
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilization of compressible hydrogels as electrolyte materials for supercapacitor applications†
Amrita Jain‡
a,
Yasamin Ziai‡a,
Kamil Bocheneka,
Sai Rashmi Manippadya,
Filippo Pierini a and
Monika Michalska*b
a and
Monika Michalska*b
aInstitute of Fundamental Technological Research, Polish Academy of Sciences, Pawińskiego 5B, 02-106 Warsaw, Poland
bDepartment of Chemistry and Physico-Chemical Processes, Faculty of Materials Science and Technology, VŠB-Technical University of Ostrava, 17. Listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic. E-mail: monika.kinga.michalska@gmail.com
First published on 12th April 2023
Abstract
Utilization of CoO@Co3O4-x-Ag (x denotes 1, 3, and 5 wt% of Ag) nanocomposites as supercapacitor electrodes is the main aim of this study. A new low-temperature wet chemical approach is proposed to modify the commercial cobalt oxide material with silver nanoparticle (NP) balls of size 1–5 nm. The structure and morphology of the as-prepared nanocomposites were investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and N2 adsorption–desorption measurements. Hydrogels known to be soft but stable structures were used here as perfect carriers for conductive nanoparticles such as carbons. Furthermore, hydrogels with a large amount of water in their network can give more flexibility to the system. Fabrication of an electrochemical cell can be achieved by combining these materials with a layer-by-layer structure. The performance characteristics of the cells were examined by electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV), and galvanostatic charge discharge (GCD). Cobalt oxide modified with 5 wt% Ag gave the best supercapacitor results, and the cell offers a specific capacitance of ∼38 mF cm−2 in two-electrode configurations.
Introduction
The rapid development of electronic gadgets such as smartwatches, smartphones, tablets, and laptops demands power sources that can have acceptable mechanical stability, reliability, safety, and environmentally friendly materials; therefore, the development of efficient energy storage (EES) systems becomes significantly essential to satisfy the above-mentioned requirements.1–4 Supercapacitors are always considered strong EES candidates due to their high power density, fast charge–discharge rate, and long cycling stability.5,6 In addition to these, flexible supercapacitors with self-healing properties attract the attention of the research community nowadays because these materials have the potential to quickly restore and effectively maintain the electrochemical performance after mechanical damage and under various compression or stretching conditions.7–10The performance of the supercapacitors strongly depends on the properties of the electrolyte. Among the three categories of electrolytes (aqueous, solid, and gel), polymeric hydrogel networks are widely used as their unique properties, such as porosity, tuneable chemical composition, ease of processing, and flexibility, can provide the desirable characteristics of a suitable electrolyte to the system.11
All these properties are attributed to the ability of the hydrogels to sustain a significant amount of water within their network.10,12 Flexible batteries are considered the old generation of supercapacitors as hydrogel electrolytes have perfect electrical performance. A significantly higher power density and volumetric capacity, alongside a longer lifetime, make them outstanding candidates for supercapacitors in different applications.13,14 Moreover, gel-like structures can overcome the limitations that come with liquid electrolytes. The technique of incorporating nanoparticles into the hydrogel matrix has recently been explored, as their network is an excellent carrier for nanoparticles.15–17 Based on the properties of the polymers and particles, hydrogels embedded with nanoparticles have been used for different applications such as biomedical,18 environmental,19 flexible electronics,20 and sensors.15 There are some drawbacks in the use of hydrogels in supercapacitors, which are mainly the low healing capability and insignificantly compressible nature.2 The conventional polymer matrixes, most commonly used to prepare polymer hydrogel electrolytes, are poly(vinyl alcohol) (PVA), poly(vinylidene fluoride) (PVdF), poly(ethylene oxide) (PEO), and poly(methyl methacrylate) (PMMA). The use of acid/alkali/salt gel electrolytes has also been studied.21–24 Therefore, there is high demand to develop and explore new materials to produce multifunctional electrolytes with self-healing properties, outstanding compressibility, and acceptable electrochemical performance that can be used in advanced flexible supercapacitors.
Depending upon the working mechanism, there are two types of supercapacitors: electrochemical double layer capacitors (EDLCs) and pseudocapacitors. EDLCs store electrical energy by building a double layer at the electrode–electrolyte interface, whereas pseudocapacitors depend on the fast faradaic redox reactions of electroactive species.4,5,25 Generally, pseudocapacitors have higher capacitance than EDLCs. There are various inorganic metal oxides or hydroxides, for example, CuO, NiO, MnO2, RuO2, and V2O5 and conducting polymers such as PEDOT, PPy, and PANI that have been tested as electrode materials.26–31 Among all, cobalt-based materials such as cobalt oxides and hydroxides are considered promising materials due to their high electrochemical activity and a theoretical capacitance of ∼4.3 F g−1.32,33 Nevertheless, the major problem in cobalt-based materials is the charge transport between the electrolyte and the electrode because the ion transportation is obstructed by the lethargic ion diffusion within the conventionally dense electrode and its insulating behaviour. To solve this problem, the surface of cobalt oxide can be modified with silver nanoparticles to increase the electrical conductivity of the material, and the prepared materials were tested as electrode materials for supercapacitor applications.32
This study reports a compressible, self-healable, multifunctional system consisting of two main elements in a layer-by-layer structure. A hydrogel layer embedded with carbon nanoparticles in between and surface-modified cobalt oxide with silver nanoparticle cloths were prepared to be used as the outer layers. A novel wet chemical approach is proposed to modify the electrode material with Ag nanoparticle (NP) balls of size 1–5 nm. In addition, this chemical technique is facile, cost-effective, and environmentally friendly. The deposition of Ag NPs was done without using any reducing agent. The overall performance of these supercapacitor cells was characterized by impedance spectroscopy, cyclic voltammetry and galvanostatic charge–discharge technique.
Experimental details
Materials used
For hydrogel preparation, acrylamide (AAm, Sigma Aldrich, Poland), N,N′-methylene bisacrylamide (BIS-AAm, 99.5%, Sigma Aldrich, Poland), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959, 98%, Sigma Aldrich, Poland), and carbon (nanopowder, Sigma Aldrich, Poland) were used and for the electrode material, cobalt(II) oxide, CoO (325 mesh) was obtained from Acros Organics, silver nitrate, AgNO3 and ethanol solution, and EtOH 96% were procured from Merck Life Science spol. s r.o. All the above-mentioned materials were used as received.Hydrogel precursor sol including carbon nanopowders
To prepare the hydrogel precursor solution at a concentration of 6 wt%, 15.625 mg BIS-AAm and 6.25 mg of Irgacure 2959 were added to 289.065 mg of AAm. This mixture was dissolved in 5 ml of deionized water. The mixture was wrapped in aluminium foil to be protected from light and stirred overnight until its complete dissolution. To optimize the final embedding of the carbon nanopowder in the solution, three different concentrations of carbon nanopowders (0.1%, 0.07%, and 0.04% wt) were added to the hydrogel precursor solution. Solutions were placed under ultrasonication for 15 minutes to assure the maximum dispersion of the carbon powders in the solution. The solution was put on the magnetic stirrer until the time of use. In this study, 0.07 wt% of carbon nanopowder was chosen as the best concentration in terms of stable solubility.Fabrication of the hydrogel layer
To obtain the hydrogel layer of the final structure, the solution was first degassed with argon for 10 min. This step is necessary due to the polymerization path, which requires evacuating all oxygen molecules prior to ultraviolet (UV) irradiation. Then, 250 μl of the solution was added to a circular mold with a diameter of 1.0 cm. The solution was put in a UV irradiation chamber to be irradiated. The polymerization was done after 90 s and after that a stable hydrogel layer was ready to be implanted into the construct.Surface modification of CoO@Co3O4 with Ag NPs
The nanocomposites of CoO@Co3O4 with 1, 3 and 5 wt% of Ag were fabricated via a facile chemical low-temperature route, in which any reducing agent was utilized. In the first step, AgNO3 (used as an Ag source) was dissolved in an EtOH solution in the weight ratio of 0.01 to 0.05 of Ag/CoO@Co3O4, respectively. This process was carried out at room temperature. In the second step, the CoO@Co3O4 powder was added to the AgNO3–EtOH–H2O solution. The mixture was stirred magnetically for a few hours to obtain a black, homogenously dispersed suspension. Subsequently, the suspension was kept in an ultrasonic water bath cleaner from 25 °C to 60 °C for 1 hour. Each suspension was air-dried overnight at 60 °C and then at 150 °C for a few hours. To obtain a fine powder, CoO@Co3O4/x-Ag (x denotes 1, 3 and 5 wt% of Ag) nanocomposites were ground in an agate mortar. That proposed chemical approach was easy to realize, cost-effective and environmentally friendly. Additionally, the CoO@Co3O4 powders were surface modified with silver nanoparticle balls of size 1–5 nm, as presented in detail in the Results and discussion section.Instrumental details
The morphologies of the hydrogel and CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag) were examined using a scanning electron microscope (SEM/FIB-Zeiss Crossbeam 350, Germany), and EDX were carried out using an Ametek EDAX, Octane Elite. The accelerating voltage used for the elemental analysis of EDX was reduced to 7 kV, which minimizes the penetration depth. High-resolution imaging of the CoO@Co3O4-5% Ag powder was performed using a FEI Talos F200X transmission microscope operating at 200 kV. The analysis was performed in the TEM and STEM modes by high-angle annular dark-field (HAADF) imaging. Energy-dispersive X-ray spectroscopy (EDX) was performed using a Brucker BD4 instrument for mapping the distribution of Co, O, and Ag elements. The colloidal particle dispersion was dropped onto a carbon film supported by a 300-mesh copper grid to create the samples for microscopy investigations. X-ray powder diffraction (XRD) for CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag) powders was performed using a Rigaku SmartLab 3 kW X-ray diffractometer equipped with a Cu X-ray tube (λXRD = 1.542 Å) and a 1D high-speed silicon semiconductor strip detector (D/teX Ultra 250). The powder diffraction patterns were acquired using the continuous scanning mode in the reflection Bragg–Brentano geometry (θ/2θ scan). The XRD patterns were collected in the 10–60° 2θ range with a step of 0.01° at a scan rate of 3° min−1. The SORPTOMATIC 1990 series (Thermo Quest (CE Instruments), Rodano (Milano), Italy) equipment was used to analyse nitrogen physisorption. The nitrogen adsorption–desorption isotherm was estimated at 77.15 K. In the p/p0 range of 0.05–0.25, the specific surface area (SSA) was determined using the Brunauer–Emmett–Teller (BET) theory.34 The Barrett–Joyner–Halenda,35 Horváth–Kawazoe,36 and Gurvich37 methodologies were used to estimate the pore-size distribution (PSD) from the adsorption branch of the nitrogen adsorption–desorption isotherm.The compression test of the hydrogel samples with and without activated carbons was carried out using a universal testing machine Zwick/Roell ProLine Z050.
Electrochemical testing
The electrochemical performance of the hydrogel as an electrolyte material and CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag) as an electrode material was investigated. For the preparation of the working electrode, 90% of the active material (CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag)) and 10 wt% of PVdF binder were mixed in a common solvent, acetone. Subsequently, the required amount of the slurry was coated over 1.0 cm2 of carbon cloth (AVCarb, USA). Finally, the coated electrodes were dried at 80 °C under vacuum for 30 min. The mass of the active material is between ∼2 mg and 4 mg. The electrochemical measurements were carried out using a BioLogic VMP3 instrument.The specific capacitance (Cs) in the two-electrode configuration was obtained from the electrochemical impedance spectroscopy in the frequency range from 100 mHz to 200 kHz. The capacitance was calculated using the following equation:38
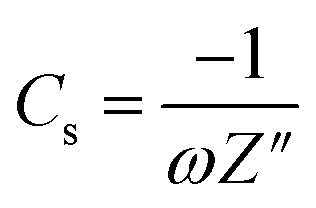 | (1) |
The specific capacitance (Cs) of the electrode in the two-electrode configuration was obtained from the galvanostatic charge–discharge (GCD) experiment using the following equation:38
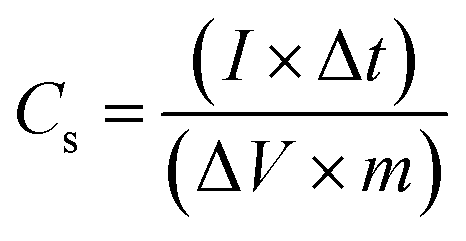 | (2) |
The specific capacitance (Cs) of the electrode in the two-electrode configuration was obtained from the cyclic voltammetry (CV) experiment using the following equation:38
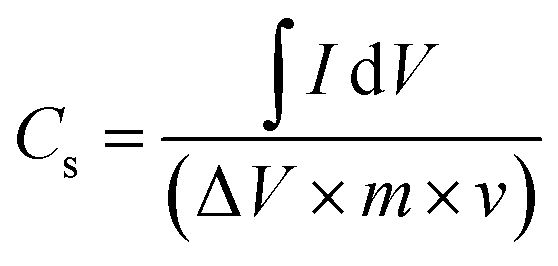 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dV represents the integral of the cathodic region traced by the CV at a scan rate of ν, ΔV is the voltage window of the two-electrode cell assembly, and m is the loading of electrode material.
dV represents the integral of the cathodic region traced by the CV at a scan rate of ν, ΔV is the voltage window of the two-electrode cell assembly, and m is the loading of electrode material.
Results and discussion
Structural and mechanical properties of the hydrogel material
Each layer of the supercapacitor was prepared and characterized separately when the whole structure was fabricated. Scheme 1 shows the step-by-step procedure of fabrication of CoO–Ag material and then electrodes described in the experimental section. Acylamide-based monomers were added to DI-water with a crosslinker and a photoinitiator, stirring the solution overnight. Activated carbon nanoparticles were added to the precursor solution, and to obtain a perfect dispersion, 30 minutes of sonication at room temperature was required. After removing the oxygen from the solution by argon bubbling, the solution was added to the mold in an ice bath. Polymerization was done with UV irradiation. The hydrogel incorporated with activated carbon can be visually observed as a free-standing, black-coloured structure (mostly opaque) and is flexible with a pretty smooth surface texture. The FE-SEM image of the hydrogel is shown in Fig. 1a. The cell-like structure of the network with pore sizes around 10–15 μm is well visible in the image. To verify the compressibility of the gels, a universal testing machine was used to examine the hydrogel with and without activated carbon and under external loading. Five compression cycles were performed for at least three identical cylindrical specimens to obtain average stress–strain and the curves are shown in ESI† (Video S1). Up to 40% of specimen strain, both hydrogels with and without carbon return to their original shape. Exceeding approximately 50% of the total strain can damage the specimen structure. The typical stress–strain curve for a hydrogel (with and without activated carbon) under loading and subsequent unloading is shown in Fig. 1b. A photograph of the as-prepared hydrogel is also shown in Fig. 1c. The mechanical properties, especially one related to the flexibility of the films, were tested by applying various deforming stresses: compression, bending, multiple twisting, folding and stretching. The gel can be folded with two or more folding and twisted multiple times (Videos S2 and S3†). It is also stretchable, confirming the high flexibility of hydrogel films. | ||
| Scheme 1 Scheme of the supercapacitor cell and characterizations. Detailed procedure of the fabrication of each layer. | ||
 | ||
| Fig. 1 (a) FE-SEM image of the hydrogel structural network. (b) Mechanical properties of the hydrogel with and without the addition of C-NPs. (c) Macroscopic demonstration of the hydrogel and mechanical flexibility. | ||
As can be seen in the graphs, the activated carbon has increased the compressive strength of the hydrogel, and it also did not influence the susceptibility to deformation. The change in specimens' dimension after cyclic loading was also not detected, which confirms the improvement in the compression strength of the hydrogel directly indicating its suitability for flexible energy storage devices.
Structural and morphological studies (XRD, SEM, TEM, and BET results)
The XRD pattern of CoOCo3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag) is presented in Fig. 2. The main diffraction lines for the sample without surface modification with Ag nanoparticles showed two characteristic diffraction lines at 35.5° and 42.4° corresponding to the planes (111) and (200) of CoO (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, ICDD: 04-8987), respectively. In all measured samples, diffraction lines at 19.1, 31.3, 36.8, 38.7, 44.8, 55.8, and 59.4° that correspond to the crystal planes (111), (220), (311), (222), (400), (422), and (511) of Co3O4 (space group: Fdm, ICDD: 42-1467) were detected.39 Furthermore, as shown in Fig. 2, the Co3O4 phase is poorly crystalline, resulting in broadened diffraction peaks. The Co3O4 content increased slightly with the increase in Ag content, and was less than 2 wt%. The modified samples also displayed two additional peaks at 38.1 and 44.3°, which demonstrated their intensity scaling up with the increase in Ag concentration and were recognized as the (111) and (200) reflections of the metallic Ag phase (space group: Fcc, 04–0783 ICDD).
m, ICDD: 04-8987), respectively. In all measured samples, diffraction lines at 19.1, 31.3, 36.8, 38.7, 44.8, 55.8, and 59.4° that correspond to the crystal planes (111), (220), (311), (222), (400), (422), and (511) of Co3O4 (space group: Fdm, ICDD: 42-1467) were detected.39 Furthermore, as shown in Fig. 2, the Co3O4 phase is poorly crystalline, resulting in broadened diffraction peaks. The Co3O4 content increased slightly with the increase in Ag content, and was less than 2 wt%. The modified samples also displayed two additional peaks at 38.1 and 44.3°, which demonstrated their intensity scaling up with the increase in Ag concentration and were recognized as the (111) and (200) reflections of the metallic Ag phase (space group: Fcc, 04–0783 ICDD).
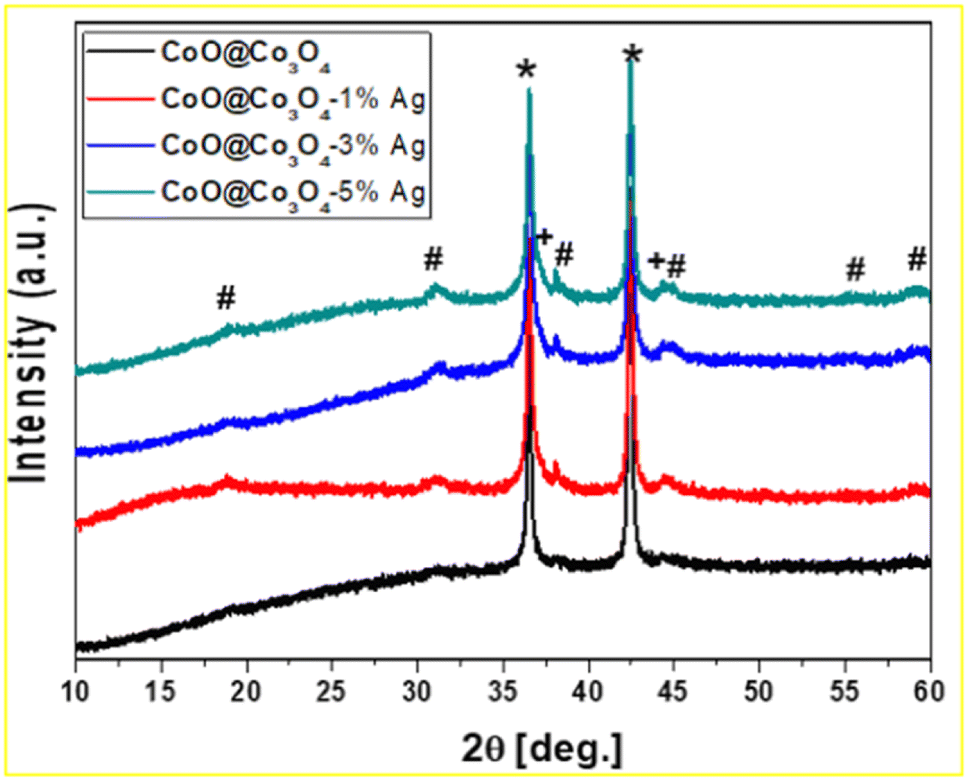 | ||
| Fig. 2 XRD pattern of CoO@Co3O4 with 1, 3, and 5 wt% Ag. (*) CoO, (#) Co3O4, (+) Ag. | ||
The morphology of all the studied samples was investigated by SEM and TEM techniques (see Fig. 3 and 4). The SEM analysis for the unmodified CoO@Co3O4 sample revealed that the material used for the surface modification with Ag NPs was not fully crystalized, the grain boundaries were not visible, and from this point of view, it seems to be very dense (Fig. 3a). After modification with Ag NPs, the difference in morphology was observed (Fig. 3b–d). The samples were composed of well-visible grains and the average particle size was 50 to 100 nm (Fig. 3b–d). All samples were strongly agglomerated into large particles. To confirm the presence of Ag NPs, the EDS analysis was also performed, which revealed the presence of Ag in metallic form (Fig. 3e). To deeply understand the morphology of the sample, the presence of Ag, and finally to get information about the average size of Ag, TEM measurements were done for the sample with the highest content of Ag: CoO@Co3O4-5% Ag (Fig. 4). HAADF-STEM imaging presents the distribution of the Ag nanoparticles on the CoO@Co3O4 particle surfaces. The EDX mapping of Co, O, and Ag shows the uniform distribution of silver nanoparticles on the surface of the CoO@Co3O4 particles. According to the recorded images (Fig. 4), the CoO@Co3O4 nanoparticle diameters vary between 10 and 20 nm. The STEM images confirmed the presence of 1 to 5 nm size of Ag nanoparticle balls.
 | ||
| Fig. 3 SEM images of (A) CoO@Co3O4 powder surface modified with (B) 1 wt%, (C) 3 wt%, (D) 5 wt% Ag, and with their registered (E) EDS spectrum. | ||
 | ||
| Fig. 4 STEM, BF-STEM, HAADF-STEM images and Co, O, Ag EDX mapping of the CoO@Co3O4-5% Ag sample. | ||
The N2-absorption–desorption experiments were examined to better understand the texture of the particles in our samples and to determine their specific surface area (SSA), and the resulting isotherms are presented in Fig. 5(A–D). The measured SSA of samples is in the range between 16 and 18 m2 g−1 (see Table 1). The results of cumulative pore volume according to the Gurvich method of meso- and micropores are presented in Table 1. After surface modification with Ag NPs, the cumulative pore volume increased from 0.043 to 0.056 cm3 g−1. In Table 2 and Table 3, the mesopores and micropores for the investigated samples are presented, respectively. The cumulative pore volume of mesopores varies from 0.132 to 0.162 cm3 g−1. All samples had mesoporous nature, in which the most dominant pore size distribution was 10–50 nm from 49.9 to 74.2%, and 50–200 nm from 17 to 34.4%. The cumulative pore volume of micropores for 4 studied samples was at the same level: 0.007 cm3 g−1. The highest number of micropores (94.8 to 96.9) was recorded for the range 0–1 nm. The rest were micropores in the range from 1 to 10 nm (from 3.4 to 5.2%). As can be seen from the pore size distribution, there is a mixture of micropores, mesopores and macropores in the prepared composites, which is very much appreciated in the case of supercapacitor applications discussed later in the manuscript.
 | ||
| Fig. 5 N2 adsorption–desorption curves of (A) CoO@Co3O4 powder surface modified with (B) 1 wt%, (C) 3 wt%, and (D) 5 wt% Ag. | ||
| Sample | SSABET, m2 g−1 | cumulative pore V, cm3 g−1, according to Gurvich |
|---|---|---|
| CoO@Co3O4 | 16 | 0.038 |
| CoO@Co3O4-1% Ag | 17 | 0.045 |
| CoO@Co3O4-3% Ag | 18 | 0.056 |
| CoO@Co3O4-5% Ag | 17 | 0.043 |
| Sample | Cumulative pore V, cm3g−1 | Pore size distribution, V, % mesopores | ||||
|---|---|---|---|---|---|---|
| 0–10 nm | 10–50 nm | 50–200 nm | 200–300 nm | 300–1000 nm | ||
| CoO@Co3O4 | 0.132 | 8 | 60 | 32 | 0 | 0 |
| CoO@Co3O4-1% Ag | 0.127 | 22.4 | 74.2 | 17 | 0 | 0 |
| CoO@Co3O4-3% Ag | 0.161 | 7.9 | 60 | 32.1 | 0 | 0 |
| CoO@Co3O4-5% Ag | 0.162 | 8.8 | 49.9 | 34.4 | 6.2 | 0.7 |
| Sample | 0–1 nm | 1–10 nm |
|---|---|---|
| CoO@Co3O4 | 96.6 | 3.4 |
| CoO@Co3O4-1% Ag | 96.2 | 3.8 |
| CoO@Co3O4-3% Ag | 96.9 | 3.1 |
| CoO@Co3O4-5% Ag | 94.8 | 5.2 |
Electrochemical results
Four different electrochemical supercapacitor cells were fabricated to test the electrochemical properties of the prepared cobalt samples as electrodes with a hydrogel as an electrolyte. Initially, four samples of cobalt oxide were prepared by varying the content of Ag using the surface modification technique (CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag)) and the hydrogel was prepared by incorporating activated carbons into the polymer network. The hydrogel was sandwiched in between each cobalt sample and hence four cells were fabricated, respectively.| Cell A: CoO@Co3O4|Hydrogel|CoO@Co3O4 |
| Cell B: CoO@Co3O4-1% Ag|Hydrogel|CoO@Co3O4-1% Ag |
| Cell C: CoO@Co3O4-3% Ag|Hydrogel|CoO@Co3O4-3% Ag |
| Cell D: CoO@Co3O4-5% Ag|Hydrogel|CoO@Co3O4-5% Ag |
The above-mentioned cells were characterized by CV, GCD and electrochemical impedance techniques, and the results are discussed below.
Fig. 6a shows the CV curves of the four tested samples, that is CoO@Co3O4-x-Ag (x denotes 1, 3 and 5 wt% of Ag) using a hydrogel as an electrolyte material recorded at a scan rate of 5 mV s−1 and in the potential range of 0 V to 1.0 V. In comparison to the CV curve of CoO@Co3O4-0 wt% of Ag, the enclosed area is increased with the increase in the percentage of Ag, indicating an enlarged capacitance. Cobalt modified with 5 wt% of Ag shows the maximum capacitance of the order of ∼30 F g−1 (calculated using eqn (2)). The signals obtained at low potentials are the effect of the redox reactions of Co2+/Co3+ and those at high potentials result from the redox conversion between Co3+ and Co4+. They are generally dominating the redox reaction of Co-based electrodes.40,41 As can be seen from the CV curves, an EDLC type of pattern was observed, which might be the reason for the surface redox reaction–controlled pseudocapacitance. In this phenomenon, the cations and anions of the electrolyte undergo adsorption and desorption over the electrode surface, and hence the process is faradaic, but the pattern of the CV curve is very much similar to EDLC.42 The scan rate-dependent CVs have been recorded for Cell D up to a maximum scan rate of 100 mV s−1, and are shown in Fig. 6b. As can be seen from the figure, with the increase in the scan rate, CV curves deviate (tilt) in voltammogram curves, which may be due to the development of resistive elements in the cell at higher scan rates. Apart from that, good interfacial compatibility was observed between the electrode and the electrolyte.
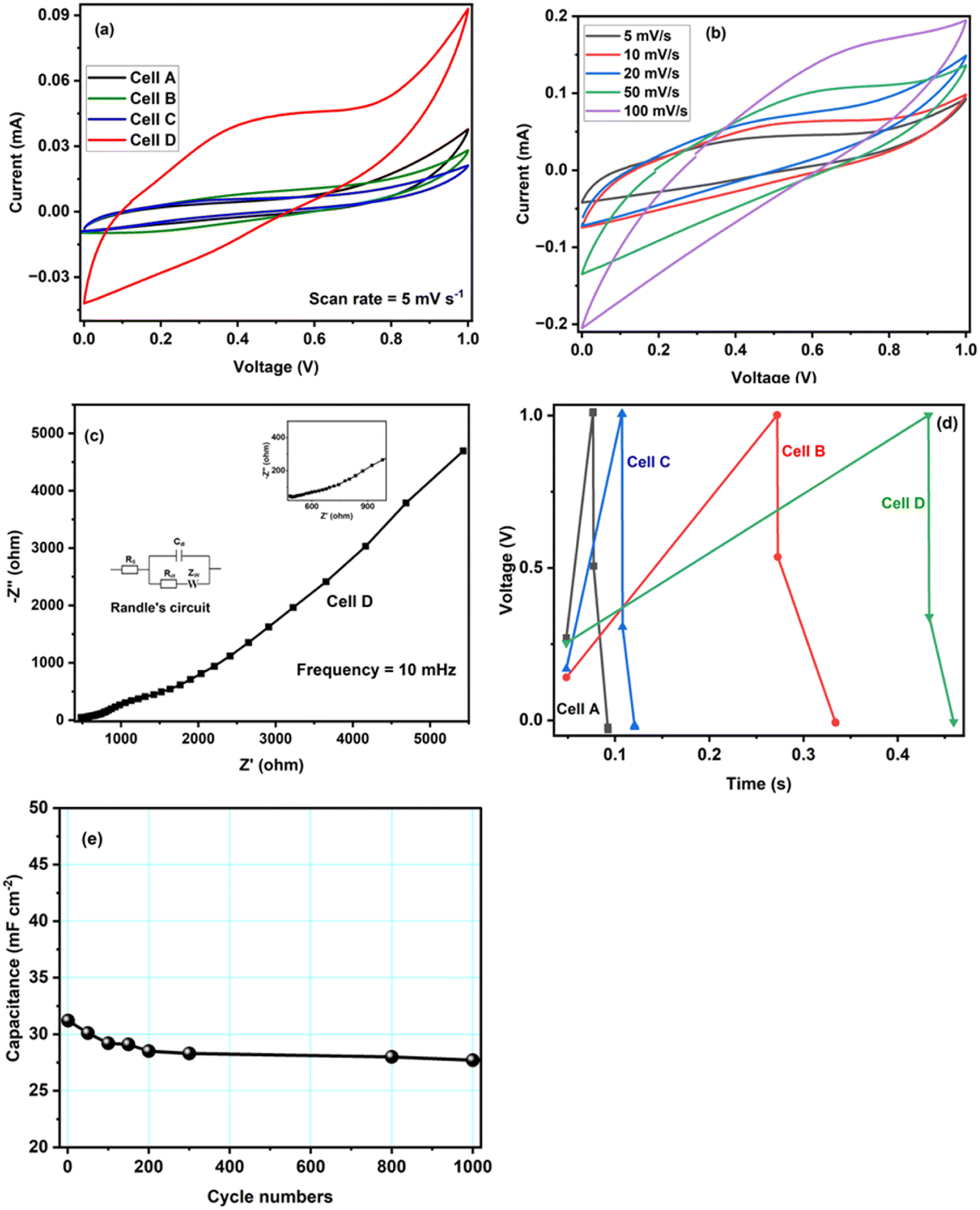 | ||
| Fig. 6 (a) CV curves of Cell A-Cell D at a scan rate of 5 mV s−1. (b) CV curves of Cell D at different scan rates. (c) Electrochemical impedance spectra of Cell D measured in the frequency range of 200 kHz to 10 mHz; the inset shows Randle's circuit and expanded impedance plots of Cell D at a higher frequency are illustrated in the inset of 6(c). (d) GCD curves of Cell A-Cell D at 1 mA cm−2. (e) Cycling stability of Cell D at a current density of 1.0 mA cm−2. | ||
To gain further insights, electrochemical impedance spectroscopy (EIS) was also performed for Cell D, and the curve is shown in Fig. 6c. It is again a very important technique that gives information about the interfacial phenomena, charge transfer resistance, bulk resistance, overall resistance, and capacitance of the device in different frequency regions, and it also investigates the kinetic and conductive properties of the electrochemical cells. EIS measurements show the complex impedance (Z) with its real (Z′) and imaginary (Z′′) components at different frequency ranges (in the present case 200 kHz to 10 mHz). The real part Z′ of the complex impedance was used to evaluate the resistance values of the cells and the imaginary part Z′′ to calculate the capacitance of the cell using eqn (1). An equivalent circuit is shown in Fig. 6c, where Rs is the resistance from the electrolyte, Rct is the charge transfer resistance and Cp is the pseudocapacitance because of the redox process of cobalt electrodes43 In the impedance plot, the intercept of the real part (Z′) in the high frequency range is the combination of the ionic resistance of the electrolyte material and contact resistance between the active material and the current collector. Bulk resistance, charge transfer resistance, overall resistance and capacitance values of the capacitor Cell D were found to be of the order of ∼483 Ω cm2, 156 Ω cm2, ∼71 Ω cm2 and ∼27 F g−1. The capacitance value was calculated using eqn (1). The resistance obtained in the high frequency region confirms that the superficial conductive path is provided by hydrogels and also no semicircle is observed in the high frequency region, which clearly means that charge resistance caused by Faradaic reactions is also minimal.44 All of these results confirm that the hydrogel synthesized with activated carbon has great potential as an electrolyte material for EES devices.
Furthermore, GCD studies also confirmed that the hydrogel and CoO-based electrodes are suitable for electrochemical applications. Fig. 6d shows the typical GCD curves of supercapacitor cells at a current density of 1.0 mA cm−2, the specific capacitance value of Cell D was obtained in the order of ∼38 mF cm−2 (calculated using eqn (3)). GCD is an important technique carried out with the variation of time and voltage at a constant current value. From Fig. 6d, it can be seen that the control sample shows the triangular feature, which shows the formation of a double layer at the electrode–electrolyte interface, whereas CoO@Co3O4 with Ag shows the deviation of triangular features due to the increase in the content of Ag, confirming the dominance of pseudocapacitance behaviour in the system.45,46 This increase in the value of capacitance also arises from the fact that the conductivity of the CoO samples increases with the increase in the content of silver particles. Additionally, the silver nanoparticle balls of size 1–5 nm were distributed uniformly on the surface of CoO@Co3O4, as confirmed by STEM studies.
Continuous charge–discharge cycles were carried out for ∼1000 cycles at a current density of 1.0 mA cm−2, and are shown in Fig. 6e. The cycling test was performed on the best cell, and as shown in the figure, there is a decrease in the early stage of cycling, which could be due to charge loss at the electrode–electrolyte interface, and then the cell was stable. The study confirms the stable cycling performance of Cell D and the possibility of using the materials for energy storage applications.
Conclusions
In this article, we demonstrated a compressible, self-healing, multifunctional system comprising two key components (electrode and electrolyte) and built up layer by layer. The successful modification of the commercial cobalt oxide material with 1–5 nm metallic Ag nanoparticles was investigated. The material was utilized as a symmetrical supercapacitor electrode in which the hydrogel layer, embedded with carbon nanoparticles, was used as an electrolyte. In the proposed two-electrode configuration, the best electrochemical performance with a specific capacitance of ∼38 mF cm−2 was delivered by cobalt oxide modified with 5 wt% Ag. The cyclic compression tests show the high flexibility of the hydrogel modified with the activated carbon, which is required for commercial applications.Data availability
The data of this study is available on request.Author contributions
AJ: conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review & editing, resources, visualization, funding acquisition. YZ: investigation, visualization, formal analysis, writing – original draft, writing – review & editing. SRM: writing – original draft, writing – review & editing. KB: investigation, writing – original draft, writing – review & editing. FP: investigation. MM: methodology, formal analysis, investigation, writing – original draft, writing – review & editing, resources, visualization, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Centre for Research and Development (NCBR, Poland); project number: V4-Japan/2/17/AtomDeC/2022 and the Ministry of Education, Youth and Sports, Czech Republic (contract no. 8F21007) under the Visegrad Group-Japan 2021 Joint Call on Advanced Materials in cooperation with the International Visegrad Fund. YZ and FP received support by the First TEAM grant number POIR.04.04.00-00-5ED7/18-00, within the framework of the First TEAM programme of the Foundation for Polish Science (FNP), co-financed by the European Union under the European Regional Development Fund.References
- J. Sun, B. Luo and H. Li, Adv. Energy Sustainability Res., 2022, 3, 2100191 CrossRef.
- E. Feng, W. Gao, J. Li, J. Wei, Q. Yang, Z. Li, X. Ma, T. Zhang and Z. Yang, ACS Sustainable Chem. Eng., 2020, 8, 3311–3320 CrossRef CAS.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- M. S. Asl, R. Hadi, L. Salehghadimi, A. G. Tabrizi, S. Farhoudian, A. Babapoor and M. Pahlevani, J. Energy Storage, 2022, 50, 104223 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- Poonam, K. Sharma, A. Arora and S. K. Tripathi, J. Energy Storage, 2019, 21, 801–825 CrossRef.
- H. Peng, Y. Lv, G. Wei, J. Zhou, X. Gao, K. Sun, G. Ma and Z. Lei, J. Power Sources, 2019, 431, 210–219 CrossRef CAS.
- Z. Wang and Q. Pan, Adv. Funct. Mater., 2017, 27, 1700690 CrossRef.
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed.
- Y. Shi, Y. Zhang, L. Jia, Q. Zhang and X. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 36028–36036 CrossRef CAS.
- N. A. Choudhury, S. Sampath and A. K. Shukla, Energy Environ. Sci., 2009, 2, 55–67 RSC.
- C. Rinoldi, Y. Ziai, S. S. Zargarian, P. Nakielski, K. Zembrzycki, M. A. Haghighat Bayan, A. B. Zakrzewska, R. Fiorelli, M. Lanzi, A. Kostrzewska-Księżyk, R. Czajkowski, E. Kublik, L. Kaczmarek and F. Pierini, ACS Appl. Mater. Interfaces, 2023, 15, 6283–6296 CrossRef CAS PubMed.
- X. Cao, C. Jiang, N. Sun, D. Tan, Q. Li, S. Bi and J. Song, J. Sci.: Adv. Mater. Devices, 2021, 6, 338–350 CAS.
- L. Jiang and X. Lu, Mater. Chem. Front., 2021, 5, 7479–7498 RSC.
- Y. Ziai, F. Petronella, C. Rinoldi, P. Nakielski, A. Zakrzewska, T. A. Kowalewski, W. Augustyniak, X. Li, A. Calogero, I. Sabała, B. Ding, L. D. Sio and F. Pierini, NPG Asia Mater., 2021, 14, 18 CrossRef.
- M. A. Campea, M. J. Majcher, A. Lofts and T. Hoare, Adv. Funct. Mater., 2021, 31, 2102355 CrossRef CAS.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem. Int. Ed., 2018, 57, 2096–2100 CrossRef CAS PubMed.
- C. Rinoldi, M. Lanzi, R. Fiorelli, P. Nakielski, K. Zembrzycki, T. Kowalewski, O. Urbanek, V. Grippo, K. Jezierska-Woźniak, W. Maksymowicz, A. Camposeo, R. Bilewicz, D. Pisignano, N. Sanai and F. Pierini, Biomacromolecules, 2021, 22, 3084–3098 CrossRef CAS.
- S. Pirsa, F. Asadzadeh and I. Karimi Sani, J. Inorg. Organomet. Polym. Mater., 2020, 30, 3188–3198 CrossRef CAS.
- Q. Peng, J. Chen, T. Wang, X. Peng, J. Liu, X. Wang and J. Wang, InfoMat, 2020, 2, 843–865 CrossRef CAS.
- A. Gupta, A. Jain and S. K. Tripathi, J. Polym. Res., 2021, 28, 235 CrossRef CAS.
- A. Poosapati, K. Negrete, M. Thorpe, J. Hutchison, M. Zupan, Y. Lan and D. Madan, J. Appl. Polym. Sci., 2021, 138, 50813 CrossRef CAS.
- J. Wen, Q. Zhao, X. Jiang, G. Ji, R. Wang, G. Lu, J. Long, N. Hu and C. Xu, ACS Appl. Energy Mater., 2021, 4, 3660–3669 CrossRef CAS.
- V. R. Jeedi, E. L. Narsaiah, M. Yalla, R. Swarnalatha, S. N. Reddy and A. Sadananda Chary, SN Appl. Sci., 2020, 2, 2093 CrossRef CAS.
- Poonam, K. Sharma, A. Arora and S. K. Tripathi, J. Energy Storage, 2019, 21, 801–825 CrossRef.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed.
- N. R. Chodankar, H. D. Pham, A. K. Nanjundan, J. F. S. Fernando, K. Jayaramulu, D. Golberg, Y. Han and D. P. Dubal, Small, 2020, 16, 2002806 CrossRef CAS PubMed.
- M.-K. Wu, J.-J. Zhou, F.-Y. Yi, C. Chen, Y.-L. Li, Q. Li, K. Tao and L. Han, Dalton Trans., 2017, 46, 16821–16827 RSC.
- L.-D. Chen, Y.-Q. Zheng and H.-L. Zhu, J. Mater. Sci., 2018, 53, 1346–1355 CrossRef CAS.
- L. Manjakkal, A. Pullanchiyodan, N. Yogeswaran, E. S. Hosseini and R. Dahiya, Adv. Mater., 2020, 32, 1907254 CrossRef CAS PubMed.
- A. Jain, S. R. Manippady, R. Tang, H. Nishihara, K. Sobczak, V. Matejka and M. Michalska, Sci. Rep., 2022, 12, 21024 CrossRef CAS.
- S. J. Uke, V. P. Akhare, D. R. Bambole, A. B. Bodade and G. N. Chaudhari, Front. Mater., 2017, 4, 21 CrossRef.
- C. Wang, P. Sun, G. Qu, J. Yin and X. Xu, Chin. Chem. Lett., 2018, 29, 1731–1740 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- G. Horváth and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef.
- P. Bertier, K. Schweinar, H. Stanjek, A. Ghanizadeh, C. R. Clarkson, A. Busch, N. Kampman, D. Prinz, A. Amann-Hildebrand, B. M. Krooss and V. Pipich, The Clay Minerals Society Workshop Lectures Series, The Clay Minerals Society, Virginia, 2016, pp. 151-161 Search PubMed.
- A. Jain, M. Michalska, A. Zaszczyńska and P. Denis, J. Energy Storage, 2022, 54, 105367 CrossRef.
- M. Michalska, H. Xu, Q. Shan, S. Zhang, Y. Dall'Agnese, Y. Gao, A. Jain and M. Krajewski, Beilstein J. Nanotechnol., 2021, 12, 424–431 CrossRef CAS PubMed.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- C.-A. Tseng, P. K. Sahoo, C.-P. Lee, Y.-T. Lin, J.-H. Xu and Y.-T. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 40426–40432 CrossRef CAS PubMed.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597 RSC.
- Q. Liao, N. Li, H. Cui and C. Wang, J. Mater. Chem. A, 2013, 1, 13715 RSC.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- M. Mirzaeian, N. Akhanova, M. Gabdullin, Z. Kalkozova, A. Tulegenova, S. Nurbolat and K. Abdullin, Energies, 2020, 13, 5228 CrossRef CAS.
- R. Kumar, A. Soam and V. Sahajwalla, Mater. Adv., 2021, 2, 2918–2923 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00893b |
| ‡ These authors are equal contributors. |
| This journal is © The Royal Society of Chemistry 2023 |