
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic interface between metal Cu nanoparticles and CoO for highly efficient hydrogen production from ammonia borane†
Hongmei Li,
Wenxue He,
Liuxin Xu,
Ya Pan,
Ruichao Xu,
Zhihu Sun * and
Shiqiang Wei
* and
Shiqiang Wei
National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, P. R. China. E-mail: zhsun@ustc.edu.cn
First published on 13th April 2023
Abstract
The development of efficient non-noble metal catalysts for the dehydrogenation of hydrogen (H2) storage materials is highly desirable to enable the global production and storage of H2 energy. In this study, Cux–(CoO)1−x/TiO2 catalysts with a Cu–CoO interface supported on TiO2 are shown to exhibit high catalytic efficiency for ammonia borane (NH3BH3) hydrolysis to generate H2. The best catalytic activity was observed for a catalyst with a Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co molar ratio of 1:1. The highest dehydrogenation turnover frequency (TOF) of 104.0 molH2 molmetal−1 min−1 was observed in 0.2 M NaOH at room temperature, surpassing most of the TOFs reported for non-noble catalysts for NH3BH3 hydrolysis. Detailed characterisation of the catalysts revealed electronic interactions at the Cu–CoO heterostructured interface of the catalysts. This interface provides bifunctional synergetic sites for H2 generation, where activation and adsorption of NH3BH3 and H2O are accelerated on the surface of Cu and CoO, respectively. This study details an effective method of rationally designing non-noble metal catalysts for H2 generation via a metal and transition-metal oxide interface.
:Co molar ratio of 1:1. The highest dehydrogenation turnover frequency (TOF) of 104.0 molH2 molmetal−1 min−1 was observed in 0.2 M NaOH at room temperature, surpassing most of the TOFs reported for non-noble catalysts for NH3BH3 hydrolysis. Detailed characterisation of the catalysts revealed electronic interactions at the Cu–CoO heterostructured interface of the catalysts. This interface provides bifunctional synergetic sites for H2 generation, where activation and adsorption of NH3BH3 and H2O are accelerated on the surface of Cu and CoO, respectively. This study details an effective method of rationally designing non-noble metal catalysts for H2 generation via a metal and transition-metal oxide interface.
Introduction
Hydrogen (H2) is a promising alternative to traditional fossil fuels due to its high energy density, sustainability, and pollution-free properties.1–3 For example, new-energy vehicles use H2 fuel cells as their power source.4 However, the wide application of H2 as a fuel is hindered by difficulties associated with its production, storage, and transportation.5–8 Exploration of efficient chemical H2 storage materials for H2 storage and release has hence attracted extensive attention in recent years. Among these materials, ammonia borane (NH3BH3) is considered to have many outstanding advantages, such as a high H2 content (19.6 wt%), high water solubility, non-toxicity, and excellent stability in aqueous solution and air under ambient conditions.9–11 More importantly, with the right catalyst, NH3BH3 can rapidly and completely release all of its H2 in aqueous solution at room temperature, as shown in the following reaction equation:| NH3BH3 + 2H2O → NH4BO2 + 3H2 | (1) |
So far, a large number of noble metal catalysts have been demonstrated to exhibit high activity in the hydrolysis of NH3BH3.12–15 Nevertheless, noble metals are costly and scarce, which significantly restricts their wide applications. Thus, cheap and abundant non-noble metal catalysts have attracted increasing attention. However, the design of a non-noble metal catalyst that exhibits high performance and long-term stability still remains a challenge.
Recent studies have shown that some transition metal (TM) catalysts (cobalt (Co), copper (Cu), iron (Fe), nickel (Ni)) exhibit catalytic activity in the hydrolytic dehydrogenation of NH3BH3.16–23 Among them, Cu is an abundant element and is one of the TMs with the most potential for NH3BH3 hydrolysis. As the catalytic performance of single metal Cu catalysts is poor,16,24 bimetallic or polymetallic Cu-based alloys and nanoparticles (NPs) have been extensively explored to improve the activity of Cu-based catalysts.25–29 The improved performance of polymetallic NPs arises from the synergistic effect of optimised composition and electron transfer between metals. Cu-based alloy catalysts also have limitations towards NH3BH3 hydrolysis as they only activate NH3BH3 molecules,30 while having only a weak effect on the adsorption and activation of H2O molecules, which is the rate-determining step in the hydrolysis reaction. Multiple studies have shown that transition-metal oxides (TMOs) accelerate the activation of water molecules.31–33 Therefore, a heterogeneous structure incorporating TMOs and metal NPs is an effective strategy for synthesising catalysts for hydrolysis reactions. Xu et al. reported that a catalyst with a Cu–Co3O4 interface greatly improves the hydrolysis performance of NH3BH3; Cu effectively activates NH3BH3 molecules and Co3O4 effectively activates H2O molecules.34 Wang and co-authors reported that a Cu/Cu0.76Co2.24O4 double active site catalyst modified with oxygen vacancies (VO) significantly increased the H2 production rate in the hydrolysis of NH3BH3.31 Zhou et al. fabricated CuNi bimetallic NPs anchored on Co3O4 nanosheets, where the synergistic effect between Cu and Ni species and the strong interactions between CuNi and Co3O4 afforded a maximum turnover frequency (TOF) of 31.5 molH2 molmetal−1 min−1 in 0.14 M NaOH at 298 K.33 However, the addition of ineffective proportions of TMOs with non-ideal compositions leads to catalysts being produced with low intrinsic activity. Therefore, the optimisation of the interfacial effect of metals and TMOs to achieve the best catalytic performance remains a challenging task. Moreover, these metal and oxide NPs tend to aggregate and lead to the deactivation of the reaction being catalysed, which results in greatly reduced stability and repeatability. An effective way to overcome this problem is to find a suitable support. Titanium dioxide (TiO2) is often used as a carrier for metal nanomaterials due to its excellent properties, such as non-toxicity and stability.35,36
In this study, it is reported that high catalytic activity for NH3BH3 hydrolysis can be achieved over catalysts with a Cu–CoO interface. CoO was selected to form an heterogeneous interface with metallic Cu NPs as Co2+ ions are in a relatively low oxidation state and therefore have the tendency to donate electrons and form active interfacial sites. A simple impregnation method was used to load Cu2+ and Co2+ on a TiO2 support, followed by the selective reduction of Cu2+ using NaBH4, thus leading to the formation of a series of Cux–(CoO)1−x/TiO2 samples containing a Cu–CoO interface. As expected, all of the catalysts significantly increase the rate of NH3BH3 dehydrogenation, and a volcano-type curve was obtained. Of the prepared catalysts, Cu0.5–(CoO)0.5/TiO2 exhibits the best catalytic activity, with a TOF of 104.0 molH2 molmetal−1 min−1 in 0.2 M NaOH at room temperature, surpassing the performance of most of the reported Cu-based catalysts for NH3BH3 hydrolysis. This outstanding performance arises from the synergistic interactions between Cu and CoO, which promote the activation of NH3BH3 and H2O molecules, respectively. The hybrid Cu0.5–(CoO)0.5/TiO2 catalyst is simple to synthesise, low cost, high efficiency, and can be widely used in a range of dehydrogenation reactions.
Experimental
Catalyst preparation
A series of Cux–(CoO)1−x/TiO2 samples were synthesised via an incipient wetness impregnation and co-reduction method. The detailed synthesis of Cu0.5–(CoO)0.5/TiO2 is used here as an example. Firstly, CuCl2·2H2O (341 mg) and CoCl2·6H2O (476 mg) were dissolved in water (14 mL), to which TiO2 powder (3.5 g) was added under magnetic stirring. The pH of the solution was kept at 7.0 and the reaction mixture was subjected to ultrasonication for 1 h. The OH groups on the surface of the TiO2 support serve as adsorption sites for Cu2+ and Co2+ cations. The pH of the solution is higher than the isoelectric point (5.8) of TiO2, which makes the surface of TiO2 negatively charged and favourable for cation adsorption. After drying at 60 °C overnight, the obtained powder was washed with a large amount of ultra-pure water to remove residual chloride ions. The impregnated intermediate was dehydrated at 60 °C to form CuO and CoO oxides, resulting in the formation of (CuO)0.5–(CoO)0.5/TiO2. Secondly, 350 mg of the above product was dispersed in 50 mL of water, to which 10 mL of an aqueous solution containing 100 mg of NaBH4 was added dropwise under vigorous magnetic stirring. When bubbles ceased to be generated in the reaction mixture, the reduction was taken to be complete. Finally, the resulting precipitate was filtered off, washed with water, and dried overnight at 60 °C. By changing the molar ratio of Cu to Co while keeping the total metal content at 4 mmol, CoO/TiO2, Cu0.1–(CoO)0.9/TiO2, Cu0.3–(CoO)0.7/TiO2, Cu0.7–(CoO)0.3/TiO2, Cu0.9–(CoO)0.1/TiO2, and Cu/TiO2 catalysts were synthesised using the same method.Catalyst characterisation
Powder X-ray diffraction (PXRD) analysis was recorded using a Rigaku Miniflex-600 diffractometer operated at a voltage of 40 kV and current of 15 mA using a Cu Kα radiation source. Inductively coupled plasma atomic emission spectroscopy (ICP-AES, Optima 7300 DV) was used to determine the exact Cu and Co content in the catalysts. X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo ESCALAB 250 spectrometer using an Al Kα (hν = 1486.6 eV) excitation source to analyse the elemental valence states of the samples. Energy-dispersive X-ray spectroscopy (EDX) mapping was conducted on a JEOLJEM-ARM200F transmission electron microscope (TEM)/scanning transmission electron microscope (STEM) equipped with a spherical aberration corrector and operated at 300 kV. TEM, high-resolution TEM (HRTEM), and EDX images were obtained on a JEM-2100F instrument operated at an accelerating voltage of 200 kV. Brunauer–Emmett–Teller (BET) specific surface areas of the samples were measured using an automatic volume adsorption device (Micrometrics ASAP 2020).X-ray absorption fine structure (XAFS) characterisation
XAFS measurements at the Cu K-edge (8979 eV) and Co K-edge (7709 eV) were performed at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF), China. The storage rings of the SSRF and BSRF were operated at 3.5 and 2.5 GeV in top-up mode, respectively. On both of the beamlines, Si(111) double-crystal monochromators were used and detuned by 30% to eliminate the higher-order harmonics. The Cu K-edge XAFS spectra were collected in transmission mode, the Co K-edge XAFS spectra in fluorescence mode. The ATHENA module implemented in the IFEFFIT software packages was used to process the obtained XAFS data.37Catalytic activity assessment
The H2 generation rate of the hydrolysis of NH3BH3 was measured via the water displacement method. In a typical process, a round-bottom flask (25 mL) containing a magneton was positioned in a water bath to control the temperature at 298 K with stirring. The flask was then sequentially filled with 10 mg of the Cu0.5–(CoO)0.5/TiO2 catalyst and 5 mL of an aqueous solution of NH3BH3 (0.065 mol L−1). Immediately, the hydrolysis reaction commenced and a large volume of H2 was produced and was collected in a water-filled inverted volumetric cylinder. The released H2 replaced the water in the cylinder, and the time was recorded after every 1 mL of H2 was collected. The activity of the other catalysts was also determined using the above method, while keeping the mass of all of the catalysts at 10 mg. For cycling experiments, the same amount of an aqueous solution of NH3BH3 was injected into the flask after the previous reaction was completed.Reactive kinetics measurements were also performed in the same reaction system. To measure the reaction order, the concentration of NH3BH3 and the mass of the catalyst were varied. Then, based on the rate equation, the rate constant, k, was obtained:
 | (2) |
|
lnr = nlnC + lnk
| (3) |
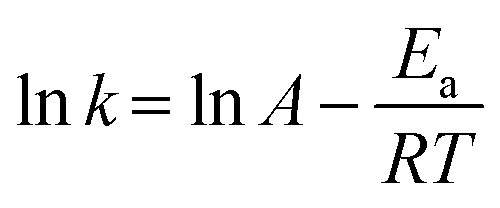 | (4) |
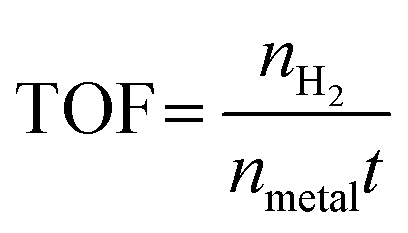 | (5) |
Results and discussion
Synthesis and characterisation of the catalysts
Several samples of Cux–(CoO)1−x/TiO2, Cu/TiO2 and CoO/TiO2 were synthesised on rutile TiO2 NPs via an incipient wetness impregnation and co-reduction method using CuCl2 and CoCl2 as raw materials. Reduced Co was coated on Cu particles, but the Co NPs were soon oxidised in the air and co-loaded on the TiO2 in the form of a CoO species. The Cu and Co content in these samples was measured by ICP-AES analysis (Table S1†). The total metal content in each catalyst was controlled at around 4 mmol. The PXRD patterns of all of the samples showed no diffraction peaks related to Cu and Co (Fig. S1†), suggesting the small particle size, high dispersion, or amorphous state of the Cu- and Co-related species. Among all the catalysts, Cu0.5–(CoO)0.5/TiO2 exhibited the highest catalytic activity for NH3BH3 hydrolysis (vide infra), and was thus selected for detailed characterisation.Fig. 1a shows the TEM images of the Cu0.5–(CoO)0.5/TiO2 catalyst. The surface of the catalyst has a rough appearance, which is very different to the smooth surface of the pristine TiO2 support (Fig. S2a†), indicating that small metal NPs are formed on the surface of the TiO2 support. Moreover, different from the severe particle agglomeration on the TiO2-free sample Cu0.5–(CoO)0.5 (Fig. S2b†), the Cu0.5–(CoO)0.5 NPs on the surface of TiO2 in Cu0.5–(CoO)0.5/TiO2 are fine and highly dispersed. The morphology of the Cu0.5–(CoO)0.5/TiO2 catalyst can be more clearly observed from its HRTEM image. As is shown in Fig. 1b, the interplanar spacing of 0.25 nm corresponds to the (101) crystal plane of rutile TiO2, and the other lattice spacing of 0.208 nm can be matched to the (111) crystal plane of face-centred cubic (fcc) Cu NPs. The crystal plane angle between two 111 planes calculated from the atomic spacing obtained by inverse Fourier transform (FT) of the selected region is 71°, which is also characteristic of Cu NPs. The light grey region surrounding the Cu NPs and TiO2 lattice is probably related to the CoO species, the lattice stripes of which cannot be observed because of its amorphous state. The corresponding elemental mapping of Cu0.5–(CoO)0.5/TiO2 shown in Fig. 1c further confirms the uniform distribution of Cu, Co, Ti, and O. As marked by the yellow circles, the Cu and Co species are closely connected and form an interface. Moreover, the HRTEM images of Cu/TiO2 and CoO/TiO2 were also recorded. The lattice stripes of Cu can be clearly observed for Cu/TiO2 (Fig. S2c†), but the amorphous CoO of CoO/TiO2 can only be observed vaguely (Fig. S2d†). This characterisation indicates that metallic Cu NPs and CoO with a uniform distribution were successfully synthesised on the TiO2 support.
 | ||
| Fig. 1 (a) TEM and (b) HRTEM images and atomic images obtained by local inverse Fourier transform of Cu0.5–(CoO)0.5/TiO2. (c) EDX elemental mapping images of Cu0.5–(CoO)0.5/TiO2. | ||
To determine the chemical composition and coordination structure of Cu0.5–(CoO)0.5/TiO2, XAFS measurements at the Cu and Co K-edges were performed. Fig. 2a shows the Cu K-edge X-ray absorption near-edge structure (XANES) spectra of Cu0.5–(CoO)0.5/TiO2, Cu foil, Cu2O, and CuO. It can be seen that the absorption edge of Cu0.5–(CoO)0.5/TiO2 is close to that of the Cu foil, indicating that the Cu NPs mainly exist in the metallic state. In addition, the stronger white line of Cu0.5–(CoO)0.5/TiO2 compared with that of the Cu foil suggests that the Cu is partially oxidised on the surface of the particles. Correspondingly, the FT k3-weighted extended XAFS (EXAFS) χ(k) curve of Cu0.5–(CoO)0.5/TiO2 shows two peaks at 1.50 and 2.20 Å (Fig. 2b). The 1.50 Å peak is located at the same position as the Cu–O peak for CuO, and the 2.20 Å peak is at the same position as the metallic Cu–Cu peak of the Cu foil. The apparent presence of metallic Cu–Cu bonds further proves that Cu metal NPs are generated in Cu0.5–(CoO)0.5/TiO2. The EXAFS FT curve of Cu0.5–(CoO)0.5/TiO2 has a distinctly different spectral shape than that of Cu2O, especially in the higher-shell R-region beyond 2 Å. Similarly, Fig. 2c–d show the Co K edge XAFS spectra of Cu0.5–(CoO)0.5/TiO2. The position of the absorption edge of Cu0.5–(CoO)0.5/TiO2 is closer to that of CoO than to those of Co foil and Co3O4, and the overall spectral shape resembles that of CoO, revealing that Co mainly exists in the form of CoO. In the EXAFS FT curve in Fig. 2d, two peaks at 1.47 and 2.76 Å can be observed, corresponding to the Co–O Co–Co bonds in CoO, respectively. No metallic Co–Co peak like that for the Co foil at 2.1 Å is visible, indicating the absence of metal Co particles.
 | ||
| Fig. 2 (a) Normalized XANES spectra and (b) Fourier transformation of EXAFS spectra at the Cu K-edge of Cu0.5–(CoO)0.5/TiO2, and reference samples of Cu foil, Cu2O and CuO. (c) Normalized XANES spectra and (d) Fourier transformation of EXAFS spectra at the Co-K edge of Cu0.5–(CoO)0.5/TiO2, and reference samples of Co foil, CoO and Co3O4. (e) The high-resolution XPS spectra of Cu 2p of Cu/TiO2 and Cu0.5–(CoO)0.5/TiO2. (f) The high-resolution XPS spectra of Co 2p of CoO/TiO2 and Cu0.5–(CoO)0.5/TiO2. | ||
To investigate the surface electronic states and interactions of the samples, XPS spectra of Cu0.5–(CoO)0.5/TiO2 and Cu/TiO2 were analysed, with their high-resolution spectra Cu 2p shown in Fig. 2e. For Cu0.5–(CoO)0.5/TiO2, the peaks at 932.0 and 952.0 eV can be attributed to the 2p3/2 and 2p1/2 of Cu0, and the peaks at 933.6 and 954.8 eV to the 2p3/2 and 2p1/2 of Cu2+, respectively. The two satellite peaks of Cu2+ 2p3/2 at 940.9 eV and 943.7 eV can also be observed.26,38 The Cu/TiO2 spectrum features apparent Cu0 and Cu2+ peaks. The peak area ratio of Cu0:Cu2+ increases from 0.6 for Cu/TiO2 to 1.2 for Cu0.5–(CoO)0.5/TiO2. Compared with Cu/TiO2, the binding energy of Cu0 2p3/2 in Cu0.5–(CoO)0.5/TiO2 shows a slight shift towards a lower energy, from 932.2 to 932.0 eV, suggesting the presence of electron-rich Cu0 in Cu0.5–(CoO)0.5/TiO2. In addition, high-resolution Co 2p spectra were recorded to determine the oxidation state of CoO in both CoO/TiO2 and Cu0.5–(CoO)0.5/TiO2 (Fig. 2f). The peaks in the spectra at around 780.7 and 796.6 eV, associated with the satellite peaks at 785.5 and 802.5 eV, are characteristic features of the Co 2p3/2 and 2p1/2 of CoO, respectively.18,39,40 Moreover, the binding energy of Co 2p3/2 in Cu0.5–(CoO)0.5/TiO2 (780.7 eV) is 0.2 eV higher than that (780.5 eV) of CoO/TiO2. The opposite trend in the Cu 2p3/2 and Co 2p3/2 binding energies is due to the transfer of electrons from CoO to Cu, indicating that the Cu–CoO heterostructure of Cu0.5–(CoO)0.5/TiO2 exhibits strong electronic interactions and interfacial synergism. Similar DFT calculations have been performed in previous studies, which have repeatedly confirmed that electron transfer occurs at the metal oxide interface.34,41
Catalytic performance for NH3BH3 hydrolysis
The catalytic performance of all of the prepared samples in the hydrolytic dehydrogenation of NH3BH3 was investigated by adding 10 mg of the catalysts to 5 mL of an aqueous solution containing NH3BH3 (0.325 mmol) at 298 K. If completely hydrolysed, 0.325 mmol of NH3BH3 yields approximately 21 mL of H2 (nH2/nAB = 3.0). Fig. 3a shows the time course data of the generation of H2 from different samples. CoO/TiO2 exhibits no activity towards the hydrolysis of NH3BH3, while Cu/TiO2 shows low activity, indicating that Cu is the effective element for the hydrolysis of NH3BH3. In contrast, all the Cux–(CoO)1−x/TiO2 (x ranges from 0.1 to 0.9) samples show much higher catalytic activity, exhibiting a strong synergistic effect between Cu and Co. The hydrolysis performance tests show that the Cu0.5–(CoO)0.5/TiO2 catalyst exhibits the highest catalytic activity, catalysing the hydrolysis reaction in just 174 s. As shown in Fig. 3b, the catalytic activity of the samples was quantified by the TOF values (Table S2†), which show volcano-type activity. Cu/TiO2 has a very low TOF of 1.0 molH2 molmetal−1 min−1. When CoO is present, the TOF rises rapidly to 14.9 molH2 molmetal−1 min−1 for the Cu0.9–(CoO)0.1/TiO2 catalyst with the lowest Co content. A maximum was reached for Cu0.5–(CoO)0.5/TiO2, the TOF of which of 40.8 molH2 molmetal−1 min−1 is higher than those of most of the reported Cu-based or other non-noble metal catalysts for NH3BH3 hydrolysis under the same conditions (Table S3†). With a further increase in Co content, the TOF declines. It is thus reasonable to infer that for Cu0.5–(CoO)0.5/TiO2 the synergistic effect of the interface between the metal Cu and CoO is the best, leading to this catalyst exhibiting the maximizing efficiency in NH3BH3 hydrolysis. When the Cu:Co ratio is higher or lower, the interfacial effect of Cu and CoO is weakened, and the catalyst activity is greatly reduced.34,42
 | ||
| Fig. 3 (a) Hydrogen production over time for the hydrolysis of ammonia borane (0.325 mmol, 5 mL) catalysed by Cux–(CoO)1−x/TiO2 samples with different Cu:Co ratios at 298 K. (b) The corresponding TOF value. | ||
As Cu0.5–(CoO)0.5/TiO2 exhibited the highest catalytic activity for the hydrolysis of NH3BH3, it was selected for further kinetic studies at different reaction temperatures, catalyst concentrations, and NH3BH3 concentrations. Fig. 4a shows the effect that temperature has on NH3BH3 hydrolysis. As the temperature increases from 298 to 313 K, the H2 evolution activity increases. According to the Arrhenius plot shows in Fig. 4b and the Arrhenius equation, the Ea value of Cu0.5–(CoO)0.5/TiO2 for the hydrolysis of NH3BH3 is 48.1 kJ mol−1. Fig. S3† shows the influence of catalyst concentration on the reaction rate, which was measured by varying the amount of Cu0.5–(CoO)0.5/TiO2 from 10 to 25 mg. As the amount of catalyst increases, the rate of H2 production increases. The slope of ln(rate) versus ln (catalyst amount) curve is 1.0, indicating that the catalytic hydrolysis of NH3BH3 is a first-order reaction relative to the catalyst concentration. The effect that NH3BH3 concentration has on the H2 production rate was measured (Fig. S4†), and the volume of H2 was found to increase in line with an increase in the NH3BH3 content, but the initial rate of H2 production remained the same. The relationship between the H2 production rate and the corresponding NH3BH3 concentration was determined on the natural logarithmic scale, and the slope of the curve was measured as 0.073, suggesting that the hydrolysis of NH3BH3 catalysed by Cu0.5–(CoO)0.5/TiO2 is a zero-order reaction with respect to the concentration of NH3BH3. In summary, the hydrolysis of NH3BH3 is facilitated by Cu0.5–(CoO)0.5/TiO2 catalyst and is independent of the NH3BH3 concentration.
 | ||
| Fig. 4 (a) H2 production over time for the hydrolysis of NH3BH3 catalysed by Cu0.5–(CoO)0.5/TiO2 at different temperatures. (b) The corresponding Arrhenius plot and the Ea. (c) Recyclability testing of Cu0.5–(CoO)0.5/TiO2 as a catalyst in the hydrolysis of NH3BH3 for five cycles. (d) N2 adsorption–desorption isotherms of Cu0.5–(CoO)0.5/TiO2 before and after five cycles. | ||
The stability of a catalyst is the key to its practical application. With this in mind, the recyclability of Cu0.5–(CoO)0.5/TiO2 was tested by adding an additional aqueous solution of NH3BH3 (0.325 mmol, 5 mL) at room temperature after the previous reaction was completed, with the results shown in Fig. 4c. It can be seen that the total H2 production does not change, but the rate of H2 generation gradually slows down. This shows that the catalyst has relatively high stability and can catalyse the complete hydrolysis of NH3BH3, but that the activity decreases slightly. After five cycles, the activity of Cu0.5–(CoO)0.5/TiO2 is still 67% that of the original value. To explore the causes behind the decrease in the catalyst activity, nitrogen (N2) adsorption–desorption isotherm experiments were conducted to determine the specific surface area of the Cu0.5–(CoO)0.5/TiO2 catalyst before and after five catalytic cycles. Fig. 4d shows the BET curves plotted over a relative pressure range of 0–1.0 (P/P0). The BET specific surface area of Cu0.5–(CoO)0.5/TiO2 is 27.22 m2 g−1, which decreased to 12.54 m2 g−1 after five cycles of hydrolysis reactions. The decrease in the specific surface area of Cu0.5–(CoO)0.5/TiO2 indicated that the support and metal NPs agglomerated, leading to a reduction in the number of active sites of the catalyst, which may have led to the decrease in its catalyst activity. In addition, the PXRD and TEM data of Cu0.5–(CoO)0.5/TiO2 were recorded after five cycles. As shown in Fig. S5a,† no diffraction peaks of Cu and Co can be observed in the PXRD pattern Cu0.5–(CoO)0.5/TiO2, indicating that the particles of Cu0.5–(CoO)0.5/TiO2 after the reaction are small or amorphous. The TEM image shown in Fig. S5b† indicates that some Cu0.5–(CoO)0.5/TiO2 aggregates into larger particles, which may be another reason its reduced catalytic efficiency after reaction. In addition, there may be some other possible reasons behind the reduced catalytic efficiency of Cu0.5–(CoO)0.5/TiO2. Firstly, an increase in the number of cycles, the concentration of the reactants is diluted, and the by-products generated accumulate, hindering the forward reaction. Secondly, the increase in the by-products gradually leads to an increase in solution viscosity, which slows the reaction. Moreover, a large number of boronised species produced in the reaction cover the surface of the catalyst, reducing the catalytic efficiency.
Study on hydrolytic mechanism of NH3BH3
It has been proposed in several studies that the cleavage of the O–H bond in the H2O molecule is the rate-determining step in the hydrolysis of NH3BH3.43–47 To further elucidate this mechanism and facilitate the rational design of the catalyst, isotopic experiments on the Cu0.5–(CoO)0.5/TiO2 catalyst were conducted using D2O instead of H2O. The kinetic isotope effect (KIE) was used reveal whether water is the rate-determining step in this reaction, with the results shown in Fig. 5a. The hydrolysis of NH3BH3 in D2O exhibits a slower rate of H2 release and a longer reaction time compared to that of NH3BH3 in H2O. The KIE value (H2O rate/D2O rate) was calculated to be 5.5. | ||
| Fig. 5 Hydrolysis of ammonia borane catalysed by Cu0.5–(CoO)0.5/TiO2 (a) in different solutions (H2O or D2O), and (b) in different concentrations of NaOH from 0.1 to 0.4 M, at 298 K. (c) Hydrogen production over time for the hydrolysis of ammonia borane (0.325 mmol, 5 mL) catalysed by different samples. | ||
To further explore the effect of OH− on NH3BH3 hydrolysis, a series of experiments were conducted at different concentrations of NaOH to test the catalytic performance of Cu0.5–(CoO)0.5/TiO2. As shown in Fig. 5b, with an increase in NaOH concentration, the H2 release rate gradually accelerates. When the concentration of NaOH is 0.2 M, the TOF reaches a maximum value of 104.0 molH2 molmetal−1 min−1, which exceeds the TOF values of most non-noble metal catalysts for the same reaction (Table S3†). In 0.2 M NaOH, the apparent activation energy, Ea, is estimated to be 24.28 kJ mol−1 (Fig. S6†), significantly lower than the Ea of 48.1 kJ mol−1 recorded in the absence of NaOH. These results indicate that OH− provided by NaOH promotes the catalytic reaction. On the one hand, OH− adsorption on the catalyst surface increases electron density, which is conducive to the attack on the reactants and accelerates the intermediate reaction. On the other hand, OH− provides more adsorbed O–H, accelerating the oxidation addition reaction of water.17,48,49 However, when the concentration of NaOH exceeds 0.2 M, the catalytic rate decreases. This may be due to the excess OH− species overlaying the active sites originally used for H coordination and H2 dissociation.15,31 The rate of generation of H2 was found to be generally accelerated by NaOH, supporting that the rate-determining step in the hydrolysis reaction is the cleavage of the O–H bond in H2O.
Based on the above results, it is reasonable to conclude that the excellent performance of Cu0.5–(CoO)0.5/TiO2 in the hydrolysis of NH3BH3 can be attributed to the strong interfacial interaction of Cu–CoO, which accelerates the rate-determining step. To further verify this conclusion, comparison experiments were conducted, and the results are shown in Fig. 5c. Such interfacial effects may also be observed upon physically mixing the two components; thus, it is of interest to observed how the catalysts behave when mixed. For this purpose, Cu/TiO2 and CoO/TiO2 were mechanically mixed and tested under the same conditions. As observed from Table S2,† the mixed Cu/TiO2 + CoO/TiO2 catalyst has a TOF value of 9.9 molH2 molmetal−1 min−1, higher those of Cu/TiO2 or CoO/TiO2 alone, but far less than that of Cu0.5–(CoO)0.5/TiO2. As expected, the interfacial contact between Cu and CoO in the mechanically mixed NPs gives rise to an interfacial synergy that increases the reaction rate. However, the interfacial contact between Cu/TiO2 and CoO/TiO2 is limited, meaning that its activity is far worse than that of Cu0.5–(CoO)0.5/TiO2 comprised of closely connected Cu–CoO. In terms of Cu–CoO, the presence of electron-rich Cu is conducive to the attack and activation of NH3BH3,19,31,50,51 and CoO enhances the adsorption of H2O, thus speeding up the rate-determining step of the reaction, leading to an improvement in the activity of the catalyst in a bifunctional way to achieve the best reaction efficiency.
As previously mentioned, some of the Cu atoms in the Cu0.5–(CoO)0.5/TiO2 catalyst are oxidised. Given that interfacial sites play essential roles in NH3BH3 hydrolysis, the enhanced H2 generation rate may be attributed to CuO–CoO interfacial sites. To clarify this point, a control experiment was conducted by synthesising a CuO–CoO/TiO2 sample with similar Cu and Co content to Cu0.5–(CoO)0.5/TiO2, and it was characterised by PXRD (Fig. S7†) and XPS (Fig. S8†). The Cu 2p and Co 2p spectra, as well as the content of each element measured by XPS (Table S4†), indicate the presence of CuO and CoO in CuO–CoO/TiO2. The catalytic performance of CuO–CoO/TiO2 was then investigated (Table S2†). The attained TOF value of 19.0 molH2 molmetal−1 min−1 is much better than those of Cu/TiO2 and CoO/TiO2, as the CuO/CoO interface activates H2O and accelerate the rate-determining step of the reaction.34,52,53 However, the TOF of CuO–CoO/TiO2 is still significantly inferior to that of Cu0.5–(CoO)0.5/TiO2, indicating that the heterogeneous structure of Cu–CoO compared with that of CuO–CoO plays a major and decisive role in promoting the reaction. In addition, to explore the role of the TiO2 support, a support-free Cu0.5–(CoO)0.5 catalyst was prepared, and performance testing was conducted under the same conditions. The H2 production rate of Cu0.5–(CoO)0.5 was observed to be poor (Fig. 5c) and its TOF value was only 2.4 molH2 molmetal−1 min−1 (Table S2†), much lower than that of Cu0.5–(CoO)0.5/TiO2. This shows that the addition of the TiO2 support improves the dispersion of Cu and CoO NPs by inhibiting NP aggregation and exposing more active sites, which is conducive to promoting the catalytic reaction.
A possible catalytic synergistic mechanism of NH3BH3 hydrolysis catalysed by Cu0.5–(CoO)0.5/TiO2 catalyst was proposed (Scheme 1). Firstly, the strong interfacial interaction results in unbalanced electron distribution on the surface of Cu–CoO, which accelerates and activates the adsorption of NH3BH3 and H2O on the surface of Cu and CoO, respectively, thus reducing the reaction energy barrier.41,54 Secondly, the O–H bond of the activated H2O molecule breaks, with the formation of H* and OH*. Then, OH* attacks and breaks the B–H bond of NH3BH3 to form NH3BH2–OH accompanied by the release of H*, which combines with another H* released by H2O to generate 1 mol of H2 at the surface of the Cu0.5–(CoO)0.5/TiO2 catalyst. At the same time, the second H2O molecule adsorbed on CoO is activated, and the above steps are repeated until 3 mol of H2 is finally released from the catalyst surface. In the catalytic process, the strong interaction between Cu and CoO makes promotes the adsorption and activation of NH3BH3 and H2O, reducing the energy barrier of the entire reaction and accelerating the release rate of H2 in the catalytic reaction. Therefore, the unique metal and metal oxide structures in the Cu0.5–(CoO)0.5/TiO2 catalyst play an important synergistic role in accelerating reactant activation. The support TiO2 ensures the dispersion and stability of the active sites during the reaction. The overall coordination of the catalyst greatly improves the catalytic performance of H2 production from NH3BH3.
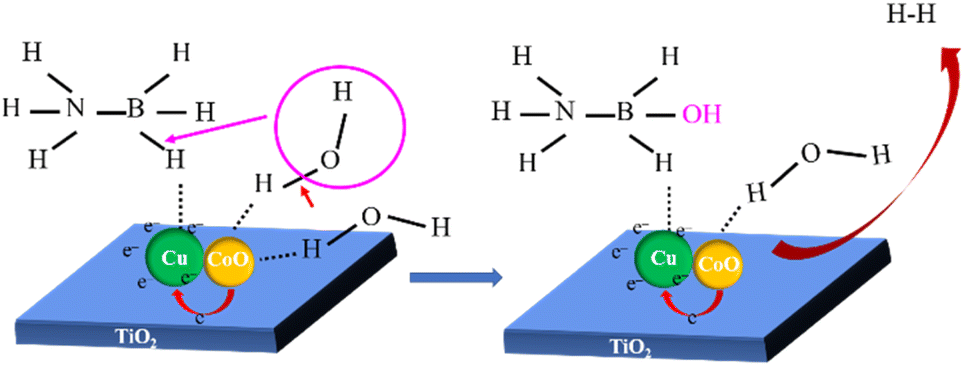 | ||
| Scheme 1 A proposed mechanism for ammonia borane hydrolysis over the Cu0.5–(CoO)0.5/TiO2 catalyst. | ||
Conclusions
In summary, metallic Cu NPs and oxidised CoO NPs were successfully loaded on a TiO2 support via simple incipient wetness impregnation and co-reduction methods. The optimum Cu0.5–(CoO)0.5/TiO2 catalyst exhibits high activity and stability in the hydrolysis of NH3BH3, with a TOF value of 104.0 molH2 molmetal−1 min−1 in 0.2 M NaOH at room temperature, which is better than the performance of most reported non-noble metal catalysts. The synergistic interface of Cu–CoO accelerates the oxidative cleavage of the O–H bonds in H2O (the rate-determining step) and the activation of NH3BH3 molecules, thus increasing the hydrolysis rate of NH3BH3. This study demonstrates the high efficiency of the interface structure of metal and transition metal oxides for hydrolytic dehydrogenation of ammonia borane, which can also be applied to many other homogeneous or heterogeneous catalytic hydrogen production reactions, such as NaBH4 hydrolysis and electrocatalytic hydrogen evolution reactions, providing a simple and reliable strategy for the development of non-precious metal catalysts.Author contributions
Hongmei Li: conceptualization, methodology, writing – original draft. Wenxue He: investigation, resources. Liuxin Xu: data curation. Ya Pan: validation, visualization. Ruichao Xu: formal analysis, data curation. Zhihu Sun: writing – review & editing, funding acquisition. Shiqiang Wei: supervision, project administration.Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
The authors thank National Key Research and Development Program of China (No. 2021YFA1500403), the National Natural Science Foundation of China (Grant No. 12075243 and 12135012) for financial support. They also greatly appreciate the synchrotron radiation beam times of SSRF and BSRF.Notes and references
- I. P. Jain, Int. J. Hydrogen Energy, 2009, 34, 7368–7378 CrossRef CAS.
- S. Sharma and S. K. Ghoshal, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- E. Antolini, RSC Adv., 2016, 6, 3307–3325 RSC.
- A. M. Abdalla, S. Hossain, O. B. Nisfindy, A. T. Azad, M. Dawood and A. K. Azad, Energy Convers. Manage., 2018, 165, 602–627 CrossRef CAS.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2021, 50, 3437–3484 RSC.
- Y. Ma, X. R. Wang, T. Li, J. Zhang, J. Gao and Z. Y. Sun, Int. J. Hydrogen Energy, 2021, 46, 27330–27348 CrossRef CAS.
- C. Lang, Y. Jia and X. Yao, Energy Storage Mater., 2020, 26, 290–312 CrossRef.
- Z. Lin, B. Xiao, Z. Wang, W. Tao, S. Shen, L. Huang, J. Zhang, F. Meng, Q. Zhang, L. Gu and W. Zhong, Adv. Funct. Mater., 2021, 31, 2102321 CrossRef CAS.
- A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed.
- C. Y. Peng, L. Kang, S. Cao, Y. Chen, Z. S. Lin and W. F. Fu, Angew. Chem., Int. Ed., 2015, 54, 15725–15729 CrossRef CAS PubMed.
- J. Manna, S. Akbayrak and S. Özkar, Appl. Catal., B, 2017, 208, 104–115 CrossRef CAS.
- X. Huang, Y. Liu, H. Wen, R. Shen, S. Mehdi, X. Wu, E. Liang, X. Guo and B. Li, Appl. Catal., B, 2021, 287, 119960 CrossRef CAS.
- S. Rej, L. Mascaretti, E. Y. Santiago, O. Tomanec, Š. Kment, Z. Wang, R. Zbořil, P. Fornasiero, A. O. Govorov and A. Naldoni, ACS Catal., 2020, 10, 5261–5271 CrossRef CAS.
- F. Fu, C. Wang, Q. Wang, A. M. Martinez-Villacorta, A. Escobar, H. Chong, X. Wang, S. Moya, L. Salmon, E. Fouquet, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2018, 140, 10034–10042 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, Y. Zhu, G. Mi, X. Du and Y. Dong, Renewable Energy, 2018, 118, 146–151 CrossRef CAS.
- C. Wang, J. Tuninetti, Z. Wang, C. Zhang, R. Ciganda, L. Salmon, S. Moya, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2017, 139, 11610–11615 CrossRef CAS PubMed.
- S. Mehdi, Y. Liu, H. Wei, H. Wen, R. Shen, Z. Peng, H. Zhang, X. Wu, C. Wang, S. Guan, T. Liu and B. Li, ACS Appl. Nano Mater., 2022, 5, 5064–5074 CrossRef CAS.
- J. Wang, Y. Chen, S. Guan, J. Shi, M. Li and B. Liu, J. Alloys Compd., 2022, 913, 165215 CrossRef CAS.
- Y. Feng, X. Zhang, Y. Shao, X. Chen, H. Wang, J. Li, M. Wu, H. Dong, Q. Liu and H. Li, ACS Appl. Mater. Interfaces, 2022, 14, 27979–27993 CrossRef CAS PubMed.
- J. Liao, Y. Shao, Y. Feng, J. Zhang, C. Song, W. Zeng, J. Tang, H. Dong, Q. Liu and H. Li, Appl. Catal., B, 2023, 320, 121973 CrossRef CAS.
- J. Liao, Y. Wu, Y. Shao, Y. Feng, X. Zhang, W. Zhang, J. Li, M. Wu, H. Dong, Q. Liu and H. Li, Chem. Eng. J., 2022, 449, 137755 CrossRef CAS.
- Y. Feng, Y. Li, Q. Liao, W. Zhang, Z. Huang, X. Chen, Y. Shao, H. Dong, Q. Liu and H. Li, Fuel, 2023, 332, 126045 CrossRef CAS.
- P. Li, X. Zhang, J. Wang, B. Xu, X. Zhang, G. Fan, L. Zhou, X. Liu, K. Zhang and W. Jiang, Colloids Surf., 2021, 629, 127383 CrossRef CAS.
- K. Guo, Y. Ding, J. Luo, M. Gu and Z. Yu, ACS Appl. Energy Mater., 2019, 2, 5851–5861 CrossRef CAS.
- P. Zhang, G. Zeng, T. Song, S. Huang, T. Wang and H. Zeng, Appl. Catal., B, 2019, 242, 389–396 CrossRef CAS.
- J. Li, Q.-L. Zhu and Q. Xu, Catal. Sci. Technol., 2015, 5, 525–530 RSC.
- J.-M. Yan, Z.-L. Wang, H.-L. Wang and Q. Jiang, J. Mater. Chem., 2012, 22, 10990 RSC.
- Y. Liu, J. Zhang, H. Guan, Y. Zhao, J.-H. Yang and B. Zhang, Appl. Surf. Sci., 2018, 427, 106–113 CrossRef CAS.
- X. Yang, Q. Li, L. Li, J. Lin, X. Yang, C. Yu, Z. Liu, Y. Fang, Y. Huang and C. Tang, J. Power Sources, 2019, 431, 135–143 CrossRef CAS.
- C. Wang, Y. Ren, J. Zhao, S. Sun, X. Du, M. Wang, G. Ma, H. Yu, L. Li, X. Yu, X. Zhang, Z. Lu and X. Yang, Appl. Catal., B, 2022, 314, 121494 CrossRef CAS.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607–2622 CrossRef CAS.
- J. Zhou, X. Feng, Y. Zhao, R. Cui, D. Wang and B. Zhang, J. Alloys Compd., 2022, 923, 166345 CrossRef CAS.
- W. Xu, S. Zhang, R. Shen, Z. Peng, B. Liu, J. Li, Z. Zhang and B. Li, Energy Environ. Mater., 2022, 1–7 Search PubMed.
- W. He, X. Zhang, K. Zheng, C. Wu, Y. Pan, H. Li, L. Xu, R. Xu, W. Chen, Y. Liu, C. Wang, Z. Sun and S. Wei, Angew. Chem., Int. Ed., 2022, e202213365 Search PubMed.
- G. Yang, S. Guan, S. Mehdi, Y. Fan, B. Liu and B. Li, Green Energy Environ., 2021, 6, 236–243 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- J. Liao, D. Lu, G. Diao, X. Zhang, M. Zhao and H. Li, ACS Sustainable Chem. Eng., 2018, 6, 5843–5851 CrossRef CAS.
- Y. Pan, L. Xu, L. Huang, W. He, H. Li, S. Wang, Z. Long and Z. Sun, ACS Appl. Energy Mater., 2021, 4, 11151–11161 CrossRef CAS.
- Y. Pan, L. Xu, W. He, H. Li, W. Chen and Z. Sun, Nanoscale, 2022, 14, 7303–7313 RSC.
- S. Guan, Y. Guo, H. Zhang, X. Liu, Y. Fan and B. Liu, Sustainable Energy Fuels, 2022, 6, 1753–1761 RSC.
- J. Manna, S. Akbayrak and S. Özkar, RSC Adv., 2016, 6, 102035–102042 RSC.
- Y. Ge, X. Qin, A. Li, Y. Deng, L. Lin, M. Zhang, Q. Yu, S. Li, M. Peng, Y. Xu, X. Zhao, M. Xu, W. Zhou, S. Yao and D. Ma, J. Am. Chem. Soc., 2021, 143, 628–633 CrossRef CAS PubMed.
- F. Tong, X. Liang, M. Liu, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, ACS Catal., 2022, 12, 3558–3565 CrossRef CAS.
- M. H. Fang, S. Y. Wu, Y. H. Chang, M. Narwane, B. H. Chen, W. L. Liu, D. Kurniawan, W. H. Chiang, C. H. Lin, Y. C. Chuang, I. J. Hsu, H. T. Chen and T. T. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 47465–47477 CrossRef CAS PubMed.
- Z. Li, T. He, L. Liu, W. Chen, M. Zhang, G. Wu and P. Chen, Chem. Sci., 2017, 8, 781–788 RSC.
- S. Chen, B. Gong, J. Gu, Y. Lin, B. Yang, Q. Gu, R. Jin, Q. Liu, W. Ying, X. Shi, W. Xu, L. Cai, Y. Li, Z. Sun, S. Wei, W. Zhang and J. Lu, Angew. Chem., Int. Ed., 2022, 61, e202211919 CAS.
- J. Deng, X. Zhou, J. Zou, Y. Qin and P. Wang, ACS Appl. Energy Mater., 2022, 5, 7408–7419 CrossRef CAS.
- Q. Yao, K. Yang, X. Hong, X. Chen and Z.-H. Lu, Catal. Sci. Technol., 2018, 8, 870–877 RSC.
- P. Li, R. Chen, S. Zhao, W. Li, Y. Lin and Y. Yu, Appl. Catal., B, 2021, 298, 120523 CrossRef CAS.
- S. Zhang, M. Li, L. Li, F. Dushimimana, J. Zhao, S. Wang, J. Han, X. Zhu, X. Liu, Q. Ge and H. Wang, ACS Catal., 2020, 10, 14903–14915 CrossRef CAS.
- K. Feng, J. Zhong, B. Zhao, H. Zhang, L. Xu, X. Sun and S. T. Lee, Angew. Chem., Int. Ed. Engl., 2016, 55, 11950–11954 CrossRef CAS PubMed.
- H. Zheng, K. Feng, Y. Shang, Z. Kang, X. Sun and J. Zhong, Inorg. Chem. Front., 2018, 5, 1180–1187 RSC.
- J. Li, X. Ren, H. Lv, Y. Wang, Y. Li and B. Liu, J. Hazard. Mater., 2020, 391, 122199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01265d |
| This journal is © The Royal Society of Chemistry 2023 |