
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the thermal stability of poly[methyl(trifluoropropyl)siloxane] by introducing diphenylsiloxane units
Yang You,
Anna Zheng,
Dafu Wei,
Xiang Xu *,
Yong Guan* and
Jianding Chen*
*,
Yong Guan* and
Jianding Chen*
School of Materials Science and Engineering, Shanghai Key Laboratory of Advanced Polymeric Materials, East China University of Science and Technology, Shanghai, 200237, China. E-mail: xiangxu@ecust.edu.cn; jiandingchen@ecust.edu.cn; yguan@ecust.edu.cn
First published on 12th April 2023
Abstract
A series of poly(methyl(trifluoropropyl)-diphenyl siloxane) (P(MTFPS-co-DPS)) was synthesized by polycondensation of diphenylsilanediol and methyltrifluoropropylsiloxanediol. Their chemical structures were investigated by gel permeation chromatography (GPC), Fourier transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR), and differential scanning calorimeter (DSC). The effect of diphenylsiloxane (DPS) units on the thermal stability of poly[methyl(trifluoropropyl)siloxane] (PMTFPS) was studied by thermogravimetric analysis (TGA), isothermal degradation tests, and pyrolysis-gas chromatography-mass spectrometry (Py-GCMS). The results showed that the thermal stability of PMTFPS improved with the introduction of DPS units into the chain. In particular, the temperature for 5% mass loss in PMTFPS increased by 72 °C under a nitrogen atmosphere. In addition, the mechanism by which the DPS units improve the thermal stability of PMTFPS was also investigated.
1. Introduction
Poly[methyl(trifluoropropyl)siloxane] (PMTFPS) products, which exhibit superior oil resistance over a wide operating temperature range, have become essential materials in the aerospace and military fields.1–5 Poly[methyl(trifluoropropyl)siloxane] is one of the few elastomers that can be utilized in fuel medium at −60–210 °C. With the rapid development of science and technology, PMTFPS products with high thermal stability are highly sought after in the aeronautics and astronautics industry. However, because of the strong electron-withdrawing effect of the fluorocarbon groups in the side chain, PMTFPS products suffer from inferior thermal stability when exposed to temperatures above 210 °C for extended periods of time. To overcome this drawback, many methods have been studied to improve the thermal resistance of PMTFPS materials.Two main thermal degradation pathways have been proposed for polysiloxanes under an inert atmosphere or in air:6–10 (a) the so-called “back-biting” reaction from the chain end and siloxane rearrangement reactions; and (b) the oxidation scission or dissociation of side chains.
The so-called “back-biting” reaction was the cyclic degradation of the main chain. In high temperature environment, the main degradation form of PMTFPS backbone was cyclization fracture.5,11 The rearrangement of –Si–O– in the main chain formed cyclic monomers as main degradation products, resulting in a rapid decrease in molecular weight.7–9 The incorporation of methyl-phenyl siloxane or diphenyl siloxane groups on the branched chains of polysiloxane can inhibit the “back-biting” degradation and improve the temperature stability of silicone resins from 200 °C to 250 °C.12 The introduction of silphenylene units into the main chains of linear polysiloxanes was found to be efficient to enhance their thermal stability.13 However, little attention has been paid to improve the thermal stability of PMTFPS with those methods.
In this study, a series of poly(methyl(trifluoropropyl)-diphenyl siloxane) (P(MTFPS-co-DPS)) was synthesized with diphenylsilanediol (DPS-diol) and methyltrifluoro-propylsiloxanediol (MTFPS-diol) using a polycondensation method. The obtained copolymer was cured by hydrosilylation reaction. The effect of diphenylsilane (DPS) unit on the thermal stability of PMTFPS was investigated via thermogravimetric analysis (TGA) and isothermal degradation tests. The mechanism by which DPS unit improved the thermal stability of PMTFPS was discussed using pyrolysis-gas chromatography-mass spectrometry (Py-GCMS).
2. Experimental
2.1 Materials
DPS-diol (>98%), dimethylvinylchlorosilane (97%), sodium hydroxide (NaOH, ≥98%), chlorodimethylvinylsilane (CDVMS) and platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane were purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). 1,3,5-tris(3,3,3-trifluoropropylmethyl) cyclotrisiloxane (F3) was purchased from Zhengjiang Huanxin Fluorine Material Co., Ltd (Zhengjiang, China). MTFPS-diol, 1,3,5-tris(3,3,3-trifluoropropyl) methylcyclotrisiloxane (D3F), 2,4,6,8-tetramethylcyclotetrasiloxane (D4H) and 2,4,6-trimethyl-2,4,6-trivinylcyclotrisiloxane (V3) were obtained from FSIR Advanced Material Co., Ltd (Zhenjiang, China).2.2 Preparation of PMTFPS and P(MTFPS-co-DPS)
The preparation processes of PMTFPS and P(MTFPS-co-DPS) were shown in Scheme 1.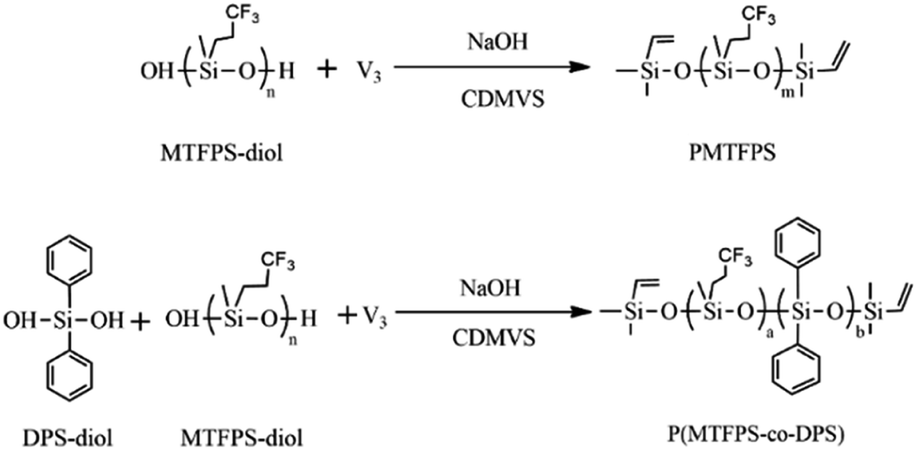 | ||
| Scheme 1 The synthesis processes of PMTFPS and P(MTFPS-co-DPS). | ||
PMTFPS and P(MTFPS-co-DPS) were synthesized by condensation polycondensation of DPS-diol and MTFPS-diol. A small amount of V3 was used to afford the crosslinking points. The mixture of DPS-diol, MTFPS-diol and V3 were added into a 250 ml three-necked flask equipped with a stirrer. This reaction was conducted in oil bath at 140 °C for 1 h and the inlet of the chamber was open to air to release the water vapor. Y. Gao et al.14 reported when the reaction temperature was higher than 120 °C, the so-called “back-biting” side reaction became more and more serious. The molecular weight and yield of PMTFPS were reduced through the “back-biting” reaction by chain ends, and there were a large number of cyclic monomers in the system. Thus, the experimental temperatures were designed to be 100, 110, and 120 °C. Then, the bath temperature was cooled to 100, 110, 120 °C, respectively, and NaOH was added to the flask. A vacuum pump was employed to remove water during polycondensation. After being stirred for 12 h, the obtained product was terminated by CDMVS. The abbreviations for each polymer systems as follows: P(MTFPS-co-DPS) (the mass ratio of DPS-diol to MTFPS-diol was 1 to 10) synthesized at 100, 110 and 120 °C were denoted as P(MTFPS-co-DPS)-1, P(MTFPS-co-DPS)-2 and P(MTFPS-co-DPS)-3, respectively; P(MTFPS-co-DPS) (the mass ratio of DPS-diol to MTFPS-diol is 1 to 5) synthesized at 120 °C were denoted as P(MTFPS-co-DPS)-4; polymer synthesized by MTFPS-diol copolymerization at 120 °C were denoted as PMTFPS. Polymer yields under these conditions were typically 72–85%, and their detailed formulations were listed in Table 1.
| Sample | Temperature (°C) | MTFPS-diol (g) | DPS-diol (g) | V3 (g) | NaOH (g) | CDMVS (g) |
|---|---|---|---|---|---|---|
| P(MTFPS-co-DPS)-1 | 100 | 30 | 3 | 0.06 | 0.01 | 0.04 |
| P(MTFPS-co-DPS)-2 | 110 | 30 | 3 | 0.06 | 0.01 | 0.04 |
| P(MTFPS-co-DPS)-3 | 120 | 30 | 3 | 0.06 | 0.01 | 0.04 |
| P(MTFPS-co-DPS)-4 | 120 | 30 | 6 | 0.06 | 0.01 | 0.04 |
| PMTFPS | 120 | 30 | — | 0.06 | 0.01 | 0.04 |
2.3 Curing of PMTFPS and P(MTFPS-co-DPS)
PMTFPS and P(MTFPS-co-DPS) were cured by hydrosilylation addition reaction using Pt-catalyst. D3F (1.6 wt%), D4H (0.3 wt%) and Pt-catalyst (a drop) were added in polymers. The resulting compounds were uniformly stirred and vulcanized at 150 °C for 1 h.2.4 Characterization
Number (Mn) and weight (Mw) average molecular weights were determined by GPC (Waters 1515 system, Waters corporation), which was equipped with an 18-angle laser scattering detector (LS signal) and a refractive detector (RI signal) using tetrahydrofuran (THF) as the eluent at a flow rate of 1.0 ml min−1 at 25 °C.FTIR were obtained from a Nicolet 6700 infrared spectrometer (Thermo Electron Scientific Instruments Corp.) using a KBr-pressed plate. Each spectrum was recorded at a resolution of 2 cm−1 between 4000 and 400 cm−1.
1H-NMR and 29Si-NMR spectra were performed on a 600 MHz NMR spectrometer (Ascend 600 NMR, BRUKER, Switzerland) using acetone-d6 as the solvent at ambient temperature.
The glass-transition temperature (Tg) of the sample was determined by DSC (214 Polyma, NETZSCH) over the temperature range from −100 to −20 °C at a heating rate of 10 °C min−1.
TGA was performed by a NETZSCH STA409PC simultaneous thermal analyzer. For each test, a cured sample of approximately 5–8 mg was placed in the crucible. The temperature range was set from 25 to 600 °C under a nitrogen atmosphere at heating rate of 10 °C min−1.
Isothermal thermogravimetric analysis was performed in a Muffle Furnace (TCXC-1700, Shanghai Tongcoo Electric Equipment Co., Ltd, China). Further, approximately 2 g of each cured sample was evaluated. The polymers were air pyrolyzed in a muffle furnace under isothermal conditions at specified temperatures for 10 min and 3 h, respectively. Each sample was evaluated thrice, and the average mass loss was calculated. All the data points were obtained from the average values of three independent results for each condition.
Pyrolysis-gas chromatography-mass spectrometry (Py-GCMS) was a CDS5150 (Shimadzu, Japan) pyrolyser coupled with a 2010PLUS gas chromatography mass spectrometer (Agilent, 7890A-5975C). Approximately 0.5–1 mg of PMTFPS sample was taken in a quartz filler tube and pyrolyzed in a helium (99.999%) carrier gas at different temperatures (400 °C and 600 °C). The volatile degradation products were separated and analyzed by the GC-MS, and the carrier gas flow velocity was set to 40 ml min−1. The programs were recorded and processed using a data processor, and the amounts of individual products were calculated using the area summation method.
3. Results and discussion
3.1 Characterization of PMTFPS and P(MTFPS-co-DPS)
The Mn, Mw, and polydispersity of PMTFPS and P(MTFPS-co-DPS) are characterized by GPC analysis, the results were shown in Table 2. By comparing P(MTFPS-co-DPS)-1, P(MTFPS-co-DPS)-2 and P(MTFPS-co-DPS)-3, it can be observed the change of temperature had an effect on the molecular weight of P(MTFPS-co-DPS). The reason was the removal of water was beneficial for the formation of copolymers, and the increase in temperature favored the expulsion of water.14–17 Therefore, the Mn and Mw of P(MTFPS-co-DPS)-3 were higher than those of P(MTFPS-co-DPS)-1 and P(MTFPS-co-DPS)-2. Moreover, the reaction temperature for P(MTFPS-co-DPS)-4 was the same as that of P(MTFPS-co-DPS)-3, but the Mn and Mw of P(MTFPS-co-DPS)-4 were lower than those of P(MTFPS-co-DPS)-3. This was because the rigidity of the molecular chain increased and the number of molecular conformations decreased after the introduction of DPS units, which led to the reaction difficult.| Sample | Mn/×103 | Mw/×103 | Polydispersity |
|---|---|---|---|
| MTFPS-diol | 1.17 | 1.21 | 1.03 |
| PMTFPS | 125.42 | 174.07 | 1.39 |
| P(MTFPS-co-DPS)-1 | 4.38 | 5.46 | 1.25 |
| P(MTFPS-co-DPS)-2 | 5.46 | 8.87 | 1.62 |
| P(MTFPS-co-DPS)-3 | 19.83 | 36.99 | 1.87 |
| P(MTFPS-co-DPS)-4 | 6.33 | 15.78 | 2.50 |
Fig. 1 showed the FTIR spectra of P(MTFPS-co-DPS) and PMTFPS. A broad and strong absorption band from 1070 to 1130 cm−1 was attributed to the Si–O–Si asymmetric stretching vibration. For PMTFPS, the bands at 2966, 2908, 1265, and 696 cm−1 were attributed to Si–CH3 groups, and 898 cm−1 was ascribed to stretching vibration of C–F of –SiCH2CH2CF3.18 The spectrum of P(MTFPS-co-DPS) was similar to that of PMTFPS, except two significant![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) differences. The new absorption bands at 3074 and 1593 cm−1 in the curves of P(MTFPS-co-DPS) were characteristic peaks of –C6H5, indicating that DPS units were successfully introduced into the chain.
differences. The new absorption bands at 3074 and 1593 cm−1 in the curves of P(MTFPS-co-DPS) were characteristic peaks of –C6H5, indicating that DPS units were successfully introduced into the chain.
 | ||
| Fig. 1 FTIR spectra of P(MTFPS-co-DPS) and PMTFPS. | ||
3.2 Analysis of the component content of the P(MTFPS-co-DPS)
The structures of P(MTFPS-co-DPS) was further validated by 1H-NMR spectra, as shown in Fig. 2. The signals at 0.3 ppm were attributed to the Si–CH3 groups, at 0.8 ppm to Si–CH2–, and at 2.2 ppm to CF3–CH2– groups. The signals at 7.0–7.6 ppm were attributed to the protons on DPS unit, which proved that DPS was introduced into the chain. This was consistent with the infrared results. Additionally, it can be observed that the peak of P(MTFPS-co-DPS) was split when the spectra was amplified from 0.20–0.35 (Fig. 2(b)). It suggested that P(MTFPS-co-DPS) most likely had a sequence consisting of –FFFFFPFFFFFF– (F was a –Si(CH3)(CH2CH2CF3)–O– unit, and P was a –Si(C6H5)2–O– unit). The existence of split peaks at 0.20–0.35 ppm indicated that the methyl siloxane unit was in a different chemical environment, and the distribution of F- and P-units in each sample followed a random distribution. | ||
| Fig. 2 1H-NMR spectra of PMTFPS and P(MTFPS-co-DPS): (a) 0–8 ppm and (b) 0.1–0.5 ppm. | ||
The quantitative analysis of the 1H-NMR spectra was performed via integrating the peaks in each spectrum. The contents of MTFPS and DPS were obtained according to the peak area of signals at 0.9 ppm and 7.4–7.8 ppm, and the results were presented in Table 2.
With respect to P(MTFPS-co-DPS),
| DPS (mol%) = [S(④,⑤,⑥)/10]/[(S(④,⑤,⑥)/10 + S(②)/2)] |
| MTFPS (mol%) = [S(②)/2]/[(S(④,⑤,⑥)/10 + S(②)/2)] |
It can be seen from Table 3 that the amount of DPS-diol involved in the reaction was less than the designed. Except for the loss of monomers during the synthesized process and the generation of the low-boiling siloxane residues, it was also due to the rigidity of the molecular chain increased and the number of molecular conformations decreased after the introduction of DPS unit, which led to the reaction difficult. In addition, by comparing P(MTFPS-co-DPS)-1, P(MTFPS-co-DPS)-2 and P(MTFPS-co-DPS)-3, it can be investigated that although the increase of temperature was conducive to the growth of the molecular chain, it has no effect on the reaction efficiency of the co-polymerization of DPS-diol and MTFPS-diol.
| Sample | Theoretical valuea | Experimental valueb | ||
|---|---|---|---|---|
| MTFPS, mol% | DPS, mol% | MTFPS, mol% | DPS, mol% | |
| a Theoretical value was calculated on the basis of the proportion of reactants.b Experimental value was calculated on the basis of the ratio of the integral area. | ||||
| P(MTFPS-co-DPS)-1 | 65 | 35 | 93 | 7 |
| P(MTFPS-co-DPS)-2 | 65 | 35 | 93 | 7 |
| P(MTFPS-co-DPS)-3 | 65 | 35 | 93 | 7 |
| P(MTFPS-co-DPS)-4 | 48 | 52 | 88 | 12 |
3.3 Analysis of cumulative sequence of P(MTFPS-co-DPS)
Previous studies have shown that 29Si-NMR spectroscopy was an effective tool for the cumulative sequence analysis of silicon copolymers.19–24 Fig. 3 showed the 29Si-NMR spectrum of P(MTFPS-co-DPS) and PMTFPS. A single signal (at −22.3 ppm) was produced for the F unit of PMTFPS, which was attributed to the structure of FF![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) FF. However, multiple split peaks appeared for P(MTFPS-co-DPS). Three mainly sets of peaks at −21.8, −22.3 and −46.2 ppm were observed in the curve of P(MTFPS-co-DPS)-3, which was attributed to the structure of F
FF. However, multiple split peaks appeared for P(MTFPS-co-DPS). Three mainly sets of peaks at −21.8, −22.3 and −46.2 ppm were observed in the curve of P(MTFPS-co-DPS)-3, which was attributed to the structure of F![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) FFF, FFFF and FFFF. It can be inferred that P(MTFPS-co-DPS) most likely had a sequence consisting of –FFFFFPFFFFFF–. The existence of split peaks confirmed effusively that the distributions of F- and P-unit in each sample followed random statistics, which was in well agreement with the results obtained from 1H-NMR analysis.
FFF, FFFF and FFFF. It can be inferred that P(MTFPS-co-DPS) most likely had a sequence consisting of –FFFFFPFFFFFF–. The existence of split peaks confirmed effusively that the distributions of F- and P-unit in each sample followed random statistics, which was in well agreement with the results obtained from 1H-NMR analysis.
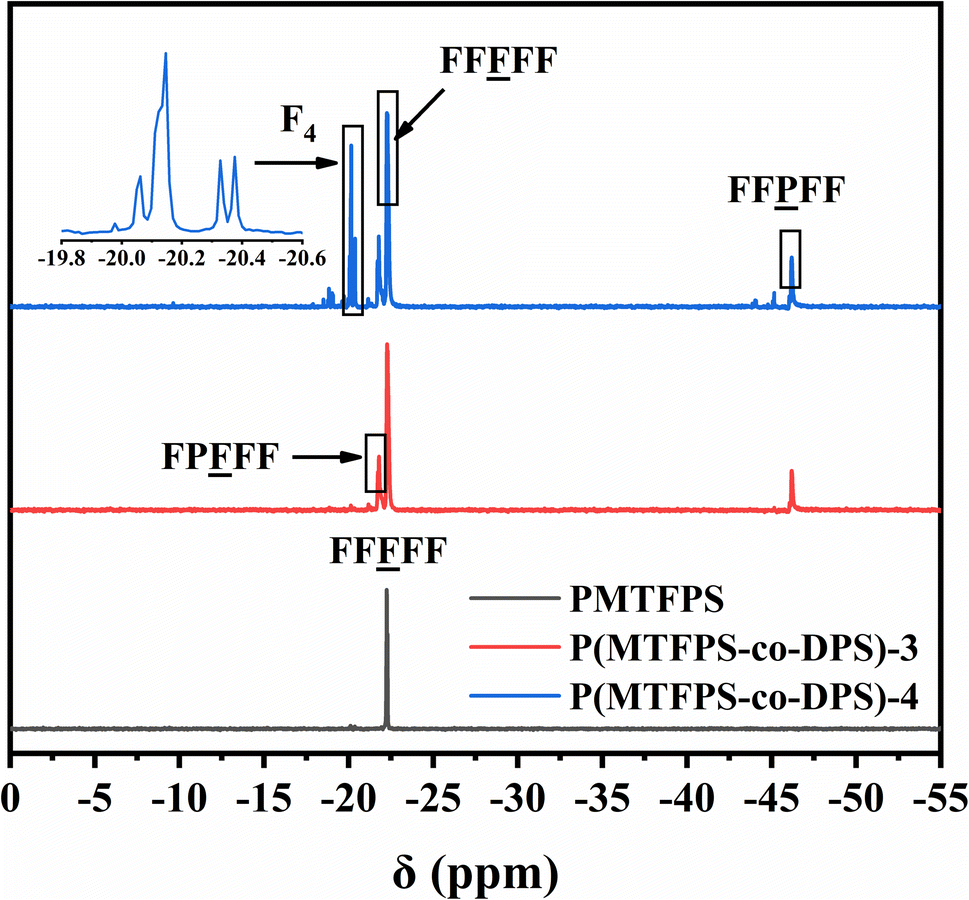 | ||
| Fig. 3 29Si-NMR spectrum for P(MTFPS-co-DPS) and PMTFPS. | ||
In addition, the curve of P(MTFPS-co-DPS)-4 exhibited additional signals, which caused by cyclic degradation products. The chemical shift around −20.0 ppm was attributed to 1,3,5,7-tetrakis(3,3,3-triflu-oropropylmethyl) cyclotetrasiloxane (F4). The two major signals at −20.1 and −20.3 ppm was attributed to the stereoisomer of F4.5 This was because the polymerization was an equilibrium reaction with a significant tendency to form the cyclic compounds, F4.25 The signals of F4 indicated that the more DPS unit was added, the more by-products (cyclosiloxane) were produced.
The glass transition temperature (Tg) of sample was determined by DSC. According to the proposal of International Confederation for Thermal Analysis (ICTA), the intersection point of the extension line of the transition line and the baseline extension line was taken as Tg. The results were shown in Fig. 4.
 | ||
| Fig. 4 DSC curves of PMTFPS and P(MTFPS-co-DPS). | ||
Cypryk et al.26 reported that the random copolymer exhibited one Tg value, and the block copolymer exhibited two Tgs values. Only one Tg was observed in the curve of P(MTFPS-co-DPS) in Fig. 4, which confirmed that P(MTFPS-co-DPS) had a uniform distribution of P and F units. Compared to the Tg of PMTFPS (−70.8 °C), the Tgs of all P(MTFPS-co-DPS) copolymers were higher. The reason was the Tg was closely related to the chain rigidity and flexibility of the polymer. According to group contribution theory, the internal friction of a material was related to its molecular structure. The more flexible the molecular chain, the smaller internal friction resistance of the chain segment movement, and the lower the Tg. Polymers incorporating DPS units exhibited greater rigidity and lower mobility, resulting in an increased Tg.
3.4 Thermal properties of cured polymer
To investigate the effect of DPS units on the thermal stability of PMTFPS, TGA analysis was used. Fig. 5 and Table 4 showed the thermogravimetric (TG) and derivative mass (DTG) results for the cured PMTFPS and P(MTFPS-co-DPS). According to the degradation mechanism of PMTFPS,27,28 there are two pathways of the weight loss: (a) the oxidation scission of side groups and (b) the “back-biting” reaction. Cause this experiment was carried out under N2 atmosphere, the oxidation scission of side groups was eliminated. Thus, the degradation stage was due to the “back-biting” of the main chain and there was only one peak in the DTG curve can be observed in the Fig. 5(b). From Table 4, it can be observed that the temperatures for 5% mass loss (Td5) and temperatures for maximum rate of mass loss (Tdmax) in all P(MTFPS-co-DPS) samples were higher than those for PMTFPS. Particularly, the Td5 and Tdmax of P(MTFPS-co-DPS)-3 were 417 °C and 494 °C, respectively, while the corresponding values of PMTFPS were 345 °C and 404 °C. This was because the introduction of DPS unit into PMTFPS increased the rigidity of the chain, making it difficult for the backbone to “back-bite”. | ||
| Fig. 5 TG (a) and DTG (b) curves of cured PMTFPS and P(MTFPS-co-DPS). | ||
| Sample | Td5 (°C) | Tdmax (°C) |
|---|---|---|
| PMTFPS | 345 | 404 |
| P(MTFPS-co-DPS)-1 | 366 | 412 |
| P(MTFPS-co-DPS)-2 | 384 | 411 |
| P(MTFPS-co-DPS)-3 | 417 | 494 |
| P(MTFPS-co-DPS)-4 | 385 | 450 |
Interestingly, although P(MTFPS-co-DPS)-4 exhibited a higher content of DPS unit than P(MTFPS-co-DPS)-3, its Td5 and Tdmax were lower than those of P(MTFPS-co-DPS)-3. The reason was the growth of the molecular chains was negatively affected by the addition of DPS unit, resulting in a low molecular weight. The lower molecular weights of P(MTFPS-co-DPS)-4 rendered its less thermally stable than P(MTFPS-co-DPS)-3.
The cured P(MTFPS-co-DPS) was ablated isothermally in an air environment for 10 min and 3 h to evaluate its thermal stability. Regardless of whether it was pyrolyzed for 10 min or 3 h, the mass losses of P(MTFPS-co-DPS) were less than those of PMTFPS. The results indicated that the thermal stability of PMTFPS was improved by the introduction of DPS units into the chain. According to the degradation pathways of PMTFPS,27 the mass loss of PMTFPS was mainly caused by the combined effect of the oxidation scission of side groups and the “back-biting” reaction when it was exposed to high temperatures in air. The introduction of DPS units into copolymers blocked the adjacent fluorosilicone units and changed the direction of the main chain, making it difficult for P(MTFPS-co-DPS) backbone to “back-bite” and rendering it more thermally stable than PMTFPS. From Fig. 6, it can be observed that P(MTFPS-co-DPS)-3 showed better thermal oxygen stability than P(MTFPS-co-DPS), which was consistent with the TGA results.
 | ||
| Fig. 6 Mass loss of cured PMTFPS and P(MTFPS-co-DPS) after air pyrolysis (a) for 10 min and (b) for 3 h. | ||
3.5 The heat resistance mechanism of P(MTFPS-co-DPS)
To obtain insight into the degradation mechanism, PMTFPS and P(MTFPS-co-DPS)-3 were pyrolyzed at 400 °C and 600 °C in a helium atmosphere, respectively. The product compositions, which were identified through the mass spectra, were presented in Table 5 and Fig. 7.| T (°C) | F3 (%) | F4 (%) | F5 (%) | |
|---|---|---|---|---|
| PMTFPS | 400 | 42.5 | 51.5 | 4.8 |
| 600 | 50.2 | 46.5 | 2.5 | |
| P(MTFPS-co-DPS)-3 | 400 | 12.0 | 49.0 | 3.3 |
| 600 | 33.1 | 40.3 | 1.7 |
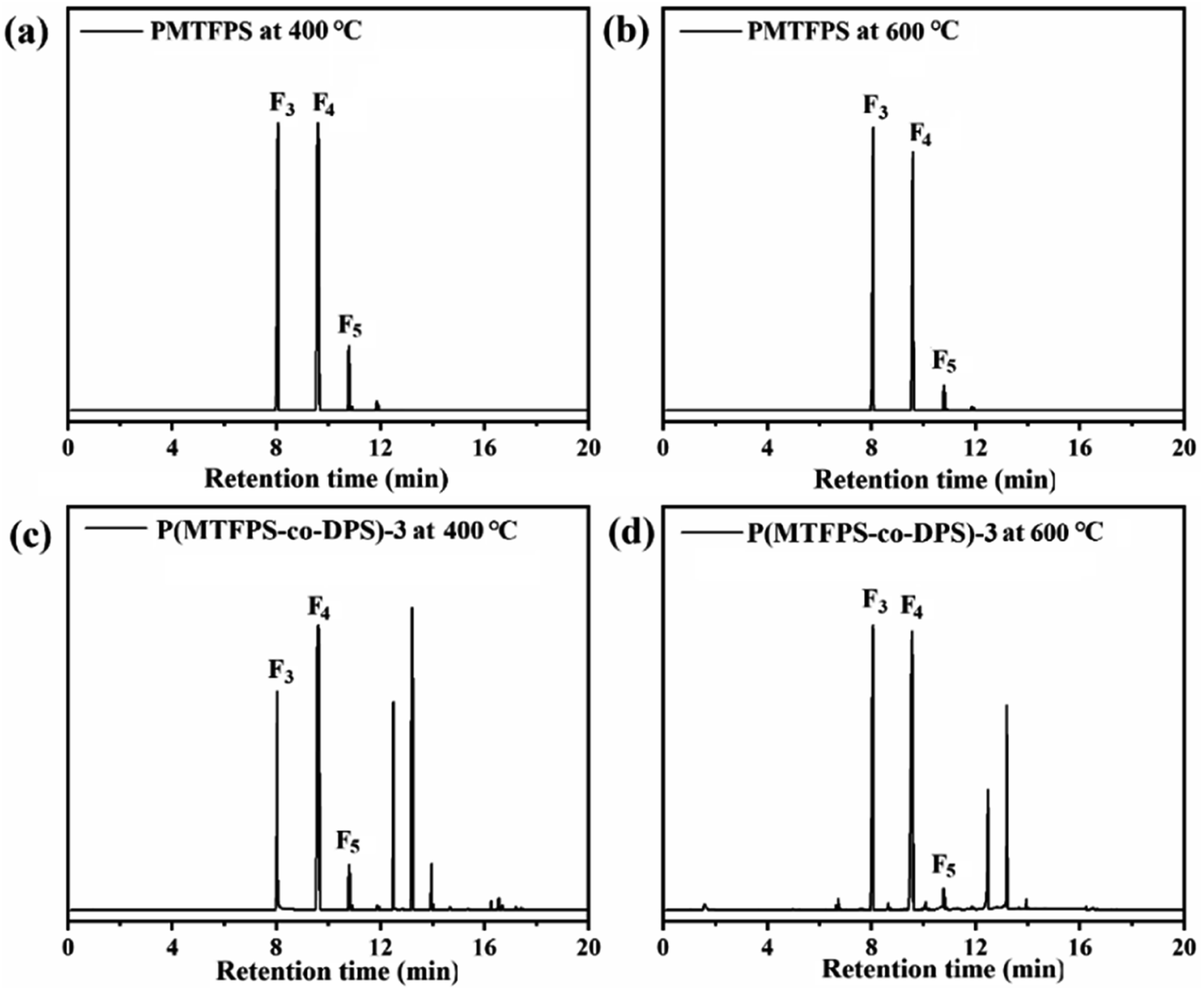 | ||
| Fig. 7 Compositions of the principal decomposition products of (a) PMTFPS pyrolyzed at 400 °C; (b) PMTFPS pyrolyzed at 600 °C; (c) P(MTFPS-co-DPS)-3 pyrolyzed at 400 °C; and (d) P(MTFPS-co-DPS)-3 pyrolyzed at 600 °C. | ||
Firstly, for both polymers, cyclic monomers, including F3, F4 and 1,3,5,7,9-penta(3,3,3-trifluoropylmethyl) cyclopentasiloxane (F5) were the primary products, and they were generated from the rearrangement of the siloxane main chain.
From the pyrolysis of P(MTFPS-co-DPS)-3, a relatively low amount of product F3 and F4 occurred compared with that from the pyrolysis of PMTFPS at pyrolysis temperatures of 400 °C and 600 °C. According to previous literature,11 it was suggested that the introduction of DPS unit in the chain significantly inhibited the “back-biting” reaction.
Once again, by comparing P(MTFPS-co-DPS)-3 with PMTFPS, the sum of cyclic monomers (i.e., F3, F4, and F5) significantly decreased. For example, the proportion was 75.1% for P(MTFPS-co-DPS)-3 at pyrolysis temperatures of 600 °C, which was 99.2% for PMTFPS. It could be inferred that when the DPS unit was introduced into the chain, the “back-biting” reaction was inhibited; thus, the amount of the cyclic monomer decreased.
4. Conclusions
In summary, a series of PMTFPS containing DPS units were successfully synthesized through the polycondensation method of DPS-diol and MTFPS-diol, and the so-obtained copolymers with contents of F and P units were uniformly distributed in the chain. 1H-NMR, 29Si-NMR and DSC results confirmed the sequence distribution of P(MTFPS-co-DPS) following random statistics. Besides, DSC curves exhibited that the Tg of different samples gradually increased with the increase of the DPS unit. TGA and isothermal degradation tests indicated that P(MTFPS-co-DPS) had higher thermal stability than PMTFPS. Analysis of Py-GCMS results indicated that the introduction of the DPS unit in the chain inhibited the “back-biting” reaction during the thermal degradation of PMTFPS, resulting in the improvement of the thermal stability of P(MTFPS-co-DPS).Author contributions
Yang You: conceptualization, investigation, data curation, writing – original draft preparation. Anna Zheng: supervision. Dafu Wei: validation. Xiang Xu: methodology, software, formal analysis. Yong Guan: writing-review & editing. Jianding Chen: conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21805086), Shanghai Leading Academic Discipline Project (B502), and the Shanghai Key Laboratory Project (Grant No. ZD20170203).References
- O. R. Pierce, G. W. Holbrook, O. K. Johannson, J. C. Saylor and E. D. Brown, Ind. Eng. Chem. Res., 1960, 52, 783–784 CrossRef CAS.
- M. A. Grunlan, J. M. Mabry and W. P. Weber, Polymer, 2003, 44, 981–987 CrossRef CAS.
- D. J. Cornelius and C. M. Monroe, Polym. Eng. Sci., 1985, 25, 467–473 CrossRef CAS.
- Y. Furukawa, S. Shinya, M. Saito, S. i. Narui and H. Miyake, Polym. Adv. Technol., 2010, 13, 60–65 CrossRef.
- H. Kahlig, P. Zollner and B. X. Mayer-Helm, Polym. Degrad. Stab., 2009, 94, 1254–1260 CrossRef.
- M. Patel and A. R. Skinner, Polym. Degrad. Stab., 2001, 73, 399–402 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lazzari, Polymer, 2001, 42, 2395–2402 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lageard, Polymer, 2002, 43, 2011–2015 CrossRef CAS.
- J. D. Jovanovic, M. N. Govedarica, P. R. Dvornic and I. G. Popovic, Polym. Degrad. Stab., 1998, 61, 87–93 CrossRef CAS.
- S. B. Zhang and H. J. Wang, J. Therm. Anal. Calorim., 2011, 103, 711–716 CrossRef CAS.
- Y. You, J. D. Chen, A. N. Zheng, D. F. Wei, X. Xu and Y. Guan, J. Appl. Polym. Sci., 2020, 137, 49347 CrossRef CAS.
- Z. Z. Yang, S. Han, R. Zhang, S. Y. Feng, C. Q. Zhang and S. Y. Zhang, Polym. Degrad. Stab., 2011, 96, 2145–2151 CrossRef CAS.
- M. Ikeda, T. Nakamura, Y. Nagase, K. Ikeda and Y. Sekine, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 2595–2607 CrossRef CAS.
- Y. Gao, W. Jiang, Y. Guan, P. Yang and A. Zheng, Polym. Eng. Sci., 2010, 50, 2440–2447 CrossRef CAS.
- T. Yu, H. B. Chang, W. P. Lai and X. F. Chen, Polym. Chem., 2011, 2, 892–896 RSC.
- G. Q. Qian and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4064–4073 CrossRef CAS.
- X. Xu, Z. X. Xu, P. Chen, X. D. Zhou, A. Zheng and Y. Guan, J. Inorg. Organomet. Polym., 2015, 25, 1267–1276 CrossRef CAS.
- D. Cai, A. Neyer, R. Kuckuk and H. M. Heise, J. Mol. Struct., 2010, 976, 274–281 CrossRef CAS.
- I. R. Herbert and A. D. H. Clague, Macromolecules, 2002, 22, 3267–3275 CrossRef.
- G. Cai and W. P. Weber, Polymer, 2004, 45, 2941–2948 CrossRef CAS.
- J. Chojnowski, S. Rubinsztajn, W. Fortuniak and J. Kurjata, Macromolecules, 2008, 41, 7352–7358 CrossRef CAS.
- B. X. Mayer, P. Zollner and H. Kahlig, J. Chromatogr. A, 1999, 848, 251–260 CrossRef CAS.
- B. X. Mayer, P. Zollner, W. Rauter and H. Kahlig, J. Chromatogr. A, 2001, 917, 219–226 CrossRef CAS PubMed.
- H. Kahlig and B. X. Mayer, J. Chromatogr. A, 2006, 1131, 235–241 CrossRef PubMed.
- H. F. Fei, X. Y. Gao, X. J. Han, Q. Wang, T. Hu, Z. J. Zhang and Z. M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1023–1031 CrossRef CAS.
- M. Cypryk, B. Delczyk, A. Juhari and K. Koynov, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1204–1216 CrossRef CAS.
- A. N. Zheng, Y. K. Huang, Y. You, J. Hu, D. F. Wei, X. Xu and Y. Guan, Polym. Degrad. Stab., 2018, 158, 168–175 CrossRef CAS.
- Y. Guan, R. Yang, Y. K. Huang, C. C. Yu, X. Li, D. F. Wei and X. Xu, Polym. Degrad. Stab., 2018, 156, 161–169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |