DOI:
10.1039/D3RA01364B
(Review Article)
RSC Adv., 2023,
13, 10958-10986
Visible-light acridinium-based organophotoredox catalysis in late-stage synthetic applications
Received
28th February 2023
, Accepted 29th March 2023
First published on 6th April 2023
Abstract
The field of photoredox catalysis has been transformed by the use of organic photocatalysts, which give access to re-activities that were previously only possible with transition-metal photocatalysts. Recent advancements in the use of an acridinium photocatalyst in organic synthesis are covered in this review. Both the late-stage functionalization of biorelevant molecules and the activation of inert chemical bonds are explored, with an emphasis on their mechanistic features.
 Praveen P. Singh | Praveen P. Singh is working as Assistant Professor in the Department of Chemistry at the United College of Engineering and Research, Prayagraj, India. He obtained his BSc, MSc in Organic Chemistry from T. D. P. G. College (V. B. S Purvanchal University) Jaunpur and DPhil. from Department of Chemistry, University of Allahabad, India. His current research interests include the development of synthetic receptors for the recognition of biological target structures and the application of visible light chemical photo-catalysis towards organic synthesis as well as nanophotocatalysis. |
 Jaya Singh | Jaya Singh is an Assistant Professor in the Department of Chemistry, LRPG College, Sahibabad, Ghaziabad, Uttar Pradesh, India. She has done her graduation and post-graduation from the University of Allahabad. She also obtained her DPhil. Degree from the University of Allahabad, India. She was the topper in BSc and gold medalist in MSc (Chemistry). She has over 17 years of teaching and research experience in varied fields. Her field of research is synthetic organic chemistry and green chemistry. |
 Vishal Srivastava | Vishal Srivastava is working as Assistant Professor, Department of Chemistry, C. M. P. College, (Constituent P. G. College of Central University of Allahabad) Prayagraj, India. He has completed BSc, MSc in Organic Chemistry and Doctoral Degree (DPhil.) from Department of Chemistry, University of Allahabad, India. His current research work involves the designing of novel biologically active photoredox catalysed synthesis of organic compounds. |
1. Introduction
Photoredox catalysis has quickly become a platform for the development of synthetic techniques over the past decade as it exhibits a pioneering approach for molecular functionalization1–3 and has made a significant impact on organic chemistry. The importance of light to form chemical bonds under benign conditions has grown significantly, and visible-light photoredox catalysis has attracted huge attention.4–11 A century ago, the visionary scientist Giacomo Ciamcian emphasised the need to develop technology that allowed the conversion of light into chemicals and fuels12 after recognizing the impact of light to initiate redox transformations that use low-energy visible light and, through photoinduced electron transfer (PET), produces highly reactive intermediates under mild operating conditions. This has enabled to the development of a wide range of reactions, including the synthesis of complex compounds.13 Iridium and ruthenium-based photocatalysts are frequently used in many of those photoreactions due to the high redox potentials and the comparatively prolonged lives of their excited states. In fact, the photophysical and electrochemical properties of the photo-catalysts can potentially be changed by a modest modification of the ligand structure. Even though, the commercially available organic dyes like eosin Y14 and flavins15–17 have demonstrated interesting photochemical properties as photocatalysts, variation of their molecular scaffolds is frequently complicated, which inhibits the use of structurally related photocatalysts with different photochemical properties. Recent efforts have concentrated on devising practically simple and modular syntheses to access new and effective organophotocatalysts18,19 in order to close this gap and create entirely organic, less expensive, long-lasting, and environmentally benign photocatalysts.
In this context, ruthenium and iridium catalysts serve as particularly adaptable scaffolds in the field of photoredox catalysis, which has been transformed by polypyridyl transition metal complexes. They are incredibly valuable catalysts for photochemical transformations because of the broad range of characteristics resulting from the tunability of their ligands. With the invention of entirely organic substitutes, opportunities for potentially sustainable approaches emerged. Acridinium salts, developed by Fukuzumi20,21 are one of them and are known to exhibit exceptional photophysical properties that complement those of polypyridyl transition metal complexes. They are excellent photocatalysts for a plethora of preparative transformations owing to their strong reduction potential in the excited state, stability, pH independence, and dissolution rate in a wide range of solvents.
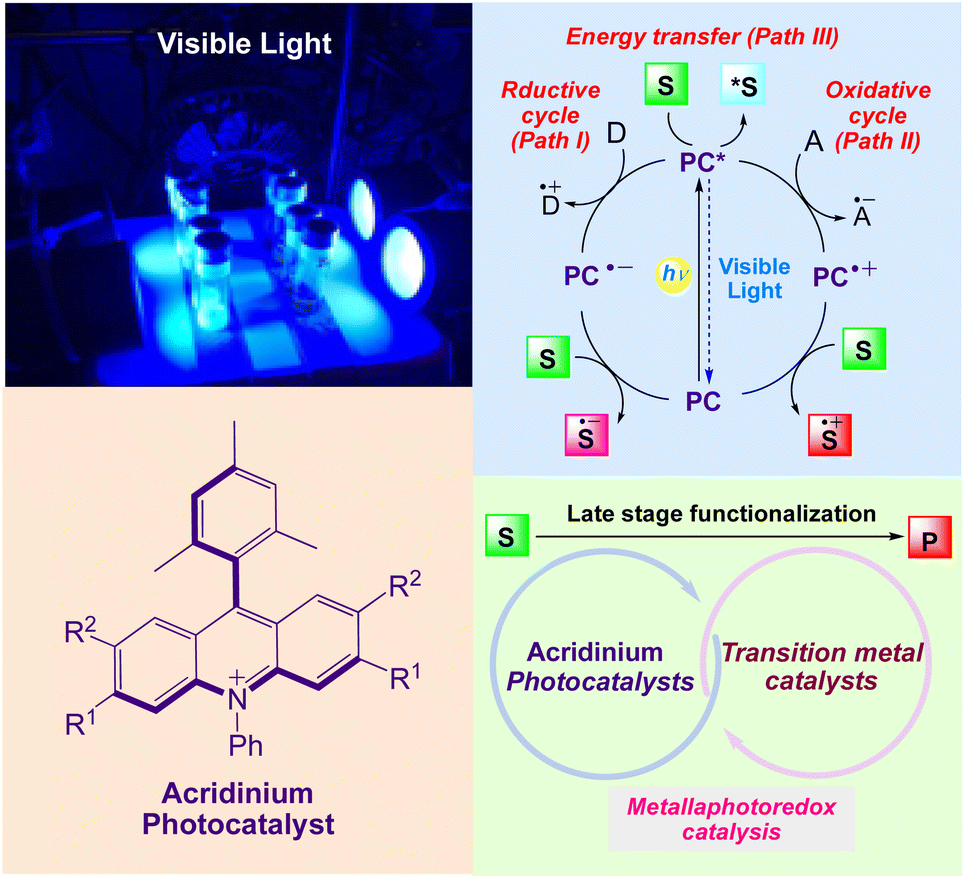
As depicted in (Scheme 1),22–25 upon irradiation, an excited photocatalyst (PC*) can act as an electron shuttle, interacting with sacrificial electron donor D (path I) or acceptors A (path II) to yield either a strongly reducing or oxidizing catalyst toward organic substrates S. PC* can also directly transfer energy to an organic substrate to yield electronically excited species (path III). Depending on the reaction conditions, the inverse events can occur to complete a reductive quenching cycle. Moreover, a PC can also transfer its excited state energy to a substrate or reagent that is not able to absorb light at the given wavelength, thereby inducing a chemical reaction. The combination of photocatalysis with transition metal catalysis generates metallaphotocatalysis,26,27 that enables selective carbon-heteroatom and carbon–carbon cross-coupling reactions under benign conditions. It also significantly affects the synthesis of small molecules and also plays a crucial role in late stage functionalization of organic molecules.
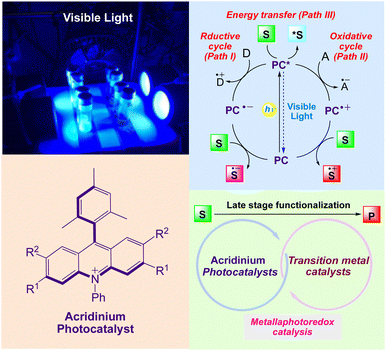 |
| | Scheme 1 General mechanism of visible light induced acridinium photocatalyst for synthesis of organic compounds. | |
Furthermore, recent synthetic studies have demonstrated that due to the distinctive modularity of acridinium salts, the structural changes made by using these catalyst preparation techniques28 can significantly alter the photophysical and electrochemical properties.29 In continuation of our work on photocatalysed organic synthesis30,31 this review aims to provide a comprehensive report on the current research, especially the role of acridinium salts in the organic synthesis and late stage chemical transformations.
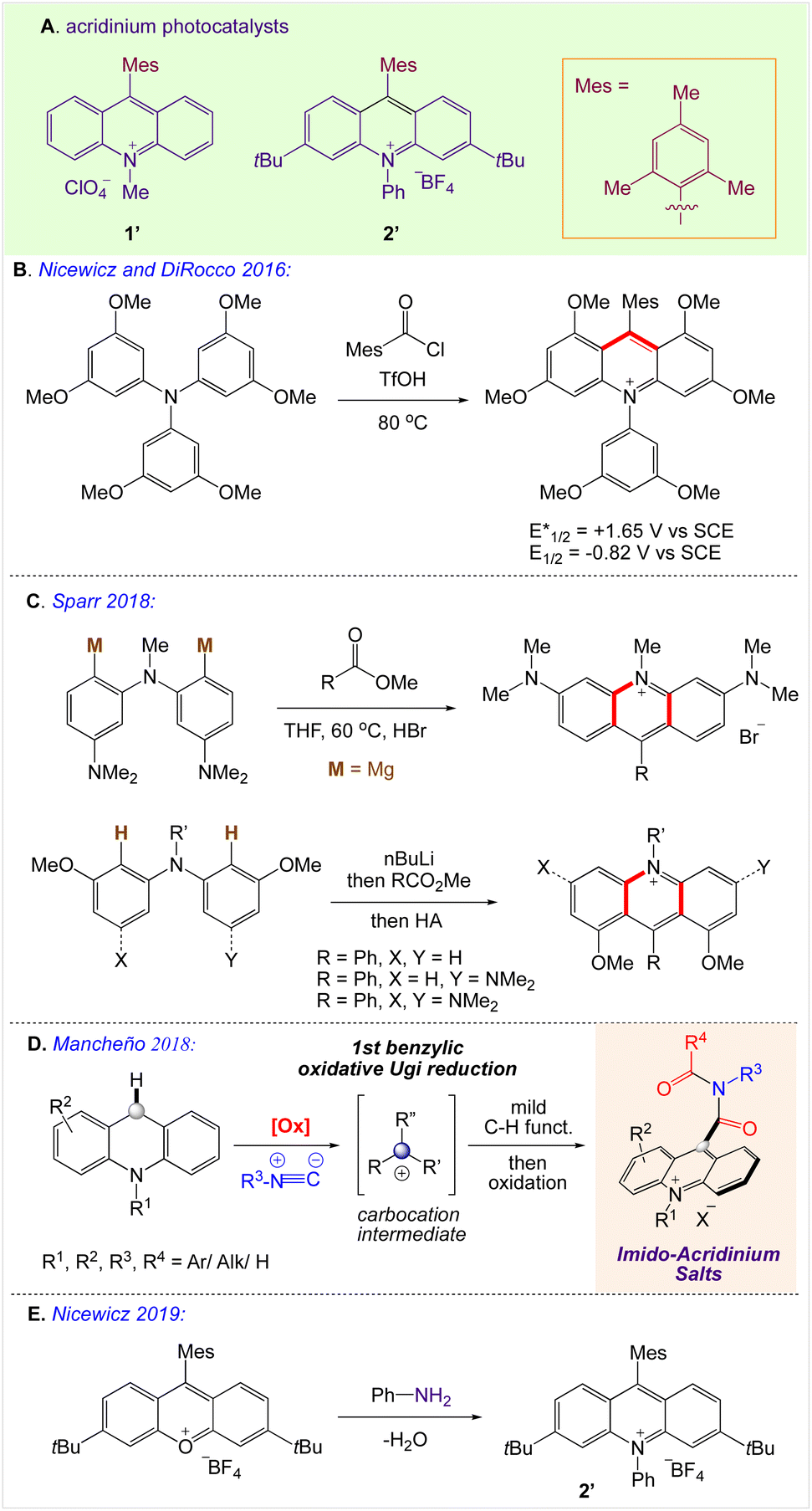
2. Synthesis of acridinium-based photocatalyst
The polypyridyl complexes of ruthenium and iridium are now the leading catalysts for PET-based catalysis because of their substantial excited-state lifetimes and adaptable redox potentials.32–34
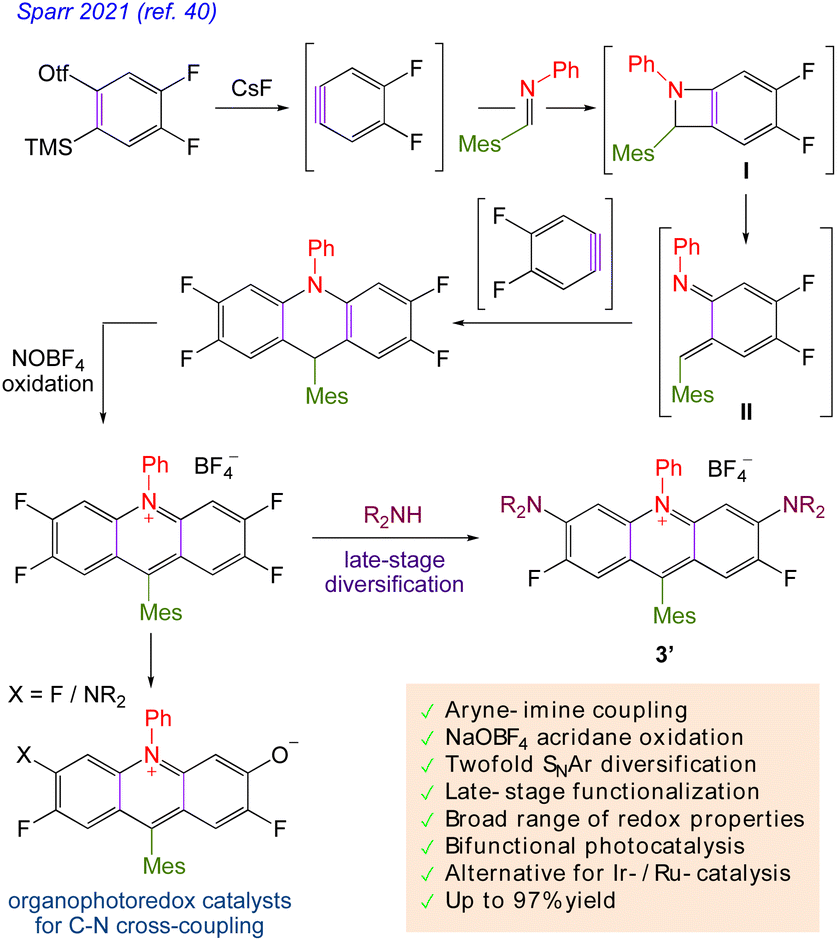
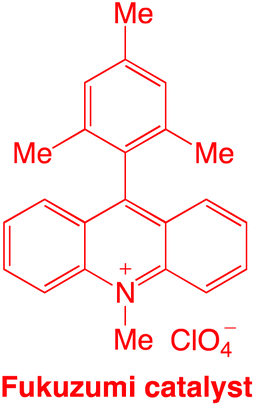
Despite their usefulness, their sustainability is a matter of concern due to their high cost and dependency on precious metals. Organic dyes offer an intriguing substitute, such as the acridinium salt that Fukuzumi (1′, Scheme 2A)35,36 initially introduced and Nicewicz (Scheme 2B)37 popularised as a photoredox catalyst. The stability and photophysical characteristics of acridinium-based photoredox catalysis have improved as a result of structural changes made to the catalyst's core. By adding 1,5-bifunctional nucleophiles to aromatic esters, Sparr and co-workers demonstrated an effective method for producing acridinium salts that are rich in electrons (Scheme 2C).38,39 Mancheño and co-workers developed a synthetic strategy based on a novel straightforward oxidative Ugi-type reaction at the benzylic position of C9-unsubstituted acridanes (Scheme 2D).35a Nicewicz also created a library of unique acridinium salts using this adaptable method and evaluated their photophysical characteristics (Scheme 2E).35b
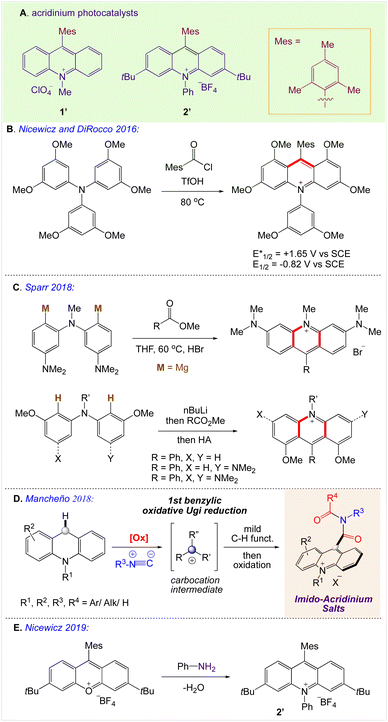 |
| | Scheme 2 Synthesis of acridinium-based photoredox catalysts. | |
3. Late-stage acridinium diversification
Sparr40 in 2021 described an ad-hoc approach for preparing acridinium salts with a particularly wide range of photoredox characteristics. The process involves connecting an aryne–imine aryne to a key component of tetrafluoro acridinium salt for nucleophilic aromatic substitution reactions that result in the formation of diaminoacridinium and unidentified aza-rhodol photo-catalysts during the late stages of diversification. The organic acridinium photocatalysts are suitable for bifunctional photoredox catalysis and organocatalytic photochemical C–N cross-couplings due to their diverse functionalities and redox characteristics (Scheme 3).
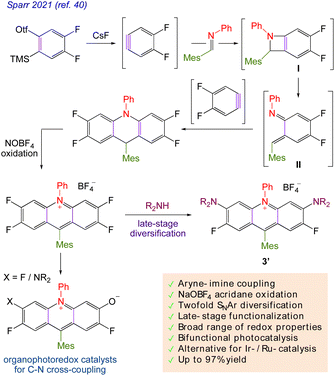 |
| | Scheme 3 Late-stage acridinium diversification. | |
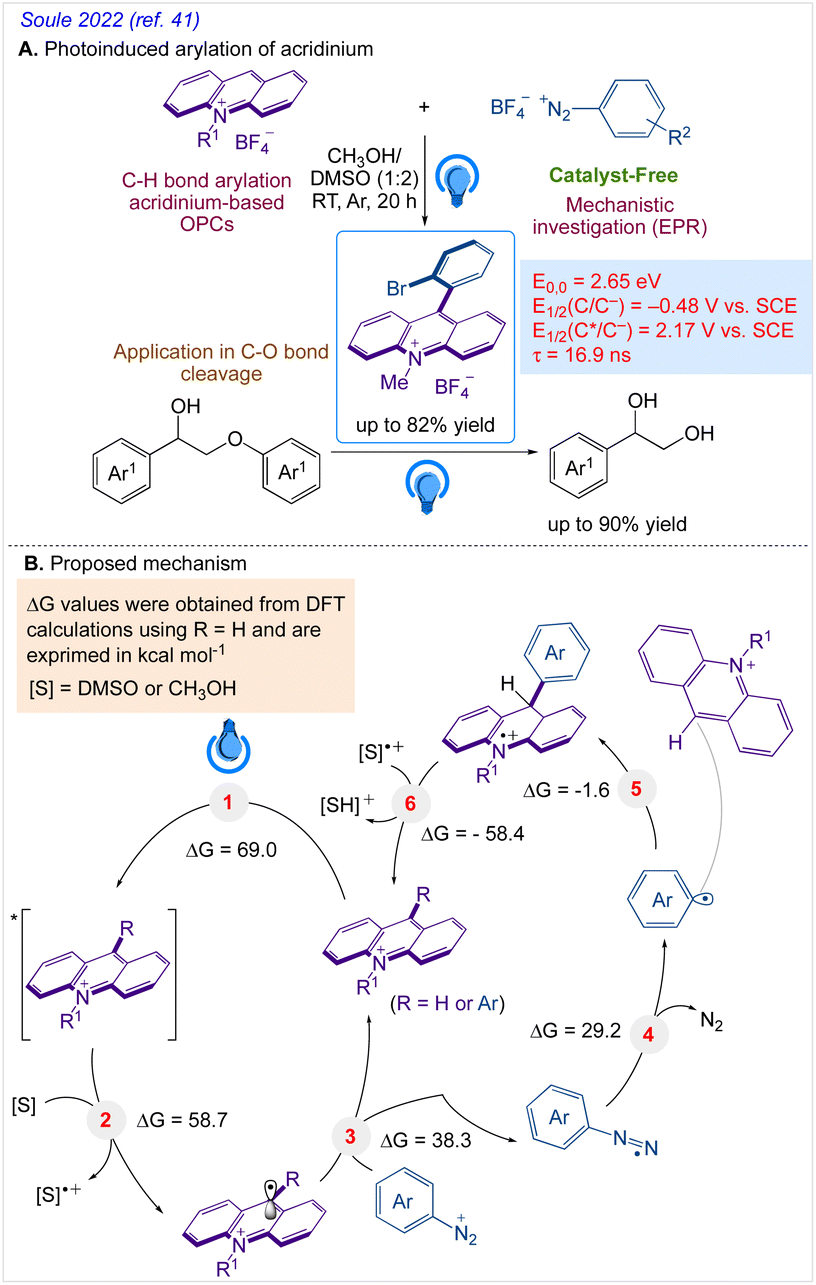
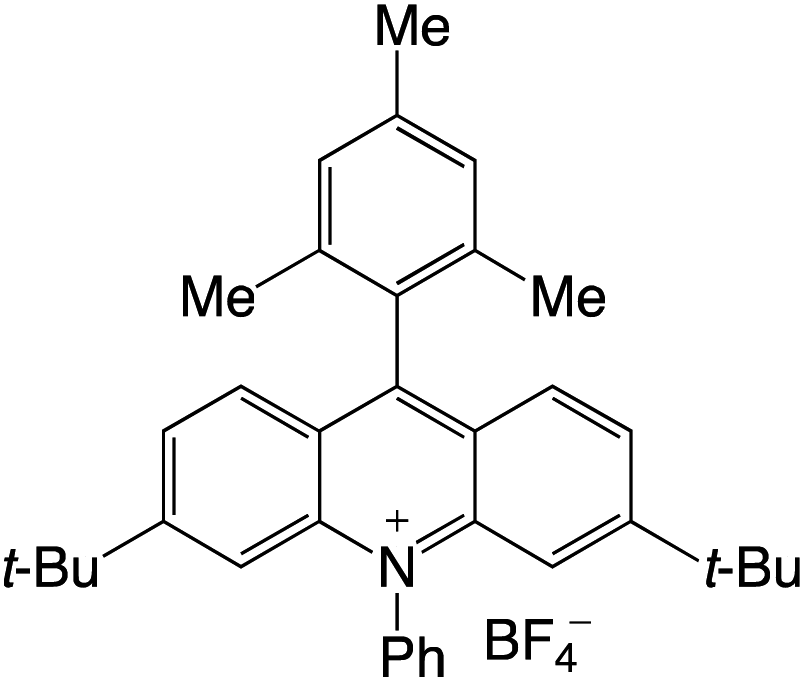
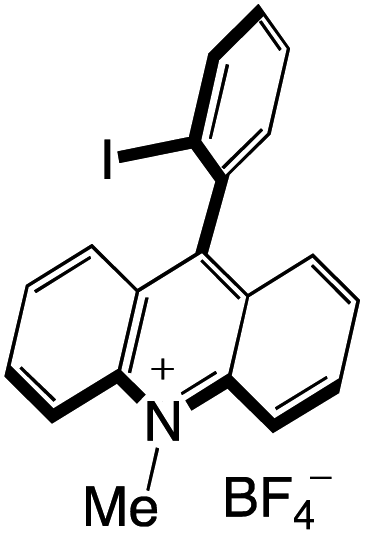
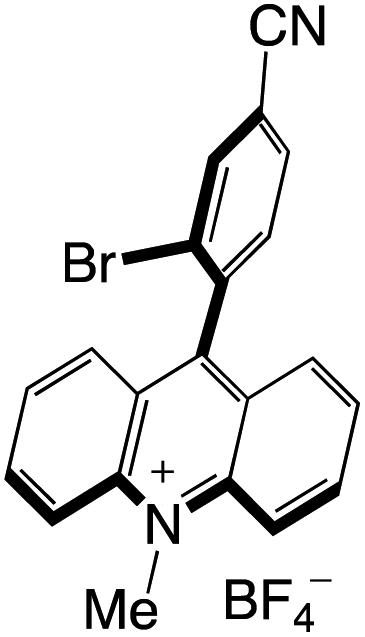
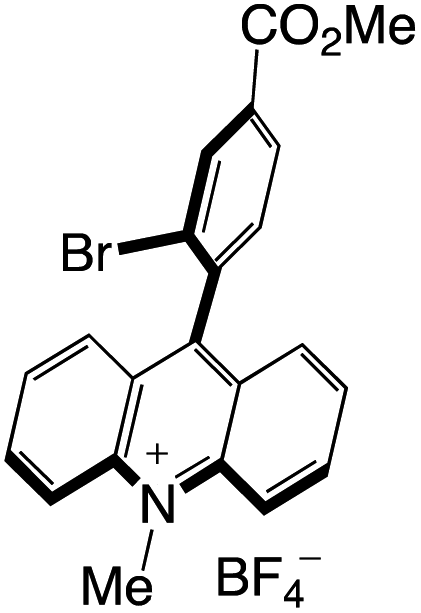
A highly functional group-tolerant (e.g., halogen, nitrile, ketone, ester, and nitro) photoinduced arylation of N-substituted acridinium salts has been devised by Soule and co-workers41 in 2022. A wide variety of C9-arylated acridinium-based catalysts with precisely calibrated excited-state lifetimes and redox potentials have been generated in a single process. Later on, the photoredox-catalyzed fragmentation of 1,2-diol derivatives using these functionalized acridinium salts was assessed (lignin models). For the selective CβO–Ar bond breakage of diol monoarylethers to produce 1,2-diols in good yields, 2-bromophenyl substituted N-methyl acridinium has excelled all photoredox catalysts, including commercial Fukuzumi's catalyst.
A reaction pathway was proposed by Soulé and co-workers41 (Scheme 4). According to them, upon irradiation of MeAcrH·BF4, (step 1) generation of the reduced acridinyl radical (step 2) was achieved. Kano et al.42 had earlier reported a comparable synthesis of an acridinyl radical from photoexcited acridinium molecules. The acridinyl radical then effectively reduced all utilized diazonium salts (step 3). These findings support the formation of an aryl radical (step 4) and thermodynamically advantageous electron transfer [−0.2 V < E1/2(diazonium/aryl radical) < +0.2 V vs. SCE,43 E1/2(acridinium/acridinyl radical)] = −0.54 V vs. SCE.44 The subsequent reaction between the aryl radical and the revived acridinium precursor produced the appropriate acridane species (step 5). Further, DFT calculations supports it's reoxidation into the final product (step 6).
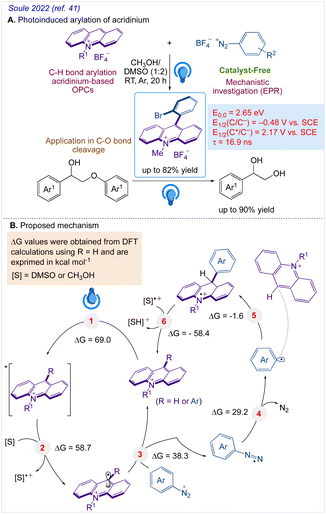 |
| | Scheme 4 (A) Late-stage photoinduced arylation of acridinium salts. (B) Proposed mechanism for the photoinduced C9 arylation of N-methyl acridinium salts with aryl diazonium salts. | |
4. Photophysical properties of acridinium-based photocatalysts
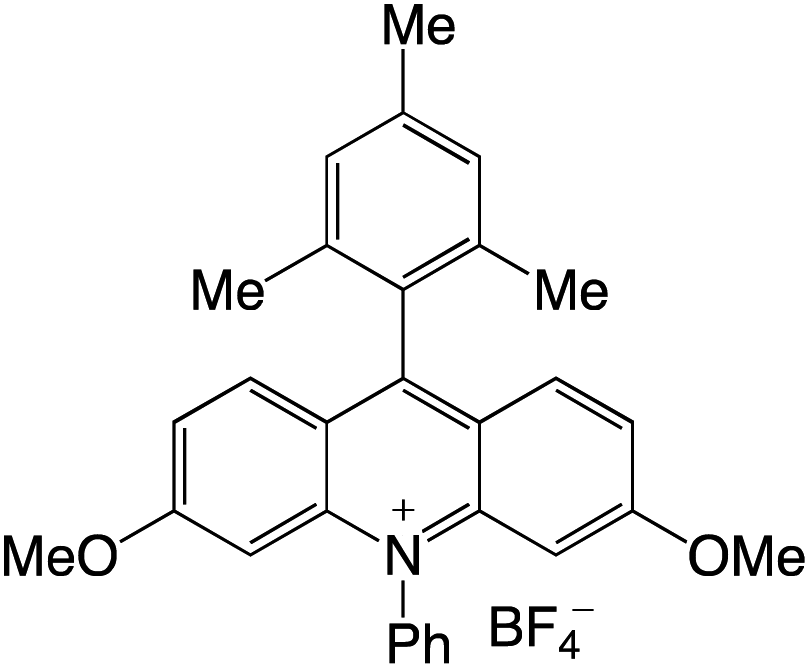
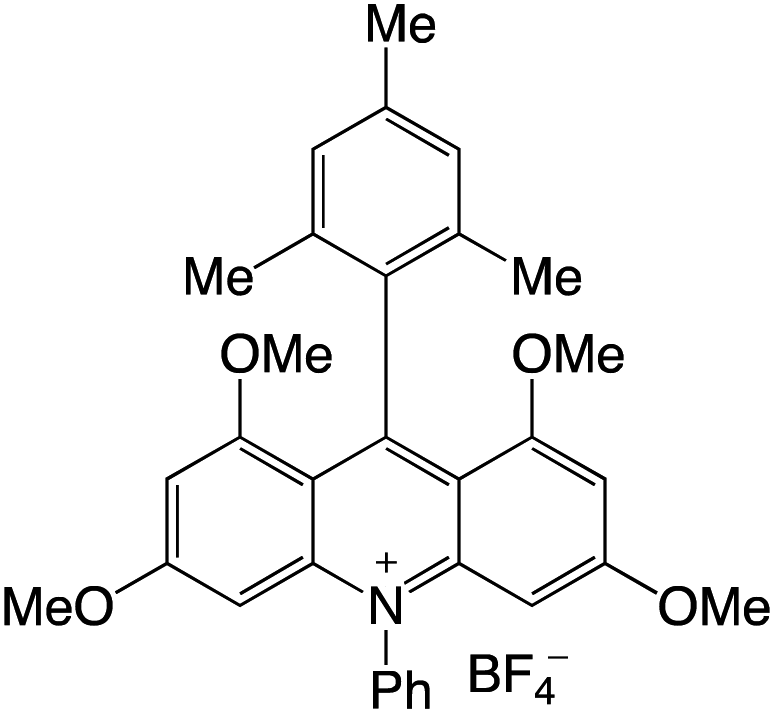
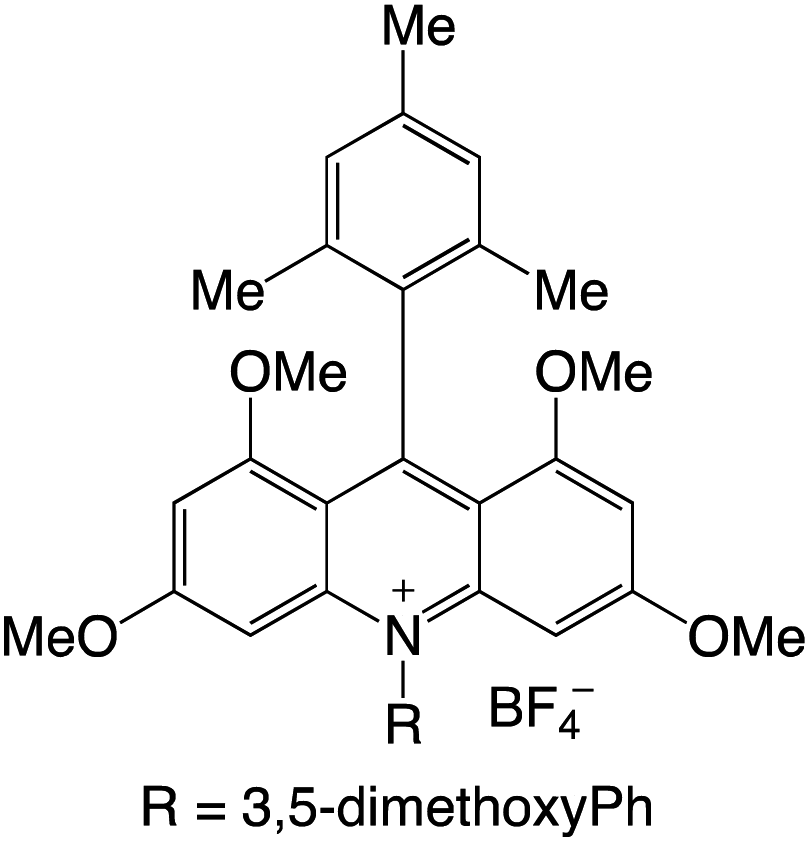
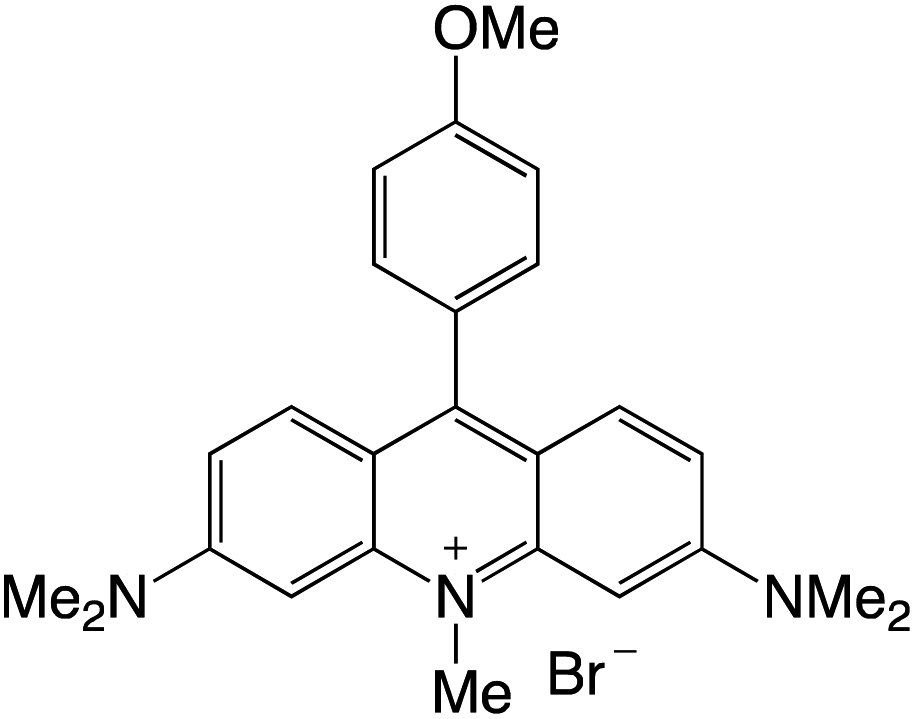
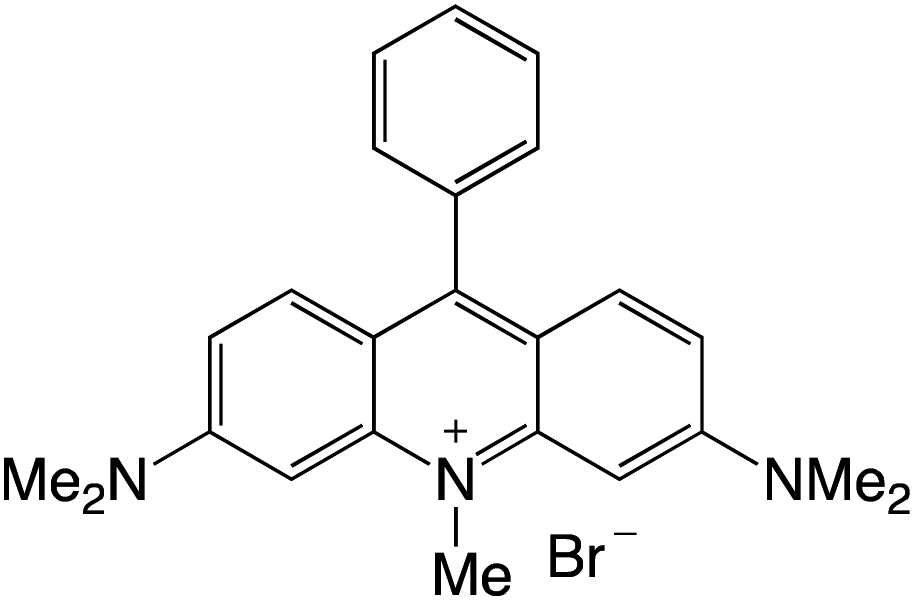
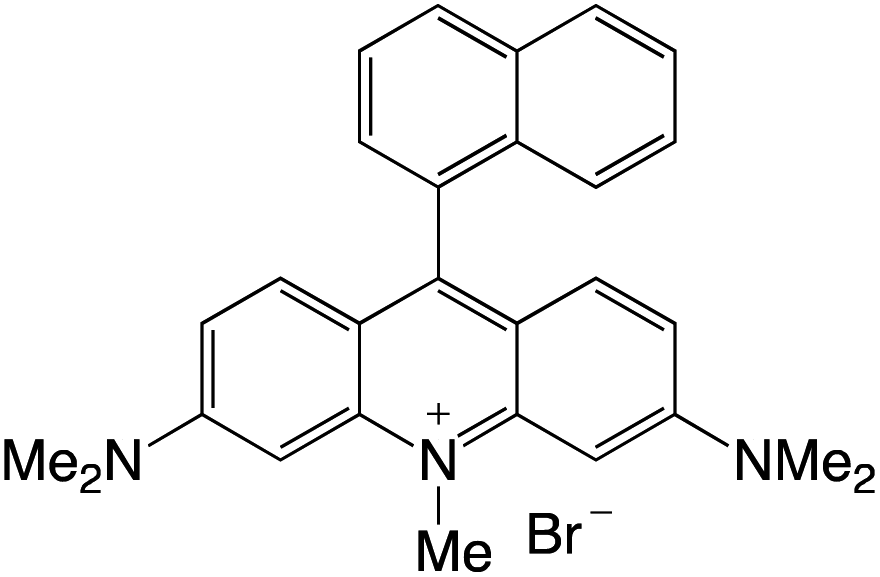
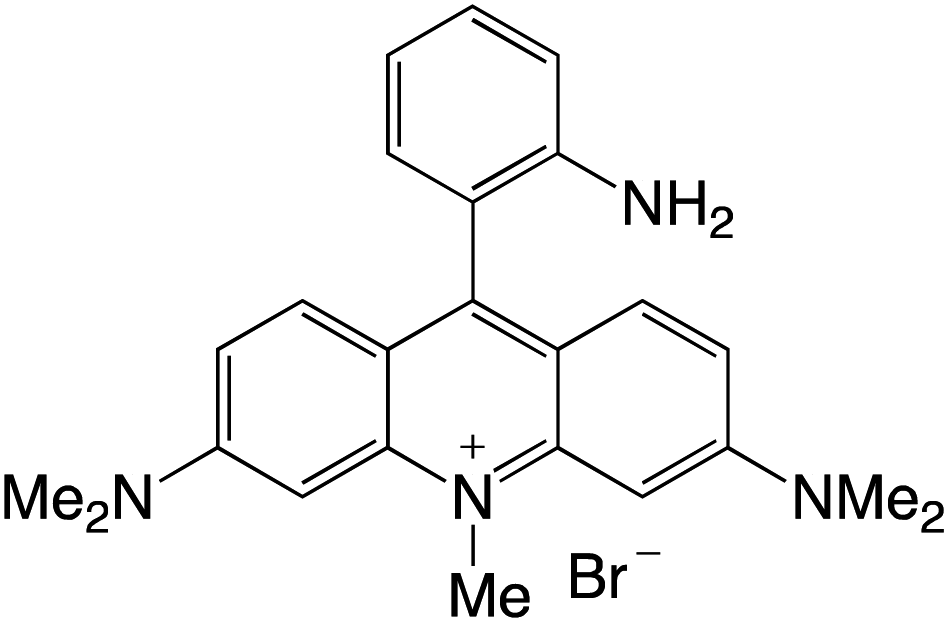
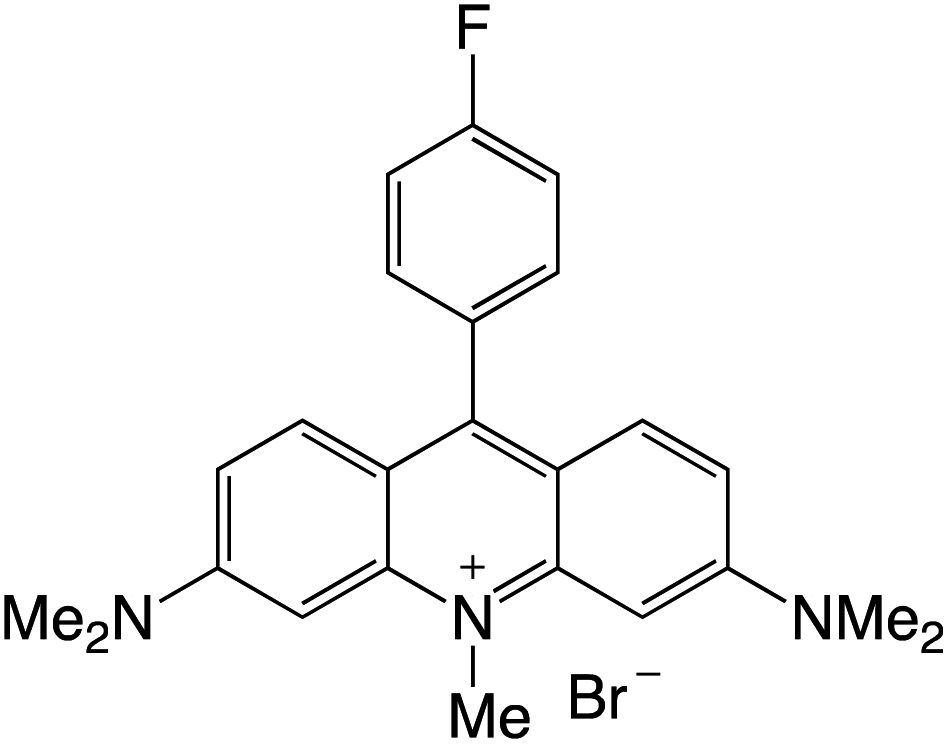
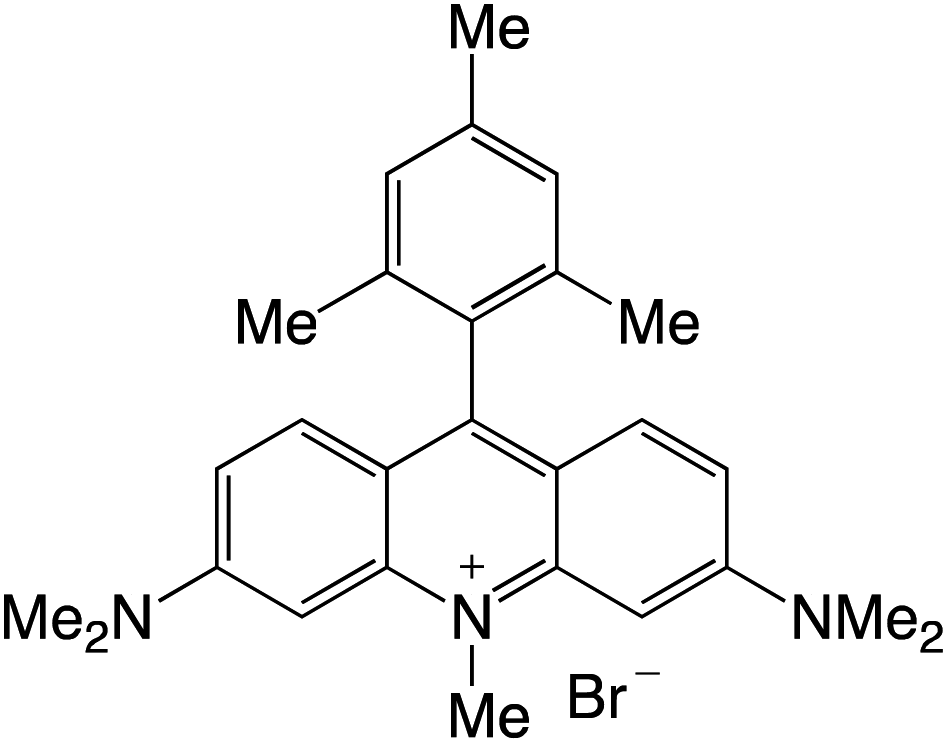
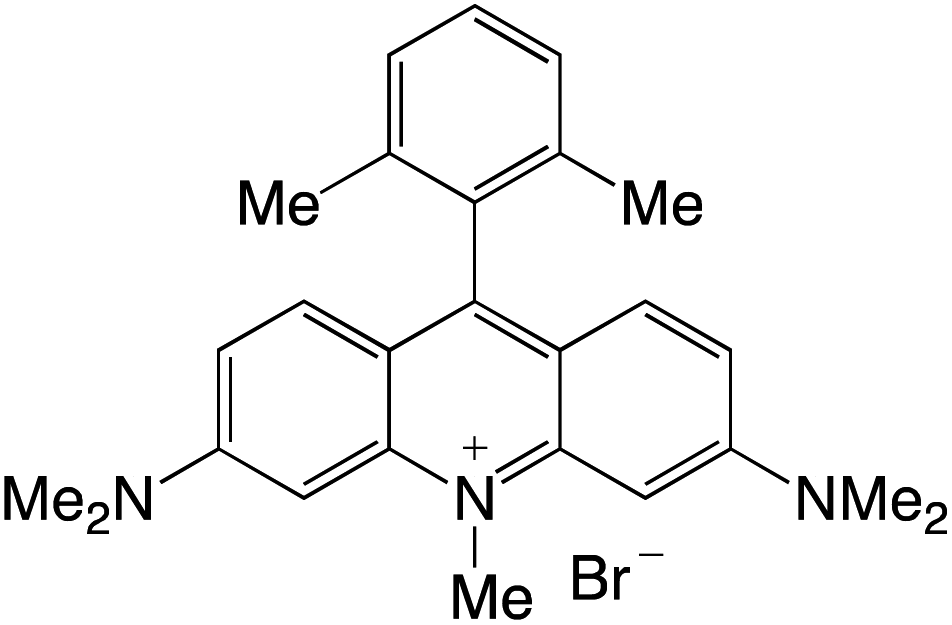
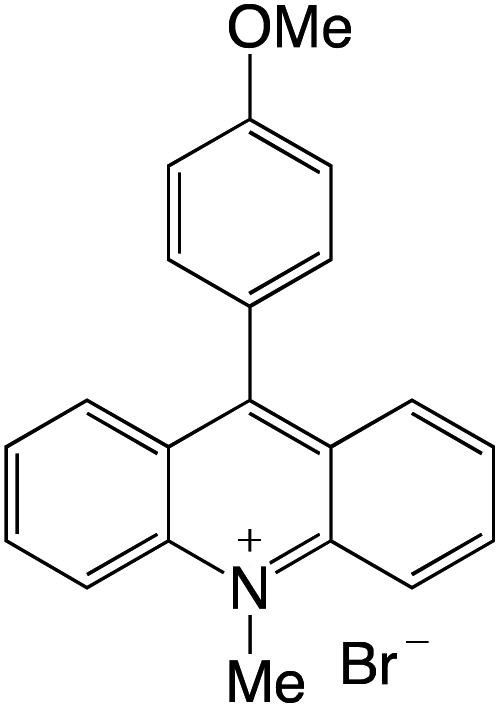
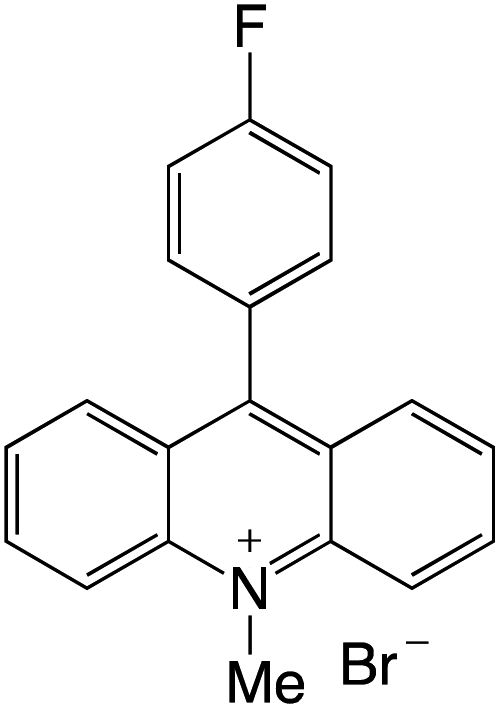
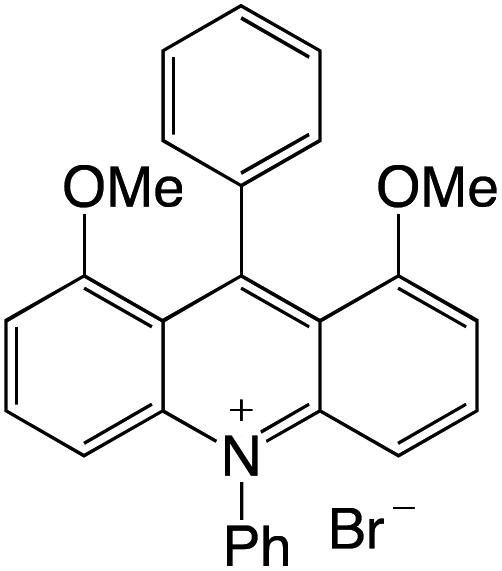
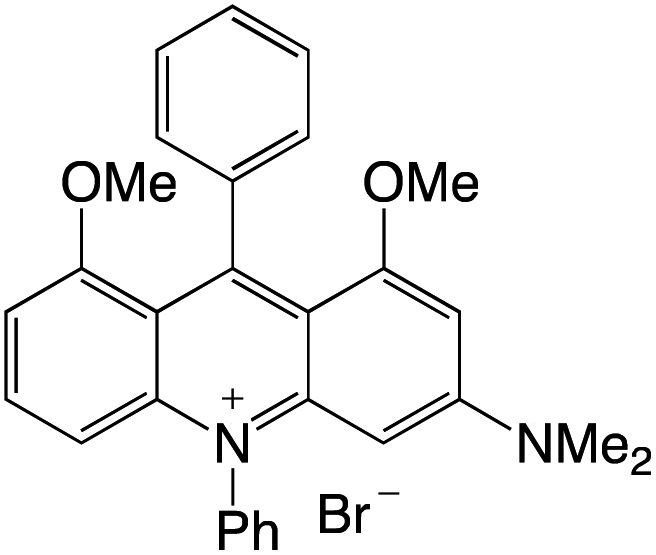
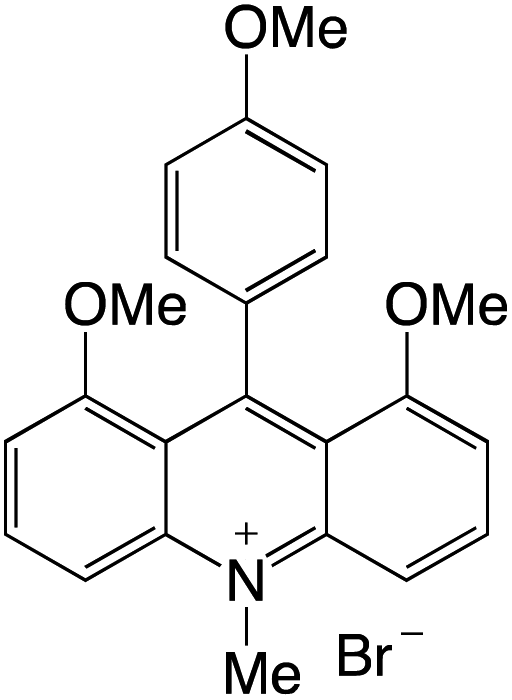
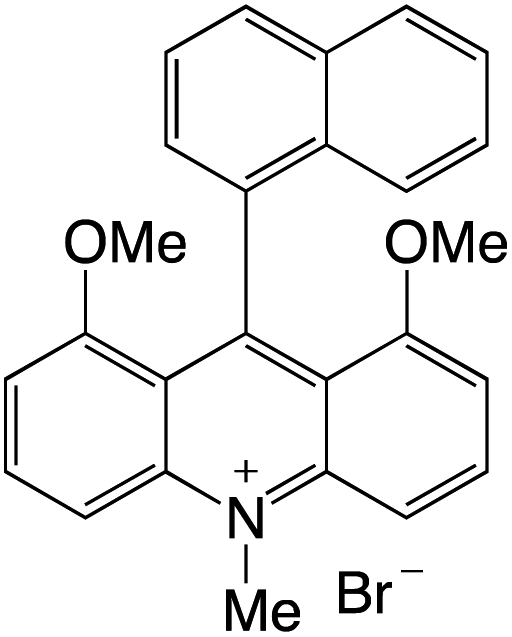
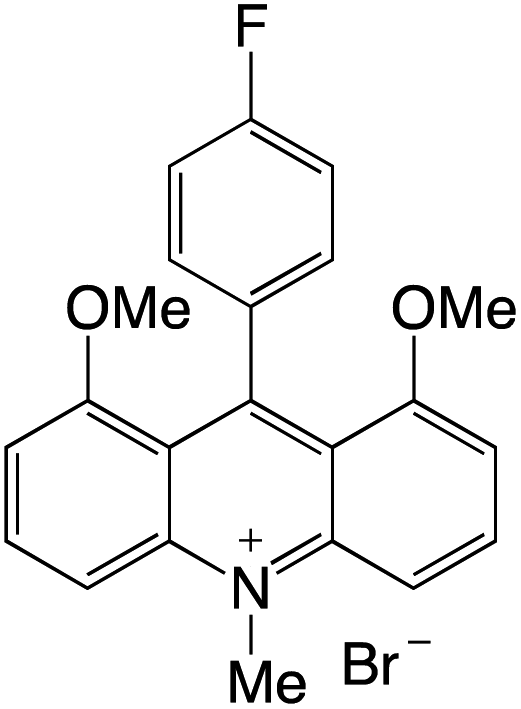
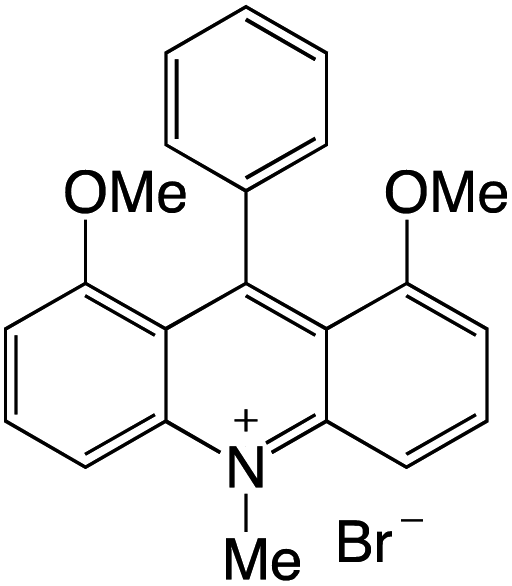
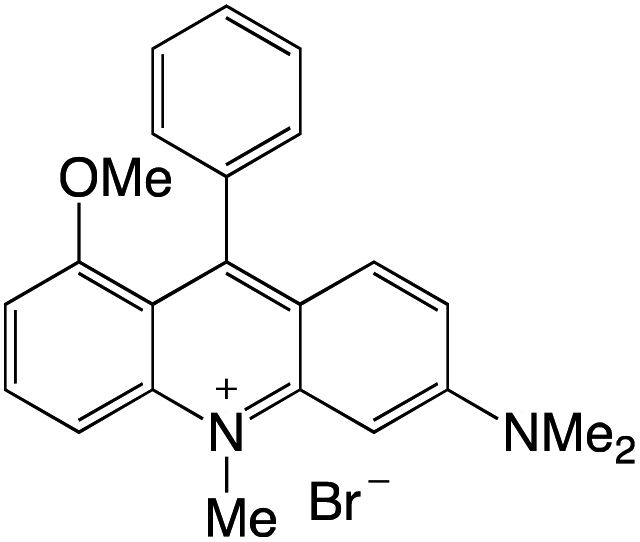
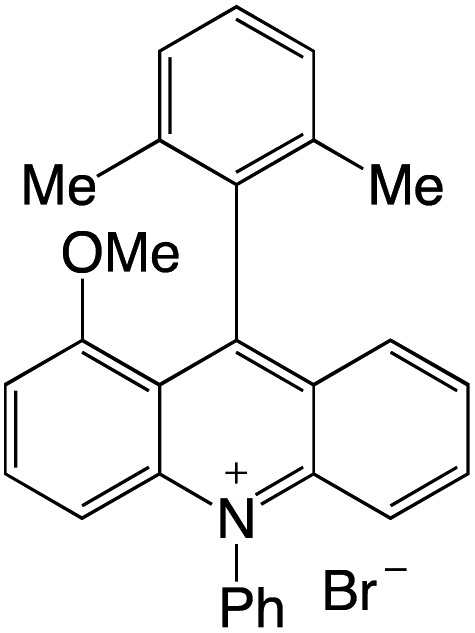
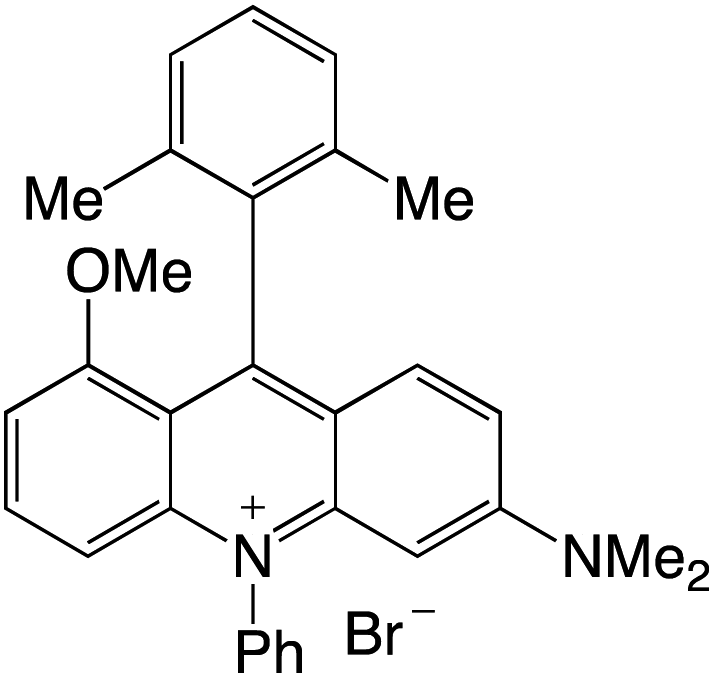
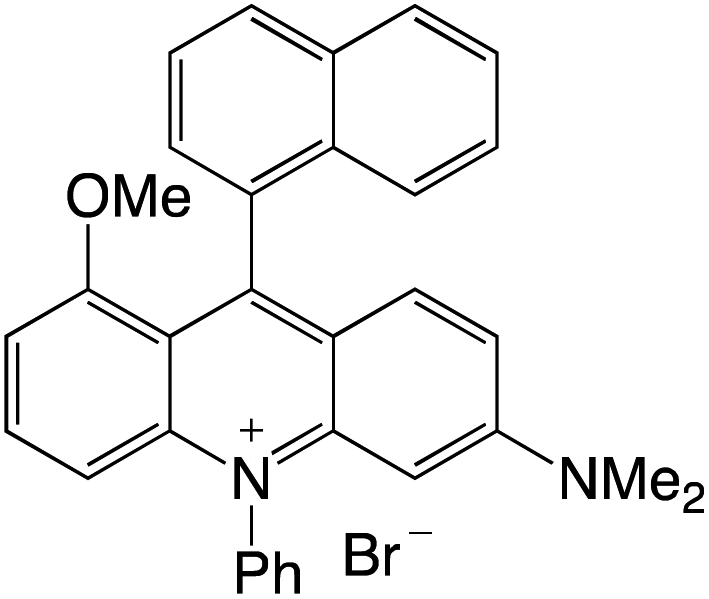
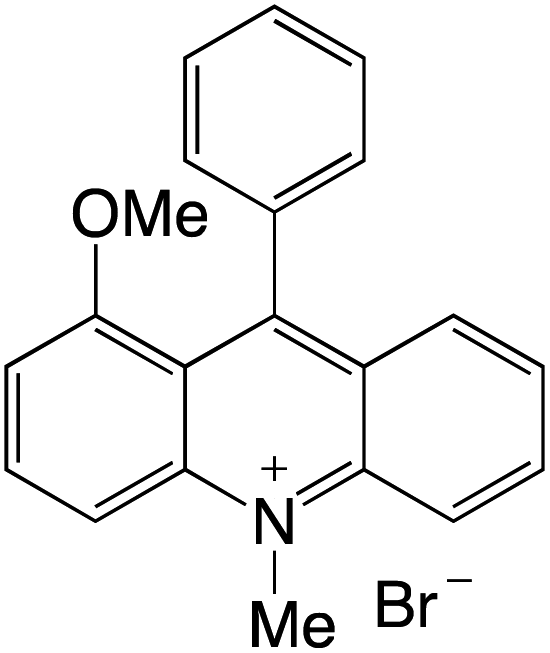
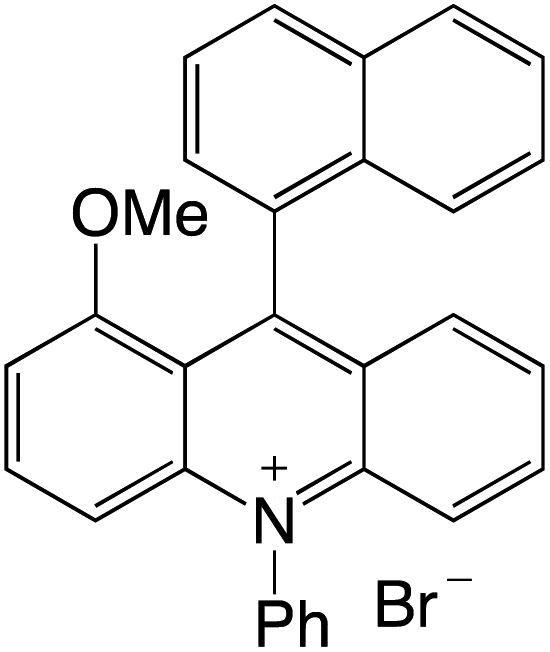
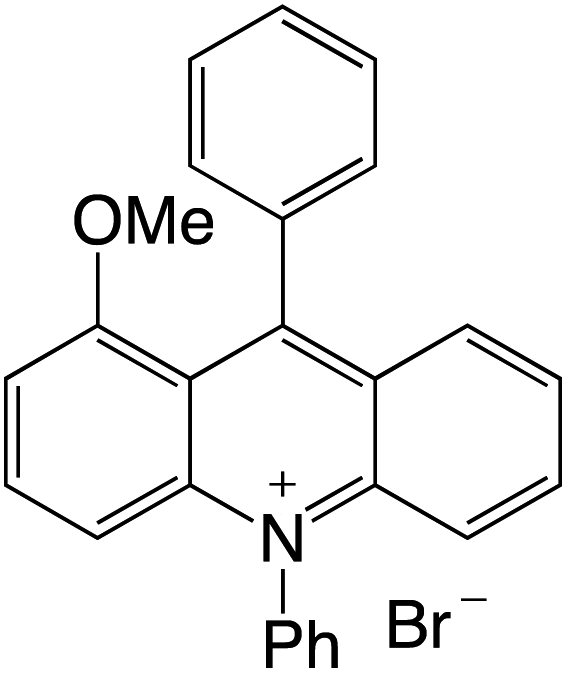
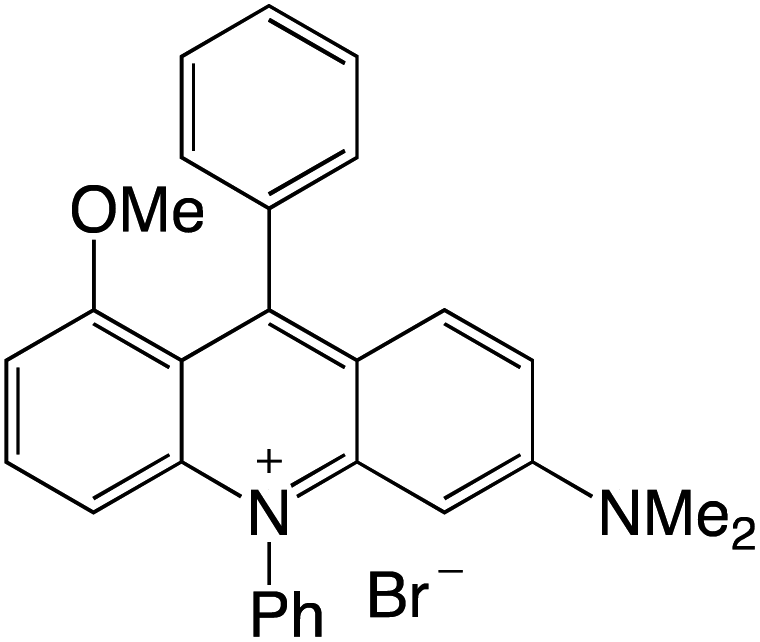
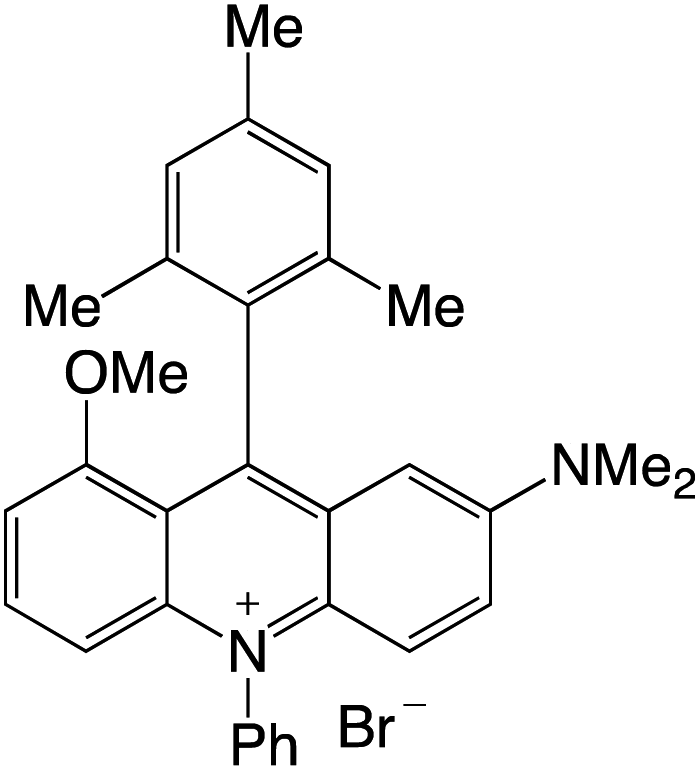
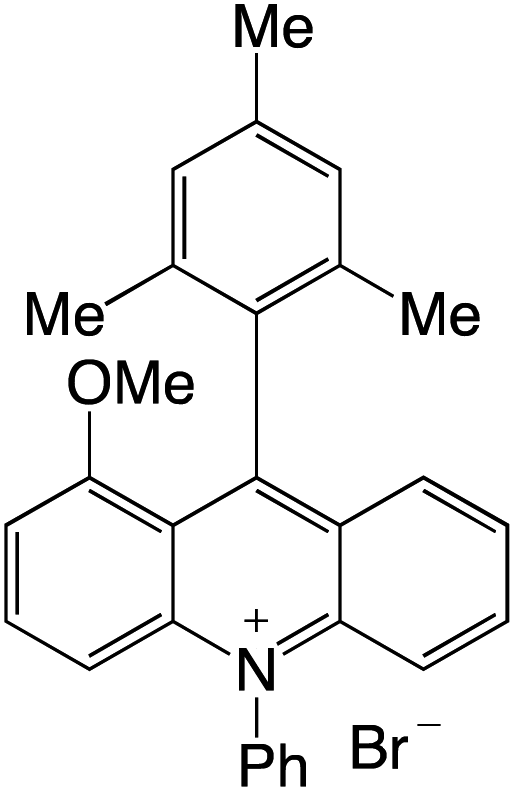
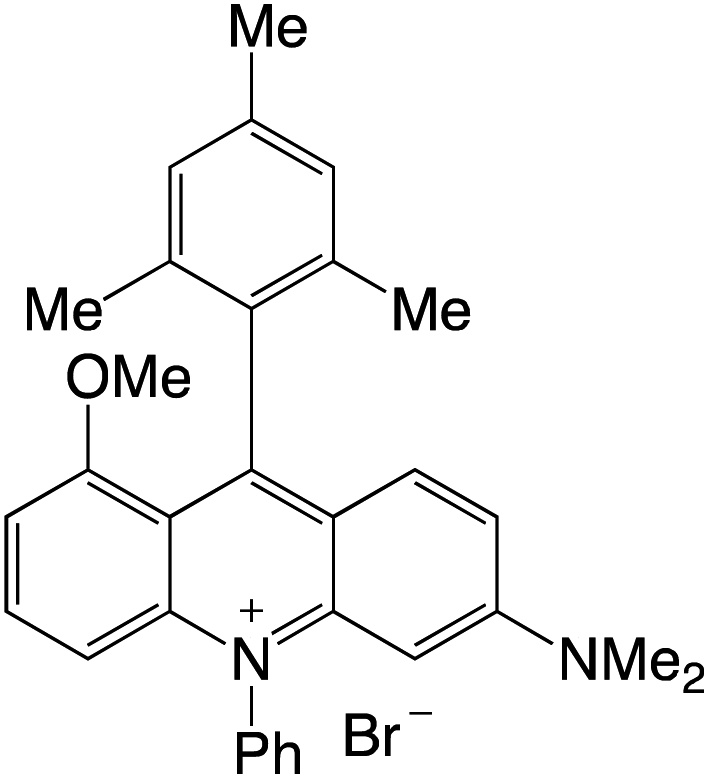
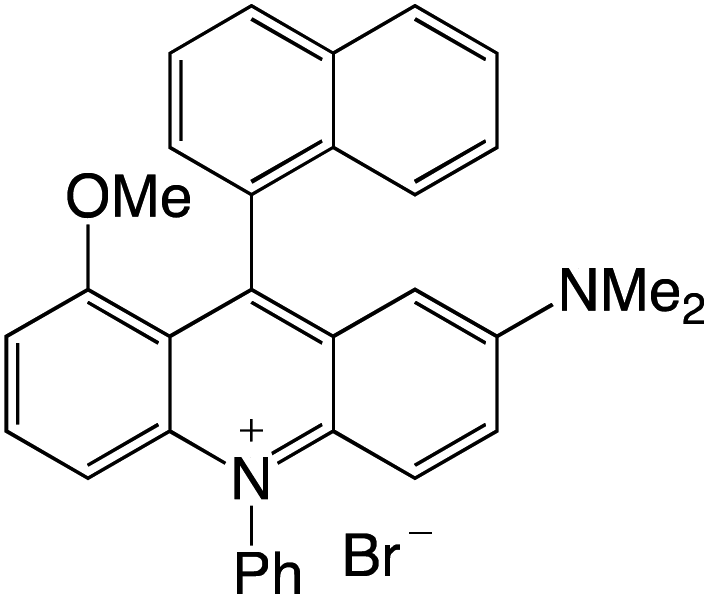
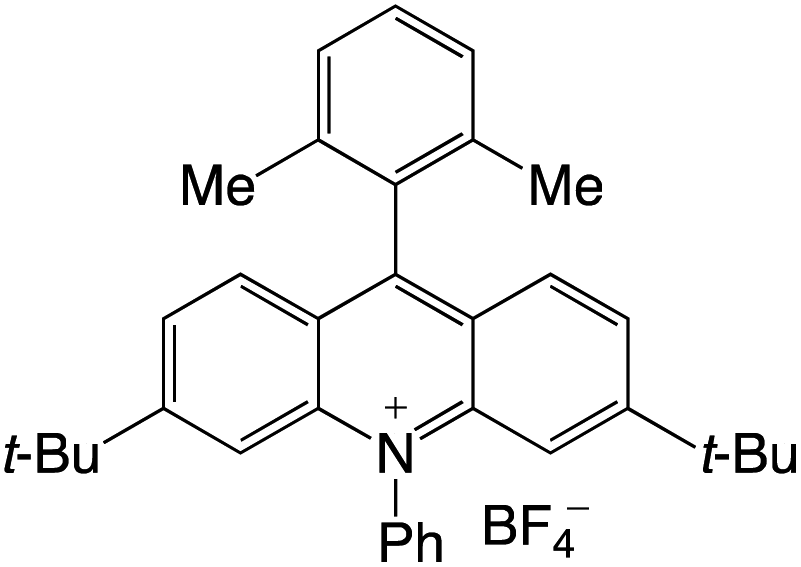
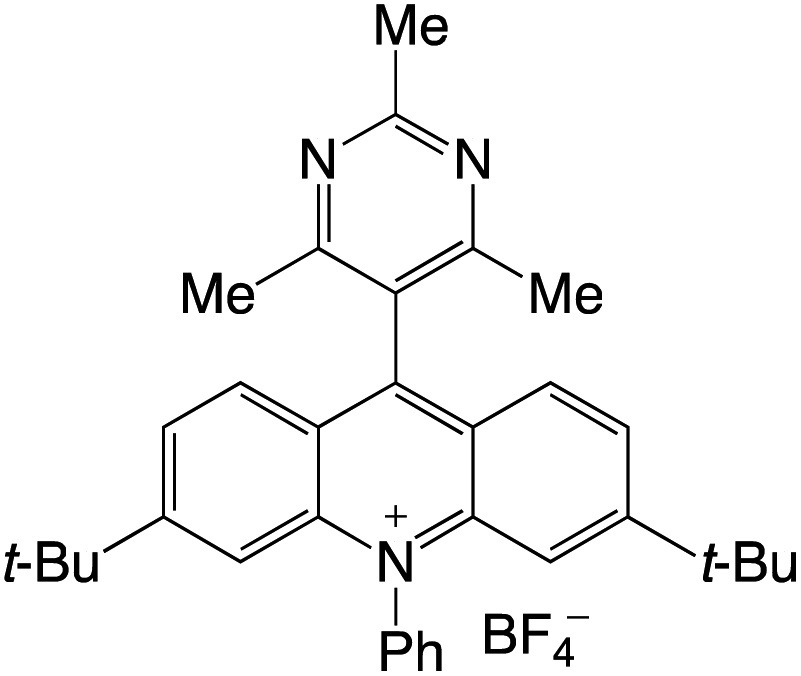
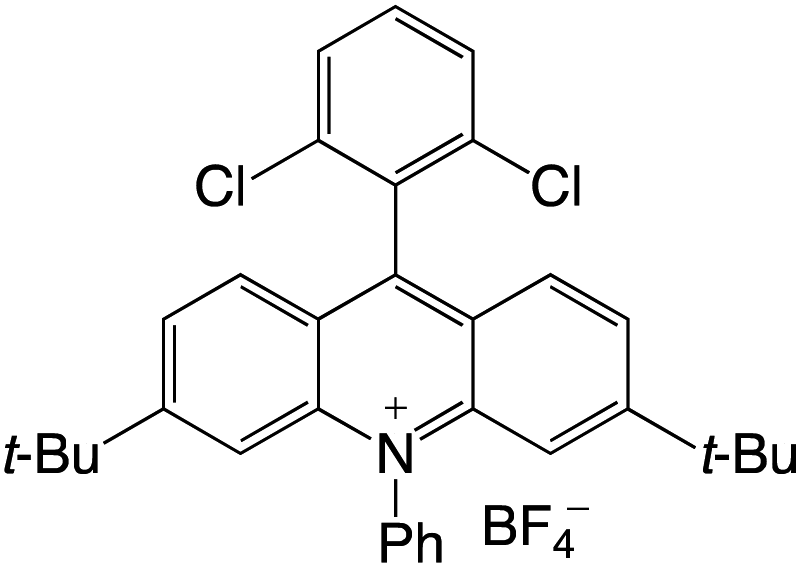
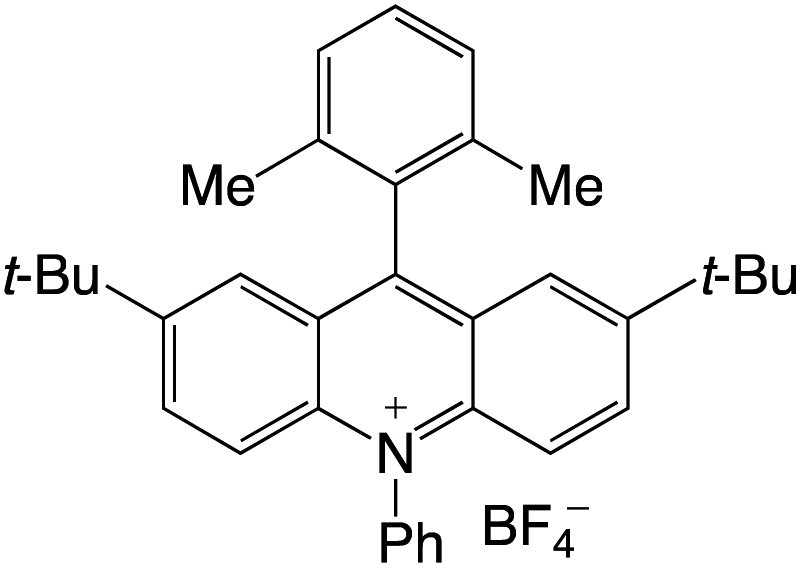
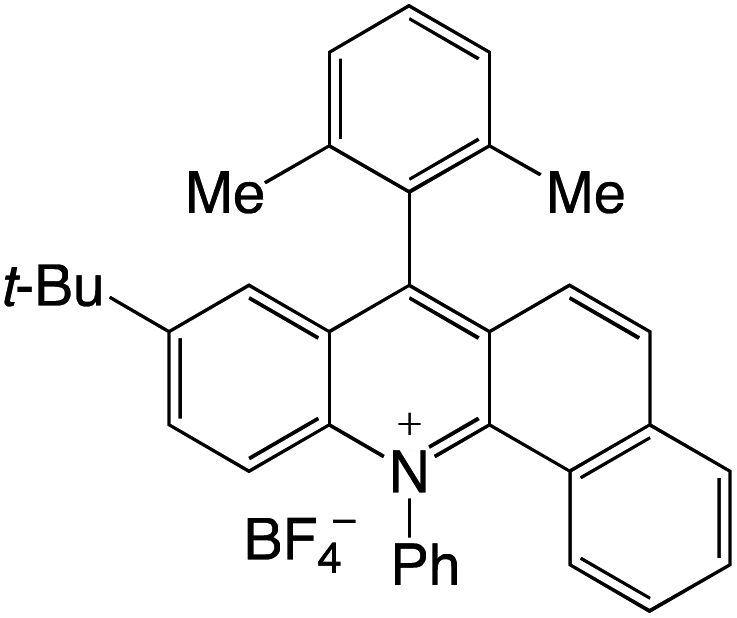
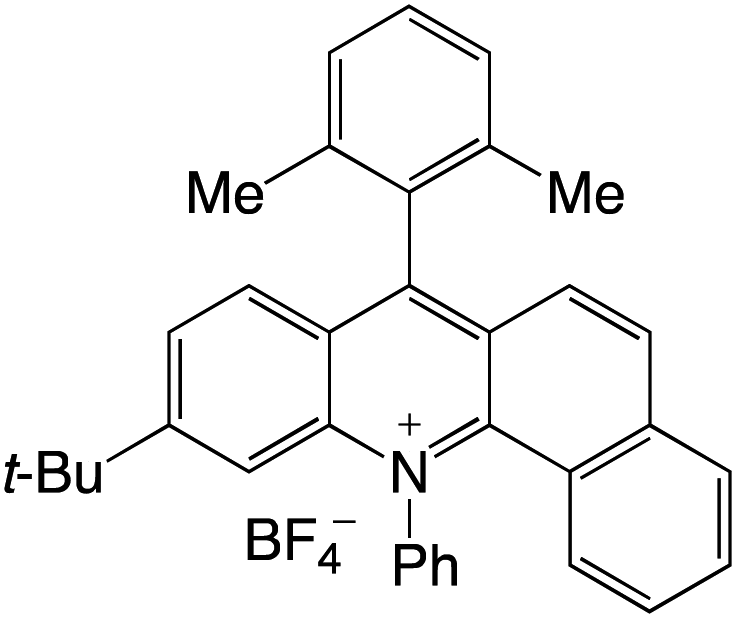
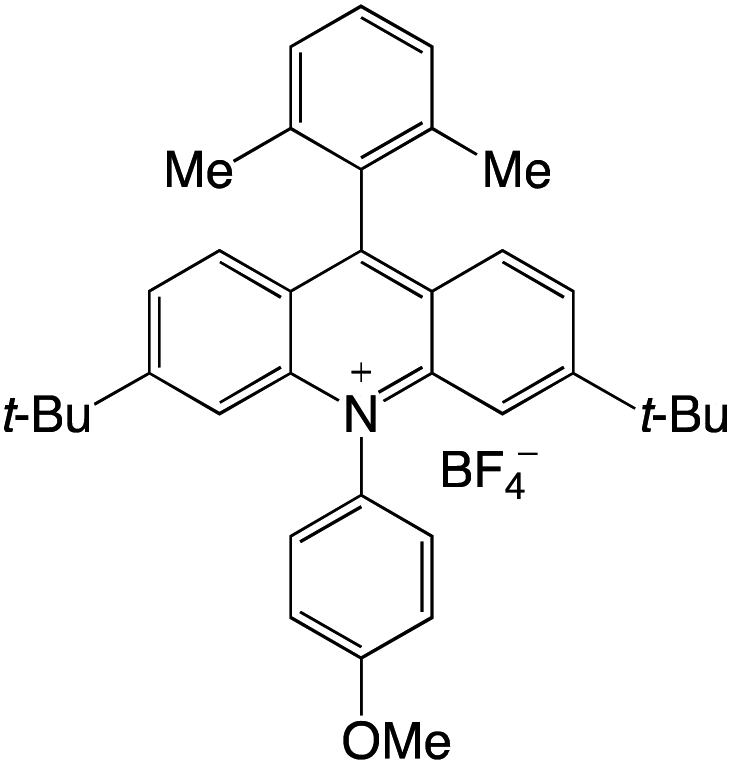
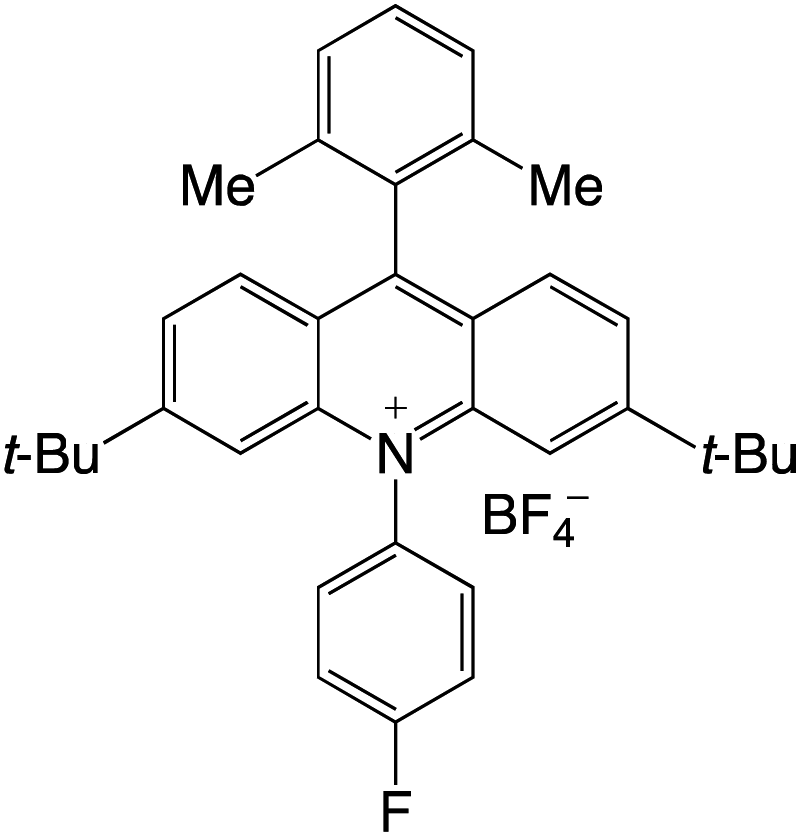
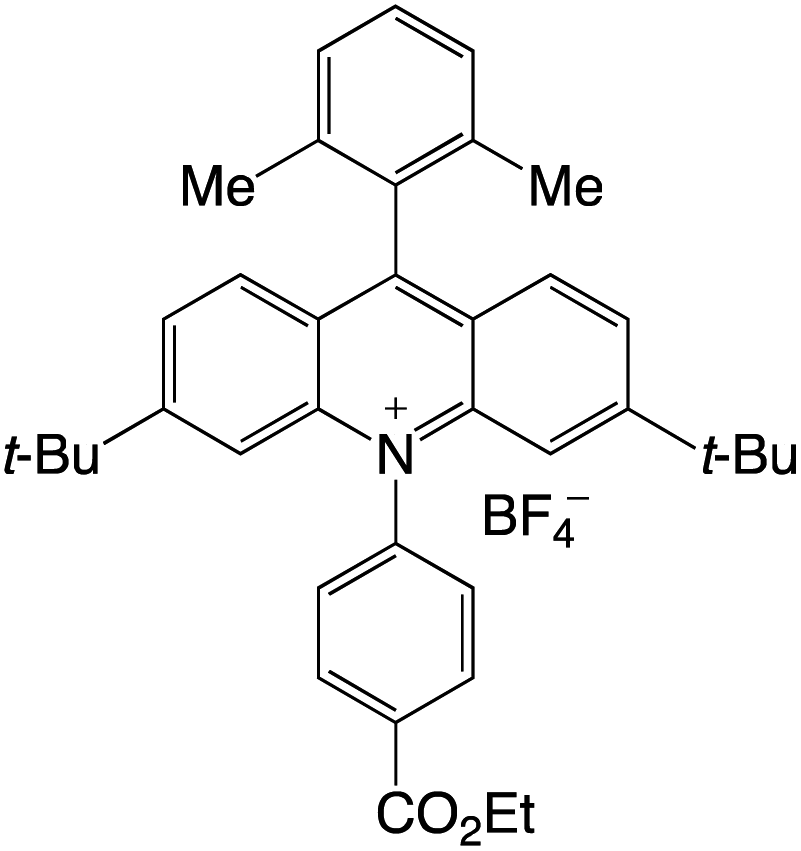
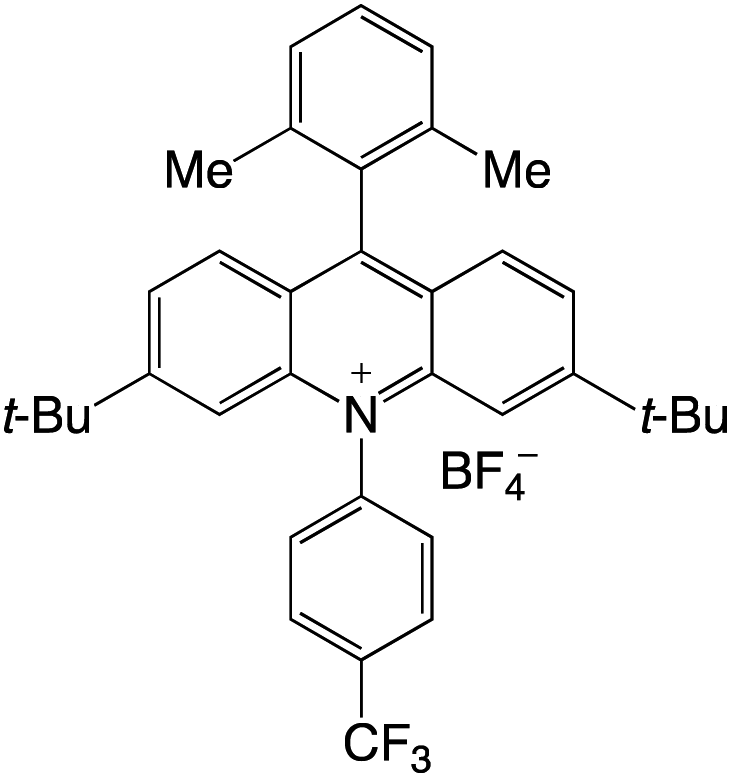
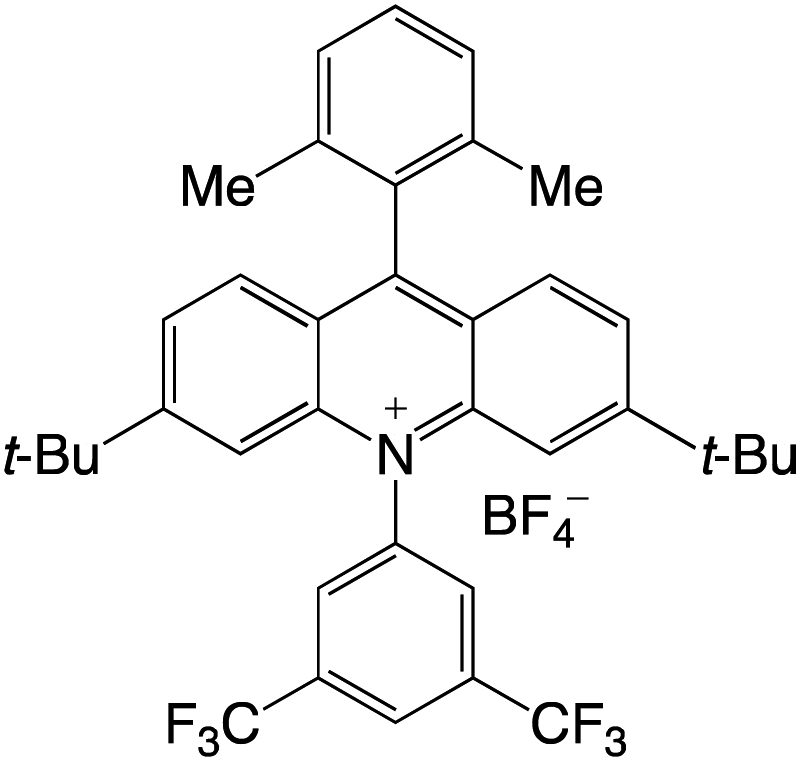
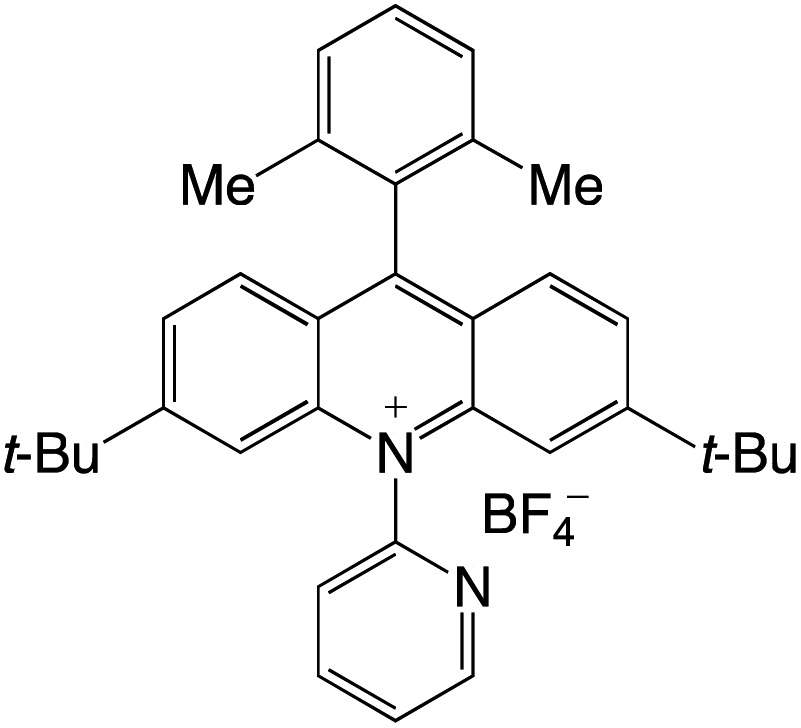
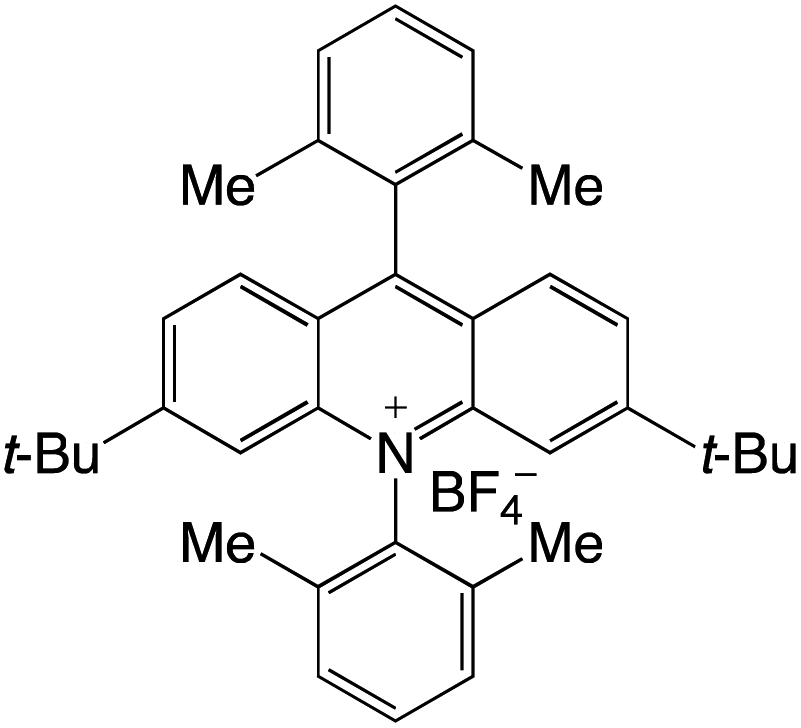
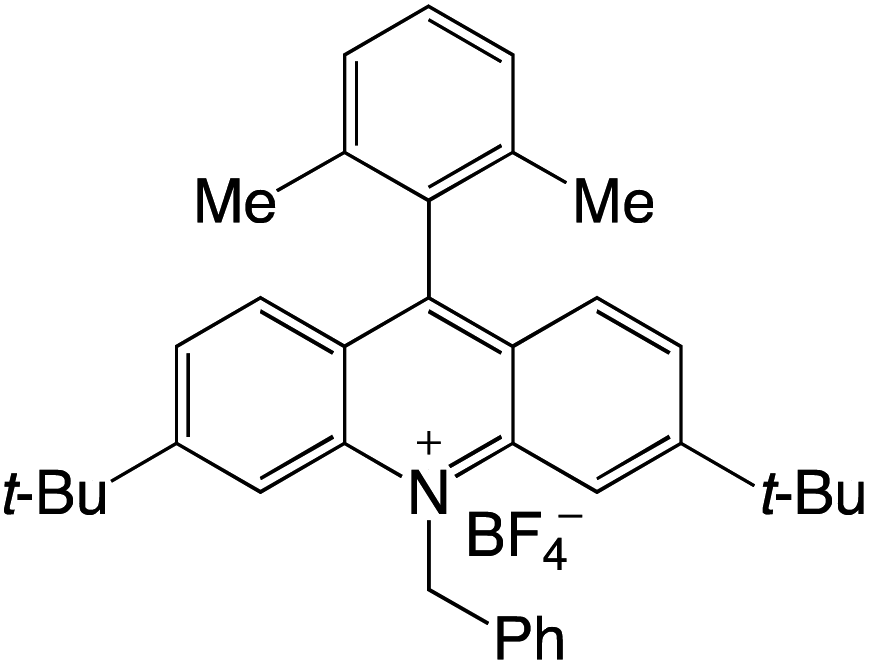
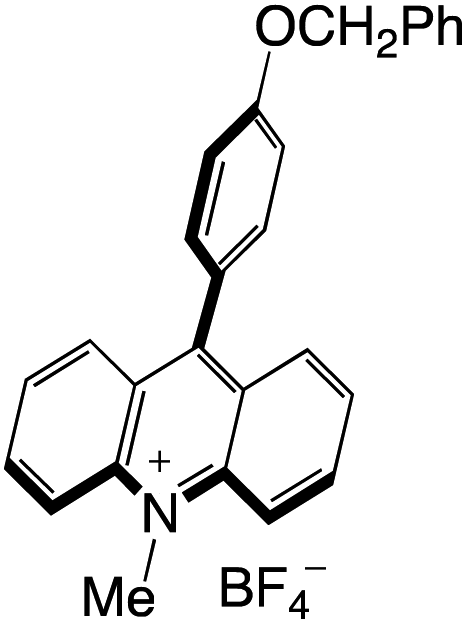
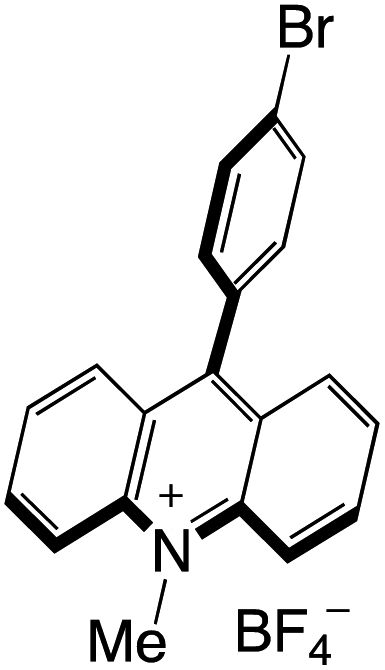
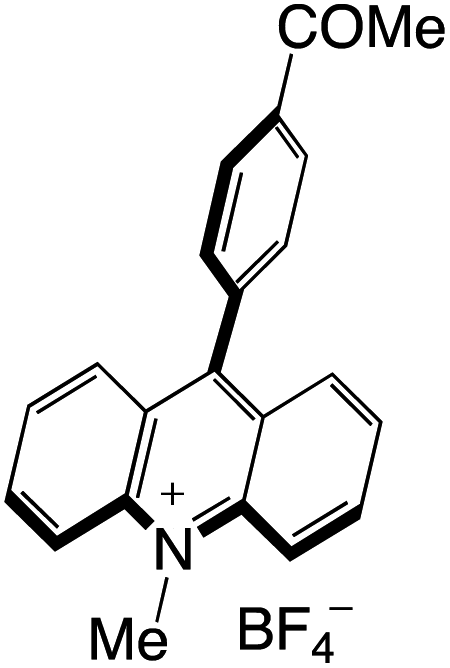
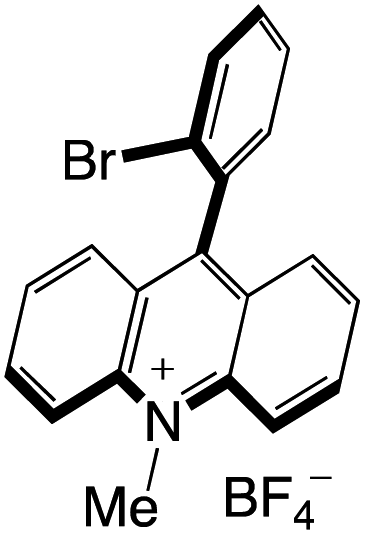
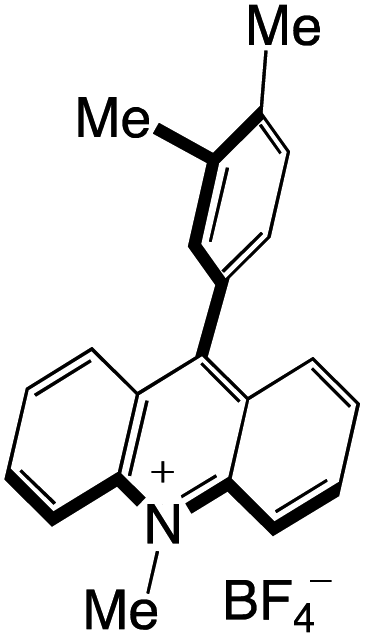
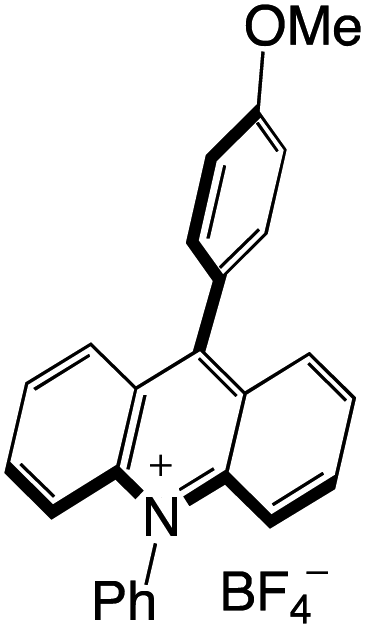
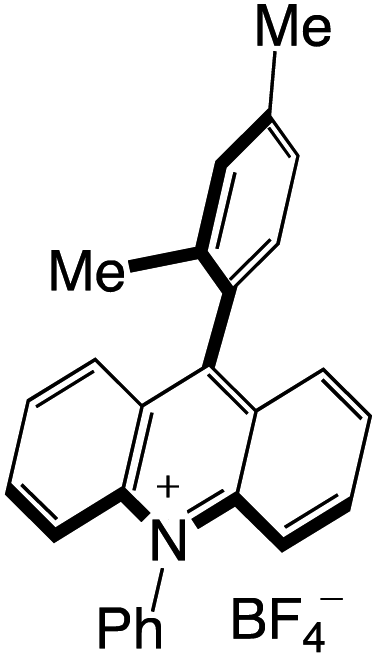
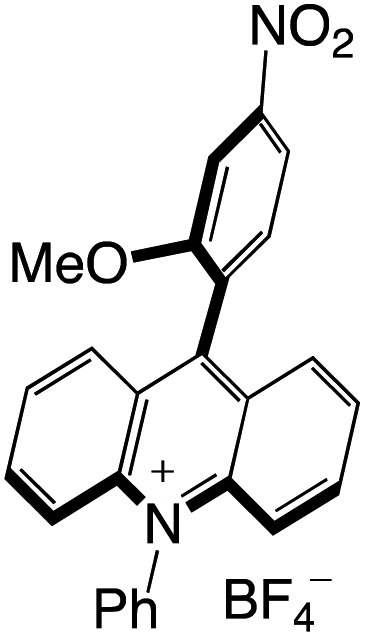
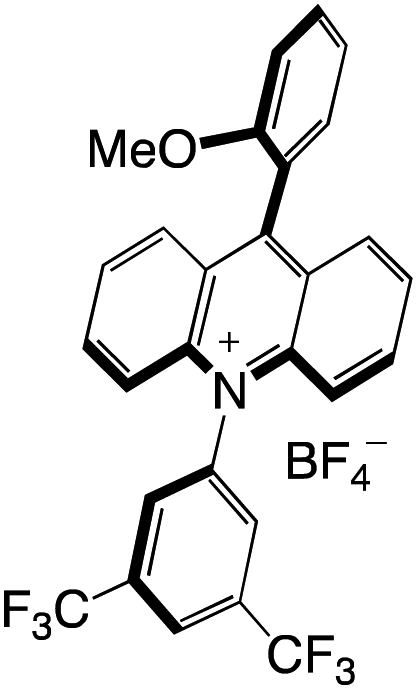
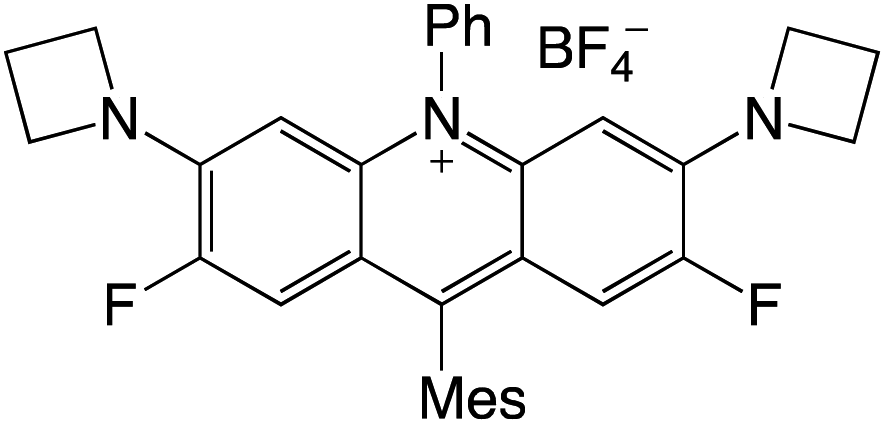
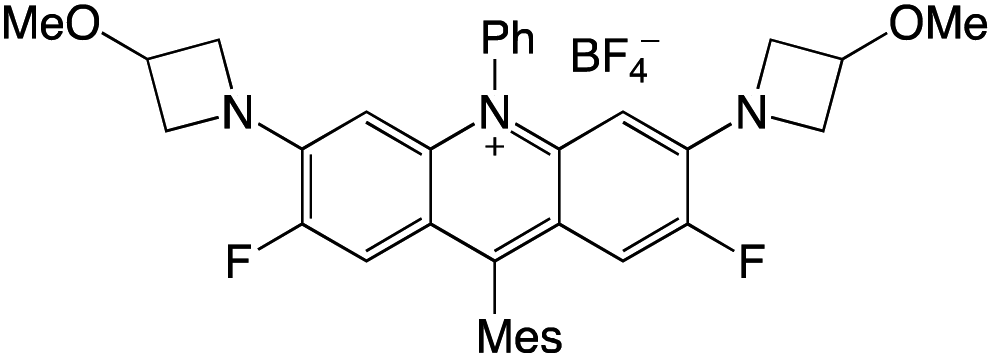
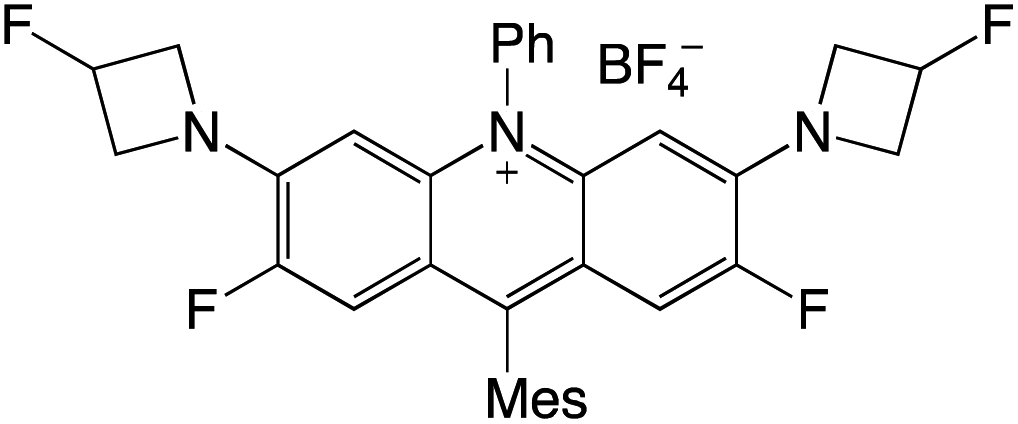
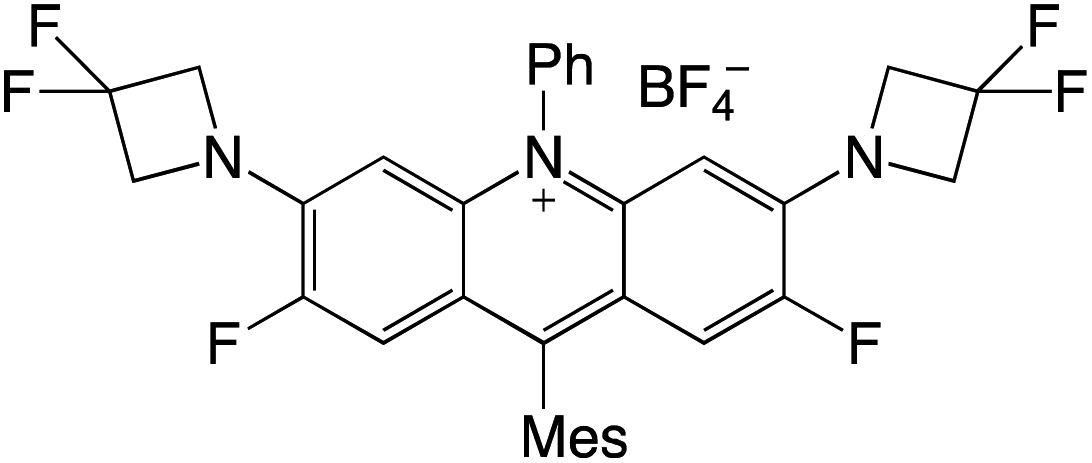
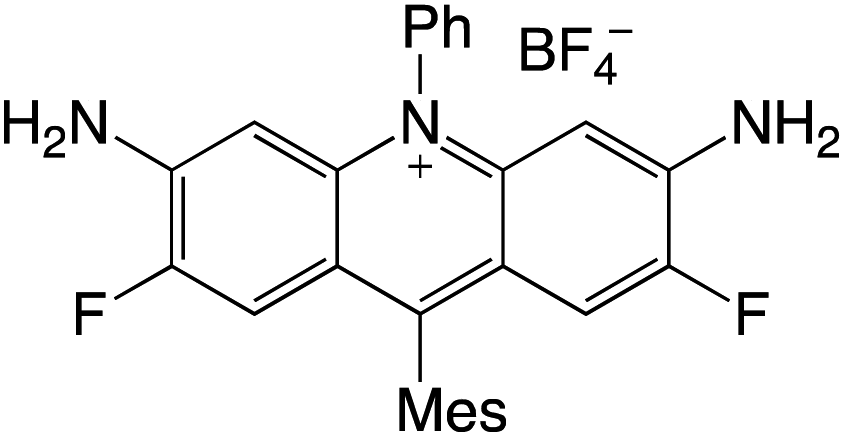
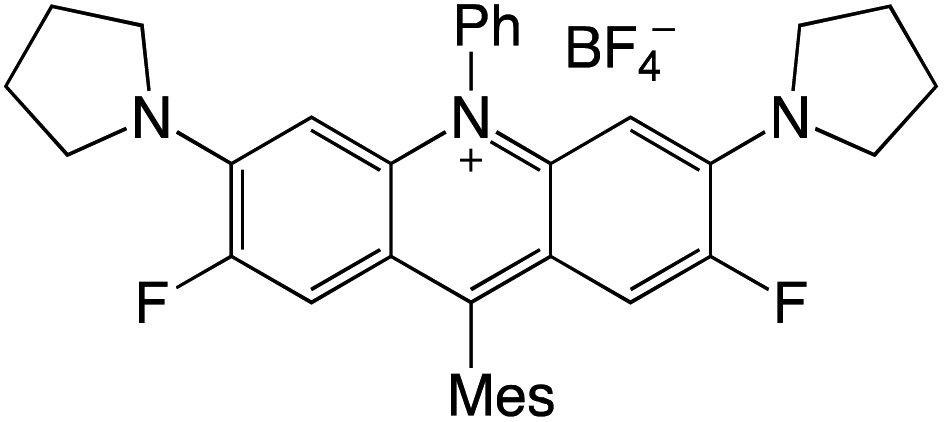
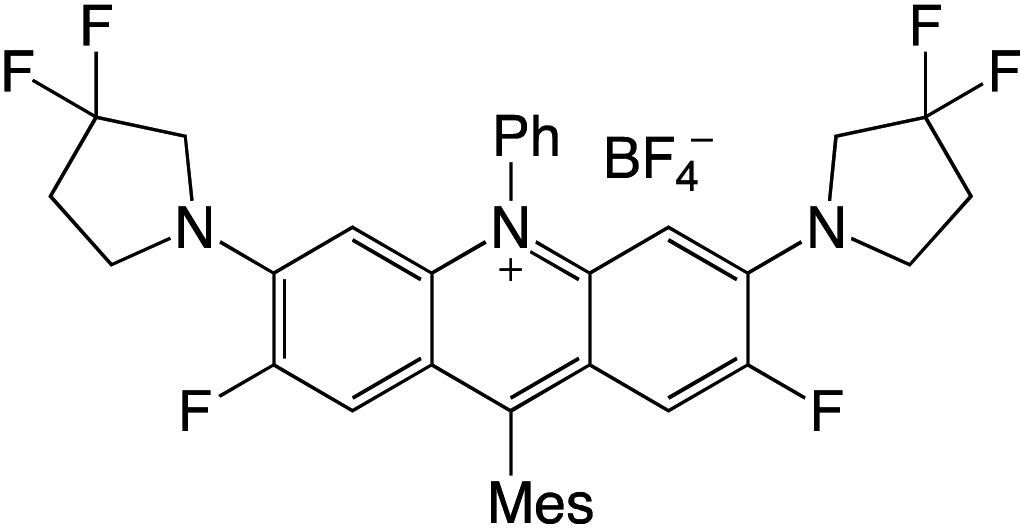
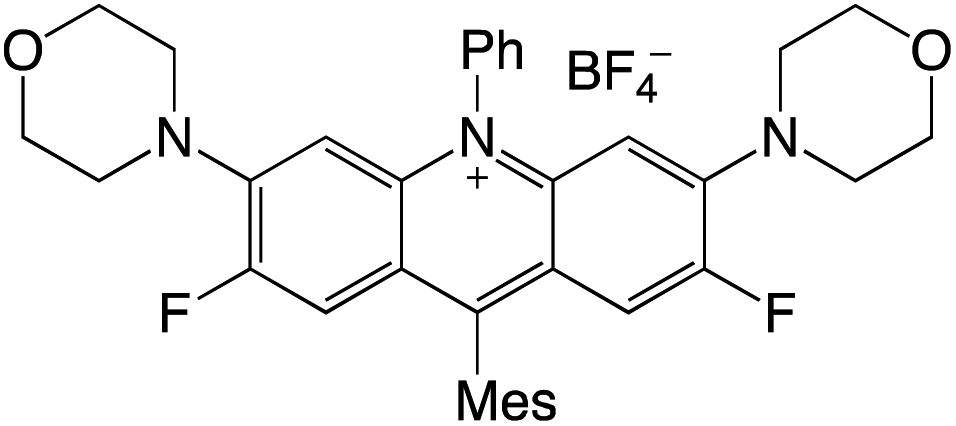
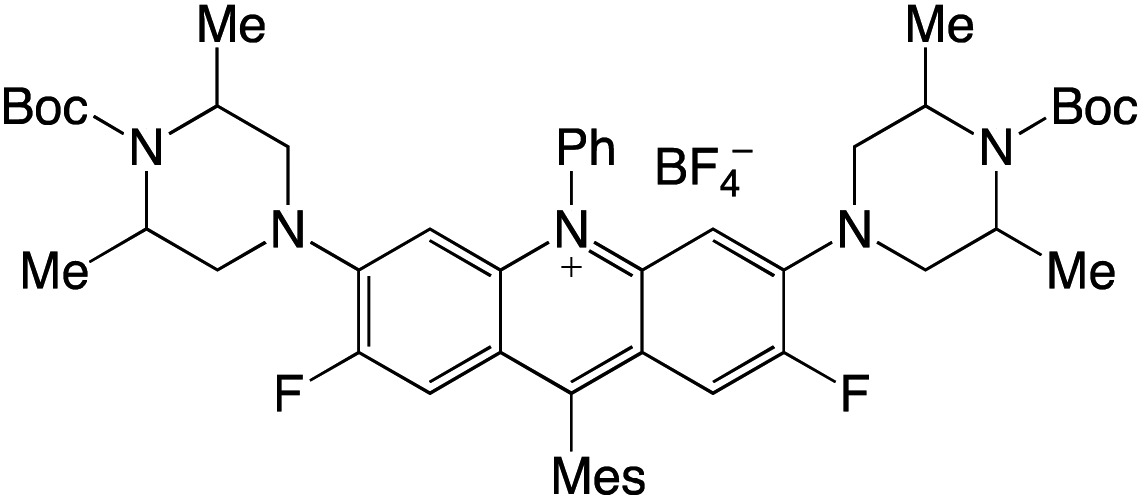
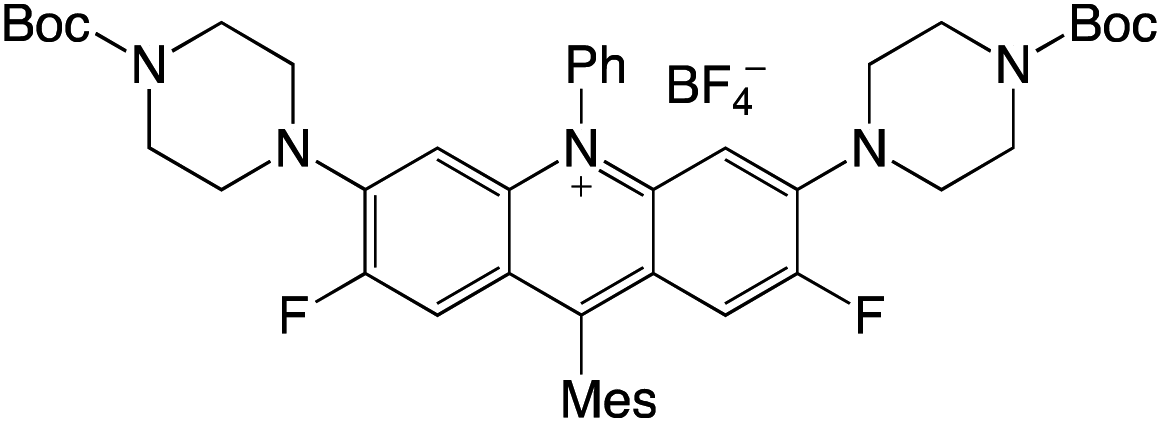
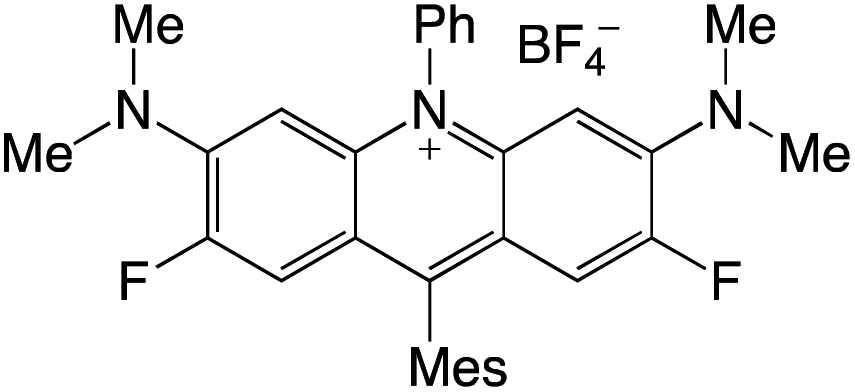
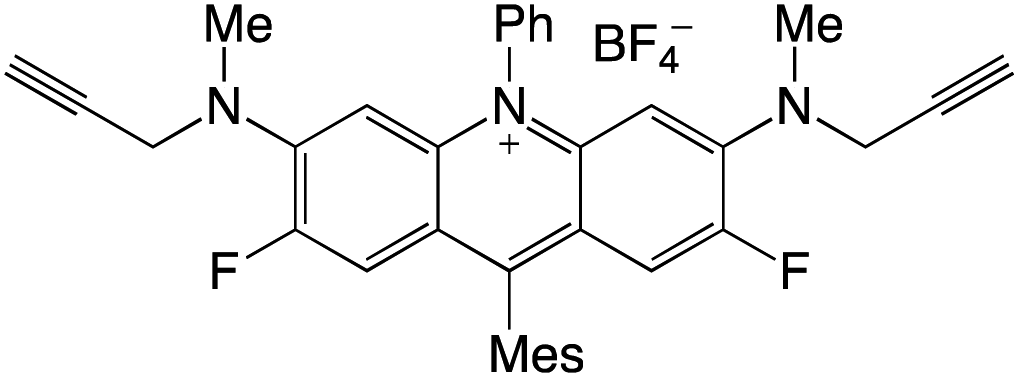
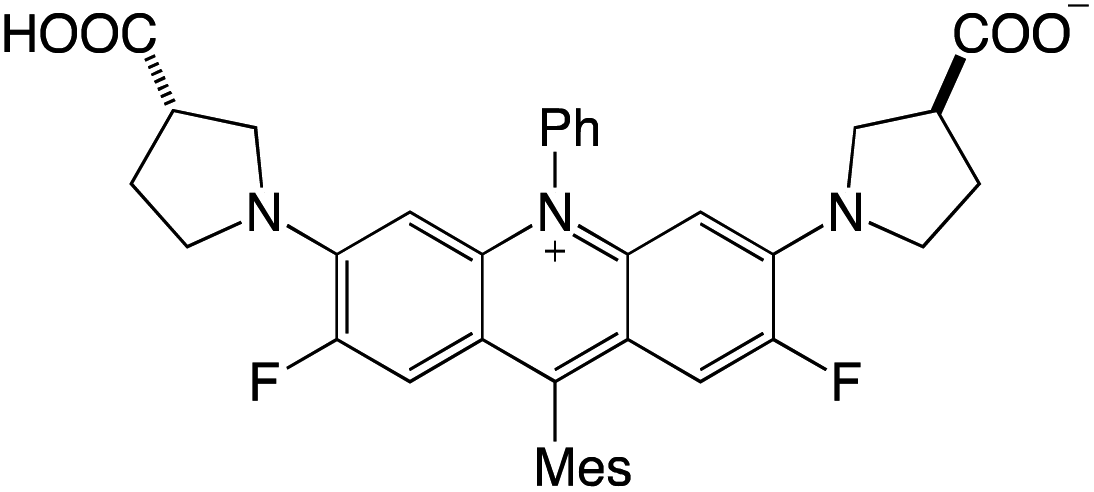
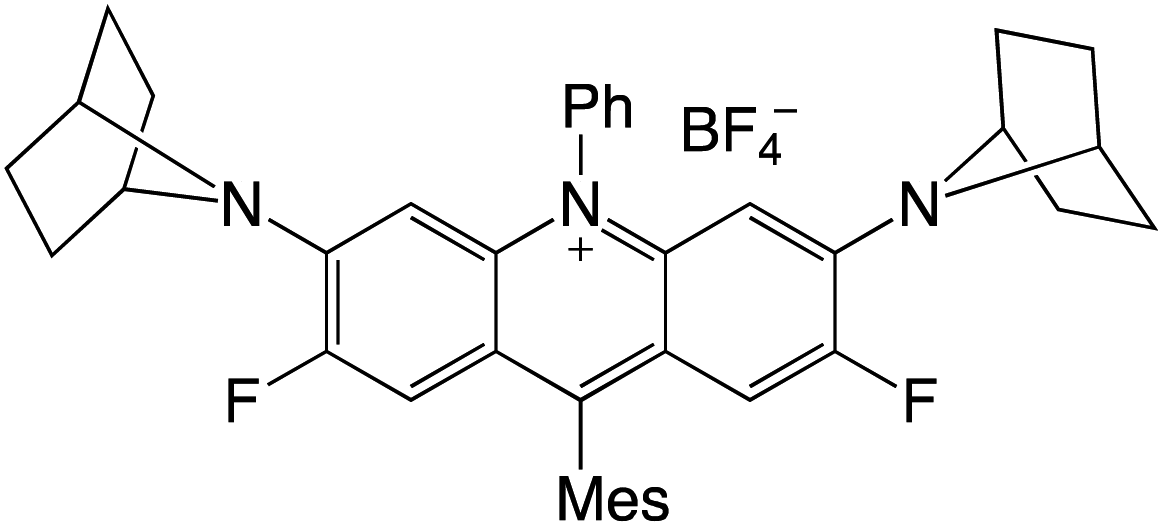
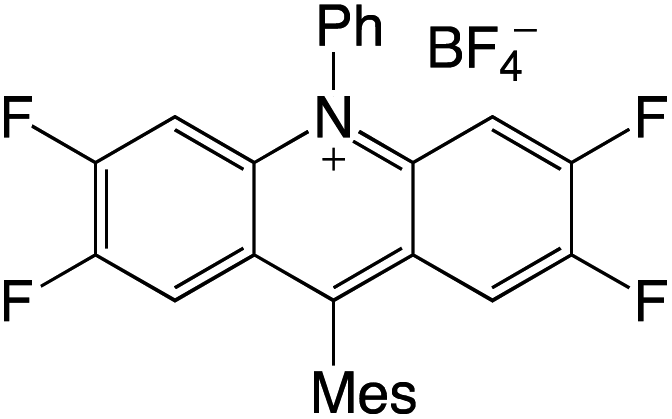
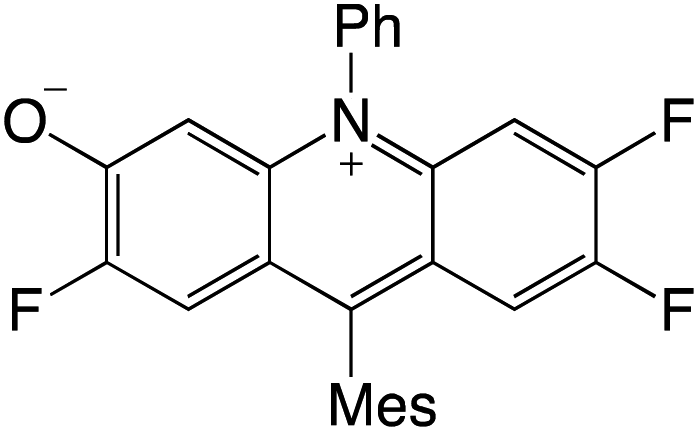
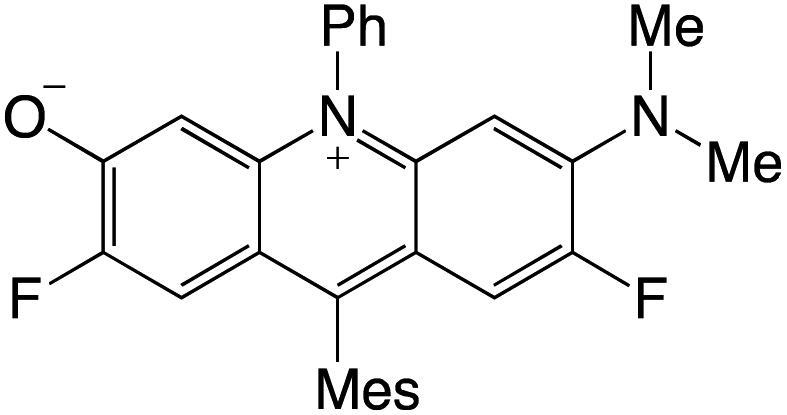
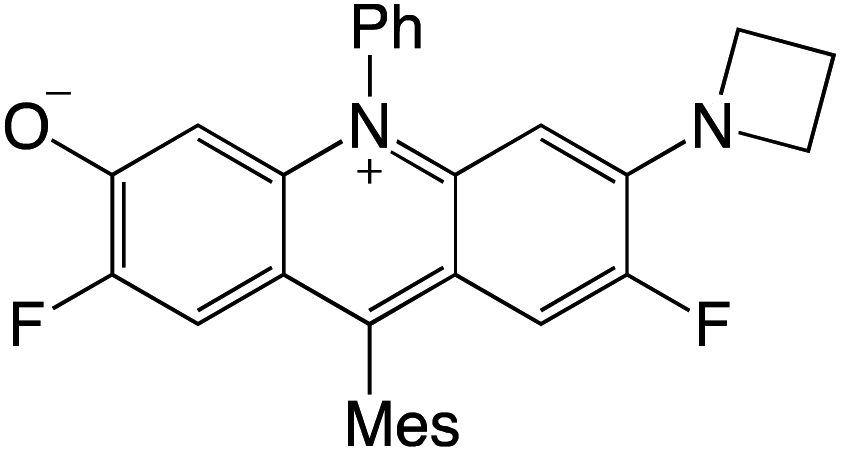
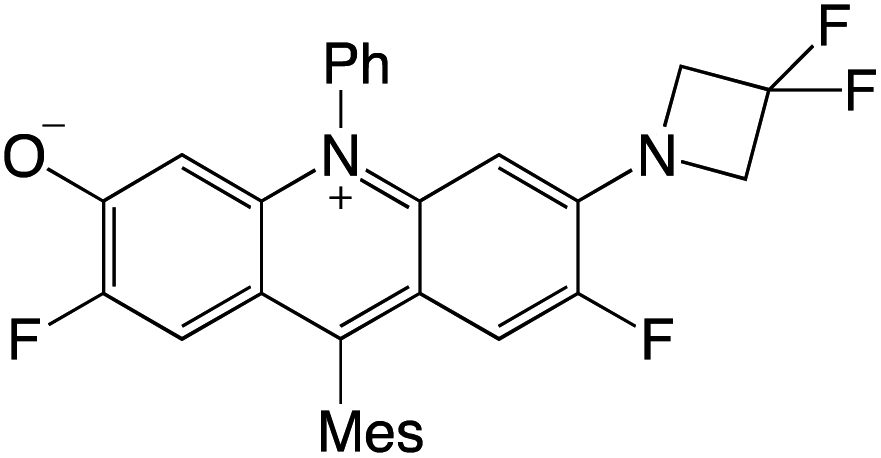
Organophotocatalysts offer the chemists for accessing to exotic chemistries and a wide variety of substrates that are typically unreactive in synthetic environments. Furthermore, the variety of these chemical compounds presents a collection that has promise for being helpful in the development and improvement of new synthetic techniques. Table 1 provides information on the photophysical and electrochemical properties of acridinium-based organophotocatalysts.
Table 1 Photophysical properties of the acridinium based photocatalyst
| Entry |
Acridinium based photocatalyst |
E0,0a [eV] |
E1/2 (C/C−)b [V vs. SCE] |
E1/2 (C*/C−)c [V vs. SCE] |
λabsd [nm] (ε × 103 [M−1 cm−1]) |
λeme [nm] |
τf[ns] |
Ref. |
| Determined at the intersection between normalized absorption and emission spectra, with E = 1240/λ. Ground-state reduction potentials determined by cyclic voltammetry (E vs. SCE). Excited-state reduction potential, estimated with ground-state reduction potentials and excited-state energies. Conc. ≈ 1.5 × 10−5 M. λem = 370 nm with ref = quinine sulfate (Φ = 0.546 in H2SO4 0.5 M). Time-correlated single-photon counting technique. |
| 1 |
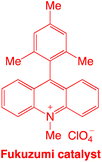 |
2.57 |
−0.57 |
2.08 |
425 |
— |
6 |
29 |
| 2 |
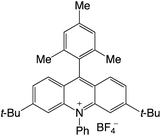 |
2.66 |
−0.59 |
2.08 |
420 |
517 |
14.4 |
29 |
| 3 |
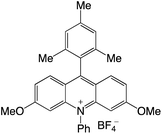 |
— |
−0.71 |
2.01 |
407 |
525 |
3.0 |
29 |
| 4 |
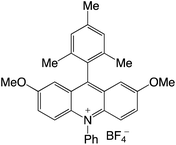 |
— |
−0.57 |
1.90 |
466 |
545 |
18.7 |
29 |
| 5 |
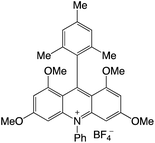 |
— |
−0.84 |
1.62 |
412 |
550 |
1.3, 8.9 |
29 |
| 6 |
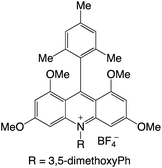 |
— |
−0.82 |
1.65 |
414 |
550 |
1.3, 12.3 |
29 |
| 7 |
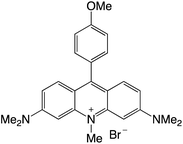 |
2.41 |
−1.15 |
1.26 |
501 |
531 |
— |
29 |
| 8 |
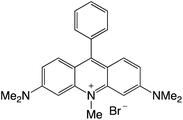 |
2.40 |
−1.10 |
1.30 |
502 |
534 |
— |
29 |
| 9 |
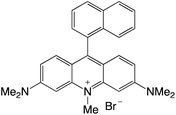 |
2.39 |
−1.13 |
1.26 |
506 |
534 |
— |
29 |
| 10 |
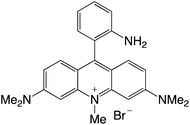 |
2.39 |
−1.14 |
1.25 |
506 |
532 |
— |
29 |
| 11 |
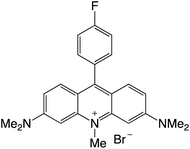 |
2.40 |
−1.12 |
1.28 |
503 |
534 |
— |
29 |
| 12 |
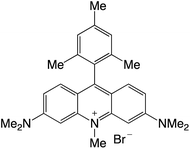 |
2.40 |
−1.15 |
1.25 |
503 |
534 |
2.2 |
29 |
| 13 |
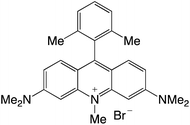 |
2.40 |
−1.15 |
1.25 |
504 |
533 |
— |
29 |
| 14 |
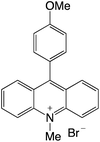 |
2.77 |
−0.56 |
2.21 |
438 |
499 |
— |
29 |
| 15 |
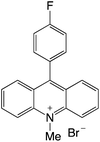 |
2.83 |
−0.51 |
2.32 |
426 |
512 |
— |
29 |
| 16 |
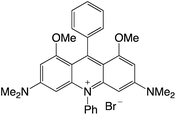 |
2.40 |
−1.19 |
1.21 |
498 |
540 |
4.4 |
29 |
| 17 |
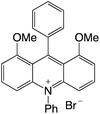 |
2.23 |
−0.47 |
1.76 |
503 |
595 |
3.1 |
29 |
| 18 |
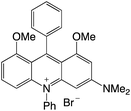 |
2.25 |
−0.94 |
1.31 |
501 |
584 |
4.7 |
29 |
| 19 |
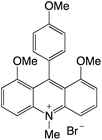 |
2.30 |
−0.62 |
1.68 |
494 |
567 |
5.9 |
29 |
| 20 |
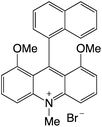 |
2.39 |
−0.51 |
1.88 |
497 |
531 |
4.1 |
29 |
| 21 |
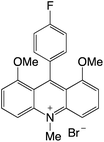 |
2.31 |
−0.51 |
1.80 |
497 |
579 |
3.0 |
29 |
| 22 |
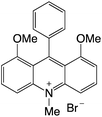 |
2.33 |
−0.52 |
1.81 |
497 |
576 |
2.7 |
29 |
| 23 |
 |
2.30 |
−0.83 |
1.47 |
506 |
575 |
0.9, 4.4 |
29 |
| 24 |
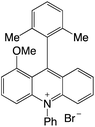 |
2.25 |
−0.56 |
1.69 |
480 |
634 |
1.2, 3.3, 16.8 |
29 |
| 25 |
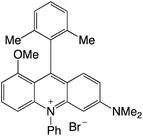 |
2.29 |
−0.90 |
1.39 |
514 |
574 |
1.1, 7.2 |
29 |
| 26 |
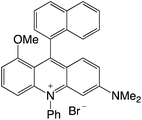 |
2.27 |
−0.87 |
1.40 |
516 |
578 |
1.0, 6.2 |
29 |
| 27 |
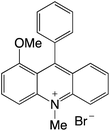 |
2.29 |
−0.48 |
1.81 |
473 |
635 |
1.0, 3.0, 17.3 |
29 |
| 28 |
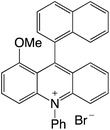 |
2.26 |
−0.53 |
1.73 |
480 |
635 |
1.0, 4.5 |
29 |
| 29 |
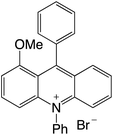 |
2.23 |
−0.54 |
1.69 |
479 |
637 |
1.0, 9.9 |
29 |
| 30 |
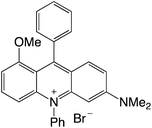 |
2.29 |
−0.89 |
1.40 |
511 |
576 |
1.0, 6.9 |
29 |
| 31 |
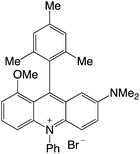 |
1.94 |
−0.71 |
1.23 |
583 |
723 |
1.5, 5.5 |
29 |
| 32 |
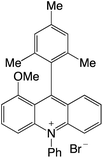 |
2.25 |
−0.57 |
1.68 |
479 |
632 |
1.4, 12.1 |
29 |
| 33 |
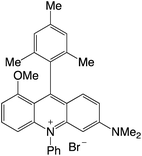 |
2.29 |
−0.89 |
1.40 |
513 |
573 |
1.1, 6.8 |
29 |
| 34 |
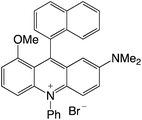 |
1.87 |
−0.68 |
1.19 |
590 |
755 |
0.9, 5.0 |
29 |
| 35 |
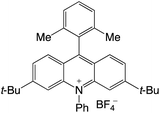 |
2.67 |
−0.56 |
2.11 |
419 |
493 |
16.4 |
35 |
| 36 |
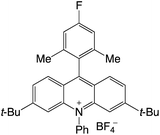 |
2.67 |
−0.54 |
2.13 |
421 |
493 |
16.8 |
35 |
| 37 |
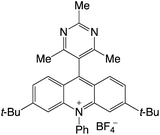 |
2.63 |
−0.47 |
2.16 |
427 |
500 |
16.1 |
35 |
| 38 |
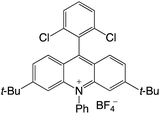 |
2.64 |
−0.43 |
2.21 |
425 |
504 |
17.1 |
35 |
| 39 |
 |
2.62 |
−0.53 |
2.09 |
431 |
499 |
19.0 |
35 |
| 40 |
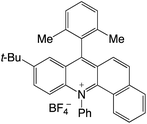 |
2.60 |
−0.53 |
2.07 |
460 |
504 |
0.3, 16.8 |
35 |
| 41 |
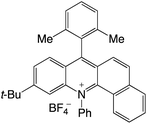 |
2.60 |
−0.54 |
2.06 |
462 |
501 |
0.3, 16.6 |
35 |
| 42 |
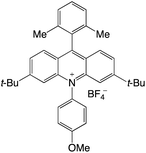 |
2.63 |
−0.58 |
2.05 |
422 |
523 |
1.1, 18.8 |
35 |
| 43 |
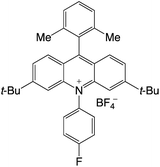 |
2.66 |
−0.55 |
2.11 |
419 |
493 |
17.6 |
35 |
| 44 |
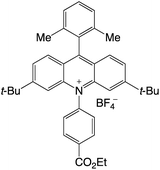 |
2.66 |
−0.54 |
2.12 |
421 |
494 |
18.4 |
35 |
| 45 |
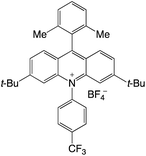 |
2.65 |
−0.51 |
2.14 |
421 |
495 |
20.7 |
35 |
| 46 |
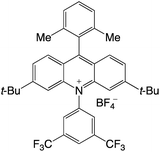 |
2.64 |
−0.45 |
2.19 |
425 |
497 |
20.8 |
35 |
| 47 |
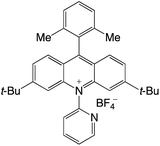 |
2.67 |
−0.50 |
2.17 |
419 |
511 |
2.7, 19.1 |
35 |
| 48 |
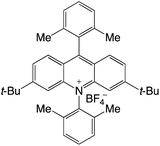 |
2.67 |
−0.53 |
2.14 |
421 |
488 |
22.8 |
35 |
| 49 |
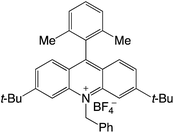 |
2.67 |
−0.54 |
2.13 |
422 |
493 |
23.7 |
35 |
| 50 |
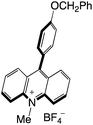 |
2.46 |
−0.58 |
1.88 |
264, 329, 345, 369, 453 |
565 |
6.4 |
41 |
| 51 |
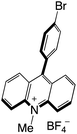 |
2.61 |
−0.51 |
2.10 |
264, 331, 347, 363, 411, 431, 460 |
485, 523 |
8.3 |
41 |
| 52 |
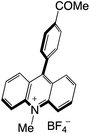 |
2.60 |
−0.49 |
2.11 |
264, 331, 346, 363, 407, 430, 458 |
486, 523 |
14.3 |
41 |
| 53 |
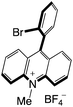 |
2.65 |
−0.48 |
2.17 |
264, 331, 347, 364, 407, 430, 457 |
481, 507 |
16.9 |
41 |
| 54 |
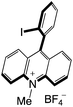 |
2.63 |
−0.48 |
2.15 |
264, 331, 348, 364, 410, 431, 459 |
507 |
<2 |
41 |
| 55 |
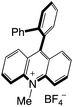 |
2.61 |
−0.52 |
2.09 |
264, 331, 347, 363, 412, 432, 459 |
487, 527 |
7.8 |
41 |
| 56 |
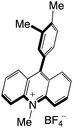 |
2.59 |
−0.56 |
2.03 |
263, 346, 360, 413, 435, 464 |
491, 524 |
<2 |
41 |
| 57 |
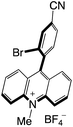 |
2.64 |
−0.42 |
2.22 |
264, 334, 349, 366, 406, 430, 458 |
482, 508, 544 |
19.2 |
41 |
| 58 |
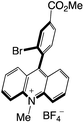 |
2.65 |
−0.44 |
2.21 |
264, 333, 349, 365, 409, 431, 458 |
481, 508, 545 |
18.0 |
41 |
| 59 |
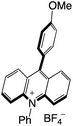 |
2.45 |
−0.52 |
1.93 |
266, 344, 361, 456 |
574 |
6.3 |
41 |
| 60 |
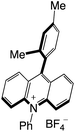 |
2.63 |
−0.51 |
2.12 |
265, 328, 345, 361, 412, 433, 462 |
491, 515 |
10.0 |
41 |
| 61 |
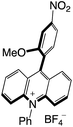 |
2.62 |
−0.43 |
2.19 |
265, 296, 334, 349, 364, 414, 434, 461 |
489, 515, 551 |
12.0 |
41 |
| 62 |
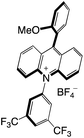 |
2.38 |
−0.42 |
1.96 |
267, 331, 347, 363, 415, 440, 468 |
436 |
6.4 |
41 |
| 63 |
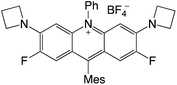 |
2.40 |
−0.98 |
1.42 |
458–513 |
494–551 |
3.7 |
40 |
| 64 |
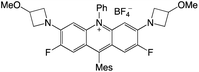 |
2.42 |
−0.94 |
1.48 |
458–513 |
494–551 |
3.4 |
40 |
| 65 |
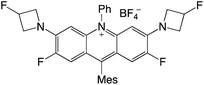 |
2.45 |
−0.89 |
1.56 |
458–513 |
494–551 |
3.4 |
40 |
| 66 |
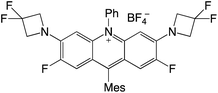 |
2.50 |
−0.80 |
1.70 |
458–513 |
494–551 |
3.2 |
40 |
| 67 |
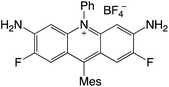 |
2.63 |
−0.99 |
1.64 |
458–513 |
494–551 |
3.9 |
40 |
| 68 |
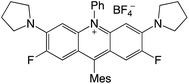 |
2.37 |
−0.97 |
1.40 |
458–513 |
494–551 |
0.6 |
40 |
| 69 |
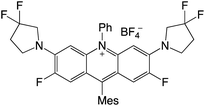 |
2.48 |
−0.85 |
1.61 |
458–513 |
494–551 |
5.1 |
40 |
| 70 |
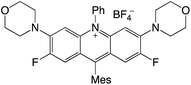 |
2.38 |
−0.73 |
1.65 |
458–513 |
494–551 |
5.4 |
40 |
| 71 |
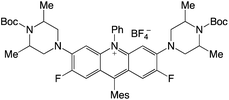 |
2.37 |
−0.73 |
1.64 |
458–513 |
494–551 |
4.7 |
40 |
| 72 |
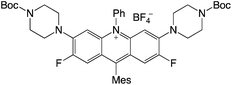 |
2.38 |
−0.73 |
1.65 |
458–513 |
494–551 |
4.3 |
40 |
| 73 |
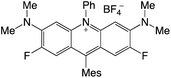 |
2.39 |
−0.89 |
1.50 |
458–513 |
494–551 |
3.1 |
40 |
| 74 |
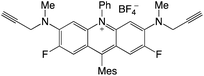 |
2.46 |
−0.75 |
1.71 |
458–513 |
494–551 |
4.4 |
40 |
| 75 |
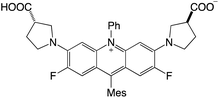 |
2.37 |
−1.06 |
1.31 |
458–513 |
494–551 |
1.4 |
40 |
| 76 |
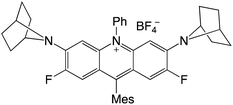 |
2.40 |
−0.81 |
1.59 |
458–513 |
494–551 |
4.2 |
40 |
| 77 |
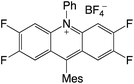 |
2.35 |
−0.33 |
2.02 |
458–513 |
494–551 |
5.9 |
40 |
| 78 |
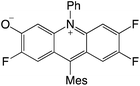 |
2.35 |
−1.20 |
1.51 |
458–513 |
494–551 |
5.4 |
40 |
| 79 |
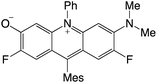 |
2.40 |
−1.36 |
1.03 |
458–513 |
494–551 |
5.02 |
40 |
| 80 |
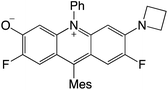 |
2.42 |
−1.41 |
1.01 |
458–513 |
494–551 |
4.8 |
40 |
| 81 |
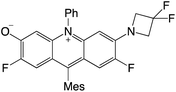 |
2.40 |
−1.36 |
1.03 |
458–513 |
494–551 |
5.4 |
40 |
5. Synthetic applications of acridinium-based photocatalysts
5.1. Amide bond formation
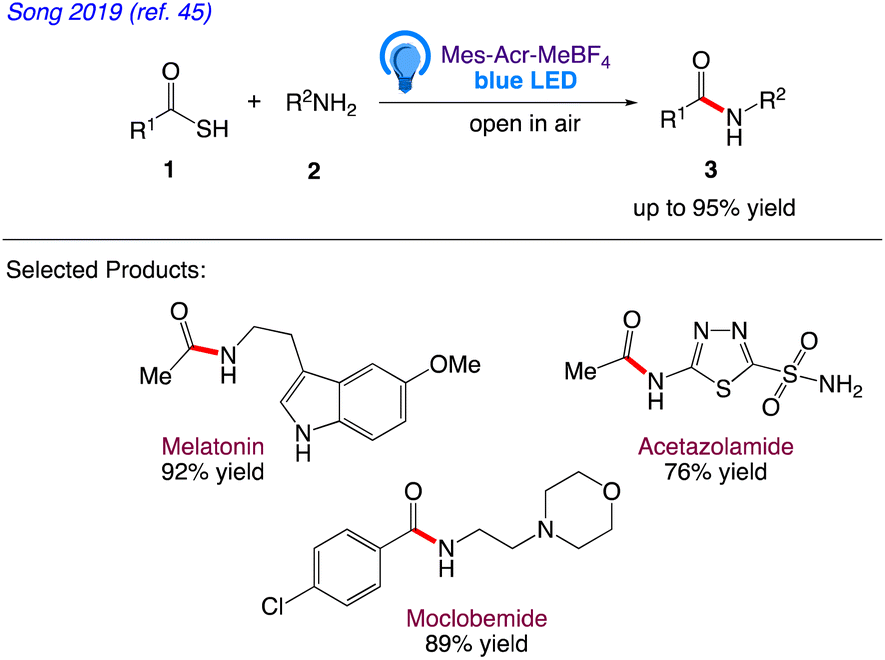
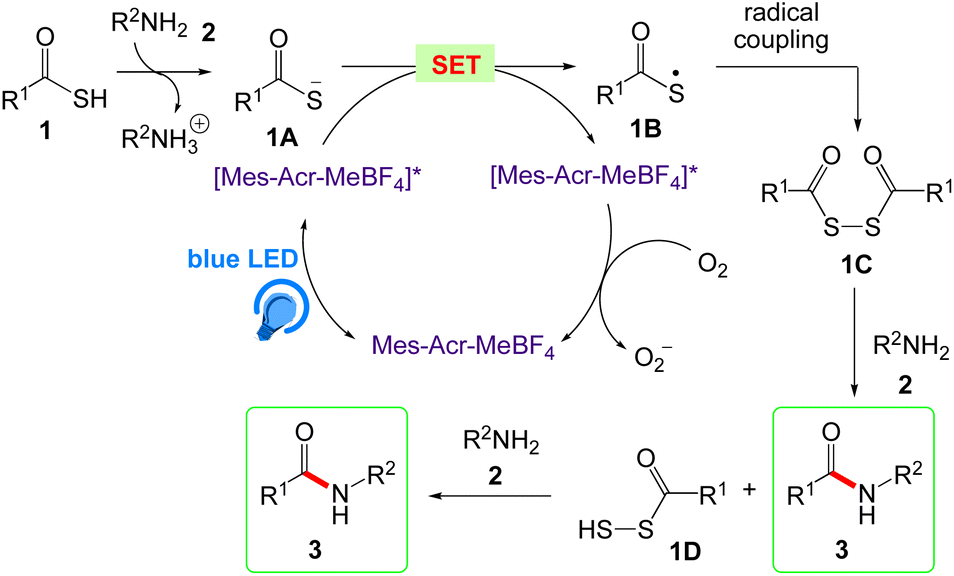
Song et al.45 reported an organic photo-redox catalysed metal, base and additive free amide bond formation. Without harming other functional groups like alcohols, phenols, ethers, esters, halogens, or heterocycles, this green technique demonstrated good functional selectivity.
This technique had a wide substrate range, was well compatible with water, air and had high yields. In this protocol, Song et al. has given the synthesis of amide bond formation by reaction between substituted thioacid 1 and primary amine 2 in presence of blue LED under an open air atmosphere (Scheme 5).
 |
| | Scheme 5 Acridinium catalyzed amide bond formation. | |
The general mechanism for the amide bond formation reaction by photoredox catalyst is proposed by Song et al. in Scheme 6. According to this mechanism, intermediate 1A was generated by deprotonation of thioacid 1 via reaction with amine 2. The blue LED photoexcite [Mes-Acr-MeBF4] to generate excited [Mes-Acr-MeBF4]*, that further reduced to the [Mes-Acr-MeBF4]˙ radical by electron-rich thioacid anion 1A. Photocatalysed thioacid anion 1A generates thioacid radical 1B via single-electron transfer (SET). The diradical coupling of thioacid radical 1B has given key disulfide 1C which followed by aminolysis with amine 2 to provide amide 3 and per thioacid 1D. The aminolysis of per thioacid 1D further results the amide 3.
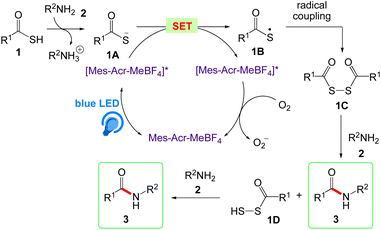 |
| | Scheme 6 Proposed mechanism of acridinium catalyzed amide bond formation. | |
5.2. Synthesis of enantioenriched α-deuterated α-amino acids
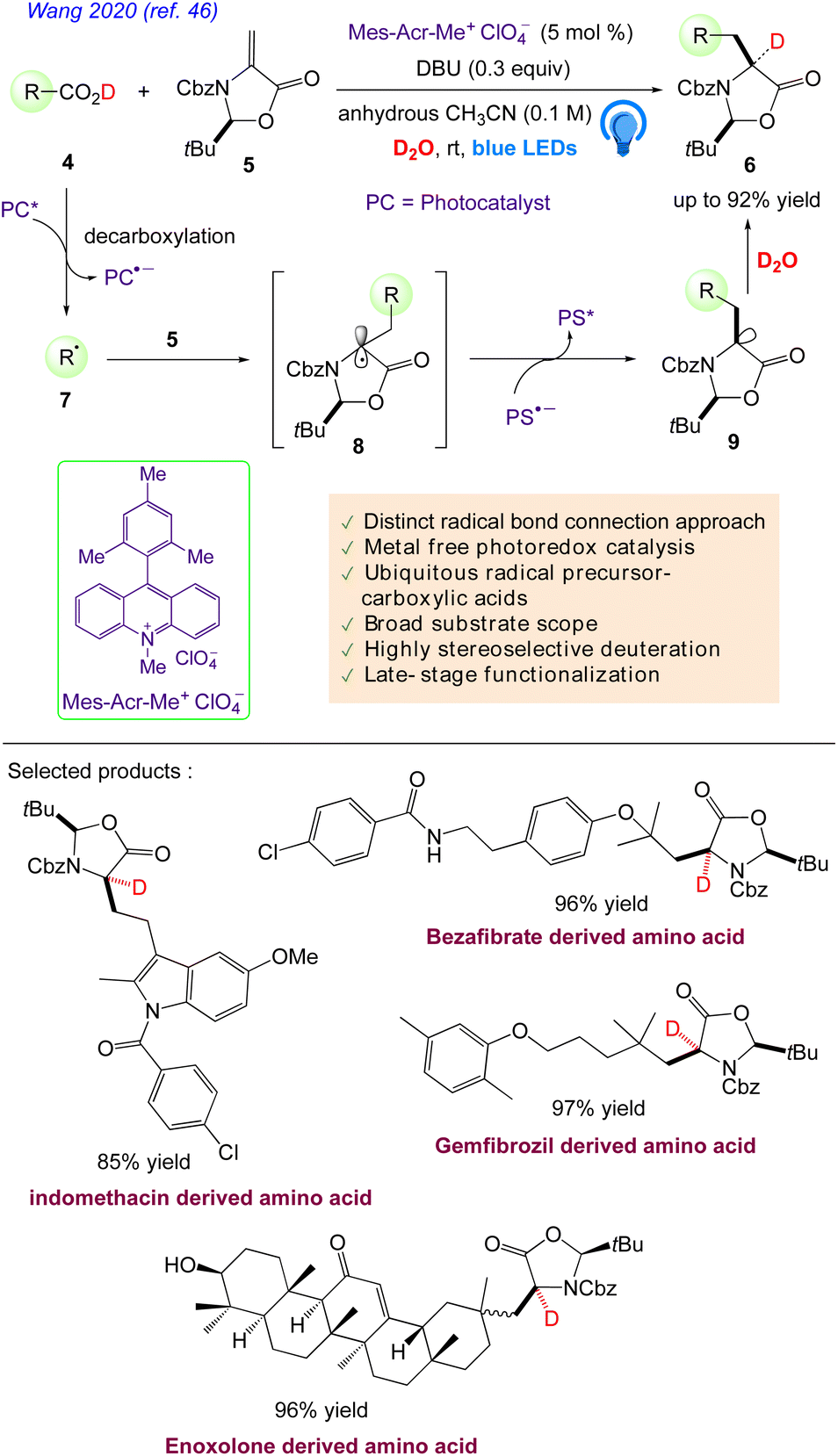
Wang et al. reported46 a mild, adaptable organophotoredox protocol for the formation of various, enantioenriched, deuterated α-amino acids.
This radical-based approach offers the unrivalled functionality of the convergent unification of readily available feedstock carboxylic acids and a chiral methyleneoxazolidinone fragment as well as the simultaneous highly diastereo, chemo, and regioselective incorporation of deuterium, which could vastly enhance the range of highly valuable deuterated amino acids for biological and medicinal purposes. They hypothesised that by using common, easily accessible alkyl carboxylic acids 4 as radical progenitors, the direct addition of a decarboxylative radical 7 to (S)-methyleneoxazolidinone 5 as a chiral inducer might result in enantioenriched amino acids 6 (Scheme 7). The synthesis of more structurally diverse amino acids is made possible by the easy accessibility of feedstock alkyl carboxylic acids 4. Additionally, researchers hypothesised that the Re-face- selective deuteration of the chiral anion intermediate 9 might offer a new method for creating enantioenriched deuterated amino acids 6. The simultaneous chemo, regio, and diastereoselective incorporation of large side chains and deuterium into amino acids is anticipated to be the driving force behind the strategy's efficacy.
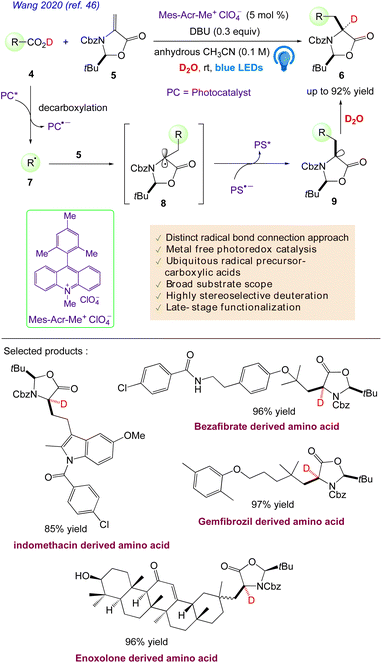 |
| | Scheme 7 Synthesis of enantioenriched α-deuterated α-amino acids. | |
5.3. Synthesis of quinolin-2(1H)-ones from quinoline N-oxides
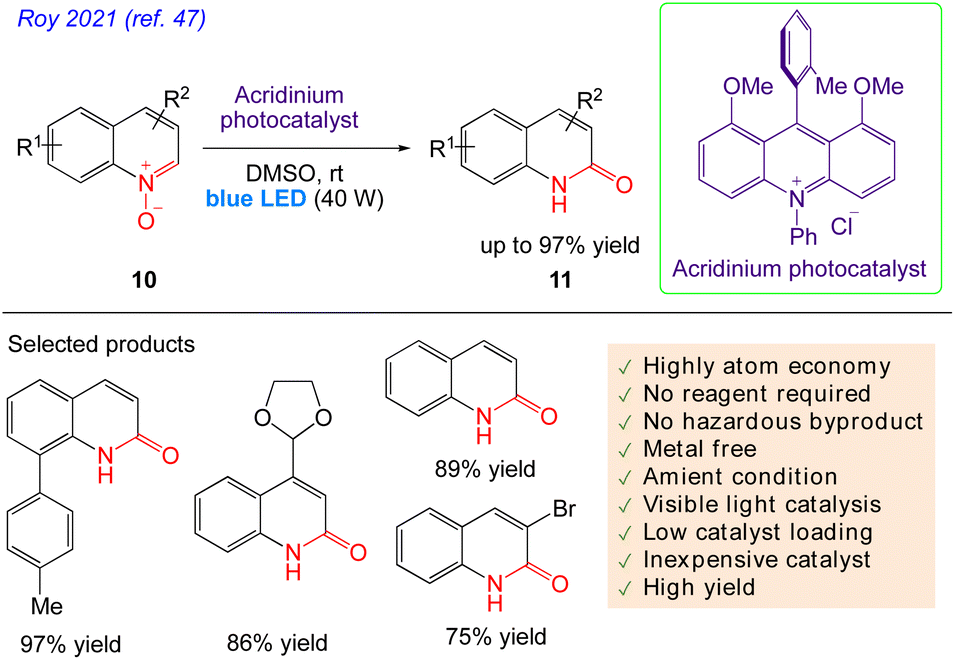
Roy et al.47 reported visible light catalysed an effective, simple procedure for the synthesis of a variety of quinolones and isoquinolones. No stoichiometric reagents are needed for this atom-efficient, environmentally benign protocol without formation of any by products. This very efficient and reagent-free photocatalytic technique involves the reaction of quinoline N-oxide 10, in presence of blue LED. The reaction successfully yielded the desired product 11 with excellent yield at room temperature (Scheme 8).
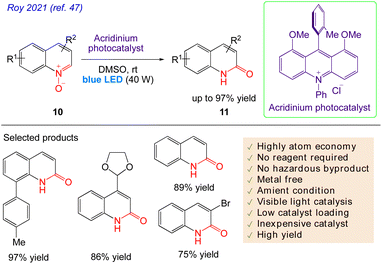 |
| | Scheme 8 Synthesis of quinolin-2(1H)-ones from quinoline N-oxides. | |
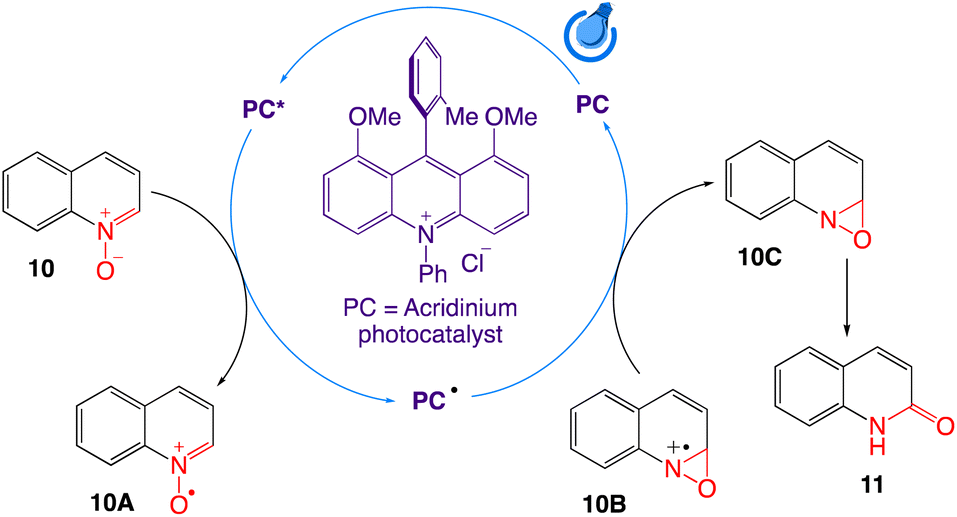
The authors have proposed a SET mechanism as shown in Scheme 9. Single electron donation of N-oxide 10 to the HOMO of the photocatalyst from the oxide resulted in a highly unstable nucleophilic quinolinium N-oxo radical 10A, which ultimately participates in attacking the vicinal electrophilic C-2 centre to form the kinetically favourable three membered oxaziridine radical cation ring 10B. The formed radical cation then acts as an acceptor of electrons from the singly occupied LUMO of the photocatalyst to form neutral but relatively unstable oxaziridine 10C. The weak N–O bond of oxaziridine 10C finally undergo isomerization to generate stable carbonyls via simultaneous hydride migration to generate the corresponding quinolin-2(1H)-ones 11 as the final product.
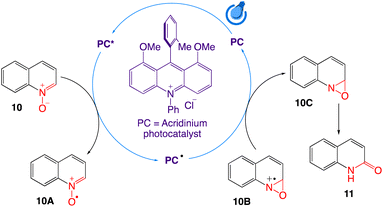 |
| | Scheme 9 Proposed mechanism for visible light photoredox catalytic synthesis of quinolin-2(1H)-ones. | |
5.4. C–H oxygenation of alkylarenes
Mancheño et al.48 reported a metals and additive free technique for the highly selective, photocatalyzed C–H oxygenation of alkylarenes 12 under air to the corresponding carbonyls 13. An imide–acridinium, which when exposed to visible light, transforms into a very potent photooxidant, that catalyses the entire reaction. The desired carbonyl compounds were obtained in good yields with excellent chemoselectivity. This process could also be applied to benzyl alcohols and alkylarenes, producing high yields of the related aldehydes and ketones. Different functional groups were tolerated well in both the aldehyde and ketone production processes because of the incredibly mild reaction conditions (Scheme 10).
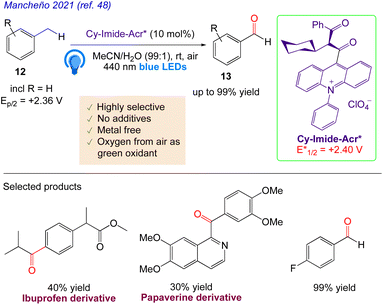 |
| | Scheme 10 C–H oxygenation of alkylarenes by photoredox catalysis. | |
5.5. Aliphatic C–H functionalization using pyridine N-oxides as H-atom abstraction agents
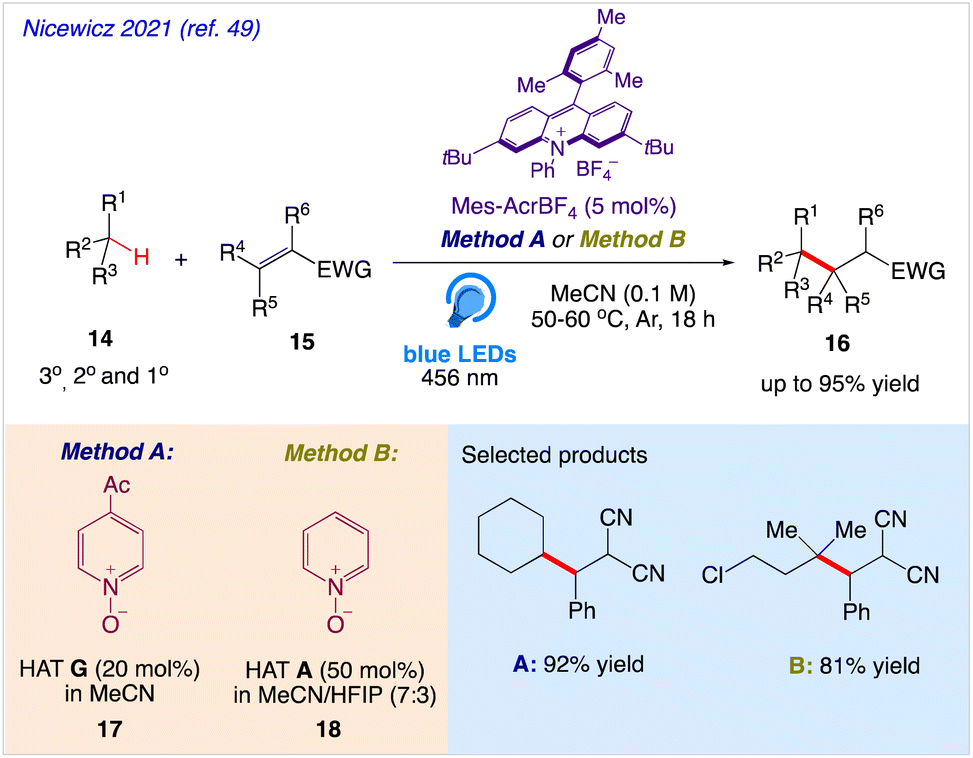
Nicewicz et al.49 reported a highly efficient approach for C–H alkylation reactions with the help of a synergistic combination of an acridinium photoredox catalyst and easily accessible pyridine N-oxides as HAT precursors. The abstraction of tertiary, secondary, and even powerful primary C–H bonds in the presence of electron donating and electron withdrawing moieties is made possible by this entirely organic method. In this protocol, unactivated tertiary, secondary, and primary C(sp3)H bonds 14 were successfully alkylated and heteroarylated with electron withdrawing olefins 15 under visible light (Scheme 11). This approach contained key intermediates that can readily have their reactivity changed by making structural changes.
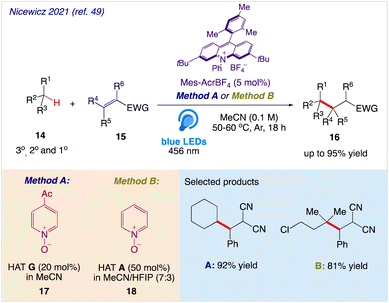 |
| | Scheme 11 C–H alkylation reaction using pyridine N-oxides as H-atom abstraction agents. | |
A plausible mechanism was proposed by authors, as depicted in Scheme 12. According to this mechanism, highly oxidizing Mes-Acr+* was generated by photoexcitation and pyridine N-oxide undergoes SET to become an N-oxy radical. A hydrogen atom can be abstracted from a C–H substrate by this electrophilic species (BDE = 99 kcal mol−1), which can subsequently produced an alkyl radical that interacts with an electron-deficient olefin. The resulting electrophilic radical alpha to the EWG was reduced by the acridine radical Mes-Acr followed by protonation from the N-hydroxy pyridinium to deliver the C–H alkylated product along with the photoredox catalyst Mes-Acr+ and pyridine N-oxide, that ultimately closing both the photo and HAT catalytic cycle.
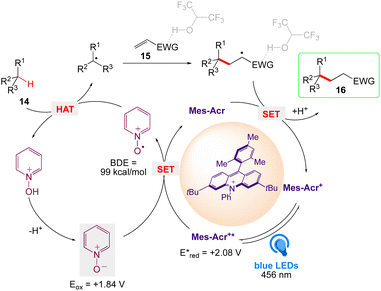 |
| | Scheme 12 Proposed mechanism of the C–H alkylation. | |
5.6. α-arylation of cyclic ketones
Gianetti et al.50 reported a photocatalytic cycle that activates a C(sp2)-X bond 19 (X = I, Br, Cl) and α -carbonyl C(sp3)-H 20 bond to produce ketones 21 from widely available aryl halides. A recently developed photoredox technique for the α-arylation of ketones has illustrated the potential value of this acridinium family. This work develops a metal-free photoredox method for-arylating ketones that is versatile and tolerant of a wide range of functional groups. On a multigram–scale reaction, the transformation has proven to be quite strong and clean. The synthesis of various economically significant building blocks for a wide range of bioactive and pharmacological substances serves as an important illustration of its value (Scheme 13).
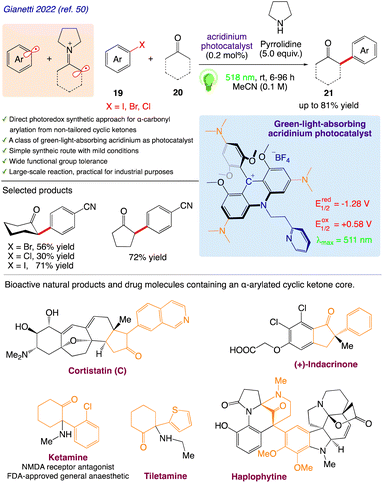 |
| | Scheme 13 Photoredox activation for the α-arylation of cyclic ketones. | |
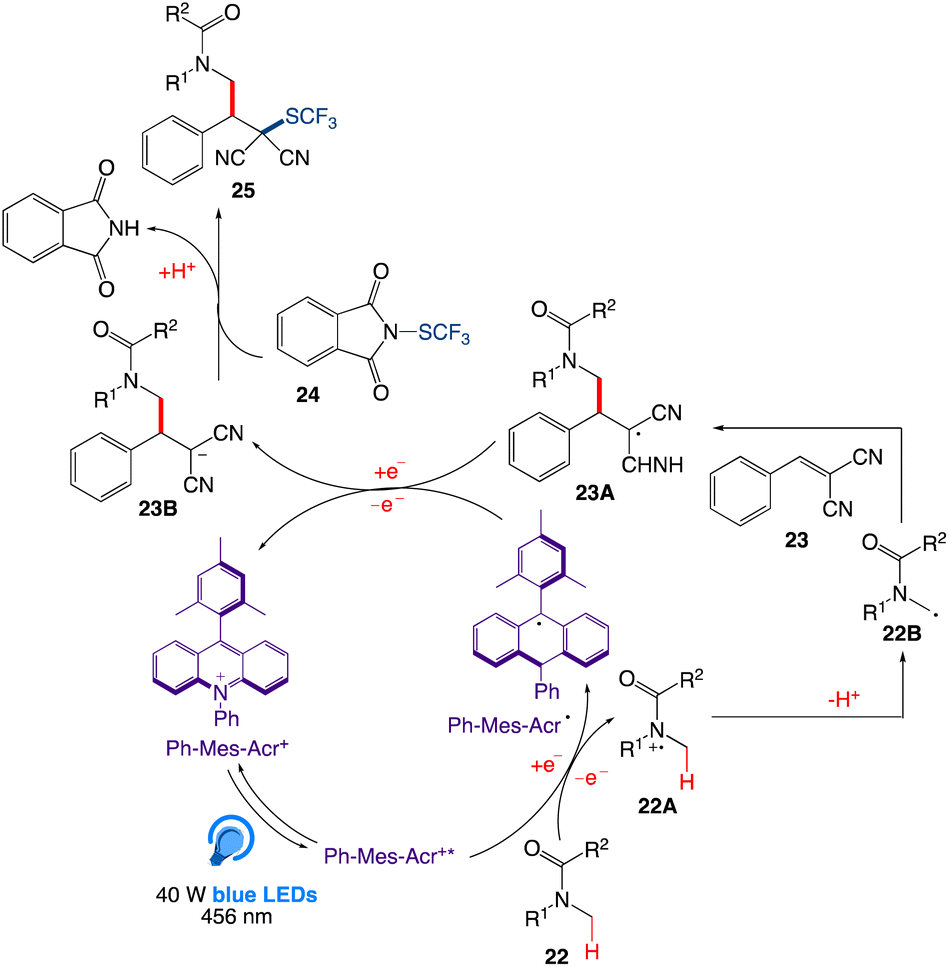
5.7. Multicomponent approach for tandem C(sp3)–H activation and alkylation followed by trifluoromethylthiolation
Sureshkumar et al.51 reported visible light driven one-pot tandem direct C(sp3)H activation and alkylation followed by trifluoromethylthiolation using an organophotocatalyzed multicomponent method. They presented a tandem, metal-free, three-component method for the photoinduced radical route for the difunctionalization of activated alkenes by utilising 5 equiv. of 22, 1 equiv of 23, 1.5 equiv. of “SCF3” source 24, acridinium photocatalyst to give the desired product 25 with excellent yield (Scheme 14).
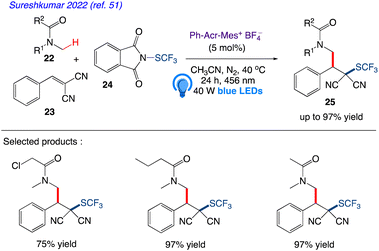 |
| | Scheme 14 Multicomponent approach for tandem C(sp3)–H activation and alkylation followed by trifluoromethylthiolation. | |
A plausible mechanism was proposed by author as depicted in Scheme 15. According to this mechanism, upon irradiation of organophotocatalyst Ph-Mes-Acr+ in presence of a blue LED, excited-state species Ph-Mes-Acr+* is generated, which emits an electron from 22 via a single-electron transfer (SET) process to generate the radical cation intermediate 22A along with the regeneration of Ph-Mes-Acr˙. After loosing a proton, intermediate 22A then generates radical intermediate 22B that reacts with alkene 23 to generate intermediate 23A. Intermediate 23A regenerates photocatalyst Ph-Mes-Acr+ by abstracting one electron from Ph-Mes-Acr˙. In the next step, intermediate 23A is converted to the anionic intermediate 23B, followed by the trifluoromehylthiolation by 24 to produce the final product 25.
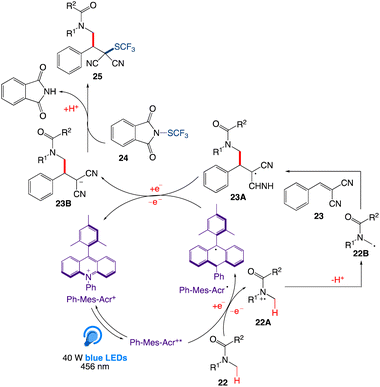 |
| | Scheme 15 Plausible reaction mechanism of C(sp3)–H activation and alkylation followed by trifluoromethylthiolation. | |
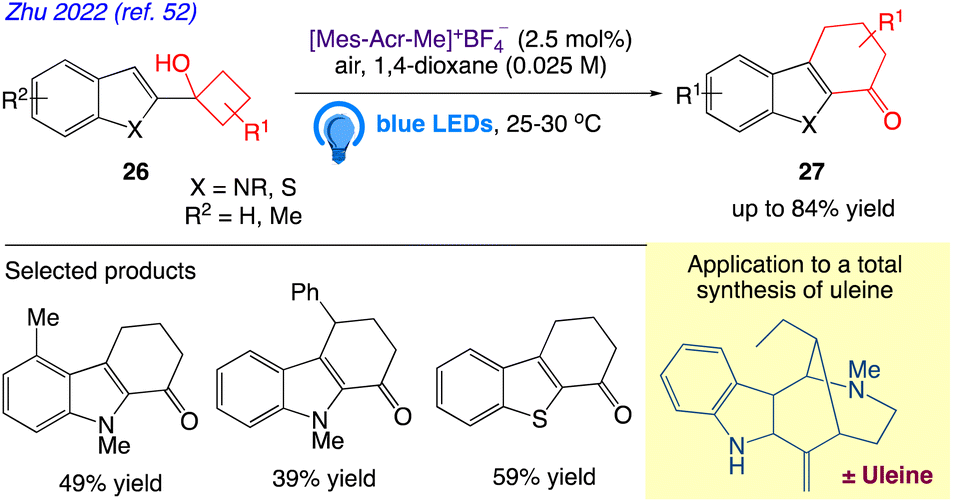
5.8. Two-carbon ring expansion of cyclobutanols to cyclohexenones
Zhu et al.52 reported visible-light photoredox catalysed an oxidative two-carbon homologation of cyclobutanols 26 to cyclohexenones 27 (Scheme 16). In this present protocol, tetrahydrocarbazol-1-ones with various functionalizations, as well as their thio-analogues, were easily produced from the commonly available 1-(1H-indol-2-yl)cyclobutan-1-ols and 1-(benzo[b]thiophen-2-yl)cyclobutan-1-ols owing to the reaction's broad substrate range. Subsequently, a straightforward complete synthesis of (±)-uleine that incorporates this two-carbon ring expansion step was developed.
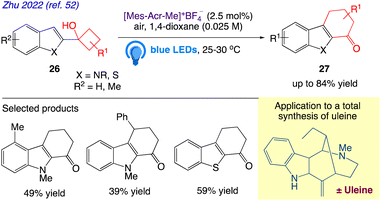 |
| | Scheme 16 Two-carbon ring expansion of cyclobutanols to cyclohexenones. | |
A plausible mechanism, proposed by author is depicted in Scheme 17. According to this mechanism a SET oxidation of indole 26 via the excited acridinium salt to radical cation 26A, produced benzylic radical 26B through a pinacol rearrangement. A second SET from 26B to either MesAcr+* afforded the benzylic cation 26C, which further undergo a Wagner–Meerwein 1,2-alkyl shift to afforded cation 26D. Finally, a rearomatization of 26D has given product 27 and oxidation of MesAcr˙ by oxygen to MesAcr+ complete the photoredox catalytic cycle.
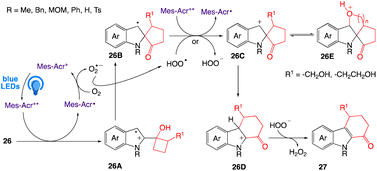 |
| | Scheme 17 Proposed mechanism of two-carbon ring expansion of cyclobutanols to cyclohexenones. | |
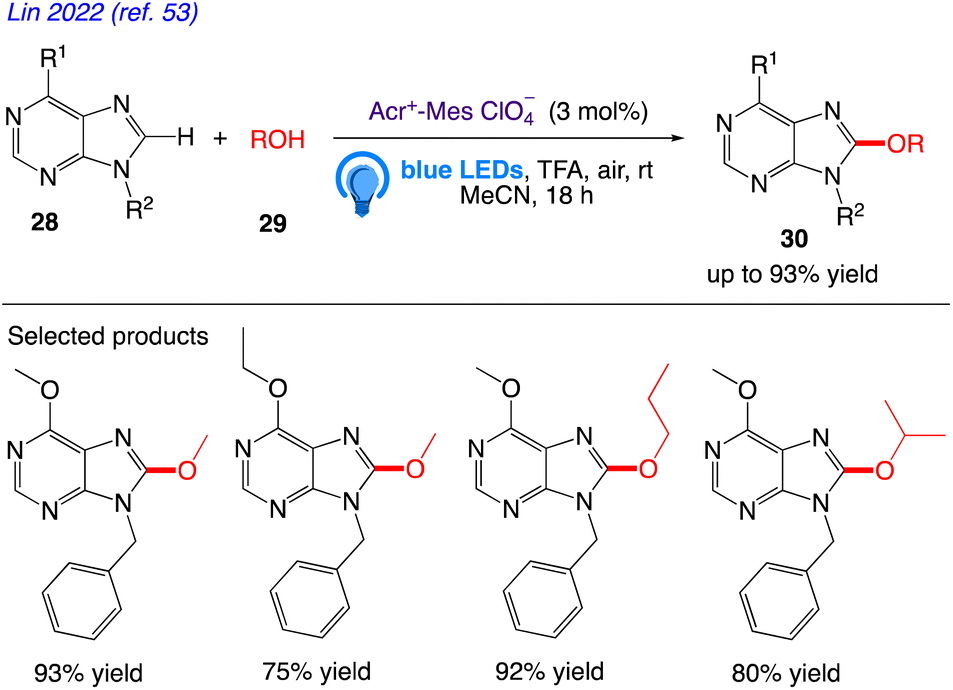
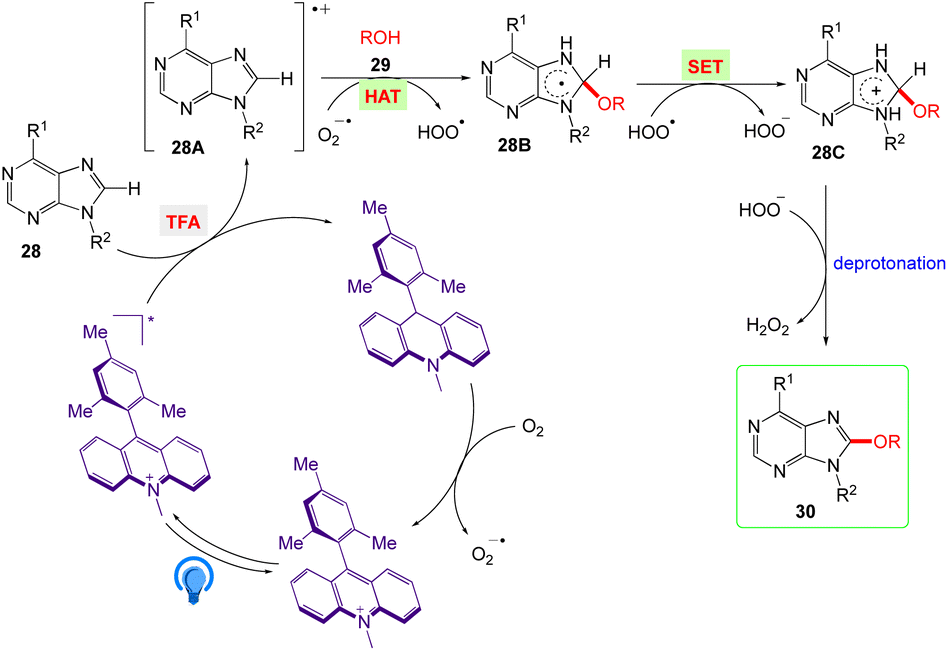
5.9. Purine C8 alkoxylation with alcohol
Lin et al.53 reported the visible light mediated and acridinium catalysed cross-dehydrogenation coupling reaction between purines 28 and alcohols 29 to synthesise a number of C8-alkoxy purine derivatives 30, under an air atmosphere, which served as the only oxidant in the reaction (Scheme 18). With good to excellent yields, these mild reaction conditions provide a wide variety of C8-alkoxylated compounds.
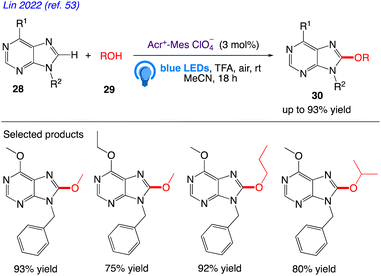 |
| | Scheme 18 Synthesis of C8 alkoxylated purine derivatives. | |
The plausible mechanism as proposed by authors are depicted in Scheme 19. According to this mechanism, The Acr+-Mes ClO4− photocatalyst is converted into it's excited [Acr+-Mes ClO4−]* via irradiation of blue LEDs. Further, the excited state via a single electron transfer (SET) process with 28 generates the purine radical cation 28A and the Acr-Mes radical. The Acr-Mes radical is then oxidized by O2 and completes the photocatalytic cycle. The alcohol (ROH) 29 and purine radical cation 28A reacts to generate the alkoxide adduct radical 28B. Subsequently, 28B undergoes an SET process with HOO˙ and generates intermediate 28C. Finally, deprotonation of 28C yields the target product 30, and HOO− acquires the proton to give H2O2.
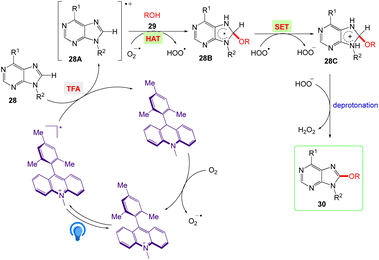 |
| | Scheme 19 Proposed reaction mechanism of synthesis of C8 alkoxylated purine derivatives. | |
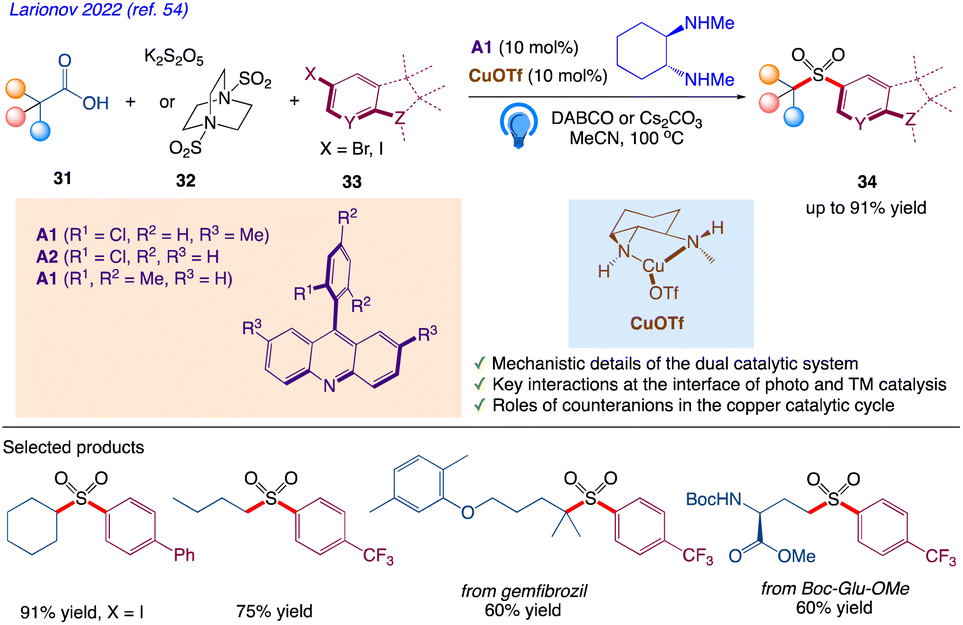
5.10. Tricomponent decarboxysulfonylative cross-coupling with aryl halides
Larionov et al.54 reported a visible light-induced, dual catalytic, direct decarboxysulfonylative cross-coupling of carboxylic acids with aryl halides. This reaction includes tricomponent decarboxysulfonylative cross-coupling of carboxylic acid 31 with aryl iodide 33 occurs readily in the dual catalytic system of acridine photocatalyst and diamine ligated copper(I) triflate, with DABCO or potassium metabisulfite 32 that results sulfone 34 with 91% yield (Scheme 20).
 |
| | Scheme 20 Tricomponent decarboxysulfonylative cross-coupling with aryl halides. | |
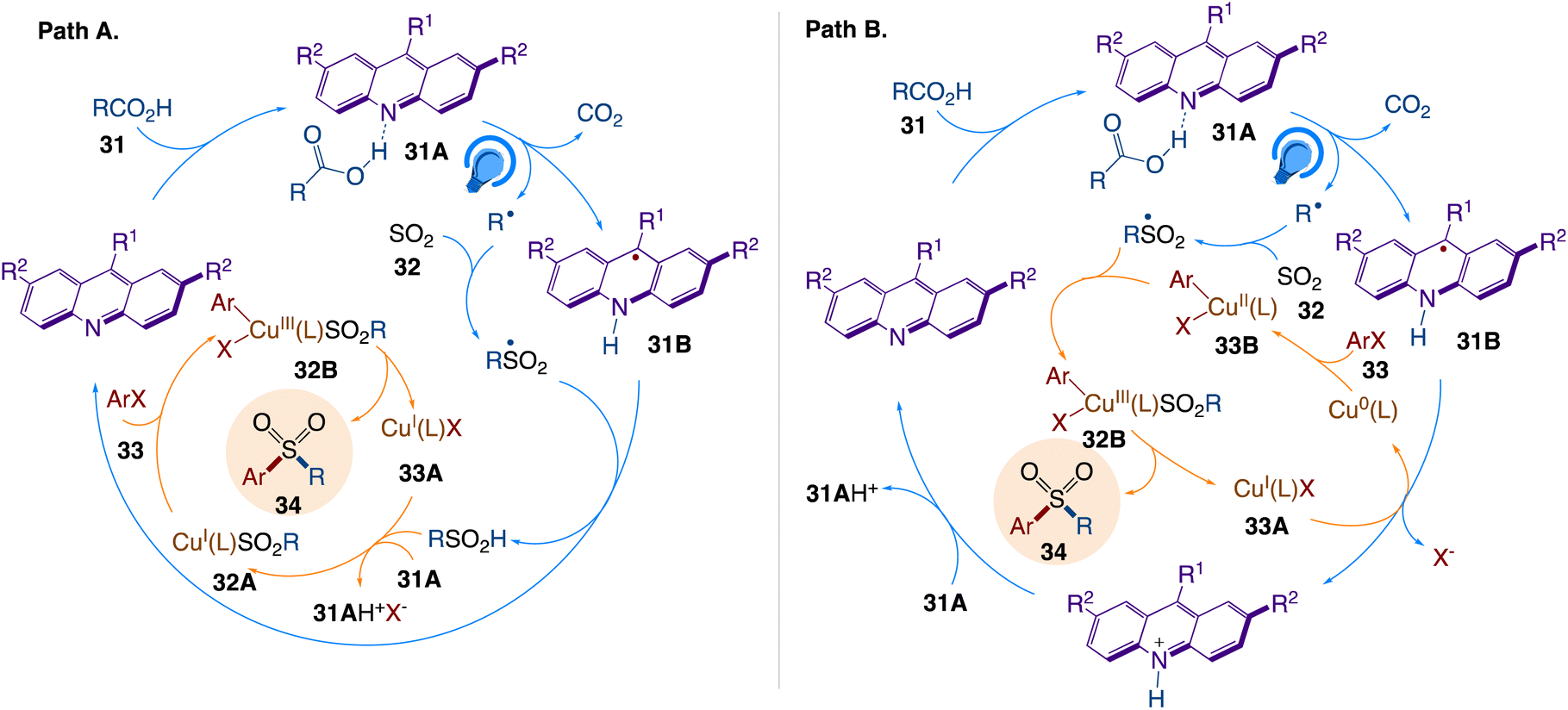
The plausible mechanism as proposed by author is depicted in Scheme 21. According to the mechanism, sulfonyl radical generates sulfinic acid via hydrogen abstraction from the acridinyl radical 31B, emerging from the photoinduced PCET-enabled decarboxylation in complex 31A (path A, Scheme 21). The base-mediated anion exchange then converts CuI complex 33A to sulfinate intermediate 32A that undergo an oxidative addition with an aryl halide via intermediate 32B, followed by reductive elimination, giving rise to the sulfone product in an overall orthogonal relay dual catalytic process. Acridinyl radical-mediated formation of Cu0 species can also enable an alternative mechanism for the aromatic decarboxysulfonylation that proceeds through an oxidative addition of the aryl halide to the Cu0 intermediate, producing arylcopper species 33B (path B, Scheme 21). The copper intermediate 32B is produced by further cross-termination with the sulfonyl radical, and the sulfone product is then produced by reductive elimination.
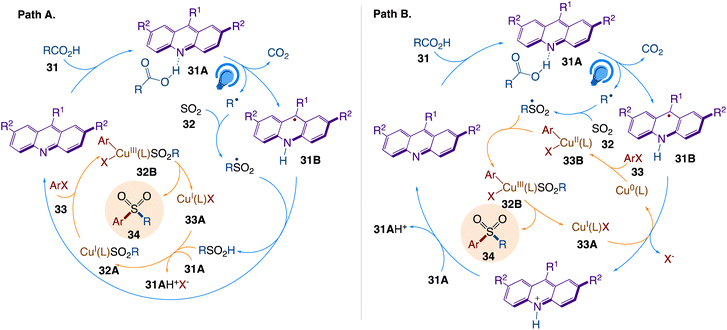 |
| | Scheme 21 Mechanistic pathways of the dual catalytic decarboxysulfonylation. | |
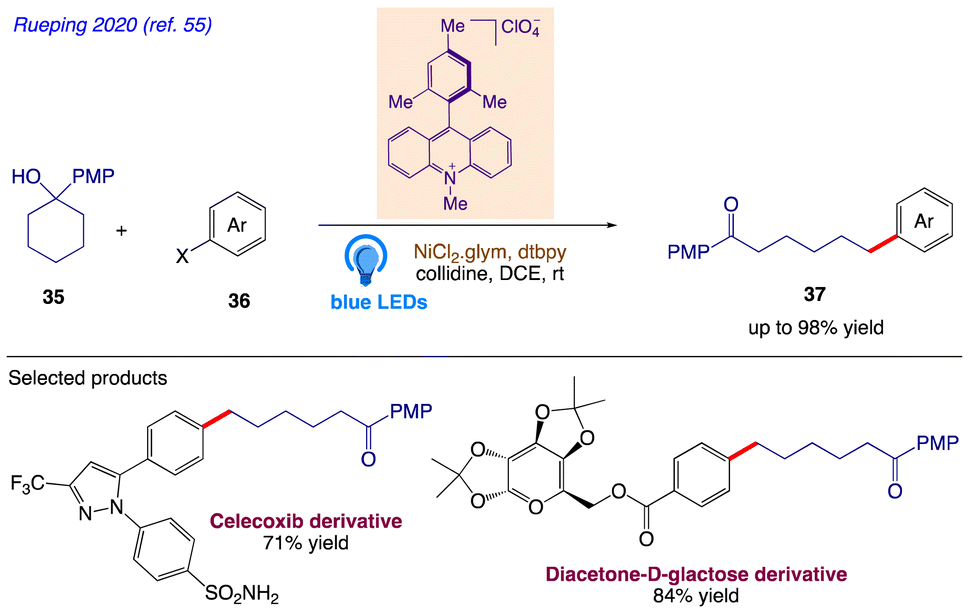
5.11. Cross-coupling arylation
Rueping et al.55 reported a synthetic protocol for the site-specific arylation of ketones 37 from tertiary alcohols 35 and 4-bromoacetophenone 36 via photoredox-enabled MS-PCET and nickel catalysis (Scheme 22).
 |
| | Scheme 22 Arylation of ketones from easily accessible tertiary alcohols through a photoredox-enabled MS-PCET and nickel catalysis. | |
The plausible mechanism as proposed by author is depicted in Scheme 23. According to the mechanism, upon irradiation with visible light, the ground-state of Mes-Acr-Me+ is promoted to its highly oxidizing singlet excited state *Mes-Acr-Me+, followed by a single electron transfer with the tertiary alcohol 35, furnishing the corresponding arene radical cation along with the reduced form of the photocatalyst Mes-Acr-Me˙. Subsequent deprotonation (PT) and intramolecular ET reaction between the alkoxide and the radical cation gives the key alkoxyl radical species, which readily cleaves into a carbonyl moiety and a distal carbon-centered radical through scission of the neighboring C–C bond at the β position. The alkyl radical is then captured by the Ni(0) and gives an alkylnickel(I) intermediate, which rapidly undergo oxidative addition with an aryl halide, generating a Ni(III) species. The cross-coupled product and the Ni(I) intermediate are then produced by reductive elimination at this point; the latter can be reduced by the reduced form of the photocatalyst and regenerate both at once, completing the catalytic cycle.
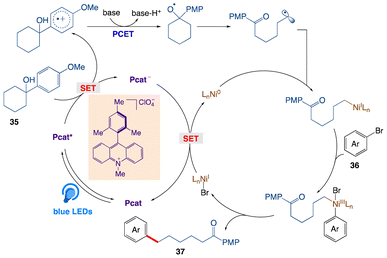 |
| | Scheme 23 Proposed mechanism for C–C activation/cross coupling. | |
5.12. Homobenzylic oxygenation
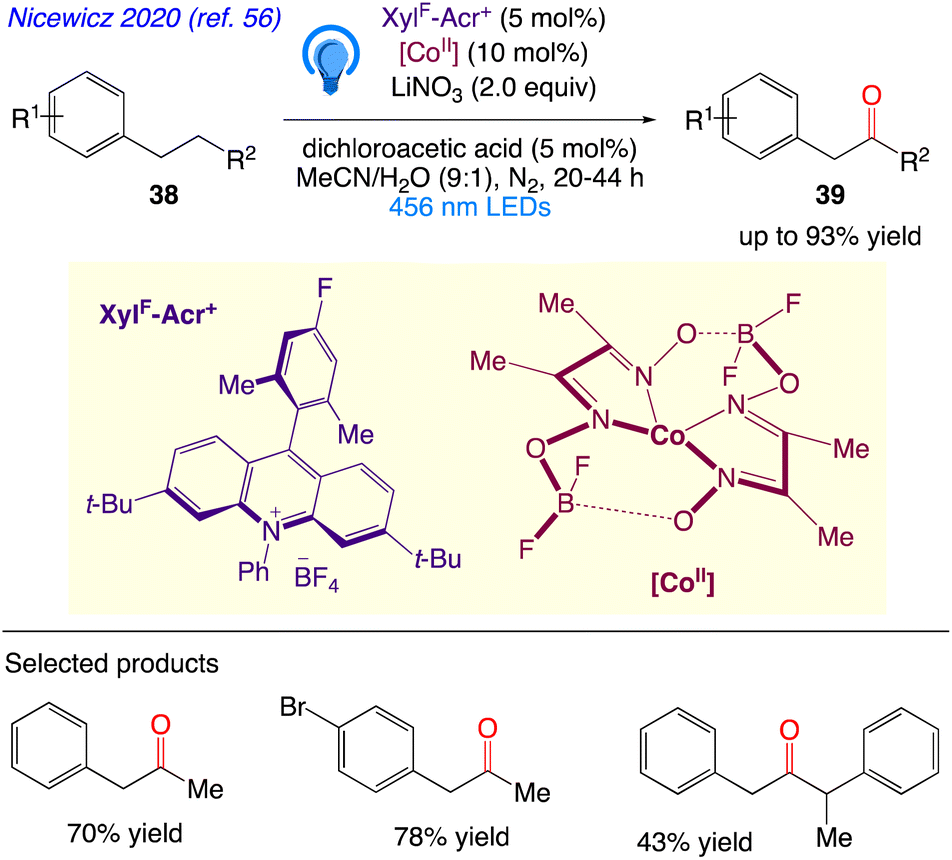
Nicewicz et al.56 reported protocol for the selective oxidation of typically inert C–H bonds. In this synthetic protocol, they demonstrated that utilising a combination of dual organic photoredox and cobalt catalysis, it is possible to utilize of the reactivity of benzylic C(sp3)H bonds at the homobenzylic site. In a two-part catalytic system, alkyl arenes 38 are dehydrogenated, followed by an anti-Markovnikov Wacker-type oxidation, to produce benzyl ketone 39 products (Scheme 24).
 |
| | Scheme 24 Site-selective homobenzylic oxidation. | |
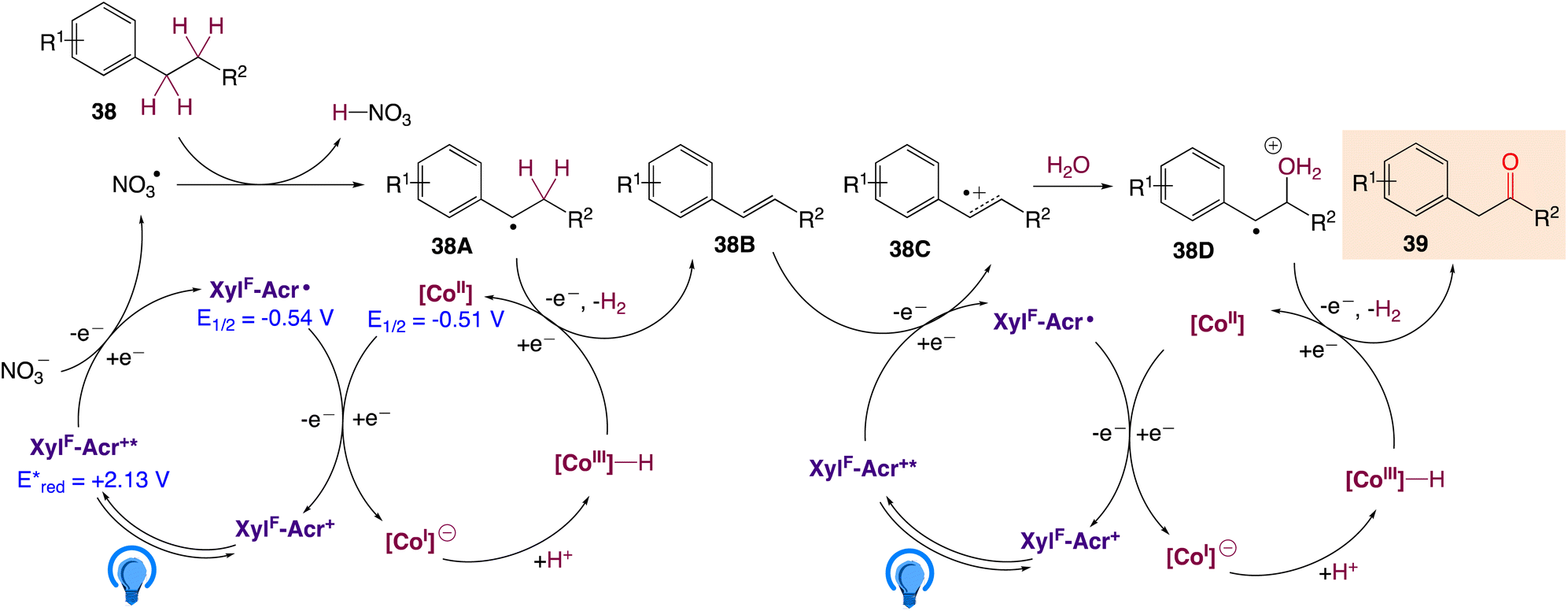
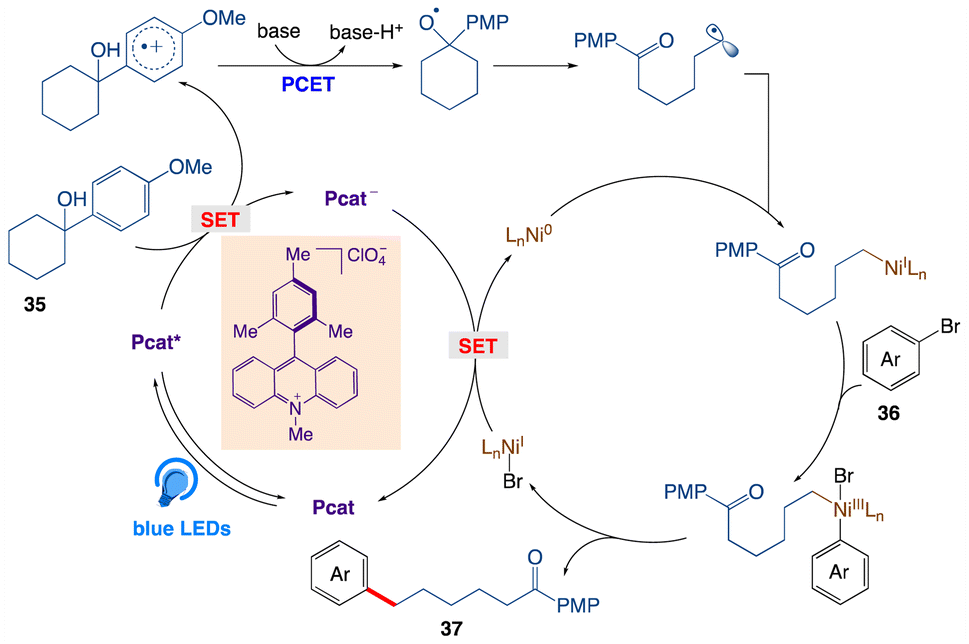
A plausible mechanism, as proposed by authors is depicted in Scheme 25. According to this mechanism, initial oxidation of the nitrate anion (Epp/2ox = +1.97 V) results from the excited state photooxidant, XylF-Acr+* 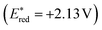 . The resulting nitrate radical is a potent H-atom abstracting agent, allowing it to excise the weak C(sp3)–H bond of 38 generating HNO3 and benzylic radical 38A. XylF-Acr˙ (E1/2ox = −0.54 V) can feasibly undergo single electron transfer (SET) with [CoII] (E1/2red = −0.51 V) to regenerate XylF-Acr+ and a reduced cobalt complex (vide infra). [CoI]− likely undergoes protonation to form [CoIII]–H assisted by the aforementioned acid additive. 38A is then intercepted by [CoIII]–H, liberating H2 and 38B. One possibility of this mechanism is the direct protonation of [CoIII]–H. This would form [CoIII]+ (E1/2red ∼ +0.2 V), which could potentially oxidize 38A (E1/2ox ∼ +0.37 V). While this SET is endergonic by about +0.2 V (4.6 kcal mol−1), rapid deprotonation of the resulting benzylic cation intermediate would render SET irreversible, ultimately generating styrene 38B. Alternatively two molecules of [CoIII]–H could undergo a bimolecular reductive elimination of H2 generating two equivalents of [CoII]. 38B would then be formed via the addition of 38A to [CoII], forming a putative [CoIII] alkyl intermediate capable of undergoing a net β-hydride elimination. 38B can then engage in a second catalytic cycle to form the olefin radical cation (38C) whereupon trapping with water would afford a distonic radical cation (38D). Subsequent deprotonation and a second dehydrogenation would furnish the desired product 39 via a sequence similar to the mechanisms proposed by Lei and Nicewicz.
. The resulting nitrate radical is a potent H-atom abstracting agent, allowing it to excise the weak C(sp3)–H bond of 38 generating HNO3 and benzylic radical 38A. XylF-Acr˙ (E1/2ox = −0.54 V) can feasibly undergo single electron transfer (SET) with [CoII] (E1/2red = −0.51 V) to regenerate XylF-Acr+ and a reduced cobalt complex (vide infra). [CoI]− likely undergoes protonation to form [CoIII]–H assisted by the aforementioned acid additive. 38A is then intercepted by [CoIII]–H, liberating H2 and 38B. One possibility of this mechanism is the direct protonation of [CoIII]–H. This would form [CoIII]+ (E1/2red ∼ +0.2 V), which could potentially oxidize 38A (E1/2ox ∼ +0.37 V). While this SET is endergonic by about +0.2 V (4.6 kcal mol−1), rapid deprotonation of the resulting benzylic cation intermediate would render SET irreversible, ultimately generating styrene 38B. Alternatively two molecules of [CoIII]–H could undergo a bimolecular reductive elimination of H2 generating two equivalents of [CoII]. 38B would then be formed via the addition of 38A to [CoII], forming a putative [CoIII] alkyl intermediate capable of undergoing a net β-hydride elimination. 38B can then engage in a second catalytic cycle to form the olefin radical cation (38C) whereupon trapping with water would afford a distonic radical cation (38D). Subsequent deprotonation and a second dehydrogenation would furnish the desired product 39 via a sequence similar to the mechanisms proposed by Lei and Nicewicz.
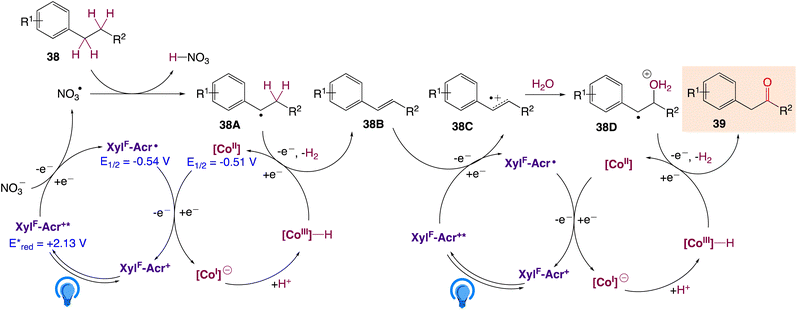 |
| | Scheme 25 Mechanistic proposal for LiNO3-mediated homobenzylic oxidation. | |
5.13. Synthesis of alkene-containing N-heterocycles
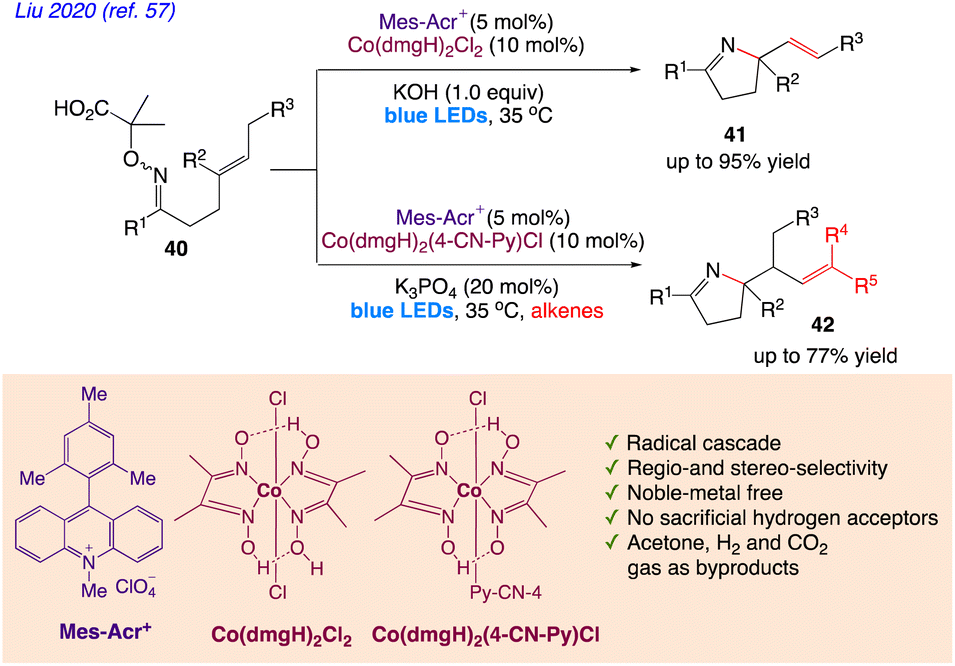
Liu et al.57 reported a synthetic protocol for radical aza-cyclization of α-imino-oxy acids 40 with pendant alkenes 41 and 42 via synergistic photoredox and cobaloxime catalysis (Scheme 26). With the fusion of cobalt catalysis and photoredox organocatalysis, they devised the iminyl-radical-mediated cyclization technique.
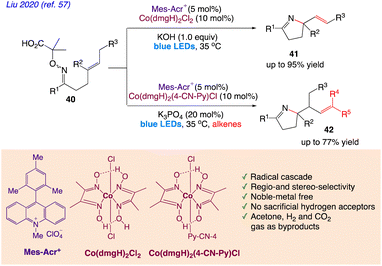 |
| | Scheme 26 Radical aza-cyclization of α-imino-oxy acids for synthesis of alkene-containing N-heterocycles. | |
The plausible mechanism as proposed by author is depicted in Scheme 27. According to this mechanism, initial step involves a single oxidation of the carboxylate derived from the α-imino-oxy acid 40 and base by Mes-Acr+*, generating an iminyl radical 40A with loss of CO2 and acetone, followed by 5-exo cyclization to deliver an alkyl radical 40B. The reduced photocatalyst Mes-Acr˙ could be single-electron oxidized by a CoIII complex to close the photoredox catalytic cycle, as well as the generation of a CoII complex. Similarly, the alkyl radical 40B can react with the CoII complex to form an organocobalt(III) complex 40C. The critical β-H elimination step readily occurs to furnish the Heck-type product 41 and a CoIII–H intermediate. The reaction of the CoIII–H complex with a proton leads to H2 extrusion and completion of the cobalt catalytic cycle.
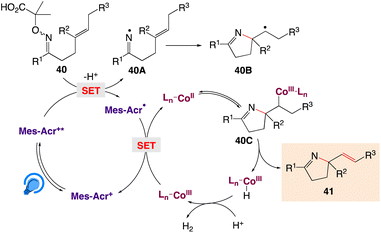 |
| | Scheme 27 Proposed Mechanism for synthesis of alkene-containing N-heterocycles. | |
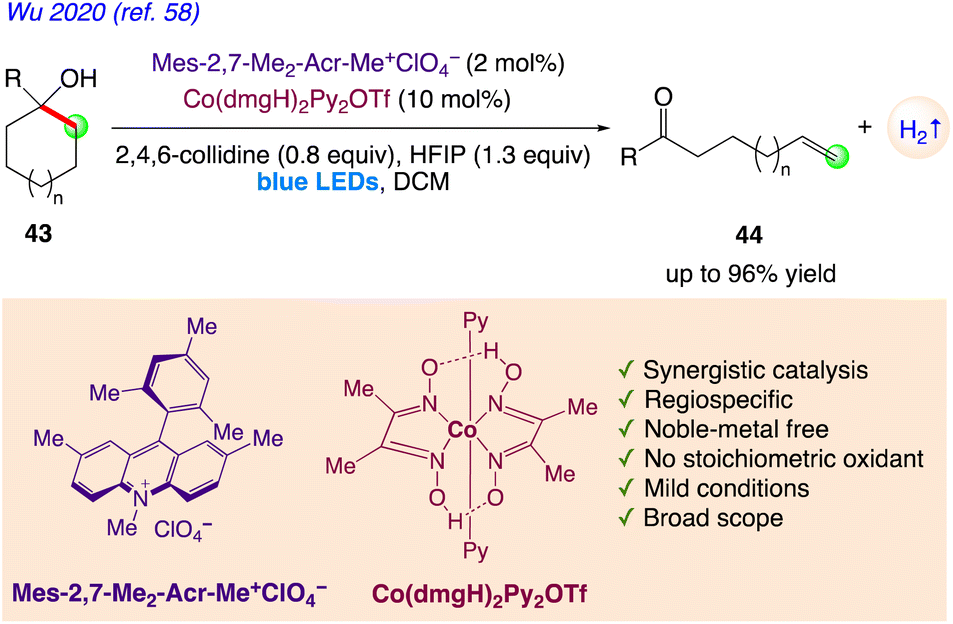
5.14. Regiospecific synthesis of distally unsaturated ketones with hydrogen evolution
Wu et al.58 reported a photoredox/cobalt dual catalytic system for the synthesis of distally unsaturated ketones 44 from dehydrogenation of nonstrained tertiary cycloalkanols 43 using the dual catalyst system of photosensitizer Mes-2,7-Me2-Acr-Me+ and cobaloxime catalyst Co(dmgH)2Py2OTf under visible-light irradiation (Scheme 28).
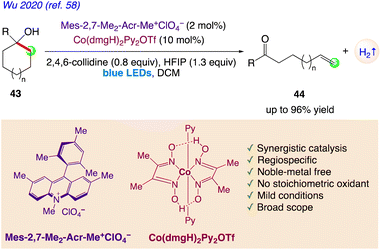 |
| | Scheme 28 Regiospecific synthesis of distally unsaturated ketones with hydrogen evolution. | |
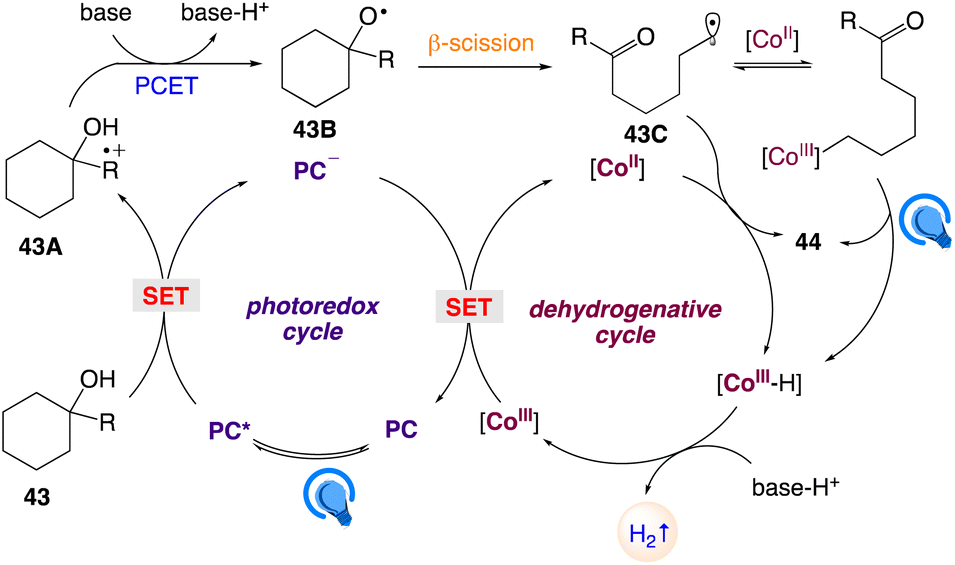
The plausible mechanism as proposed by author is depicted in Scheme 29. According to this mechanism, visible-light-excitation of Mes-2,7-Me2-Acr-Me+ followed by SET with para-methoxyphenyl group of the tertiary alcohol 43 gave rise to the corresponding arene radical cation intermediate 43A along with the reduced form of the photocatalyst Mes-2,7-Me2-Acr-Me˙. This reduced photocatalyst Mes-2,7-Me2-Acr-Me˙ further oxidized by the CoIII catalyst to regenerate the photocatalyst and generates CoII. In presence of an exogenous Bronsted base alkoxyl radical 43B is generated by subsequent intramolecular PCET between the arene radical cation 43A and the adjacent hydroxyl group, which undergoes C–C bond β-scission to form the ring-opened distal-carbonyl alkyl radical 43C. The alkyl radical 43C is then trapped by the CoII species to give an alkyl–CoIII intermediate, which undergoes photoinduced homolytic cleavage of the Co–C bond followed by β-H abstraction via CoII to gives a CoIII hydride and the desired product 44. On the other hand, a direct HAT on the alkyl radical 43C by CoII, without the intermediacy of alkyl–CoIII, cannot be excluded. Protonation of the CoIII hydride emits H2 and regenerates the CoIII catalyst to complete the cycle.
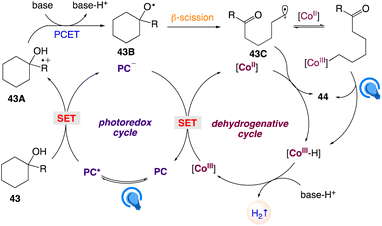 |
| | Scheme 29 Proposed reaction mechanism for regiospecific synthesis of distally unsaturated ketones with hydrogen evolution. | |
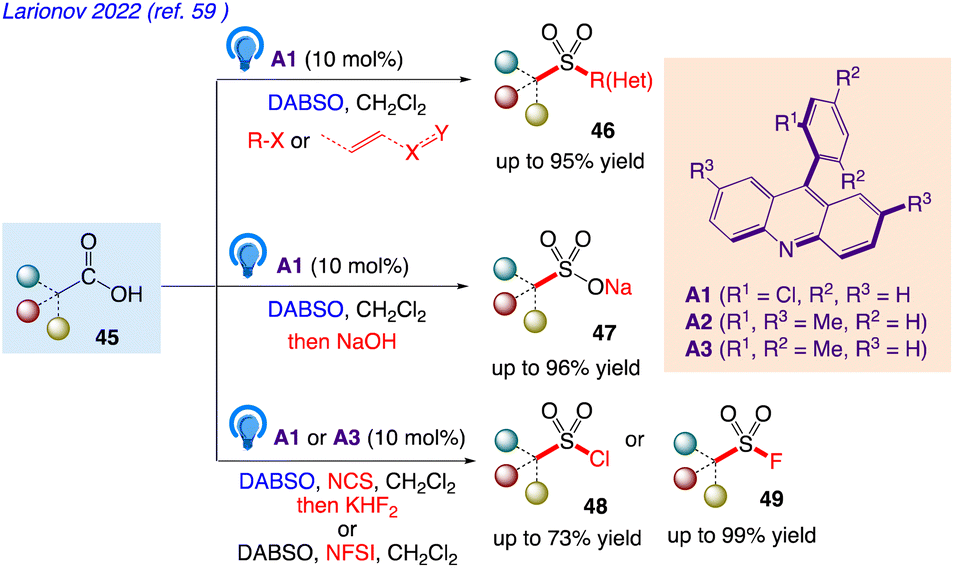
5.15. Functional group-divergent decarboxysulfonylation by acridine photocatalysis
Larionov et al.59 reported a photocatalytic system for direct decarboxylative conversion of carboxylic acids 45 to sulfones 46 and sulfinates 47, as well as sulfonyl chlorides 48 and fluorides 49 in single step and in a multicomponent fashion (Scheme 30).
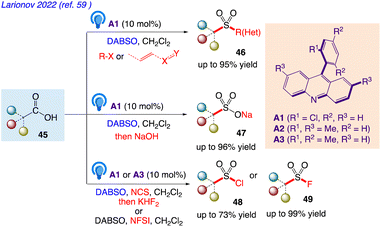 |
| | Scheme 30 Functional group-divergent decarboxysulfonylation by acridine photocatalysis. | |
This synthetic protocol includes visible light-induced, organophotocatalytic reaction which includes conversion into sulfones, sulfinates, sulfonyl chlorides, and sulfonyl fluorides via direct tricomponent decarboxylative coupling of carboxylic acids.
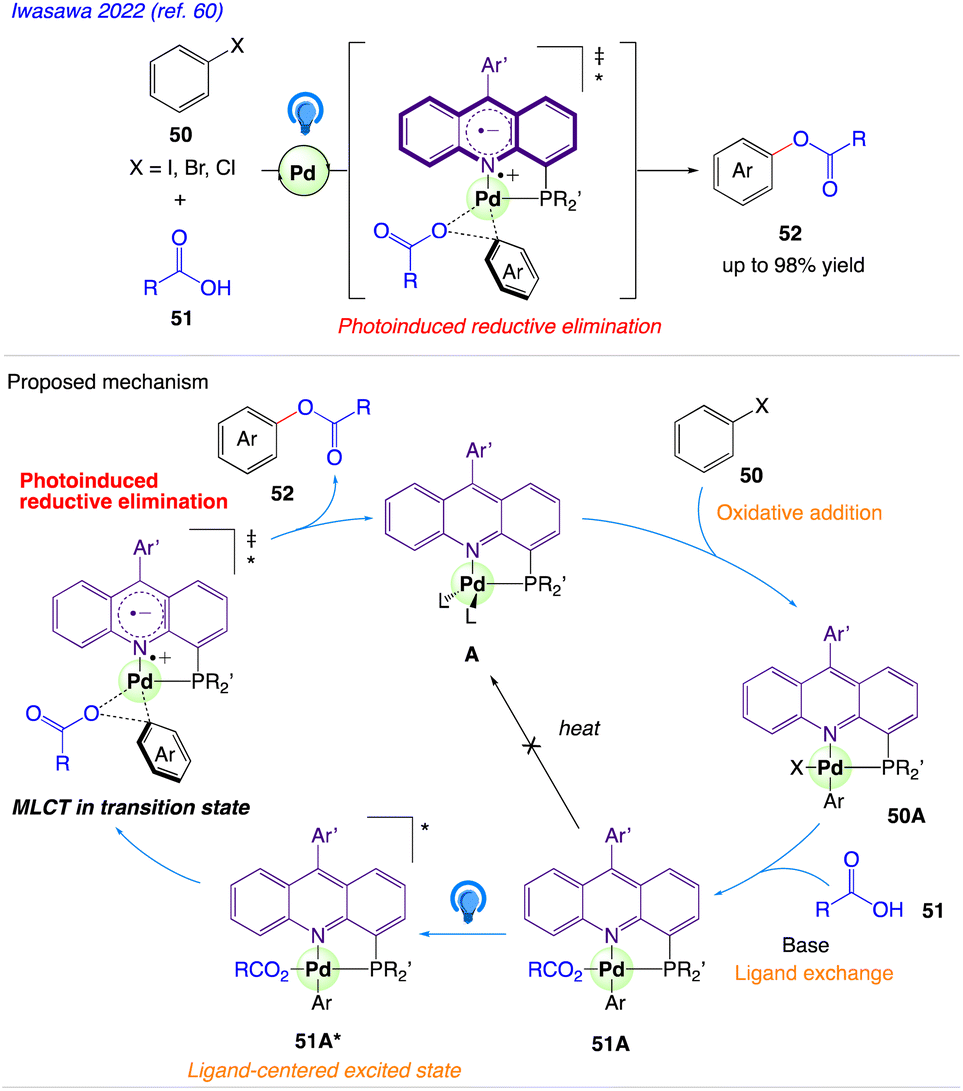
5.16. Reductive elimination
Iwasawa et al.60 reported a 4-phosphinoacridine ligands catalysed synthetic protocol for visible-light-induced transition-metal catalysis and developed a Pd-catalyzed cross-coupling reaction of aryl halides 50 with carboxylic acids 51 under irradiation with blue LEDs to give the aryl esters 52 (Scheme 31).
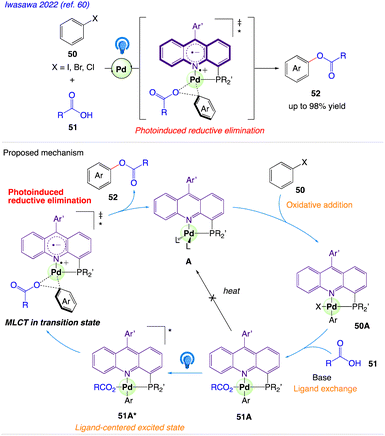 |
| | Scheme 31 Proposed Mechanism for acridine–Pd-catalyzed cross-coupling of aryl halides with carboxylic acids. | |
The plausible mechanism as proposed by author is depicted in Scheme 31. According to this mechanism, Pd(0) complex A undergoes oxidative addition of an aryl halide 50 to give ArPd(II)X complex 50A, which reacts with carboxylic acids 51 and further converted to ArPd(II)O2CR complex 51A via ligand exchange. Complex 51A is excited by visible light to form an acridine-centered excited state 51A* in T1, which undergoes reductive elimination of an aryl ester 52 and regenerate A. This step is assisted by an MLCT (metal to ligand charge transfer) character of the transition state in the excited state.
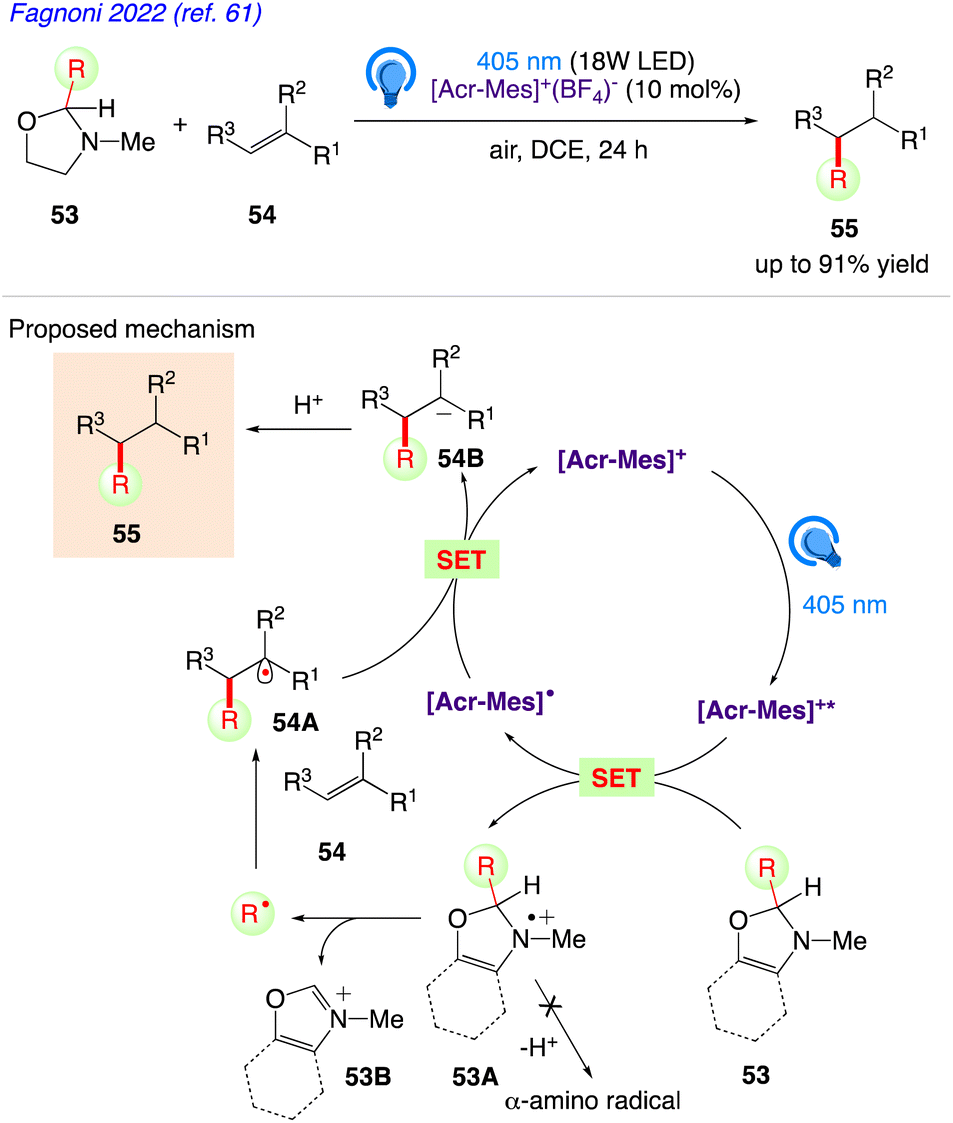
5.17. Alkyl radical generation via C–C bond cleavage in 2-substituted oxazolidines
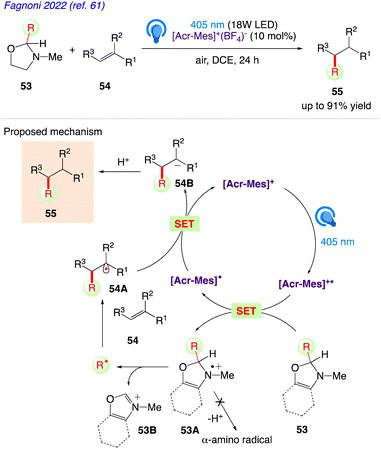
Under photoredox catalysed conditions, Fagnoni et al.61 reported a class of easily synthesised uncharged precursors for the quick release of alkyl radicals (tertiary, α-oxy, and α-amido). Using photo-organoredox catalysis, in presence of a commercially available and widely employed organic dye (Acr-Mes+BF4−) as the photoredox catalyst, 2-substituted-1,3-oxazolidines 53 have been successfully used to these radicals in alkylation of olefins 54 to give the desired alkylated product 55.
The plausible mechanism as proposed by author is depicted in Scheme 32. According to this mechanism, compound 53 is radical precursor and the monoelectronic oxidation of 53 by the photoexcited acridinium catalyst Acr-Mes+* gives the corresponding radical cation 53A.
 |
| | Scheme 32 Proposed Mechanism for alkyl radical generation via C–C bond cleavage in 2-substituted oxazolidines. | |
At this step, an unprecedented C–C cleavage takes place in 53A, releasing a carbon-centered radical and a stable iminium ion 53B. The unique structure of the oxazolidines prevents potential α-deprotonation from positions 2 and 4 as well as from the N-Me group to give a α-amino radical during the radical cation stage. The alkyl radicals are in turn, trapped by electron-poor olefins or vinyl (hetero)aromatics 54 to give adduct radical 54C. Back electron transfer from Acr-Mes˙ to the adduct radical 54C followed by protonation gave the desired alkylated product 55 followed by restoring the photoredox catalyst.
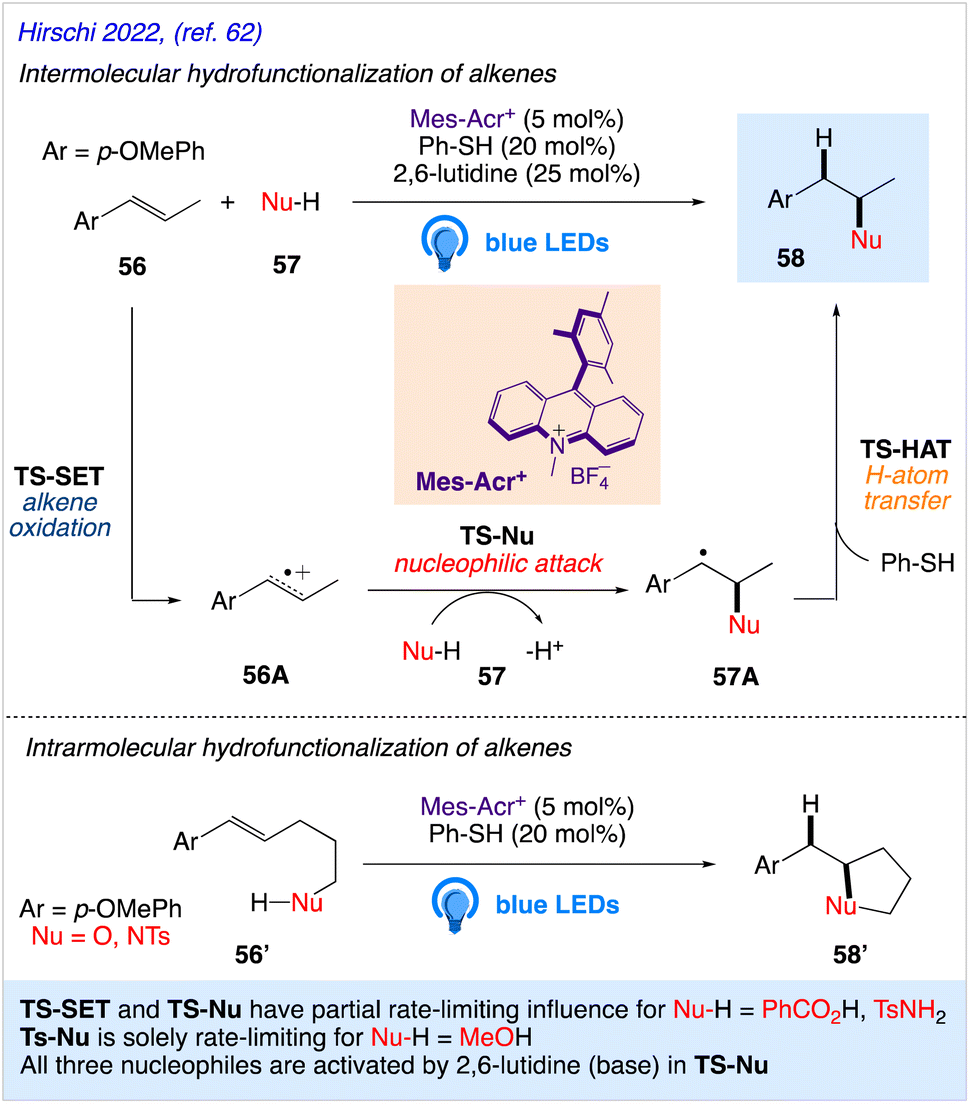
5.18. Anti-Markovnikov hydrofunctionalization of alkenes
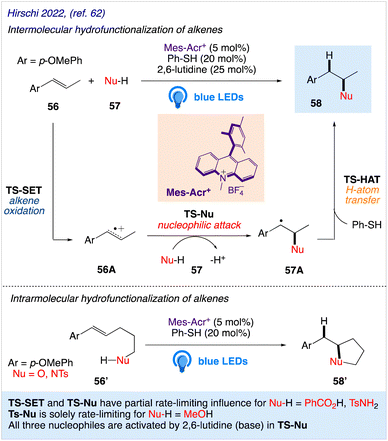
Nicewicz group have reported both intermolecular and intramolecular alkene hydrofunctionalization reactions utilizing acridinium salts as photocatalysts. Hirschi et al.62 reported a mechanistic evaluation of intermolecular alkene hydrofunctionalization reactions for the same. They proposed the mechanism of the intermolecular hydrofunctionalization using model alkene (anethole) 56 that undergo oxidation via single electron transfer (SET) by a photoexcited acridinium catalyst (Mes-Acr+*) to form cation radical intermediate 56A and the reduced acridine radical Mes-Acr˙. Further, it is followed by attack of the nucleophile (Nu-H) 57, resulting in the generation of carbon-centered radical intermediate 57A. H-atom transfer (HAT) from the thiol co-catalyst to 57A afforded the anti-Markovnikov product 58 (Scheme 33). Qualitative evidence for rate-limiting nucleophilic attack in all three reactions is provided by a typical 13C KIE on the olefinic carbon that is subject to nucleophilic assault.
 |
| | Scheme 33 Acridinium catalyzed anti-markovnikov hydrofunctionalization of alkenes. | |
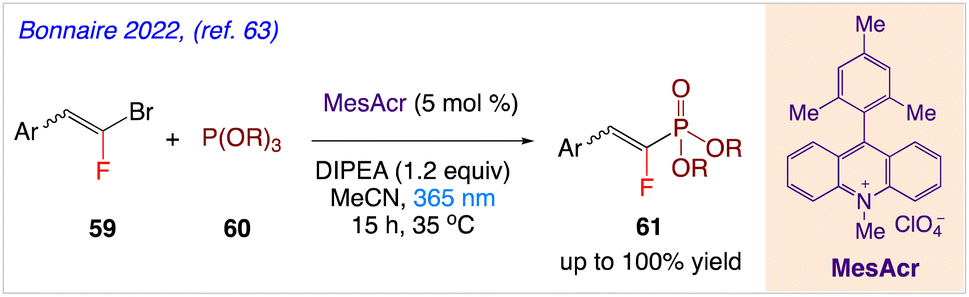
5.19. Synthesis of gem-fluorophosphonate alkenes
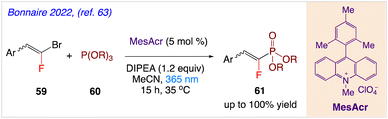
Bonnaire et al.63 reported an effective method for synthesizing fluorovinylphosphonates using acridinium photocatalysis in moderate and metal-free conditions. In this synthetic protocol, in presence of Fukuzumi's catalyst (9-mesityl-10-methylacridinium perchlorate), gem-bromo fluoroalkenes 59 have been coupled with phosphite 60 to produce valuable gem-fluorophosphonate derivatives 61 (Scheme 34). The intended products were produced in yields that ranged from good to excellent, and the reaction exhibited high chemical stability.
 |
| | Scheme 34 Synthesis of gem-fluorophosphonate alkenes. | |
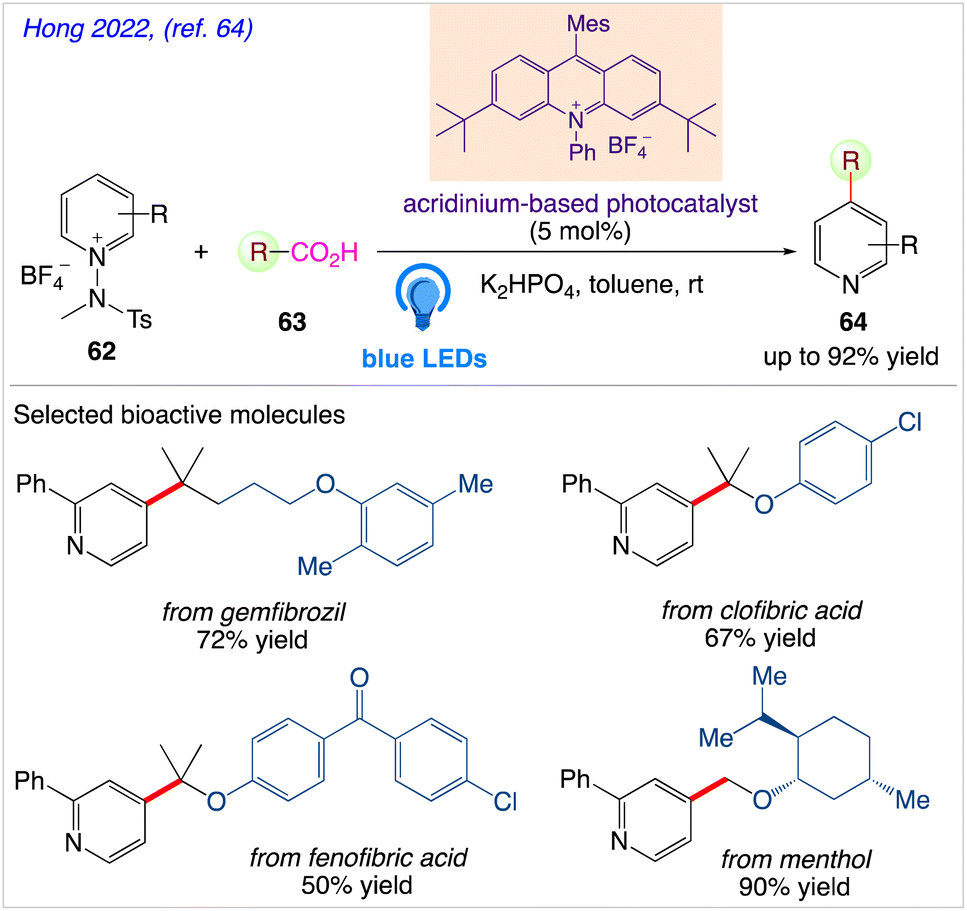
5.20. Photocatalytic decarboxylative pyridylation of carboxylic acids
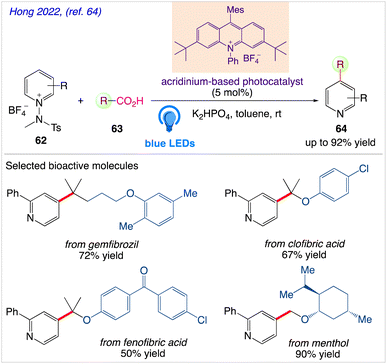
Through radical-mediated decarboxylation and the simultaneous inclusion of pyridyl groups, Hong et al.64 reported a highly potent catalytic system for visible-light-induced reactions using N-amidopyridinium salts 62 and diverse carboxylic acids 63 to give the desired pyridine based organic compound 64 (Scheme 35). Due to the prevalence and abundance of carboxylic acids, they anticipated that this catalytic system can directly enable the C–H pyridylation of carboxylic acids upto a greater amount as well as scope of its wide spread applicability in synthetic and therapeutic chemistry.
 |
| | Scheme 35 Photocatalytic decarboxylative pyridylation of carboxylic acids. | |
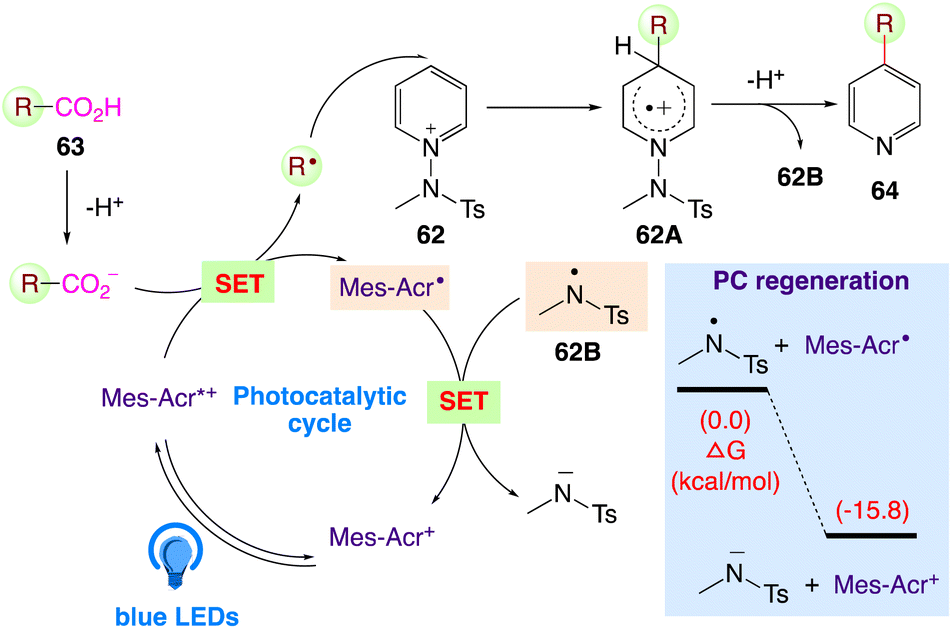
The plausible mechanism as proposed by author is depicted in Scheme 36. According to this mechanism, the carboxylate, generated under basic conditions, undergoes SET with the excited state of Mes-Acr+ in presence of visible light. The generated carboxylic radical generates the alkyl radical via decarboxylation, which is assured for radical addition at the C4 position of the N-amidopyridinium salt 62. Intermediate 62A undergoes facile deprotonation and homolytic cleavage to furnish amidyl radical 62B. Furthermore, the desired product 64 is generated, with the in situ-generated N-centered amidyl radical 62B regenerating the photocatalyst via direct SET.
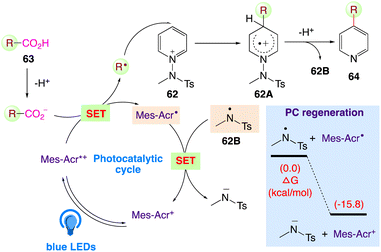 |
| | Scheme 36 Proposed reaction mechanism for photocatalytic decarboxylative pyridylation of carboxylic acids. | |
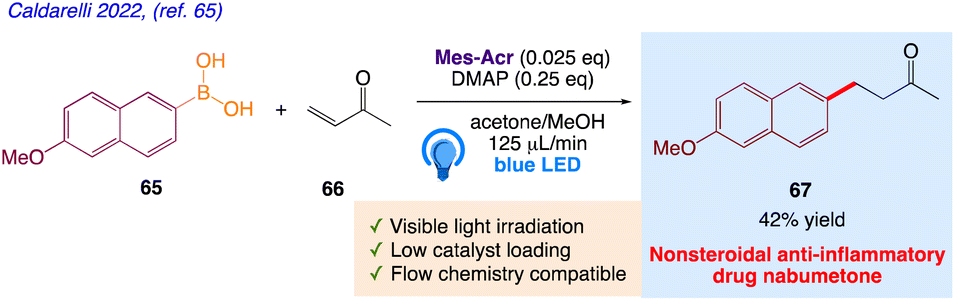
5.21. Giese-type coupling of arylboronic acids with electron poor olefins (synthesis of nabumetone)
Caldarelli et al.65 reported the synthetic protocol involving arylboronic acids 65 with different methyl vinyl ketone 66 to give the desired product nabumetone 67 in a Giese-type reaction under photochemical conditions (Scheme 37). This protocol utilizes use of inexpensive metal-free photocatalysts and DMAP as a Lewis base activator with broader and an easy set-up. This reaction process has been used to create a straightforward anti-inflammatory medication and can be executed in-batch or in-flow.
 |
| | Scheme 37 Acridinium-based photocatalyst in the Giese-type coupling of arylboronic acids with electron poor olefins (synthesis of nabumetone). | |
5.22. Acridine radical photoreductant induced organic transformations
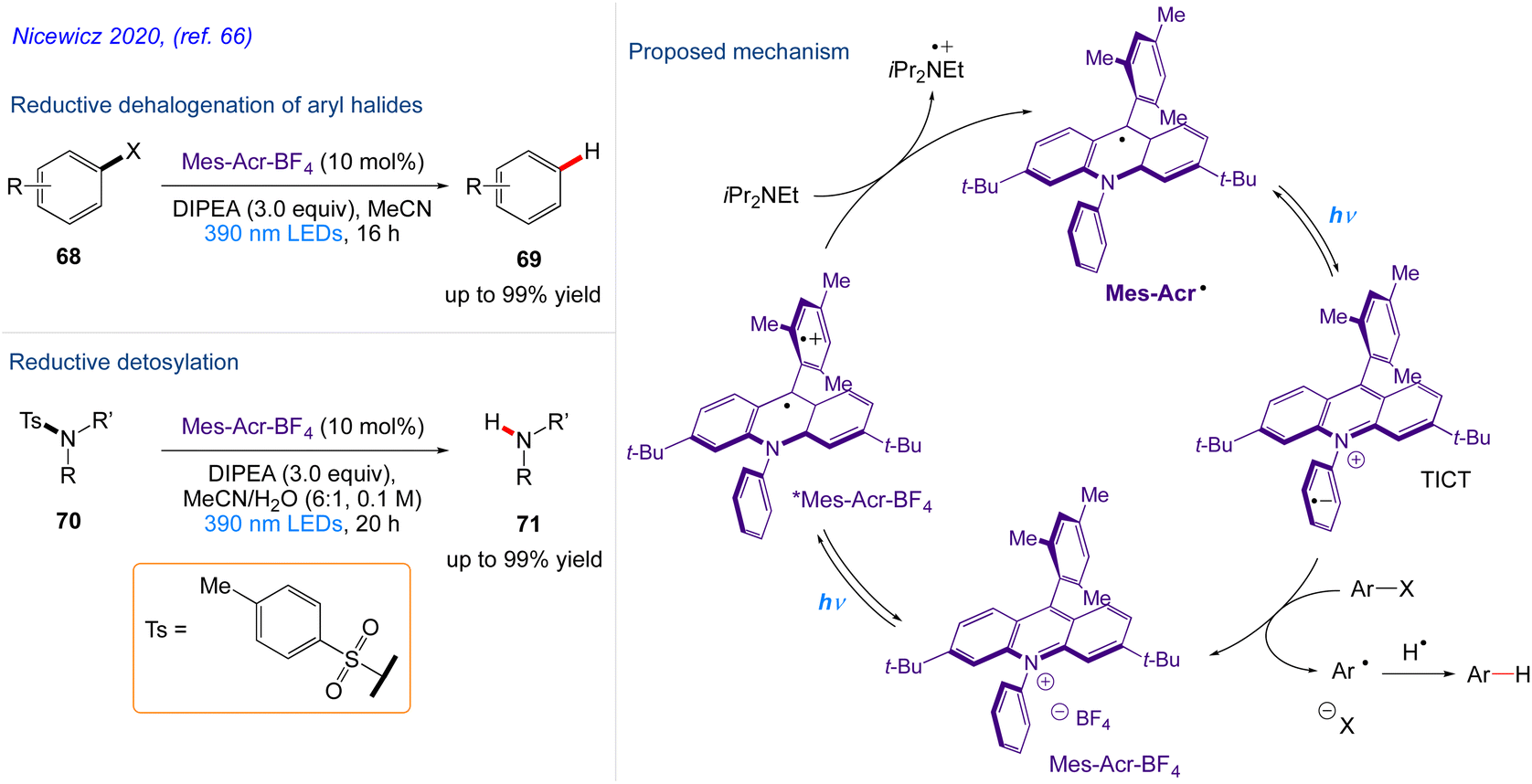
Nicewicz and co-workers66 repored the in situ generation of acridine radical that may act as a potent single-electron reductant produced by single-electron reduction of an acridinium derivative upon excitation with 390 nm light. In addition to enhancing the well-known oxidative chemistry associated with acridinium salts, the development of chemoselective dehalogenation reaction carried out from aryl halide 68 to give the desired substituted aryl compound 69 and detosylation reaction including tosylated amine derivatives 70 were smoothly converted to the desired free amines using Mes-Acr˙. This protocol also highlights the potential for the development of a variety of reactions based on the excitation of organic radicals.
The plausible mechanism as proposed by author is depicted in Scheme 38. According to this mechanism, upon excitation, Mes-Acr+BF4− engages in single electron transfer with the tertiary amine reductant DIPEA, generating Mes-Acr˙ and the corresponding amine cation radical. Mes-Acr˙, which is then excited by 390 nm light and undergoes electron transfer with an electronically matched aryl halide, producing an arene radical anion and regenerating Mes-Acr+BF4−. The aryl radical is produced as a result of the splitting of arene radical anion. The resultant aryl radical extracts a hydrogen atom from the amine cation radical, producing the desired product as well as the corresponding iminium salt.
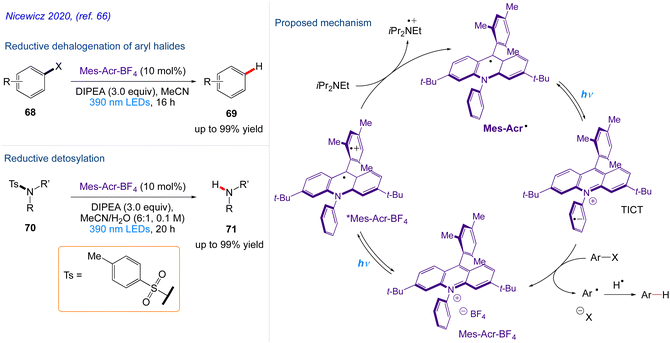 |
| | Scheme 38 Acridine radical photoreductant induced organic transformations. | |
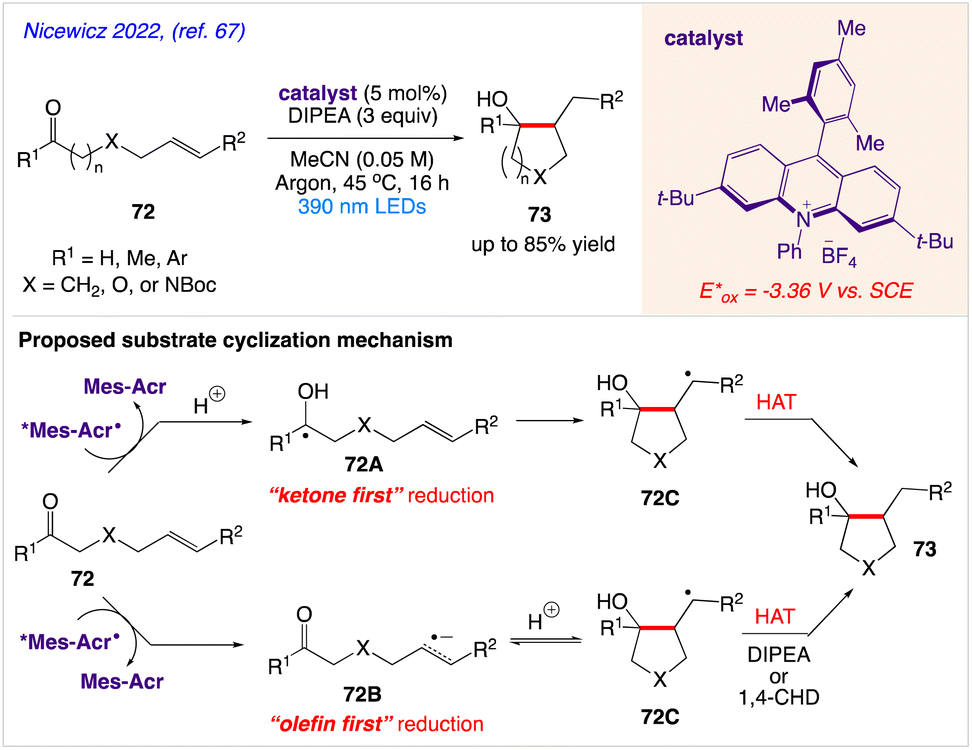
5.23. Excited-state acridine radicals catalysed ketone−olefin coupling of aliphatic and aromatic carbonyls
Nicewicz and co-workers67 described a mild, metal-free ketone−olefin coupling reaction using an excited-state acridine radical super reductant as a photoredox catalyst. They demonstrated both intramolecular and intermolecular ketone−olefin couplings of aliphatic and aromatic ketones and aldehydes 72 to give the desired cycloadduct 73.
The plausible mechanism as proposed by author is depicted in Scheme 39. According to this mechanism, If “ketone first” reduction is operative, a ketyl radical 72A is formed, which can undergo a radical 5-exo-trig cyclization with the corresponding olefin to provide a carbon-centered radical 72C. Then, terminal HAT can occur to give the corresponding cyclized product 73. Alternatively, when “olefin first” reduction occurs, they proposed that the olefin radical anion 72B formed, can undergo a two-electron attack at the carbonyl to generate 72C. Subsequent HAT from either DIPEA or 1,4-CHD can trap out the corresponding cycloadduct 73.
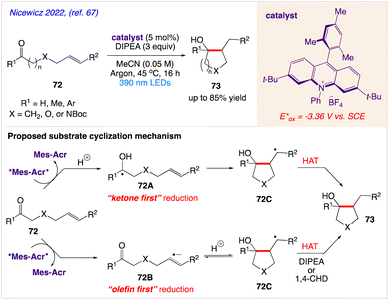 |
| | Scheme 39 Excited-state acridine radicals catalysed ketone−olefin coupling of aliphatic and aromatic carbonyls. | |
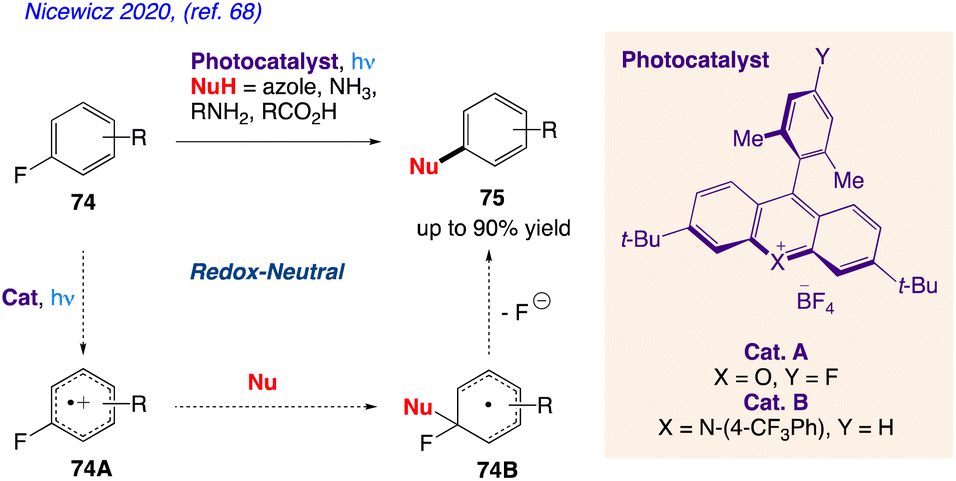
5.24. Nucleophilic aromatic substitution of unactivated fluoroarenes
Nicewicz and co-workers68 proposed a method for the cation radical-accelerated nucleophilic aromatic substitution, which enables for the nucleophilic defluorination of inactive fluoroarenes. Under benign conditions, this method can be rendered easy to use by application of organic photoredox catalysis and is suitable for a variety of nucleophile classes, including azoles, amines, and carboxylic acids. Using this technique, specific fluorinated heterocycles can be functionalized. The functionalization of pharmaceuticals in its late stages is also described. The organic transformation, as proposed by them has been represent in Scheme 40 indicating nucleophilic substitution of fluoroarenes 74 desired compound 75 via intermediate 74A and 74B.
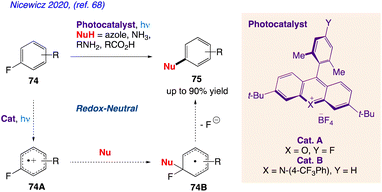 |
| | Scheme 40 Nucleophilic aromatic substitution of unactivated fluoroarenes. | |
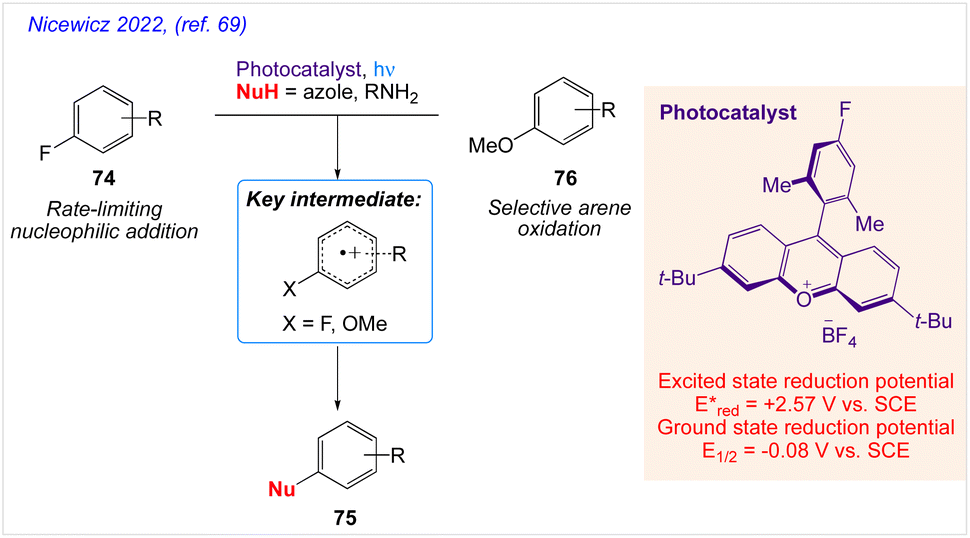
Further, Nicewicz and co-workers69 also reported mechanistic investigations into site-selective arene amination of unactivated arenes via cation radical accelerated nucleophilic aromatic substitution (Scheme 41).
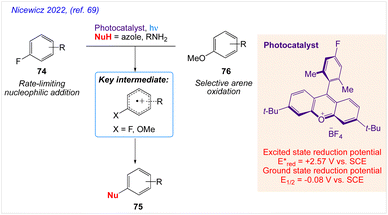 |
| | Scheme 41 Mechanistic investigations into amination of unactivated arenes via cation radical accelerated nucleophilic aromatic substitution. | |
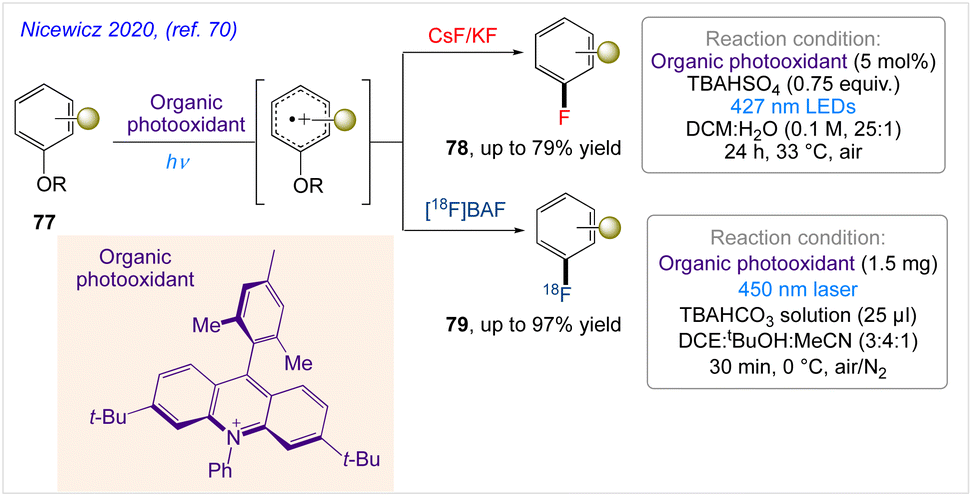
5.25. 19F- and 18F–arene deoxyfluorination
Nicewicz and co-workers70 demonstrated a polarity-reversed photoredox-catalysed arene deoxyfluorination that operates throughcation-radical-accelerated aromatic nucleophilic substitution that enables the fluorination of electron-rich arenes with 19F– and 18F– under mild conditions. In this protocol, selective (radio)fluorination of electron-rich arenes with CsF or KF and [18F]TBAF has obtained under mild conditions(Scheme 42).
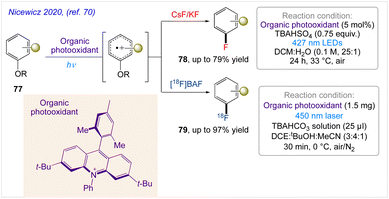 |
| | Scheme 42 19F- and 18F–arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. | |
6. Conclusions
A considerable number of acridinium catalysts have been developed in recent years and applied in a variety of visible-light photocatalytic reactions. This made it possible to tailor the redox attributes to a range that most suited for photosynthetic procedures. Recent advances in synthetic chemistry have made it possible to access a variety of photocatalysts with flexible photophysical and electrochemical properties, leading to new reactivities including the functionalization of inert bonds and late stage synthetic applications. In this review, recent applications of acridine in organic synthesis are discussed. As a result, we think that, the field of acridinium based organophotocatalysis will develop, that open up a new avenue for investigation and certainly it has a bright future ahead of it.
Conflicts of interest
“There are no conflicts to declare”.
References
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. S. Wang, P. H. Dixneuf and J. F. Soul, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- A. Wimmer and B. Konig, Beilstein J. Org. Chem., 2018, 14, 54–83 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Mei and H. Wu, Chin. J. Org. Chem., 2019, 39, 3040–3050 CrossRef CAS.
- Y. Chen, L. Q. Lu, D. G. Yu, C. J. Zhu and W. J. Xiao, Sci. China Chem., 2019, 62, 24–57 CrossRef CAS.
- L. Marzo, K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC.
- Z. Zhang, J. H. Ye, T. Ju, L. L. Liao, H. Huang, Y. Y. Gui, W. J. Zhou and D. G. Yu, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS.
- F. F. Tan, X. Y. He, W. F. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS PubMed.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
-
(a) J. Eberhard, K. Peuntinger, R. Fröhlich, D. M. Guldi and J. Mattay, Eur. J. Org. Chem., 2018, 2682–2700 CrossRef CAS;
(b) A. Gini, M. Uygur, T. Rigotti, J. Alemán and O. García Mancheño, Chem. – Eur. J., 2018, 24, 12509–12514 CrossRef CAS PubMed;
(c) K. Durka, M. Urban, M. Dabrowski, P. Jankowski, T. Klis and S. Lulinśki, ACS Omega, 2019, 4, 2482–2492 CrossRef CAS PubMed;
(d) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed.
- D. P. Haria and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
- J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef CAS PubMed.
- J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS PubMed.
- J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., 2019, 131, 13789–13800 CrossRef.
-
(a) Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 2020, 6028–6043 CrossRef CAS;
(b) K. C. Cartwright, S. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84, 2933–2940 CrossRef CAS PubMed;
(c) F. F. Tan, X. Y. He, W. F. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS PubMed;
(d) Y. X. Cao, G. Zhu, Y. Li, N. L. Breton, C. Gourlaouen, S. Choua, J. Boixel, H. P. J. Rouville and J. F. Soulé, J. Am. Chem. Soc., 2022, 144, 5902–5909 CrossRef CAS PubMed;
(e) K. C. Cartwright, S. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84(5), 2933–2940 CrossRef CAS PubMed.
- A. Vega-Pecaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
-
(a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed;
(b) Y. C. Lin and C. T. Chen, Org. Lett., 2009, 11, 4858–4861 CrossRef CAS PubMed;
(c) C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2019, 10, 210–215 CrossRef;
(d) S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS PubMed.
-
(a) K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC;
(b) I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed;
(c) H. Yan, J. Song, S. Zhu and H. C. Xu, CCS Chem., 2021, 3, 317–325 CrossRef CAS.
- L. Marzo, S. K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Dual catalysis strategies in photochemical synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
-
(a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS;
(b) S. Reischauer and B. Pieber, iScience, 2021, 24, 102209 CrossRef CAS PubMed.
-
(a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122(2), 1485–1542 CrossRef CAS PubMed;
(b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
-
(a) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC;
(b) V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. F. Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9485–9498 CrossRef CAS PubMed.
- C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2020, 10, 210–215 CrossRef CAS.
- B. Zilate, C. Fischer and C. Sparr, Chem. Commun., 2020, 56, 1767–1775 RSC.
-
(a) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2014, 87, 91–95 CrossRef;
(b) V. Srivastava, P. K. Singh and P. P. Singh, Chem. Heterocycl. Compd., 2014, 50, 573–578 CrossRef CAS;
(c) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88, 59–65 CrossRef CAS;
(d) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88, 227–233 CrossRef CAS;
(e) V. Srivastava, P. K. Singh and P. P. Singh, Asian J. Chem., 2016, 28, 2159–2163 CrossRef CAS;
(f) V. Srivastava, P. K. Singh and P. P. Singh, Rev. Roum. Chim., 2016, 61, 755–761 Search PubMed;
(g) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2017, 90, 435–441 CrossRef CAS;
(h) V. Srivastava, P. K. Singh, S. Kanaujia and P. P. Singh, New J. Chem., 2018, 42, 688 RSC;
(i) P. K. Singh, P. P. Singh and V. Srivastava, Croat. Chem. Acta, 2018, 91, 383–387 CrossRef;
(j) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 40–43 CrossRef CAS;
(k) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 1333–1336 CrossRef CAS;
(l) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 151041 CrossRef CAS;
(m) V. Srivastava, P. K. Singh and P. P. Singh, Rev. Roum. Chim., 2020, 65, 221–226 CrossRef;
(n) S. Tivari, P. K. Singh, P. P. Singh and V. Srivastava, RSC Adv., 2022, 12, 35221 RSC;
(o) M. Mishra, V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2022, 95(1), 25–30 CrossRef;
(p) M. Z. Beg, P. K. Singh, P. P. Singh and V. Srivastava, Mol. Diversity, 2023 DOI:10.1007/s11030-022-10595-2;
(q) S. P. Singh, V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron, 2023, 132, 133245 CrossRef CAS.
-
(a) V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC;
(b) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2020, 10, 20046 RSC;
(c) A. Srivastava, P. K. Singh, A. Ali, P. P. Singh and V. Srivastava, RSC Adv., 2020, 10, 39495 RSC;
(d) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2021, 11, 14251–14259 RSC;
(e) V. Srivastava and P. P. Singh, Org. Biomol. Chem., 2021, 19, 313–321 RSC;
(f) P. P. Singh, P. K. Singh, M. Z. Beg, A. Kashyap and V. Srivastava, Synth. Commun., 2021, 51, 3033–3058 CrossRef CAS;
(g) V. Srivastava, P. K. Singh, A. Srivastava, S. Sinha and P. P. Singh, Photochem, 2021, 1, 237–246 CrossRef;
(h) V. Srivastava, P. K. Singh, S. Tivari and P. P. Singh, Org. Chem. Front., 2022, 9, 1485 RSC;
(i) V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol. C, 2022, 50, 100488 CrossRef CAS;
(j) V. Srivastava and P. P. Singh, RSC Adv., 2022, 12, 18245 RSC;
(k) P. P. Singh, S. Sinha, G. Pandey and V. Srivastava, RSC Adv., 2022, 12, 29826 RSC;
(l) P. P. Singh, P. K. Singh and V. Srivastava, Org. Chem. Front., 2023, 10, 216 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
-
(a) A. Gini, M. Uygur, T. Rigotti, J. Alemán and O. G. Mancheño, Chem. – Eur. J., 2018, 24, 12509–12514 CrossRef CAS PubMed;
(b) A. R. White, L. Wang and D. A. Nicewicz, Synlett., 2019, 30, 827–832 CrossRef CAS PubMed.
- H. Kotani, K. Ohkubo and S. J. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999–16006 CrossRef CAS PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- C. Fischer and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 2436 CrossRef CAS PubMed.
- C. Fischer and C. Sparr, Tetrahedron, 2018, 74, 5486 CrossRef CAS.
- V. Hutskalova and C. Sparr, Org. Lett., 2021, 23, 5143–5147 CrossRef CAS PubMed.
- Y. X. Cao, G. Zhu, Y. Li, N. L. Breton, C. Gourlaouen, S. Choua, J. Boixel, H. P. J. Rouville and J. F. Soulé, J. Am. Chem. Soc., 2022, 144, 5902–5909 CrossRef CAS PubMed.
- K. Kano, B. Zhou and S. Hashimoto, Bull. Chem. Soc. Jpn., 1987, 60, 1041–1047 CrossRef CAS.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J. M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- N. W. Koper, S. A. Jonker, J. W. Verhoeven and C. van Dijk, Recl. Trav. Chim. Pays-Bas, 1985, 104, 296–302 CrossRef CAS.
- W. Song, K. Dong and M. Li, Org. Lett., 2020, 22, 371–375 CrossRef CAS PubMed.
- P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557–1562 CrossRef CAS PubMed.
- S. Mandal, S. Bhuyan, S. Jana, J. Hossain, K. Chhetria and B. G. Roy, Green Chem., 2021, 23, 5049 RSC.
- M. Uygur, J. H. Kuhlmann, M. C. Pérez-Aguilar, D. G. Piekarski and O. G. Mancheño, Green Chem., 2021, 23, 3392 RSC.
- M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2020, 12, 10499–10505 CrossRef.
- M. M. Hossain, A. C. Shaikh, J. Moutet and T. L. Gianetti, Nat. Synth., 2022, 1, 147–157 CrossRef.
- K. G. Ghosh, D. Das, S. Garai, P. Chandu and D. Sureshkumar, J. Org. Chem., 2022, 87, 8611–8622 CrossRef CAS PubMed.
- A. Leclair, Q. Wang and J. Zhu, ACS Catal., 2022, 12, 1209–1215 CrossRef CAS.
- Z. Luo, C. Lu, G. Histand and D. Lin, J. Org. Chem., 2022, 87, 11558–11564 CrossRef CAS PubMed.
- V. D. Nguyen, R. Trevino, S. G. Greco, H. D. Arman and O. V. Larionov, ACS Catal., 2022, 12, 8729–8739 CrossRef CAS PubMed.
- L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532–3539 CrossRef CAS PubMed.
- J. B. McManus, J. D. Griffin, A. R. White and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 10325–10330 CrossRef CAS PubMed.
- J. L. Tu, J. L. Liu, W. Tang, M. Su and F. Liu, Org. Lett., 2020, 22, 1222–1226 CrossRef CAS PubMed.
- X. Wang, Y. Li and X. Wu, ACS Catal., 2022, 12, 3710–3718 CrossRef CAS.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, G. B. Karki, H. D. Armana and O. V. Larionov, Chem. Sci., 2022, 13, 4170 RSC.
- N. Toriumi, T. Inoue and N. Iwasawa, J. Am. Chem. Soc., 2022, 144, 19592–19602 CrossRef CAS PubMed.
- A. L. Ruiz, M. L. Mantia, D. Merli, S. Protti and M. Fagnoni, ACS Catal., 2022, 12, 12469–12476 CrossRef PubMed.
- S. C. Mallojjala, V. O. Nyagilo, S. A. Corio and J. S. Hirschi, J. Am. Chem. Soc., 2022, 144, 17692–17699 CrossRef CAS PubMed.
- S. Morand, W. Lecroq, P. Jubault, S. Lakhdar, J. P. Bouillon and S. C. Bonnaire, Org. Lett., 2022, 24, 8343–8347 CrossRef CAS PubMed.
- C. Kim, J. Jeong, M. Vellakkaran and S. Hong, ACS Catal., 2022, 12, 13225–13233 CrossRef CAS.
- M. Caldarelli, L. Laze, L. Piazza, G. Caputo, M. D. Amici and G. Papeo, Tetrahedron Lett., 2022, 103, 153978 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- N. J. Venditto, Y. S. Liang, R. K. E. Mokadem and D. A. Nicewicz, J. Am. Chem. Soc., 2022, 144, 11888–11896 CrossRef CAS PubMed.
- V. A. Pistritto, M. E. S. Horton and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 17187–17194 CrossRef CAS PubMed.
- V. A. Pistritto, S. Liu and D. A. Nicewicz, J. Am. Chem. Soc., 2022, 144, 15118–15131 CrossRef CAS PubMed.
- N. E. S. Tay, W. Chen, A. Levens, V. A. Pistritto, Z. Huang, Z. Wu, Z. Li and D. A. Nicewicz, Nat. Catal., 2020, 3, 734–742 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jaya Singh
b and
Vishal Srivastava
a,
Jaya Singh
b and
Vishal Srivastava












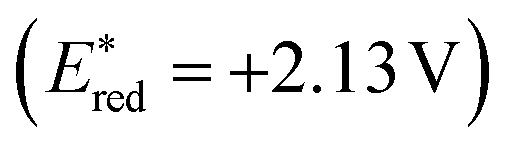 . The resulting nitrate radical is a potent H-atom abstracting agent, allowing it to excise the weak C(sp3)–H bond of 38 generating HNO3 and benzylic radical 38A. XylF-Acr˙ (E1/2ox = −0.54 V) can feasibly undergo single electron transfer (SET) with [CoII] (E1/2red = −0.51 V) to regenerate XylF-Acr+ and a reduced cobalt complex (vide infra). [CoI]− likely undergoes protonation to form [CoIII]–H assisted by the aforementioned acid additive. 38A is then intercepted by [CoIII]–H, liberating H2 and 38B. One possibility of this mechanism is the direct protonation of [CoIII]–H. This would form [CoIII]+ (E1/2red ∼ +0.2 V), which could potentially oxidize 38A (E1/2ox ∼ +0.37 V). While this SET is endergonic by about +0.2 V (4.6 kcal mol−1), rapid deprotonation of the resulting benzylic cation intermediate would render SET irreversible, ultimately generating styrene 38B. Alternatively two molecules of [CoIII]–H could undergo a bimolecular reductive elimination of H2 generating two equivalents of [CoII]. 38B would then be formed via the addition of 38A to [CoII], forming a putative [CoIII] alkyl intermediate capable of undergoing a net β-hydride elimination. 38B can then engage in a second catalytic cycle to form the olefin radical cation (38C) whereupon trapping with water would afford a distonic radical cation (38D). Subsequent deprotonation and a second dehydrogenation would furnish the desired product 39 via a sequence similar to the mechanisms proposed by Lei and Nicewicz.
. The resulting nitrate radical is a potent H-atom abstracting agent, allowing it to excise the weak C(sp3)–H bond of 38 generating HNO3 and benzylic radical 38A. XylF-Acr˙ (E1/2ox = −0.54 V) can feasibly undergo single electron transfer (SET) with [CoII] (E1/2red = −0.51 V) to regenerate XylF-Acr+ and a reduced cobalt complex (vide infra). [CoI]− likely undergoes protonation to form [CoIII]–H assisted by the aforementioned acid additive. 38A is then intercepted by [CoIII]–H, liberating H2 and 38B. One possibility of this mechanism is the direct protonation of [CoIII]–H. This would form [CoIII]+ (E1/2red ∼ +0.2 V), which could potentially oxidize 38A (E1/2ox ∼ +0.37 V). While this SET is endergonic by about +0.2 V (4.6 kcal mol−1), rapid deprotonation of the resulting benzylic cation intermediate would render SET irreversible, ultimately generating styrene 38B. Alternatively two molecules of [CoIII]–H could undergo a bimolecular reductive elimination of H2 generating two equivalents of [CoII]. 38B would then be formed via the addition of 38A to [CoII], forming a putative [CoIII] alkyl intermediate capable of undergoing a net β-hydride elimination. 38B can then engage in a second catalytic cycle to form the olefin radical cation (38C) whereupon trapping with water would afford a distonic radical cation (38D). Subsequent deprotonation and a second dehydrogenation would furnish the desired product 39 via a sequence similar to the mechanisms proposed by Lei and Nicewicz.