
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracing the transition from covalent to non-covalent functionalization of pyrene through C-, N-, and O-based ionic and radical substrates using quantum mechanical calculations†
Anwesh Pandey * and
Nandan Kumar*
* and
Nandan Kumar*
Advanced Computation and Data Sciences Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India. E-mail: apdapbbau@gmail.com; mr.kumarnandan@gmail.com
First published on 12th May 2023
Abstract
Pyrene is one of the widely investigated aromatic hydrocarbons given its unique optical and electronic properties. Modulating inherent characteristics of pyrene via covalent or non-covalent functionalization has been attractive for a wide variety of advanced biomedical and other device applications. In this study, we have reported the functionalization of pyrene via C, N, and O based ionic and radical substrates, and emphasized the transition of covalent to non-covalent functionalization through making the modulation in the substrate. As expected, strong interactions were observed for cationic substrates, however, anionic substrates also exhibited a competitive binding strength. For instance, methyl and phenyl substituted CH3 complexes exhibited IEs in the range of −17 kcal mol−1 to −127 kcal mol−1 and −14 kcal mol−1 to −95 kcal mol−1 and for cationic and anionic substrates, respectively. The analysis of topological parameters showed that un-substituted cationic, anionic, and radical substrates interact with pyrene via covalent interactions, and further become non-covalent upon methylation and phenylation of the substrates. In cationic complexes, the polarisation component is observed to be dominating the interactions, whereas highly competitive contributions from polarization and exchange components were observed in anionic and radical complexes. The contribution of the dispersion component increases with an increase in the degree of methylation and phenylation of the substrate, and starts dominating once the interactions become non-covalent in nature.
Introduction
Polyaromatic hydrocarbons (PAHs) are a ubiquitous family of aromatic compounds obtained by fusing two or more benzene rings. PAHs are of tremendous research interest due to their higher stability, rigid and planar structure, and their characteristic spectra.1–7 Pyrene is a peri-fused PAH consisting of four benzene structures and possesses excellent emission properties, a long excited-state lifetime, and successful electron hole-pair dissociation.8–11 The functionalization studies via reversible and irreversible means (non-covalent and covalent respectively) have been in tremendous surge over the past decade, especially for carbon-based nanostructures.12–15The functionalization of pyrene through various functional groups via traditional synthesis approaches viz., formylation/acetylation, bromination, alkylation, oxidation, and borylation reactions have been studied well highlighting its optical properties for device applications such as organic light-emitter diodes, as well as organic photovoltaics, solar cells, and production of metal–organic frameworks.16–18 The structural variability and aromatic characteristics of pyrene allow the covalent and non-covalent functionalization with different reactants to enhance the optical properties, electron transfer efficiency, and others.19–23 The covalent functionalization employs specific chemically active functional groups to convert sp2-hybridized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds into sp3-hybridized C–C bonds, which would induce defects in pyrene. However, the non-covalent functionalization has little damage on the structure of pyrene and can maintain most of the original properties.24
C bonds into sp3-hybridized C–C bonds, which would induce defects in pyrene. However, the non-covalent functionalization has little damage on the structure of pyrene and can maintain most of the original properties.24
The π–π, XH–π, cation–π, anion–π and other non-covalent interactions have been identified to play an important role in the non-covalent functionalization of pyrene and other PAHs.25–27 Sastry et al. has comprehensively studied a range of PAHs interacting with different substrates via non-covalent interactions and has emphasized the nature of interactions,28–30 and their modulation based on the size and nature of PAHs and substrates.31–35 Interaction of different substrates with pyrene is also undertaken in several other studies highlighting the functionalization of pyrene or other pyrene-like model systems for a range of applications.25–27,36,37
Recently, the interaction of pyrene with biomolecules and other systems have also been studied. Sengottiyan et al. has reported the binding studies of plasma proteins pyrene derivatives addressing the antimicrobial resistance.36 Another study reports the role of pyrene in probing different protein conformations thereby highlighting its importance in molecular organization of biological molecules37 and in elucidating how mechanochromic luminescence behaviours can be modulated by side-chain substituents.38 Several studies have reported pyrene as a model system for studying the properties of graphene for various applications, viz., field effect transistors, hydrogen chemisorption, reactions in interstellar medium, etc.39–43 However, the nature of interactions as well as the transition between covalent and non-covalent interactions and vice versa has not yet been thoroughly examined.
In the present work, we have comprehensively studied the functionalization of pyrene via different cationic, anionic, and radical substrates such as H, CH3, NH2, and OH, and their methylated and phenylated analogs. The modulation of the substrate and its effects on the pyrene structures followed by the transition from covalent to non-covalent functionalization is studied in detail. The energy decomposition analysis of these complexes was carried out to quantify the nature of interactions and to explain the role of energy components such as electrostatic, polarization, and dispersion in the stabilization of pyrene-substrate complexes. This study will be helpful in providing insights into the functionalization of pyrene-based materials for organic and advanced biomedical applications.
Computational details
The geometries of pyrene-substrate complexes were optimized at M06-2X/6-31G* level of theory, and the energetics, such as, interaction energy (IE) and deformation energy (DE) was calculated at M06-2X/6-311G** level of theory. The choice of M06-2X functional for the study is based on its suitability for main-group chemistry, molecular structure prediction, and quantification of non-covalent interactions.44,45 However, we also carried out benchmark calculations using 21 different levels of theory (Table S1†) in order to verify the performance of the M06-2X for pyrene-substrate complexes, and it was observed to be performing in concordance, as shown in ESI (Tables S2 and S3†).IE was obtained by calculating the difference between the total energy of the pyrene–substrate complex (EPy_S) and the sum of the individual energies of the pyrene (EPy) and substrate (ES), as mentioned in eqn (1).46 DE was calculated as the difference in the energy of pyrene before (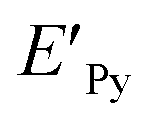 ) and after complexation (EPy), as given in eqn (2).47
) and after complexation (EPy), as given in eqn (2).47
| IE = EPy_S − (EPy + Es) | (1) |
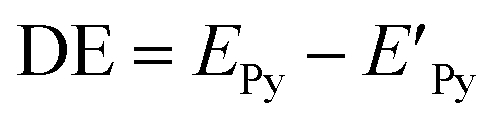 | (2) |
Natural population analysis (NPA)48 was performed to inspect the charge transfer among pyrene and substrate in complexes at M06-2X/6-311G**//M06-2X/6-31G* level of theory. All these calculations were carried out using Gaussian16 package.49 We further decomposed the interaction energies into different energy components by carrying out energy decomposition analysis using the localized molecular orbital energy decomposition analysis (LMO-EDA) scheme.50,51
| IE = Eele + Eexc + Erep + Epol + Edis | (3) |
In LMO-EDA schema, the interaction energy is segregated into five components viz., electrostatic (Eele), exchange (Eexc), repulsion (Erep), polarization (Epol), and dispersion (Edis) energies (eqn (3)). The energy decomposition analysis of all the pyrene-substrate complexes was performed using GAMESS-US52 at M06-2X/6-311G** level of theory.
Further, to characterize the covalent and non-covalent interactions,53 quantum theory of atoms in molecules (QTAIM) was employed to analyse the topological parameters at bond critical points for the pyrene-substrate complexes at M06-2X/6-311G** level. The kinetic energy density (G(r)), and potential energy density (V(r)) was examined to calculate the Laplacian of electron density (∇2ρ) and total energy density (H(r)) using eqn (4) and (5), respectively, these analyses were carried out using AIM2000 software.54 Further, we analysed the regions of non-covalent interactions using Multiwfn software55 and represented them using NCI plot, and related figures were generated using VMD.56
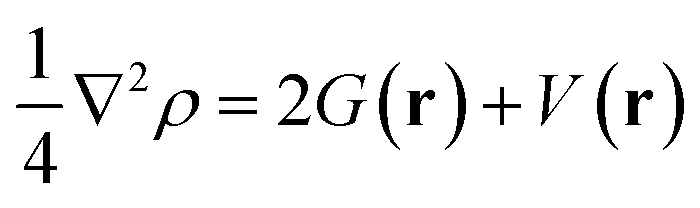 | (4) |
| H(r) = G(r) + V(r) | (5) |
Results and discussion
In the present work, the covalent and non-covalent functionalization of pyrene (C16H10) was studied by considering the C, N, and O based cationic, anionic, and radical substrates. The considered substrates are *H, *CH3, *NH2, and *OH, and their methylated (–CH3) and phenylated (–C6H5) analogs, as shown in Scheme 1. Asterisk (*) represents the atom of substrate interacting with pyrene.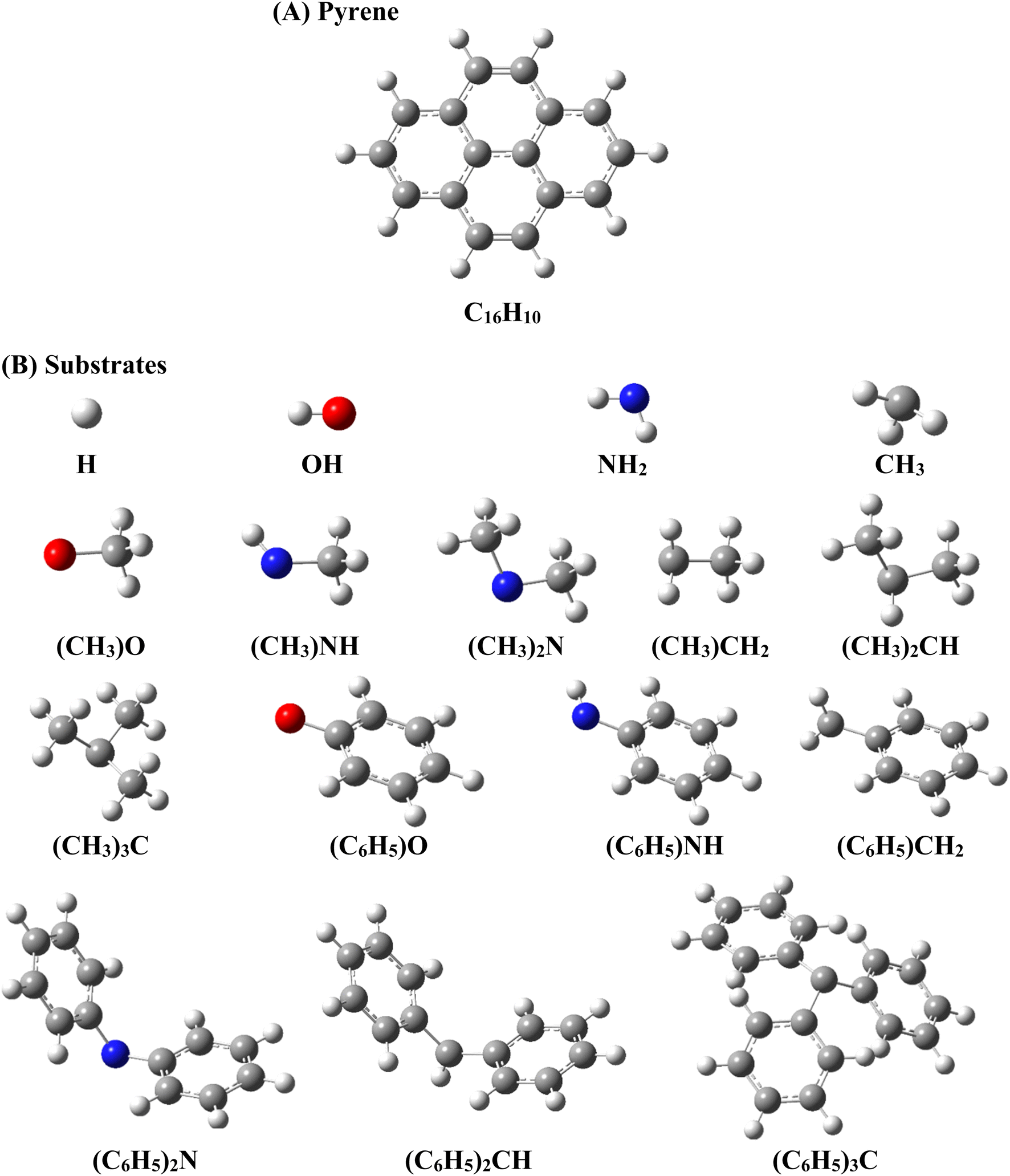 | ||
| Scheme 1 Schematic representation of (A) pyrene and (B) substrates considered in this study. | ||
This section is divided into five subsections. In the first subsection, we have discussed the geometrical parameters of pyrene-substrate complexes. The second subsection gives an account on the variation of interaction and deformation energies with respect to the substrate. Then, the charge transfer between pyrene and substrate during complexation was elucidated followed by the energy decomposition analysis. Finally, the nature of substrate binding to pyrene was estimated based on QTAIM parameters, i.e., ρ, ∇2ρ, H(r), and (−[G(r)/V(r)]), and the transition from covalent to non-covalent complexes was presented in detail. Additionally, NCI surfaces were analysed to highlight the non-covalently bound regions via colour coded representations.
Analysis of geometrical parameters
The geometrical parameters of pyrene-substrate complexes were calculated at M06-2X/6-31G* level of theory, as shown in Fig. 1, 2 and S1–S6.† We further calculated the geometrical parameters at 21 different methods to examine the consensus among the values calculated at the aforementioned methods, which suggested that M06-2X/6-31G* seems to be performing well, as shown in Fig. S7 and Table S2.†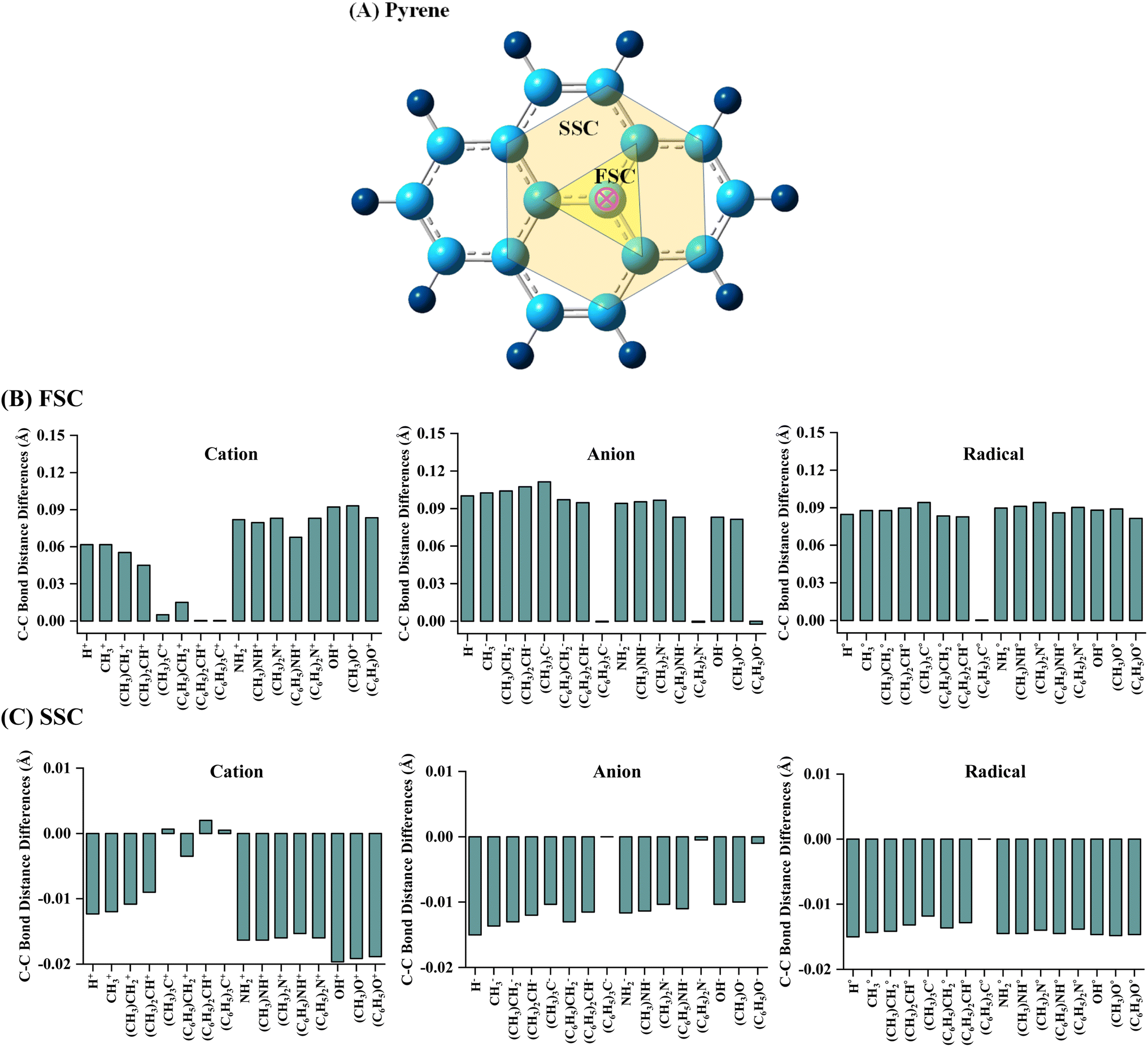 | ||
| Fig. 1 The representation of (A) pyrene (C16H10) structure with first sphere carbons (FSC) and second sphere carbons (SSC) of interacting site carbon (pink circle), and variation in C–C bond distance of pyrene (B) FSC and (C) SSC while interacting with cationic, anionic, and radical substrates. Calculations are done at M06-2X/6-31G* level of theory. Bond distance differences are calculated with respect to the pristine structure. Positive and negative values show lengthening and shortening of the C–C bond of pyrene while interacting with substrates. | ||
The C–C bond distances of pyrene were analysed in order to inspect the effect of substrate binding to the pyrene. The carbons forming the C–C bond with the carbon atom of the interacting site of pyrene are labelled as first sphere carbons (FSC) and the carbons forming the C–C bond with FSC carbons are labelled as second sphere carbons (SSC), as shown in Fig. 1A. A cursory view of the calculated C–C bond distance of pyrene showed an alternate lengthening and shortening of bonds irrespective of the type of substrates (Table S3†). However, no noticeable changes were observed for C–C bond length of pyrene beyond SSC. In comparison to the pristine structure, a lengthening of 0.01 Å to 0.12 Å was observed for cationic and anionic complexes and 0.01 Å to 0.09 Å for radical complexes. The highest C–C bond lengthening in cationic complexes was observed for OH and NH2 and their –CH3 and –C6H5 substituted analogs, whereas, a maximum increase was observed for CH3 and its –CH3 substituted analogs in the case of anionic complexes. Besides, it was observed that the lengthening of C–C bonds of FSC decreases upon –CH3 and –C6H5 substitution in cationic complexes. In contrast, it is noteworthy that an increase in the lengthening of C–C bonds was observed in anionic and radical complexes (Fig. 1B).
The C–C bond distance of SSC seemed to be inversely proportional to the lengthening of the C–C bond of FSC (Fig. 1C). Further, we analysed the bond distance between the interacting atom of pyrene and substrate (R0). The shortest R0 was observed for cationic, anionic, and radical H substrate, i.e., ∼1.20 Å, and the longest R0 was observed for tri-methylated methyl ((C6H5)3C) cation, anion, and radical substrates, i.e., ∼3.60 Å to ∼4.10 Å, as shown in Fig. 2 and S1–S6.† The bond distance between pyrene and substrate (R0) of un-methylated and un-phenylated substrate complexes followed the trend; H < OH < NH3 < CH3.
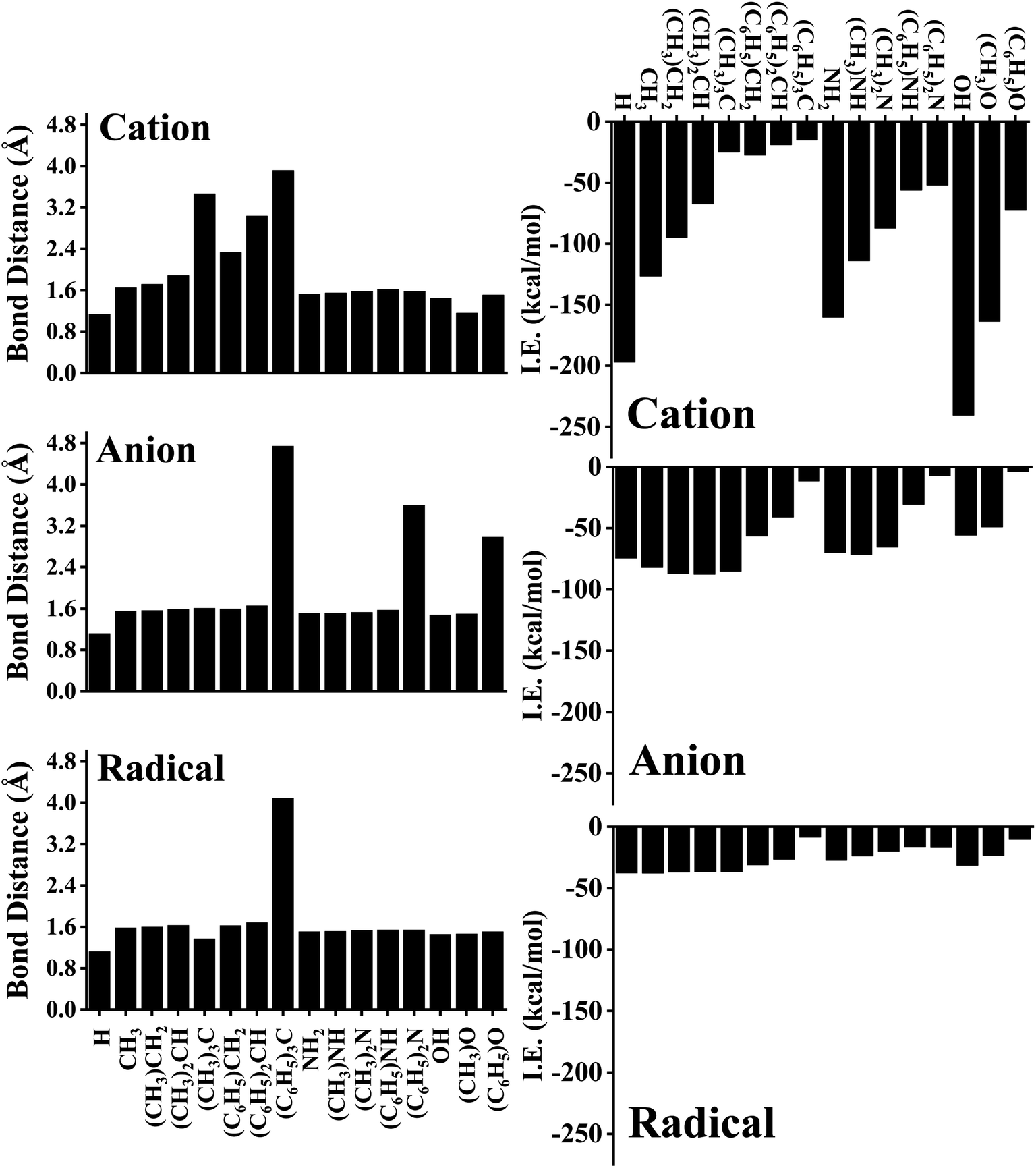 | ||
| Fig. 2 The variation of the bond length (R0; Å) with corresponding interaction energy (IE; kcal mol−1) for each pyrene–substrate complex. Calculations are done at M06-2X/6-311G**//M06-2X/6-31G* level of theory. | ||
Upon methylation and phenylation of these substrates exhibited a significant increment in R0, i.e., ∼1.20 Å to ∼4.10 Å, suggesting that the interaction between pyrene and substrates may undergo a transition between covalent to non-covalent interactions upon increasing the methylation and phenylation. The transition of covalent and non-covalent interactions was inspected by examining the different energetics, and the topological parameters of pyrene-substrate complexes.
Energetics analysis
The BSSE corrected interaction energy of pyrene-substrate complexes calculated at M06-2X/6-311G**//M06-2X/6-31G* level of theory is shown in Fig. 2 and Table 1. In order to verify performance of M06-2X/6-311G**//M06-2X/6-31G*, we further carried out the benchmark study for H, CH3 and (CH3)3C complexes using 21 different levels of theory, and calculated relative energy (RE) (absolute value) between the interaction energies obtained at M06-2X/6-311G**//M06-2X/6-31G* and the considered 21 methods, as shown in Table S4.† The calculated RE was observed to be varying between 0.08 kcal mol−1 to 2.77 kcal mol−1 for pyrene-H complexes, 0.00 kcal mol−1 to 3.62 kcal mol−1 for pyrene-CH3 complexes, and 0.00 kcal mol−1 to 2.86 kcal mol−1 for pyrene-(CH3)3C complexes, except for the geometries obtained at HF and MP2. These benchmark results suggested that the values obtained at M06-2X/6-311G**//M06-2X/6-31G* level of theory are in good accordance with the majority of the considered 21 different levels of theory, and hence we have considered it for the further discussion.| System | Interaction energy | Deformation energy | Charge transfer | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cation | Anion | Radical | Cation | Anion | Radical | Cation | Anion | Radical | |
| H | −197.03 | −74.44 | −37.65 | 12.09 | 39.73 | 26.13 | 1.393 | −1.147 | −0.252 |
| CH3 | −126.43 | −82.07 | −37.69 | 15.61 | 41.94 | 29.91 | 0.758 | −0.990 | −0.087 |
| (CH3)CH2 | −94.62 | −87.03 | −36.87 | 14.31 | 43.16 | 30.77 | 0.708 | −0.991 | −0.094 |
| (CH3)2CH | −67.45 | −87.67 | −36.52 | 11.64 | 45.82 | 33.23 | 0.605 | −0.987 | −0.100 |
| (CH3)3C | −24.96 | −85.11 | −36.52 | 0.32 | 49.32 | 36.87 | 0.086 | −0.985 | −0.099 |
| (C6H5)CH2 | −27.28 | −56.45 | −30.96 | 1.20 | 40.06 | 28.96 | 0.290 | −0.920 | −0.062 |
| (C6H5)2CH | −18.91 | −40.99 | −26.42 | 0.24 | 40.75 | 30.55 | 0.067 | −0.854 | −0.098 |
| (C6H5)3C | −14.96 | −11.62 | −8.45 | 0.35 | 0.35 | 0.18 | 0.018 | −0.803 | −0.171 |
| NH2 | −160.26 | −69.74 | −27.27 | 24.08 | 38.04 | 30.82 | 1.007 | −0.799 | 0.113 |
| (CH3)NH | −113.99 | −71.45 | −23.75 | 24.24 | 39.12 | 32.17 | 0.977 | −0.789 | 0.115 |
| (CH3)2N | −87.21 | −65.35 | −19.81 | 27.35 | 41.88 | 35.36 | 0.962 | −0.769 | 0.118 |
| (C6H5)NH | −56.07 | −30.53 | −16.53 | 19.89 | 34.07 | 29.98 | 0.902 | −0.702 | −0.154 |
| (C6H5)2N | −51.95 | −7.04 | −16.95 | 22.74 | 0.26 | 33.59 | 0.914 | −0.039 | 0.188 |
| OH | −240.47 | −55.67 | −31.39 | 27.58 | 33.24 | 29.42 | 1.193 | −0.651 | 0.268 |
| (CH3)O | −163.45 | −48.92 | −23.27 | 28.66 | 33.14 | 30.51 | 1.194 | −0.622 | 0.280 |
| (C6H5)O | −72.00 | −3.80 | −10.31 | 24.92 | 0.23 | 27.22 | 1.161 | −0.037 | 0.324 |
As expected, cationic complexes exhibited a higher value of IE than anionic and radical complexes except for methylated CH3 substrates which exhibits a competitive binding strength. The higher and lower values of IE of cationic, anionic, and radical complexes are as follows: OH+ (−240.47 kcal mol−1) and (C6H5)3C+ (−14.96 kcal mol−1) for cationic, (CH3)CH2− (−87.67 kcal mol−1) and (C6H5)O− (−3.80 kcal mol−1) for anionic, and CH3˙ (−37.69 kcal mol−1) and (C6H5)3C˙ (−8.45 kcal mol−1) for radical complexes respectively. Since R0 showed a significant increment (∼1.20 Å to ∼4.10 Å) upon methylation and phenylation of CH3, NH3, and OH substrates, we examined the variation of IE of complexes upon methylation and phenylation of the substrate. The IE of cationic complexes decreased from −126.43 kcal mol−1 to −14.96 kcal mol−1 (for CH3-based substrates), −160.26 kcal mol−1 to −51.95 kcal mol−1 (for NH2-based substrates), and 240.47 kcal mol−1 to −72.00 kcal mol−1 (for OH-based substrates) with increasing methylation and phenylation. In contrast, in the case of anionic complexes, a significant variation of IE was not observed upon methylation of the CH3 substrate (i.e., from −82.07 kcal mol−1 to −87.67 kcal mol−1), and exhibits a competitive binding strength to cationic complexes, and the same was observed for methylated NH2 complexes, i.e., −69.74 kcal mol−1 to −71.45 kcal mol−1 and OH complexes, i.e., −55.67 kcal mol−1 to −48.92 kcal mol−1. However, a significant decrement in the IEs was observed upon phenylation of CH3 (−82.07 kcal mol−1 to −11.62 kcal mol−1), NH2 (−69.74 kcal mol−1 to −7.04), and OH complexes (−55.67 kcal mol−1 to −3.80 kcal mol−1).
Although methylation and phenylation of anionic CH3, NH3, and OH substrates lead to the weakening of pyrene-substrate complexes (except for methylated complexes), they have shown comparable interaction energies to typical covalent and other strong non-covalent interactions. The radical substrates exhibited the IE in the range of −8.45 kcal mol−1 to −37.69 kcal mol−1, and similarly, IE was not observed to be significant varying upon methylation of substrates, however, phenylated complexes exhibited 2 to 5 folds decrement in the IEs with the higher degree of phenylation. We further examined deformation energy (DE) in order to explain the effect of cationic, anionic, and radical substrates over on the surface of pyrene. A cursory view of Table 1 suggested that complexes having higher IEs tend to undergo high deformations. In the case of cationic complexes, maximum deformation was observed for OH complexes followed by NH2 complexes, and minimum deformation was observed for methylated and phenylated CH3 complexes.
Similar observation was made for radical complexes, as shown in Table 1. In contrast, in the case of anionic complexes, high deformation was observed for methylated and phenylated CH3 complexes. We further compared the DE with C–C bond lengths in FSC, SSC, and beyond SSC regions of pyrene in cationic, anionic, and radical complexes, as shown in Fig. S8.† DE exhibited a positive correlation which implied lengthening of the C–C bond of FSC region of cationic, anionic, and radical complexes i.e., R2 = 0.96, R2 = 0.99, and R2 = 0.95, respectively. However, in the case of anionic complexes, a negative correlation was observed which implied shortening the C–C bond of SSC region of cationic, anionic, and radical complexes, i.e., R2 = 0.95, R2 = 0.89, and R2 = 0.74, respectively. Besides, a good correlation for radical (R2 = 0.82), fair correlation for cationic (R2 = 0.67), and poor correlation for anionic (R2 = 0.28) complexes was observed for the C–C bond lengths of pyrene beyond SSC region. Hence, these results suggested that deformation of the pyrene structure is primarily due to the changes in the C–C bond lengths of FSC followed by SSC while interacting with the substrates.
Charge analysis
The variation in the charge transfer between pyrene and substrate was examined by calculating the NPA charges at M06-2X/6-311G**//M06-2X/6-31G* level of theory, as shown in Table 1.Charge transfer observed to be directly proportional to the IEs of pyrene-substrate complexes with a few exceptions. Cationic phenylated and methylated complexes exhibited the minimum charge transfer, however, un-methylated or un-phenylated complexes exhibited higher charge transfer. Among the cationic complexes, OH substrate complexes exhibited the high charge transfer, since the IEs of OH was higher than the other cationic substrates.
A similar type of observation was made for anionic and radical complexes, as shown in Table 1. Further, we examined the variation of charge transfer with respect to degree of methylation and phenylation of the substrate, as shown in Fig. 3. A good correlation was observed between IE and CT of methyl cation and its methylated analogs (R2 = 0.86), while such correlation was not observed for anionic and radical complexes due to negligible variation in the CT and IE of methylated complexes. In the case of phenylated analogs of methyl cation, anion, and radical, a positive correlation was observed for cationic (R2 = 0.93) and radical (R2 = 0.81) complexes, and a negative correlation (R2 = 0.97) was observed for anionic complexes. Similarly, a good correlation was observed for methylated (R2 = 0.99) and phenylated (R2 = 0.98) NH2 cations, and a reasonable negative correlation (R2 = 0.98) was observed for phenylated NH2 cation. Overall, the variation in IE of pyrene-substrate complexes is found to be linearly dependent on CT between pyrene and substrate. Overall, the charge transfer variation for the –N substrates with phenyl groups is larger than those observed in other subfigures of Fig. 3. This difference may be attributed to the electronic properties of the phenyl group, which can affect the charge transfer between the substrate and the molecule. Additionally, the size and shape of the phenyl group may also play a role in the observed charge transfer variations.
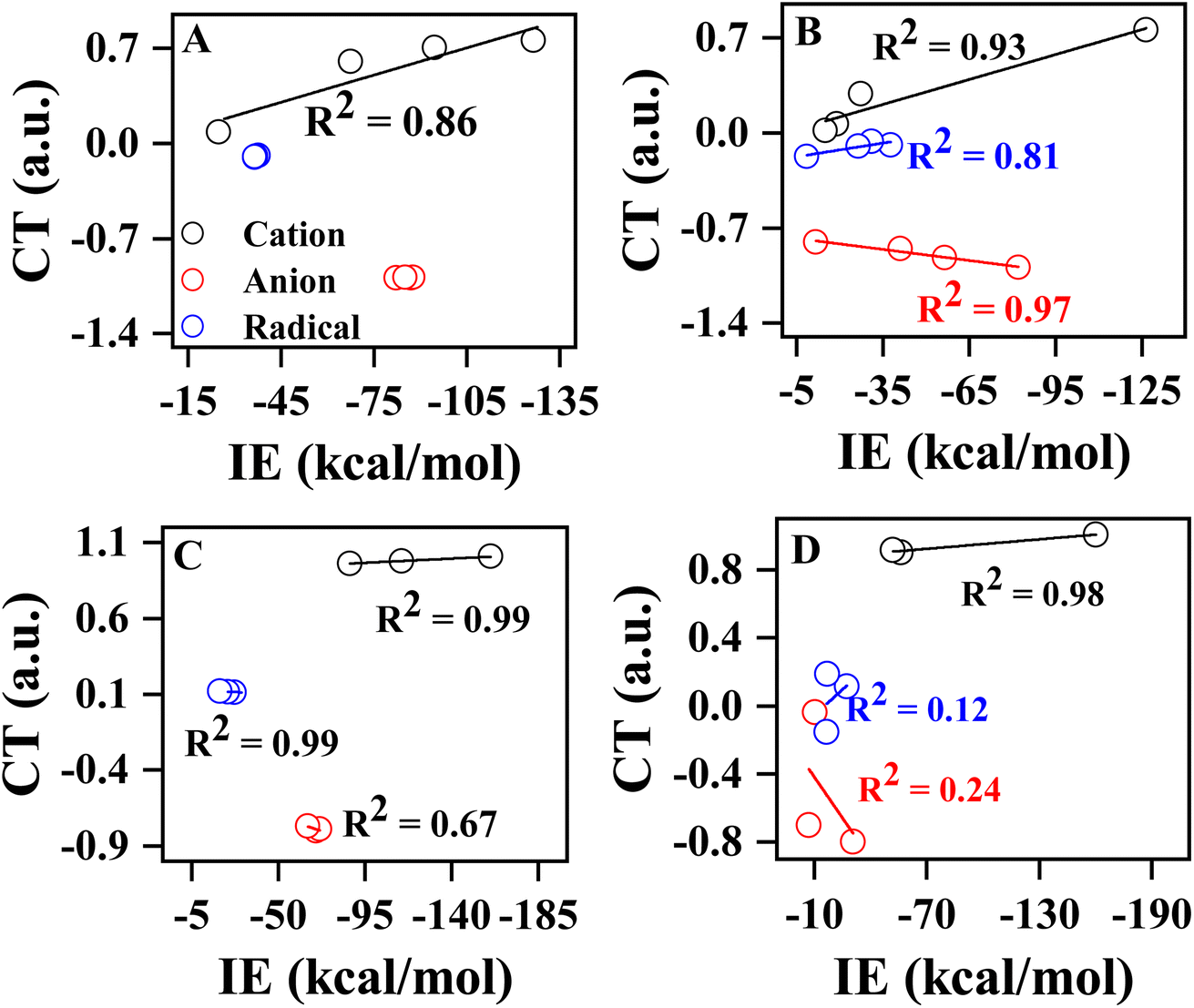 | ||
| Fig. 3 The variation of charge transfer (CT; a.u.) with respect to interaction energy (IE; kcal mol−1) for CH3 and C6H5 substituted substrates interacting with C16 H10 (A) CH3 to C(CH3)3 (B) CH3 to C(C6H5)3 (C) NH2 to N(CH3)2 and (D) NH2 to N(C6H5)2. Calculations are done at M06-2X/6-311G**//M06-2X/6-31G* level of theory. | ||
Contribution from different energy components
We performed localized molecular orbital-based energy decomposition analysis in order to elucidate the contribution of different energy components into the IE of pyrene-substrate complexes, as shown in Fig. 4, Tables S4 and S5.† A cursory view of percentage contribution of energy components such as electrostatic, exchange, polarization and dispersion showed that cationic complexes are predominantly polarization driven, followed by closely competing electrostatic and exchange components except for fully-methylated and phenylated CH3 complexes.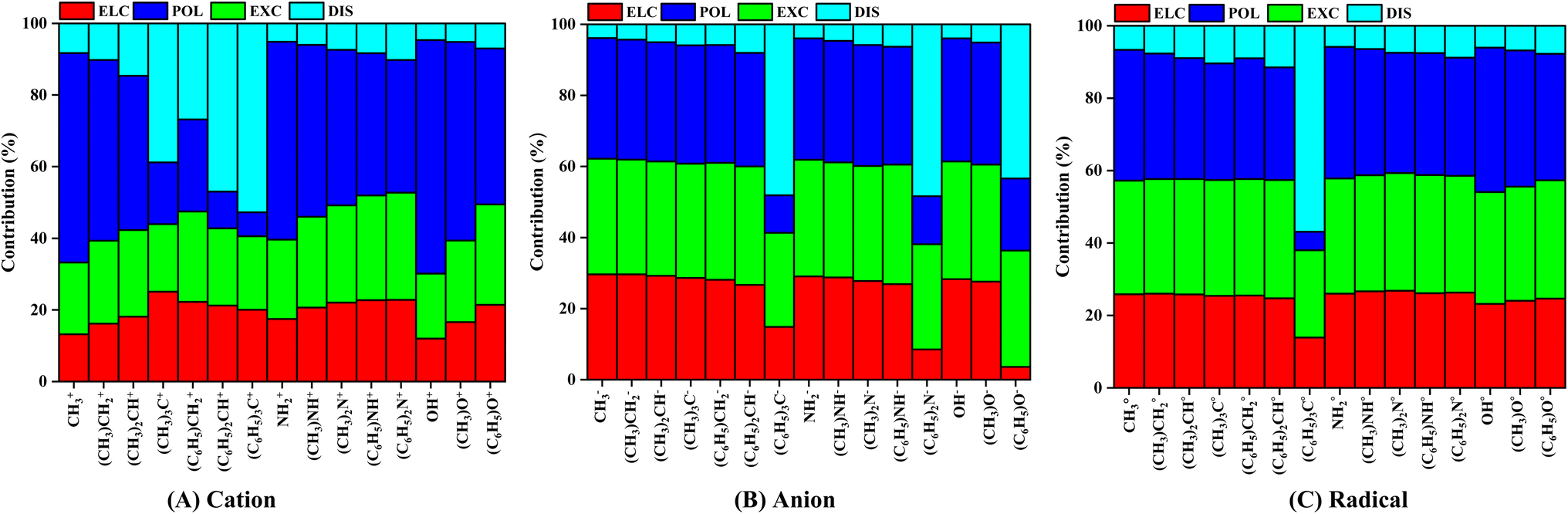 | ||
| Fig. 4 The variation in the percentage contribution of energy components such as electrostatic (ELC), polarization (POL), exchange (EXC), and dispersion (DIS) into the interaction energy of (A) cation, (B) anion, and (C) radical substrates with pyrene. Calculations are done at M06-2X/6-311G**//M06-2X/6-31G* level of theory. | ||
Highest contribution of polarization component was observed for un-substituted complexes, viz., CH3+ (58.48%), NH2+ (55.22%), and OH+ (65.24%). As the degree of methylation and phenylation increases, the contribution of polarization component minutely decreases, however, the contribution of other components minutely increases, except for phenylated CH3 complexes. In the case of fully-methylated and phenylated CH3+ complexes, the contribution of dispersion component increased by 5 to 7 folds, i.e., from CH3+ (8.28%) to (CH3)3C+ (38.81%), and (C6H5)3C+ (52.74%). However, such significant increases in the contribution of dispersion component were not observed for N-based, and O-based cationic substrate complexes, as shown in Fig. 4, Tables S5A and S6A.†
In the case of anionic complexes, the contribution of polarization and exchange components are highly competitive followed by the contribution of electrostatic component, except for fully phenylated complexes, as shown in Fig. 4, Tables S5B and S6B.† The contribution of these components remains steady irrespective of increasing the degree of methylation on the substrates, i.e., these components vary between 26% to 28% (electrostatic), 29% to 34% (exchange), 31% to 35% (polarization), and 3% to 5%. However, a sudden two-to-three-folds drop in the contribution of electrostatic and polarization components, and eight to nine folds increases in the contribution of dispersion component was observed for fully phenylated complexes, and it becomes dominated by dispersion component. Hence, it was observed that dispersion plays an important role in stabilizing the fully phenylated anionic complexes, viz., (C6H5)3C− (48.11%), (C6H5)2N− (48.39%), and (C6H5)O− (43.36%) respectively. The increment in the contributions of dispersion component is seemed to be compensated by the decrement in the contribution of electrostatic and polarization components (Fig. 4, Tables S5B and S6B†).
Similar to the anionic complexes, radical complexes were also to be dominated by highly competitive polarization and exchange components followed by electrostatic component, as shown in Fig. 4C, Tables S5C and S6C.† As the degree of methylation or phenylation increases on the substrates, the contribution of polarization component decreases minutely, i.e., 36.12% to 31.04% for CH3˙ to (C6H5)2CH˙ whereas no noticeable variation was observed for the exchange component, i.e., 31.39% to 32.68% for CH3˙ to (C6H5)2CH˙. And the contribution of dispersion component increases by eight-folds, i.e., 6.65% to 56.87% for CH3˙ to (C6H5)3C˙ respectively. In the case of NH2 and OH-based substrates, a noticeable variation in the contribution of polarization and exchange components was not observed, as shown in Fig. 4, Tables S5C and S6C.† Overall, this analysis suggested that the interaction of cationic substrates with pyrene is polarization driven, however, a highly competitive contributions from polarization and exchange components were observed for anionic and radical substrate complexes except for few methylated and phenylated complexes, as these are driven by dispersion component.
The contribution of dispersion component was observed to be increasing in the multiple folds with increases the degree of methylation and phenylation of substrate, and dominates the interaction energy. From Fig. 4, we may see that as the degree of methylation and phenylation is increased for CH3+, the dispersion contribution also increases gradually. However, in the case of fully phenylated (C6H5)3C+, (C6H5)3C−, and (C6H5)3C˙, a rapid increase in the dispersion contribution is observed, viz., 54.74%, 48.11%, and 56.87%, respectively, which may be attributed to the fact that an increment in the surface area of the substituent groups with respect to that of the parent molecule. Further, methyl and phenyl groups being much larger in size as compared to H-atom, leads to an increment in the van der Waals forces between the pairs of the pairs of molecules. Van der Waals forces being a type of dispersive forces add to the increased dispersive contributions. Furthermore, methyl and phenyl groups are non-polar in nature and with gradual increment of the substations in their contributions significantly tends to dominate towards dispersion and hence reflects in the relative contribution of the dispersion component in IE for these complexes.
Topological parameter analysis
Quantum theory of atoms in molecules analysis was carried out to elucidate the nature of interactions involves in pyrene-substrate complexes. A cursory view of the topological parameters such as ρ, H(r) and (−[G(r)/V(r)]) suggest the occurrence of both covalent and non-covalent interactions (Fig. 5A). The value of ρ is fairly high (≥0.15 a.u.) for the majority of the complexes with (−[(G(r)/V(r)]) < 1 and a negative value of H(r), suggesting a moderate to the strong type of covalent interaction while the remaining complexes are either partially covalent or non-covalent in nature (Table S7†). The representative of each complex which is covalent, partially covalent, and non-covalent is shown in Fig. 5A.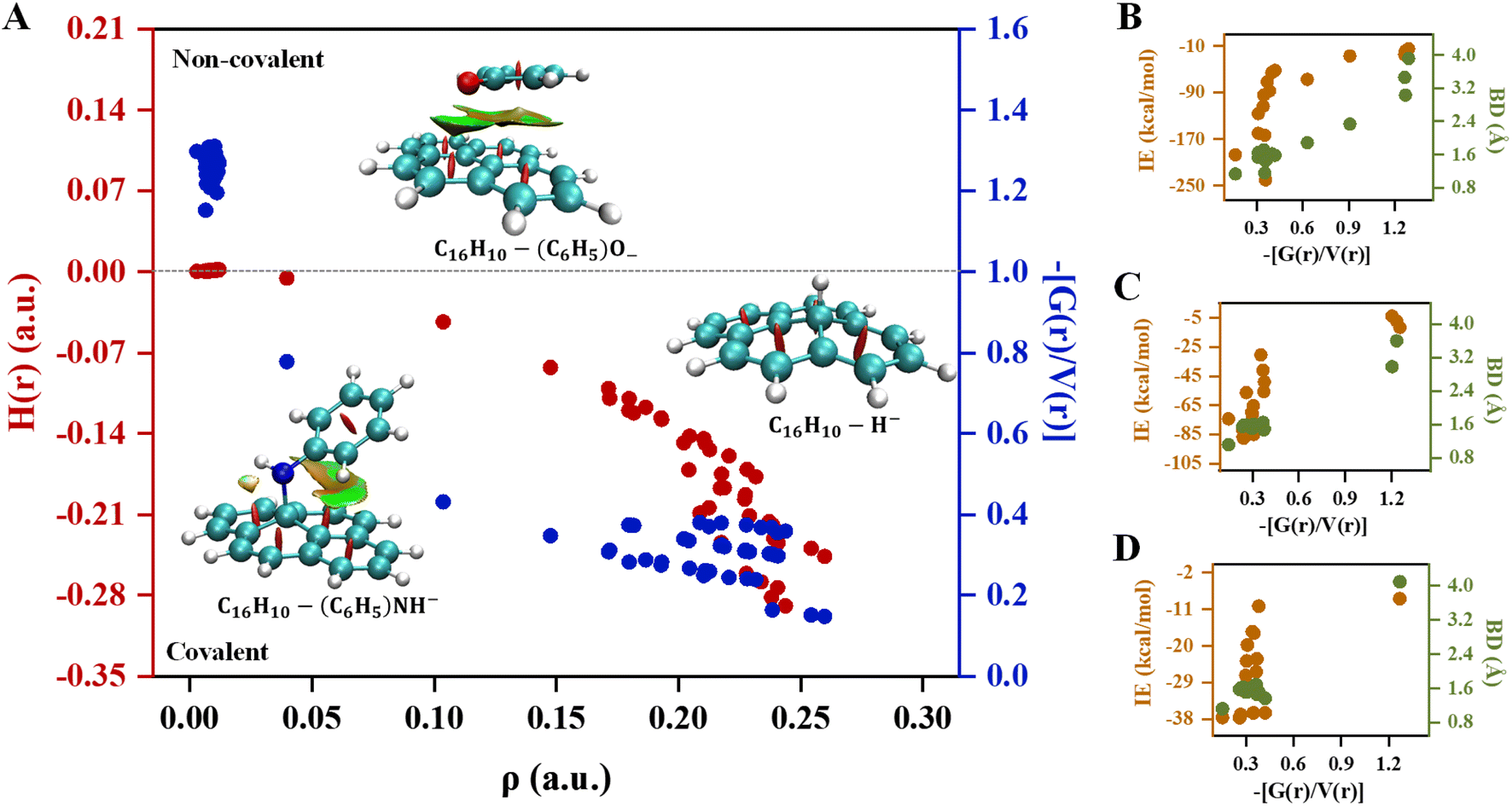 | ||
| Fig. 5 The variation in (A) topological parameters; electron density (ρ; a.u.), total energy density (H(r); a.u.) and (−[G(r)/V(r)]) of C16H10-substrate complexes and relation between (−[G(r)/V(r)]), interaction energy (IE; kcal mol−1) and bond distance (BD; Å) for the individual (B) cationic, (C) anionic, and (D) radical complexes. Calculations are done at M06-2X/6-311G**//M06-2X/6-31G* level of theory. | ||
In cationic complexes, a positive value for H(r) was obtained for (CH3)3C+ (C6H5)2CH+ and (C6H5)3C+ complexes with a small value of ρ, while the value for (−[G(r)/V(r)]) was > 1, suggesting non-covalent interactions (Table S7A†). However, the other cationic complexes showed a negative value of H(r) with ρ of 0.14 a.u. to 0.24 a.u., suggesting covalent in nature. Besides, the (−[(G(r)/V(r)]) value for (CH3)2CH+ and (C6H5)CH2+ complexes were obtained to be between 0.5 and 1 along with a negative value of H(r), suggesting interactions to be partially covalent in nature (Table S7A†). Thus, a transition from covalent to non-covalent interactions was observed by increasing the degree of methylation and phenylation on the CH3 substrate. In contrast, covalent to non-covalent transition was not observed for cationic methylated and phenylated NH2+ and OH+ complexes.
The un-substituted CH3+ substrate complexes are covalent in nature, whereas mono-phenylated and di-methylated methyl complexes are partially covalent in nature, and, other methylated and phenylated methyl substrate complexes are non-covalent in nature. Interestingly, in the cationic complexes, covalent interactions exhibited IE in the range of −51 kcal mol−1 to −240 kcal mol−1 and BD in the range of 1.156 Å to 1.610 Å, non-covalent interactions exhibited IE in the range of −14 kcal mol−1 to −24 kcal mol−1 and BD in the range of 1.610 Å to 3.919 Å, whereas the partial–covalent interactions exhibited values lie between covalent and non-covalent interactions, as shown in Fig. 5B and 6. We have identified a clear transition from the covalent to non-covalent interactions in the anionic and radical complexes except for methylated anionic and radical complexes, and phenylated NH2 and OH radical complexes (Tables S7B and C†). It may be due to the +I effect of the methyl group which increases the electron density of the anionic substrate leading to strong methylated anionic and radical complexes.
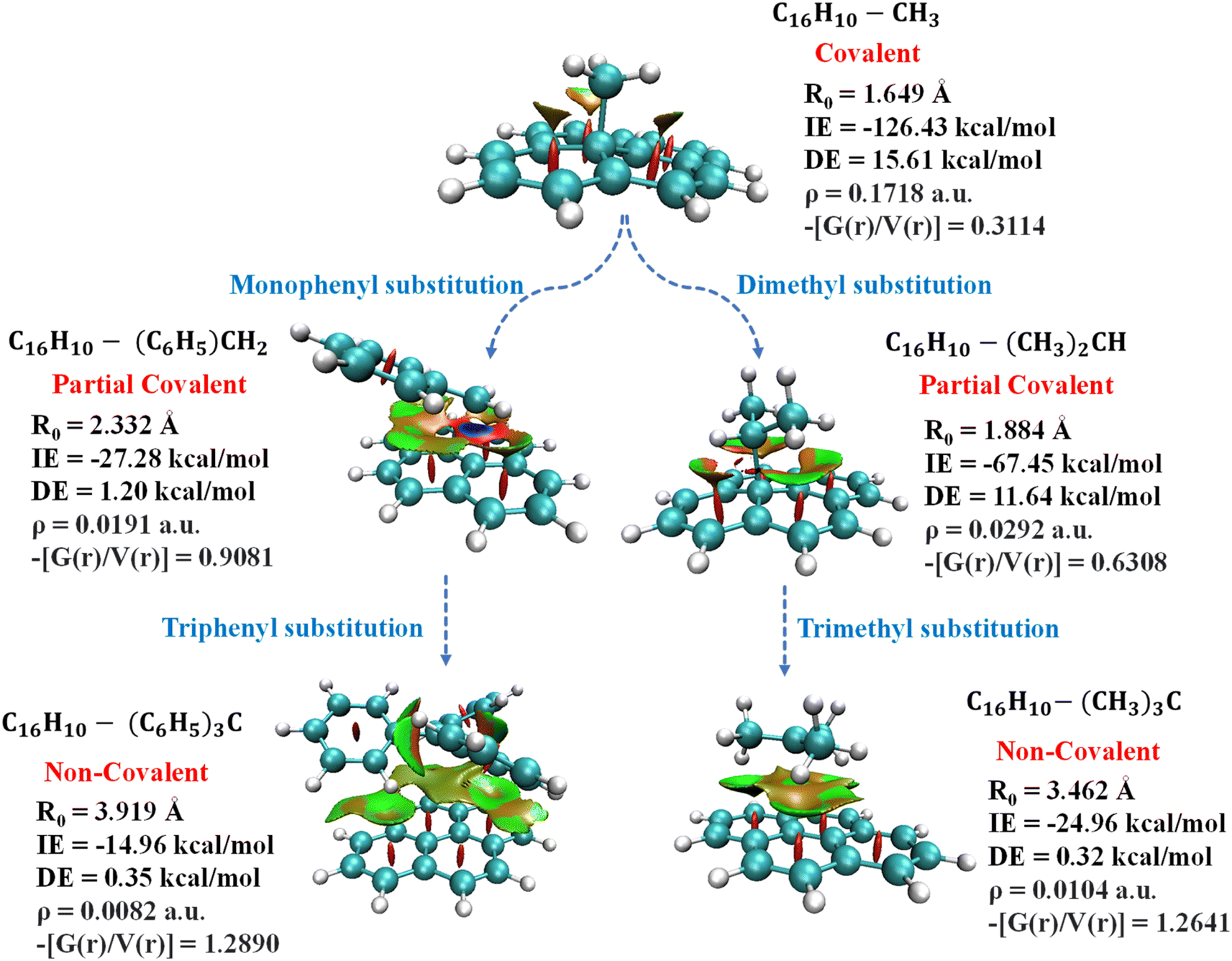 | ||
| Fig. 6 Depiction of transition of pyrene-substrate complexes from covalent to non-covalent functionalization with the increase in the substitution of cationic substrate; CH3 to C(C6H5)3 and C(CH3)3. | ||
The un-substituted as well as mono- and di-phenylated anionic complexes are covalent in nature whereas fully phenylated CH3−, NH−, and OH− complexes were observed to be non-covalent in nature (Table S7B and C†). In these cases, IE of covalent interactions ranges between −30 kcal mol−1 to −87 kcal mol−1 and BD in the range of 1.119 Å to 1.653 Å for anionic, however, for the radical complexes, IE lies between −10 kcal mol−1 to −37 kcal mol−1 and BD in the range of 1.120 Å to 1.680 Å, and non-covalent interactions exhibited IE < −11 kcal mol−1 and BD > 2.980 Å for anionic and radical complexes.
Overall, these results suggested that un-substituted cationic substrates bound to pyrene very strongly via covalent interactions, and the methylated and phenylated methyl substrate bound via non-covalent interactions with IE comparable to other covalent and strongest non-covalent interactions, as shown in Fig. 6. However, un-substituted anionic and radical substrates bind to pyrene via moderately strong covalent interaction and it becomes weak non-covalent interactions upon phenylation, as shown in Fig. 7. Furthermore, the NCI maps for different regions of the pyrene-substrate complexes have been represented in Fig. S9–S14,† which provide a clear picture of the extent and nature of the interactions through coloured regions.
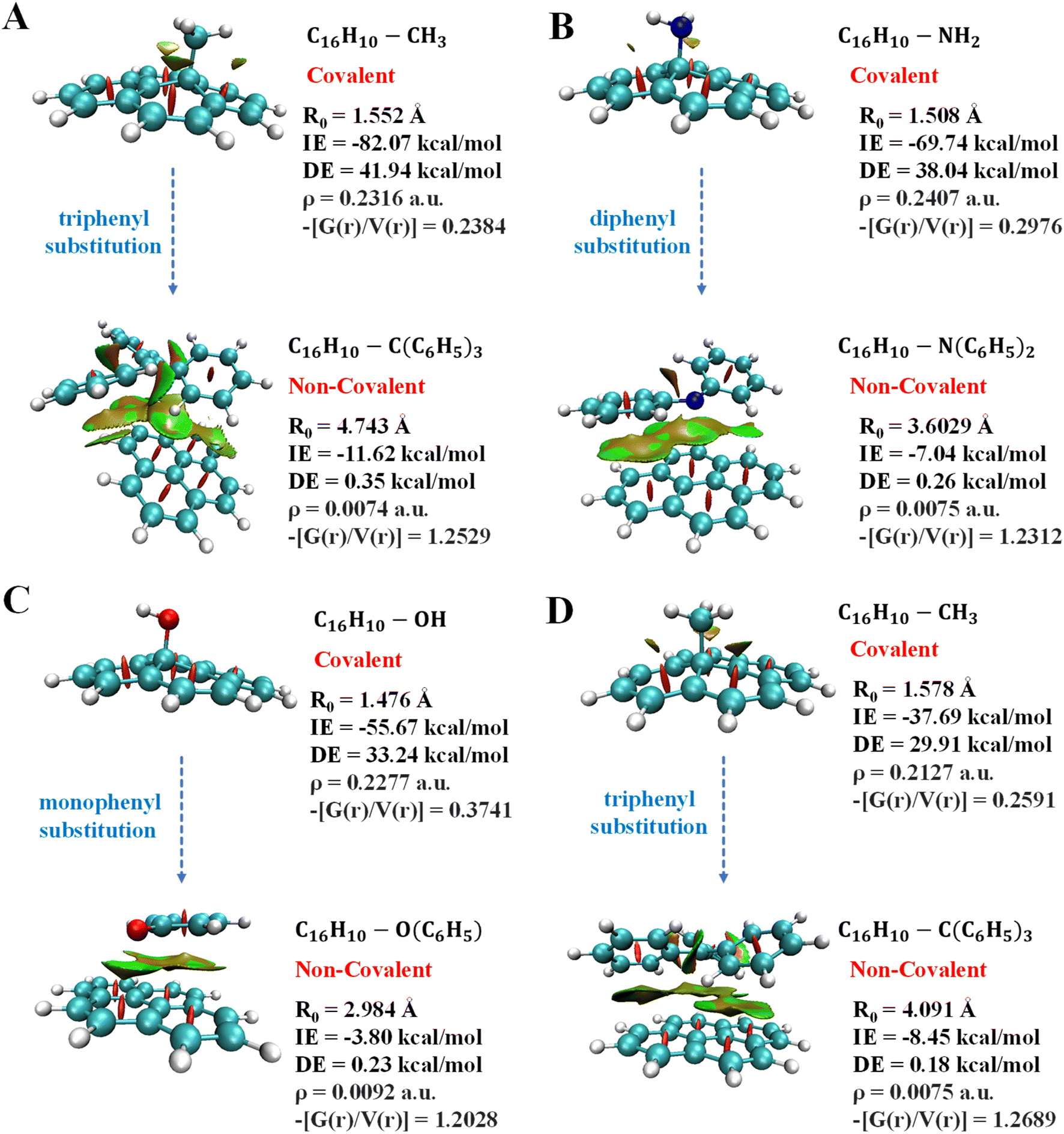 | ||
| Fig. 7 Depiction of transition of pyrene-substrate complexes from covalent to non-covalent functionalization with the increase in substitution of anionic substrates (A) CH3 to C(C6H5)3 (B) NH2 to N(C6H5)2 (C) OH to O(C6H5) and radical substrate (D) CH3 to C(C6H5)3. | ||
Conclusions
In this article, comprehensive quantum mechanical calculations on the functionalization of pyrene via C, N, and O based ionic and radical substrates were carried out at M06-2X/6-311G**//M06-2X-631G* level of theory. We further calibrated the geometrical parameters and interaction energies through benchmark calculations performed at twenty-one different levels of theories. This study reveals the transition of strong covalent interactions to moderately strong or weak non-covalent interactions for cationic and anionic substrates interacting with pyrene.The analysis of interaction energy showed that cationic substrates interact strongly with pyrene, however, anionic substrates also exhibited a competitive binding strength. In the case of cationic complexes, a higher interaction energy was observed for OH (−240.47 kcal mol−1) followed by H (−197.03 kcal mol−1), NH2 (−160.26 kcal mol−1), and CH3 (−126.43 kcal mol−1). However, strong interaction was observed in the case of anionic complexes of CH3 and its methylated analogues (−82.07 kcal mol−1 to −85.11 kcal mol−1) followed by methylated NH2 and OH substrates (−48.92 kcal mol−1 to −71.45 kcal mol−1). The interaction energy of cationic substrates decreases by 2 to 9 folds with increasing the degree of phenylation and methylation whereas it improves slightly in case of methylated anionic substrates (CH3 and NH2) and exhibits a competitive binding strength. The radical substrates exhibited the interaction energy in the range of −8.45 kcal mol−1 to −37.69 kcal mol−1, and similarly, minimum energy was observed for the fully phenylated followed by methylated substrates.
Energy decomposition analysis revels that the interaction of cationic substrates with pyrene is polarization driven, however, a highly competitive contributions from polarization and exchange components were observed for anionic and radical substrate complexes except for few methylated and phenylated complexes which are driven by dispersion component.
The analysis of topological parameters elucidated that un-substituted cationic substrates bound to pyrene via covalent interactions, and the methylated and phenylated methyl substrate bound via non-covalent interactions. However, un-substituted anionic and radical substrates bind to pyrene via moderately strong covalent interaction and becomes a weak non-covalent interaction upon phenylation. The contribution of dispersion component was observed to be increasing with increases the degree of methylation and phenylation of substrate, and later dominates the interaction once it becomes non-covalent in nature.
These findings on the nature of interactions and the transition of strong covalent interactions to moderately strong or weak non-covalent interactions for C, N, and O based cationic and anionic substrates interacting with pyrene are disclosed for the first time, and are hoped to strengthen the understanding of pyrene with different substrates of different nature, and may be applicable for the design of new functional materials with diverse and advanced applications.
Conflicts of interest
The authors have no conflict of interest.Acknowledgements
Authors would like to thank Prof. G. Narahari Sastry, Director, CSIR-NEIST for his invaluable contribution to this research project through his continuous suggestions, and guidance during the course of this work. Ministry of Petroleum & Natural Gas, Govt. of India for providing the funding for the research project (GPP-373). Department of Biotechnology is thanked for the financial support in the form of a Centre of Excellence in Advanced Computation and Data Sciences (BT/PR40188/BTIS/137/27/2021).References
- R. G. Harvey, Polycyclic Aromatic Hydrocarbons, Wiley-VCH, Weinheim, 1997 Search PubMed.
- Q. Miao, Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Application, Wiley-VCH, Weinheim, 2016 Search PubMed.
- A. Kuc, T. Heine and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085430 CrossRef.
- R. Rieger and K. J. Mullen, Phys. Org. Chem., 2010, 23, 315 CAS.
- Y. T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS PubMed.
- L. T. Scott, Chem. Soc. Rev., 2015, 44, 6464 RSC.
- D. Umadevi and G. N. Sastry, J. Phys. Chem. C, 2011, 115, 9656 CrossRef CAS.
- F. P. Kinik, A. O. Guerrero, D. Ongari, C. P. Ireland and B. Smit, Chem. Soc. Rev., 2021, 50, 3143 RSC.
- T. M. Figueira-Duarte and K. Mullen, Chem. Rev., 2011, 111, 7260 CrossRef CAS PubMed.
- D. Jana and S. Jana, ACS Omega, 2020, 5, 9944 CrossRef CAS PubMed.
- M. I. Nan, E. Lakatos, G. I. Giurgi, L. Szolga, R. Po, A. Terec, S. Jungsuttiwong, I. Grosu and J. Roncali, Dyes Pigm., 2020, 181, 108527 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, P. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- Y. B. Kumar, R. K. Rawal, A. Thakur and G. N. Sastry, Comput. Theor. Chem., 2022, 21, 157 Search PubMed.
- J. H. Jeong, S. Kang, N. Kim, R. Joshi and G. H. Lee, Phys. Chem. Chem. Phys., 2022, 24, 10684 RSC.
- D. Umadevi, S. Panigrahi and G. N. Sastry, Acc. Chem. Res., 2012, 47, 2574 CrossRef PubMed.
- X. Feng, J. Hu, C. Redshaw and T. Yamato, Chem.–Eur. J., 2016, 22, 11898 CrossRef CAS PubMed.
- D. Zych, Molecules, 2019, 24, 2551 CrossRef PubMed.
- A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282, 139 CrossRef.
- M. V. Chumillas, X. Liu, A. L. Pérez, D. Armentano, J. F. Soria and E. Pardo, Coord. Chem. Rev., 2022, 451, 214273 CrossRef.
- A. Karmakar, A. Paul, E. P. Sabatini, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Molecules, 2021, 26, 1101 CrossRef CAS PubMed.
- H. Lei, C. Liu, Z. Wang, Z. Zhang, M. Zhang, M. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429 CrossRef CAS.
- T. Yoshii, D. Umemoto, M. Yamamoto, Y. Kuwahara, H. Nishihara, K. Mori, T. Kyotani and H. Yamashita, Chem. Cat. Chem., 2020, 12, 5880 CAS.
- B. Zhang, L. Fan, R. B. Ambre, T. Liu, Q. Meng, B. J. J. Timmer and L. Sun, Joule, 2020, 4, 1408 CrossRef CAS.
- Y. Zhang, H. Li, Q. Y. Wu and L. Gu, Front. Chem., 2022, 10, 970033 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
- H. Schneider, J. Phys. Org. Chem., 2022, 35, 4340 CrossRef.
- N. Kumar, A. S. Gaur and G. N. Sastry, J. Chem. Sci., 2021, 133, 1 CrossRef.
- Y. B. Kumar, A. Pandey, N. Kumar and G. N. Sastry, J. Comput. Chem., 2023, 44, 432 CrossRef CAS PubMed.
- N. Kumar, Y. B. Kumar, H. Sarma and G. N. Sastry, Front. Chem., 2021, 9, 738852 CrossRef CAS PubMed.
- S. Panigrahi and G. N. Sastry, RSC Adv., 2014, 4, 14557 RSC.
- T. Dinadayalane and G. N. Sastry, J. Org. Chem., 2002, 67, 4605 CrossRef CAS PubMed.
- D. Vijay and G. N. Sastry, Phys. Chem. Chem. Phys., 2008, 10, 582 RSC.
- U. D. Priyakumar and G. N. Sastry, Tetrahedron Lett., 2003, 44, 6043 CrossRef CAS.
- D. Umadevi and G. N. Sastry, Chem. Phys. Lett., 2012, 549, 39 CrossRef CAS.
- S. Sengottiyan, K. Malakar, A. Kathiravan, M. Velusamy, A. Mikolajczyk and T. Puzyn, J. Phys. Chem. B, 2022, 21, 3831 CrossRef PubMed.
- Z. Qian, W. Deng, X. Zhang, H. Miao and G. Zhang, RSC Adv., 2017, 7, 46721 RSC.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909 CrossRef CAS PubMed.
- V. Mishyn, A. Hugo, T. Rodrigues, P. Aspermair, H. Happy, L. Marques, C. Hurot, R. Othmen, V. Bouchiat, R. Boukherroub, W. Knoll and S. Szunerits, Sens. Diagn., 2022, 1, 235 RSC.
- Y. Wang, H. J. Qian, K. Morokuma and S. Irle, J. Phys. Chem. A, 2012, 116, 7154 CrossRef CAS PubMed.
- R. M. Ferullo, C. E. Zubieta and P. G. Belelli, Phys. Chem. Chem. Phys., 2019, 21, 12012 RSC.
- F. B. C. Machado, A. J. A. Aquino and H. Lischka, Phys. Chem. Chem. Phys., 2015, 17, 12778 RSC.
- C. B. Martínez, D. C. Arriagada and S. G. Oliva, Mon. Not. R. Astron. Soc., 2018, 481, 3052 CrossRef.
- Y. Wang, P. Verma, X. Jin, D. G. Truhlar and X. He, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10257 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- Y. I. Neela and G. N. Sastry, Mol. Phys., 2015, 113, 137 CrossRef.
- D. Vijay, H. Sakurai and G. N. Sastry, Int. J. Quantum Chem., 2011, 111, 1893 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et. al., Gaussian 16, Gaussian Inc, Wallingford, 2016 Search PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- G. F. Caramori, I. Østrøm, A. O. Ortolan, G. R. Nagurniak, V. M. Besen, A. M. Castro, R. P. Orenha, R. L. T. Parreira and S. E. Galembeck, Dalton Trans., 2020, 49, 17457 RSC.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. A. Gordon, J. J. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- N. Kumar, S. Saha and G. N. Sastry, Phys. Chem. Chem. Phys., 2021, 23, 8478 RSC.
- AIM2000, J. Comput. Chem., 2001, 22, 545 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01457f |
| This journal is © The Royal Society of Chemistry 2023 |