 | 55 | 7 |
| 4 | NPs-SiO2–NH2 | 0.20 | 36 | 29 | 34 |
| 5 | NPs-SiO2–C18–SO3H | 1.6 | 31 | 69 | a |
| 6 | NPSi–SiO2–C18–SO3H | 0.20 | 26 | 62 | 12 |
| 7 | NPs-SiO2–C18–NH2 | 1.6 | 38 | 50 | 12 |
| 8 | NPs-SiO2–C18–NH2 | 0.20 | 34 | 41 | 25 |
| 9 | NPs-SiO2–C18–SO3H–NH2 | 0.20 | 18 | 48 | 39 |
Moreover, it is important to highlight the increase on the organic incorporation (atoms in T position) when the particle size is reduced (Table 2, entries 3–8), corroborating the results obtained from elemental analysis. When adding more than one organic group, a higher functionalization was reached. Thus, the number of T-type silicon atoms was higher in bi-functional materials and even highest in the tri-functional one (Table 2, entries 3, 7 and 9). For multi-functional NPs-SiO2 materials containing all three groups, up to 39% of organic incorporation was obtained in ∼30 nm particle size, according to 29Si NMR spectra (Fig. 6b and Table 2, entry 9), being this the highest value reached. With these spectroscopic results, the successful synthesis of functional silica nanoparticles was confirmed.
It is important to remark that for all materials synthetized containing thiol groups, an oxidation of these previously incorporated moieties was carried out in order to obtain the active sulfonic groups (–SO3H). The effectiveness of the procedure was followed by 13C NMR spectroscopy. The results showed that the chemical shift at 24 ppm, corresponding to thiol groups, was present in the materials prior to the oxidation process, and disappeared after the oxidation (Fig. 5a). In addition, a new chemical shift at 54 ppm appeared after the oxidation step, corresponding to the sulfonic groups, confirming the success of the oxidation process.
Thus, the present study shows the effective synthesis of spherical organosilica nanoparticles. Different organic fragments, both isolated and combined, have been successfully incorporated and preserved on its surface. These functionalized materials offer different hydrophobic/hydrophilic, acidic and basic properties that will be explored below for catalysed biomass conversion.
Catalytic activity
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO, C–OH, furan ring) allows the production of fuel additives, monomers (polymers), solvents, surfactants, herbicides, macrocycles, and other valuable chemicals by catalytic oxidation, hydrogenation, hydrogenolysis, amination and cascade reactions (Scheme 3).29,35 The most widely described route for the production of HMF is the acid catalysed dehydration of a hexose, in which three water molecules are consecutively eliminated (Scheme 4). From thermodynamic and kinetic standpoints,36 the production of HMF by fructose dehydration in presence of acid catalysts is an attractive alternative, and the fructose can be derived from the isomerization of glucose in a basic medium (Scheme 4). Glucose is a cheaper alternative to fructose and represents between 35 and 50% of the composition of the lignocellulosic biomass.37
C, CO, C–OH, furan ring) allows the production of fuel additives, monomers (polymers), solvents, surfactants, herbicides, macrocycles, and other valuable chemicals by catalytic oxidation, hydrogenation, hydrogenolysis, amination and cascade reactions (Scheme 3).29,35 The most widely described route for the production of HMF is the acid catalysed dehydration of a hexose, in which three water molecules are consecutively eliminated (Scheme 4). From thermodynamic and kinetic standpoints,36 the production of HMF by fructose dehydration in presence of acid catalysts is an attractive alternative, and the fructose can be derived from the isomerization of glucose in a basic medium (Scheme 4). Glucose is a cheaper alternative to fructose and represents between 35 and 50% of the composition of the lignocellulosic biomass.37
 | ||
| Scheme 3 HMF as a platform molecule. | ||
 | ||
| Scheme 4 Formation of HMF from fructose catalysed in acid medium and fructose from glucose basic isomerization. | ||
Therefore, the benefits of C18 hydrocarbon chains (–C18) over the catalytic performance of mono-functionalized silica nanoparticles with propyl-sulfonic groups (–SO3H) were investigated, using a combination of methyl isobutyl ketone (MIBK) and 1-butanol in a ratio of 70:30 as suitable solvent for fructose dehydration. This mixture, immiscible with water, allows the formation of a biphasic system that facilitates the continuous extraction of the HMF formed in the aqueous phase.
The catalytic activity of catalyst containing isolated sulfonic groups and both –C18 and –SO3H moieties were firstly tested for production of HMF from fructose (Fig. 7). The catalytic tests were carried out in a sealed vial under microwave irradiation. Microwave heating has numerous advantages over conventional heating, such as higher reaction rate, better yields and higher selectivity, high reproducibility due to the homogeneity of microwave irradiation and, finally, lower energy consumption.38,39 The data from Fig. 7 for the fructose conversion and HMF yield showed the benefits of the incorporation of the hydrophobic chain over the catalytic activity of the acid catalysts. Higher HMF yield and fructose conversion were obtained in the presence of the amphiphilic catalysts. These results were attributed to the hydrophobic properties of the alkyl chain that enhance reactivity due to better dispersion and organization of the amphiphilic catalyst with hydrophobic alkyl chain and polar sulfonic groups, at the interface of the organic phase and water, favouring mass transfer and migration of HMF to the organic phase after its formation. On the other hand, the catalytic performance of the NPs-SiO2 with particle sizes of ∼300 and ∼30 nm was previously tested and the yield of HMF was <1%, while the degradation of fructose was observed. These results allowed attributing the catalytic activity of the different functionalized organosilica nanoparticles for the dehydration of fructose to HMF to the presence of sulfonic groups.
 | ||
Fig. 7 HMF yield (■) and fructose conversion ( ) obtained for the fructose dehydration in the presence of solid acid catalysts: (a) NPs-SiO2–SO3H, (b) NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼400 nm, (c) NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼30 nm. Reaction composition: 300 mg fructose solution, 900 mg MIBK:BuOH 70:30 and 0.63 S mol%, 160 °C, 10 min. ) obtained for the fructose dehydration in the presence of solid acid catalysts: (a) NPs-SiO2–SO3H, (b) NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼400 nm, (c) NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼30 nm. Reaction composition: 300 mg fructose solution, 900 mg MIBK:BuOH 70:30 and 0.63 S mol%, 160 °C, 10 min. | ||
However, when the catalytic performance of NPs-SiO2–C18–SO3H catalysts with 1:4 C18/SH molar ratio and with different particle size of ∼400 nm and ∼30 nm of particle size were compared, only slightly superior catalytic data of glucose conversion and HMF yield were reached in the presence of the catalyst with lower particle size (Fig. 7). These results may suggest that selectivity and activity are correlated only with the number of acidic centres. Thus, NPs-SiO2–C18–SO3H catalyst with ∼30 nm of particle size was considered the best catalyst for further study to optimize the reaction parameters. The Fig. 8 displayed the trend of fructose conversion and HMF yield when the reaction temperature and time were varied. A progressive increase in conversion is observed under harsher reaction conditions until the maximum at which fructose is completely converted is reached. Complete conversion involves thermal degradation of fructose, which takes place from 100–105 °C to produce oligomerization products, hydration and aromatic compounds (Fig. 8).40 Progressive increase in HMF yield was observed with increasing reaction time and temperature, reaching a maximum at 180 °C and 3 min. This increase was more intense from 150 °C. An increase of 40 °C (from 100 to 140 °C) is observed to result in a 20% increase in fructose conversion, but had hardly any effect on the HMF yield, which only increased from 0% to 2.8%. In contrast, an increase in temperature from 140 °C to 150 °C resulted in a yield increase of 10%. At 150 °C, a total yield increase of 30% was achieved by increasing the reaction time from 1.5 to 11 min. At that point, the greatest change occurred with increasing reaction temperature, with a maximum yield of 60% HMF being achieved at 180 °C and after 3 min of reaction. Further increase in temperature induced higher HMF degradation leading to lower HMF yields.29
 | ||
Fig. 8 Trends of HMF yield ( ), fructose conversion ( ), fructose conversion ( ), HMF aqueous phase ( ), HMF aqueous phase ( ) and HMF organic phase (•) as a function of reaction conditions. Reaction composition: 300 mg fructose solution, 900 mg MIBK:BuOH 70:30 and NPs-SiO2–C18–SO3H (1:4), particle size of ∼30 nm, 3.8 wt%. ) and HMF organic phase (•) as a function of reaction conditions. Reaction composition: 300 mg fructose solution, 900 mg MIBK:BuOH 70:30 and NPs-SiO2–C18–SO3H (1:4), particle size of ∼30 nm, 3.8 wt%. | ||
In all cases, the HMF yield analysed in organic phase was higher than yield remaining in aqueous phase. These trends supported the efficiency of the continuous extraction process of HMF from aqueous phase regardless of the reaction conditions used. Under the optimized reaction conditions, practically complete conversion of fructose was reached, with 61% HMF selectivity, the HMF partition coefficient (R) was 3.1 (organic phase/water).
Next, the effect of the amount of catalyst on the yield and selectivity of HMF was explored, varying in the range of 2–9.5 wt% with respect to fructose, at 180 °C and for 3 min TOS. Practically no variation of HMF yield and fructose conversion were noted and the maximum data were achieved with 3.8 wt% of catalyst (Fig. S6†).
Different studies explored the effect of solvents over the preparation of HMF from fructose, including water,41 alcohols,42 DMSO (aprotic solvent),17–19 ionic liquids23 and solvent mixtures.20 Aqueous systems are the more attractive but HMF yields are generally <65% due to HMF rehydration to levulinic, formic acids and humins production. The addition of DMSO, a polar aprotic solvent to aqueous systems allowed improving fructose conversion from 91% to 94% and HMF selectivity from 60% to 67%.20 The effect of solvent on the rate and selectivity of fructose dehydration have been clearly reported. However, the use of DMSO, due to the complex recovery of HMF, requires a lot of energy and also leads to yield losses, which means that on an industrial level it is unaffordable.21 Therefore, the effects of the nature of solvent and the presence of two phases were explored using water, 1,4-dioxane, methanol and DMSO instead of the mixture MIBK:BuOH, employing the amphiphilic NPs-SiO2–C18–SO3H catalyst (Fig. 9). For all systems, a single-phase reaction system was obtained. The reactions were carried out at 160 °C for 10 min. The maximum HMF yield and fructose conversion were achieved using biphasic system MIBK:BuOH/H2O, enabling the continuous extraction of HMF from aqueous phase and lowering secondary reactions. Among the studied single-phase systems, DMSO showed the best results, while the worst result was obtained in water due to the secondary reactions favoured in this medium (rehydration and humins formation).
 | ||
Fig. 9 Influence of the solvent on the yield to HMF (■) and fructose conversion ( ) at 160 °C and 10 min of TOS. Reaction composition: 300 mg fructose solution, 900 mg solvent and catalyst NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼30 nm, 3.8 wt%. ) at 160 °C and 10 min of TOS. Reaction composition: 300 mg fructose solution, 900 mg solvent and catalyst NPs-SiO2–C18–SO3H 1:4 C18/SH molar ratio, particle size of ∼30 nm, 3.8 wt%. | ||
Taking into account previously published works,43,44 the reaction of fructose dehydration to HMF was considered as a first order process, and the rate of the reaction can be expressed as:

Finally, an attempt of reuse of the catalyst revealed a decrease in HMF yield and selectivity in comparison to fresh catalyst, achieving 42% HMF yield and 52% HMF selectivity, at 180 °C after 3 min of TOS (Table S3†). This result can be attributed to a decrease in the number of active acid sites since elemental analysis showed a decrease in sulphur content after the first run, from 3.9 to 2.7 wt% (Table S4†).
A comparison of the results with recent literature (Table S5†) revealed that similar HMF yield and selectivity data were achieved.47–57 The best results were obtained using DMSO as solvent, due to its great ability to suppress side reactions, being able to reach yields close to 100%.
Thus, we can conclude that the catalytic system described here using a biphasic medium (H2O/MIBK:BuOH) and an amphiphilic and acidic catalyst (NPs-SiO2–C18–SO3H) showed similar results to those described in a shorter reaction time.
 | ||
Fig. 10 Data of fructose yield (■), glucose conversion ( ) and selectivity ( ) and selectivity ( ) for the glucose isomerization in the presence of (a) NPs-SiO2–NH2, particle size of ∼400 nm, (b) NPs-SiO2–C18–NH2, particle size of ∼400 nm, (c) NPs-SiO2–NH2, particle size of ∼30 nm, (d) NPs-SiO2–C18–NH2, particle size of ∼30 nm. Reaction conditions: 100 °C, 10 min, 5 mol% N respect to glucose. ) for the glucose isomerization in the presence of (a) NPs-SiO2–NH2, particle size of ∼400 nm, (b) NPs-SiO2–C18–NH2, particle size of ∼400 nm, (c) NPs-SiO2–NH2, particle size of ∼30 nm, (d) NPs-SiO2–C18–NH2, particle size of ∼30 nm. Reaction conditions: 100 °C, 10 min, 5 mol% N respect to glucose. | ||
Reaction temperature and time were varied in the presence of the NPs-SiO2–NH2 catalyst (particle size of ∼30 nm) so as to find a maximum in fructose yield and selectivity. Fig. S9† shows a progressive increase in glucose conversion with increasing temperature up to 170–180 °C, where a maximum of 49% conversion and a maximum fructose yield of 12% was reached. Above 140 °C, fructose selectivity decreased due to its conversion to HMF. However, varying the reaction time did not improve either glucose conversion or fructose selectivity (Fig. S9†). Under optimum reaction conditions, at 180 °C, 3 min of TOS and 5 mol% N (respect to glucose), a 12% fructose yield with 23% fructose selectivity were achieved. It is important to note that the isomerization of glucose to fructose can be catalysed by both Lewis and Brønsted base and acid sites. However, the yield in the presence of bases is typically less than 10%,30 a yield similar to that obtained with NPs-SiO2–NH2 catalyst and compared to other results reported in the literature (Table S6†). A previous work compared the catalytic performance of amines with different chemical structure (acyclic/cyclic amine), nature of the functional group (primary, secondary, tertiary amine), and pKa.28 The results revealed that trimethylamine could isomerize glucose to fructose with similar selectivity (54%) and yield (31%) as Sn-Beta.58 Also, the authors concluded the higher catalytic properties of tertiary amines to isomerization process due to its no participation in side reactions (Maillard reaction). Subsequently, as future work, organosilica nanoparticles would be functionalized with secondary and tertiary amines to maximize the catalytic properties towards the isomerization of glucose to fructose.
Conclusions
Using an optimized Stöber method, the synthesis of organic–inorganic hybrid silica nanoparticles including one, two and/or three different accessible functionalities, being octadecyl, alkyl-thiol and alkyl-ammonium groups, has been efficiently carried out. These different organic moieties provide the silica nanoparticles with lipophilic, Brønsted acidic (after oxidation to sulfonic groups), Lewis-type and Brønsted-type basic properties, respectively. All materials have been fully characterized showing thermal stability up to 250–300 °C and a spherical and homogeneous morphology which is partially lost both upon introduction of alkyl-amino functional groups and upon functionalization of the smaller nanoparticles. Furthermore, the influence of the NH4OH concentration used during the synthesis process on the nanoparticle size is shown, allowing to reduce nanoparticles size down to 30 nm.The possible uses of the prepared materials as amphiphilic catalysts and with acid or base properties were evaluated. The material with sulfonic groups and C18 alkyl chains, i.e. with Brønsted acidic centres and hydrophilic/hydrophobic properties, has been used for the production of HMF by dehydration of fructose in a biphasic system (H2O/MIBK:BuOH), obtaining a 60% HMF yield. On the other hand, nanoparticles functionalized with amino groups were used for the production of fructose by glucose isomerization, with a yield of 12%. Both reactions took place in short reaction times, owing to the use of microwave heating, demonstrating the potential applicability of the synthesized materials as future multifunctional hybrid materials with controllable acid–base and hydrophobicity/hydrophilicity properties to carry out different catalysed reactions.
Author contributions
A. Coloma: investigation, data curation, formal analysis, methodology, visualization, and writing – original draft; A. Velty: conceptualization, data curation, formal analysis, methodology, validation, supervision, and writing – review & editing; U. Díaz: conceptualization, data curation, validation, funding acquisition, project administration, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from Grant PID2020-112590GB-C21 and from Severo Ochoa excellence program CEX2021-001230-S, both funded by MCIN/AEI/10.13039/501100011033. This study forms part of the Advanced Materials program and was supported by MICIN with funding from European Union NextGeneration (PRTR-C17.I1) and by Generalitat Valenciana (MFA/2022/003). A. C. acknowledge financial support from JAE-Intro 2020 – CSIC (JAEINT20_EX_0986).References
- J. S. Yuan, K. H. Tiller, H. Al-Ahmad, N. R. Stewart and C. N. Stewart Jr, Trends Plant Sci., 2008, 13, 421–429 CrossRef CAS PubMed.
- S. Elumalai, B. Agarwal, T. M. Runge and R. S. Sangwan, Biorefining of Biomass to Biofuels, 2018, pp. 87–116 Search PubMed.
- E. Farnetti, R. Di Monte and J. Kaspar, Inorganic and Bio-inorganic Chemistry – Vol. II – Homogeneous and Heterogeneous Catalysis, 2006 Search PubMed.
- P. Barbaro and F. Liguori, Heterogenized Homogeneous Catalysts for Fine Chemical Production, Springer, 2010 Search PubMed.
- U. Díaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 RSC.
- H. Knözinger and K. Kochloefl, Heterogeneous Catalysis and Solid Catalysts, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co., 2009 Search PubMed.
- X. Ma, L. Li, L. Yang, C. Su, K. Wang, S. Yuan and J. Zhou, J. Hazard. Mater., 2012, 209–210, 467–477 CrossRef CAS PubMed.
- M. C. Hernández-Soto, A. Erigoni, C. Segarra, F. Rey, U. Díaz, E. Gianotti, I. Miletto and M. Pera-Titus, Appl. Catal., A, 2022, 643, 118710 CrossRef.
- P. G. Jeelani, P. Mulay, R. Venkat and C. Ramalingam, Silicon, 2020, 12, 1337–1354 CrossRef CAS.
- S. L. Greasley, S. J. Page, S. Sirovica, S. Chen, R. A. Martin, A. Riveiro, J. V. Hanna, A. E. Porter and J. R. Jones, J. Colloid Interface Sci., 2016, 469, 213–223 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- B. G. Cha and J. Kim, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, 1–22 Search PubMed.
- M. Fuji and C. Takai, Superior Thermal Insulation Film with Transparency Achieved by Hollow Silica Nanoparticles, Elsevier B.V., 2018 Search PubMed.
- M. Manzano and M. Vallet-Regí, J. Mater. Sci.: Mater. Med., 2018, 29, 65 CrossRef PubMed.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- W. J. Zhou, L. Fang, Z. Fan, B. Albela, L. Bonneviot, F. De Campo, M. Pera-Titus and J. M. Clacens, J. Am. Chem. Soc., 2014, 136, 4869–4872 CrossRef CAS PubMed.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitz, Appl. Catal., B, 2016, 181, 874–887 CrossRef CAS.
- A. S. Amarasekara, L. T. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021 CrossRef CAS PubMed.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591–5600 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097–2128 RSC.
- Y. N. Li, J. Q. Wang, L. N. He, Z. Z. Yang, A. H. Liu, B. Yu and C. R. Luan, Green Chem., 2012, 14, 2752–2758 RSC.
- K. Buchholz and J. Seibel, Carbohydr. Res., 2008, 343, 1966–1979 CrossRef CAS PubMed.
- J. M. Carraher, C. N. Fleitman and J. P. Tessonnier, ACS Catal., 2015, 5, 3162–3173 CrossRef CAS.
- M. Fischer, P. Drabo and I. Delidovich, React. Kinet., Mech. Catal., 2022, 135, 2357–2377 CrossRef CAS.
- S. Zhao, X. Guo, P. Bai and L. Lv, Asian J. Chem., 2014, 26, 4537–4542 CrossRef CAS.
- C. Liu, J. M. Carraher, J. L. Swedberg, C. R. Herndon, C. N. Fleitman and J. P. Tessonnier, ACS Catal., 2014, 4, 4295–4298 CrossRef CAS.
- C. Bispo, K. De Oliveira Vigier, M. Sardo, N. Bion, L. Mafra, P. Ferreira and F. Jérôme, Catal. Sci. Technol., 2014, 4, 2235–2240 RSC.
- C. Liu, J. M. Carraher, J. L. Swedberg, C. R. Herndon, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2014, 4, 4295–4298 CrossRef CAS.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. Sing, Adsorption by powders and porous solids: principles, methodology and applications, Accademic Press, 2013 Search PubMed.
- Ö. Tüncel, E. Kahraman, G. Bağci, N. Atabey and S. Özçelik, Mater. Sci. Eng. C, 2020, 119, 111585 CrossRef PubMed.
- J. Gao, X. Zhang, Y. Lu, S. Liu and J. Liu, Chem.–Eur. J., 2015, 21, 7403–7407 CrossRef CAS PubMed.
- A. Idris, Z. Man, A. S. Maulud, M. A. Bustam, H. A. Mannan and I. Ahmed, Appl. Surf. Sci., 2020, 506, 144978 CrossRef CAS.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2008, 9, 2244–2249 CrossRef CAS.
- M. Gaba and N. Dhingra, Indian J. Pharm. Educ. Res., 2011, 45, 175–183 Search PubMed.
- N. Sharma, U. K. Sharma and E. V. Van der Eycken, Green Techniques for Organic Synthesis and Medicinal Chemistry, 2018, pp. 441–468 Search PubMed.
- A. Golon and N. Kuhnert, J. Agric. Food Chem., 2012, 60, 3266–3274 CrossRef CAS PubMed.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- M. H. Tucker, R. Alamillo, A. J. Crisci, G. M. Gonzalez, S. L. Scott and J. A. Dumesic, ACS Sustainable Chem. Eng., 2013, 1, 554–560 CrossRef CAS.
- Y. Li, X. Lu, L. Yuan and X. Liu, Biomass Bioenergy, 2009, 33, 1182–1187 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- J. Wang, H. Cui, J. Wang, Z. Li, M. Wang and W. Yi, Chem. Eng. J., 2021, 415, 128922 CrossRef CAS.
- Y. Zhu, K. Song, X. Xu, J. He and J. Guo, Catalysts, 2022, 12, 984 CrossRef CAS.
- C. Antonetti, A. M. Raspolli Galletti, S. Fulignati and D. Licursi, Catal. Commun., 2017, 97, 146–150 CrossRef CAS.
- E. I. García-López, F. R. Pomilla, B. Megna, M. L. Testa, L. F. Liotta and G. Marcì, Nanomaterials, 2021, 11, 1–20 Search PubMed.
- M. L. Testa, G. Miroddi, M. Russo, V. La Parola and G. Marcì, Materials, 2020, 13, 1178 CrossRef CAS PubMed.
- R. Tomer and P. Biswas, New J. Chem., 2020, 44, 20734–20750 RSC.
- S. Karimi, F. Seidi, M. Niakan, H. Shekaari and M. Masteri-Farahani, Renewable Energy, 2021, 180, 132–139 CrossRef CAS.
- S. Wang, T. L. Eberhardt and H. Pan, Fuel, 2022, 316, 123255 CrossRef CAS.
- I. S. Omodolor, S. Kalidindi, S. A. Walz, M. R. Coleman, R. Gogar, S. Viamajala, M. López Granados and A. C. Alba-Rubio, Catal. Sci. Technol., 2023, 13, 132–146 RSC.
- J. Wang, H. Zhao, B. Zhu, S. Larter, S. Cao, J. Yu, M. G. Kibria and J. Hu, ACS Catal., 2021, 11, 12170–12178 CrossRef CAS.
- P. Dugkhuntod, N. Maineawklang, C. Rodaum, P. Pornsetmetakul, K. Saenluang, S. Salakhum and C. Wattanakit, ChemPlusChem, 2022, 87, 1–9 CrossRef PubMed.
- X. Ye, X. Shi, B. Jin, H. Zhong, F. Jin and T. Wang, Sci. Total Environ., 2021, 778, 146276 CrossRef CAS PubMed.
- M. S. Rahaman, S. Tulaphol, M. A. Hossain, J. B. Jasinski, N. Sun, A. George, B. A. Simmons, T. Maihom, M. Crocker and N. Sathitsuksanoh, Fuel, 2022, 310, 122459 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01486j |
| This journal is © The Royal Society of Chemistry 2023 |