
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer therapeutic potential of benzofuran scaffolds
Ashraf A. Abbas
 and
Kamal M. Dawood
*
and
Kamal M. Dawood
*
Department of Chemistry, Faculty of Science, Cairo University, Giza, 12613, Egypt. E-mail: kmdawood@sci.cu.edu.eg; Fax: +20-2-35727556; Tel: +20-2-35676602
First published on 11th April 2023
Abstract
Benzofuran moiety is the main component of many biologically active natural and synthetic heterocycles. These heterocycles have unique therapeutic potentials and are involved in various clinical drugs. The reported results confirmed the extraordinary inhibitory potency of such benzofurans against a panel of human cancer cell lines compared with a wide array of reference anticancer drugs. Several publications about the anticancer potencies of benzofuran-based heterocycles were encountered. The recent developments of anticancer activities of both natural and synthetic benzofuran scaffolds during 2019–2022 are thoroughly covered. Many of the described benzofurans are promising candidates for development as anticancer agents based on their outstanding inhibitory potency against a panel of human cancer cells compared with reference anticancer drugs. These findings encourage medicinal chemists to explore new areas to improve human health and reduce suffering.
 Ashraf A. Abbas | Professor Ashraf A. Abbas was born in Egypt in September 1968 and he is presently Professor of Organic Chemistry, Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt since 2009. He graduated from Cairo University in 1990. He received his MSc and PhD degrees in 1994 and 1997, respectively, from Cairo University. He spent Six months in the Fakultat fur Chemie, universitat of Konstanz (Germany) on a DAAD fellowship (1996) to finish his PhD thesis and one year in Tokyo Institute of technology (TIT, Japan) as UNESCO fellow (postdoctoral fellowship) from Oct 2001 to Sept 2002. He received several research prizes in chemistry; (1) he was awarded the prize of Prof. Dr Mohamed Abdel Salam in chemistry (2001) for young scientists provided by Academy of Scientific Research and Technology (Egypt). (2) He was awarded the Cairo university encouragement prize in Chemistry in 2004. (3) He was awarded the Third Word Academy of Science (TWAS) prize in chemistry for young scientists in 2004 provided by ICTP-Strada, Triesta-Italy, (4) he was awarded the State Award in Chemistry, Egypt 2005. He published many papers in the field of synthesis and applications of macrocycles and bis-heterocycles chemistry. |
 Kamal M. Dawood | Kamal M. Dawood graduated from Cairo University, Egypt in 1987 then carried out his MSc and PhD studies under the supervision of Professor Ahmad Farag, Cairo University and received his PhD in 1995. In 1997 he was awarded the UNESCO Fellowship at TIT for one year and in 1999 he was awarded the JSPS Fellowship for two years and in both fellowships he worked with Professor T. Fuchigami at Tokyo Institute of Technology (TIT) in the field of “Anodic Selective Fluorination of Heterocycles”. Further, he was awarded the Alexander von Humboldt (AvH) Fellowship at Hanover University in 2004–2005 with Prof. A. Kirschning (in the area of polymer supported palladium catalyzed cross coupling reactions) and as AvH three short visits in 2007, 2008 and 2012 with Prof. P. Metz at TU-Dresden (in the field of Metathesis Reactions in Domino Processes). Since May 2007 – to date, he has been appointed as full Professor of Organic Chemistry, Faculty of Science, Cairo University. He worked as Professor of Organic Chemistry at Chemistry Department, Kuwait University from Sept. 2013 till Aug 2017. He received a number of National Awards: Cairo University Award in Chemistry (2002), the State-Award in Chemistry (2007), Cairo University Award for Academic Excellence (2012) and Cairo University Merit Award (2017). He published about 150 scientific papers and reviews in distinguished international journals. There are more than 3300 citations of his work (Scopus h-index 30). |
1. Introduction
Heterocyclic fragments widely exist in a broad array of natural products. Many such versatile natural products have unique pharmacological properties and constitute the main core of several drugs in the market.1,2 In particular, the benzofuran moiety is commonly available in a large number of promising natural products and natural drugs.3,4 Thus, great interest has been focused on the synthesis and extraction of benzofuran-based heterocycles due to their pronounced importance in pharmaceutical and agricultural fields.5,6 Natural benzofurans are characterized by their distinctive wide range of biological properties. For example, the naturally occurring benzofuranones (Aurones) (Fig. 1) showed promising anticancer properties.7–10 Salvianolic acid B and Ganodone are two examples of naturally occurring benzofuran derivatives that are used as potential natural sources of anticancer medicines (Fig. 1).11–14 The natural isobenzofuran derivative is found in edible mushroom.15,16 Interestingly, some natural and synthetic approved drugs contain a benzofuran unit, such as Griseofulvin (Antibiotics drug), Dronedarone and Amiodarone (Antiarrhythmic drugs), Ramelteon (Sedative drug), Vilazodone and Citalopram (Anti-depressive drugs), Saprisartan (anti-hypertension drug) and Benzbromarone (for gut treatment) are depicted in Fig. 2. Moracin (hydroxylated 2-arylbenzofuran derivatives) is another class of highly bioactive natural products that contains a benzofuran moiety (Fig. 1).17,18 Such benzofuran members are lead structures in drug development and some of them have promising anticancer activities.19,20 In addition, among the natural products that contain benzofuran moiety is rocaglamide and its analogues, which are of great interest as highly potent anticancer agents.21,22 Moreover, many synthetic benzofuran compounds are reported to have significant anticancer activities.23,24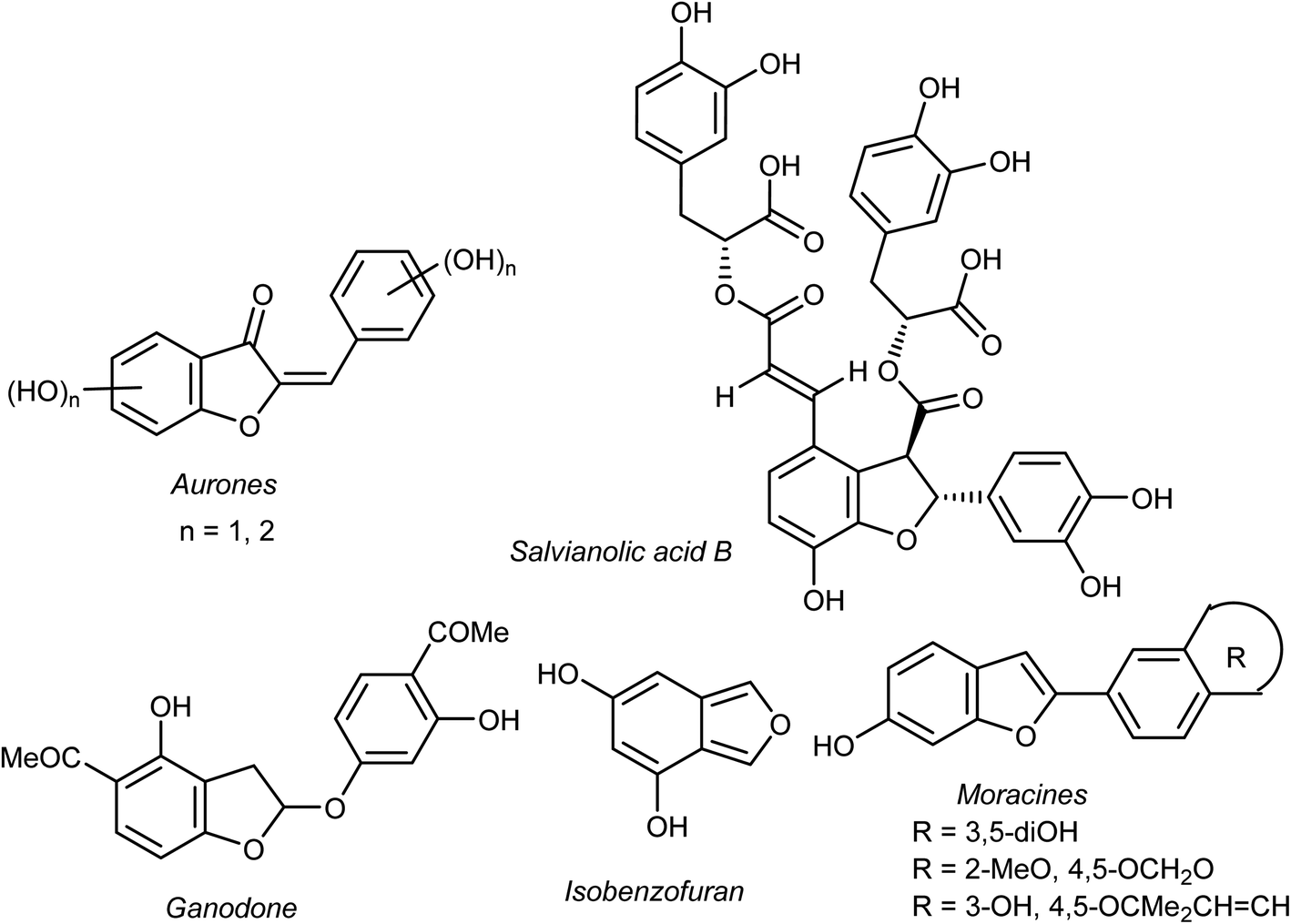 | ||
| Fig. 1 Structures of some natural benzofuran derivatives with anticancer activities. | ||
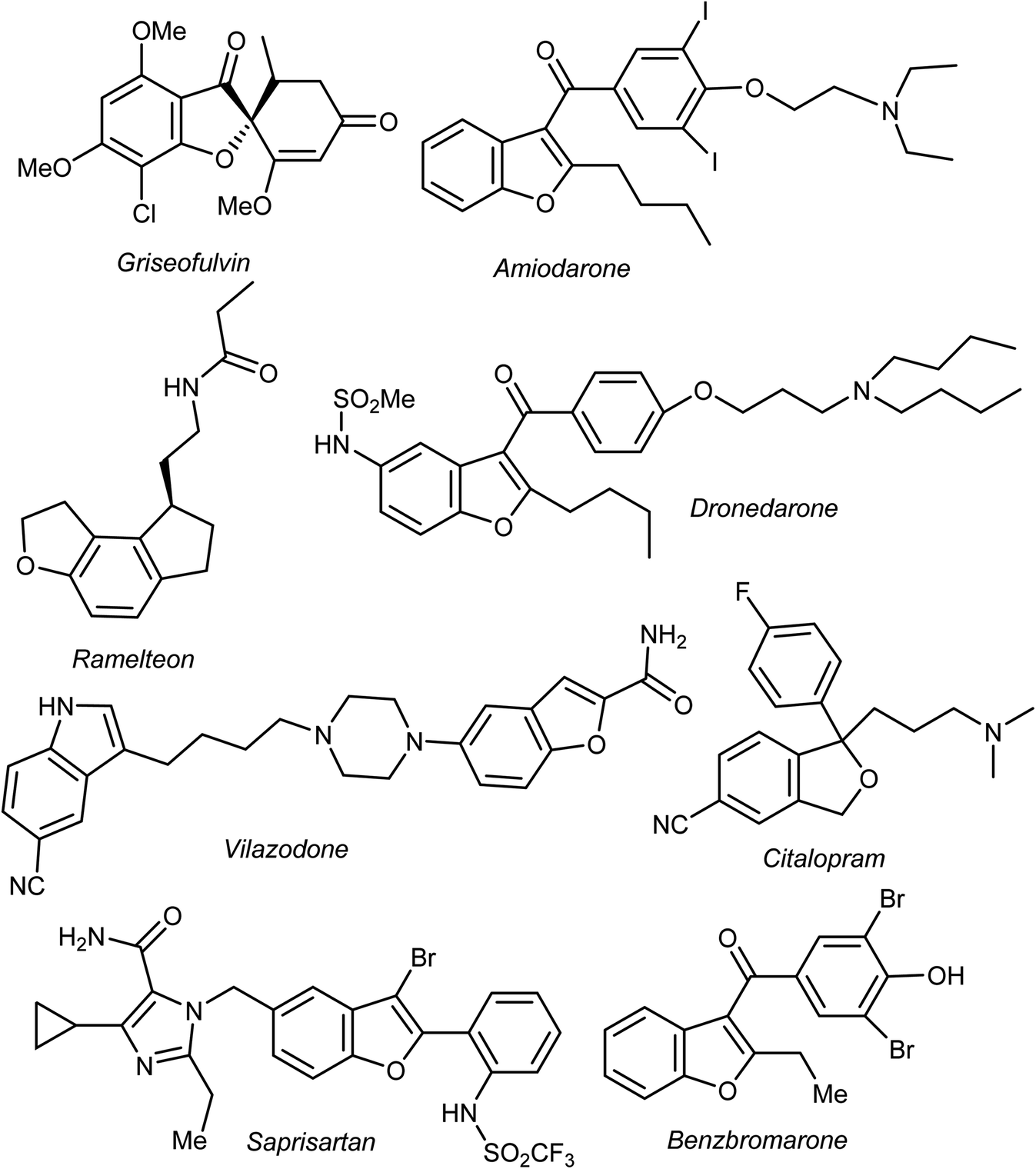 | ||
| Fig. 2 Some clinical drugs containing benzofuran scaffolds. | ||
Recently, our research interest was focused on the biological applications of benzofuran derivatives and other heterocycles of potent anticancer activities.25–29 Herein, we report an updated comprehensive outlines of the recent developments (2019–2022) of the anticancer potentiality of both natural and synthetic bioactive benzofuran-based compounds as a highly significant source for drug development and discovery.
2. Anticancer activity of benzofuran derivatives
2.1. Anticancer activity of benzofuran-3-carboxaldehyde derivatives
Chen et al. reported the two benzofuran derivatives 1 and 2, naturally isolated from mangrove endophytic fungus Fusarium sp. 2ST2, and tested their cytotoxicity against five human cancer cell lines; A549 (lung), HeLa (cervical), KYSE150 (esophageal squamous), PC-3 (pancreatic), and MDA-MB-435 (breast) cancers. Unfortunately, the reported compounds showed non-promising activity against all of the studied cancer cell lines at 50 μM concentration.30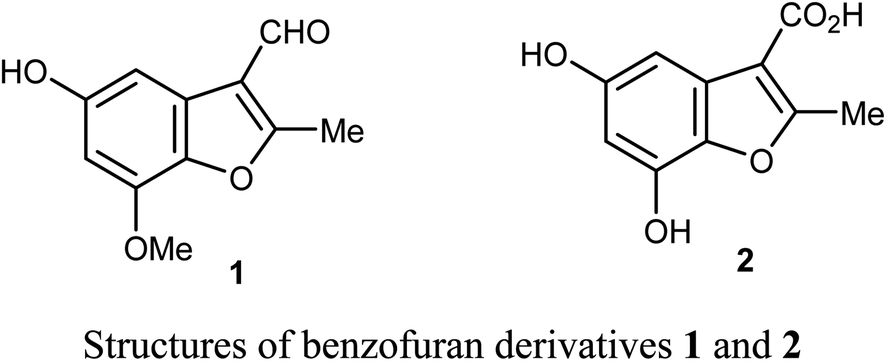
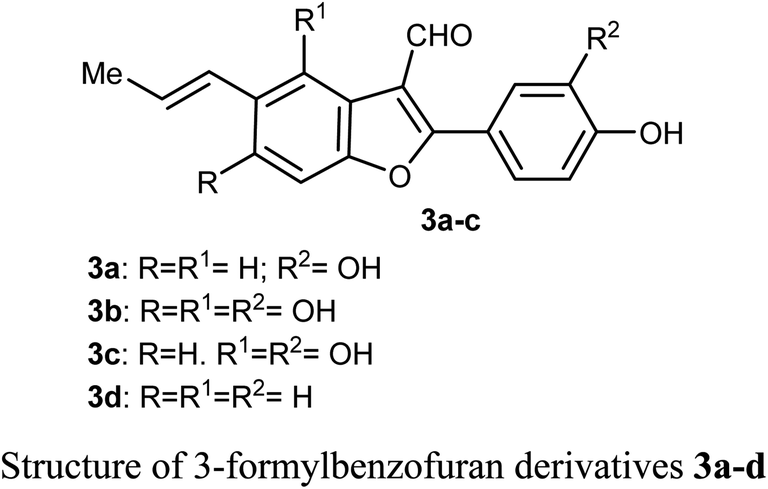
Lang et al. isolated five derivatives of 3-formylbenzofuran 3a-d from the 95% EtOH extract of Itea yunnanensis, and screened their anti-hepatocellular carcinoma (HCC) activity by MTT assay [MTT is 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide]. The derivatives 3b and 3c displayed promising growth inhibition potency on SK-Hep-1 cells (with IC50 = 5.365 and 6.013 μM, respectively) compared with the standard control sorafenib (IC50 = 9.452 μM). The mechanistic study proved that compound 3b significantly reduced the phosphorylation level of ERK (extracellular signal-related kinase), which suggested that 3b stopped cell growth and promoted apoptosis of SK-Hep-1 cells by blocking RAS/RAF/MEK/ERK signaling pathway. Thus, the 3-formylbenzofuran derivatives 3b and 3c had the potential to be lead structures as anti-hepatocellular carcinoma agents.31
2.2. Anticancer activity of 2(3)-phenylbenzofuran derivatives
Some benzofuran scaffolds 4–6 were found to be good inhibitors of Pin1 (peptidyl-prolyl cis–trans isomerase NIMA-interacting 1); Pin1 had an oncogenic role in multiple human cancers particularly hepatocellular carcinoma (HCC). Particularly, 4,6-di(benzyloxy)-3-phenylbenzofuran 5a revealed an excellent selectivity towards Pin1 (with IC50 = 0.874 μM) with 75.38% inhibition. This was a stronger activity compared to juglone. A cell-based biological study proved that 5a appreciably suppressed cell proliferation of HCC cells via restoring the nucleus-to-cytoplasm export of XPO5 and upregulating mature miRNAs expression. The Pin1 inhibitor 5a increased the biogenesis of tumor suppressor miRNAs (miR-122 and miR-29b) and reduced the HCC cell proliferation. Thus, 5a was considered as a promising guide structure for Pin1 inhibition with the therapeutic potential of miRNA-based treatment for human cancers.32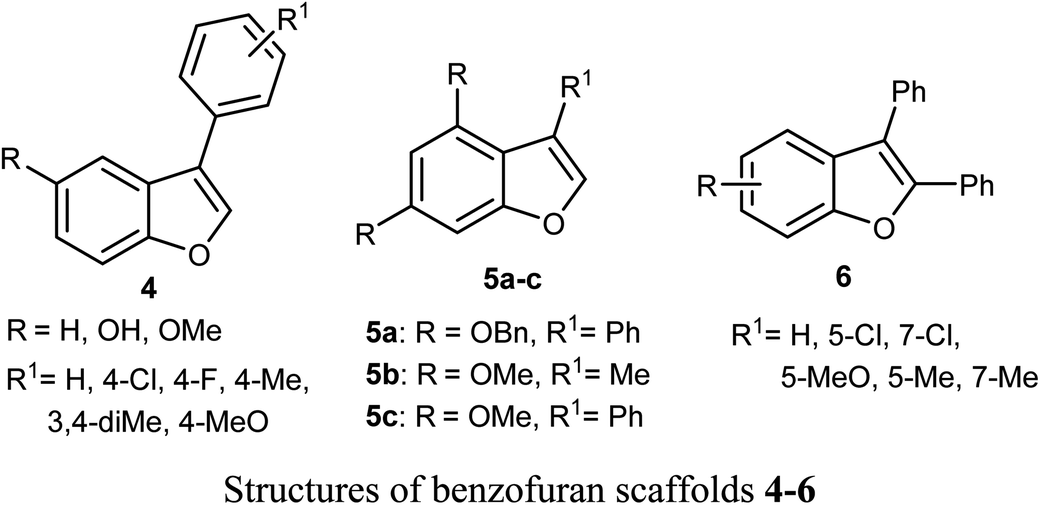
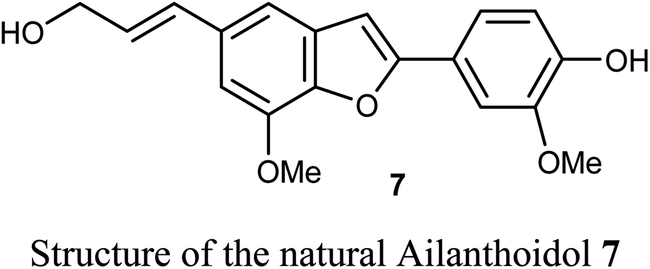
The anticancer activity of the naturally isolated benzofuran derivative (ailanthoidol) 7 (isolated from the barks of Zanthoxylum ailanthoides) was discussed by Tseng et al. Compound 7 displayed more potent cytotoxicity in Huh7 (mutant p53) hepatoma cells than in liver carcinoma (HepG2) cells (wild-type p53). The benzofuran derivative 7 decreased the cell viability in Huh7 cells, with IC50 of 45 μM and 22 μM at 24 h and 48 h, respectively, while for HepG2 it had IC50 > 80 μM. This compound induced G1 arrest and reduced the expression of cyclin D1 and CDK2 (Cyclin Dependent Kinase 2). This compound induced significant apoptosis in Huh7 cells as examined by flow cytometry analysis with annexin-V/PI staining. Compound 7 also increased the expression of cleaved PARP (poly-ADP ribose polymerase) and Bax (Bcl-2-associated X protein) and decreased the expression of procaspase 3/8 and Bcl-xL/Bcl-2. Thus, the mechanism of action revealed that compound 7 exhibited a more potent antiproliferation potential on mutp53 HCC than on wtp53 HCC cells due to the downregulation of mutp53 and inactivation of the STAT3.33
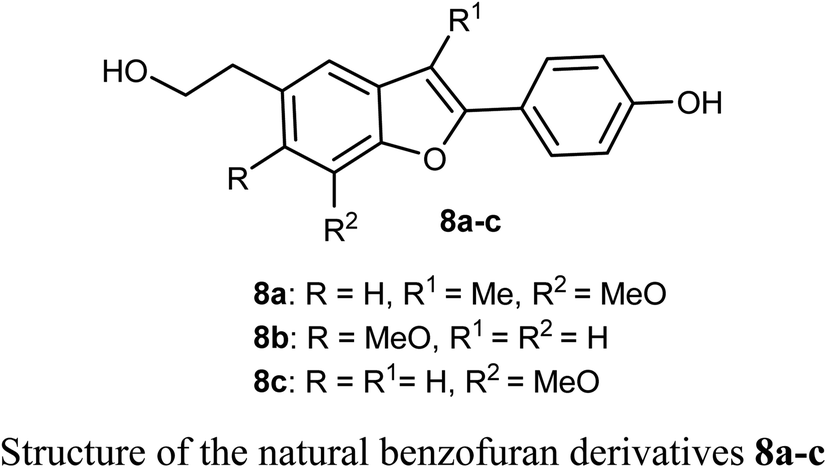
The biological activities of the natural compound 8a and its synthetic derivatives 8b and 8c against five human cancer cell lines; HepG2, MCF-7, A549, PC3 (prostate carcinoma), and Hep3B (hepatocellular carcinoma) and various protein levels were reported. The experiments were conducted at 6 μM which was close to the IC50 values of the tested compounds. The derivatives 8a–c induced apoptotic cell death related PARP cleavage in HepG2, A549, MCF74, PC3, and Hep3B cancer cell lines. In addition, the tumor inhibitor protein p53 was also induced in p53 wild-type cancer cells.34
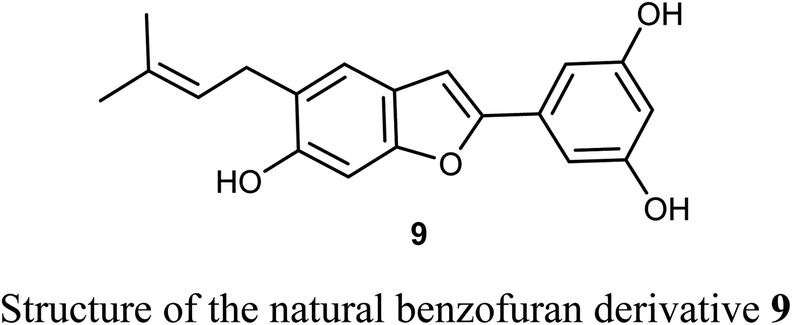
The naturally isolated benzofuran derivative 9, isolated from the leaves of Morus alba L., was described as an inhibitor of cell proliferation that induced cell apoptosis in human non-small-cell lung carcinoma (NSCLC) cells, PC9 and A549 using MTT bioassay. Compound 9 treatment resulted in mitochondrial apoptosis in PC9 and A549 cells and also increased autophagy flux by the increase of autophagosome formation. Interestingly, the molecular mechanism of compound 9 against lung cancer through apoptosis and autophagy confirmed its importance as a therapeutic agent for NSCLC treatment. Compound 9 inhibited the growth of A549 and PC9 cells in a dose-dependent manner with IC50 values of 48.4 μM and 6.6 μM, respectively. The great perspective of compound 9 as an anti-lung cancer agent, made it possible to be developed as a novel therapeutic agent for NSCLC treatment in the future.35
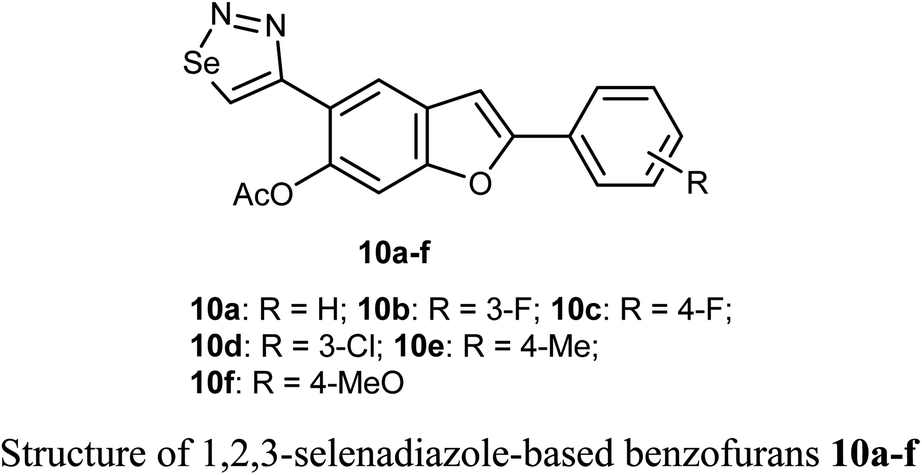
Selenium-containing compounds exhibit a wide range of biological potencies including anticancer activity.36,37 In particular, 1,2,3-selenadiazole-based benzofuran derivatives 10a–f were prepared by Olomola et al. and screened for antiproliferation activity against the breast cancer (MCF-7) and the human embryonic kidney derived (HEK-293-T) cell lines using the MTT assay. Most of the derivatives 10a–f exhibited low to moderate inhibition except compound 10f which showed promising antigrowth action against MCF-7 cells with IC50 = 2.6 μM compared to doxorubicin (IC50 = 0.8 μM). Compound 10f was relatively less cytotoxic against the normal cell line (IC50 = 1.2 μM). Compound 10a exhibited, however, moderate effect against the MCF-7 cell line (IC50 = 7.1 μM), but it was relatively more cytotoxic towards the Hek293-T cell line (IC50 = 3.5 μM) compared to doxorubicin (IC50 = 1.2 μM).38
2.3. Anticancer activity of 2-benzoylbenzofuran derivatives
A series of 2-benzoylbenzofuran derivatives 11a–i were synthesized and screened for in vitro anticancer activity against human breast cancer (MCF-7, MDA-MB-231), human embryonic kidney cancer (HEK-293), and human epidermal cancer (HaCaT) cell lines employing MTT bioassay. The benzofuran derivative 11e demonstrated an excellent potency against anti-oestrogen receptor-dependent breast cancer cells with low toxicity, compared with the positive controls tamoxifen and raloxifene. Compound 11e might be an interesting lead for further development as a potential inhibitor of oestrogen-receptor. Compound 11e also significantly inhibited the growth of ER-dependent MCF-7 breast cancer cells, but had a minor effect on normal cells (HaCaT), leading to a remarkable selectivity in the treatment of breast cancer. The increased inhibitory and selectivity potency of compound 11e were referred to its high hydrogen-bond interactions with ARG394 with a distance of 2.9 Å.39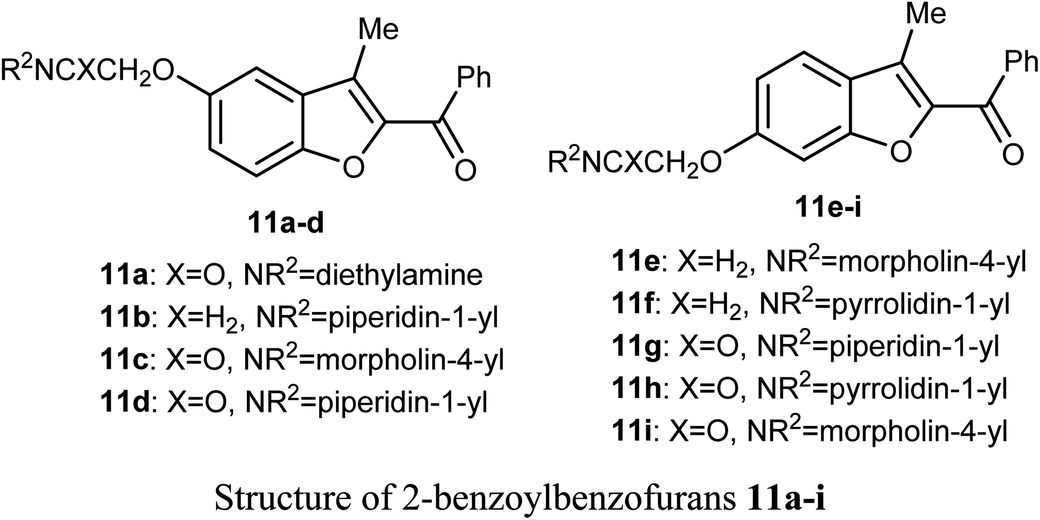
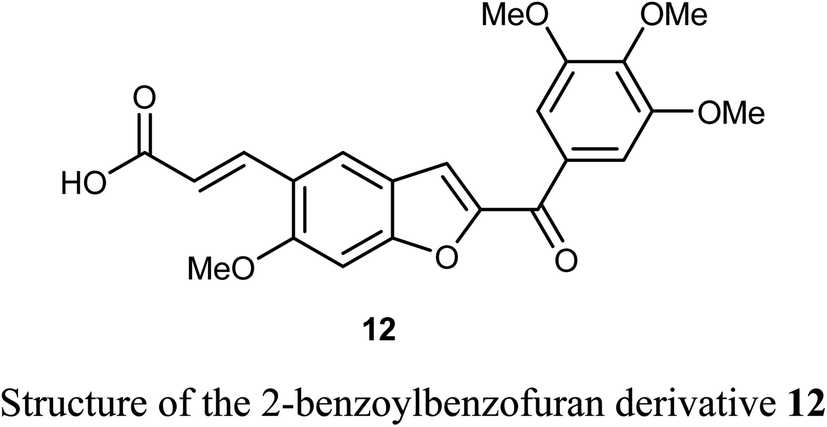
The benzofuran derivative 12 was described as a potent inhibitor against cervical cancer cells, SiHa and HeLa. Treatment with 12 enhanced the growth inhibitory rate in a concentration-dependent manner, (with IC50 values 1.10 and 1.06 μM) against SiHa and HeLa cells, respectively, which were higher than the positive reference combretastatin (CA-4) (with IC50 values 1.76 and 1.86 μM), respectively. The mechanistic studies showed that benzofuran derivative 12 induced G2/M phase arrest and apoptosis in SiHa and HeLa cells.40
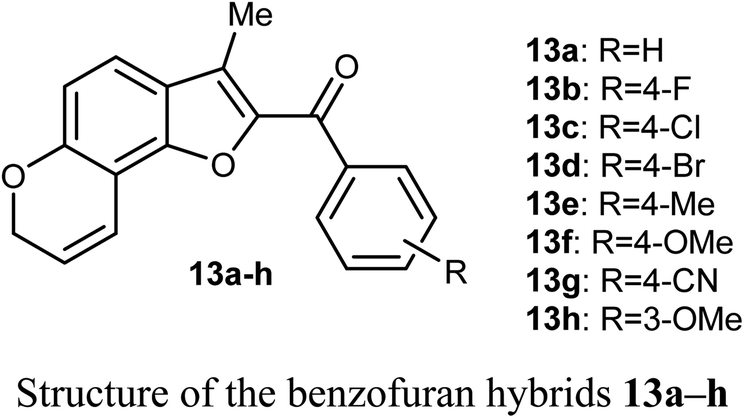
The in vitro antiproliferative activity of the benzofuran hybrids 13a–h was reported by Dharavath et al. against two cell lines; C-6 (nerve cells) and MCF-7 (human breast adenocarcinoma cells) using the MTT method. The bioassay data of the in vitro antiproliferative activity disclosed that the benzofuran derivatives 13b and 13g demonstrated superior inhibition against MCF-7 cell line (with IC50 1.875 and 1.287 μM, respectively), much better than the cisplatin drug (IC50 2.184 μM). Compounds 13b and 13g showed also excellent inhibitory activity against C-6 cell line (with IC50 = 1.980 and 1.622 μM, respectively) with more potency than the standard drug cisplatin (IC50 = 2.258 μM). The other compounds showed moderate antiproliferative activity with IC50 ranging between 2.207–5.278 μM and 2.635–4.826 μM against MCF-7 and C-6 cell lines, respectively. All compounds showed strong binding interactions with calf thymus DNA (CT-DNA) through an intercalation mode when measured by UV-vis spectroscopy and fluorescence. Thus, compounds 13b and 13g were considered as promising candidates for further exploration of new therapeutics.41
2.4. Anticancer activity of 3-oxadiazolylbenzofuran derivatives
A set of benzofuran-based oxadiazole conjugates 14a–c were reported to have promising anticancer activities. Evaluation of the in vitro cytotoxic activity of compounds 14a–c against the pancreatic cancer cells (MIA PaCa2) and human colon cancer cells (HCT116) was discussed, where the IC50 values ranged between 3.27–11.27 μM. The bromo derivative 14c was the most efficient one against HCT116 with IC50 value 3.27 μM. The decrease of glycogen synthase kinase-3β (GSK3β) induced apoptosis via suppression of basal NF-κB activity in MIA-PaCa2 and HCT116.42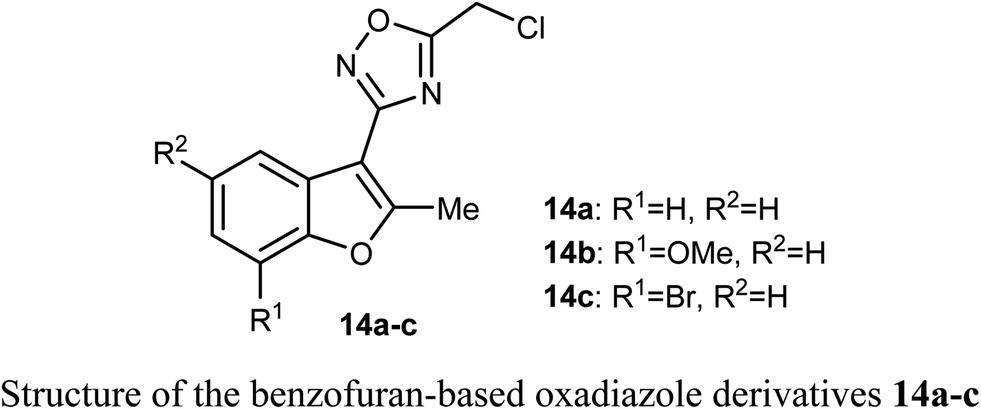
2.5. Anticancer activity of benzofuran-2-carbohydrazide derivatives
Synthesis of 3-methylbenzofuran derivatives 16a–h and 17a–c and 3-(morpholinomethyl)-benzofuran derivatives 18a–e and 19a–b as anticancer agents towards the cell lines NCI-H23 and A549, with VEGFR-2 inhibitory potency was reported. All the benzofuran compounds greatly inhibited both the cancer cell lines NCI-H23 and A549. The 3-methylbenzofuran derivative 16b, having p-methoxy group, exerted the most antiproliferative activity against A549 cell line (IC50 = 1.48 μM) closer to the reference drug staurosporine (IC50 = 1.52 μM). Furthermore, the benzofurans 16b, 18a and 18d displayed good VEGFR-2 inhibitory activity with IC50 equal 77.97, 132.5 and 45.4 nM, respectively. Moreover, the three benzofurans 16b, 18a and 18d had high cytotoxic impact towards normal lung WI-38 cells showing mean tumor selectivities equal 5.3, 8.4 and 12.9, respectively. A molecular docking study showed that the derivatives 16b, 18a and 18d had different types of hydrophobic and hydrogen bonding interactions through their carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and NH functions with the key ASP-1046 and GLU-885 amino acids residues.43
O) and NH functions with the key ASP-1046 and GLU-885 amino acids residues.43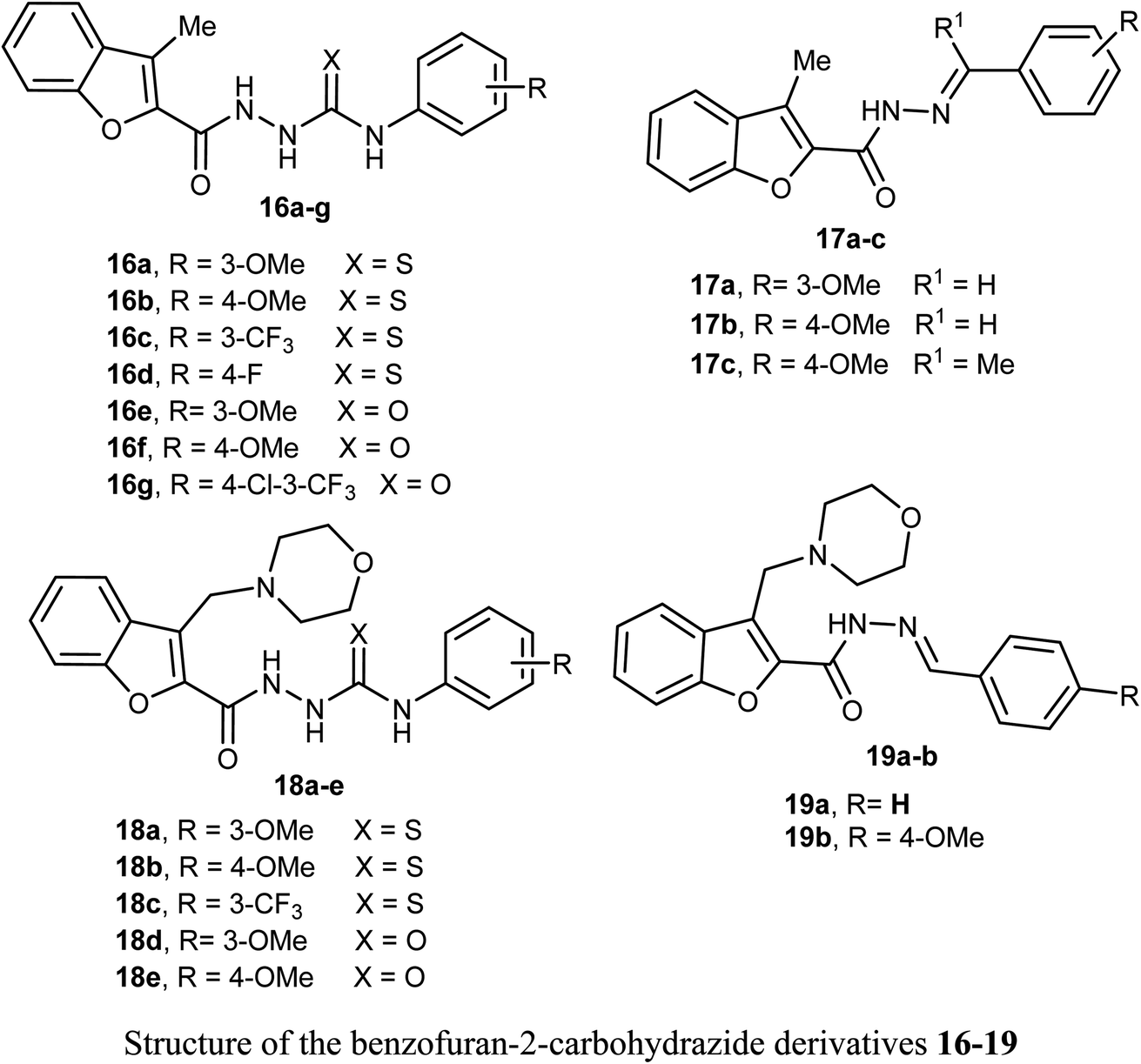
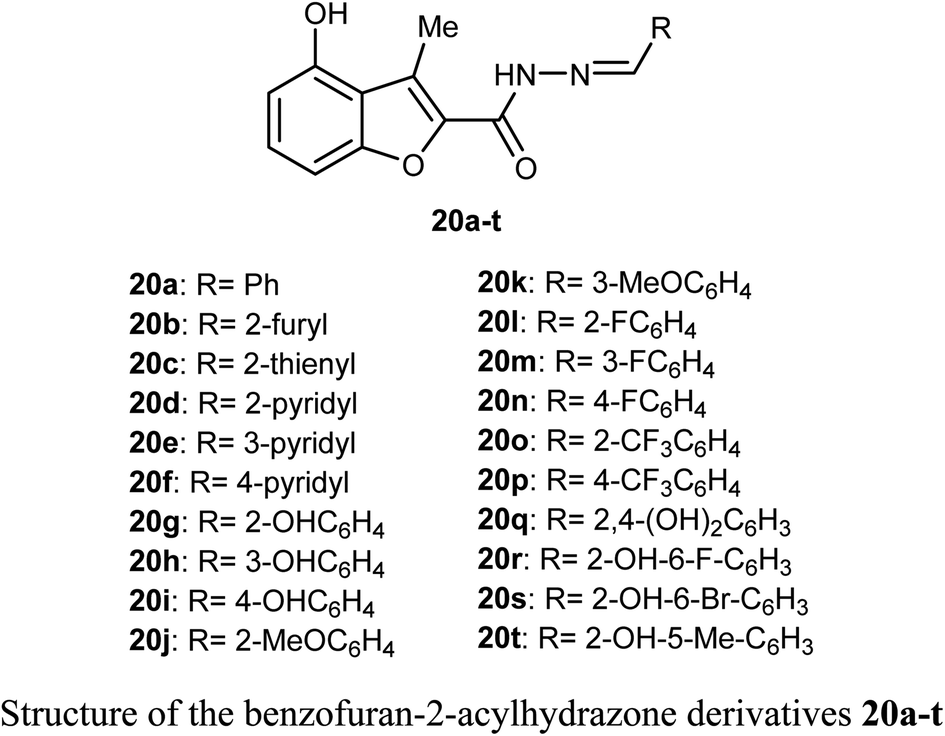
A library of benzofuran acylhydrazone scaffolds 20a–t were reported as potent inhibitors of LSD1 (histone lysine specific demethylase 1); a therapeutic target in oncology with over-expression in several human tumors. Among the reported benzofurans, fifteen compounds demonstrated excellent inhibitory activity against LSD1 with IC50 varying between 3.5–89 nM when carried at a concentration 5 μM. The SAR results confirmed that the compounds having two hydroxyl functions played a vital role in enhancing the inhibitory activity against LSD1 enzyme where compounds 20q, 20r, 20s and 20t had IC50 values in the order of 14, 15, 27 and 24 nM, respectively.44
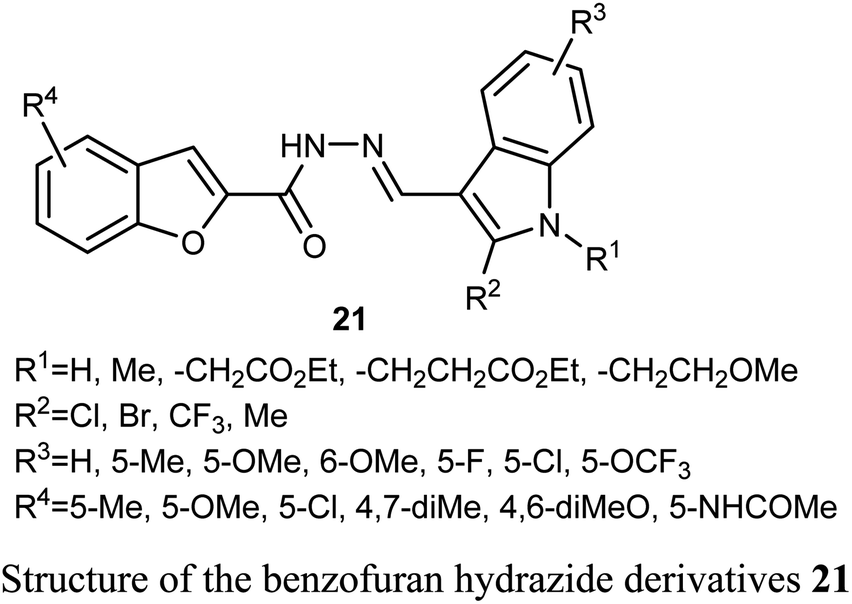
A series of benzofuran-based hydrazide derivatives 21 were synthesized and patented by Kim et al. for evaluation of their antiproliferative activity using MTT method. Most of the reported hydrazides exhibited an excellent anticancer potency in a nanomolar scale (IC50 values ranged between 82–600 nM) with low toxicity and excellent solubility. The most potent derivative, among the reported series, against human cervical HeLa cancer cell line was the derivative 21 (where R1 = CH2CH2CO2Et; R2 = Cl; R3 = 5-OMe; R4 = 5-Me) (with IC50 = 82 nM).45
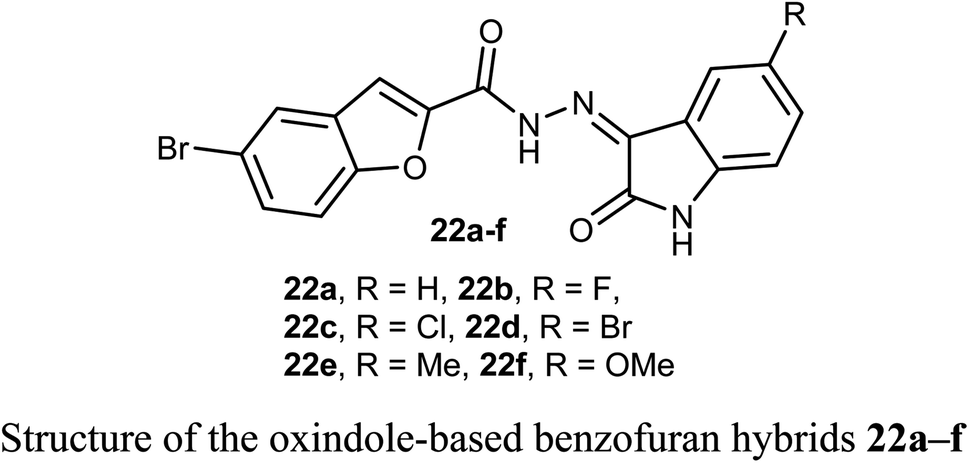
Some oxindole-based benzofuran hybrids 22a–f were designed and synthesized as dual CDK2/GSK-3β inhibitors targeting breast cancer. All derivatives showed moderate to potent activity on both MCF-7 (IC50 = 2.27–12.9 μM) and T-47D (IC50 = 3.82–9.7 μM) breast cancer cell lines. Compounds 22d, f were the most potent inhibitors with IC50 values (3.41, and 2.27 μM) and (3.82 and 7.80 μM) against MCF-7 and T-47D cell lines, compared with the reference drug staurosporine (IC50 = 4.81 and 4.34 μM), respectively. Compounds 22d, f also exhibited potent dual CDK2/GSK-3β inhibitory activity with IC50 values (37.77 and 52.75 nM) and (32.09 and 40.13 nM) on CDK2 and GSK-3β, respectively. The derivatives with the highest activity led to cell cycle arrest in the G2/M phase due to their dual CDK2/GSK-3β inhibition.46
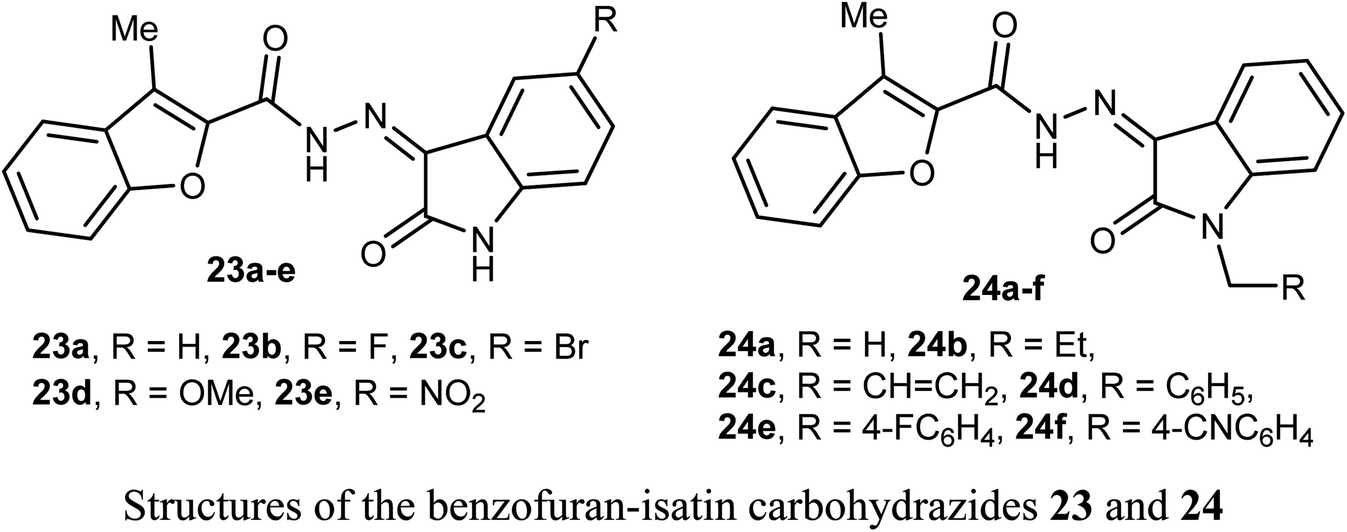
The anticancer activity of two sets of benzofuran-isatin carbohydrazide hybrids 23a–e and 24a–f were screened against 55 human cancer cell lines with single dose assay (10−5M concentration). All derivatives 23a–e and 24a–i demonstrated a good anti-proliferative activity against colorectal cancer SW-620 and HT-29 cell lines. The highest the most inhibitory activity was noticed for compounds 23a and 23d (compound 23a had IC50 values 8.7 and 9.4 μM, and that of compound 23d were 6.5 and 9.8 μM, respectively). Compounds 23a and 23d exhibited selective cytotoxicity with a good safety study. In addition, compounds 23a and 23d greatly decreased the expression of the anti-apoptotic Bcl2 protein, and increased the level of the cleaved PARP and resulted in SW-620 cells apoptosis. Compounds 23a and 23d with such significant potency and high selective cytotoxicity suggested that they might serve as potential anticancer agents and apoptotic inducers.47
2.6. Anticancer activity of 2-acetylbenzofuran molecular hybrids
A number of benzofuran-based hybrids 25-26 were synthesized and their in vitro antiproliferative activity was evaluated against HePG2, HCT-116, MCF-7, PC3, and HeLa cancer cell lines. Compounds 25 and 26 exhibited promising antiproliferative activity. The most active derivative 26 was then examined for its inhibition potency against EGFR kinase and the results showed that it had a remarkable EGFR TK inhibitory activity with IC50 value 0.93 μM, very close to the reference drug gefitinib (IC50 = 0.9 μM). The apoptosis results showed that compound 26 induced apoptosis of cancer cells and increased caspase-3 by 5.7-folds. Molecular docking results of 26 showed good fitting and suitable interactions with the key amino residues in the binding site of the EGFR kinase, where the cyano group enabled the hydrogen bonding interactions with the Met769 amino acid and benzofuran moiety showed van der Waals interactions with the EGFR binding site.48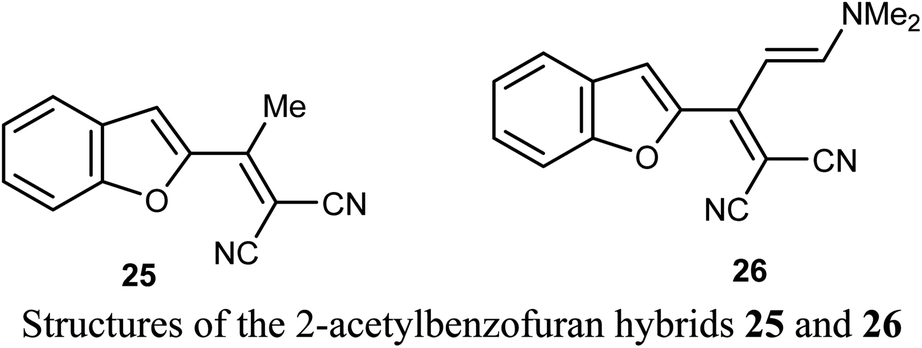
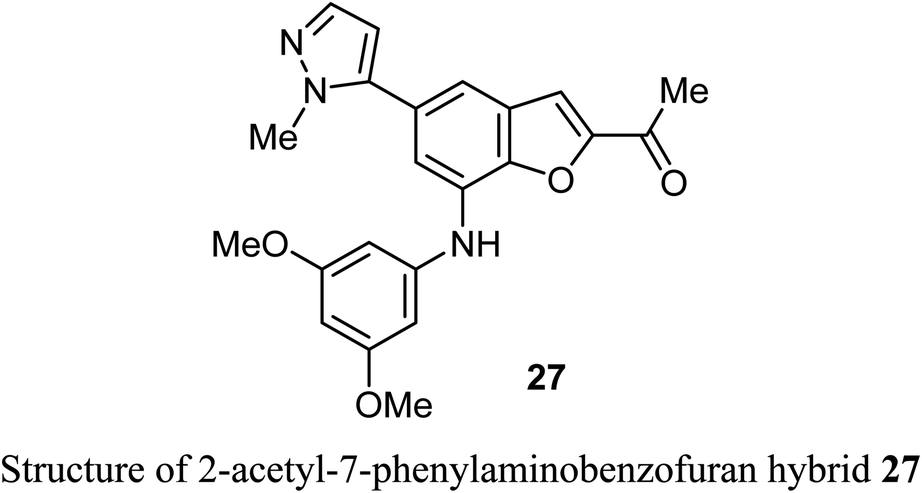
The 2-acetyl-7-phenylaminobenzofuran hybrid 27 was reported as a promising STAT3 inhibitor (signal transducer and activator of transcription 3). Interestingly, compound 27 displayed very potent antiproliferation activity against MDA-MB-468 cells with IC50 value of 0.16 μM. On the other hand, compound 27 exhibited moderate to good anti-proliferation activities against HepG2, MDA-MB-231 and A549 cancer cell lines with IC50 = 1.63–5.80 μM. The results proved that compound 27 caused a notable G2/M cycle-arresting and early apoptosis in a concentration-dependent manner in MDA-MB-468 cells.49
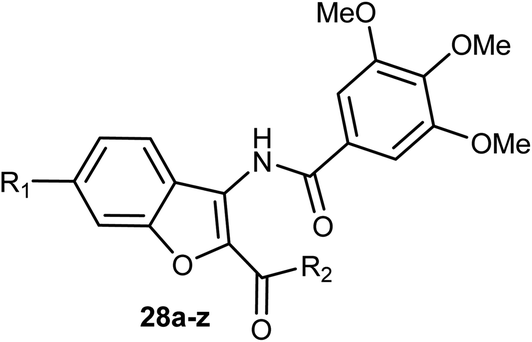
3-Amidobenzofuran derivatives 28a–z were synthesized and their antiproliferative activity against four cancer cell lines; HCT-116 (human colon carcinoma cells), HeLa (human cervical cancer cell), HT-29 (human colon cancer cell lines), and MDA-MB-231 (human breast cancer cell) were tested by MTT assay. Most of the benzofurans showed promising antiproliferative activity against the tested cancer cell lines. Compound 28g was the most active inhibitor and showed significant antiproliferative efficacy toward MDA-MB-231, HCT-116, and HT-29 with IC50 values of 3.01, 5.20, and 9.13 μM, respectively (Table 1). The flow cytometric analysis of the cell cycle showed that 28g could induce cell cycle arrest at G2/M phase in a dose-dependent manner.50
|
||||||
|---|---|---|---|---|---|---|
| In vitro cytotoxicity (IC50, μM)a | ||||||
| Comp. | R1 | R2 | HCT-116 | HT-29 | HeLa | MDA-MB-231 |
| a IC50 values are presented as mean values of three independent experiments done in quadruplicates. Coefficients of variation were <10%. | ||||||
| 28a | H | Ph | >30 | >30 | >30 | >30 |
| 28b | H | 4-MeC6H4 | 3.70 | 13.21 | >30 | 26.10 |
| 28c | H | 4-BrC6H4 | 6.02 | 19.61 | >30 | >30 |
| 28d | H | 4-ClC6H4 | 11.22 | 9.71 | 25.16 | 9.70 |
| 28e | H | 4-MeOC6H4 | 9.01 | 11.64 | 19.70 | 12.15 |
| 28f | H | Me | >30 | 14.60 | >30 | >30 |
| 28g | H | 4-FC6H4 | 5.20 | 9.13 | 11.09 | 3.01 |
| 28h | Cl | 4-BrC6H4 | 18.20 | >30 | >30 | 29.17 |
| 28i | Cl | 4-MeOC6H4 | 10.98 | 14.09 | >30 | 15.06 |
| 28j | Cl | 4-MeC6H4 | 20.96 | 19.70 | >30 | 27.04 |
| 28k | Cl | 4-ClC6H4 | 21.98 | 20.18 | >30 | >30 |
| 28l | Cl | Ph | >30 | >30 | >30 | >30 |
| 28m | Cl | 4-FC6H4 | 20.10 | 26.80 | >30 | 24.07 |
| 28n | Cl | 4-BrC6H4 | >30 | 24.12 | >30 | >30 |
| 28o | Me | 4-FC6H4 | 26.01 | >30 | >30 | >30 |
| 28p | Me | 4-ClC6H4 | 22.36 | >30 | >30 | >30 |
| 28q | Me | 4-BrC6H4 | >30 | 29.02 | >30 | 16.41 |
| 28r | Me | Ph | >30 | >30 | >30 | >30 |
| 28s | Me | 4-MeC6H4 | >30 | 11.02 | >30 | 12.88 |
| 28t | Me | 4-MeOC6H4 | >30 | 17.90 | >30 | 20.75 |
| 28u | Me | 4-BrC6H4 | >30 | 10.29 | >30 | 7.80 |
| 28v | MeO | 4-ClC6H4 | >30 | 17.63 | >30 | 22.14 |
| 28w | MeO | 4-MeOC6H4 | >30 | 8.28 | >30 | 16.58 |
| 28x | MeO | 4-MeC6H4 | >30 | >30 | >30 | >30 |
| 28y | MeO | 4-BrC6H4 | >30 | 26.01 | >30 | >30 |
| 28z | MeO | Ph | >30 | 7.02 | >30 | >30 |
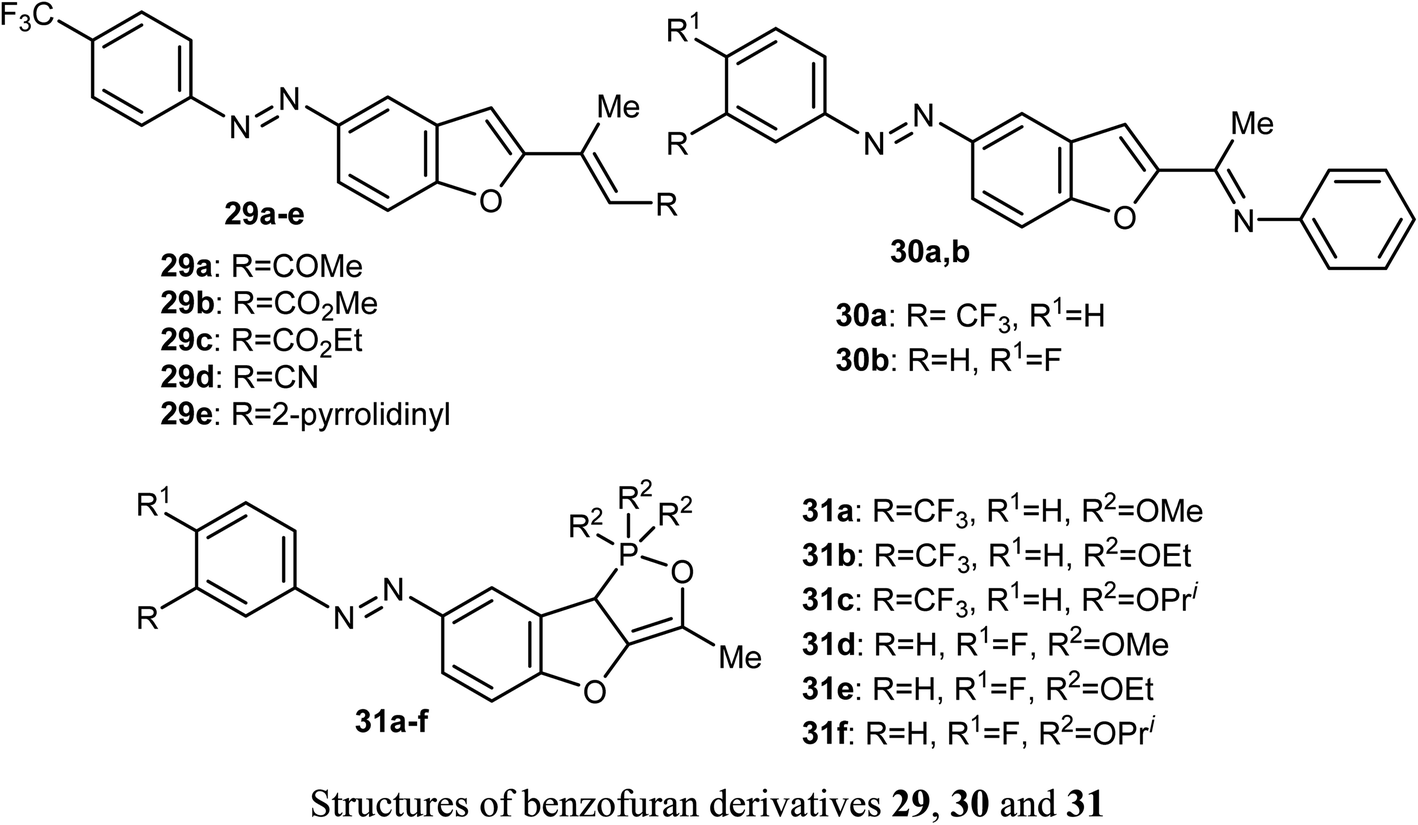
El-Sayed et al. discussed the synthesis and anticancer activity of the benzofuran derivatives 29, 30 and 31. The biological activity was assessed by evaluating their antiproliferative activities against two human cancer cell lines: MCF-7 and HepG2 cells. The benzofuran molecules 29b, 30a, and 31c demonstrated the best antiproliferative potency against the two cell lines compared to the reference drug doxorubicin. The mechanism of action study revealed that derivative 30a displayed promising inhibition of tubulin polymerization which resulted in cell cycle arrest in the G2/M phase, mitotic spindle formation disruption, and apoptosis of HepG2 cells. The benzofuran derivatives 30a, 31c, and 29b, were also found to inhibit tubulin polymerization in both cell lines. Docking study established the in vitro results confirming that compound 30a was a promising anticancer agent.51
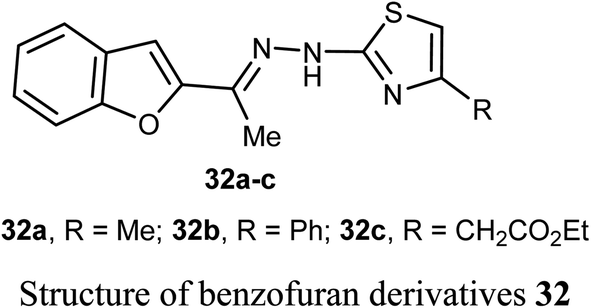
El-Khouly et al. reported the anticancer activity of the benzofuran derivatives 32a–c against four cancer cell lines; HePG2, HeLa, MCF-7 and PC3. The bioassay results confirmed that most of the derivatives had promising inhibitory potency against the defined cell lines with IC50 values ranging between 8.49–16.72 μM, 6.55–13.14 μM and 4.0–8.99 μM, respectively, when compared with doxorubicin reference drug (4.17–8.87 μM). SAR study revealed that compound 32a, having methyl group at thiazole scaffold, showed higher inhibition compared to all other derivatives.52
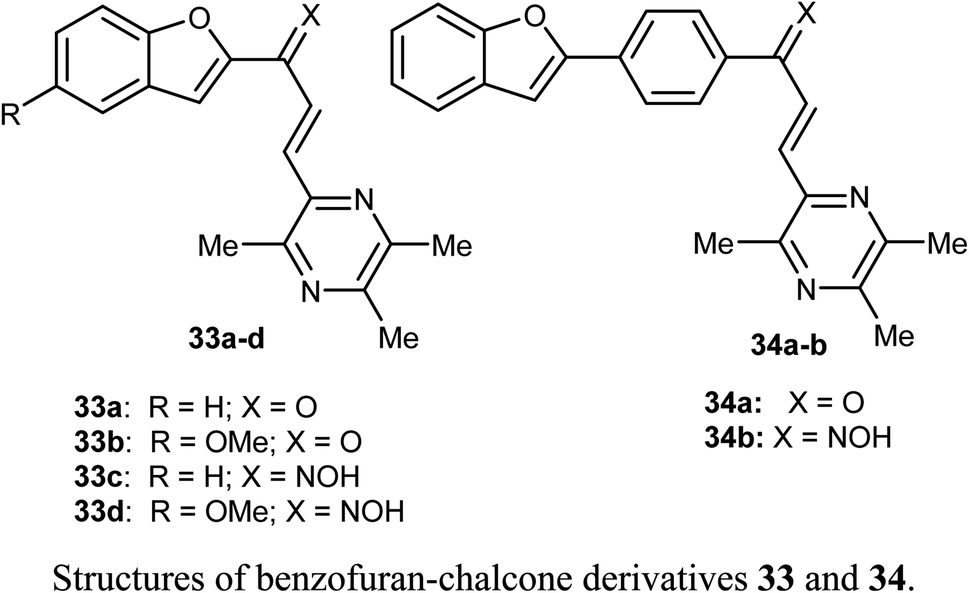
Bukhari et al. reported the synthesis of some benzofuran-chalcone derivatives 33 and 34 and screened for their in vitro anticancer activity against A-375, MCF-7, A-549, HT-29 and H-460 human cancer cell lines. All of the synthesized benzofuran-chalcone derivatives showed good to moderate activity against all of the tested cell lines compared with cisplatin as a positive control. Compound 33d showed the highest inhibition activity with IC50 values 4.15, 3.22, 2.74, 7.29, 3.81 μM against four different cell lines; A-375, MCF-7, A-549, HT-29 and H-460, respectively, compared with cisplatin (IC50 values 9.46, 12.25, 5.12, 25.4, 6.84 μM, respectively).53
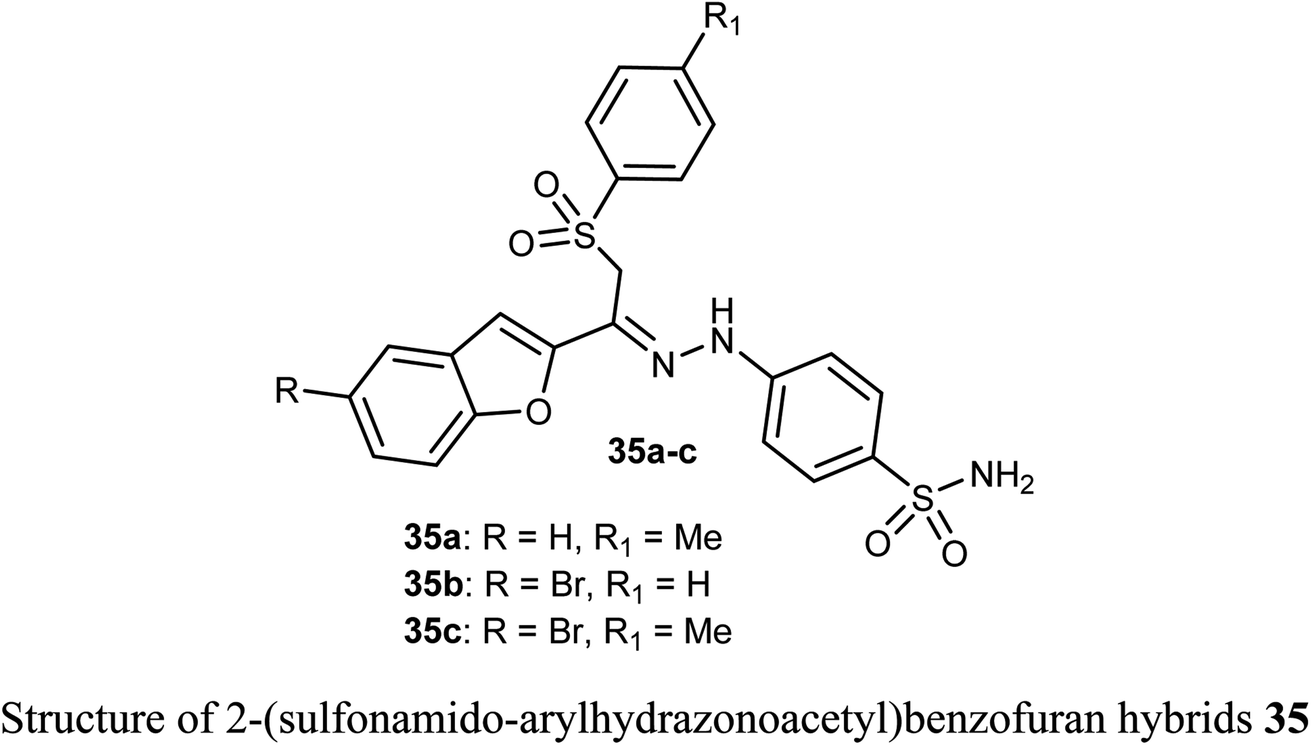
The sulfonamide-arylhydrazones of benzofuran derivatives 35a–c were tested for their inhibitory actions toward the target tumor-associated hCA-I, hCA-II, hCA-IX and XII isoforms. The reported compounds were effective inhibitors of hCA-IX and XII isoforms with Ki spanning values 10.0–97.5 and 10.1–71.8 nM, respectively. They demonstrated the highest selectivity toward hCA IX and XII over hCA I (SIs: 39.4–250.3 and 26.0–149.9, respectively), and over hCA II (SIs: 19.6–57.1 and 13.0–34.2, respectively).54
2.7. Anticancer activity of piperazine-based benzofuran derivatives
The benzofuran scaffolds 36 were synthesized and their inhibitory activities against LSD1 (lysine-specific demethylase-1) were examined. Compound 36b was the most potent inhibitor of LSD1 with an IC50 value 0.065 μM. Compound 36c with methylpiperazine moiety was less potent (IC50 = 0.420 μM) than 36a (unsubstituted piperazine), showing that hydrogen-bond donor groups at R2 moiety were favorable. The in vivo inhibitory potency of compound 36b was evaluated at a dose of 10 and 20 mg kg−1 day−1, where after 15 days of treatment at a dose of 20 mg kg−1 day−1, compound 36b remarkably inhibited the tumor growth and decreased the average tumor weight and volume by 64% and 68%, respectively, in comparison with the control. All tumor studies revealed that compound 36b was a promising lead structure, where it effectively inhibited H460 cell growth in vivo without significant side effects.55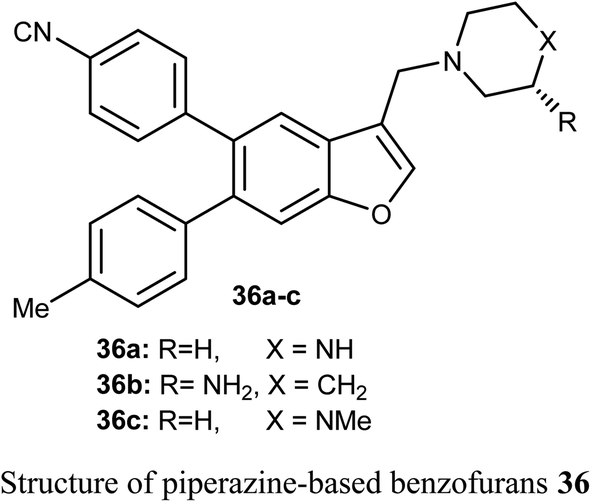
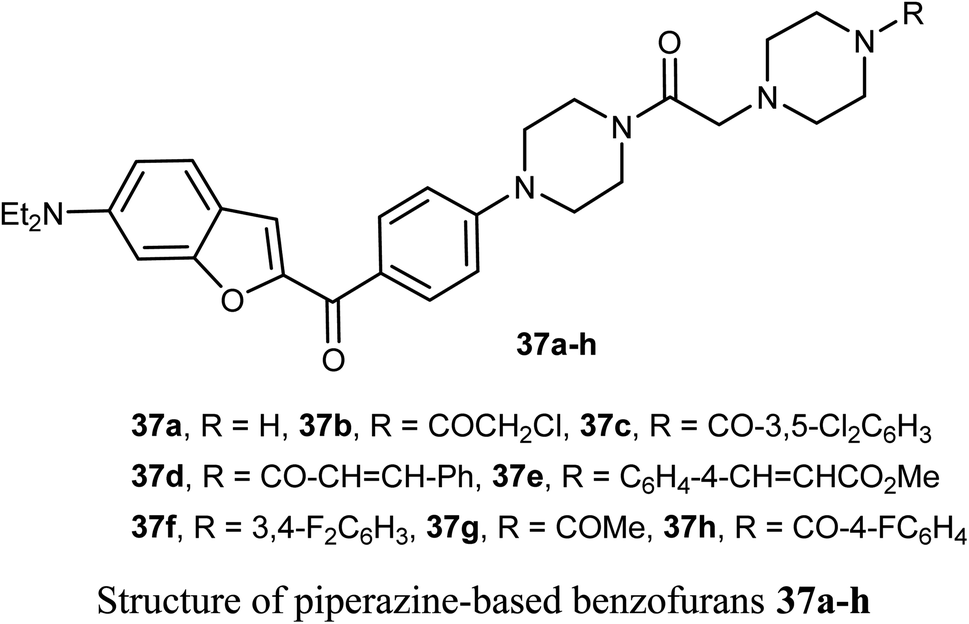
The piperazine-based benzofurans 37a–h were synthesized and their in vitro anticancer activities were tested against five human cancer cell lines; MCF-7, A549, HeLa, HCT116 and human gastric carcinoma (SGC7901) by MTT assay. Compounds 37a–h showed highly selective cytotoxic activity against the five cancer cells (IC50 < 10 μM), better or equal to the inhibitory activity of 5-fluorouracil (5-FU). The SAR study declared that electron-donor groups on the phenyl ring (such as CH3, CH3O, Cl and F) resulted in more promising anticancer activities, however, electron-withdrawing groups (such as CN and CF3) minimized the activity. Compound 37e demonstrated a promising anticancer activity against A549, HeLa, SGC7901, HCT116 and MCF-7 cell lines. Apoptosis study revealed that compound 37e significantly induced apoptosis on A549 cell.56
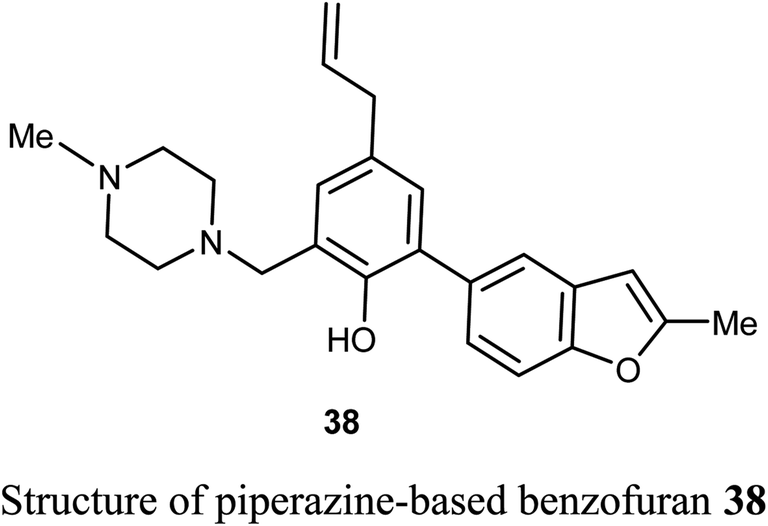
The piperazine-based benzofuran derivative 38 was designed, synthesized and evaluated as antitumor agent using MTT colorimetry method. Compound 38 was characterized by its high in vitro inhibitory activity against A549 and K562 cancer cells with IC50 values of 25.15 μM and 29.66 μM, respectively. The experiment was conducted at five concentrations (ranging between 128–8 μM), and each concentration was six parallel samples in triplicates. Thus, compound 38 was useful for further development as an anticancer drug.57
2.8. Anticancer activity of 2-aminobenzofuran derivatives
The 2-aminobenzofuran derivatives 39, 40 and benzodifurans 41, 42 were designed, synthesized and their anticancer potentials against prostatic tumor cells (PC-3) were evaluated. The bioassay results showed that the anticancer potential was affected by lipophilicity, where the anticancer activity of 2-aminobenzofuran derivatives was more promising when compared to benzodifurans. The benzofuran 39 was the most promising molecule. The benzofuran 39-treatment decreased PC-3 cell viability in a dose-dependent manner with IC50 = 33 μM as determined by CCK-8 assay (cell counting kit-8).58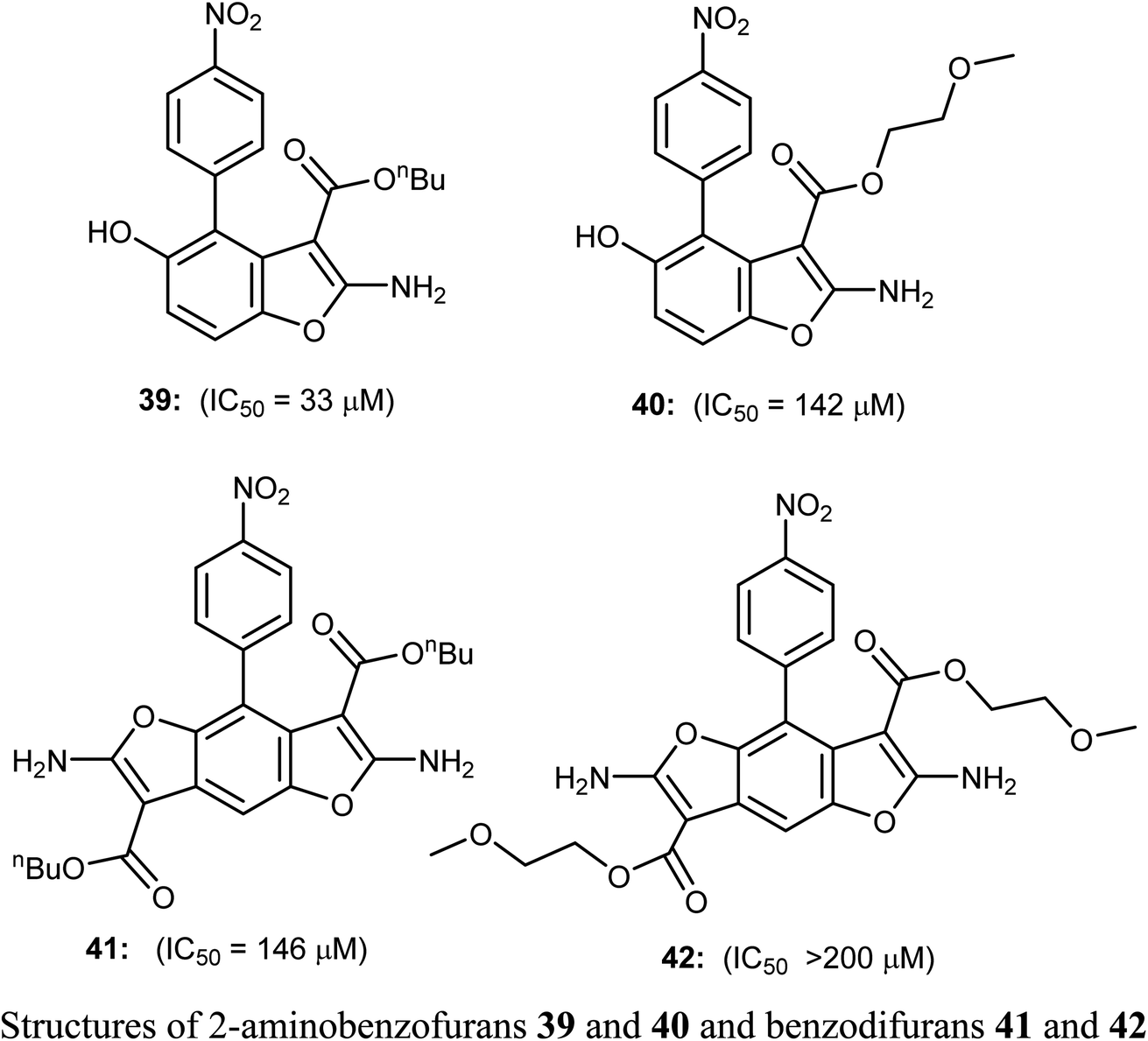
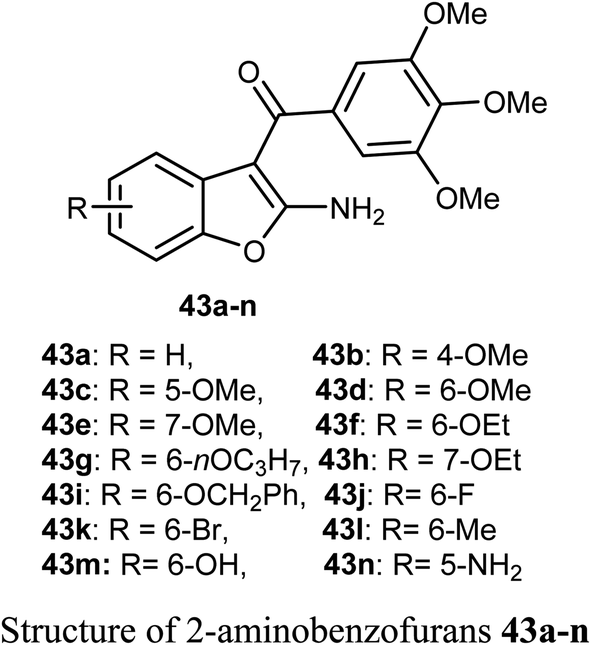
A series of biologically active 2-aminobenzofuran derivatives 43a–n were synthesized and evaluated in vivo and in vitro as inhibitors of tubulin polymerization. The in vitro antiproliferative activities against the six human cell lines; HT-29, HL-60, HeLa, medulloblastoma (Daoy), B-cell leukemia (SEM) and T-cell leukemia (Jurkat) was examined using combretastatin A-4 (CA-4) phosphate as a reference standard. The SAR study showed that the position of electron-donating or electron-withdrawing groups at the benzene ring of benzofuran had a great effect on the inhibitory potency. The presence of 6-methoxy group on benzofuran caused the maximum inhibitory activity while the presence of 4-methoxy group caused the least inhibitory activity. A similar effect was also noticed in the case of the presence of ethoxy, methyl or bromo substituents at position-6 of benzofuran, particularly the 6-ethoxybenzofuran derivative 43f was the most potent compound of the prepared series. The derivatives 43f and 43l were the most potent compounds of the series (with IC50 values 0.005–2.8 and 0.3–3.5 nM, respectively) against the six cancer cell lines, as compared with CA-4 (with IC50 = 0.8–3100 nM). More specifically, compound 43f exhibited the most excellent antiproliferative activity (IC50 = 0.005 nM) against the daoy medulloblastoma cell line, compared with IC50 > 10 μM against normal human astrocytes, confirming low toxicity in normal cells compared to tumor cells. The antitumor effect in vivo of compound 43f was studied in a syngeneic mouse tumor model. Compound 43f was treated by the intraperitoneal route at two different doses (5.0 or 15 mg kg−1) every other day. The tumor mass was reduced by 16.9% and 45.7% with 43f at 5 and 15 mg kg−1, respectively, and there was no sign of toxicity and no decrease in the animal body weight at the higher dose (15 mg kg−1). The in vivo activity of compound 43f showed better activity at 15 mg kg−1 than the reference CA-4 phosphate at 30 mg kg−1 against a syngeneic murine mammary tumor, where treatment with CA-4 reduced tumor growth by 26.5% only.59
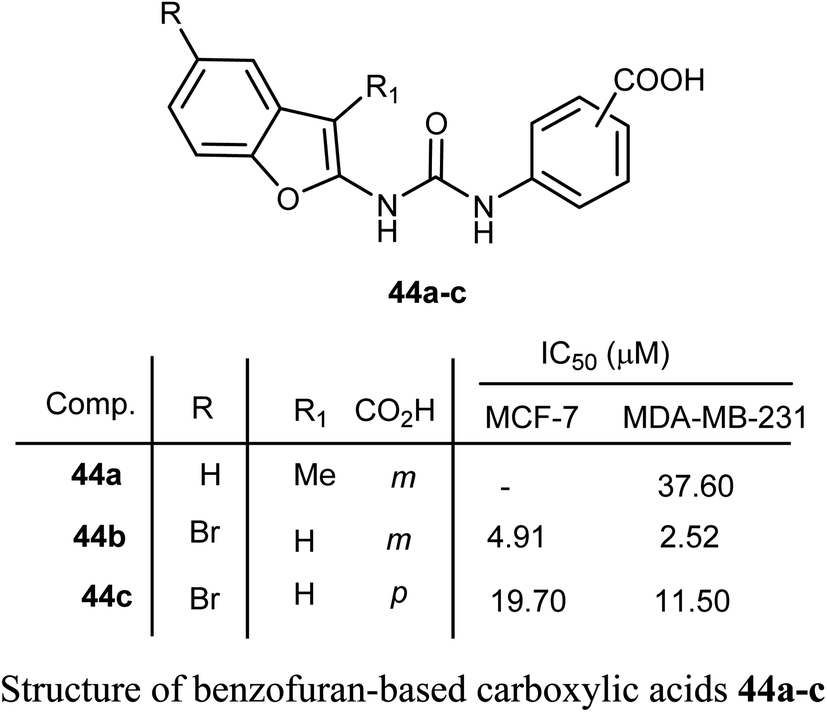
Benzofuran-based carboxylic acids 44a–c were evaluated for their anti-proliferative action against human breast cancer (MCF-7 and MDA-MB-231) cell lines. The derivatives showed variable inhibitions towards MCF-7 and MDA-MB-231 cell lines. In particular, the benzofuran derivative 44b was the most effective toward MDA-MB-231 (IC50 = 2.52 μM) with close potency to the reference drug doxorubicin (IC50 = 2.36 μM). The derivative 44b (which treated MDA-MB-231 cells) was remarkably arrested at the G2-M phase by increasing the cell population to 32.30% compared to 10.80% (for control). The number of cells in the sub-G1 phase was altered to 25.53% (for 44b-treated MDA-MB-231 cells) compared with 1.43% (for the control cells).60
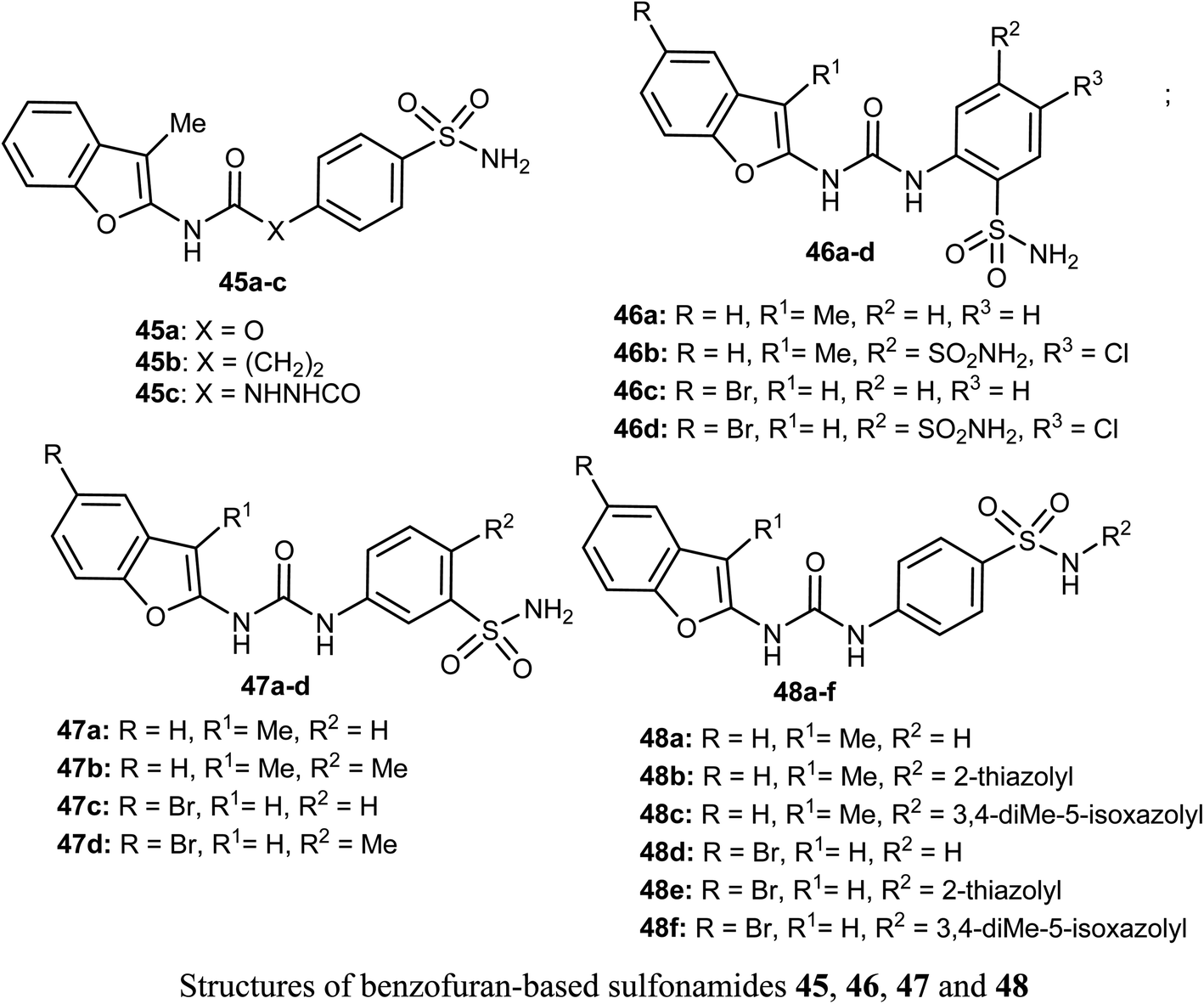
Different series of benzofuran-based sulfonamides 45, 46, 47 and 48 were designed, synthesized, and were found to be potent inhibitors against four tumor-related human-carbonic anhydrase (hCA) isoforms; hCA I, II, IX and XII. The bioassay results showed that most of the derivatives showed moderate to high inhibitory activity and in particular compounds 47b and 47d were the most effective hCA-IX inhibitors (with inhibition constants KIs = 8.4 and 5.5 nM, respectively), and were significantly selective toward hCA-IX isoform over the off-target hCA II isoform (S. I. = 26.4 and 58.9, respectively). The antiproliferative activity of 47b and 47d towards two breast cancer cell lines; MDA-MB-231 and MCF-7, was measured using MTT bioassay and staurosporine as the reference drug. The biological results proved that 47b had excellent antiproliferative activity against both cancer cell lines; MDA-MB-231 and MCF-7 (with IC50 = 6.27 and 6.45 μM, respectively), very close to staurosporine (IC50 = 6.75 and 3.67 μM, respectively), however, compound 47d had moderate anti-proliferation action against MDA-MB-231 and MCF-7 cancer cell lines (with IC50 = 14.16 and 13.79 μM, respectively). In addition, the up-regulation of the expression levels of pro-apoptotic Bax and Caspase-3 proteins and down-regulation of the expression level of anti-apoptotic Bcl-2 protein were reported upon treatment of both MDA-MB-231 and MCF-7 cells with compound 47b.61
2.9. Anticancer activity of benzofuran-2-carboxamide derivatives
Two sets of the 1-(benzofuran-3-yl)-1H-1,2,3-triazole derivatives 49a–i and 50a–i were synthesized, and their antiproliferative activities were screened against four cancer cell lines; HCT116, HeLa, HepG2, and A549 cells. The benzofuran-2-carboxamide derivative 50g showed the highest anti-proliferation potency against HCT-116, HeLa, HepG2, and A549 cells (IC50 = 0.87, 0.73, 5.74 and 0.57 μM, respectively) (Table 2). The SAR study proved that the presence of 6-methoxy group (as in structures 50a–i) was essential for high antiproliferative activity compared with the unsubstituted analogues 49a–i. The mechanistic studies demonstrated that compound 50g diminished the polymerization of tubulin, resulting in disruption of mitotic spindle formation, cell cycle arrest in the G2/M phase, and apoptosis of A549 cells. Thus, compound 50g was worthy to be a lead compound of further development as an anticancer agent.62
|
||||||
|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | (IC50, μM)a,b | |||
| HCT-116 | HeLa | HepG2 | A549 | |||
| a IC50 values are presented as the means ± SD of triplicate experiments.b Drug treatment for 72 h. | ||||||
| 49a | H | OEt | >100 | >100 | 30.97 ± 6.24 | >100 |
| 49b | H | OCH(CH3)2 | 41.71 ± 8.94 | 59.30 ± 5.15 | 58.00 ± 9.48 | >100 |
| 49c | H | HN(CH2)2CH3 | 16.43 ± 4.19 | 6.84 ± 0.34 | 2.92 ± 0.22 | 9.29 ± 3.66 |
| 49d | H | 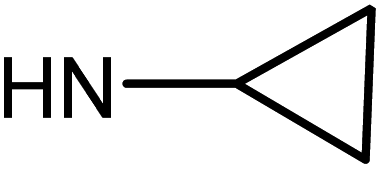 |
>100 | >100 | >100 | >100 |
| 49e | H | 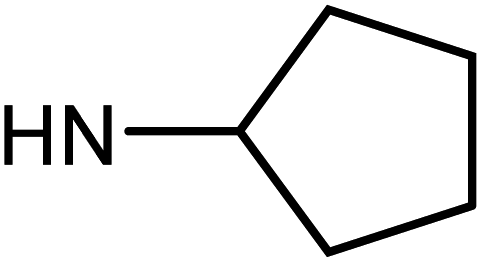 |
6.07 ± 0.38 | 43.37 ± 7.55 | 36.82 ± 5.43 | 22.52 ± 2.65 |
| 49f | H | 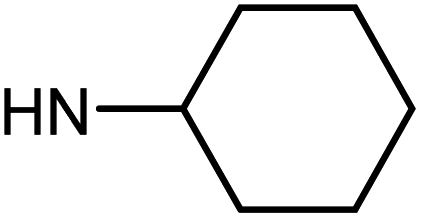 |
8.59 ± 5.47 | 24.99 ± 7.71 | 51.12 ± 9.75 | 94.74 ± 8.93 |
| 49g | H | 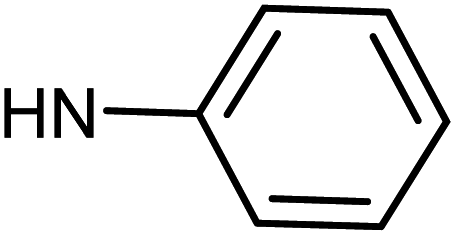 |
1.02 ± 0.31 | 1.83 ± 0.52 | 8.34 ± 2.53 | 0.83 ± 0.34 |
| 49h | H | 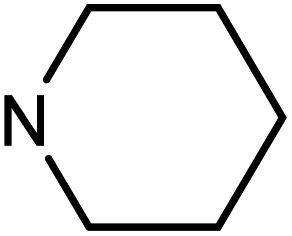 |
73.52 ± 17.17 | >100 | 65.23 ± 5.42 | 73.99 ± 7.86 |
| 49i | H | 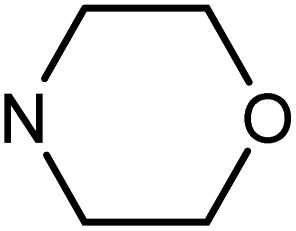 |
>100 | >100 | 64.85 ± 2.98 | >100 |
| 50a | OMe | OEt | 36.64 ± 7.30 | >100 | 11.57 ± 3.59 | >100 |
| 50b | OMe | OCH(CH3)2 | 40.21 ± 4.23 | 29.48 ± 0.67 | 14.79 ± 5.02 | 50.41 ± 7.88 |
| 50c | OMe | HN(CH2)2CH3 | 52.46 ± 8.74 | 2.84 ± 0.95 | 6.29 ± 2.93 | >100 |
| 50d | OMe | 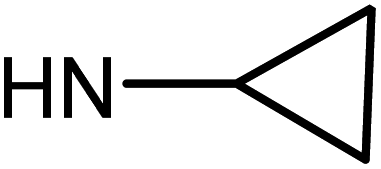 |
>100 | 78.38 ± 7.48 | >100 | >100 |
| 50e | OMe | 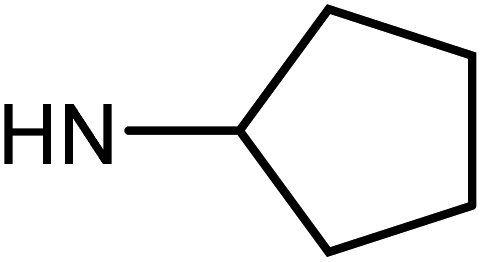 |
1.27 ± 0.38 | 7.66 ± 1.26 | 20.26 ± 9.05 | 18.95 ± 3.17 |
| 50f | OMe | 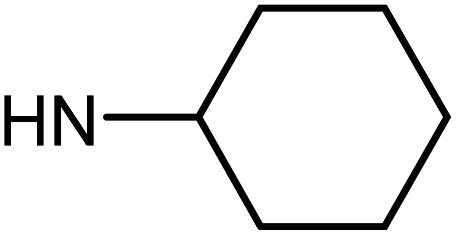 |
5.56 ± 1.76 | 29.99 ± 5.59 | 41.65 ± 6.01 | >100 |
| 50g | OMe | 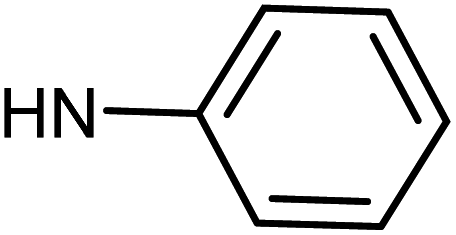 |
0.87 ± 0.79 | 0.73 ± 0.67 | 5.74 ± 1.21 | 0.57 ± 0.31 |
| 50h | OMe | 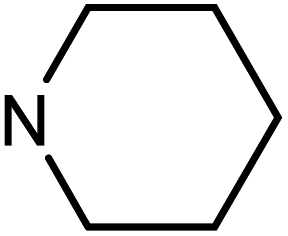 |
47.79 ± 7.56 | >100 | 6.83 ± 3.21 | 38.79 ± 16.90 |
| 50i | OMe | 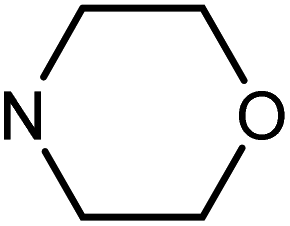 |
80.08 ± 0.86 | 49.75 ± 11.74 | 10.92 ± 4.65 | 37.85 ± 6.96 |
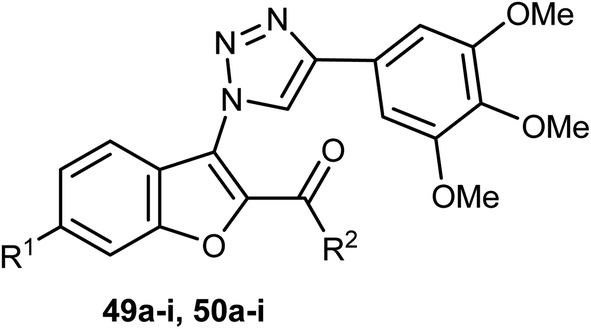
The benzofuran-carboxamide derivatives 51a–j and 52a–d were synthesized and their antiproliferative activities were conducted against four human cancer cell lines; HT-29, MCF-7, Panc-1 and epithelial cancer (A-549) using MTT assay method and doxorubicin as a reference drug. Some compounds exhibited good antiproliferative activities against the cancer cells and the 4-(morpholin-4-yl)phenethyl derivative 51d was the most active one against all of the four cell lines (with IC50 values ranging between 0.70–1.8 μM), with almost equal potency to doxorubicin reference (with IC50 values ranging between 0.90–1.41 μM). The most active caspase-3 enhancers were compounds 51d and 51h which led to an increase in the levels of caspase 8 and 9 declaring activation of both intrinsic and extrinsic pathways. Both compounds 51d and 51h showed marked induction of Bax, and down-regulation of Bcl-2 protein levels in MCF-7 cell lines. Compound 51d also demonstrated cell cycle arrest at the Pre-G1 and G2/M phases in the cell cycle of MCF-7 cell line. The SAR analysis revealed that the presence of the N-phenethyl carboxamide moiety considerably improved the antiproliferative activity of compounds 51 and such activity was further enhanced by the presence of a morpholinyl group at the para-position of the N-phenethyl ring causing compound 51d to have antiproliferative activity similar to the reference drug doxorubicin. In addition, the effect of para substitution at the phenethyl moiety on the antiproliferative activity of compounds 51 was in the order: morpholinyl > piperidinyl ≥ pyrrolidinyl > dimethylamino > H.63
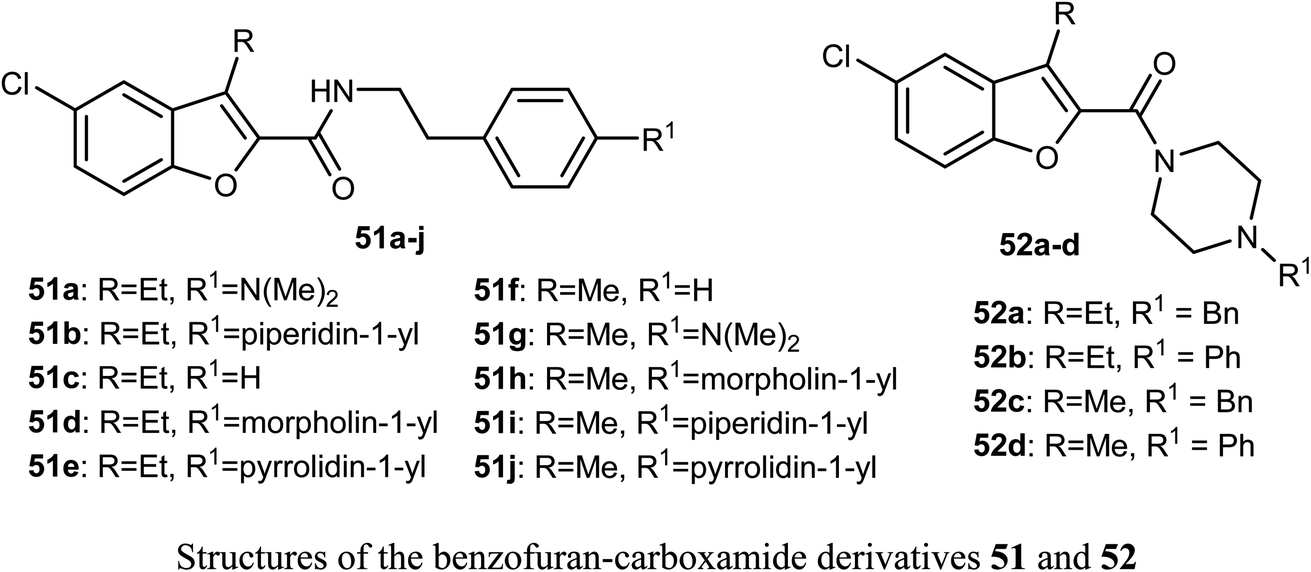
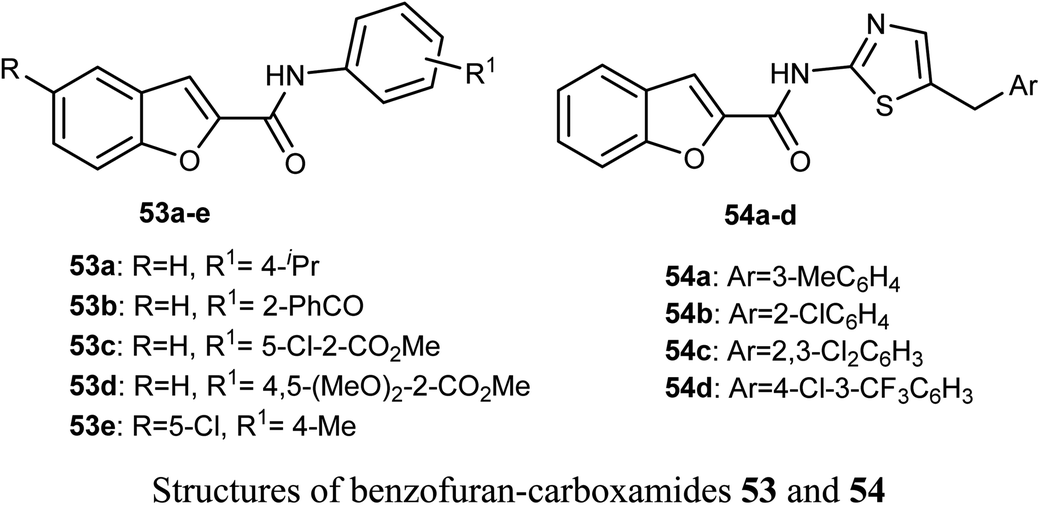
The in vitro anticancer activity of the benzofuran-carboxamides 53 and 54 were screened by Matiichuk et al. against sixty human tumor cell lines obtained from nine neoplastic diseases. The results showed that all of the tested compounds displayed different levels of activity. The mean graph mid-points (MG-MID) were determined for GI50 and TGI (TGI is the molar concentration of a compound leading to total inhibition), to get an average activity factor over all cell lines for the tested compound. Compound 54a disclosed high inhibition potency (GI50 < 10 μM) against all of the 58 tested tumor cell lines (with MG-MID = 2.03 μM), 54c showed high inhibition potency against 56 cell lines (MG-MID = 3.26 μM) and 54d exhibited high inhibition potency against 53 human tumor cell lines (MG-MID = 3.26 μM). The colon cancer subpanel exhibited the best sensitivity to compounds 54a and 54c (with a mean GI50 = 0.87 and 0.84 μM, respectively). The most sensitive cell line was T-47D (Breast Cancer, GI50 = 0.088 μM). In contrast, compounds 53a–e showed weak to moderate activity on the tested cell lines.64
2.10. Anticancer activity of dihydrobenzofuran derivatives
The naturally isolated benzofuran derivatives 55a–c (isolated from the ethyl acetate fraction of Polygonum barbatum weed) were examined for their anticancer potency against lung cancer (NCI-H460) and oral cancer (CAL-27) cell lines, employing MTT assay. Compound 55a had more anticancer potency with IC50 of 53.24 and 48.52 μM against NCI-H460 and CAL-27, respectively, when compared with the standard drug 5-fluorouracil (IC50 = 97.76 μM). Compound 55a induced apoptosis after 24–48 h treatment as confirmed by morphological and flow cytometry analysis.65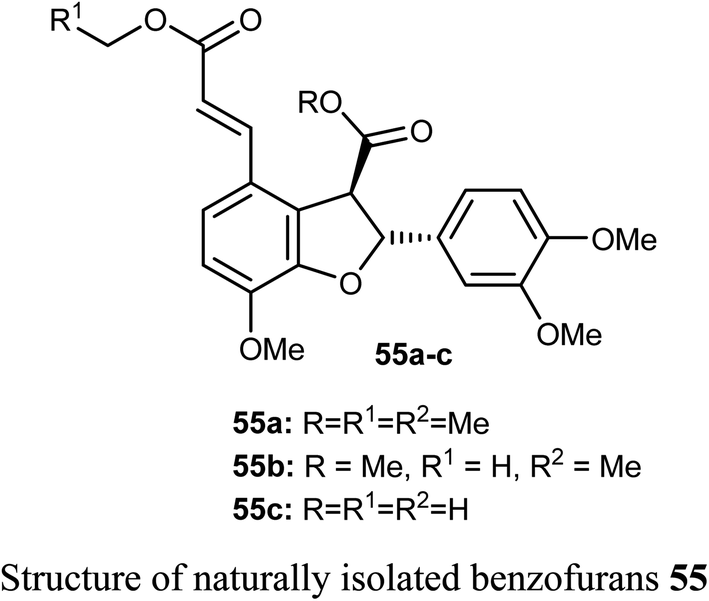
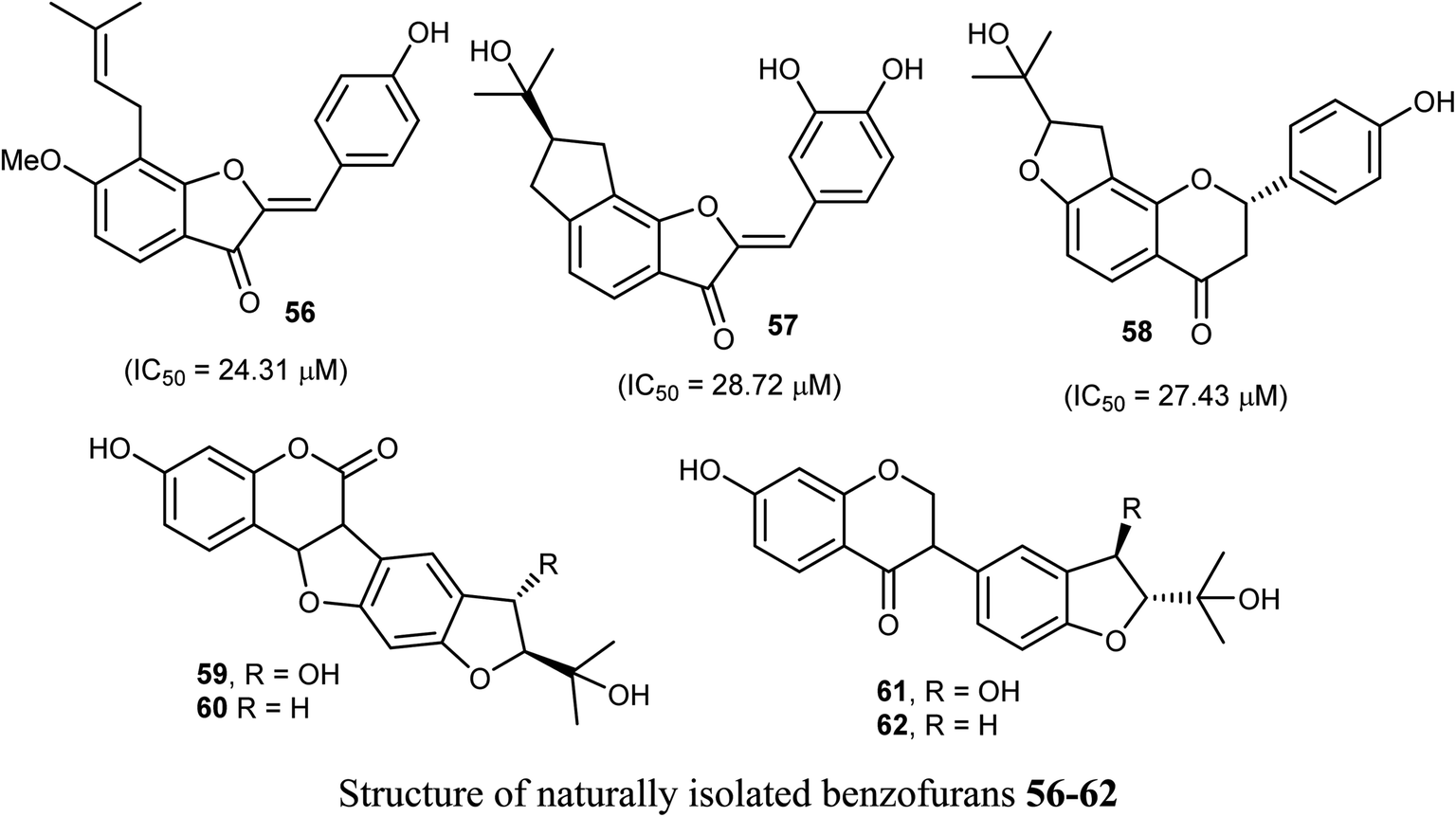
Some benzofuran derivatives 56–62 were isolated from the seeds of Psoralea corylifolia and were screened for in vitro inhibitory activity against the human colon cancer (SW620) cell line. Compounds 56–62 were found to have reasonable cytotoxicity against the SW620 cells with IC50 = 24.31–28.72 μM compared with 5-fluorouracil (IC50 = 3.19 μM). Among these compounds, 56 was the most cytotoxic one but compounds 59–62 did not exhibit cytotoxicity toward the SW620 cell line.66
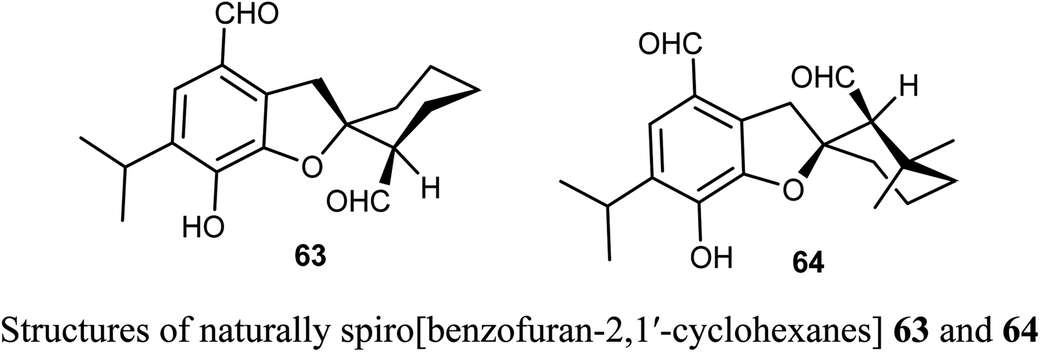
The naturally extracted spiro[benzofuran-2,1′-cyclohexane] scaffolds 63-64, were screened for their cytotoxicity against five cancer cell lines (A-549, SMMC-7721, HL-60, MCF-7, and SW-480) and a noncancerous (BEAS-2B) cell lines. The bioassay was measured using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) method, where compound 63 exhibited significant cytotoxicity against A-549, SMMC-7721, and MCF-7 cell lines (with IC50 values 6.60, 7.13, and 11.50 μM) more potent than the reference cisplatin (13.84, 7.82, 13.46 μM, respectively).67
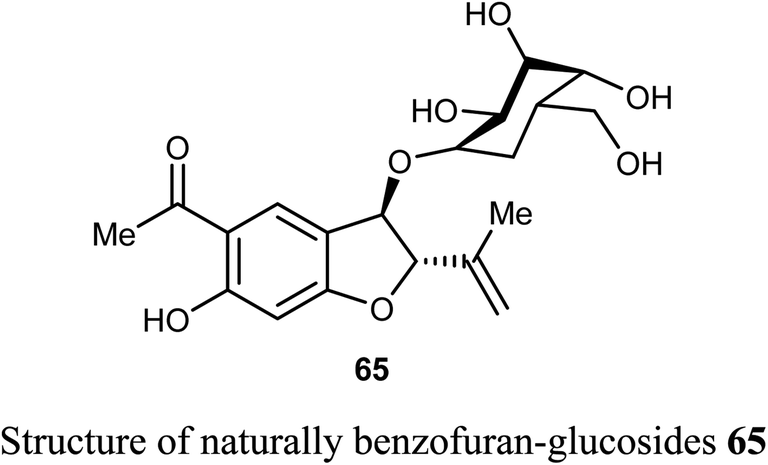
The naturally benzofuran-glucoside derivative 65 was isolated from Senecio glaucus L. by Zaher et al. and evaluated for its cytotoxic activity. Compound 65 exhibited potent cytotoxic activity against the pancreatic cancer cell line (PANC-1) under glucose deficient medium (IC50 = 7.5 μM) using antimycin as anticancer reference compound.68
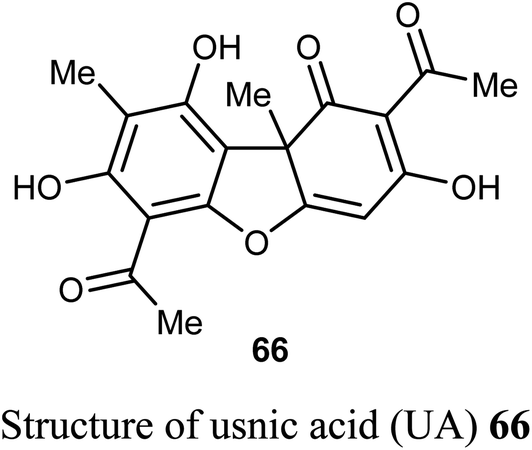
The natural product usnic acid (UA) 66 was reported as a potent anticancer agent against human gastric adenocarcinoma (AGS), gastric carcinoma (SNU-1) and squamous carcinoma (A-431) cells. The bioassay study was carried out by treatment of AGS cells with 10–50 μM of UA in DMSO for 12, 24 and 48 h using MTT technique. UA promoted cell death in A-431 cells with IC50 values 98 and 39 μM after 48 and 72 h of UA treatment, respectively. UA reduced the cell viability of AGS cells by 17–45%, 20–65%, and 52–80% (P < 0.05–0.001) after 12, 24, 48 h, respectively. On the other hand, UA treatment had no remarkable effect on the cell viability of human HaCaT and HEK-293 cells after 48 h. Thus, UA induced cytotoxic effects on human gastric cancer cells but not on normal gastric cells. Furthermore, UA 66 demonstrated selective apoptotic effect (10–25 μM) through the generation of ROS and led to DNA damage on human gastric cancer cells accompanied by upregulation of γH2A.X (Ser139) phosphorylation, DNA-PKcs and p53.69,70
Usnic acid (UA) 66 encapsulated heparin-modified gellan gum (HAG) nanoparticles (NPs) was synthesized and its in vitro cytotoxicity screening against human lung cancer cell line A549 was performed using SRB bioassay. The IC50 values were 12 and 14 μM for the free and encapsulated forms of usnic acid, respectively. The in vitro cytotoxicity study, apoptosis assay and cell cycle assay determined that the developed nanoparticulate formulation was safe and effective and that the HAG NPs were more suitable for inhibition of cancer cell growth and as a drug delivery carrier than the free form of the drug.71
Wang et al. reported the synthesis and anticancer activity of some usnic acid (UA) derivatives 67a–g against leukemia cells using (+)-UA parent compound as a positive control. The biological assay revealed that the UA-derivatives 67a–g inhibited the proliferation of the leukemia cells; HL-60 and K562 with IC50 ranging between 2.15–3.25 and 2.70–6.20 μM, respectively. Particularly, the 4-fluoro and 4-chloro substituted derivatives 67c and 67g exhibited the highest potency against both cells HL-60 and K562 with IC50 = 2.64–2.82 μM. The mechanism of the study proved that compound 67g induced apoptosis in HL-60 and K562 cell lines, and affected the expression of MNK/eIF4E axis-related proteins, such as Mcl-1, p-eIF4E, p-4E-BP1. The bioassay results confirmed that 67g was a potent apoptosis inducer in leukemia cells, where compound 67g caused significant apoptosis occurrence in HL-60 cells at 6 μM (63.5%) in comparison with control cells (2.6%). In contrast, K562 cells were less sensitive to 67g, and the apoptosis was observed at a prolonged period at 48 h and higher concentrations.72
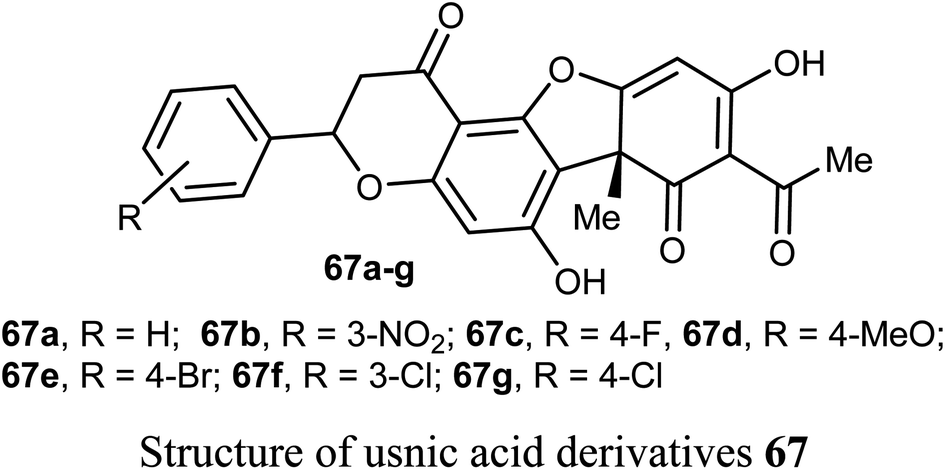
A new series of the usnic acid derivatives 68a–r having hydrazonothiazole pharmacophores were described and tested as Tdp1 inhibitors. Most of the prepared compounds were found to have high inhibitory potency with IC50 values in a nanomolar scale (IC50 = 20–200 nM). The SAR study confirmed that the size of the heterocyclic moiety and the nature of heteroatom had no significant effect on the inhibitory activity, however, the presence of a bromine atom or bulky aromatic motif attached to the heterocyclic moiety 68f–h and 68r enhanced the inhibitory activity. The presence of pyrrole moiety led to the most potent inhibition such as in compounds 68m and 68n.73
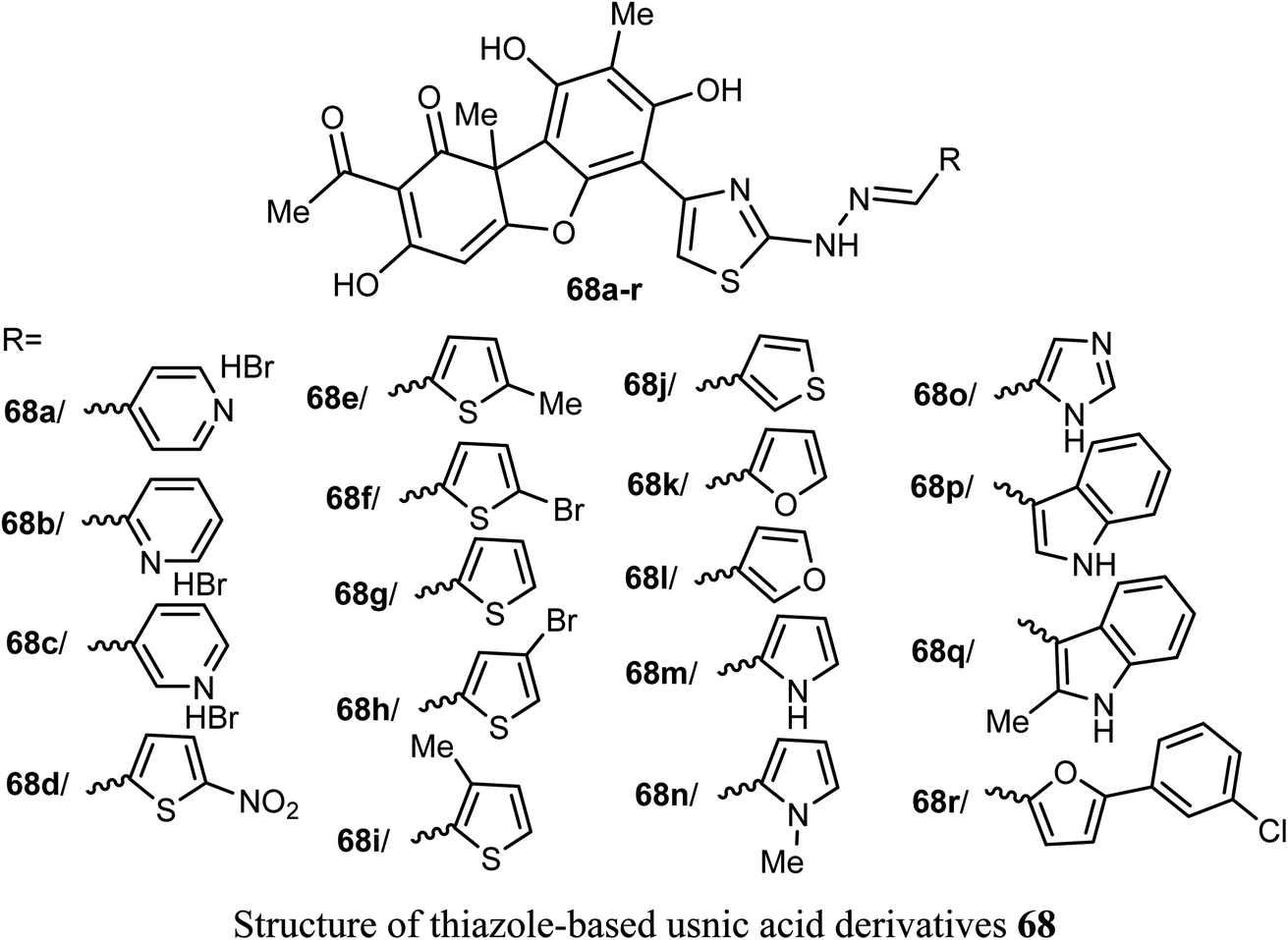
Pyrczak-Felczykowska et al. reported the synthesis of the usnic acid-isoxazole derivative 69 and assessed its activity against cancer cells of different origins. The viability of breast cancer or normal cells was evaluated by MTT method. The in vivo anticancer activity was also examined using the mice xenograft technique. Compound 69 inhibited tumor growth upon injection to nude mice with xenografted breast cancer cells and vacuolization was selectively noticed in tumor cells but not in other organs. Thus, compound 69 induced Endoplasmic Reticulum (ER) stress in breast cancer cells but not in healthy cells. Compound 69 also effectively stopped tumor growth in vivo.74
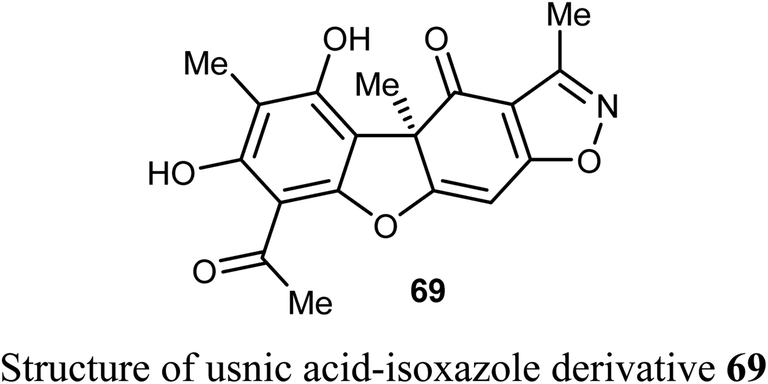
The anticancer activity of the amidino-rocaglates 70a–l against MDA-MB-231 breast cancer cells was evaluated in a sulforhodamine B (SRB) assay. The benzofuran derivatives 70c, 70d, and 70l were the most inhibitory active compared with the reference rocaglate-hydroxamate 71. Compound 70d was found to have excellent cytotoxicity with IC50 = 0.97 nM, an almost a 2-fold increase in potency over the lead compound 71 (IC50 = 1.9 nM). Furthermore, the dimethylamide 60c and methyl ester 70l were found to have IC50 values 1.2 and 3.6 nM, respectively. The bioassay results disclosed that amidino-rocaglates might be valuable cancer chemotherapeutic agents.75,76
The natural benzofuran product (rocaglamide) 72 was reported to significantly promote the natural killer (NK) infiltration of the non-small cell lung cancer (NSCLC) cells by autophagy inhibition. Compound 72 specifically damaged mtDNA, then enhanced mtDNA leakage into the cytoplasm and activated the cGAS-STING signaling, resulting in increased infiltration of NK cells and enhanced antitumor immunity in NSCLC. Thus, compound 72 had significant potential in cancer immunotherapy.77
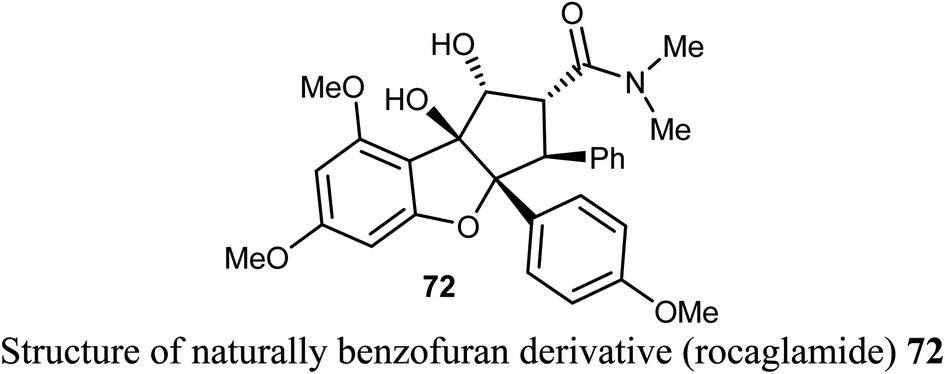
A number of p-azidobenzoyl-tetrahydrobenzofuran-4(5H)-one hybrids 73a–j were described by Pandit et al. for anticancer activity against renal cancer panel UO-31 cell lines. The results of the anticancer evaluation of the hybrids 73a–j showed promising responses against UO-31 cell lines (<80%). The type of substituents at the phenyl moiety had a crucial effect on the activity of the compounds. Compounds 73a and 73e, having electron-withdrawing functions (4-NO2 and 4-Br, respectively) effectively reduced the viability percentage of UO-31 cells at 10 μM concentrations and resulted in the highest potency against UO-31 cell lines in renal cancer panel. Compounds 73a and 73e showed high anticancer activity and prominent selectivity against UO-31 with growth inhibition of 69.36% and 80.66%, respectively.78
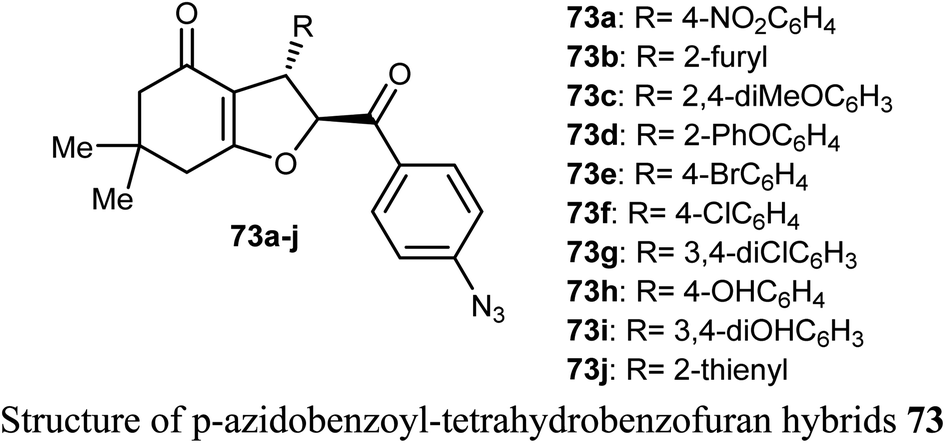
2.11. Anticancer activity of indole-based benzofuran derivatives
The anticancer activity of various indole-based benzofuran scaffolds 74a–i were determined against two cervical cancer cell lines; SiHa and C33a. Four of the prepared derivatives (74a, 74d, 74e and 74h) demonstrated IC50 values < 40 μM. Compounds 74e and 74h were found to be the most effective and induced autophagy as established by the progressive conversion of LC3I to LC3II and down-regulation of p62 in cervical cancer cells C33a and SiHa.79
2.12. Anticancer activity of benzofuran-2-carboxylate derivatives
The antiproliferative activities of a number of shikonin-benzofuran substrates 75a–q against five cancer cell lines; MDA-MB-231, HepG2, HT29, HCT116 and A549 using the MTT assay and shikonin and colchicine as the positive drugs were evaluated by Shao et al. Many of the reported compounds exhibited promising antiproliferation activities against the cancer cell lines compared to that of shikonin and had low cytotoxicity to non-cancer cells. Particularly, compounds 75c, 75f and 75i (IC50 = 0.18, 1.03 and 0.82 μM, respectively) demonstrated much higher antiproliferation potency against HT29 cells than the standard drugs, shikonin (IC50 = 2.80 μM) and colchicine (IC50 = 1.81 μM). The most powerful inhibition was observed for compound 75c (IC50 = 0.18 μM) against HT29 cells. Compound 75c induced cell apoptosis, regulated the expression of apoptosis-related proteins in HT29 cells and stimulated the HT29 cell cycle arrest at G2/M phase.80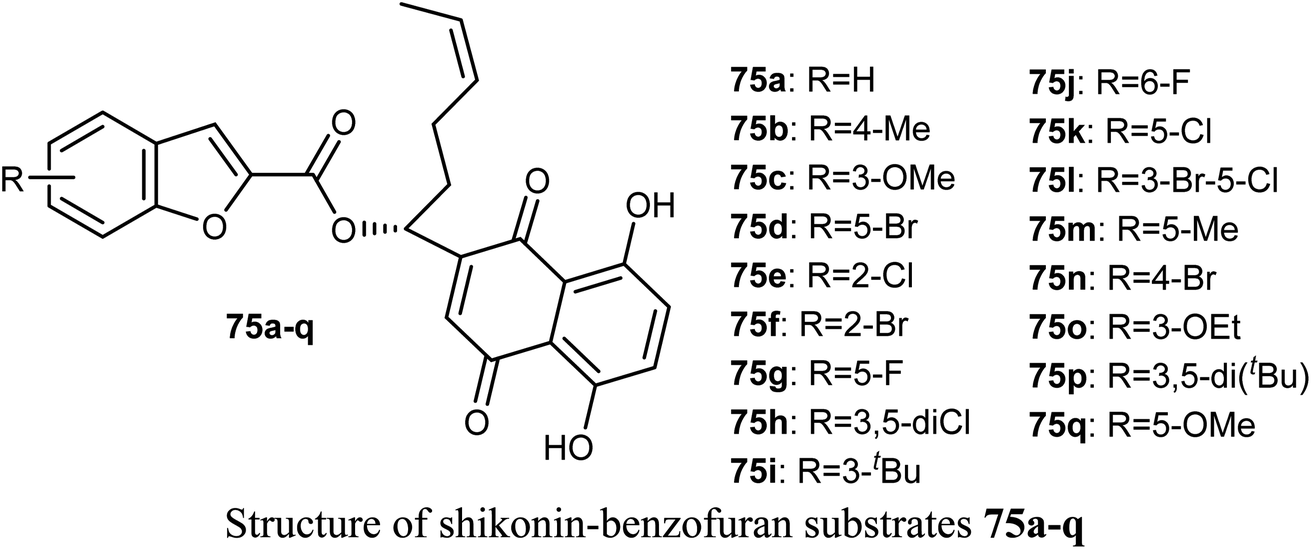
2.13. Anticancer activity of 4(5)-hetaryl-benzofuran derivatives
Synthesis of a large number of benzofuran derivatives 76–79 and 80–83 and their in vitro anticancer activity against the breast cancer cell lines; MCF-7 and MDA231 were reported. Most of the reported compounds demonstrated very potent antiproliferative activities (IC50 = 1.19–2.78 μM) against MCF-7 much better than lapatinib reference drug (IC50 = 4.69 μM). Moreover, the derivatives 80a and 83 exhibited promising cytotoxic activity against both MCF-7 and MDA231. The inhibitory potencies of the derivatives 80a and 83 against p38α MAP kinase were also highly significant with IC50 = 0.04 μM superior to the reference standard SB203580 (IC50 = 0.50 μM). In addition, the behavior of the benzofuran derivative 83 on the apoptotic induction and cell cycle progression of MCF-7 cell line was found to induce preG1 apoptosis and cell growth arrest at G2/M phase preventing the mitotic cycle. Compound 83 also exhibited very good drug-like properties, good ADME (absorption, distribution, metabolism and excretion) properties with moderate solubility, high GI permeation and very good bioavailability scores.81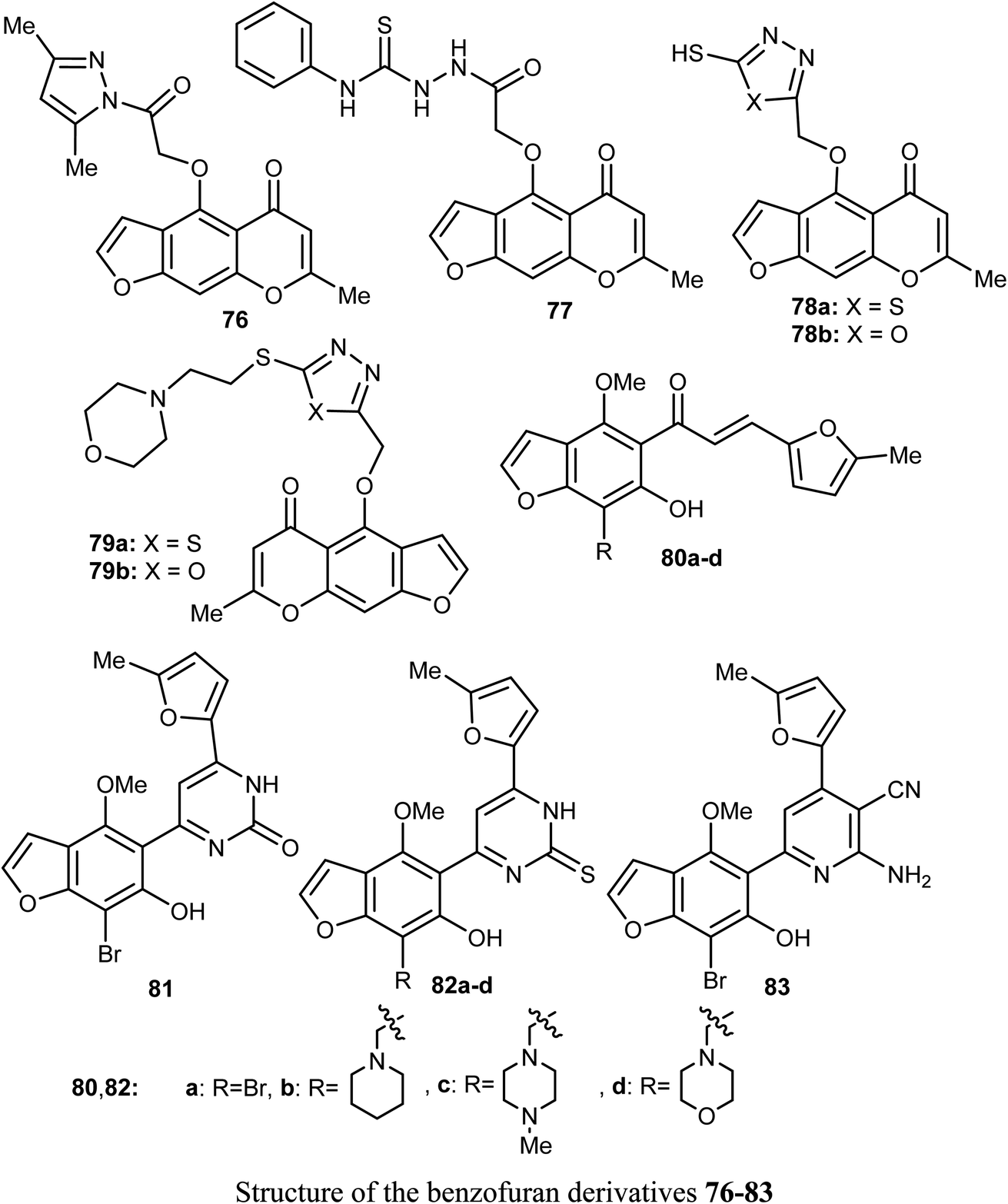
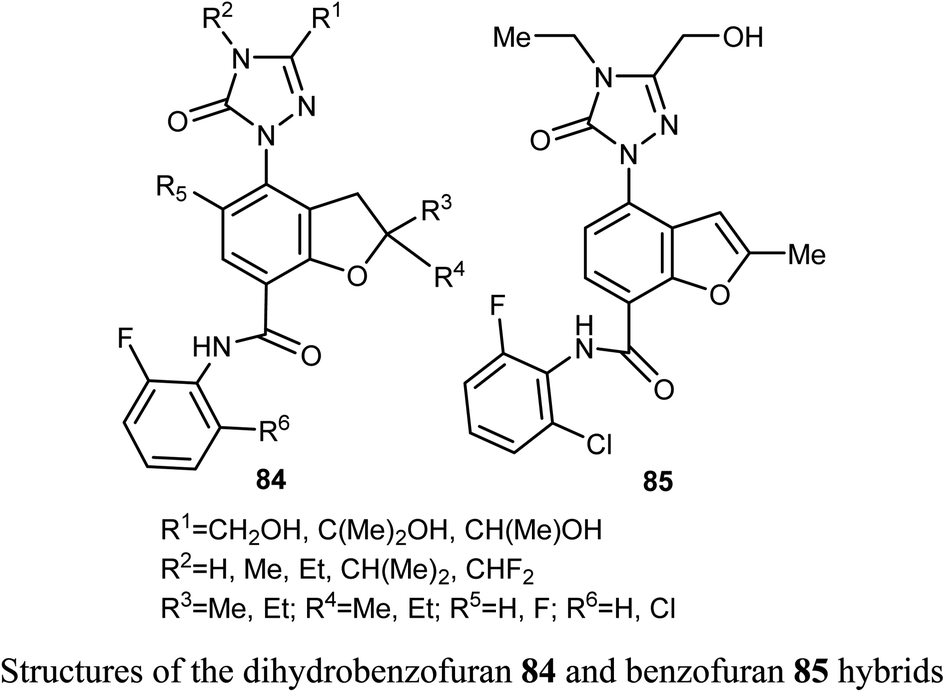
A series of dihydrobenzofuran hybrids 84 and the benzofuran derivative 85 were invented and patented by Kuduk et al. and then evaluated for in vitro inhibition of human leukemia cell line MOLM-13. The reported compounds exhibited moderate to high inhibition effect against MOLM-13 in nano-molar scale with IC50 values ranging from 0.3–700 nM. Among them, four derivatives had the highest potency with IC50 = 0.3–7 nM. The best anti-leukemia cancer substrate was found to be compound 84 (R1 = C(OH)Me2, R2 = Me, R3 = R4 = Me, R5 = F, R6 = Cl) with IC50 = 0.3 nM, suggesting that this candidate could be further developed as anti-leukemia cancer agent.82
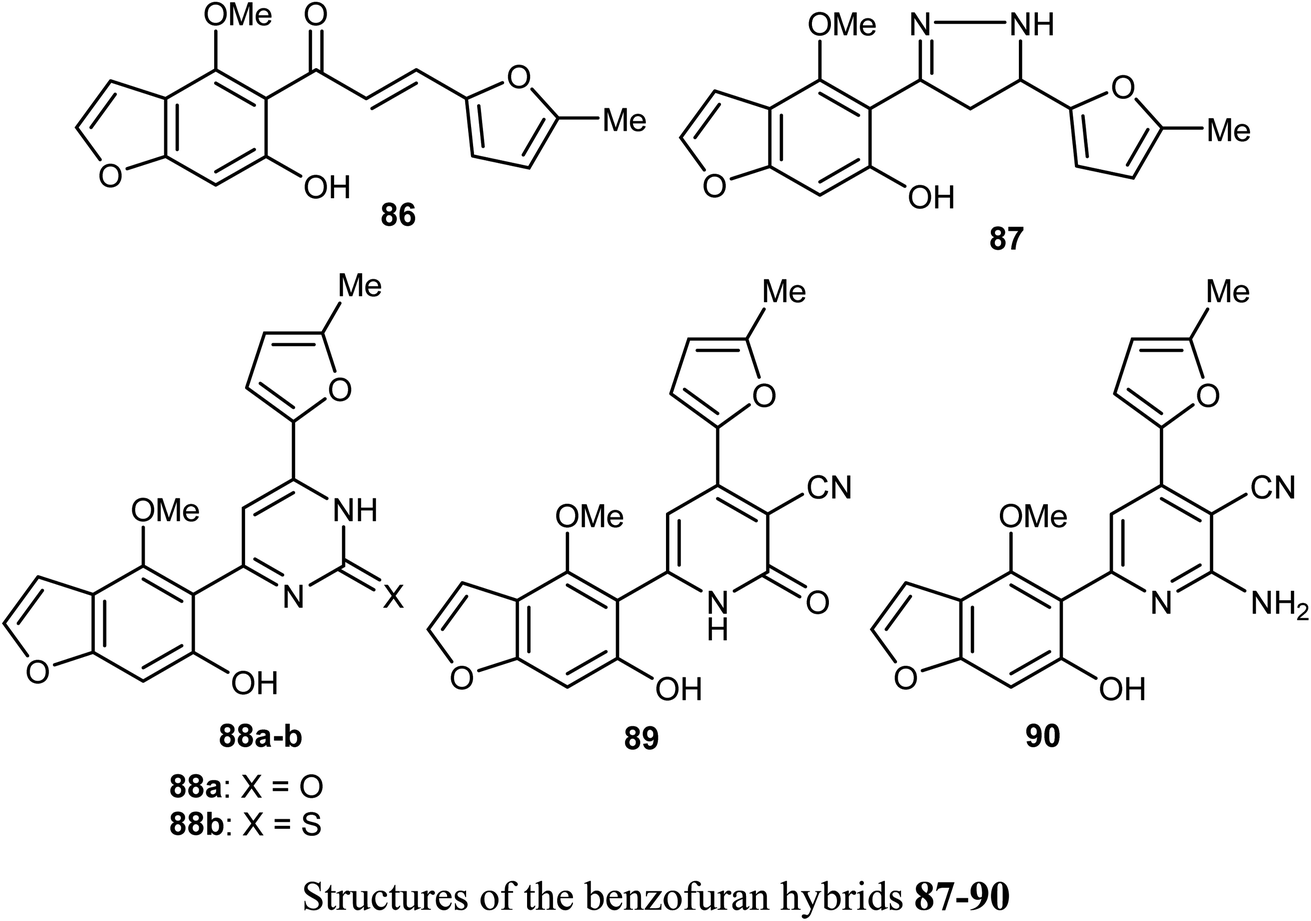
Ahmed et al. reported the synthesis of a set of benzofuran hybrids 87–90, from the chalcone derivative 86, and assessed their antitumor activities against breast cancer. The in vitro and in vivo bioassay results disclosed that the derivatives 86, 88a, and 89 revealed the highest antiproliferative activities against the MCF-7 cell line with IC50 values 0.07, 2.94, and 2.48 μM, respectively, compared with the standard lapatinib (IC50 = 4.69 μM). Particularly, the benzofuran molecule 86 (IC50 = 0.07 μM), presented the most cytotoxic potency against MCF-7 BC cell line, caused inhibition of a panel of 22 kinases in the range of −39% to −97%, and arrested cell cycle at G2/M phase. In addition, for the in vivo experiments, where male swiss mice were selected as the animal model to induce solid ehrlich tumor, compound 86 reduced the tumor cells in sections of ehrlich tumor, displayed a strong positive caspase-3 and negative EGFR immuno-reaction.83
2.14. Anticancer activity of thiazole-based benzofuran derivatives
A set of 2-(pyrazolyl)benzofuran derivatives 91a–s were designed, synthesized and evaluated for antiproliferative action against HeLa cell line of EGFR PK inhibitory potency. Seven compounds 91c, 91d, 91h, 91j, 91k, 91m and 91n demonstrated promising sensitivity against the cancer cells. They were more potent or equipotent to the reference drug doxorubicin. For example, the derivative 91m was the most potent anticancer agent with IC50 value 0.60 μM, which was twice that of the reference drug doxorubicin (IC50 = 1.10 μM). The EGFR protein kinase inhibitory activity of the synthesized compounds showed that compound 91k was the most active one and had higher EGFR PK inhibitory activity (IC50 = 0.07 μM) than that of the reference drug erlotinib (IC50 = 0.08 μM). In addition, cell cycle analysis and apoptosis assay of compound 91k were performed and it caused G1/S phase arrest and apoptosis in HeLa cancer cells, in addition to its activation of the caspases-7 and -3. The molecular docking study showed that compound 91k had a good fitting and proper interaction with the key amino residues in the binding site of EGFR kinase compared to erlotinib. SAR study revealed that electron-withdrawing substituents at the benzylidene moiety greatly affected the activity, where the 2,4-dichloro derivative 91m had twice the activity of doxorubicin, the 4-bromo derivative 91n had similar activity to that of doxorubicin. Thus, enhanced lipophilicity of the molecules, due to the presence of the halogen atoms, led to substantial improvement in the binding affinity and caused a marked increase in the anticancer activity of the compounds 91m and 91n. Similarly, 3- and 4-methoxybenzylidene derivatives 91c and 91d exhibited significant cytotoxic activity (IC50 = 0.63, 0.82 μM, respectively) much higher than that of doxorubicin, due to the oxygen atom of the methoxy group that acted as H-bond acceptor with the target protein kinase.84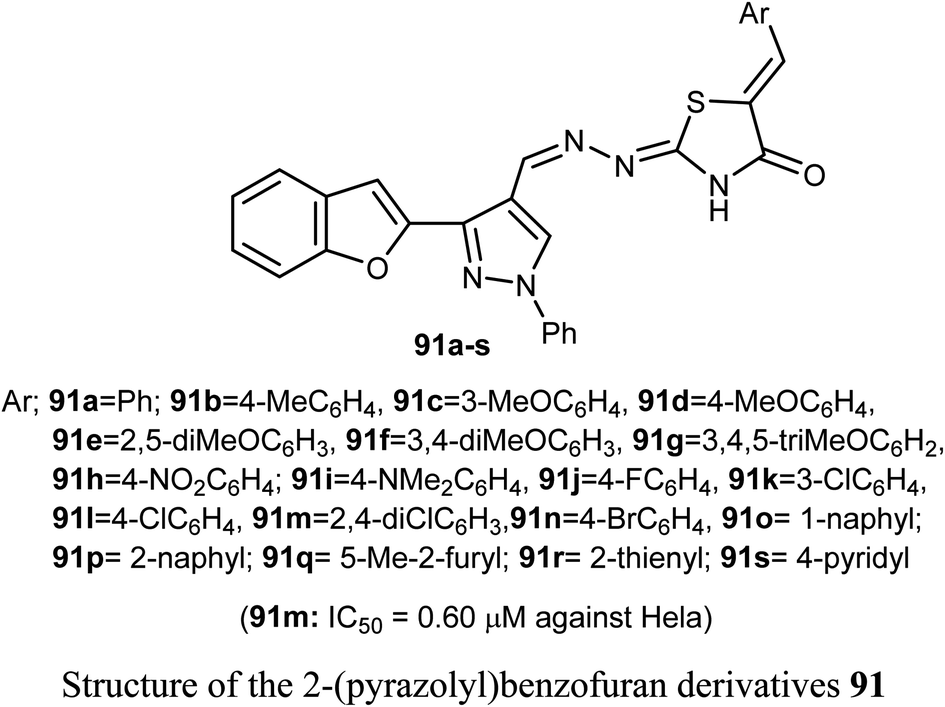
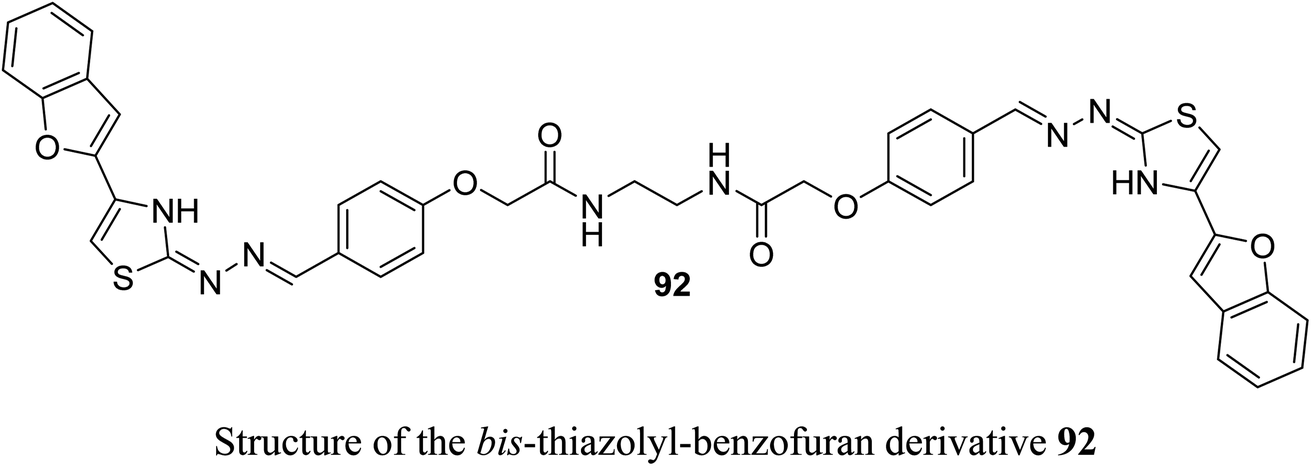
Recently, we reported the synthesis of the bis-amide-based bis-thiazole derivative having benzofuran moiety 92 and was tested for its anticancer activity against colorectal (HCT-116), and breast (MCF-7) cancer cell lines using the MTT assay. The results disclosed that compound 92 had a promising cytotoxicity against HCT-116 cells (with IC50 = 14.26 μM) and a moderate activity against MCF-7 (with IC50 = 37.3 μM) compared with 5-fluorouracil (5-FU) reference drug (with IC50 values of 11.78 μM and 5.81 μM against HCT-116 and MCF-7), respectively.85
3. Molecular targeting for benzofurans as anticancer agents
In medicinal chemistry, benzofuran scaffolds are central pharmacophores and favored structures, where they have been useful as active ingredients in several medications with proven clinical efficacy. Recent studies have demonstrated that benzofuran analogues can endure a wide range of pharmacological activities. The remarkable progress of benzofuran derivatives in a variety of disorders in a very short amount of time demonstrates the importance of this area of study to medicinal chemistry. The high demand to explore new and highly efficient anticancer medicines in the human fight against cancers has not diminished. There is a high need for both novel anticancer medications and improved approaches to treating the disease. Recently, the powerful cytotoxic effects of benzofurans against human breast and ovarian cancer cells have been discovered.86,87 A panel of human tumor cell lines were tested in vitro after the researchers developed various new benzofuran derivatives.88 It was discovered that the presence of a 2-methylimidazole or 2-ethylimidazole ring, as well as the substitution of the imidazolyl-3-position with a naphthylacyl or methoxyphenacyl group, were necessary for adjusting the level of cytotoxic activity. As benzofuran derivatives have pharmacological activities against cancer, their molecular targets for anticancer activity are summarized in Fig. 3.89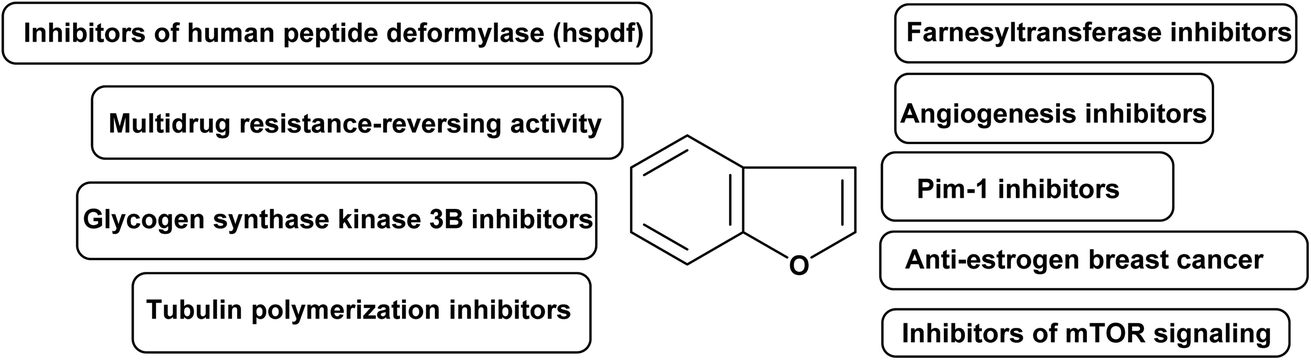 | ||
| Fig. 3 Summarized molecular targets for benzofurans as anticancer compounds. | ||
3.1. Farnesyltransferase inhibitors
Membrane-bound GTP binding proteins (G-proteins) govern cell development by cycling between an inactive GDP-bound state and an active GTP-bound state, functioning as molecular switches. Some G-proteins were activated constantly in tumor cells, promoting their cancerous growth.90,91 A series of benzofuran-based farnesyltransferase inhibitors have been designed and synthesized as antitumor agents. One of these derivatives showed the most potent enzyme inhibitory activity with IC50 of 1.1 nM and antitumor activity in human cancer xenografts in mice.923.2. Angiogenesis inhibitors
The formation of a new blood supply system, known as angiogenesis, is a hallmark of newly developing tumors. To put it simply, angiogenesis is the driving force behind not only the initial development of a tumor but also its subsequent development, spread, and recurrence.93 For these reasons, preventing tumor angiogenesis can halt tumor development, growth, and metastasis to other organs.94 A series of benzofuran derivatives were synthesized and evaluated against cancer proliferation. One of these derivatives, exhibited good inhibitory activity and remarkable selectivity to HUVEC. Hence, it represented a promising structural core to discover a new class of active and selective angiogenesis inhibitors.953.3. PIM-1 inhibitors
Several carcinogenic processes have been linked to Pim-1, Pim-2, and Pim-3, which are all members of the same family of serine/threonine kinases.96 The Pim-1 proto-oncogene was first discovered to be a common integration site for slowly converting Maloney murine leukemia virus in murine T cell lymphomas.97 Human lymphomas and acute leukemias with Pim-1 overexpression are found throughout a broad spectrum of disease types.98 A series of benzofuran derivatives were reported to have potent inhibition against Pim-1 and Pim-2 in enzyme assays. Xiang et al. 2011 designed new, structurally diverse and more potent benzofuran-based derivatives for the active treatment of prostate cancer and other related diseases caused by deregulation of Pim-1 kinase.993.4. Anti-estrogen breast cancer agents
Breast cancer accounts for 23% of all cancer diagnoses in women and 14% of all cancer deaths, making it the most common cancer in women and the second largest cause of cancer deaths among women (after lung cancer). Treatments for breast cancer often focus on blocking the effects of estrogen by inhibiting aromatase or blocking the ERα. Targeting the ERα subtype, which is overrepresented in breast cancer cells, is crucial. Antagonists for ERα work by blocking the receptor's active site, preventing oestrogen from binding, and blocking the hormone's effect. Estrogen binds to ER at its active site, causing conformational changes that open the complex to attachment of co-activators in breast cancer cells. Therefore, a number of actions were activated, leading to an increase in breast cell proliferation.100 Leow et al. 2013 identified important functional groups present on 5,6-dihydroxybenzofuran, which could be a promising scaffold for designing novel ER ligands chemotherapeutic anticancer.1013.5. Tubulin polymerization inhibitors
Eukaryotic cells' microtubule system is crucial for controlling cell structure. Due to their critical role in cell division, microtubules are a crucial part of the mitotic spindle.102 Since microtubules play a crucial role in mitosis and cell division, they could be a good drug target for treating cancer. As inhibitors of tubulin polymerization, small compounds like benzofurans are appealing. Dihydrobenzofuran ligands constitute a new group of antimitotic and potential antitumor agents that inhibit tubulin polymerization.1033.6. Glycogen synthase kinase 3β inhibitors
Cell cycle regulation, proliferation, death, signaling, and transcription are just some of the many cellular activities that GSK-3 (glycogen synthase kinase) can affect through phosphorylation of a wide number of substrates. In mammals, GSK-3 consists of two distinct isoforms, α and β.104 Research in recent years has shown that glycogen synthase kinase 3 (GSK-3) is overexpressed in human colon and pancreatic carcinomas, and that this contributes to cancer cell proliferation and survival. Eldehna et al. 2020 described and synthesized benzofuran hybrids as dual CDK2/GSK-3β inhibitors targeting breast cancer.1053.7. Multidrug resistance-reversing activity
Both in antimicrobial therapy and in the treatment of cancer, the emergence of resistance to several drugs is a major concern. Several mechanisms of pleiotropic drug resistance in tumour cells have been uncovered in the past decade. P-glycoprotein is an energy-dependent, membrane-bound efflux pump implicated as a mediator of multidrug resistance (MDR). PGP is an ATP binding cassette 3 member with limited substrate specificity.106 A series of benzofuryl-ethanolamine analogs of propafenone have been prepared and evaluated for multidrug resistance-reversing activity in two in vitro assays.1073.8. Inhibitors of human peptide deformylase (HsPDF)
Actinodin, actinodin analogues, or targeted siRNA suppression of expression of human peptide deformylase (HsPDF) has been linked to an antiproliferative effect in cancer cells.108 HsPDF inhibitors are a promising new class of cancer drugs. Series of benzofuran-4,5-diones derivatives were synthesized as selective inhibitors of human peptide deformylase (HsPDF) as a new class of antitumor agents.1093.9. Inhibitors of mTOR signaling
Cell proliferation, metabolism, autophagy, and angiogenesis are all regulated by the mammalian Target of Rapamycin (mTOR).110 Therefore, mTOR is a significant target in oncology since its signaling is commonly changed in cancer cells. Salomé et al. 2014 designed novel benzofuran derivatives as inhibitors of mTOR pathway against a panel of human cancer cells.111,1124. Conclusions
The occurrence of benzofuran scaffold in many highly bioactive natural products and synthetic compounds is of a great value for researchers working in the area of drug design, discovery and development. The benzofuran nucleus is a key structural motif for a wide number of approved anticancer drugs that are currently in the market. The mechanistic study proved that some benzofuran derivatives caused significant apoptosis in leukemia HL-60 cell line at 6 μM (63.5%) in comparison with control cells (2.6%), without significant side effects. The in vivo anticancer activity of several benzofuran derivatives was tested at different doses by the intraperitoneal route, where some benzofurans significantly inhibited lung tumor growth and decreased the average weight and volume without significant side effects. Based on the aforementioned findings; benzofuran derivatives have better anticancer activity than several anticancer reference drugs against panels of more than 40 human cancer cell lines (with IC50 values in nanomolar scales at low concentrations). Benzofuran scaffolds are highly promising candidates for further development as anticancer agents. Finally, the anticancer efficacies were affected by the electronic nature of the substituents on the benzofuran skeleton where studying the SAR of benzofuran derivatives was reported, in several examples, to enable designing and developing novel, potent and safe anticancer therapeutics. Therefore, much research efforts are still required to be focused on studying the substituents effects that highly enhance the drug-like properties of the benzofuran scaffolds to reach the outmost challenges and prominent benefits to improve human health and reduce suffering.Author contributions
Both authors (A. A. A. and K. M. D.) collected the material, wrote, revised and prepared the review for publication.Conflicts of interest
There are no conflicts to declare.References
- B. Gao, B. Yang, X. Feng and C. Li, Nat. Prod. Rep., 2022, 39, 139–162 RSC.
- L. Cossy and A. Guerinot, Adv. Heterocycl. Chem., 2016, 119, 107–142 CrossRef.
- K. Chand, A. Hiremathad, M. Singh, M. A. Santos and R. S. Keri, Pharmacol. Rep., 2017, 69, 281–295 CrossRef CAS.
- Y. H. Miao, Y. H. Hu, J. Yang, T. Liu, J. Sun and X. J. Wang, RSC Adv., 2019, 9, 27510–27540 RSC.
- A. Radadiya and A. Shah, Eur. J. Med. Chem., 2015, 97, 356–376 CrossRef CAS PubMed.
- R. J. Nevagi, S. N. Dighe and S. N. Dighe, Eur. J. Med. Chem., 2015, 97, 561–581 CrossRef CAS PubMed.
- G. S. Hassan, H. H. Georgey and R. F. George, Bull. Fac. Pharm. Cairo Univ., 2018, 56, 121–127 Search PubMed.
- S. Demirayak, L. Yurttas, N. Gundogdu-Karaburun, A. C. Karaburun and I. Kayagil, J. Enzyme Inhib. Med. Chem., 2015, 30, 816–825 CrossRef CAS PubMed.
- X. Zheng, H. Wang, Y. M. Liu, X. Yao, M. Tong, Y. H. Wang and D. F. Liao, J. Heterocycl. Chem., 2015, 52, 296–301 CrossRef CAS.
- A. Alsayari, A. B. Muhsinah, M. Z. Hassan, M. J. Ahsan, J. A. Alshehri and N. Begum, Eur. J. Med. Chem., 2019, 166, 417–431 CrossRef CAS PubMed.
- L. Ma, L. Tang and Q. Yi, Front. Pharmacol., 2019, 10, 97 CrossRef CAS PubMed.
- T. Qin, A. Rasul, A. Sarfraz, I. Sarfraz, G. Hussain, H. Anwar, A. Riaz, S. Liu, W. Wei, J. Li and X. Li, Int. J. Biol. Sci., 2019, 15, 2256–2264 CrossRef CAS PubMed.
- W. Sha, Y. Zhou, Z. Q. Ling, G. Xie, X. Pang, P. Wang and X. Gu, Oncotarget, 2018, 9, 36331–36343 CrossRef PubMed.
- J. J. La Clair, A. L. Rheingold and M. D. Burkart, J. Nat. Prod., 2011, 74, 2045–2051 CrossRef CAS.
- Y. Li, L. Bao, B. Song, J. Han, H. Li, F. Zhao and H. Liu, Food Chem., 2013, 141, 1614–1618 CrossRef CAS PubMed.
- B. S. Yun, H. C. Kang, H. Koshino, S. H. Yu and I. D. Yoo, J. Nat. Prod., 2001, 64, 1230–1231 CrossRef CAS PubMed.
- Y. Yang, T. Gong, C. Liu and R. Y. Chen, Chem. Pharm. Bull., 2010, 58, 257–260 CrossRef CAS PubMed.
- (a) R. Naik, D. S. Harmalkar, X. Xu, K. Jang and K. Lee, Eur. J. Med. Chem., 2015, 90, 379–393 CrossRef CAS PubMed; (b) X. L. Wang, X. X. Di, T. Shen, S. Q. Wang and X. N. Wang, Chin. Chem. Lett., 2017, 28, 37–40 CrossRef CAS; (c) Z. Wang, Y. Wang, B. Wang, W. Li, L. Huang and X. Li, J. Med. Chem., 2015, 58, 8616–8637 CrossRef CAS PubMed.
- T. Promchai, P. Janhom, W. Maneerat, R. Rattanajak, S. Kamchonwongpaisan, S. G. Pyne and T. Limtharakul, Nat. Prod. Res., 2020, 34, 1394–1398 CrossRef CAS.
- E. W. C Chan, S. K. Wong, J. Tangah, T. Inone and H. T. Chan, J. Integr. Med., 2020, 18, 189–195 CrossRef.
- C. Benz, Novel pathways in the etiology of cancer, US Pat., US20060024691A1, 2006 Search PubMed.
- S. Santagata, M. L. Mendillo, Y. C. Tang, A. Subramanian, C. C. Perley, S. P. Roche, B. Wong, R. Narayan, H. Kwon, M. Koeva and A. Amon, Science, 2013, 341, 1238303 CrossRef PubMed.
- W. Li, Z. H. Yang, A. X. Hu and X. W. Yan, Chem. Biol. Drug Des., 2015, 86, 1339–1350 CrossRef CAS PubMed.
- X. L. Xu, Y. R. Yang, X. F. Mo, J. L. Wei, X. J. Zhan and Q. D. You, Eur. J. Med. Chem., 2017, 137, 45–62 CrossRef CAS PubMed.
- K. M. Dawood, Expert. Opin. Therap. Pat., 2013, 23, 1133–1156 CrossRef CAS.
- K. M. Dawood, Expert. Opin. Therap. Pat., 2019, 29, 841–870 CrossRef CAS PubMed.
- F. M. Thabet, K. M. Dawood, E. M. Ragab, M. S. Nafie and A. A. Abbas, Design and synthesis of new bis(1,2,4-triazolo[3,4-b][1,3,4]thiadiazines) and bis(quinoxalin-2-ylphenoxy)alkanes as anti-breast cancer agents through dual PARP-1 and EGFR target inhibition, RSC Adv., 2022, 12, 23644–23660 RSC.
- H. Behbehani, F. Aryan, K. M. Dawood and H. Ibrahim, Sci. Rep., 2020, 10, 21691 CrossRef CAS PubMed.
- A. A. Abbas and K. M. Dawood, Expert. Opin. Drug Discov., 2022, 17, 1357–1376 CrossRef CAS PubMed.
- Y. Chen, G. Wang, Y. Yuan, G. Zou, W. Yang, Q. Tan, W. Kang and Z. She, Front. Chem., 2022, 10, 842405 CrossRef CAS PubMed.
- T. Q. Lang, G. Y. Luo, W. C. Pu, Z. W. Wang, J. Wang, X. L. Tian, P. Zhang, N. W. Zhao, W. D. Yang and H. F. Chai, Nat. Prod. Res., 2022, 36, 1205–1214 CrossRef CAS PubMed.
- X. Fan, H. He, J. Li, G. Luo, Y. Zheng, J. K. Zhou, J. He, W. Pu and Y. Zhao, Bioorg. Med. Chem., 2019, 27, 2235–2244 CrossRef CAS PubMed.
- T. H. Tseng, C. J. Wang, Y. J. Lee, Y. C. Shao, C. H. Shen, K. C. Lee, S. Y. Tung and H. C. Kuo, Internat. J. Mol. Sci., 2022, 23, 5102 CrossRef CAS PubMed.
- A. Sivaraman, J. S. Kim, D. S. Harmalkar, K. H. Min, J. W. Park, Y. Choi, K. Kim and K. Lee, J. Nat. Prod., 2020, 83, 3354–3362 CrossRef CAS PubMed.
- C. Gao, X. Sun, Z. Wu, H. Yuan, H. Han, H. Huang, Y. Shu, M. Xu, R. Gao, S. Li and J. Zhang, Front. Pharmacol., 2020, 11, 391 CrossRef CAS PubMed.
- H. Cao, R. Ma, S. Chu, J. Xi, L. Yu and R. Guo, Chin. Chem. Lett., 2021, 32, 2761–2764 CrossRef CAS.
- M. Liu, X. Zhang, S. Chu, Y. Ge, T. Huang, Y. Liu and L. Yu, Chin. Chem. Lett., 2022, 33, 205–208 CrossRef CAS.
- T. O. Olomola, M. J. Mphahlele and S. Gildenhuys, Bioorg. Chem., 2020, 100, 103945 CrossRef CAS PubMed.
- L. P. Jin, Q. Xie, E. F. Huang, L. Wang, B. Q. Zhang, J. S. Hu, D. C. C. Wan, Z. Jin and C. Hu, Bioorg. Chem., 2020, 95, 103566 CrossRef CAS PubMed.
- Y. Gao, C. Ma, X. Feng, Y. Liu and X. Haimiti, Chem. Biodivers., 2020, 17, e1900622 CrossRef CAS PubMed.
- R. Dharavath, M. Sarasija, M. Ram Reddy, K. Naga Prathima, N. Nagarju, K. Ramakrishna, D. Ashok and S. Daravath, Med. Chem. Res., 2022, 31, 993–1002 CrossRef CAS.
- S. Mokenapelli, G. Thalari, N. Vadiyaala, J. R. Yerrabelli, V. K. Irlapati, N. Gorityala, S. R. Sagurthi and P. R. Chitneni, Arch. Pharm., 2020, 353, 2000006 CrossRef CAS.
- M. M. Al-Sanea, G. H. Al-Ansary, Z. M. Elsayed, R. M. Maklad, E. B. Elkaeed, M. A. Abdelgawad, S. N. A. Bukhari, M. M. Abdel-Aziz, H. Suliman and W. M. Eldehna, J. Enzyme Inhib. Med. Chem., 2021, 36, 987–999 CrossRef CAS PubMed.
- X. He, Y. Gao, Z. Hui, G. Shen, S. Wang, T. Xie and X. Y. Ye, Bioorg. Med. Chem. Lett., 2020, 30, 127109 CrossRef CAS PubMed.
- B. Y. Kim, N. K. Soung and M. Ahn, PCT WO2021010731A1, 2021.
- W. M. Eldehna, S. T. Al-Rashood, T. Al-Warhi, R. O. Eskandrani, A. Alharbi and A. M. El Kerdawy, J. Enzyme Inhib. Med. Chem., 2021, 36, 270–285 CAS.
- W. M. Eldehna, R. Salem, Z. M. Elsayed, T. Al-Warhi, H. R. Knany, R. R. Ayyad, T. B. Traiki, M. H. Abdulla, R. Ahmad and H. A. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2021, 36, 1423–1434 CrossRef CAS PubMed.
- S. Fares, K. B. Selim, F. E. Goda, M. A. El-Sayed, N. A. AlSaif, M. M. Hefnawy, A. A. M. Abdel-Aziz and A. S. El-Azab, J. Enzyme Inhib. Med. Chem., 2021, 36, 1488–1499 Search PubMed.
- F. Wang, K. R. Feng, J. Y. Zhao, J. W. Zhang, X. W. Shi, J. Zhou, D. Gao, G. Q. Lin and P. Tian, Bioorg. Med. Chem., 2020, 28, 115822 CrossRef CAS PubMed.
- Q. Li, X. E. Jian, Z. R. Chen, L. Chen, X. S. Huo, Z. H. Li, W. W. You, J. J. Rao and P. L. Zhao, Bioorg. Chem., 2020, 102, 104076 CrossRef CAS PubMed.
- N. F. El-Sayed, M. El-Hussieny, E. F. Ewies, M. F. El Shehry, H. M. Awad and M. A. Fouad, Drug Develop. Res., 2022, 83, 485–500 CrossRef CAS PubMed.
- O. A. El-Khouly, M. A. Henen, A. A. Magda, M. I. Shabaan and S. M. El-Messery, Bioorg. Med. Chem., 2021, 31, 115976 CrossRef CAS PubMed.
- S. N. A. Bukhari, RSC Adv., 2022, 12, 10307–10320 RSC.
- M. A. Abdelrahman, W. M. Eldehna, A. Nocentini, H. S. Ibrahim, H. Almahli, H. A. Abdel-Aziz, S. M. Abou-Seri and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2020, 35, 298–305 CrossRef CAS.
- X. Zhang, H. Huang, Z. Zhang, J. Yan, T. Wu, W. Yin, Y. Sun, X. Wang, Y. Gu, D. Zhao and M. Cheng, Eur. J. Med. Chem., 2021, 220, 113501 CrossRef CAS PubMed.
- H. Gao, X. Zhang, X. J. Pu, X. Zheng, B. Liu, G. X. Rao, C. P. Wan and Z. W. Mao, Bioorg. Med. Chem. Lett., 2019, 29, 806–810 CrossRef CAS PubMed.
- A. Hu, J. He, D. Lin, X. Wei and J. Ye, CN110950825A, 2020.
- A. Carella, V. Roviello, R. Iannitti, R. Palumbo, S. La Manna, D. Marasco, M. Trifuoggi, R. Diana and G. N. Roviello, Internat. J. Biol. Macromol., 2019, 121, 77–88 CrossRef PubMed.
- P. Oliva, R. Romagnoli, S. Manfredini, A. Brancale, S. Ferla, E. Hamel, R. Ronca, F. Maccarinelli, A. Giacomini, F. Rruga and E. Mariotto, Eur. J. Med. Chem., 2020, 200, 112448 CrossRef CAS PubMed.
- W. M. Eldehna, A. Nocentini, Z. M. Elsayed, T. Al-Warhi, N. Aljaeed, O. J. Alotaibi, M. M. Al-Sanea, H. A. Abdel-Aziz and C. T. Supuran, ACS Med. Chem. Lett., 2020, 11, 1022–1027 CrossRef CAS PubMed.
- M. Shaldam, W. M. Eldehna, A. Nocentini, Z. M. Elsayed, T. M. Ibrahim, R. Salem, R. A. El-Domany, C. Capasso, H. A. Abdel-Aziz and C. T. Supuran, Eur. J. Med. Chem., 2021, 216, 113283 CrossRef CAS PubMed.
- Z. Y. Qi, S. Y. Hao, H. Z. Tian, H. L. Bian, L. Hui and S. W. Chen, Bioorg. Chem., 2020, 94, 103392 CrossRef CAS PubMed.
- B. G. Youssif, A. M. Mohamed, E. E. A. Osman, O. F. Abou-Ghadir, D. H. Elnaggar, M. H. Abdelrahman, L. Treamblu and H. A. Gomaa, Eur. J. Med. Chem., 2019, 177, 1–11 CrossRef CAS.
- Y. Matiichuk, Y. Ostapiuk, T. Chaban, M. Sulyma, N. Sukhodolska and V. Matiychuk, Biointerface Res. Appl. Chem., 2020, 10, 6597–6609 CAS.
- U. Farooq, S. Naz, A. Shams, Y. Raza, A. Ahmed, U. Rashid and A. Sadiq, Biol. Res., 2019, 52, 1–12 CrossRef PubMed.
- A. H. Liu, L. H. Liu and C. Y. Liu, Nat. Prod. Commun., 2020, 15, 1–4 Search PubMed.
- G. Zheng, A. Kadir, X. Zheng, P. Jin, J. Liu, M. Maiwulanjiang, G. Yao and H. A. Aisa, Org. Chem. Front., 2020, 7, 3137–3145 RSC.
- A. M. Zaher, R. Sultan, T. Ramadan and A. Amro, Nat. Prod. Res., 2021, 36, 136–141 CrossRef PubMed.
- K. Kumar, J. P. Mishra and R. P. Singh, Chem. Biol. Interact., 2020, 315, 108898 CrossRef CAS PubMed.
- M. Kumari, S. Kamat and C. Jayabaskaran, Spectrochim. Acta, Part A, 2022, 274, 121098 CrossRef CAS PubMed.
- A. Garg, S. Garg, N. K. Sahu, S. Rani, U. Gupta and A. K. Yadav, Internat. J. Pharm., 2019, 557, 238–253 CrossRef CAS.
- S. Wang, J. Zang, M. Huang, L. Guan, K. Xing, J. Zhang, D. Liu and L. Zhao, Bioorg. Chem., 2019, 89, 102971 CrossRef CAS PubMed.
- A. S. Filimonov, A. A. Chepanova, O. A. Luzina, A. L. Zakharenko, O. D. Zakharova, E. S. Ilina, N. S. Dyrkheeva, M. S. Kuprushkin, A. V. Kolotaev, D. S. Khachatryan and J. Patel, Molecules, 2019, 24, 3711 CrossRef CAS PubMed.
- A. Pyrczak-Felczykowska, T. A. Reekie, M. Jąkalski, A. Hać, M. Malinowska, A. Pawlik, K. Ryś, B. Guzow-Krzemińska and A. Herman-Antosiewicz, Internat. J. Mol. Sci., 2022, 23, 1802 CrossRef CAS PubMed.
- W. Zhang, J. Chu, A. M. Cyr, H. Yueh, L. E. Brown, T. T. Wang, J. Pelletier and J. A. Porco Jr, J. Am. Chem. Soc., 2019, 141, 12891–12900 CrossRef CAS PubMed.
- G. Schulz, C. Victoria, A. Kirschning and E. Steinmann, Nat. Prod. Rep., 2021, 38, 18–23 RSC.
- X. Yan, C. Yao, C. Fang, M. Han, C. Gong, D. Hu, W. Shen, L. Wang, S. Li and S. Zhu, Internat. J. Biol. Sci., 2022, 18, 585 CrossRef CAS PubMed.
- C. Pandit and K. M. Kapadiya, Folia Med., 2019, 61, 551–558 CrossRef CAS PubMed.
- S. K. Siddiqui, V. J. SahayaSheela, S. Kolluru, G. N. Pandian, T. R. Santhoshkumar, V. M. Dan and C. V. Ramana, Bioorg. Med. Chem. Lett., 2020, 30, 127431 CrossRef CAS.
- Y. Y. Shao, Y. Yin, B. P. Lian, J. F. Leng, Y. Z. Xia and L. Y. Kong, Eur. J. Med. Chem., 2020, 190, 112105 CrossRef.
- O. M. Abdelhafez, E. Y. Ahmed, N. A. A. Latif, R. K. Arafa, Z. Y. Abd Elmageed and H. I. Ali, Bioorg. Med. Chem., 2019, 27, 1308–1319 CrossRef CAS PubMed.
- S. Kuduk and X. Zhang, PCT WO2021240429A1, 2021.
- E. Y. Ahmed, O. M. Abdelhafez, D. Zaafar, A. M. Serry, Y. H. Ahmed, R. F. A. El-Telbany, Z. Y. Abd Elmageed and H. I. Ali, Arch. Pharm., 2022, 355, 2100327 CrossRef CAS PubMed.
- H. A. S. Abbas and S. S. Abd El-Karim, Bioorg. Chem., 2019, 89, 103035 CrossRef CAS PubMed.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed and M. S. Nafie, Anticancer Agents Med. Chem., 2023, 23, 328–345 CrossRef CAS PubMed.
- G. N. Zhang, L. Y. Zhong, S. A. Bligh, Y. L. Guo, C. F. Zhang, M. Zhang, Z. T. Wang and L. S. Xu, Phytochem, 2005, 66, 1113–1120 CrossRef CAS.
- S. Van Miert, S. Van Dyck, T. J. Schmidt, R. Brun, A. Vlietinck, G. Lemière and L. Pieters, Bioorg. Med. Chem., 2005, 13, 661–669 CrossRef CAS PubMed.
- W. C. Wan, W. Chen, L. X. Liu, Y. Li, L. J. Yang, X. Y. Deng, H. B. Zhang and X. D. Yang, Med. Chem. Res., 2014, 23, 1599–1611 CrossRef CAS.
- H. Khanam, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed.
- R. Bar-Shavit, M. Maoz, A. Kancharla, J. K. Nag, D. Agranovich, S. Grisaru-Granovsky and B. Uziely, Int. J. Mol. Sci., 2016, 17, 1320 CrossRef PubMed.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Steroids, 2019, 152, 108485 CrossRef CAS PubMed.
- K. Asoh, M. Kohchi, I. Hyoudoh, T. Ohtsuka, M. Masubuchi, K. Kawasaki, H. Ebiike, Y. Shiratori, T. A. Fukami, O. Kondoh, T. Tsukaguchi, N. Ishii, Y. Aoki, N. Shimma and M. Sakaitani, Bioorg. Med. Chem. Lett., 2009, 19, 1753–1757 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- D. T. Debela, S. G. Muzazu, K. D. Heraro, M. T. Ndalama, B. W. Mesele, D. C. Haile, S. K. Kitui and T. Manyazewal, SAGE Open Med., 2021, 9, 20503121211034366 Search PubMed.
- Y. Chen, S. Chen, X. Lu, H. Cheng, Y. Ou, H. Cheng and G.-C. Zhou, Bioorg. Med. Chem. Lett., 2009, 19, 1851–1854 CrossRef CAS PubMed.
- N. S. Magnuson, Z. Wang, G. Ding and R. Reeves, Future Oncol., 2010, 6, 1461–1478 CrossRef CAS PubMed.
- H. T. Cuypers, G. Selten, W. Quint, M. Zijlstra, E. R. Maandag, W. Boelens, P. van Wezenbeek, C. Melief and A. Berns, Cell, 1984, 37, 141–150 CrossRef CAS PubMed.
- R. Amson, F. Sigaux, S. Przedborski, G. Flandrin, D. Givol and A. Telerman, Proceed. Nat. Acad. Sci., 1989, 86, 8857–8861 CrossRef CAS PubMed.
- Y. Xiang, B. Hirth, G. Asmussen, H. P. Biemann, K. A. Bishop, A. Good, M. Fitzgerald, T. Gladysheva, A. Jain, K. Jancsics, J. Liu, M. Metz, A. Papoulis, R. Skerlj, J. D. Stepp and R. R. Wei, Bioorg. Med. Chem. Lett., 2011, 21, 3050–3056 CrossRef CAS PubMed.
- J. Russo and I. H. Russo, J. Steroid Biochem. Mol. Biol., 2006, 102, 89–96 CrossRef CAS PubMed.
- M. L. Leow, H. L. C. Chin, P. S. Yu, K. K. Pasunooti, R. X. Z. Tay, D. Zhang, H. S. Yoon and X. W. Liu, Curr. Med. Chem., 2013, 20, 2820–2837 CrossRef CAS PubMed.
- S. Honore, E. Pasquier and D. Braguer, Cell. Mol. Life Sci., 2005, 62, 3039–3056 CrossRef CAS PubMed.
- L. Pieters, S. Van Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemière, J. Med. Chem., 1999, 42, 5475–5481 CrossRef CAS PubMed.
- J. R. Woodgett, EMBO J., 1990, 9, 2431–2438 CrossRef CAS PubMed.
- W. M. Eldehna, S. T. Al-Rashood, T. Al-Warhi, R. O. Eskandrani, A. Alharbi and A. M. El Kerdawy, J. Enzyme. Inhib. Med. Chem., 2021, 36, 271–286 CrossRef PubMed.
- R. L. Juliano and V. Ling, Biochim. Biophys. Acta, Biomembr., 1976, 455, 152–162 CrossRef CAS PubMed.
- G. Ecker, P. Chiba, M. Hitzler, D. Schmid, K. Visser, H. P. Cordes, J. Csöllei, J. K. Seydel and K. J. Schaper, J. Med. Chem., 1996, 39, 4767–4774 CrossRef CAS PubMed.
- M. D. Lee, Y. She, M. J. Soskis, C. P. Borella, J. R. Gardner, P. A. Hayes, B. M. Dy, M. L. Heaney, M. R. Philips, W. G. Bornmann and F. M. Sirotnak, J. Clin. Invest., 2004, 114, 1107–1116 CrossRef CAS PubMed.
- C. Antczak, D. Shum, B. Bassit, M. G. Frattini, Y. Li, E. de Stanchina, D. A. Scheinberg and H. Djaballah, Bioorg. Med. Chem. Lett., 2011, 21, 4528–4532 CrossRef CAS PubMed.
- M. E. Welker and G. Kulik, Bioorg. Med. Chem., 2013, 21, 4063–4091 CrossRef CAS PubMed.
- C. Salomé, V. Narbonne, N. Ribeiro, F. Thuaud, M. Serova, A. de Gramont, S. Faivre, E. Raymond and L. Désaubry, Eur. J. Med. Chem., 2014, 74, 41–49 CrossRef PubMed.
- C. Salomé, N. Ribeiro, T. Chavagnan, F. Thuaud, M. Serova, A. de Gramont, S. Faivre, E. Raymond and L. Désaubry, Eur. J. Med. Chem., 2014, 81, 181–191 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |