
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient Cr(VI) removal by expanded graphite synergized with oxalic acid under UV irradiation†
Ling Zhang ,
Yanqing Sun*,
Jie Sun and
Fengming Cao
,
Yanqing Sun*,
Jie Sun and
Fengming Cao
School of Environmental and Chemical Engineering, Shanghai University, 99 Shangda Road, Shanghai 200444, PR China. E-mail: wqbiss@163.com
First published on 13th April 2023
Abstract
Expanded graphite (EG), an easily-obtained carbon material with the potential of transferring electrons, was utilized successfully in the removal of hazardous hexavalent chromium (Cr(VI)) by environment-friendly oxalic acid (Ox) under UV irradiation. EG with a unique worm-like structure was obtained via a facile microwave treatment. The results showed that the EG + Ox + UV system had optimum performance, removing 99.32% of the Cr(VI) (1 mM) within 60 min at pH = 3, and the kinetic rate constant of Cr(VI) elimination was 7.95 mol L−1 min−1. Three components are potentially involved in the Cr(VI) elimination mechanism by the EG + Ox + UV system: (1) the direct electron transfer (DET) pathway of the EG-Ox-Cr(VI) through the acceleration effect of EG caused the majority removal of Cr(VI) under UV; (2) ·CO2− generated from Ox photolysis was used to reduce some Cr(VI); (3) ·CO2− created from Cr(VI)–Ox complexes in the solution through the photoinduced electron transfer (PET) pathway also reduced a little Cr(VI). Overall, the efficient removal of Cr(VI) by the EG + Ox + UV system provided new ideas for future research on Cr(VI) treatment.
1. Introduction
Most recently, chromium-containing wastewater has become one of the most threatening water pollutants worldwide.1 The main existences of chromium in chromium-containing wastewater are both hexavalent (Cr(VI)) and trivalent (Cr(III)) chromium. Cr(III) is barely harmful or even non-toxic, while Cr(VI) is famous for its serious carcinogenesis or gene mutation damage to the human body due to its high toxicity.2 In order to control chromium pollution, it is particularly crucial to eliminate Cr(VI) from water.The process of adsorption can effectively remove Cr(VI) from water considering the benefits of simple operation, low price, and less secondary pollution,3,4 but subsequent desorption processes are complicated, and Cr(VI) still has a high level of toxicity because adsorption simply transfers Cr(VI) from the solution to the adsorbate. The method of reduction can effectively remove hazardous Cr(VI) from water by converting Cr(VI) to Cr(III). Especially, the chemical reduction method is widely used in the industry, in which traditional inorganic reductants (such as SO2, NaHSO3, and FeSO4) are commonly used.5 Although this method is easy and efficient to operate, it is still limited to the requirement of an excess amount of the reductant and the needed treatment of the large amounts of the by-produced sludge.
Oxalic acid (Ox), the most widely distributed and highest-content small-molecule organic acid in nature, is effective at removing Cr(VI) from the environment.6 As an organic reductant, Ox can be oxidized and released as CO2 after participating in the reaction, which is identified as a promising green reductant with good environmental friendliness. However, because it often takes months to years for Ox to directly reduce Cr(VI),7 it is vital to improving the reduction effect of Ox. The introduction of ultraviolet (UV),8 Fe(III),9,10 and catalysts11,12 can speed up the efficiency at which Ox reduces Cr(VI) to some extent, but the effect is still not satisfactory enough due to various problems.
In recent years, the development of carbon materials has received widespread attention. Due to abundant conjugated structures, carbon materials are confirmed to act as conductors to transfer electrons between two redox components.13 Xu et al.14 used a lactic acid/biochar system to achieve the reductive elimination of Cr(VI). Although lactic acid is a weak electron donor, biochar can not only effectively enhance the direct electron transfer of lactic acid to Cr(VI) through conjugated structures and surface functional groups, but also act as a certain electron donor, resulting in the efficient Cr(VI) elimination. Kong et al.15 successfully activated peracetic acid to remove micropollutants using the similar electron shuttle effect of reduced graphene oxide. Therefore, it is feasible to use carbon materials to mediate the reduction of Cr(VI) by Ox. Expanded graphite (EG), an easily-obtained functional carbon material, has received attention in water pollution treatment due to its unique loose and porous structure, low density, and good stability.16,17 The study of EG mainly focuses on adsorption, though the Cr(VI) adsorption capacity of EG is only about 0.28 mg g−1.18 Similar to other graphitic materials, EG has abundant interlayer electrons,19 which makes it a potential electron donor for Cr(VI) reduction. Besides, under the effect of conjugated structures, EG is likely to act as a shuttle to enhance the electron transfer from Ox to Cr(VI) under UV,20 forming an effective synergistic Cr(VI) removal effect. Differently, because of the special physical structure, EG can float on water and receive more light,21 which may be more favorable for Cr(VI) photoreduction However, reports on the Cr(VI) photochemical reduction by EG synergized with Ox have not yet appeared.
In this study, EG was chosen to cooperate with Ox in the photoreduction of Cr(VI) in an aqueous solution with UV exposure. On the elimination of Cr(VI) by the EG + Ox + UV system, the impacts of the initial concentration of Ox and Cr(VI), EG dosage, co-existing ions, and the reusability of EG were explored. The reduction product of Cr(VI) was also validated by UV-vis spectroscopy. By studying the impact of the solution's starting pH, observing the trend variation (pH and Ox concentration) of the solution during the reaction, and performing radical scavenging experiments, the mechanism of Cr(VI) elimination by the EG + Ox + UV system was also discussed. Investigations using scanning electron microscopy (SEM), X-ray diffractometry (XRD), Fourier transform infrared (FT-IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), Mott–Schottky, and electrochemical impedance spectroscopy (EIS) curves further confirmed the unique role of EG in Cr(VI) elimination via the EG + Ox + UV system.
2. Materials and methods
2.1. Chemicals and materials
Expandable graphite (50 mesh) was obtained from Qingdao Kingzhilai Graphite Co., Ltd. Potassium dichromate (K2Cr2O7), sodium hydroxide (NaOH), hydrochloric acid (HCl), oxalic acid (H2C2O4), diphenyl carbazide (C13H14N4O), sulfuric acid (H2SO4), phosphoric acid (H3PO4), acetone (C3H6O), potassium nitrate (KNO3), sodium chloride (NaCl), sodium carbonate (Na2CO3), sodium phosphate monobasic (NaH2PO4), isopropanol (IPA), carbon tetrachloride (CCl4), titanium trichloride (TiCl3) (Sinopharm Chemical Reagent Co., Ltd.) and naphthoquinone (BQ) (Shanghai Linen Technology Development Co., Ltd.) were analytical purity.2.2. Preparation of EG
EG was obtained from expandable graphite by microwave treatment: 5 g of 50 mesh expandable graphite was placed within the ceramic bowl and placed into the microwave oven (WP800TL23-K5, Galanz). After heating for 30 s at a working power of 800 W, the worm-like fluffy particles were obtained and sealed for use.2.3. Characterizations
The specific morphologies of materials were examined by a Hitachi SU-1510 SEM. XRD (Rigaku/max-2550V, Hitachi) with Cu Kα radiation (40 kV, 40 MA) was used to characterize the crystal structure of materials. FT-IR spectra were recorded in a region from 500–4000 cm−1 by an FT-IR spectrometer (Nicolet-380, Thermo Fisher) to determine the functional groups in materials. XPS (K-Alpha, Thermo Fisher) was obtained under a monochromatic Al Kα X-ray source (1486.60 eV). A UV-Vis spectrophotometer (8453, Agilent) and a visible spectrophotometer (722, Tairen) were used to monitor the reaction process. A pH meter (PHS-25, Yueping) was used to determine the solution's pH. An electrochemical workstation was used to measure the Mott–Schottky and EIS curves of materials (CHI660E, CH Instruments).2.4. Experiments of Cr(VI) removal
In a typical experiment, Cr(VI) (1.0 mM) and Ox (5.0 mM) were added to a 40 ml solution containing 20 mg of EG to disperse, and the pH of the solution was adjusted to 3 using 1 M NaOH or HCl. The mixture was placed into a photochemical reactor for the reaction after being magnetically stirred in the dark for 30 min (a 250 W high-pressure mercury lamp was applied as the UV light source). Following the extraction of 1 ml of the reaction solution at various time intervals for measurements, the Cr(VI) concentration was measured using diphenylcarbazide spectrophotometry at 540 nm. The efficiency of Cr(VI) removal was determined as eqn (1).| E% = 100% × (C0 − Ct)/C0 | (1) |
In different univariate experiments, the initial Ox concentration levels were set to 0.5, 1.0, 3.0, and 5.0 mM, the initial EG dosage level were 0.25, 0.5, 0.75, and 1 g L−1 with the initial Cr(VI) concentration level set to 0.5, 1.0, 1.5, and 2.0 mM. Cl−, CO32−, NO3−, and H2PO4− at a concentration of 1 mM were chosen as co-existing ions. All the experiments were examined at room temperature.
The Ox concentration during the reaction was measured by a TiCl3 color development method.22
To further understand the process of Cr(VI) elimination within the EG + Ox + UV system, the pseudo-zero-order, pseudo-first-order, and pseudo-second-order kinetic models were used to model the experimental results.
To verify the reusability of EG in the system, the used EG was obtained by filtration, washing thoroughly with 1 M HCl, and drying for recycling experiments and characterization.
3. Results and discussion
3.1. Removal of Cr(VI) by the EG + Ox + UV system
Fig. 1(a) shows the elimination of Cr(VI) in various systems. Cr(VI) concentration was little changed after 60 min with Ox, showing that it was difficult to reduce Cr(VI). 2.56% of Cr(VI) was removed when EG was used, indicating the slow and weak adsorption of EG for Cr(VI). When EG and Ox were used together, 46.68% of Cr(VI) was removed. This showed that the EG + Ox system enhanced the removal of Cr(VI) to a certain extent, which may be caused by a specific electron transfer between EG, Ox, and Cr(VI). However, the effectiveness of eliminating Cr(VI) still needed to be enhanced.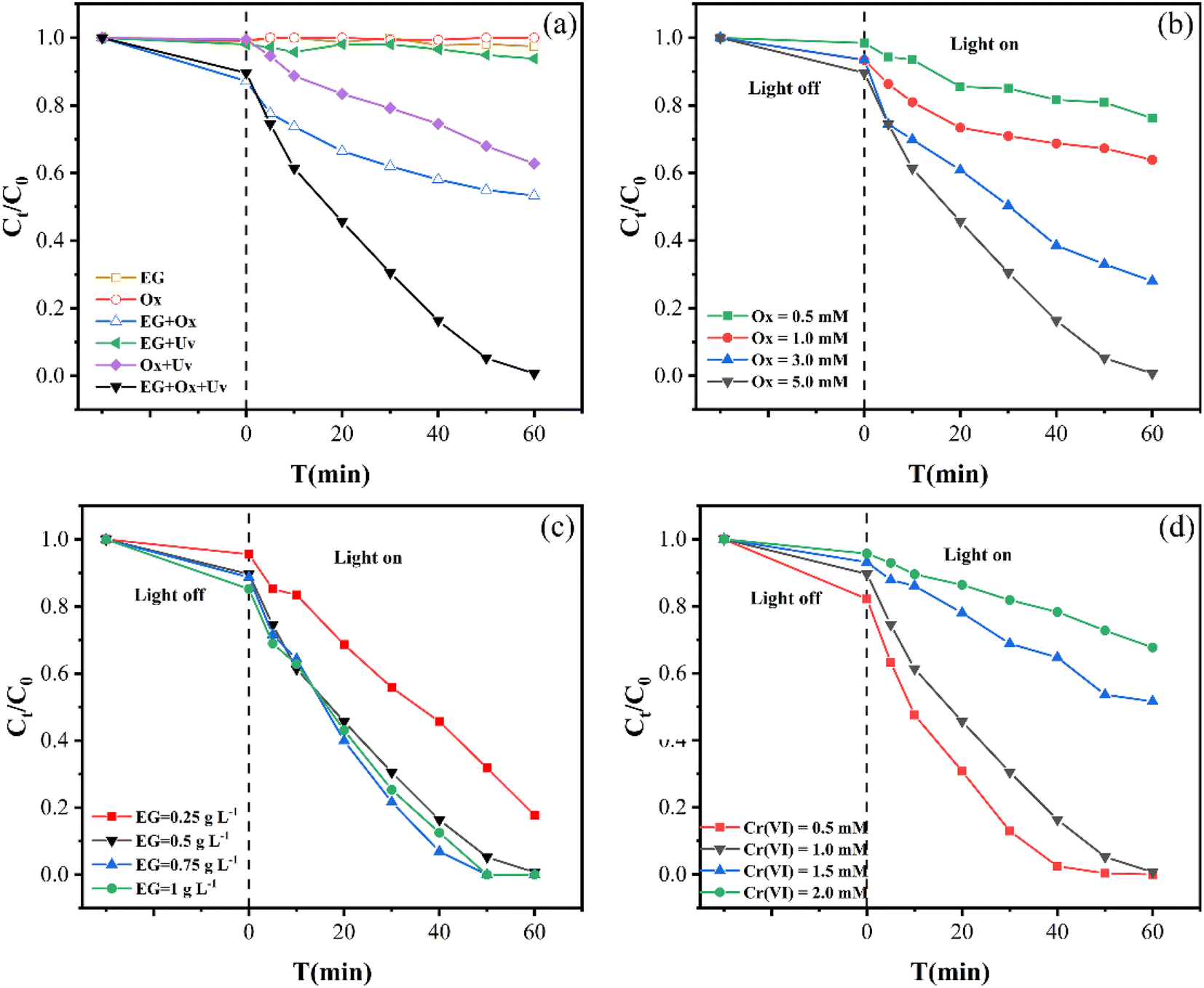 | ||
| Fig. 1 (a) The elimination of Cr(VI) in various systems, (b) the impact of the initial Ox concentration in EG + Ox + UV system, (c) the impact of the EG dosage in EG + Ox + UV system, (d) the impact of the initial Cr(VI) concentration in EG + Ox + UV system. | ||
Under UV irradiation, 37.19% of Cr(VI) was removed by Ox, which was mainly attributed to the Cr(VI) photoreduction through ·CO2− generated from the photolysis of Ox.23 The EG + UV system showed a small improvement in Cr(VI) removal (6.19%) compared with EG, indicating the weak light response of EG. Compared to other systems, the EG + Ox + UV system eliminated 99.32% of the solution's Cr(VI) content. This suggested that the addition of EG could efficiently and synergistically improve Ox's ability to remove Cr(VI). Compared with the Cr(VI) elimination performance of the reported graphite-based material and Ox-involved system (Table S1†), the EG + Ox + UV system exhibited an excellent comprehensive elimination efficiency.
A series of thorough analyses of the initial concentration of Ox and Cr(VI), EG dose, co-existing ions, and the reusability of EG in the system were carried out with the goal of eliminating Cr(VI) in the EG + Ox + UV system. The reduction products of Cr(VI) were also confirmed.
The process of removing Cr(VI) from the EG + Ox + UV system was evaluated for the reaction kinetics by fitting the pseudo-zero-order, pseudo-first-order, and pseudo-second-order rate equations. The obtained rate constant (k) and correlation coefficient (R2) are displayed in Table 1.
| Dosage of EG | Pseudo-zero-order | Pseudo-first-order | Pseudo-second-order | |||
|---|---|---|---|---|---|---|
| k (mol L−1 min−1) | R2 | k (min−1) | R2 | k (L mol−1 min−1) | R2 | |
| 0.25 g L−1 | 6.78 × 10−1 | 0.998 | 2.53 × 10−2 | 0.944 | 1.14 × 10−3 | 0.79 |
| 0.5 g L−1 | 7.95 × 10−1 | 0.965 | 6.98 × 10−2 | 0.883 | 2.88 × 10−2 | 0.471 |
| 0.75 g L−1 | 8.54 × 10−1 | 0.941 | 5.97 × 10−2 | 0.948 | 5.09 × 10−3 | 0.745 |
| 1 g L−1 | 7.96 × 10−1 | 0.964 | 4.61 × 10−2 | 0.976 | 2.85 × 10−3 | 0.850 |
The EG + Ox + UV system's Cr(VI) elimination was most consistent with the pseudo-zero-order kinetic model, according to the highest R2 value. From Table 1 and the pseudo-zero-order kinetic fitting curves in Fig. 2, the rate constant k increased from 6.78 mol L−1 min−1 to 7.95 mol L−1 min−1 to 8.54 mol L−1 min−1 as the EG dosage was raised from 0.25 g L−1 to 0.5 g L−1 to 0.75 g L−1, respectively. However, the rate constant was reduced to 7.96 mol L−1 min−1 when the EG dosage was raised to 1 g L−1, which was consistent with the experimental findings. This suggested that a specific EG dosage was crucial for the EG + Ox + UV system to remove Cr(VI). A moderate increase in the EG dose would aid in the elimination of Cr(VI) by increasing the reaction site, whereas an excessive amount of EG would decrease the removal of Cr(VI) by occluding the irradiation of part UV light.
 | ||
| Fig. 2 The pseudo-zero-order kinetic fitting curves for Cr(VI) elimination with different EG dosage. | ||
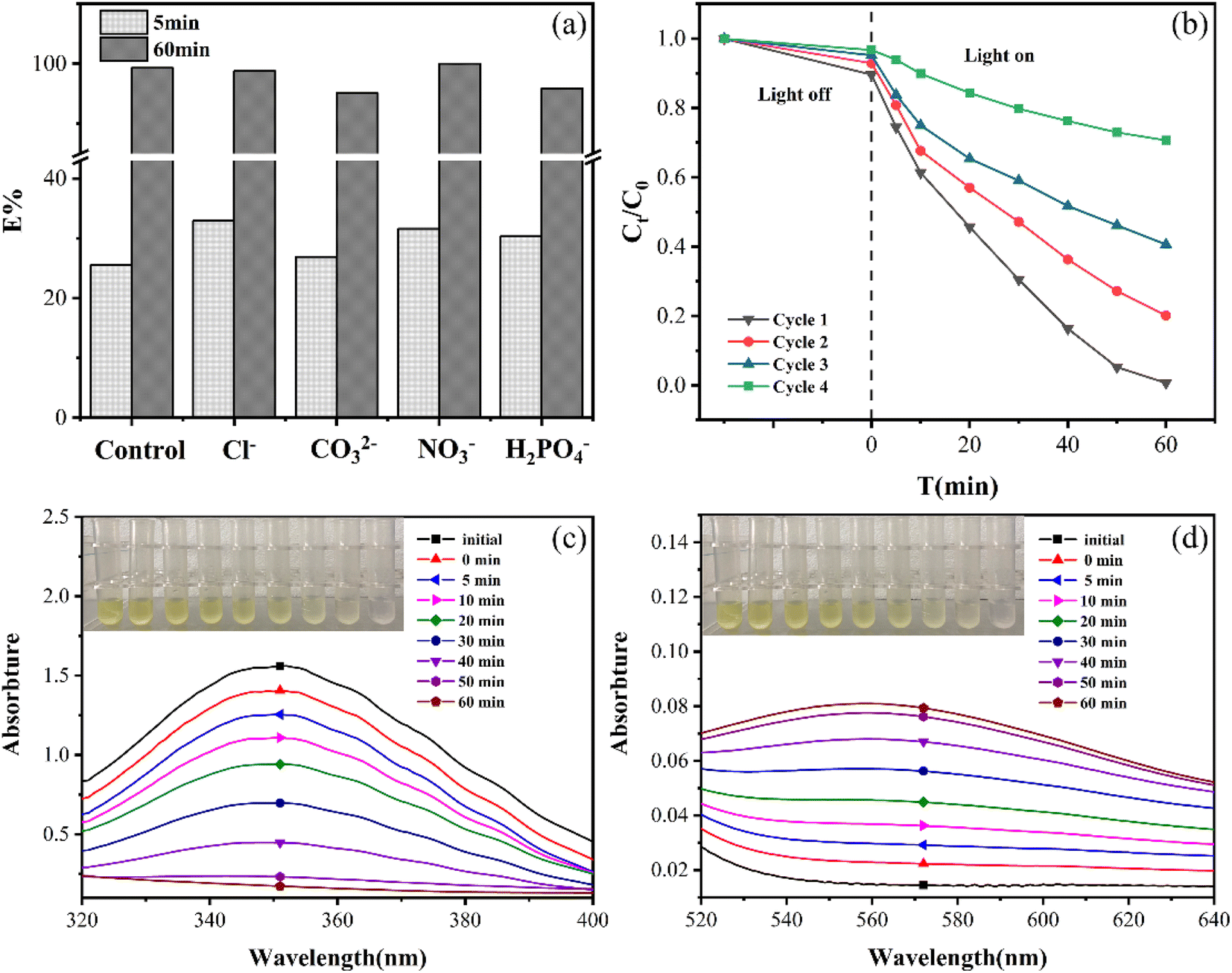 | ||
| Fig. 3 (a) The impact of co-existing ions in EG + Ox + UV system, (b) the reusability of EG in EG + Ox + UV system, the UV-Vis spectra of the solution at (c) 350 nm and (d) 570 nm during the reaction. | ||
Additionally, how coexisting ions affected the EG + Ox + UV system at 5 min was different from that at 60 min: at 5 min, the Cr(VI) removal was boosted by the inclusion of the four co-existing ions. These might be explained by the short-term increase in conductivity of the solution brought about by the addition of co-existing ions, which favoured the elimination of Cr(VI). The little rise in Cr(VI) elimination with CO32− addition over the other three ions might be due to the substantial competitiveness of CO32−, for H+ consumption in the solution.
3.2. Mechanism of Cr(VI) reduction in EG + Ox + UV system
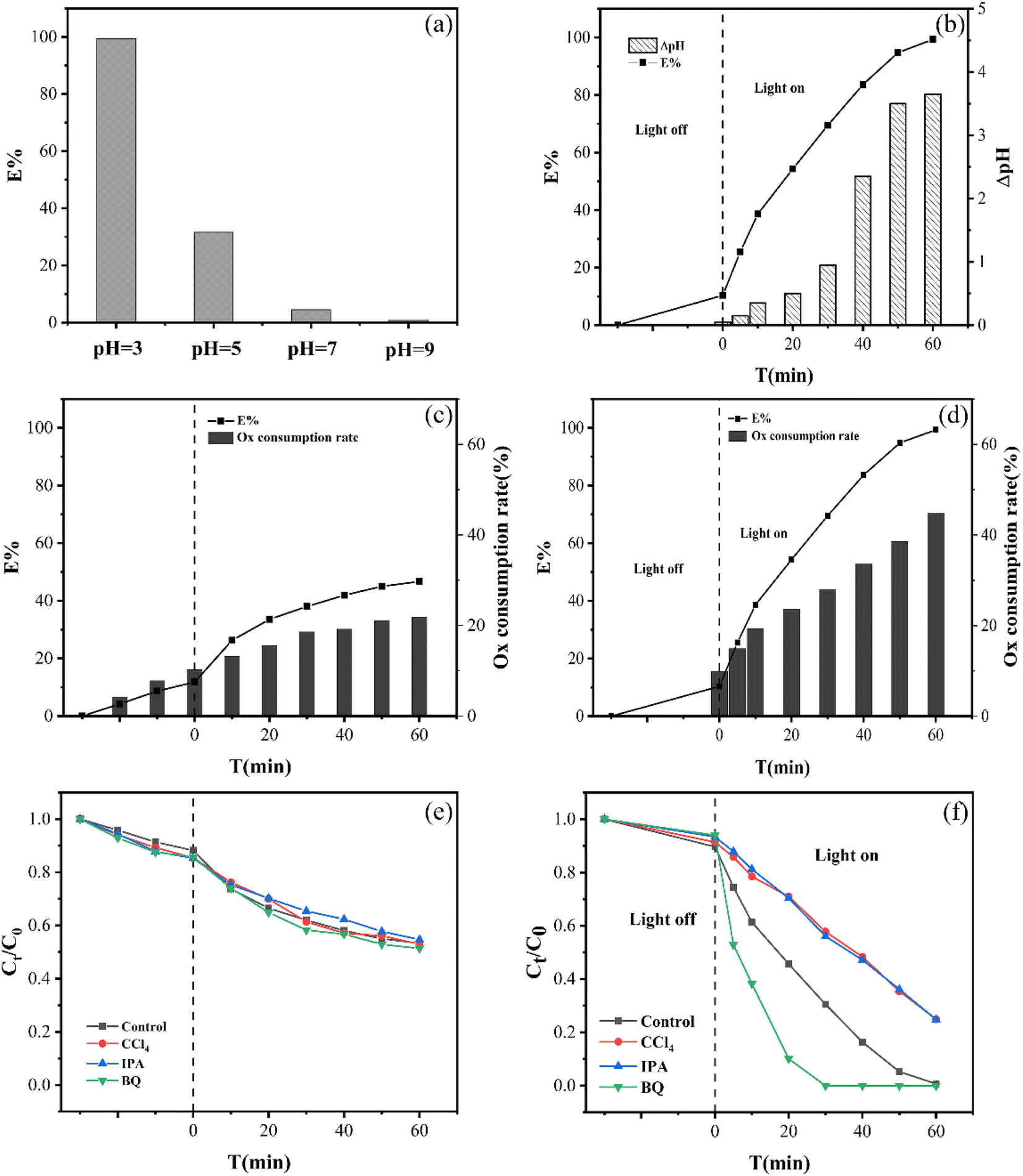 | ||
| Fig. 4 (a) The impact of the starting pH in the EG + Ox + UV system, (b) pH trend variation in solution of the EG + Ox + UV system, Ox concentration trend variation of (c) the EG + Ox system and (d) the EG + Ox + UV system at pH = 3, effects of free radical scavenging in (e) the EG + Ox system and (f) the EG + Ox + UV system. | ||
On the one hand, the major existence of Cr(VI) and Ox would vary with the starting pH of the solution, which had different reactivity in the redox reaction. Cr(VI) species were present as HCrO4− at pH < 6 or CrO42− at pH > 6, respectively. Due to the higher electrode potentials, it was simpler to convert HCrO4− to Cr(III) than CrO42− (E0(HCrO4−/Cr3+) = 1.35 VNHE, E0(CrO42−/Cr3+) = 0.56 VNHE).27 At the same time, the predominated formation of Ox at pH < 4.2 was HC2O4− and C2O42− at pH > 4.2. It could be seen that HC2O4− was more reactive than C2O42− because the power required to rupture the C–C bond in HC2O4− (99.48 kJ mol−1) was substantially lower than that in C2O42− (186.40 kJ mol−1).27 Because of this, the HC2O4− in the system was simpler to reduce HCrO4− at pH = 3 to reach the highest Cr(VI) removal efficiency.
Moreover, HC2O4− (the main existence of Ox at pH = 3) was easily underwent photolysis to ·CO2− for Cr(VI) photoreduction removal (eqn (2) and (3)).28
| HC2O4− + O2 + hν → H+ + ·C2O4− + ·O2− | (2) |
| ·C2O4− → ·CO2− + CO2 | (3) |
Additionally, at pH = 3, the oxygen-containing functional groups of EG would be protonated. Then, by electrostatic attraction, part of Cr(VI) and Ox were easily attracted to the surface of EG. After then, a highly active structure among EG, Ox, and Cr(VI) probably formed, which was favorable for electron transport and eventually contributed to lowering Cr(VI).
| Cr(VI) + ·CO2− + H+ → Cr(III) + CO2 + H2O | (4) |
| Cr(VI)–Ox + hν → Cr(III)–Ox + ·C2O4− | (5) |
According to Fig. 4(f), the EG + Ox + UV system's ability to remove Cr(VI) decreased with the addition of CCl4 from 99.32% to 75.06%, demonstrating the presence of ·CO2− active species in the Cr(VI) removal process. ·CO2− played an active species role in Cr(VI) elimination by the system, which was beneficial to Cr(VI) reduction. When ·OH was caught by IPA, the elimination of Cr(VI) decreased to 75.20%, demonstrating the presence of ·OH in the system. ·OH, as the products of Ox photolysis (eqn (6)),29 could oxidize Ox to ·CO2− and thus improve the system's ability to eliminate Cr(VI) (eqn (7)).30 The capture of ·OH would limit the generation of ·CO2−, which prevented the EG + Ox + UV system from photo-reducing Cr(VI). The Cr(VI) elimination in the system rose greatly (100% at 50 min) when BQ was added to the system to capture ·O2−, demonstrating that ·O2− impeded the Cr(VI) elimination. The reaction between the oxygen in the air and the ·CO2− in the EG + Ox + UV system resulted in the generation of ·O2− (eqn (8)).31 A part of ·CO2− would be consumed in the production of ·O2−, and ·O2− as oxidative radicals would also expand reducing species within the system. When ·O2− was captured, the amount of reducing species would increase, then the efficiency of Cr(VI) reduction would be improved.
| Ox + hν → ·OH + ·OCCOOH | (6) |
| Ox + ·OH → ·CO2− + H2O | (7) |
| ·CO2− + O2 + H+ → CO2 + HO2/·CO2− | (8) |
While in Fig. 4(e), the scavenging of radicals had little impact with regard to the EG + Ox system's elimination of Cr(VI), demonstrating that the system was not affected by free radical action. The EG + Ox system's Cr(VI) elimination might have been largely caused by the spontaneous DET pathway in the formed EG–Ox–Cr(VI).
3.3. The function of EG in EG + Ox + UV system's elimination of Cr(VI)
Data using SEM, XRD, FT-IR, XPS, Mott–Schottky as well as EIS curves of EG and EG after reaction were collected to further explore the function of EG in the EG + Ox + UV system's elimination of Cr(VI).Surface morphologies of EG and EG after the reaction were analyzed by SEM. At low magnification, EG after the reaction in Fig. 5(c) maintained the same worm-like structure as well as the EG in Fig. 5(a). At higher magnification, the surface appearance of EG after the reaction in Fig. 5(d) was different from that of EG in Fig. 5(b). EG appeared as a flatter layered-graphite structure, while EG after the reaction presented the stripped graphite layer with the bumpy and irregular morphology structure. This might be due to the graphite layers of EG being changed after participating in the process of Cr(VI) elimination.
 | ||
| Fig. 5 SEM images of EG (a, b) and EG after reaction (c, d). | ||
XRD was also analyzed on EG and EG after the reaction in Fig. 6. Both EG and EG after the reaction showed a characteristic diffraction peak of graphite crystal face (002) at the position of 2θ = 26.48°, corresponding to the formation of curling, overlapping, or stacking of the multilayer graphene structure in EG.32 While the intensity of the diffraction peak of EG after the reaction decreased compared with EG, which indicated the disorder in the crystal structure inside EG after the reaction.19 This might be due to the exfoliation of the internal flake graphene structure during the reaction process.
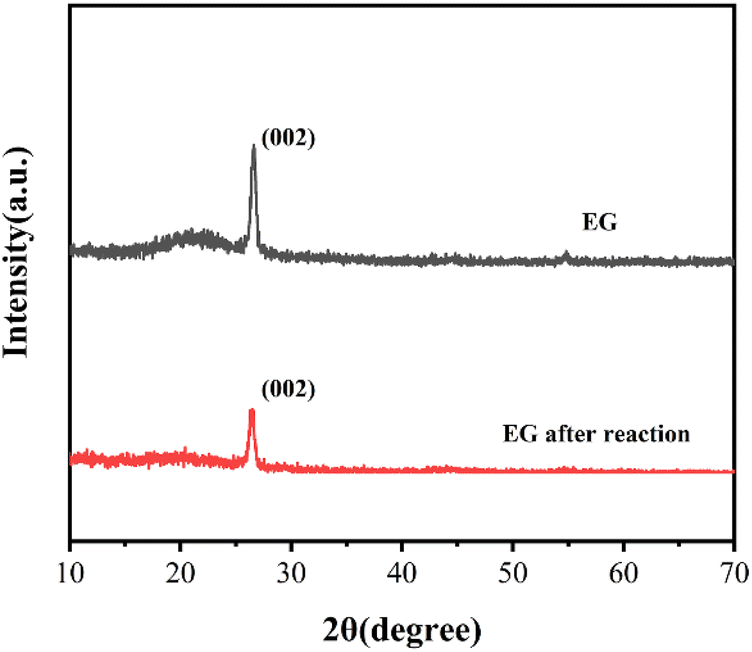 | ||
| Fig. 6 XRD patterns of EG and EG after the reaction. | ||
Fig. 7 depicts the FT-IR patterns of EG and EG after the reaction. Compared to EG, the intensity of the peak ascribed to the C–C stretch at 1634 cm−1 decreased33 and that of the peak belonging to C–O at 1063 cm−1 increased in the EG after the reaction, which could be explained by the participation of C–C of EG and the formed C–O in Cr(VI) reduction. A new peak at 583 cm−1 appeared in EG after the reaction was credited to the Cr–O stretching vibration,34 which might be due to the creation of EG–Ox–Cr(VI).
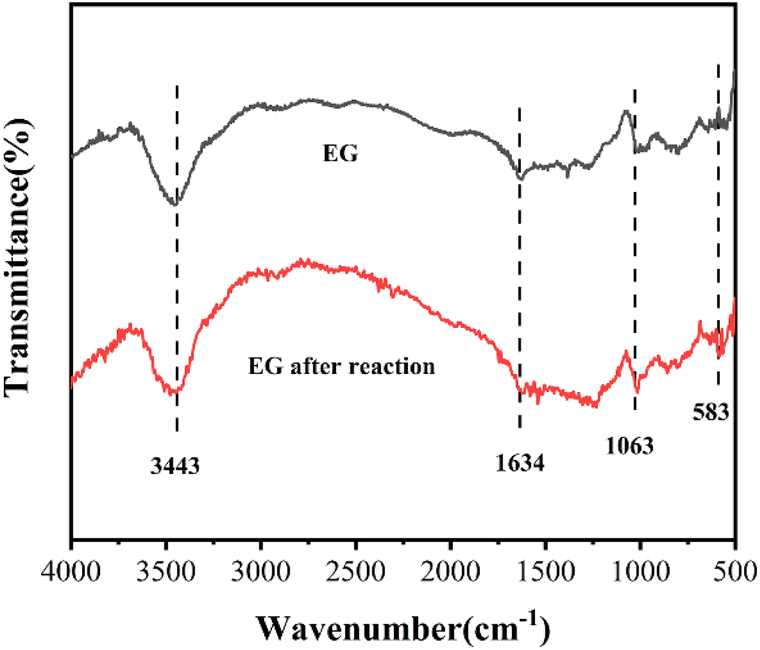 | ||
| Fig. 7 FT-IR patterns of EG and EG after reaction. | ||
XPS survey spectra of EG and EG after the reaction are displayed in Fig. 8(a). The C 1s (binding energy = 284.80 eV) and O 1s (binding energy = 531.08 eV) peaks were dominant in the XPS spectrum of EG and EG after the reaction.35 Compared with EG, C 1s in EG after the reaction increased from 81.7% to 91.1%, and O 1s decreased from 18.6% to 9.1%, which might be mainly caused by the consumption of EG.
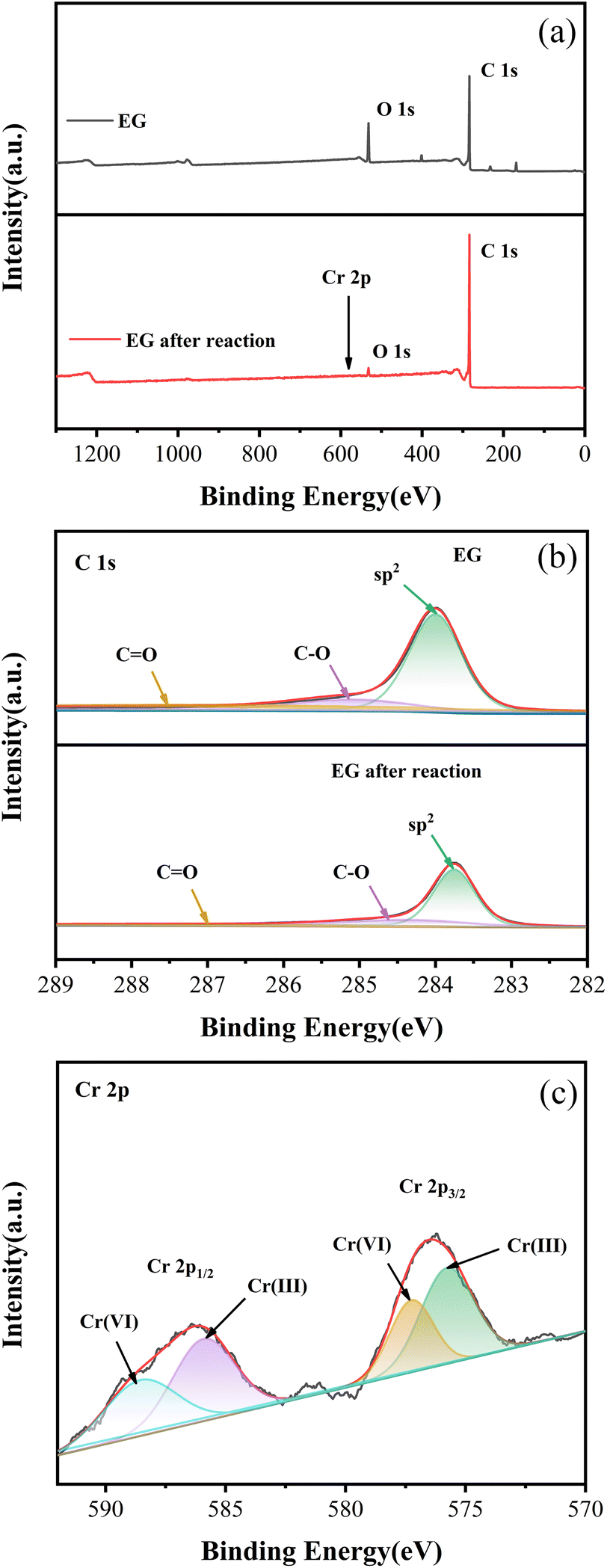 | ||
| Fig. 8 XPS spectra of EG and EG after reaction: (a) survey, (b) C 1s in EG and EG after reaction, (c) Cr 2p in EG after the reaction. | ||
In the C 1s spectra of EG and EG after the reaction (Fig. 8(b)), the peak centered at 284.0 eV could be assigned to a C–C bond with an sp2 orbital,36 and peaks located at 285.1 eV and 287.4 eV could be indexed to the characteristics of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.37 Compared to EG, C–C with sp2 orbital in EG after reaction decreased from 69.9% to 57.5%, while C–O and CO increased from 12.7% and 17.4% to 21.2% and 21.3%, respectively. This might indicate that C–C with sp2 orbital was the main group of EG involved in the reaction, which was oxidized to C–O and CO after donating electrons,38 which was consistent with the conclusion obtained in the FT-IR spectrum.
O.37 Compared to EG, C–C with sp2 orbital in EG after reaction decreased from 69.9% to 57.5%, while C–O and CO increased from 12.7% and 17.4% to 21.2% and 21.3%, respectively. This might indicate that C–C with sp2 orbital was the main group of EG involved in the reaction, which was oxidized to C–O and CO after donating electrons,38 which was consistent with the conclusion obtained in the FT-IR spectrum.
Additionally, the survey spectrum of EG after the reaction also showed a minor peak of Cr 2p. In the Cr 2p pattern of EG after the reaction in Fig. 8(c), four peaks at 577.26 eV, 588.56 eV, and 575.77 eV, 585.92 eV could be fitted,39 indicating strong evidence for the existence of Cr(VI) and Cr(III). Among them, the contents of Cr(VI) and Cr(III) were 40.6% and 59.4%, respectively. The presence of Cr(VI) and Cr(III) species on EG after the reaction might be due to the residue of the formed EG–Ox–Cr(VI) during the EG + Ox + UV system's elimination of Cr(VI).
Mott–Schottky and EIS curves were collected to confirm that EG possessed the ability to give electrons for Cr(VI) reduction. Fig. 9(a) indicated that EG's flat band potential was around −0.90 eV (vs. Ag/AgCl, pH = 7), which was comparable to −0.67 V (vs. NHE, pH = 7).40 So the ECB of EG was estimated to be around −0.67 V (vs. NHE, pH = 7). A negative value of ECB indicated that EG possessed the potential ability to give its electrons. In the EIS plot, as shown in Fig. 9(b), EG after the reaction had a larger arc radius than EG, implying the higher interfacial charge transfer resistance of EG after the reaction than EG, which resulted in the decrease for EG + Ox + UV system's removal of Cr(VI) after the reuse of EG.
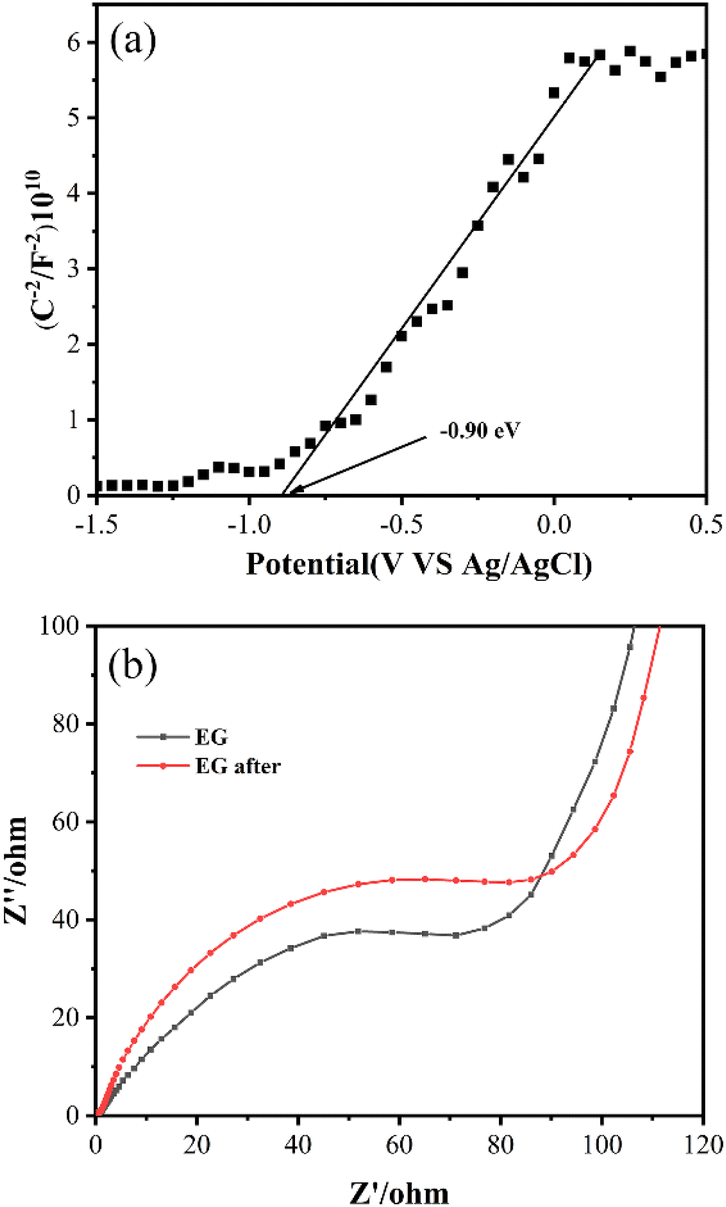 | ||
| Fig. 9 (a) Mott–Schottky curve of EG, (b) EIS curves of EG and EG after the reaction. | ||
According to the aforementioned investigation, the EG + Ox + UV system's Cr(VI) reduction possible mechanism was mostly made up of three components (Fig. 10): (1) with EG's acceleration effect of the UV exposure, the formed EG–Ox–Cr(VI) with highly active electrons among EG, Ox, and Cr(VI) could effectively achieve the reduction of Cr(VI) within the system through the spontaneous DET pathway; (2) Ox could generate ·CO2− when exposed to UV to reduce Cr(VI); (3) Cr(VI)-Ox complexes could likewise produce ·CO2− with UV to eliminate a small portion Cr(VI) by the PET pathway.
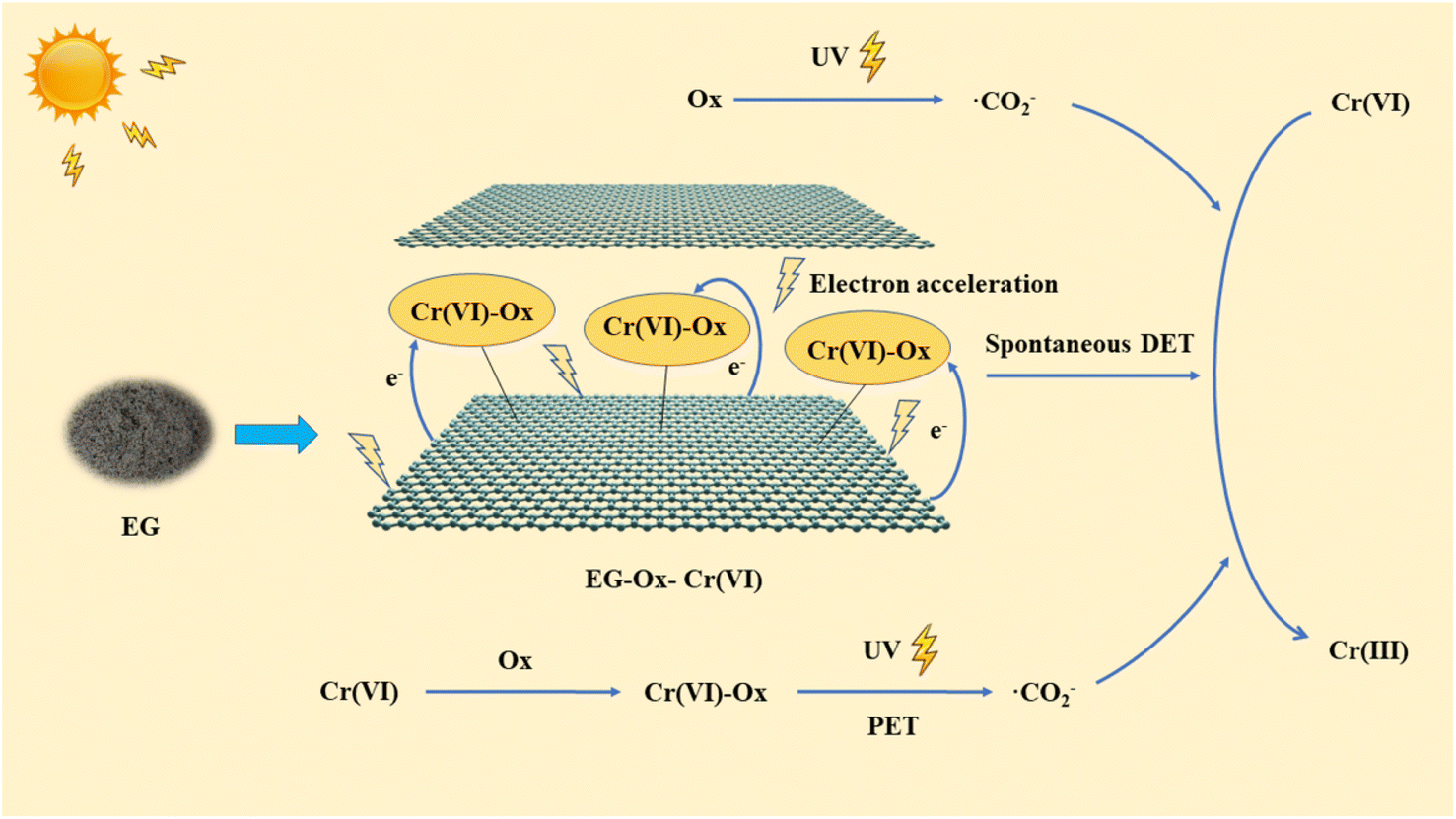 | ||
| Fig. 10 The possible mechanism for the Cr(VI) elimination in the EG + Ox + UV system. | ||
4. Conclusions
In this study, we report that the easily-obtained EG with the potential of transferring electrons successfully improved the Cr(VI) photoreduction elimination by environmentally-acceptable Ox. The moderate dosage of EG, the lower pH, and the greater Ox concentration in the EG + Ox + UV system contributed to the removal of Cr(VI). The mechanism of the EG + Ox + UV system's elimination of Cr(VI) was mainly attributed to the synergetic effects of self-reactive reduction (the spontaneous DET pathway in Cr(VI)–Ox–EG with acceleration effect of EG) and photochemical reduction (·CO2− produced by photolysis of Ox and the PET pathway in Cr(VI)–Ox complexes). The results of our research provided new ideas for future research on carbon materials in the field of Cr(VI) treatment.Author contributions
Ling Zhang: conceptualization, resources, writing – review and editing, supervision. Yanqing Sun: investigation, data curation, writing – original draft, writing – review and editing, formal analysis. Jie Sun: investigation, data curation. Fengming Cao: investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.Notes and references
- Y. Zhang, K. Yan, F. Ji and L. Zhang, Adv. Funct. Mater., 2018, 28, 1806340 CrossRef
.
- P. Johnson, C. Loganathan, V. Krishnan, P. Sakayanathan, V. Raji, S. Vijayan, P. Sathishkumar, K. Murugesan and T. Palvannan, Environ. Technol., 2018, 39, 1376–1383 CrossRef CAS PubMed
- L. Du, P. Gao, Y. Liu, T. Minami and C. Yu, Nanomaterials, 2020, 10, 686 CrossRef CAS PubMed
- F. Cao, Y. Sun, L. Zhang and J. Sun, Appl. Surf. Sci., 2022, 575, 151583 CrossRef CAS
- Y. Liu, S. Xin and B. Jiang, Environ. Technol., 2020, 41, 430–439 CrossRef CAS PubMed
- G. Sun, W. Yang and F. Wang, Int. J. Environ. Res. Public Health, 2019, 16, 2771 CrossRef PubMed
- B. Deng and A. T. Stone, Environ. Sci. Technol., 1996, 30, 2484–2494 CrossRef CAS
- S. Xia, L. Zhang, G. Pan, P. Qian and Z. Ni, Phys. Chem. Chem. Phys., 2015, 17, 5345–5351 RSC
- J. Sun, J. D. Mao, H. Gong and Y. Lan, J. Hazard. Mater., 2009, 168, 1569–1574 CrossRef CAS PubMed
- W. Niu, J. Sun, L. Zhang and F. Cao, Chemosphere, 2021, 262, 127806 CrossRef CAS PubMed
- S. M. Lee, I. H. Cho, Y. Y. Chang and J. K. Yang, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 543–548 CrossRef CAS PubMed
- Y. Li, C. Chen, Y. Wu, Y. Han and Y. Lan, Water, Air, Soil Pollut., 2017, 228, 363 CrossRef
- C. Gu, M. Cai, P. He, J. Zhu and M. Gan, Chemosphere, 2023, 313, 137557 CrossRef CAS PubMed
- X. Xu, H. Huang, Y. Zhang, Z. Xu and X. Cao, Environ. Pollut., 2019, 244, 423–430 CrossRef CAS PubMed
- D. Kong, Y. Zhao, X. Fan, X. Wang, J. Li, X. Wang, J. Nan and J. Ma, Environ. Sci. Technol., 2022, 56, 11707–11717 CrossRef CAS PubMed
- X. Wu, S. Qi, J. He and G. Duan, J. Mater. Sci., 2010, 45, 483–489 CrossRef CAS
- L. Zhang, Y. Wang, S. Jin, Q. Lu and J. Ji, Environ. Technol., 2017, 38, 2629–2638 CrossRef CAS PubMed
- H. Jin, J. Yuan, H. Hao, Z. Ji, M. Liu and S. Hou, Mater. Lett., 2013, 110, 69–72 CrossRef CAS
- R. Lan, W. Su and J. Li, Catalysts, 2019, 9, 280 CrossRef
- W. Ren, C. Cheng, P. Shao, X. Luo, H. Zhang, S. Wang and X. Duan, Environ. Sci. Technol., 2022, 56, 78–97 CrossRef CAS PubMed
- J. Zhang, X. Wang, P. Xia, X. Wang, J. Huang, J. Chen, B. Louangsouphom and J. Zhao, Res. Chem. Intermed., 2016, 42, 5541–5557 CrossRef CAS
- J.-x. Wang, J.-k. Huang and T.-t. Yan, J. Integr. Agric., 2013, 12, 1267–1278 CrossRef
- L. Zhang, F. Cao, J. Sun and Y. Sun, Environ. Res., 2021, 197, 111070 CrossRef CAS PubMed
- A. Kumar, A. Rana, G. Sharma, M. Naushad, A. H. Al-Muhtaseb, C. Guo, A. Iglesias-Juez and F. J. Stadler, ACS Appl. Mater. Interfaces, 2018, 10, 40474–40490 CrossRef CAS PubMed
- N. Chen and B. Pan, Chem. Eng. J., 2020, 387, 124079 CrossRef CAS
- P. Mytych, P. Cieśla and Z. Stasicka, Appl. Catal., B, 2005, 59, 161–170 CrossRef CAS
- Y. Mu, X. Jiang, Z. Ai, F. Jia and L. Zhang, J. Hazard. Mater., 2018, 343, 356–363 CrossRef CAS PubMed
- D. Jiang, Y. Li, Y. Wu, P. Zhou, Y. Lan and L. Zhou, Chemosphere, 2012, 89, 832–837 CrossRef CAS PubMed
- G. E. Adams and E. J. Hart, J. Am. Chem. Soc., 1962, 84, 3994–3999 CrossRef CAS
- B. An, H. He, B. Duan, J. Deng and Y. Liu, Chemosphere, 2021, 278, 130388 CrossRef CAS PubMed
- K. H. Ng, Y.-T. Liu, C. T. Chang, P.-N. Chiang, H. Y. Teah, P. H. Chang and Y.-M. Tzou, J. Cleaner Prod., 2021, 313, 127726 CrossRef CAS
- M. Qiang, H. Xiaomin, L. Ke, D. Rui, H. Zhang, X. Bo and Z. Kewen, Sep. Purif. Technol., 2021, 259, 118131 CrossRef CAS
- W. C. Hung, K. H. Wu, D. Y. Lyu, K. F. Cheng and W. C. Huang, Mater. Sci. Eng., C, 2017, 75, 1019–1025 CrossRef CAS PubMed
- X. Huang, Q. Hu, L. Gao, Q. Hao, P. Wang and D. Qin, RSC Adv., 2018, 8, 27623–27630 RSC
- R. Liu, H. Li, L. Duan, H. Shen, Q. Zhang and X. Zhao, Appl. Surf. Sci., 2018, 462, 263–269 CrossRef CAS
- Y. Du, Z. Tao, J. Guan, Z. Sun, W. Zeng, P. Wen, K. Ni, J. Ye, S. Yang, P. Du and Y. Zhu, RSC Adv., 2015, 5, 81438–81444 RSC
- M. Yu, J. Shang and Y. Kuang, J. Mater. Sci. Technol., 2021, 91, 17–27 CrossRef CAS
- Z. Guo, M. Cheng, W. Ren, Z. Wang and M. Zhang, J. Hazard. Mater., 2022, 430, 128416 CrossRef CAS PubMed
- H. R. Yang, S. S. Li, C. Yang, Q. D. An, S. R. Zhai and Z. Y. Xiao, J. Colloid Interface Sci., 2022, 607, 556–567 CrossRef CAS PubMed
- J. Zheng and Z. Lei, Appl. Catal., B, 2018, 237, 1–8 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01207g |
| This journal is © The Royal Society of Chemistry 2023 |