
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine doping induced activation of covalent organic framework cathodes for Li-ion batteries†
Guoying Rena,
Fengshi Cai *a,
Shoucheng Wanga,
Zhiqiang Luo*b and
Zhihao Yuanc
*a,
Shoucheng Wanga,
Zhiqiang Luo*b and
Zhihao Yuanc
aTianjin Key Lab for Photoelectric Materials and Devices, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: caifs@tjut.edu.cn
bKey Laboratory of Display Materials and Photoelectric Devices (Tianjin University of Technology), Ministry of Education of China, Tianjin 300384, China. E-mail: zhqluo@email.tjut.edu.cn
cNational Demonstration Center for Experimental Function Materials Education, Tianjin University of Technology, Tianjin 300384, China
First published on 22nd June 2023
Abstract
Covalent organic frameworks (COFs) are considered as promising candidate organic electrode materials for lithium-ion batteries (LIBs) because of their relatively high capacity, ordered nanopores, and limited solubility in electrolyte. However, the practical capacity of COF materials is mainly affected by their low electronic/ionic conductivity and the deep-buried active sites inside the COFs. Here, we synthesize an iodine doped β-ketoenamine-linked COF (2,6-diaminoanthraquinone and 1,3,5-triformylphloroglucinol, denoted as COF-I) by a facile one-pot solvothermal reaction. The introduction of iodine can make the COF more lithiophilic inside and exhibit high intrinsic ion/electron transport, ensuring more accessible active sites of the COFs. Consequently, when used as the cathode of LIBs, COF-I demonstrates a high initial discharge capacity of 140 mA h g−1 at 0.2 A g−1, and excellent cycling stability with 92% capacity retention after 1000 cycles. Furthermore, a reversible capacity of 95 mA h g−1 at 1.0 A g−1 is also achieved after 300 cycles. Our study provides a facile way to develop high-performance COF electrode materials for LIB applications.
1. Introduction
Organic materials have received significant attention as electrode materials for high-performance energy storage systems owing to their advantages of structural diversity, resource sustainability, environmental friendliness and easy functionalization.1,2 Two-dimensional covalent organic frameworks (COFs) are an emerging class of crystalline porous polymers with great structural stability and regular open channels. Besides, the backbone structure of COFs can be self-assembled by π–π stacking the pre-designed block units with active sites for high capacity, and their abundant host structure facilitate the infiltration of electrolyte ions.3 Therefore, COFs are attracting extensive interest in energy storage like rechargeable Li+, Na+, K+ and Zn2+ batteries.4–7 However, the strong π–π interaction causes close stacking between COF layers, resulting in the interior active sites buried in bulk COF, which limits accessibility and utilization of redox-active sites of COFs.8 In addition, poor intrinsic conductivity is one of the major drawbacks of COF electrodes for high efficient energy storage devices, which restricts the charge to adequately interact with the active group in the framework.9 These issues inevitably lead to low capacity and poor rate performance of COF electrodes in rechargeable batteries.Recently, several strategies are attempted to overcome these above-mentioned issues in COFs. For example, hybridization with conductive carbon materials such as carbon nanotube and graphene can effectively increase their conductivity and specific surface area, thus enhancing capacity and rate performance.10–13 Nevertheless, the introduction of a large amount of conductive materials reduces the amount of active material COFs in the electrode, generating practically low capacity and utilization.14 In addition, an exfoliation strategy has been developed to expose the buried redox-active sites in the pores of COF, and thus enhancing storage capacity of metal ions in rechargeable batteries.15–17 However, using the exfoliation technology to produce COF few-layer nanosheets also faces some problems such as low yield and restacking, which can hinder the further practical application of COF materials for energy-storage batteries.
Introducing active guest species is another effective method to improve the electrical conductivity and the storage performance of COF materials.18–22 For example, Dichtel et al. introduced poly(3,4-ethylenedioxythiophene) (PEDOT) into the pores of COF to improve the electrical conductivity by secondary polymerizations.23 Liu et al. significantly improved the electrical conductivity of TTF-based COFs via doping with iodine or tetracyanoquinodimethane (TCNQ).24 Besides, NiPc-CoTAA framework,25 2D ZnPc-pz COF,26 fluorene-based COF,27 and TANG-COF28 were also reported to enhance the electrical conductivity by absorbing iodine strategy. However, introducing a large amount of guest molecules into the COF pores can hinder ion transport pathway toward redox active sites of the COF, resulting in low capacity and poor rate performance. Therefore, it is still desirable to develop promising COF materials with high inherent ion/electron transport capability for rechargeable batteries.
The β-ketoenamine-linked COF (DAAQ-TFP COF), firstly reported for the energy storage in 2013, has showed the promise for electrical energy storage devices due to its inherent high surface areas and high density of redox-active sites.29,30 The reported results show that the low utilization of anthraquinone redox units is still the main factor limiting the application of bulk DAAQ-TFP COF materials. In a later study, it would be desirable to develop high-performance devices from bulk COF powders. Herein, iodine doped β-ketoenamine-linked COF (COF-I) was prepared through a facile one-pot solvothermal reaction between 1,3,5-triformylphloroglucinol (TFP) and 2,6-diaminoanthraquinone (DAAQ) by adding iodine into the synthetic process of COF. Iodine doping and rod-flower structure endow COF-I more accessible active sites and high intrinsic ion/electron transport capability. Consequently, COF-I cathode demonstrates a significantly increased discharge capacity of 140 mA h g−1 at 0.2 A g−1 and good rate performance in LIBs. This work provides a facile approach for the development of high electrochemical performance COF electrode materials without complicate fabrication and the need for the use of conductive substrates.
2. Experimental section
2.1 Material synthesis
The COF-I was synthesized by a one-pot Schiff-base condensation, as shown in Scheme 1. In a typical process, TFP (0.2 mmol), DAAQ (0.4 mmol), 5 mL of mesitylene, 5 mL of 1,4-dioxane, and 6 M acetic acid (3 mL) were mixed and grinded for 1 hour. Subsequently, 80 mg solid iodine was added, and the mixture was ultrasonically treated for 10 min to obtain uniform dispersion. And then the obtained mixture was transferred into a Teflon-lined stainless autoclave and heated at 120 °C for 72 h. The precipitate was collected and washed several times with deionized water, N,N-dimethylformamide, ethanol, acetone, and dichloromethane. The obtained product was dried at 80 °C for 24 h in a vacuum oven. The pristine COF was also prepared by the above process without iodine adding.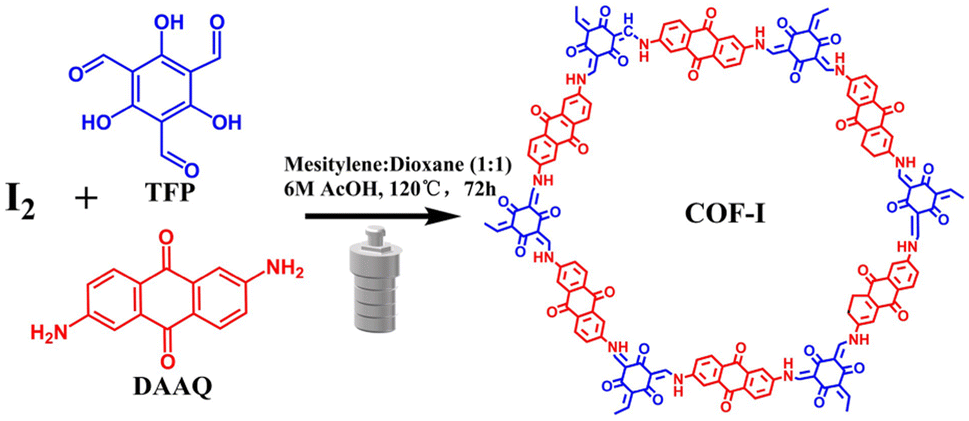 | ||
| Scheme 1 The synthetic route for COF-I. | ||
2.2 Material characterization
Fourier Transform Infrared Spectroscopy (FT-IR) was recorded on a PerkinElmer spectrometer (FTIR/STA6000-TL9000-Clarus SQ8). X-ray photoelectron spectrometer (XPS) was used to analyze the element status on a Thermo ESCALAB 250 instrument. The solid-state 13C-MAS NMR spectra were recorded on a Brucker AVANCE HD 400 MHz spectrometer. Powder X-ray diffractometer (PXRD) analysis was performed on a D/Max-2500PC diffractometer with Cu Kα radiation. Nitrogen adsorption/desorption measurements were conducted on a surface area and pore size analyzer (Autosorb-IQ3+ChemStar) at 77 K. The specific surface area was calculated by the multi-point Brunauer–Emmett–Teller (BET) method. The pore size distribution was obtained via using the nonlocal density flooding theory (NLDFT) method with N2 as the adsorbent. Electron paramagnetic resonance (EPR) was conducted by using a Bruker EMXplus-6/1 paramagnetic resonance spectrometer. Transmission electron microscopy (TEM) and Energy dispersive spectroscopy (EDS) were studied by using a Quanta FEG 250 microscope (JSM-2100F). The morphology was investigated by a scanning electron microscopy (SEM, JSM-7500F). Thermogravimetric analysis (TGA) was carried out under nitrogen atmosphere by a NETZSCH 209F3A TG-DSC analyzer with 5 °C min−1 from room temperature to 800 °C. The Hall mobility, carrier concentration and Hall coefficient of the samples were measured by a Hall tester (HMS-505). The resistivity of the sample was determined by using four-probe method with a semiconductor powder resistivity tester (ST2722). UV-vis spectra were recorded on a spectrophotometer (Lambda 750 UV/VIS/NIR). Zeta potential was measured via a Zeta potential analyzer (NanoBrook 90Plus Zeta) using dilute suspensions in ethanol.2.3 Electrochemical measurements
Cathode slurry was fabricated by mixing the active materials, acetylene black and carboxymethylcellulose sodium (CMC) with a mass ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 in styrene butadiene rubber (Sbr) aqueous solution. The slurry was uniformly coated on a titanium foil and dried at 70 °C in a vacuum oven overnight. Subsequently, the as-prepared electrode was punched and pressed, and the mass loading of active materials is about 1.5 mg cm−2. The CR2032 coin cells were assembled in the argon-filled glove box with H2O and O2 contents of both <1 ppm. Li foil was used as anode, Celgard-2325 PP membranes as separator, and 1 M LiTFSI in 1,3-ioxolane (DOL) and dimethoxyethane (DME) (v/v = 1:1) containing 1 wt% of LiNO3 as electrolyte. The galvanostatic discharge/charge tests were carried out at room temperature within the potential range of 1.3–3.2 V by LAND-CT2001A battery test system. Cyclic voltammetry (CV) measurements were performed on a Zahner electrochemical workstation at various scan rates. Electrochemical impedance spectroscopy (EIS) was tested on a CHI660 electrochemical workstation (0.1 Hz to 100 kHz; applied voltage 10 mV). The galvanostatic intermittent titration technique (GITT) was carried out by LAND-CT2001A battery test system.
:3:1 in styrene butadiene rubber (Sbr) aqueous solution. The slurry was uniformly coated on a titanium foil and dried at 70 °C in a vacuum oven overnight. Subsequently, the as-prepared electrode was punched and pressed, and the mass loading of active materials is about 1.5 mg cm−2. The CR2032 coin cells were assembled in the argon-filled glove box with H2O and O2 contents of both <1 ppm. Li foil was used as anode, Celgard-2325 PP membranes as separator, and 1 M LiTFSI in 1,3-ioxolane (DOL) and dimethoxyethane (DME) (v/v = 1:1) containing 1 wt% of LiNO3 as electrolyte. The galvanostatic discharge/charge tests were carried out at room temperature within the potential range of 1.3–3.2 V by LAND-CT2001A battery test system. Cyclic voltammetry (CV) measurements were performed on a Zahner electrochemical workstation at various scan rates. Electrochemical impedance spectroscopy (EIS) was tested on a CHI660 electrochemical workstation (0.1 Hz to 100 kHz; applied voltage 10 mV). The galvanostatic intermittent titration technique (GITT) was carried out by LAND-CT2001A battery test system.
3. Results and discussion
The FT-IR in Fig. 1a was carried out to confirm the formation of COF and COF-I. After condensation, the amino (–NH2) stretching bands of DAAQ (3100–3500 cm−1) and aldehyde groups (–CHO) stretching vibration of TFP (1659 cm−1) disappear completely for both COF and COF-I. The new characteristic peaks for C–N (1259 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1566 cm−1) groups can be observed in both COF and COF-I. This indicates that the aldehyde-ammonia condensation reaction had completed.31,32 Notably, after iodine doping, the characteristic peaks belonging to C–N and CC groups shift to the lower wave number, indicating the possible interaction between the enamine linkage and phenyl ring of framework and iodine.33 Solid-state 13C NMR spectroscopy (Fig. S1†) of COF was also investigated. The chemical shift at ∼145 ppm can be attributed to the enamine carbon of β-ketoenamine species, while the signals at ∼180 ppm correspond to carbonyl carbons of anthraquinone species. The NMR result further indicates the successful fabrication of β-ketoenamine-linked materials.
C (1566 cm−1) groups can be observed in both COF and COF-I. This indicates that the aldehyde-ammonia condensation reaction had completed.31,32 Notably, after iodine doping, the characteristic peaks belonging to C–N and CC groups shift to the lower wave number, indicating the possible interaction between the enamine linkage and phenyl ring of framework and iodine.33 Solid-state 13C NMR spectroscopy (Fig. S1†) of COF was also investigated. The chemical shift at ∼145 ppm can be attributed to the enamine carbon of β-ketoenamine species, while the signals at ∼180 ppm correspond to carbonyl carbons of anthraquinone species. The NMR result further indicates the successful fabrication of β-ketoenamine-linked materials.
 | ||
| Fig. 1 (a) FT-IR spectra of DAAQ, TFP, COF, and COF-I; (b) XPS survey spectra of COF and COF-I; (c) high-resolution I 3d XPS curve of COF-I; (d) powder X-ray diffraction patterns of the COF and COF-I; (e) N2 adsorption–desorption isotherm curves (inset: pore size distributions); (f) EPR spectra of COF and COF-I. | ||
X-ray photoelectron spectroscopy (XPS) was conducted to characterize the changes of COF surface information before and after iodine doping. The full spectra of both samples (Fig. 1b) show that the elements C, N and O are the main surface chemical components, while the signal from iodine element can be detected in the COF-I sample. The high-resolution I 3d curve (Fig. 1c) presents a couple of fitting peaks at 618.7 eV and 632.0 eV for I 3d5/2 and I 3d3/2, respectively, which are ascribed to iodine anions chemical state.25,34 For pristine COF (Fig. S2†), the high-resolution C 1s XPS curve reveals three peaks at 284.7, 286.1, and 288.7 eV for CC, C–N, and CO bonds,35 respectively. The N 1s XPS indicates that the peak position at 399.5 and 400.1 eV are assigned to imine N and sp3 N in COF,36 respectively. In high-resolution O 1s XPS spectrum of COF, it can observe two peaks at 531.2 and 533 eV, corresponding to CO and C–O bonds,17 respectively. After iodine doping, these peak positions of C, N, O elements undergo slight shift, suggesting the charge-transfer between iodine and various C, N, O species in COF.37,38 Raman spectroscopy of the COF-I sample was measured and depicted in Fig. S3.† In the Raman spectrum, the COF-I exhibits two characteristic peaks at 113 and 167 cm−1, which can be assigned to the symmetric stretching vibration of I3− and I5−, respectively, which further indicates that the charge transfer occurred between the guest iodine and the electron-rich host network at COFs.39
Powder X-ray diffraction (PXRD) pattern of COF exhibits the obvious peaks at 3.5° and 26.9° (Fig. 1d), corresponding to the (100), and (001) crystal planes, respectively, indicating a highly crystalline honeycomb frame and the ordered porous structure. This result is in accordance with previous reports.5,31,40 Pawley refinement of COF using a P6/m unit cell (Fig. S4†) was performed, and the simulated PXRD pattern is in good agreement with the experimental data. It should be noted that the broad diffraction peak position at 26.5° in COF-I has a slight downshift compared to pristine COF (Fig. 1d), ascribed to the slightly enlarged distance of two COF layers along the c direction after iodine doping. Besides, a weaker diffraction peak intensity at 3.5° for COF-I is observed, implying the decreased crystallinity of COF structure after the induction of iodine species.41
The porosity of COF and COF-I was evaluated at 77 K by N2 adsorption–desorption measurements. Both COFs show reversible isotherms with a steep uptake at low pressures (P/P0 < 0.1) (Fig. 1e), indicating their microporous nature. The Brunauer–Emmett–Teller (BET) specific surface area of COF-I is 1056 m2 g−1, larger than that of pristine COF (517 m2 g−1). The pore size distributions of both COFs are analyzed based on nonlocal density functional theory (NLDFT). COF-I has an average pore size distribution of about 2.39 nm, while COF shows small pore size distribution of about 1.56–2.29 nm (Fig. 1e). The high specific surface area and large pore size distribution is probably originated from the aggregation state change induced by the iodine doping. In addition, Fig. 1f shows the electron paramagnetic resonance (EPR) spectra. A strong EPR signal at g = 2.0045 can be observed for COF-I, implying the presence of TFP radical cations from oxidation by iodine.42,43 The charged nanochannels can create a benign environment for capturing numerous Li+ ions from the electrolyte, which is conducive to Li+ moving toward redox-active sites in the channels of COF.
The morphology and microstructure of materials were analyzed by SEM and TEM. It can be seen from Fig. 2a and b that pristine COF has the flower-like morphology with nanorods (diameter of ∼400 nm and length of ∼3.5 μm). Comparatively, the COF-I (Fig. 2d and e) maintains the flower-like morphology, but the nanorods have smaller diameter (∼300 nm) and coarser surface. This helps to provide a larger contact area with the electrolyte and improve Li+/electron transfer channels on the nanorods. High-resolution TEM (HRTEM) images (Fig. 2c and f) show that pristine COF presents the distinct lattice fringes with interlayer distance of 2.1 nm, while COF-I exhibits weak lattice fringe and the slightly extended interlayer distance of 2.3 nm after the iodine introduction. This result is consistent with the previous PXRD analysis. Energy-dispersive spectroscopy (EDS) mapping images further exhibit that the COF-I framework has a uniform distribution of C, N, O, and I elements (Fig. 2g), and the content of iodine is about 2 wt%. (Fig. S5†). The elemental mapping images of pristine COF are also shown in Fig. S6.† To evaluate the reproducibility of iodine doped COF material synthesis, we prepared ten different batches of COF-I samples. The SEM images and the corresponding EDS for ten different batches of COF-I were measured and depicted in Fig. S7.† The morphology and the iodine content of each batch display the similar results.
 | ||
| Fig. 2 SEM images of COF (a) and COF-I (d); TEM and HRTEM images of COF (b and c) and COF-I (e and f); (g) corresponding EDS elemental mappings of iodine (yellow), carbon (red), nitrogen (blue) and oxygen (green) of COF-I. | ||
The thermodynamic stability of both COF and COF-I was further investigated by thermogravimetric analysis (TGA) (Fig. S8†). The TGA curves of COF and COF-I exhibit a similar thermal decomposition process. Before 250 °C, the weight loss is attributed to the evaporation of water and solvent remaining in the nano-pore of samples. In the range of 250–520 °C, the weight loss can result from the breaking of chemical bonds and the decomposition of some oligomers. Obviously, both COF and COF-I show high thermal stability. In addition, the chemical stability of COF-I was also assessed in five solvents (1,3-dioxolane/dimethoxyethane, dichloromethane, tetrahydrofuran, water, hydrogen chloride). The digital images (Fig. S9†) show that solution color does not change after soaking the COF-I in different solvents for two months at room temperature. The FT-IR spectra (Fig. S10†) and UV-vis spectra (Fig. S11†) display that there are no dissolution of COF-I. This also further indicates that there is a strong interaction between the doped iodine and the COF skeleton, showing the high chemical stability of COF-I.
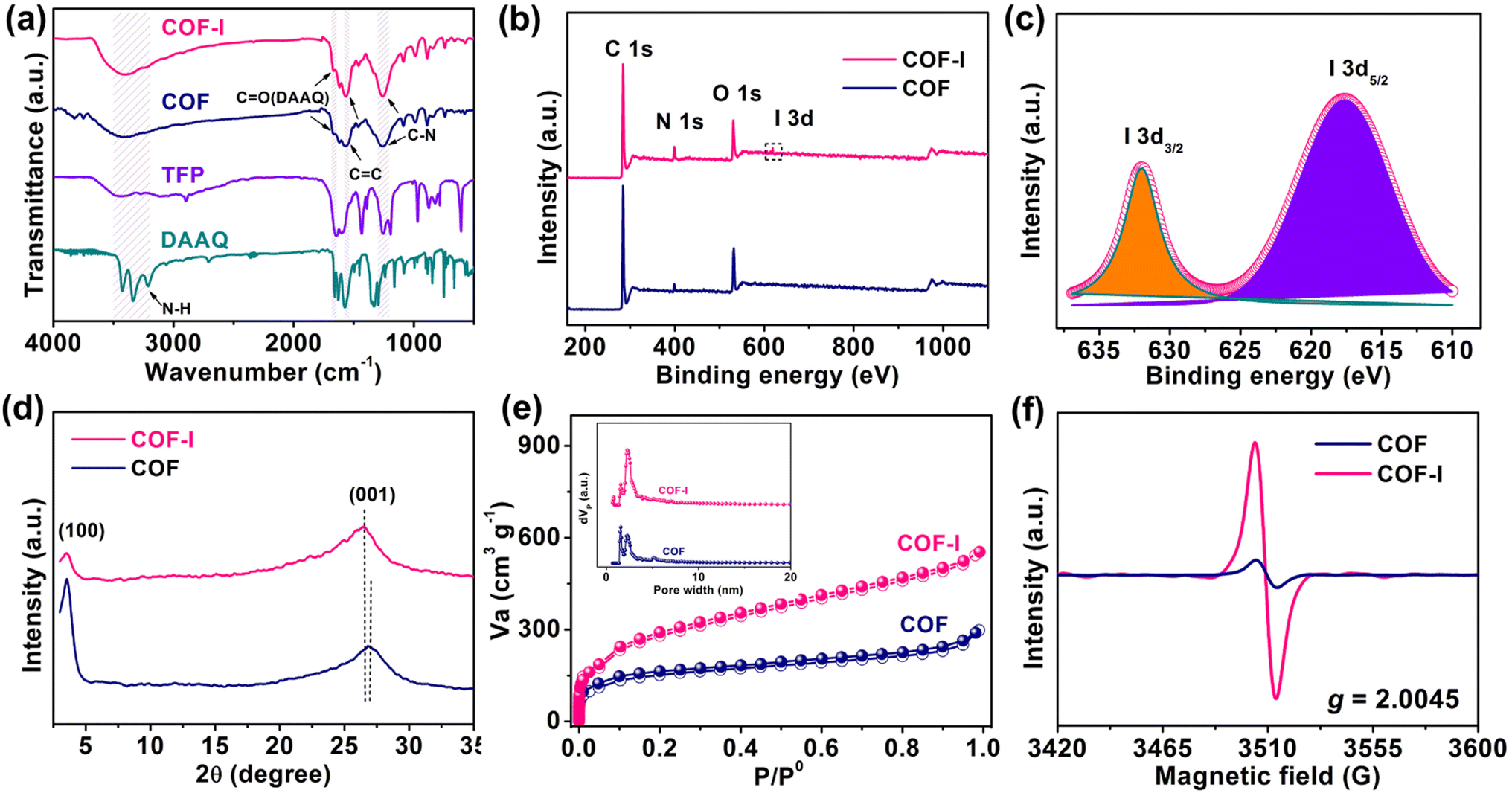
Before the electrochemistry test, Hall effect measurement was employed to investigate the electrical properties of COF-I in vacuum condition at 298 K. Fig. 3a shows that COF-I is a n-type semiconductor with carrier density of 1.3 × 1013 cm−3 and the direct-current Hall electron mobility of 11.05 cm2 V−1 s−1, which represents relatively high values compared to some previously reported 2D-COFs materials (Table S1†). The DC bulk conductivity was evaluated using the four-point probe measurement on a pressed powder pellet, and the corresponding electrical conductivity of COF-I is 0.034 S m−1 at 298 K (Fig. 3b). For comparison, the electrical conductivity of COF-I samples containing 1% and 5% of iodine was also measured (Fig. S12†). The results exhibit that COF containing 2% iodine (COF-I-2%) has the highest electrical conductivity. To further understand the enhanced electronic conductivity in COF-I, solid-state UV-vis spectroscopy (Fig. S13†) measurement was conducted and the results reveal that the absorption edge of COF-I has a red shift, which shows a narrower optical band gap of 1.9 eV compared with that of COF (2.0 eV) determined by the Kubelka–Munk (K–M) function, conducing to rapid electron transport in the COF. As a result, the utilization of active groups in the framework can improve with enhancing electron transport of the COF electrode.31,44
 | ||
| Fig. 3 (a) Hall effect measurement of COF-I; (b) relationship between pressure and resistivity of COF and COF-I. | ||
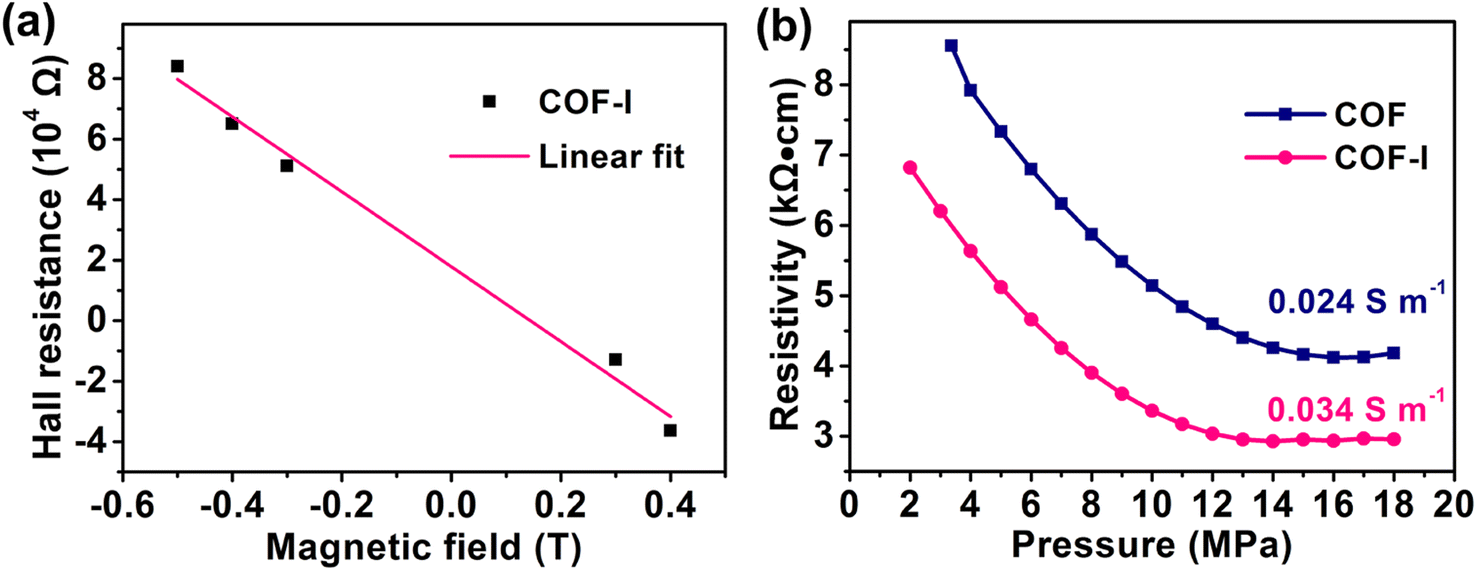
The electrochemical behavior of COF and COF-I as cathode materials for LIBs was evaluated by assembling the CR2032 coin cells. Fig. 4a exhibits the cyclic voltammetry (CV) profiles of COF and COF-I, where a pair of redox peaks can be observed, corresponding to the reversible lithiation/delithiation reaction on the CO active sites belonging to AQ.15 Compared with the redox peak current of COF, the COF-I shows dramatically enhanced peak current, indicating its high electrochemical activity.45 It should be noted that the redox peaks from iodine are not observed in CV profiles (2.5 to 3.2 V).46 The galvanostatic discharge/charge curves of COF-I cathode in Fig. 4b show a higher capacity of 140 mA h g−1 at 0.2 A g−1 compared with that of the COF (62 mA h g−1). The rate performances of COF and COF-I were evaluated at different current densities (Fig. 4c). COF-I has a reversible capacity of 164, 140, 121, 104, and 95 mA h g−1 at 0.1, 0.2, 0.4, 0.8, and 1.0 A g−1, respectively, significantly exceeding those of the pristine COF at the same current density. After the current density backing to 0.1 A g−1 successively, the discharge capacities well recover to 164 mA h g−1 with about 100% capacity restorations, demonstrating good rate capability of COF-I. To evaluate the cycling stability of COF and COF-I, long-term cycling tests were carried out (Fig. 4d). The COF-I delivers a high initial discharge capacity of 140 mA h g−1 at 0.2 A g−1 and retains the reversible capacity of 129 mA h g−1 after 1000 cycles. More impressively, the COF-I cathode yields a respectable discharge capacity of 95 mA h g−1 at 1.0 A g−1 (Fig. S14†). From the Ragone plot in Fig. 4e, the rate capability of COF-I is comparable with the some previously reported COF cathode materials, such as 2D-PAI,47 HATN-AQ-COF,48 DAPQ-COF,11 E-TP-COF,17 DAPH-TFP COF,32 DAAQ-TFP COF,32 PI-COF,49 Tp-DANT COF.4 Besides, the cycling performances of the six different batches of COF-I samples were measured over 300 cycles at 0.2 A g−1 and depicted in Fig. S15.† It indicates that the Li+-storage behavior of all batches of COF-I is similar, further demonstrating the good repeatability in preparing COF-I materials.
 | ||
| Fig. 4 Electrochemical performances of COF and COF-I. (a) CV curves at 0.3 mV s−1; (b) galvanostatic charge–discharge curves; (c) rate performance; (d) long-term cycling stability at 0.2 A g−1; (e) comparison of the rate performance to some previously reported COF cathodes. | ||
To further understand electrochemical kinetics of COF and COF-I, CV measurement was conducted at different scan rates. As shown in Fig. 5a, the shift of redox peaks (denoted as Peaks R and O) can be observed within the scan rate of 0.3–3.0 mV s−1. According to the equation: i = avb (peak current i, scan rate v and variable coefficients a and b), the b-value of 0.5 means a diffusion controlled process, whereas the b-value of 1.0 presents a capacitive-controlled behaviour.50 The b-value was determined by the slope of the linear fit of the log(i)–log(v) plot (Fig. 5b). COF-I shows the b-value of 0.719 and 0.768 for the oxidation process (bO) and the reduction process (bR), respectively, slightly lower than that of COF (0.77 and 0.841), indicating both capacitive and diffusion-limited redox process. Furthermore, the contribution ratio of diffusive (k2v1/2) and capacitive (k1v) component was calculated according to the equation: i = k1v + k2v1/2, where k1 and k2 are constants, v is scan rate.51 As the scan rate increases from 0.3 to 3.0 mV s−1, the capacitance proportion in COF-I progressively enhances from 53.47% to 91.68% (Fig. 5c), revealing the fast pseudocapacitive behavior at high rates, which stabilizes the cycling performance. The average lithium ion diffusion coefficients for COF and COF-I were calculated to be 2.92 × 10−12 and 3.42 × 10−10 cm2 s−1, respectively, by galvanostatic intermittent titration technique (GITT) testing (Fig. 5d, and Fig. S16†), indicating the faster Li+ diffusion kinetics in COF-I.52 Electrochemical impedance spectroscopy (EIS) was carried out to better understand the kinetic process. As shown in Fig. 5e, the Nyquist plots show a semicircle (high-medium frequency region) and an inclined line (low-frequency region), representing the system resistance (including the solution resistance, SEI resistance and charge transfer resistance) and the diffusion resistance of ions, respectively.6 The smaller semicircle and the higher the slope of COF-I reveal the lower charge transfer resistance and faster Li-ion diffusion compared with pristine COF, representing the faster redox reaction kinetics.53 Particularly, zeta potential test (Fig. 5f) shows that iodine-doped COF has negative zeta potential value (−36 mV), whereas zeta potential of pristine COF is close to 0 mV with neutral characteristics. This indicates the negative polarity on porous surfaces of the COF-I, which is beneficial to improve the lithium ion affinity of COF pore walls through electrostatic interaction.54 The abundant lithiophilic sites (anion I3− and I5− species) in the channels of COF-I can capture numerous Li+ ions from the electrolyte, which is conducive to elevating the Li+ concentration near the surface of COF channels.18 Meanwhile, the carbonyl oxygen atoms of COF could also serve as Li-anchoring sites effectively coordinating Li ions. Thus, Li+ could transport by site-to-site hopping along the 1D pore walls with the aid of cation–dipole interactions, which is conducive to Li+ moving toward redox-active sites in the channels of COF, as shown in Fig. S17.†
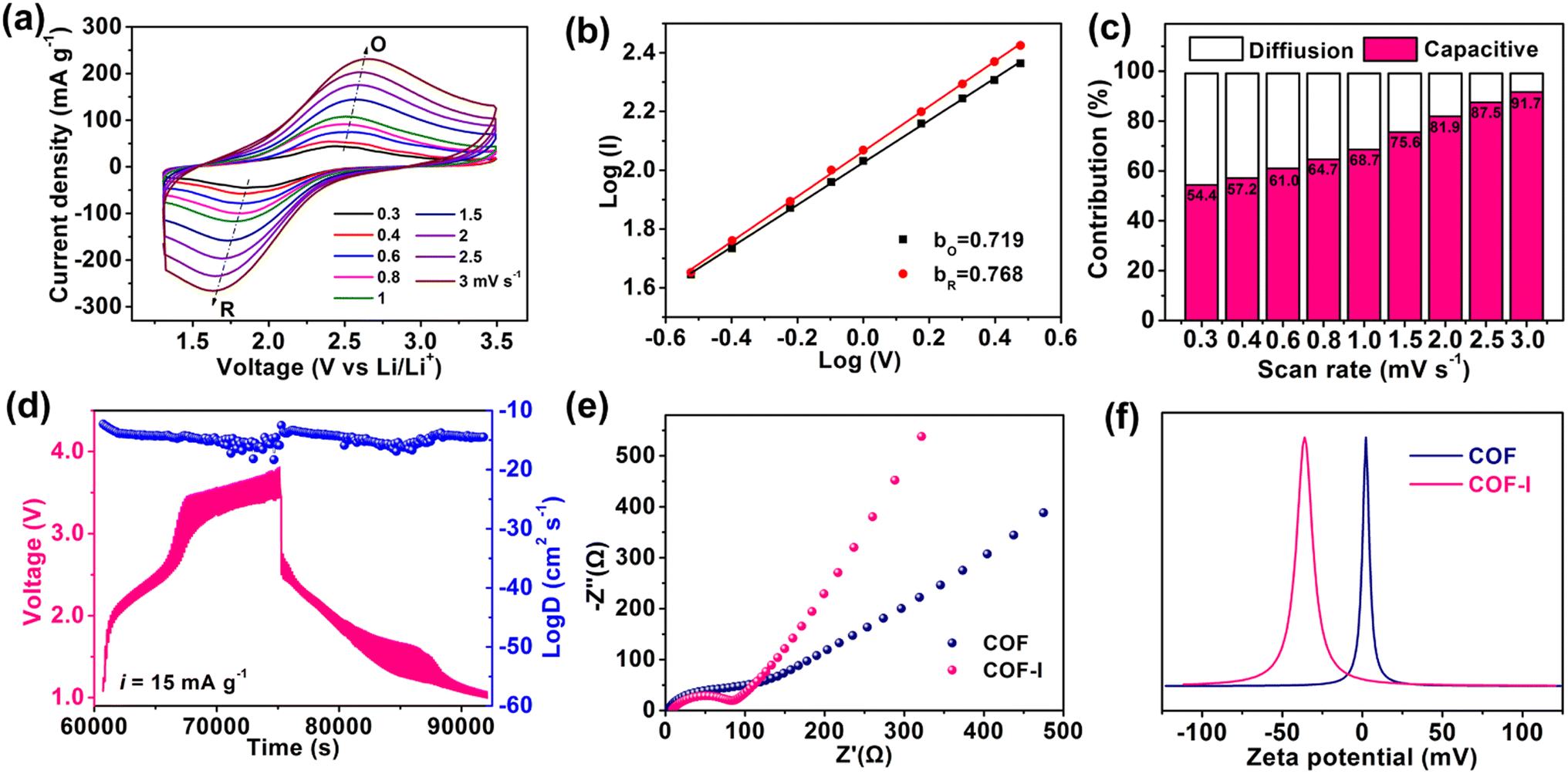 | ||
| Fig. 5 Kinetic analysis of COF-I. (a) CV curves of COF-I collected under different scan rates; (b) log(i) versus log(v) plots to determine the b values; (c) diffusion and capacitive contribution ratio at different scan rates; (d) GITT tests and corresponded ion diffusion coefficient of COF-I; (e) Nyquist plots of COF and COF-I; (f) zeta potential curves of COF and COF-I. | ||
4. Conclusions
In summary, the iodine doped β-ketoenamine-linked COF was successfully prepared through a simple one-pot solvothermal reaction by adding iodine into the synthetic process of COF. The introduction of iodine improves the lithium ion affinity of COF pore walls, endows COF more accessible active sites and high electronic/ionic conductivity. Consequently, the COF-I cathode delivers a high discharge capacity of 140 mA h g−1 at 0.2 A g−1 with 92% capacity retention after 1000 cycles and good rate performance in LIBs. This work provides a facile approach to make possible the development of high-performance COF electrode without complicate fabrication and the need for the use of conductive substrates.Conflicts of interest
There are no conflicts for this work to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Tianjin City (No. 20JCYBJC00380).References
- T. Schon, B. McAllister, P. Li and D. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- S. Cao, H. Zhang, Y. Zhao and Y. Zhao, eScience, 2021, 1, 28–43 CrossRef.
- D. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- D. Yang, Z. Yao, D. Wu, Y. Zhang, Z. Zhou and X. Bu, J. Mater. Chem. A., 2016, 4, 18621–18627 RSC.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- X. Luo, W. Li, H. Liang, H. Zhang, K. Du, X. Wang, X. Liu, J. Zhang and X. Wu, Angew. Chem., Int. Ed., 2022, 61, 202117661 Search PubMed.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 134, 202117511 Search PubMed.
- Y. Cao, M. Wang, H. Wang, C. Han, F. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CrossRef CAS.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, J. Phys. Chem. Lett., 2021, 12, 8061–8071 CrossRef CAS.
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed.
- H. Gao, Q. Zhu, A. Neale, M. Bahri, X. Wang, H. Yang, L. Liu, R. Clowes, N. Browning, R. Sprick, M. Little, L. Hardwick and A. Cooper, Adv. Energy Mater., 2021, 11, 2101880 CrossRef CAS.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- S. Xu, G. Wang, B. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- C. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. Xu, D. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31, 1901640 CrossRef PubMed.
- G. Zhao, H. Li, Z. Gao, L. Xu, Z. Mei, S. Cai, T. Liu, X. Yang, H. Guo and X. Sun, Adv. Funct. Mater., 2021, 2101019 CrossRef CAS.
- H. Chen, H. Tu, C. Hu, Y. Liu, D. Dong, Y. Sun, Y. Dai, S. Wang, H. Qian, Z. Lin and L. Chen, J. Am. Chem. Soc., 2018, 140, 896–899 CrossRef CAS PubMed.
- Q. Zhang, M. Dai, H. Shao, Z. Tian, Y. Lin, L. Chen and X. Zeng, ACS Appl. Mater. Interfaces, 2018, 10, 43595–43602 CrossRef CAS PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Kim and H. Byon, Adv. Energy Mater., 2021, 11, 2003735 CrossRef CAS.
- J. Park, C. Lee, J. Park, S. Joo, S. Kwak, S. Ahn and S. Kang, Adv. Sci., 2018, 5, 1801365 CrossRef PubMed.
- Z. Guo, Y. Zhang, Y. Dong, J. Li, S. Li, P. Shao, X. Feng and B. Wang, J. Am. Chem. Soc., 2019, 141, 1923–1927 CrossRef CAS PubMed.
- C. Mulzer, L. Shen, R. Bisbey, J. McKone, N. Zhang, H. Abruña and W. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- S. L. Cai, Y. B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S. R. Zheng, W. G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- Y. Yue, P. Cai, X. Xu, H. Li, H. Chen, H. Zhou and N. Huang, Angew. Chem., Int. Ed., 2021, 60, 10806–10813 CrossRef CAS PubMed.
- M. Wang, M. Wang, H. Lin, M. Ballabio, H. Zhong, M. Bonn, S. Zhou, T. Heine, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 21622–21627 CrossRef CAS PubMed.
- L. Wang, B. Dong, R. Ge, F. Jiang and J. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 7108–7114 CrossRef CAS PubMed.
- V. Lakshmi, C. Liu, M. Rajeswara Rao, Y. Chen, Y. Fang, A. Dadvand, E. Hamzehpoor, Y. Sakai-Otsuka, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 2155–2160 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- Y. He, N. An, C. Meng, K. Xie, X. Wang, X. Dong, D. Sun, Y. Yang and Z. Hu, J. Mater. Chem. A, 2022, 10, 11030–11038 RSC.
- F. Lv, S. Xiong, J. Zhang, X. Wang, J. Chu, R. Zhang, M. Gong, B. Wu, G. Liu and W. Luo, Electrochim. Acta, 2021, 398, 139301 CrossRef CAS.
- E. Vitaku, C. Gannett, K. Carpenter, L. Shen, H. Abruña and W. Dichte, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS PubMed.
- Y. Yang, X. Xiong, Y. Fan, Z. Lai, Z. Xu and F. Luo, J. Solid State Chem., 2019, 279, 120979 CrossRef CAS.
- M. Li, D. Yang, J. Biendicho, X. Han, C. Zhang, K. Liu, J. Diao, J. Li, J. Wang, M. Heggen, R. Dunin-Borkowski, J. Wang, G. Henkelman, J. Morante, J. Arbiol, S. Chou and A. Cabot, Adv. Funct. Mater., 2022, 2200529 CrossRef CAS.
- M. Li, Y. Wang, S. Sun, Y. Yang, G. Gu and Z. Zhang, Chem. Eng. J., 2022, 429, 132254 CrossRef CAS.
- S. Li, Y. Liu, L. Dai, S. Li, B. Wang, J. Xie and P. Li, Energy Storage Mater., 2022, 48, 439–446 CrossRef.
- M. Wang, M. Wang, H. Lin, M. Ballabio, H. Zhong, M. Bonn, S. Zhou, T. Heine, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 21622–21627 CrossRef CAS PubMed.
- B. Nath, W. Li, J. Huang, G. Wang, Z. Fu, M. Yao and G. Xu, CrystEngComm, 2016, 18, 4259–4263 RSC.
- Y. Xie, T. Pan, Q. Lei, C. Chen, X. Dong, Y. Yuan, J. Shen, Y. Cai, C. Zhou, I. Pinnau and Y. Han, Angew. Chem., Int. Ed., 2021, 60, 22432–22440 CrossRef CAS PubMed.
- Q. Geng, H. Wang, J. Wang, J. Hong, W. Sun, Y. Wu and Y. Wang, Small Methods, 2022, 6, 2200314 CrossRef CAS PubMed.
- X. Chen, H. Zhang, P. Yan, B. Liu, X. Cao, C. Zhan, Y. Wang and J. Liu, RSC Adv., 2022, 12, 11484–11491 RSC.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. Addicoat, M. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- S. Wang, X. Li, L. Da, Y. Wang, Z. Xiang, W. Wang, Y. Zhang and D. Cao, J. Am. Chem. Soc., 2021, 143, 15562–15566 CrossRef CAS PubMed.
- Z. Lei, X. Chen, W. Sun, Y. Zhang and Y. Wang, Adv. Energy Mater., 2019, 9, 1801010 CrossRef.
- D. Lei, W. Shang, X. Zhang, Y. Li, S. Qiao, Y. Zhong, X. Deng, X. Shi, Q. Zhang, C. Hao, X. Song and F. Zhang, ACS Nano, 2021, 15, 20478–20488 CrossRef CAS PubMed.
- F. Cai, Y. Duan and Z. Yuan, J. Mater. Sci.: Mater. Electron., 2018, 29, 11540–11545 CrossRef CAS.
- G. Wang, N. Chandrasekhar, B. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. Yu, R. Berger and X. Feng, Adv. Mater., 2019, 31, 1901478 CrossRef PubMed.
- X. Yang, L. Gong, X. Liu, P. Zhang, B. Li, D. Qi, K. Wang, F. He and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, 2022070 Search PubMed.
- Z. Wang, Y. Li, P. Liu, Q. Qi, F. Zhang, G. Lu, X. Zhao and X. Huang, Nanoscale, 2019, 11, 5330–5335 RSC.
- J. Ruan, F. Mo, Z. Chen, M. Liu, S. Zheng, R. Wu, F. Fang, Y. Sun and D. Song, Adv. Energy Mater., 2020, 10, 1904045 CrossRef CAS.
- J. Yang, Y. Zhai, X. Zhang, E. Zhang, H. Wang, X. Liu, F. Xu and S. Kaskel, Adv. Energy Mater., 2021, 11, 2100856 CrossRef CAS.
- Y. Cao, M. Wang, H. Wang, C. Han, F. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CrossRef CAS.
- W. Choi, H. Shin, J. Kim, J. Choi and W. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 1–13 CrossRef CAS.
- S. Liu, J. Zhao, F. Li, Y. Zhao and G. Li, J. Mater. Chem. A, 2022, 10, 5221–5229 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01414b |
| This journal is © The Royal Society of Chemistry 2023 |