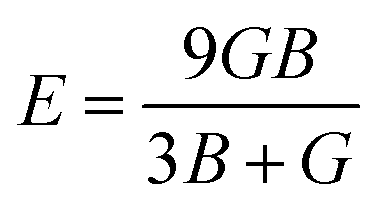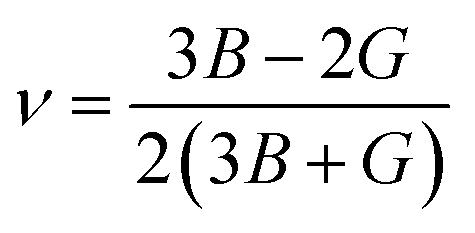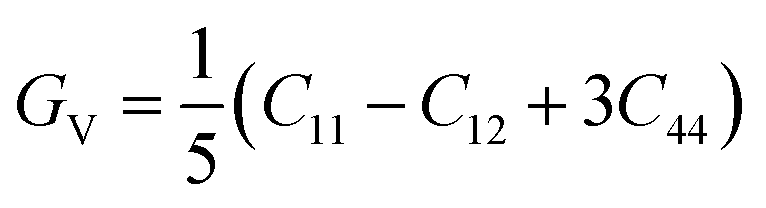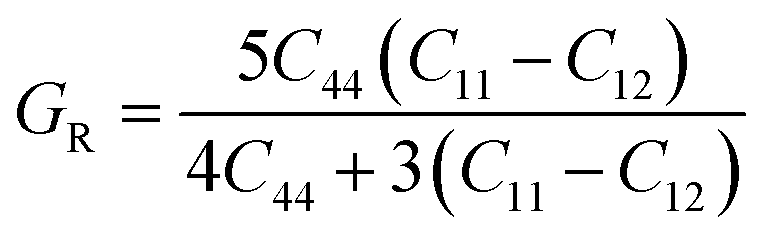DOI:
10.1039/D3RA01451G
(Paper)
RSC Adv., 2023,
13, 15457-15466
Probing the physical properties of M2LiCeF6 (M = Rb and Cs) double perovskite compounds for prospective high-energy applications employing the DFT framework
Received
6th March 2023
, Accepted 9th May 2023
First published on 1st June 2023
Abstract
Herein, the optoelectronic, structural, thermoelectric, and elastic characteristics of M2LiCeF6 (M = Rb and Cs) double perovskite compounds were investigated using ab initio modeling in the DFT framework. The Birch–Murnaghan fitting curve used for the optimization showed that these two compounds are structurally stable. The elastic properties of the M2LiCeF6 (M = Rb and Cs) double perovskite compounds were examined using the IRelast code. The results showed that these two compounds possess mechanical stability, anisotropy, and toughness, and offer resistance to plastic deformation. The precise and accurate determination of their electronic properties was achieved via the Trans-Blaha-modified Becke-Johnson (TB-mBJ) approximation. The Rb2LiCeF6 and Cs2LiCeF6 compounds are narrow band gap semiconductors with band gaps of 0.6 eV and 0.8 eV at the high symmetrical points from (Γ–M), respectively, exhibiting an indirect nature. To further understand how the various states contribute to the different band structures, total and partial density of state (DOS) computations were performed. The optical properties in the energy range of 0–40 eV for Rb2LiCeF6 and Cs2LiCeF6 were explored. The selected materials show transparency in the low incident photon energy range and have large light absorption and transmission at higher photon energies. Thus, it can be concluded that Rb2LiCeF6 and Cs2LiCeF6 can be used in high-frequency UV devices based on their optical characteristics. Both materials exhibit high electrical conductivity, power factors, and figures of merit (ZT) and act as effective thermoelectric resources. To the best of our knowledge, this is the first theoretical research on the optoelectronic, structural, thermoelectric, and elastic features of M2LiCeF6 (M = Rb and Cs).
1. Introduction
Due to their exceptional stability, double perovskites (DPs), A2B′B′′X6, can be applied in a variety of sustainable and renewable devices.1–3 In recent years, exciting research on perovskite materials has been conducted in the fields of photovoltaics and photoluminescence.4,5 Consequently, DPs have been widely implemented in optoelectronics6–9 such as in solar cells10,11. As an alternative to MAPBI3, the newly estimated Pb-free DPs are based on Ag and Bi, which are expected to be beneficial to the environment and have stability.12–16 Currently, switching between organic and inorganic cations has increased the thermal stability of DPs.17 In this context, many different substances have been tested, among which Pb-free DPs based on “Ag” and “In” are currently being explored. Various approaches have been employed to describe Cs2AgInCl6 for band gap calculations.18–20 The next generation of photovoltaic technologies will benefit from the perovskites developed for solar cells employing metal halide perovskites. Multi-junction solar cells have achieved a high power conversion efficiency (PCE), which has significantly increased over the last few years from 3.8% (ref. 21) to more than 24% according to the literature.22,23 Recently, in 2019, multi-junction thin film solar cells attained a conversion efficiency of 47.1%.24,25 The excellent performance of halide perovskites is attributed to their distinctive characteristics, including high charge carrier mobility, strong absorption, and direct and appropriate band gaps.26–29 Halide double perovskites are constructed based on the physical characteristics of a variety of metal combinations. Cooling causes an octahedral tilt and increased electron–phonon interactions, which convert the potential cubic structure to a monoclinic structure. Halide perovskite semiconductors for transport and optoelectronic applications have been extensively explored despite their drawback of phase changes.30,31 Organic or inorganic perovskites have suitable and stable photovoltaic (PV) properties and are widely applied in photoelectric devices.32 PV systems are beneficial due to their non-polluting nature, gas-free carbon emissions, and abundant natural life compared to other types of perovskites.33 However, their toxicity to humans and the environment is one of their major future limitations. Furthermore, their long-term instability on exposure to sunlight and atmospheric humidity is another challenge.34,35 Perovskites have made significant advancements in thin-film devices such as photodetectors, lasers, and thin-film transistors. Numerous factors make perovskites a good choice for these types of devices. For example, perovskites can be created at low temperatures using dependable roll-to techniques for roll deposition.36 Herein, DFT applied to WIEN2K and the BoltzTrap2 code was used to analyze the mechanical, optoelectronic, and thermoelectric properties of DPs. The computed results provide experimental researchers with guidelines and predictions for developing solar cell applications, which will help them find effective materials. Because of their superior structural, mechanical, and thermodynamic stability, the researched materials are exceptional for use in renewable energy applications. For the construction of devices, ductile behavior is crucial.
2. Computational details
DFT calculations using the full-potential linearized augmented plane wave plus local orbital (FPLAPW + lo) scheme utilized in the WIEN2K code were performed to screen the physical features of the materials.37 For the investigation of the structural and elastic parameters at the ground state, the exchange-correlation interactions were considered through the GGA-PBE functional (generalized gradient approximation of Perdew Burke and Ernzerhof).38 To compensate for the GGA-PBE functional underestimating the band gaps, the modified TB-mBJ potential was employed.39 To gain insight into the electronic character of the materials, their electronic structure was computed. The extended spherical harmonic electronics in the wavefunction with lmax (maximum value of angular momentum) were set to 10 inside the muffin-tin sphere regimes. The wavefunctions were created on a plane wave basis set with a cut-off reciprocal lattice vector, Kmax, equal to 10/RminMT, where RminMT is the smallest muffin-tin sphere radius (RMT). To avoid overlapping spheres, the RMTs were chosen to be as large as possible, i.e., RRbMT = 2.5, RLiMT = 1.67, RMTCe = 2.17, and RFMT = 1.96 Bohr. In an attempt to optimize the cell structure, more precise calculations with a higher cutoff energy and denser k-point sampling were carried out. The cutoff energy was raised to 41.2 Ry, and the irreducible Brillouin zone (IBZ) was considered in a 50 × 50 × 50 k-point mesh for the computation of the additional features (physical) that were considered, as well as 2000 Monkhorst–Pack k-points mesh for investigating the structural and other parameters.40 To confirm the total energy converged to 10−5 Ry and that the force acting on each atom was less than 0.5 Ry Bohr−1, the computational parameters were used. The equilibrium structural parameters, comprising the lattice parameter, were determined through the use of total energy minimization and internal coordinates. The elastic constants were computed utilizing the total energy vs. strain technique, as employed by Thomas Charpin37 in the WIEN2K program. This method involved applying three different deformation types to the equilibrium lattice, including tetrahedral strain, hydrostatic stress, and rhombohedra strain, and then calculating the total energy changes consistent with the smeared deformations. The three single-crystal elastic constants, i.e., C11, C12, and C44, were the results for the cubic system. The dielectric function, ε(ω), which is related to the band gap found in the electronic structure, was the most crucial parameter to consider when analyzing the optical behavior of the materials. It was also necessary to derive relationships for the refractive index, n(ω), absorption and extinction coefficients, I(ω) and K(ω), respectively, energy loss function, L(ω), reflectivity R(ω), and optical conductivity, σ(ω). ε(ω) has real and imaginary components, i.e., "ε1(ω)" and "ε2 (ω)", respectively, in the form ε(ω) = ε1(ω) + iε2(ω),41 in which the imaginary component of the dielectric function represents the absorption of light by the material on its surface, while the real part represents the dispersion of light from the surface of the material.42
3. Results and discussion
3.1. Structural optimization
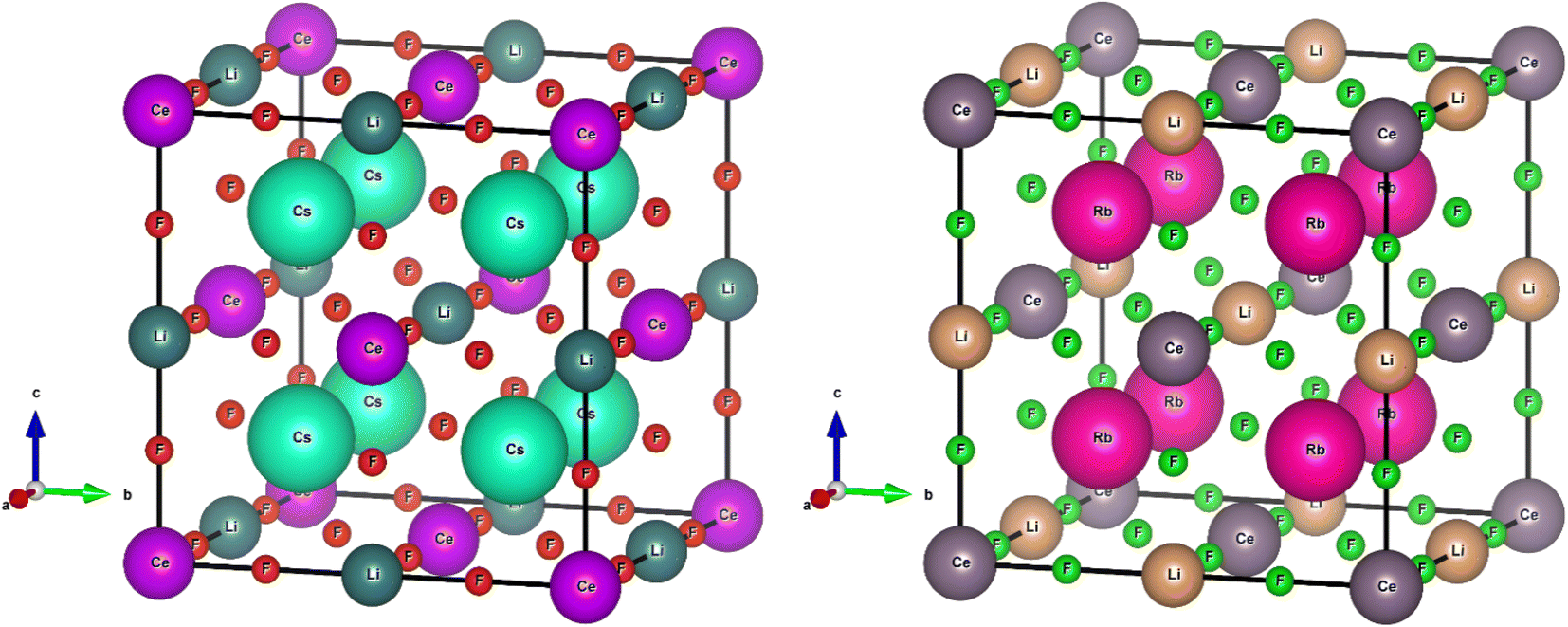
M2LiCeF6, where “M” is equal to Rb and Cs, crystallizes as a cubic perovskite with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (#225). The M (M = Rb and Cs) atoms were shown to be at (0.25, 0.25, 0.75), Li atoms at (1/2, 0, 0), Ce atoms exists on (0, 0, 0) and F atoms at (0.26, 0, 0) and (0.25, 0, 0), respectively. Fig. 1 illustrates the cubic crystalline structure of the M2LiCeF6 (M = Rb and Cs) double perovskites. The optimized volume measured the structural parameters through the Birch–Murnaghan EoS.43
m space group (#225). The M (M = Rb and Cs) atoms were shown to be at (0.25, 0.25, 0.75), Li atoms at (1/2, 0, 0), Ce atoms exists on (0, 0, 0) and F atoms at (0.26, 0, 0) and (0.25, 0, 0), respectively. Fig. 1 illustrates the cubic crystalline structure of the M2LiCeF6 (M = Rb and Cs) double perovskites. The optimized volume measured the structural parameters through the Birch–Murnaghan EoS.43
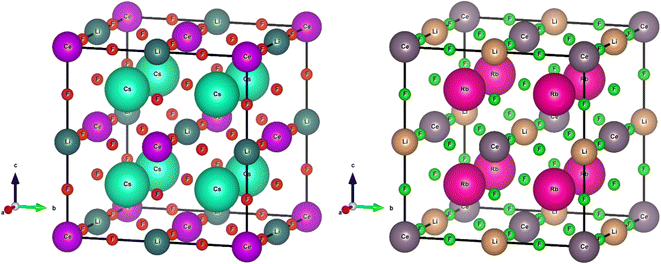 |
| | Fig. 1 Crystal unit cell of M2LiCeF6 (M = Rb and Cs) compounds. The atoms of the M (M = Rb and Cs) element are shown to be at (0.25, 0.25, 0.75), Li atoms at (1/2, 0, 0), Ce atoms at (0, 0, 0) and F atoms at (0.26, 0, 0) and (0.25, 0, 0), respectively. | |
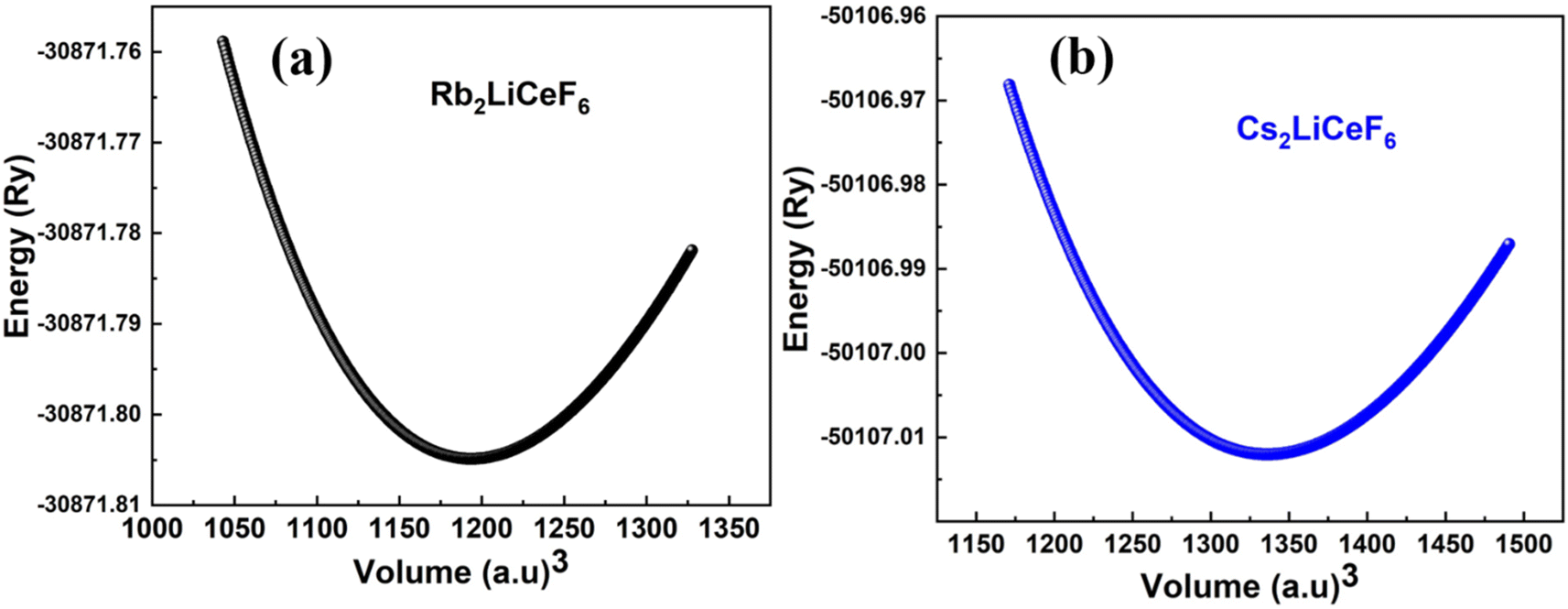
The calculated approximations of the evaluated points from the optimization curves depict the variation of volume vs. total energy and estimate the ground-state parameters, including the bulk modulus (B), pressure derivative of B (B/), and optimized lattice constant (a0), and the minimum total energy of the unit cell with the corresponding volume, as presented in Fig. 2, will yield the unit cell with the lowest energy. In this section, the energy (total) was evaluated vs. unit cell volume (V0).
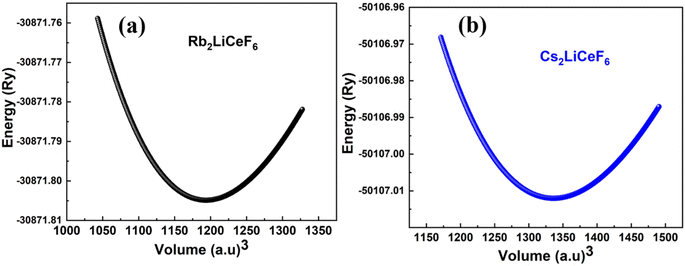 |
| | Fig. 2 Energy vs. volume plots of M2LiCeF6 (M = Rb and Cs) compounds. | |
The optimum energy, E0, was obtained by plotting the total energy against volume, where the volume is denoted as V0. The compound with the highest optimal energy is thought to have the most stable structure. The optimal structural data are shown in Table 1, including the equilibrium lattice constant (a0), E0, B0, V0, and B/0. These findings are consistent with the overall pattern of this estimation because B is frequently reduced when a0 is larger, indicating that the evaluated results are precise and useful.
Table 1 Parameters of the M2LiCeF6 (M = Rb and Cs) ternary compound crystal unit cell that were tuned structurally. E0 in Ry, B0 and B/0 in GPa, and the lattice constant a0 in Å
| Structural specifications |
Rb2LiCeF6 |
Cs2LiCeF6 |
| a0 (Å) |
8.91 |
9.25 |
| V0 (a.u3) |
1193.70 |
1336.21 |
| B0 (GPa) |
55.20 |
50.11 |
| B/0 (GPa) |
4.72 |
4.37 |
| E0 (Ry) |
−3087.80 |
−50107.01 |
3.2. Electronic properties
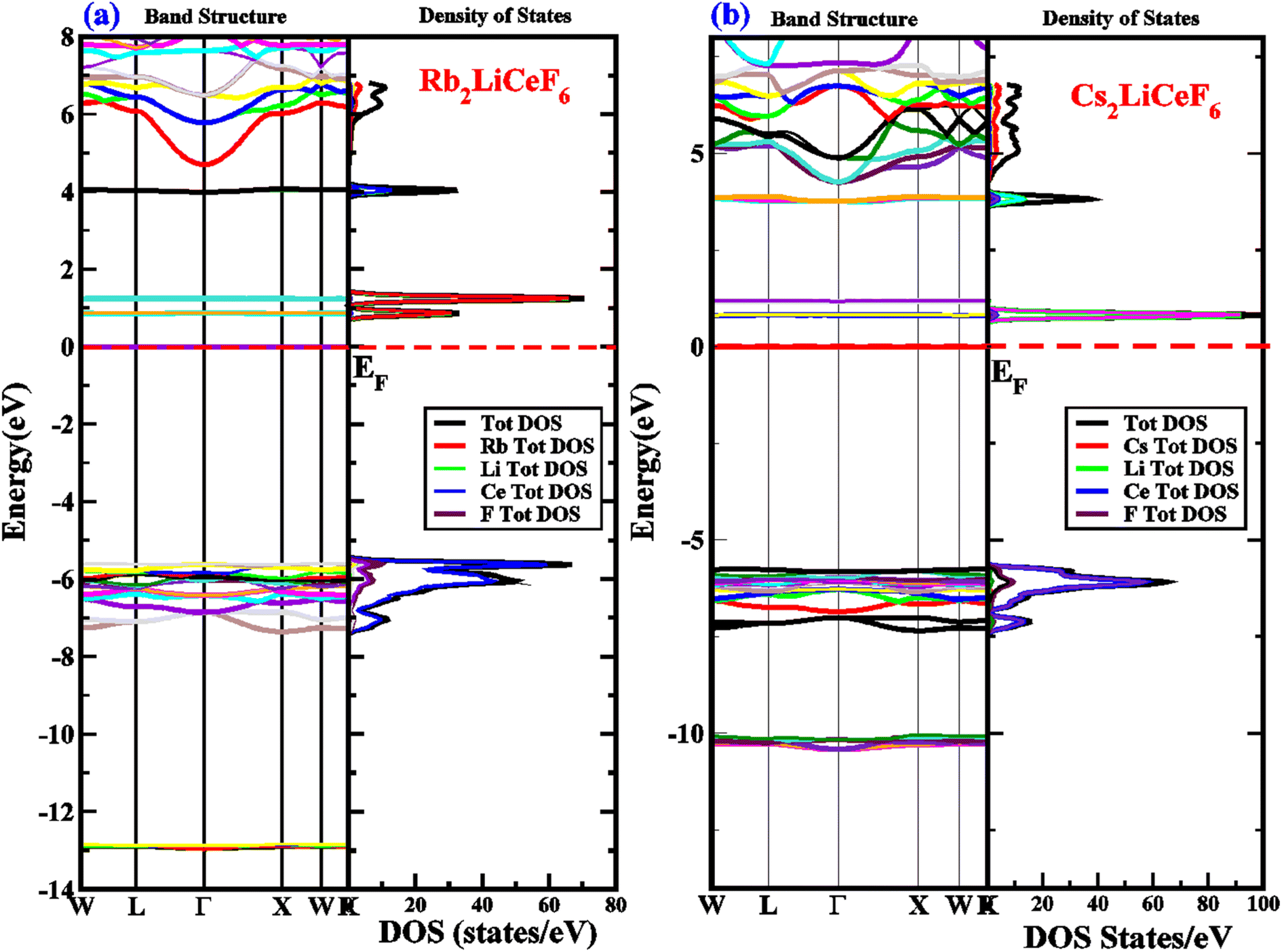
With the aim to analyze the electronic characteristics of the M2LiCeF6 (M = Rb and Cs) compounds, we investigated their real energy band structure, TDOS, and PDOS, as shown in Fig. 3. LDA and GGA calculations often find that the band gap (fundamental) between a semiconductor and insulator is underestimated.44,45 Generally, this is due to the fact that the exchange-correlation energies and charges derivative cannot be consistently generated by their fundamental geometries. Thus, the TB-mBJ approximation was successfully used in some recent studies to correct the underestimated troublesome band gaps.46–49 The optimal geometries of the M2LiCeF6 (M = Rb and Cs) compounds with assessed energy band structures over larger symmetries are displayed in Fig. 3a and b, respectively.
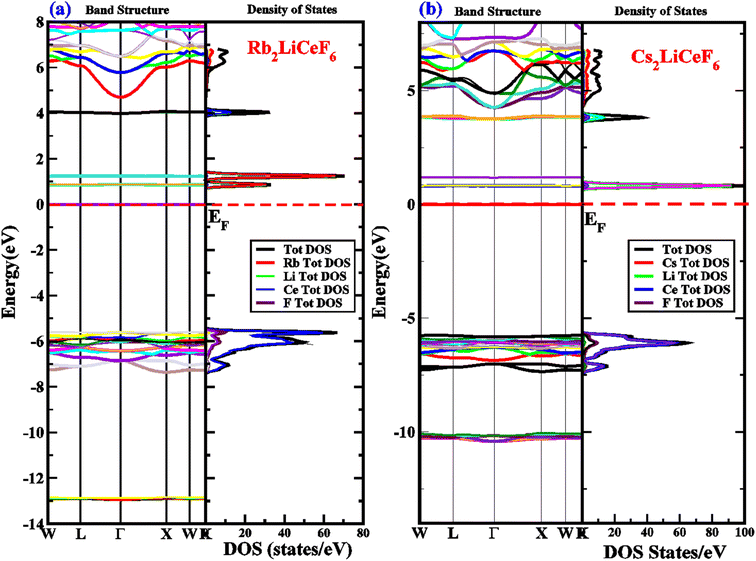 |
| | Fig. 3 (a) and (b) Electronic energy bands structure and DOS of M2LiCeF6 (M = Rb and Cs) compounds using TB-mBJ approximation, respectively. | |
The band gaps of Rb2LiCeF6 and Cs2LiCeF6 in TB-MBJ are 0.6 eV and 0.8 eV, respectively, with an indirect band gap along the Γ–X points of symmetry. The conduction band maxima are flat and move from Γ–X, confirming the narrow-band semiconducting nature of both compounds. To gain a thorough understanding, the TDOS, PDOS and band structures were examined, and their spectra are shown in Fig. 3a and b in the energy range of −14 eV to 8 eV. The role of the electronic states in the VB and CB is displayed in the DOS plots. The Fermi energy level (EF), as illustrated by the vertical dashed line at 0 eV, is used to indicate the DOS in the energy range of −14 eV to 8 eV. The part above EF shows the VB, whereas that below specifies the CB. In the case pf Rb2LiCeF6, the energy range is split into two groups based on the PDOS distribution, with the energy range of BaCuF3 being 0 eV to 0.8 eV (VB) and 0 eV to −6.8 eV (CB), while that for Cs2LiCeF6 is 0 eV to 1 eV (VB) and 0 eV to −5.5 eV, respectively. The band structure and narrow band gap semiconducting properties of these compounds are supported by the DOS plots. The “Rb” atom, as shown in red, makes up the majority of the density of states in the VB for Rb2LiCeF6, whereas the Ce atom, as shown in blue, makes up the majority of the density of states in CB. In contrast, for Cs2LiCeF6, the Li atom, as shown in green in the VB, and the Ce atom, as shown as blue in the CB, both contribute significantly to the density of states.
3.3. Elastic properties
To predict the mechanical parameters of materials, EC (elastic constant) is significant and determines how crystals respond to stimuli. The hardness and stability of a material can be evaluated using the measured EC. In this case, examining the EC of a material under zero pressure involves calculating the tensor and stress characteristics for very small strains as well as employing energy to track the lattice strains that persist to the volume.50 The IRelast package, incorporated in WIEN2K developed chiefly for cubic structures, was used to compute the ECs. Only three ECs, i.e., C11, C12, and C44, were utilized in place of Cij owing to the cubic lattice symmetry. Table 2 presents the computed ECs used to produce the assessed ECs and other elastic property characteristics. The cubic crystal mechanical stability criteria, which place restrictions on the elastic constants, are C11 > 0, C44 > 0, C11 − C12 > 0, C11 + 2C12 > 0, and B > 0. These stability requirements show how elastic stability is present in particular materials.
Table 2 Investigated parameters of elastic properties, which include EC (elastic constant), B (bulk modulus), anisotropy factor (A), Young's modulus (E), Poisson's ratio (v), Pugh ratio (B/G), B/C44, C11 − C12, shear modulus (G), and C11 − C44 for Rb2LiCeF6 and Cs2LiCeF6 quaternary double perovskites compounds
| Elastic parameter |
Rb2LiCeF6 |
Cs2LiCeF6 |
| C11 (GPa) |
104.11 |
98.97 |
| C12 (GPa) |
32.79 |
26.48 |
| C44 (GPa) |
20.36 |
44.64 |
| B (GPa) |
56.69 |
50.70 |
| A |
0.57 |
1.23 |
| E (GPa) |
68.74 |
97.42 |
| v |
0.29 |
0.27 |
| B/G |
2.22 |
1.89 |
| B/C44 |
2.78 |
1.13 |
| C11 − C12 (GPa) |
75.43 |
47.82 |
| G (GPa) |
7.18 |
7.79 |
| C11 − C44 (GPa) |
111.43 |
95.4 |
Rb2LiCeF6 has a C11 value of 104.11 GPa compared to that of Cs2LiCeF6 of 98.97 GPa, demonstrating a minor hardness advantage for Rb2LiCeF6. The parameter “A" of the crystal, which is critical in engineering research, is closely related to the propensity of a material to produce microcracks.
For fully isotropic materials, A = 1, whereas any value below or above “1” denotes anisotropy. Here, both compounds are anisotropic given that “A” varies between “1” and 1.5, and the degree of anisotropy is constrained by the level of variation. As can be observed from the examined values of A for Rb2LiCeF6 (0.57) and Cs2LiCeF6 (1.23), both of these compounds display a significant amount of anisotropy. Subsequently, it was necessary to compute the other elastic components, i.e., G, E, and v, employing the elastic constants and the equations below.51,52
| |
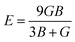 | (2) |
| |
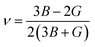 | (3) |
| |
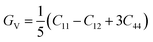 | (4) |
| |
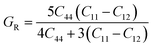 | (5) |
The results of applying the aforementioned equations to determine the values of A, E, v, and G are displayed in Table 2. The Cauchy pressure is represented as C11 − C44, which serves to identify ductile materials.53 If C11 – C44 is positive, the materials exhibit a ductile nature, whereas if it is negative, the materials will be brittle. The positive Cauchy pressure values of 83.75 GPa for Rb2LiCeF6 and 54.33 GPa for Cs2LiCeF6 indicate their ductile character. Another way to determine brittleness or ductility is the Pugh ratio (B/G). Materials are thought to be more ductile if their B/G ratio is high, which must be greater than 1.75.54 The critical points in this study were different for both compounds, which were 2.22 for Rb2LiCeF6 and 1.89 for Cs2LiCeF6. By using “v” to distinguish between the ductility and brittleness of the material, Frantsevich et al.55 discovered that a critical value of 0.26 was mandatory. Brittle materials have values below 0.26, whereas ductile materials have values over 0.26. Based on the value of 0.29 for Rb2LiCeF6 and 0.27 for Cs2LiCeF6, both of compounds are ductile in nature, as demonstrated in Table 2. The corresponding material has great lubricating capabilities if B/C44 has large values. Young's modulus (Y) and the values of (C11 − C12) can also be used for evaluating plasticity. Greater flexibility of interested compounds is shown by lower values of (C11 − C12) and “Y”. Thus, the Rb2LiCeF6 and Cs2LiCeF6 compounds have mechanical stability, anisotropy, toughness, strength, and good crack resistance.
3.4. Optical properties
To examine all the optical properties, the theoretical equilibrium lattice constants were used, i.e., ε (ω) was examined.
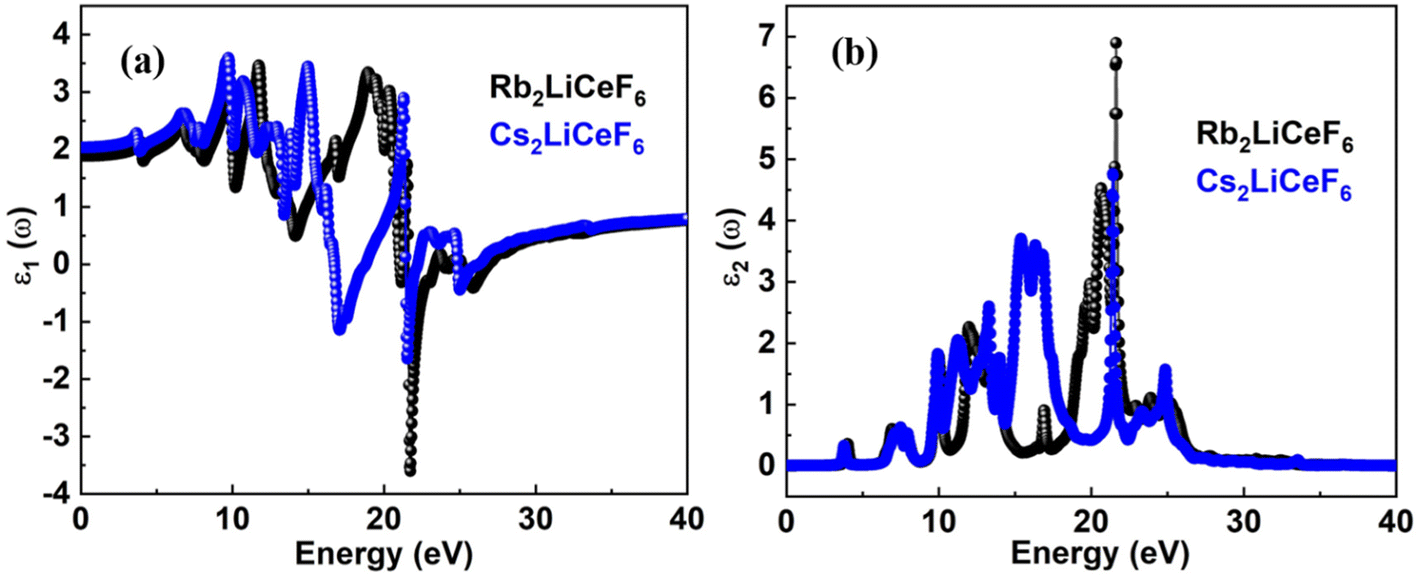
3.4.1. Dielectric function. The computed real part, ε1(ω), for both materials is shown in Fig. 4a, and the imaginary portion, ε2(ω), is shown in Fig. 4b for the incident photon energy range of up to 40 eV. In contrast to the imaginary part, ε2(ω), which is related to the polarization and is responsible for the capacity of a material to store energy, ε1(ω) defines the energy dissipation and wave damping. The predicted static dielectric function, ε1(0), for both materials is shown in Fig. 4a, which shows that Rb2LiCeF6 wastes more energy than Cs2LiCeF6, with ε1(0) = 3.6 for Rb2LiCeF6 and 3.4 for Cs2LiCeF6. The Penn model predicts that materials possessing higher band gaps have low ε1(0) and vice versa. The results show that these materials adhere to the Penn model. The figure illustrates ε2(ω), which depicts the polarizability of a material, as shown in Fig. 4b. No polarization was present in either material at an incident photon energy of 0 eV.
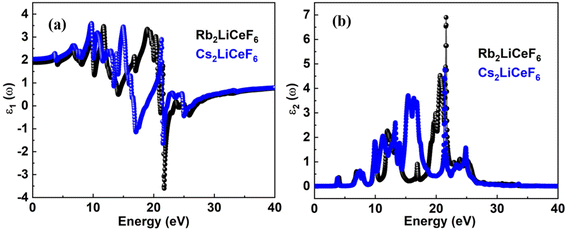 |
| | Fig. 4 ε1(ω) and ε2(ω) for (a) Rb2LiCeF6 and (b) Cs2LiCeF6. | |
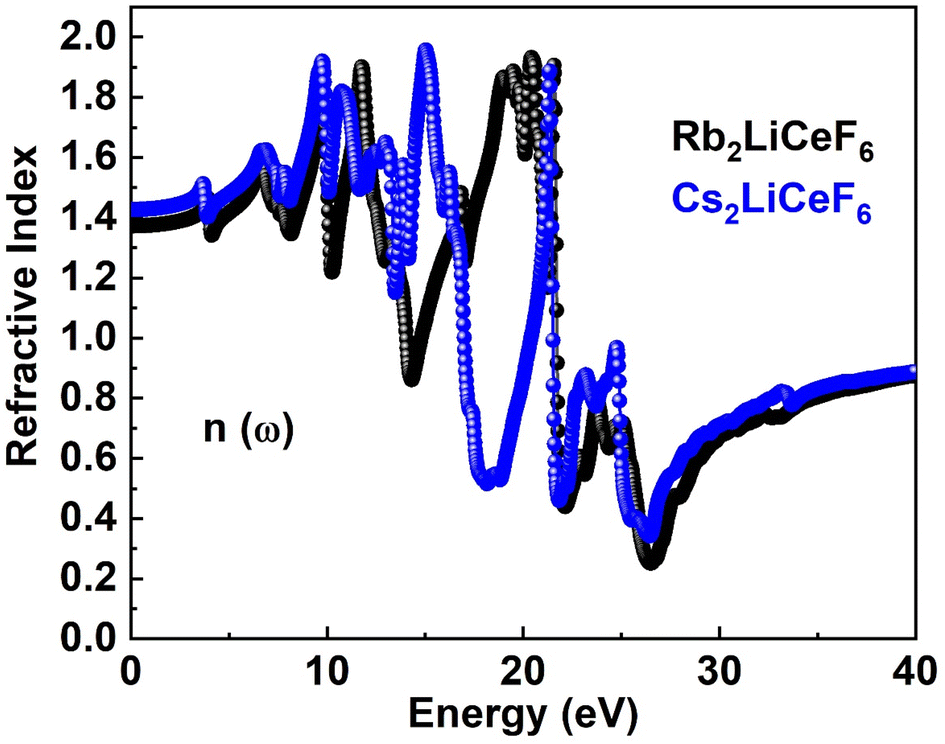
3.4.2. Refractive index. σ(ω), I(ω), and R(ω) can be computed using the ε1(ω) and ε2(ω) of the refractive index. The examined n(ω) is illustrated in Fig. 5. The spectrum of n(ω) for Rb2LiCeF6 and Cs2LiCeF6 each has a static refractive index, n(0), of 1.4 and 1.42, respectively. The peak value of n(ω) for Cs2LiCeF6 is 1.92 at approximately 15 eV and Rb2LiCeF6 has a maximum peak n(ω) of 1.9 at approximately 20 eV photon energy. To calculate the light refraction, the statistic refractive index, n(ω), is helpful, especially in photoelectric applications. Given that n(ω) >1 because of the reduction in the speed of photons when they enter matter, they interact with the electrons. More photons are delayed when they pass through a substance with a higher n(ω). Every technique that increases the electronic density of a material also increases its n(ω).
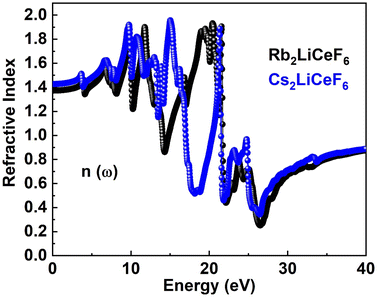 |
| | Fig. 5 n(ω) of M2LiCeF6 (M = Rb and Cs) compounds. | |
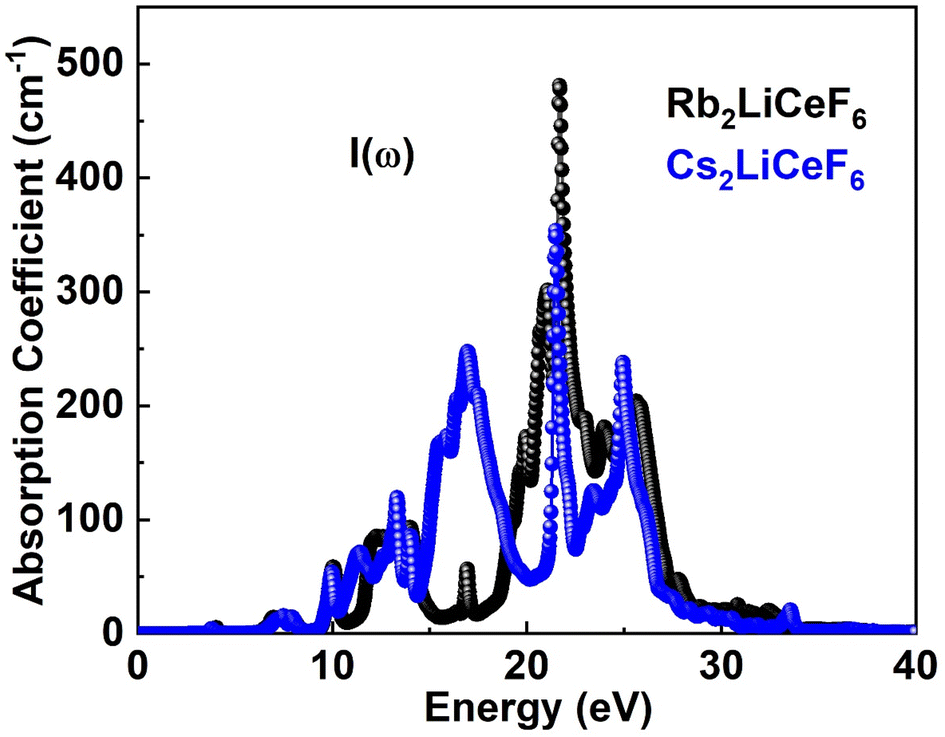
3.4.3. Absorption coefficient. Fig. 6 displays the I(ω) curves obtained using the ε(ω) approach. Fig. 6 shows that the chosen compounds have a significant I(ω) at energies between 0 eV and 40 eV.
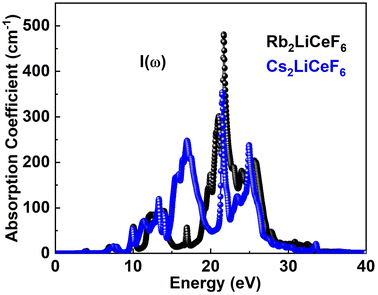 |
| | Fig. 6 I(ω) of M2LiCeF6 (M = Rb and Cs) compounds. | |
The threshold point of Rb2LiCeF6 and Cs2LiCeF6 is 0 eV, which is the point where these substances begin to absorb electromagnetic radiation. At the photon energy of 22 eV, Rb2LiCeF6 has a maximum absorption of 490, whereas that of Cs2LiCeF6 is 370.44.
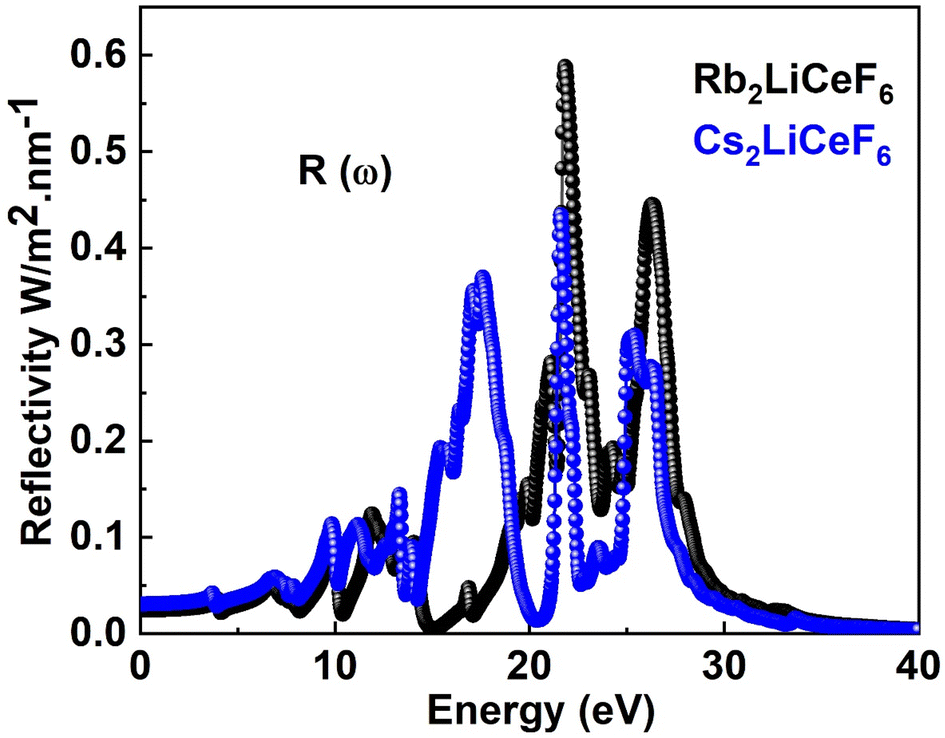
3.4.4. Reflectivity. The computed R(ω) for both double perovskites compounds is depicted in Fig. 7. Rb2LiCeF6 has a zero-frequency R(0) of 0.03, while Cs2LiCeF6 possesses a value of 0.02, and with an increase in photon energy, their reflectivity increases to a maximum of 0.59 and 0.43 at about 23 eV, respectively.
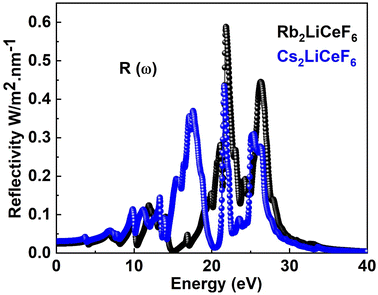 |
| | Fig. 7 R(ω) of M2LiCeF6 (M = Rb and Cs) compounds. | |
Thus, these materials have a very low reflectivity in the measured energy range, which is consistent with their band gap, making them transparent to incoming photons. Consequently, they can be applied in solar cells and lenses.
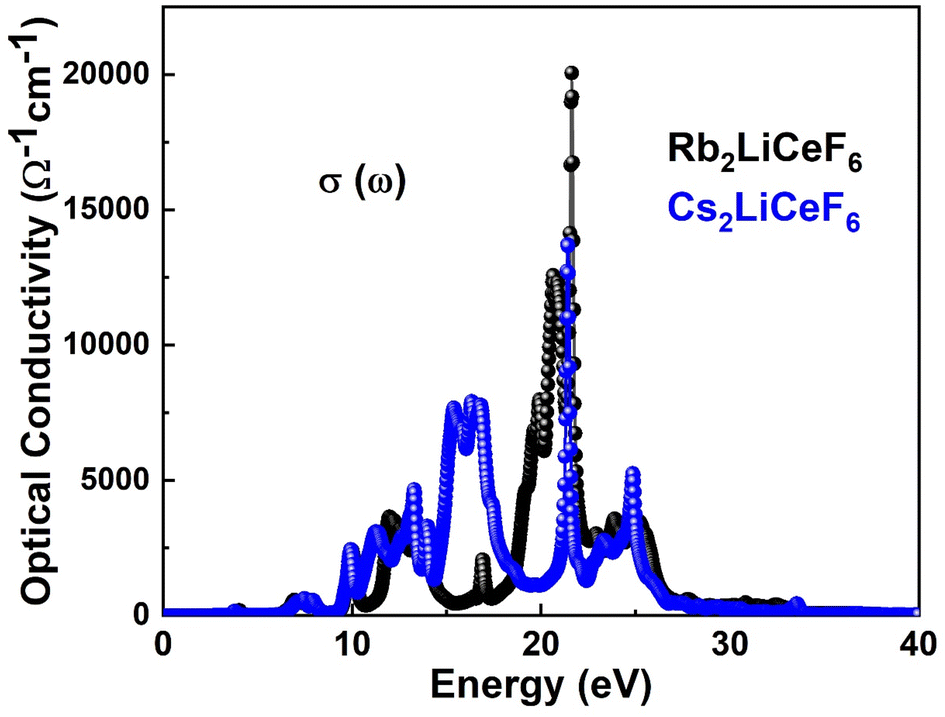
3.4.5. Optical conductivity. With the application of an electromagnetic field, electrons behave in a manner known as σ(ω). The outcome of assessing σ(ω) using dielectric functions is depicted in Fig. 8.
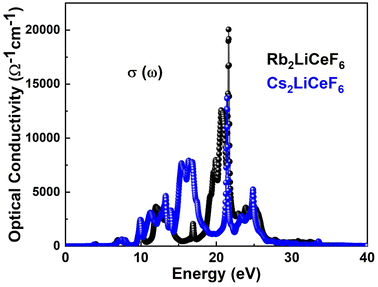 |
| | Fig. 8 σ(ω) of M2LiCeF6 (M = Rb and Cs) compounds. | |
According to Fig. 8, the optical conductivity, σ(ω), of the materials starts to develop close to their band gap energy, which is between 8–10 eV. In contrast to Rb2LiCeF6, which has a maximum σ(ω) of about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Ω−1 cm−1, the Cs2LiCeF6 compound has a major σ(ω) of about 12500 Ω−1 cm−1, as seen in Fig. 8. Thus, as a result of their excellent optical conductivity at high energies, these compounds may find application in numerous contemporary optoelectronic devices.
000 Ω−1 cm−1, the Cs2LiCeF6 compound has a major σ(ω) of about 12500 Ω−1 cm−1, as seen in Fig. 8. Thus, as a result of their excellent optical conductivity at high energies, these compounds may find application in numerous contemporary optoelectronic devices.
3.5. Thermoelectric properties
The Rb2LiCeF6 and Cs2LiCeF6 compounds are thought to exhibit strong thermoelectric capabilities because of their high estimated band gap energy and electronic state degeneracy. The transport properties of the Rb2LiCeF6 and Cs2LiCeF6 compounds vs. temperature are described in detail and the computed spectra of several parameters are shown in Fig. 9–12.
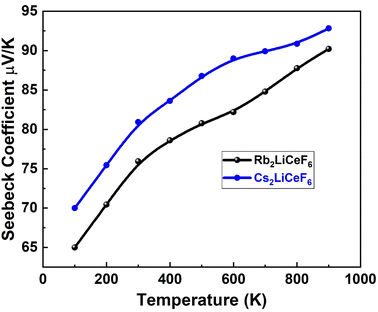 |
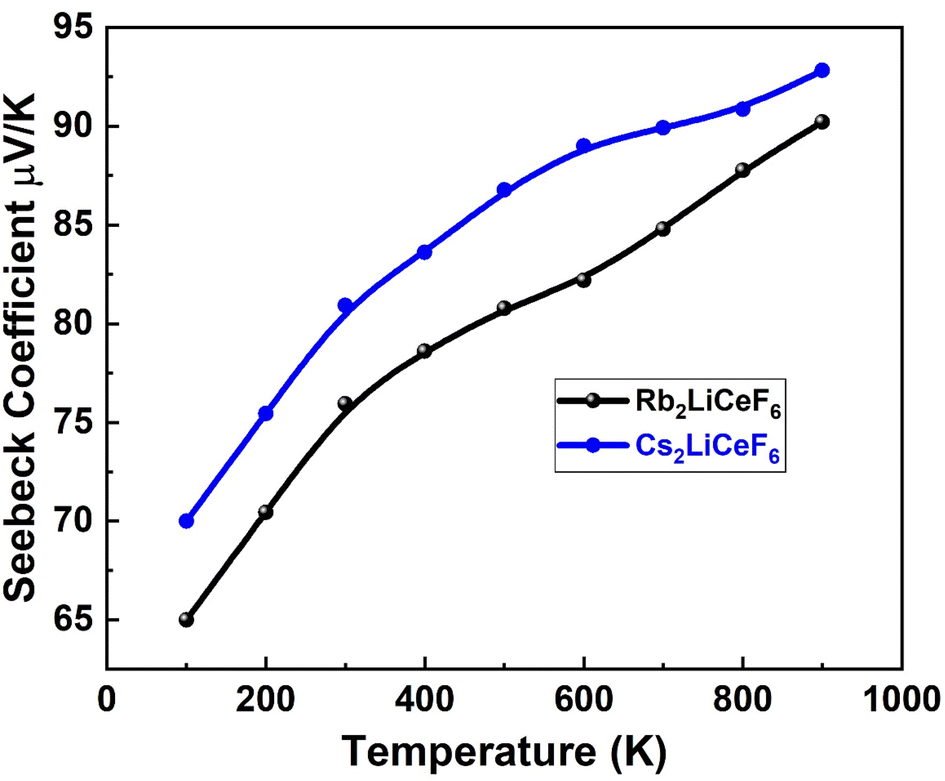
| | Fig. 9 Seebeck coefficient vs. temperature for M2LiCeF6 (M = Rb and Cs) compounds. | |
3.5.1. Seebeck coefficient “S”. Fig. 9 displays the estimated “S” for the M2LiCeF6 (M = Rb and Cs) compounds.The emergence of an electric potential through a temperature gradient in various electrical conductors or semiconductors is known as the Seebeck effect. Based on the concentration of charge carriers, the value of “S” was computed starting roughly at 100 K, which for both compounds increased exponentially with temperature, reaching a maximum of 90 V K−1 for Rb2LiCeF6 and 94 V K−1 for Cs2LiCeF6. The fact that both compounds have positive “S” values demonstrates that they are p-type compounds, making them active thermoelectric materials at higher temperatures. The greater size of the hole effective mass is the reason why the Seebeck coefficient (S) for p-type is higher than for n-type throughout the whole range of selected carrier concentrations.
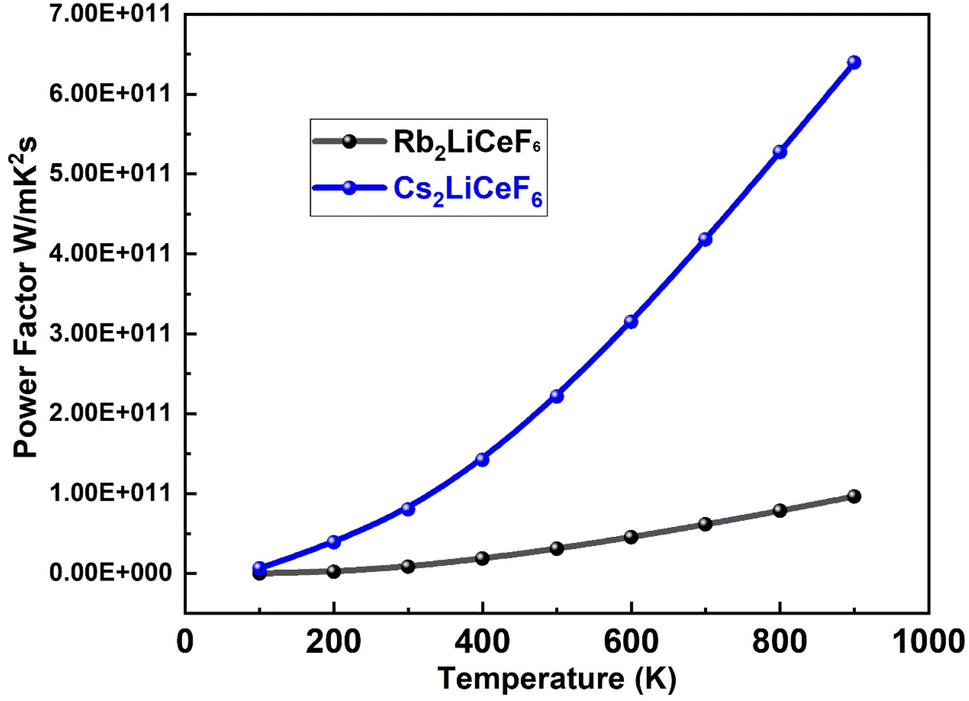
3.5.2. Thermal power factor (PF). The PF (S2σ/τ) for both compounds was plotted against relaxation time, as shown in. Fig. 10. PF is denoted by the term “S2σ” in the expression for “ZT”. Rb2LiCeF6 has a maximum “PF” value of 1 × 1011 W mK−2 s, while that of Cs2LiCeF6 reaches 6.5 × 1011 W mK2 s at around 850 K. Due to its higher “S2σ” value, Cs2LiCeF6 is anticipated to have stronger thermoelectric capabilities than Rb2LiCeF6 at high temperatures. Although it is frequently asserted that thermoelectric (TE) devices made of materials with higher power factors can ‘generate’ more energy (move more heat or extract more energy from the temperature difference), this can be only true for a thermoelectric device with fixed geometry and an unlimited heat source and cooling.
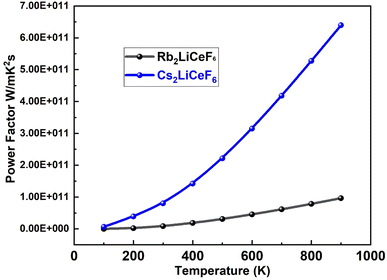 |
| | Fig. 10 Power factor “PF” vs. temperature for M2LiCeF6 (M = Rb and Cs) compounds. | |
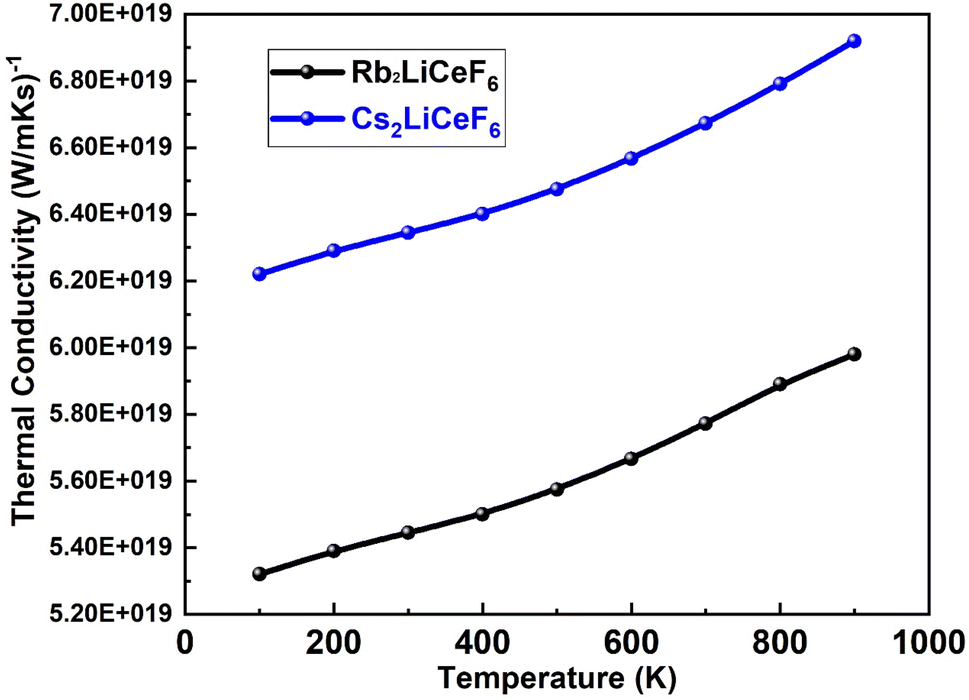
3.5.3. Thermal conductivity. Fig. 11 displays the investigated thermal conductivity versus temperature of Rb2LiCeF6 and Cs2LiCeF6. Low thermal conductivity and maximum electric carrier transport are characteristics of good thermoelectric materials. For many technological and temperature-critical applications, such as materials for thermoelectric conversion and thermal barrier coatings with higher temperature capabilities, thermal conductivity is an important parameter in the identification and development of alternative materials.
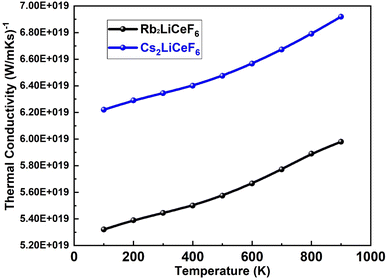 |
| | Fig. 11 Thermal conductivity vs. temperature of M2LiCeF6 (M = Rb and Cs) compounds. | |
Good thermoelectric behavior is shown by the linear increase in thermal conductivity at low temperatures as the temperature increases. Rb2LiCeF6 is a suitable thermoelectric material because its thermal conductivity values are lower than that of Cs2LiCeF6.
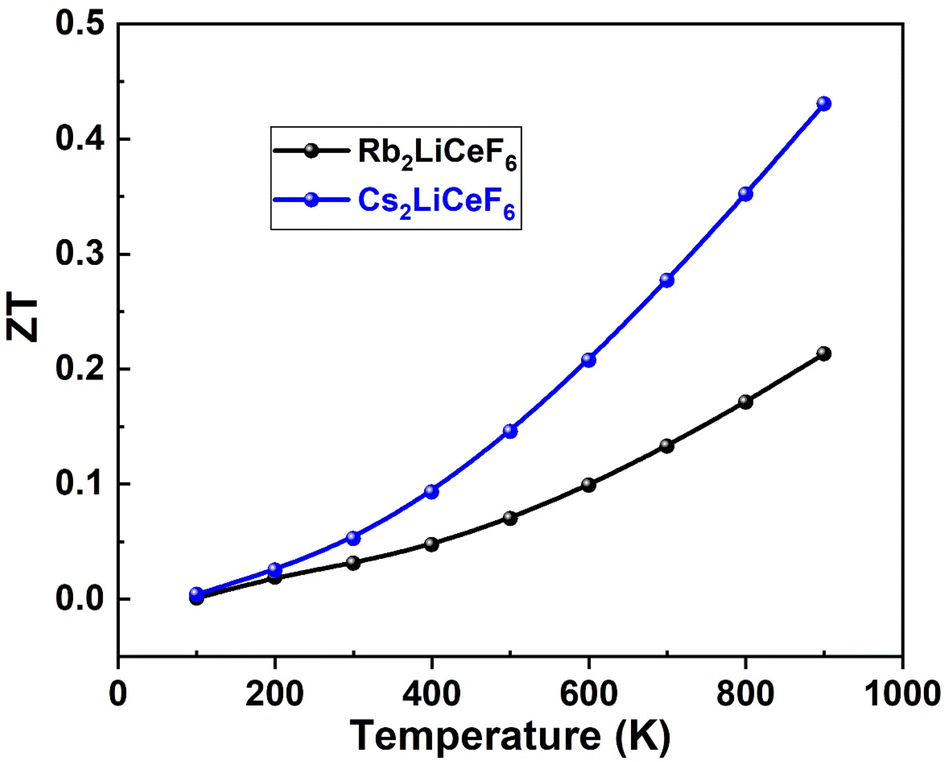
3.5.4. Figure of merit “ZT”. The dimensionless parameter “ZT” is used to rate thermoelectric materials and is significant for the quality of a material. As the “ZT” value increases, the thermoelectric efficiency, i.e., the heat conversion to electricity, increases. The formula for the figure-of-merit is ZT = S2σT/λ, where S2σ is the PF in the numerator. The highest value of “ZT” for an ideal thermoelectric material is 1, and thus achieving a value of “ZT” of ∼1 is the goal of materials scientists. Fig. 12 illustrates the greatest values of “ZT” achieved for Rb2LiCeF6 and Cs2LiCeF6 in this scenario, which were 0.20 and 0.45, respectively, demonstrating that they are both effective thermoelectric materials.
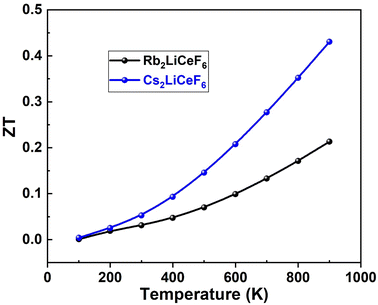 |
| | Fig. 12 Figure-of-merit “ZT” vs. temperature of Rb2LiCeF6 and Cs2LiCeF6. | |
These findings imply that the compounds under investigation possess intriguing thermoelectric capabilities and that Cs2LiCeF6 exhibits superior thermoelectric properties to Rb2LiCeF6.
4. Conclusion
Using the WIEN2K and BoltzTrap2 codes, the elastic, structural, optoelectronic, and thermoelectric features of M2LiCeF6 (M = Rb and Cs) double perovskites were examined computationally. Based on the results of some physical properties, the following conclusions were drawn.
Both M2LiCeF6 (M = Rb and Cs) double perovskites crystallize as stable cubic structures. M2LiCeF6 (M = Rb and Cs) exhibits an indirect band gap along the (Γ–X) symmetry points, which depicts that both materials are semiconductors. According to the DOS report, the Rb element dominates the contribution of the states in the band structures in the valence bands and the Ce atom in the conduction band for Rb2LiCeF6, whereas the Li element contributes more in the valence bands and Ce contributes in the CB for Cs2LiCeF6. The mechanical properties of the M2LiCeF6 (M = Rb and Cs) double perovskites reveal that these compounds are ductile, anisotropic, and elastically stable. The optical characteristics for incident photon energies are in the range of 0–40 eV. These materials can be active compounds in photovoltaic and optoelectronic devices because of their estimated maximum optical conductivity and absorption at a higher energy of incident photons. In addition, these compounds possess a considerably high “S” and “ZT” (figure of merit), signifying their prospective thermoelectric applications.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University (Abha, Saudi Arabia) for funding this work through Research Groups Program under grant number (RGP.2/379/44).
References
- G. Li, et al., Near-infrared responsive Z-scheme heterojunction with strong stability and ultra-high quantum efficiency constructed by lanthanide-doped glass, Appl. Catal., B, 2022, 311, 121363 CrossRef CAS.
- F. Igbari, Z. Wang and L. Liao, Progress of lead-free halide double perovskites, Adv. Energy Mater., 2019, 9(12), 1803150 CrossRef.
- X.-G. Zhao, D. Yang, J.-C. Ren, Y. Sun, Z. Xiao and L. Zhang, Rational design of halide double perovskites for optoelectronic applications, Joule, 2018, 2(9), 1662–1673 CrossRef CAS.
- H. Wang, et al., Interface engineering with a novel n-type small organic molecule for efficient inverted perovskite solar cells, Chem. Eng. J., 2020, 392, 123677 CrossRef CAS.
- Q. A. Akkerman, et al., Tuning the optical properties of cesium lead halide perovskite nanocrystals by anion exchange reactions, J. Am. Chem. Soc., 2015, 137(32), 10276–10281 CrossRef CAS PubMed.
- X. Li, Q. Kong, X. An, J. Zhang, Q. Wang and W. Yao, Enhanced cycling stability and storage performance of Na0. 67Ni0. 33Mn0. 67-xTixO1. 9F0. 1 cathode materials by Mn-rich shells and Ti doping, J. Colloid Interface Sci., 2023, 633, 82–91 CrossRef CAS PubMed.
- L. C. Schmidt, et al., Nontemplate synthesis of CH3NH3PbBr3 perovskite nanoparticles, J. Am. Chem. Soc., 2014, 136(3), 850–853 CrossRef CAS PubMed.
- D. P. McMeekin, et al., A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells, Science, 2016, 351(6269), 151–155 CrossRef CAS PubMed.
- J. Chen, M. E. Messing, K. Zheng and T. Pullerits, Cation-dependent hot carrier cooling in halide perovskite nanocrystals, J. Am. Chem. Soc., 2019, 141(8), 3532–3540 CrossRef CAS PubMed.
- Y. S. Park, S. Guo, N. S. Makarov and V. I. Klimov, Room temperature single-photon emission from individual perovskite quantum dots, ACS Nano, 2015, 9, 10386–10393 CrossRef CAS PubMed.
- J. Liu, J. Leng, K. Wu, J. Zhang and S. Jin, Observation of internal photoinduced electron and hole separation in hybrid two-dimensional perovskite films, J. Am. Chem. Soc., 2017, 139(4), 1432–1435 CrossRef CAS PubMed.
- Y. Wei, et al., High-Performance Visible to Near-Infrared Broadband Bi2O2Se Nanoribbon Photodetectors, Adv. Opt. Mater., 2022, 10(23), 2201396 CrossRef CAS.
- X. Chen, C. Wang, Z. Li, Z. Hou and W.-J. Yin, Bayesian optimization based on a unified figure of merit for accelerated materials screening: A case study of halide perovskites, Sci. China Mater., 2020, 63(6), 1024–1035 CrossRef CAS.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, A bismuth-halide double perovskite with long carrier recombination lifetime for photovoltaic applications, J. Am. Chem. Soc., 2016, 138(7), 2138–2141 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Cs2AgBiX6 (X= Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors, Chem. Mater., 2016, 28(5), 1348–1354 CrossRef CAS.
- M. R. Filip, S. Hillman, A. A. Haghighirad, H. J. Snaith and F. Giustino, Band gaps of the lead-free halide double perovskites Cs2BiAgCl6 and Cs2BiAgBr6 from theory and experiment, J. Phys. Chem. Lett., 2016, 7(13), 2579–2585 CrossRef CAS PubMed.
- R. J. Sutton, et al., Bandgap-tunable cesium lead halide perovskites with high thermal stability for efficient solar cells, Adv. Energy Mater., 2016, 6(8), 1502458 CrossRef.
- M. Fu, W. Chen, Y. Lei, H. Yu, Y. Lin and M. Terrones, Biomimetic Construction of Ferrite Quantum Dot/Graphene Heterostructure for Enhancing Ion/charge Transfer in Supercapacitors, Adv. Mater., 2023, 2300940 CrossRef CAS PubMed.
- T. T. Tran, J. R. Panella, J. R. Chamorro, J. R. Morey and T. M. McQueen, Designing indirect–direct bandgap transitions in double perovskites, Mater. Horiz., 2017, 4(4), 688–693 RSC.
- J. Luo, et al., Efficient and stable
emission of warm-white light from lead-free halide double perovskites, Nature, 2018, 563(7732), 541–545 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal halide perovskites as visible-light sensitizers for photovoltaic cells, J. Am. Chem. Soc., 2009, 131(17), 6050–6051 CrossRef CAS PubMed.
- L. Shi, et al., Accelerated lifetime testing of organic–inorganic perovskite solar cells encapsulated by polyisobutylene, ACS Appl. Mater. Interfaces, 2017, 9(30), 25073–25081 CrossRef CAS PubMed.
- W. S. Yang, et al., High-performance photovoltaic perovskite layers fabricated through intramolecular exchange, Science, 2015, 348(6240), 1234–1237 CrossRef CAS PubMed.
- C. Han, H. Wang, Z. Wang, X. Ou and Y. Tang, Solvation Structure Modulation Of High-Voltage Electrolyte For High-Performance K-Based Dual-Graphite Battery, Adv. Mater., 2013, 2300917 Search PubMed.
- J. F. Geisz, M. A. Steiner, K. L. Schulte, M. Young, R. M. France and D. J. Friedman, Six-junction concentrator solar cells, AIP Conf. Proc., 2018, 2012(1), 40004 CrossRef.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Chemical management for colorful, efficient, and stable inorganic–organic hybrid nanostructured solar cells, Nano Lett., 2013, 13(4), 1764–1769 CrossRef CAS PubMed.
- S. D. Stranks, et al., Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber, Science, 2013, 342(6156), 341–344 CrossRef CAS PubMed.
- Q. Dong, et al., Electron-hole diffusion lengths> 175 μm in solution-grown CH3NH3PbI3 single crystals, Science, 2015, 347(6225), 967–970 CrossRef CAS PubMed.
- D. Shi, et al., Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals, Science, 2015, 347(6221), 519–522 CrossRef CAS PubMed.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H.-M. Cheng, Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage, Nat. Chem., 2018, 10(6), 667–672 CrossRef CAS PubMed.
- A. E. Maughan, A. M. Ganose, M. A. Almaker, D. O. Scanlon and J. R. Neilson, Tolerance factor and cooperative tilting effects in vacancy-ordered double perovskite halides, Chem. Mater., 2018, 30(11), 3909–3919 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites, Science, 2012, 338(6107), 643–647 CrossRef CAS PubMed.
- T. Kirchartz, J. Bisquert, I. Mora-Sero and G. Garcia-Belmonte, Classification of solar cells according to mechanisms of charge separation and charge collection, Phys. Chem. Chem. Phys., 2015, 17(6), 4007–4014 RSC.
- X. Zhang, Y. Tang, F. Zhang and C. Lee, A novel aluminum–graphite dual-ion battery, Adv. Energy Mater., 2016, 6(11), 1502588 CrossRef.
- N. J. Jeon, et al., Compositional engineering of perovskite materials for high-performance solar cells, Nature, 2015, 517(7535), 476–480 CrossRef CAS PubMed.
- F. Di Giacomo, A. Fakharuddin, R. Jose and T. M. Brown, Progress, challenges and perspectives in flexible perovskite solar cells, Energy Environ. Sci., 2016, 9(10), 3007–3035 RSC.
- T. Charpin, A Package for Calculating Elastic Tensors of Cubic Phase Using WIEN, Laboratory of Geometrix, Paris, 2001 Search PubMed.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, and J. Luitz, wien2k,” An Augment. Pl. wave+ local orbitals Progr. Calc. Cryst. Prop., vol. 60, 2001 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- F. Tran and P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential, Phys. Rev. Lett., 2009, 102(22), 226401 CrossRef PubMed.
- S. Saha, T. P. Sinha and A. Mookerjee, Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3, Phys. Rev. B, 2000, 62(13), 8828–8834 CrossRef CAS.
- M. Husain, et al., Exploring the exemplary structural, electronic, optical, and elastic nature of inorganic ternary cubic XBaF3 (X = Al and Tl) employing the accurate TB-mBJ approach, Semicond. Sci. Technol., 2022, 37(7), 075004 CrossRef.
- F. D. Murnaghan, The compressibility of media under extreme pressures, Proc. Natl. Acad. Sci. U. S. A., 1944, 30(9), 244 CrossRef CAS PubMed.
- P. Dufek, P. Blaha and K. Schwarz, Applications of Engel and Vosko's generalized gradient approximation in solids, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(11), 7279 CrossRef CAS PubMed.
- Z. Charifi, H. Baaziz, F. E. H. Hassan and N. Bouarissa, High pressure study of structural and electronic properties of calcium chalcogenides, J. Phys.: Condens. Matter, 2005, 17(26), 4083 CrossRef CAS.
- N. Rahman, et al., First principle study of structural, electronic, optical and mechanical properties of cubic fluoro-perovskites:(CdXF3, X= Y, Bi), Eur. Phys. J. Plus, 2021, 136(3), 1–11 Search PubMed.
- M. Sohail, et al., First-principal investigations of electronic, structural, elastic and optical properties of the fluoroperovskite TlLF 3 (L= Ca, Cd) compounds for optoelectronic applications, RSC Adv., 2022, 12(12), 7002–7008 RSC.
- M. A. Ali, et al., A theoretical study of the structural, hermoelectric, and spin-orbit coupling influenced optoelectronic properties of CsTmCl3 halide perovskite, Int. J. Quantum Chem., 2020, 120(7), e26141 CrossRef CAS.
- A. Mohamed, A. El Houssine, F. Nejmaa, and B. Ibrahim, Ab-initio Study of Electronic, Optical and Thermoelectric properties of TiO2 phases using mBJ approximation, in 2020 IEEE 6th International Conference on Optimization and Applications, ICOA, 2020, pp. 1–5 Search PubMed.
- M. J. Mehl, Pressure dependence of the elastic moduli in aluminum-rich Al-Li compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(5), 2493 CrossRef CAS PubMed.
- R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc. A, 1952, 65(5), 349 CrossRef.
- W. Voigt, Lehrbuch der Kristallphysik (Textbook of crystal physics), BG Teubner, Leipzig und Berlin, 1928 Search PubMed.
- D. G. Pettifor, Theoretical predictions of structure and related properties of intermetallics, Mater. Sci. Technol., 1992, 8(4), 345–349 CrossRef CAS.
- S. F. Pugh, XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45(367), 823–843 CrossRef CAS.
- I. N. Frantsevich, Elastic constants and elastic moduli of metals and insulators, Ref. B., 1982 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Abdur Rauf†bc,
Mudasser Husain*a,
Nourreddine Sfinad,
Vineet Tirth
*a,
Abdur Rauf†bc,
Mudasser Husain*a,
Nourreddine Sfinad,
Vineet Tirth