
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCaesium carbonate promoted regioselective O-functionalization of 4,6-diphenylpyrimidin-2(1H)-ones under mild conditions and mechanistic insight†
Vijay Kumara,
Praval Pratap Singhb,
Ashish Ranjan Dwivediac,
Naveen Kumara,
Rakesh kumara,
Subash Chandra Sahoo d,
Sudip Chakrabortyb and
Vinod Kumar*a
d,
Sudip Chakrabortyb and
Vinod Kumar*a
aLaboratory of Organic and Medicinal Chemistry, Department of Chemistry, Central University of Punjab, Bathinda, Punjab 151401, India. E-mail: vpathania18@gmail.com; vinod.kumar@cup.edu.in; Tel: +911642864214
bDepartment of Computational Sciences, School of Basic Sciences, Central University of Punjab, 151401, India
cGitam School of Pharmacy, Hyderabad, Telangana 502329, India
dDepartment of Chemistry, Panjab University, Chandigarh, 160014, India
First published on 5th June 2023
Abstract
A facile one-step catalyst free methodology has been developed for the regioselective functionalization of 4,6-diphenylpyrimidin-2(1H)-ones under mild conditions. Selectivity towards the O-regioisomer was achieved by using Cs2CO3 in DMF without use of any coupling reagents. A total of 14 regioselective O-alkylated 4,6-diphenylpyrimidines were synthesized in 81–91% yield. In the DFT studies it was observed that the transition state for the formation of the O-regioisomer is more favourable with Cs2CO3 as compared to K2CO3. Furthermore, this methodology was extended to increase the O/N ratio for the alkylation of 2-phenylquinazolin-4(3H)-one derivatives.
1. Introduction
Pyrimidine is a versatile scaffold found in a number of bioactive compounds including drug molecules. Several active pharmaceutical agents containing the pyrimidine scaffold are in various phases of clinical trials and uses (Fig. 1).1–4 A range of substituents/functional groups have been explored at different positions of pyrimidinones to evaluate their impact on the activity.5–11 In particular, specific attention has been paid to the selective N/O-alkylation of the pyrimidinones.12 Conventionally, functionalisation of the N and O sites of pyrimidinones has been achieved through two different approaches. The selective O-alkylation of pyrimidin-2(1H)-ones was achieved by chlorination of pyrimidin-2(1H)-ones followed by substitution reaction with an alcohol.13 On the other hand, selective N-alkylated adducts were synthesised from pyrimidin-2-one using alkyl halide, aqueous sodium hydroxide and a phase transfer catalyst such as triethylbenzylammonium chloride.14 Similarly, reaction of alkyl halide with K2CO3 as a base under reflux conditions provided N-alkylated products in moderate yields.15 Recently, Mansouri et al. (2019) reported a microwave-assisted single-pot reaction for the selective N-functionalisation of pyrimidine derivatives using (bis(trimethylsilyl)amide)/KI (HMDS/KI) as a catalyst.16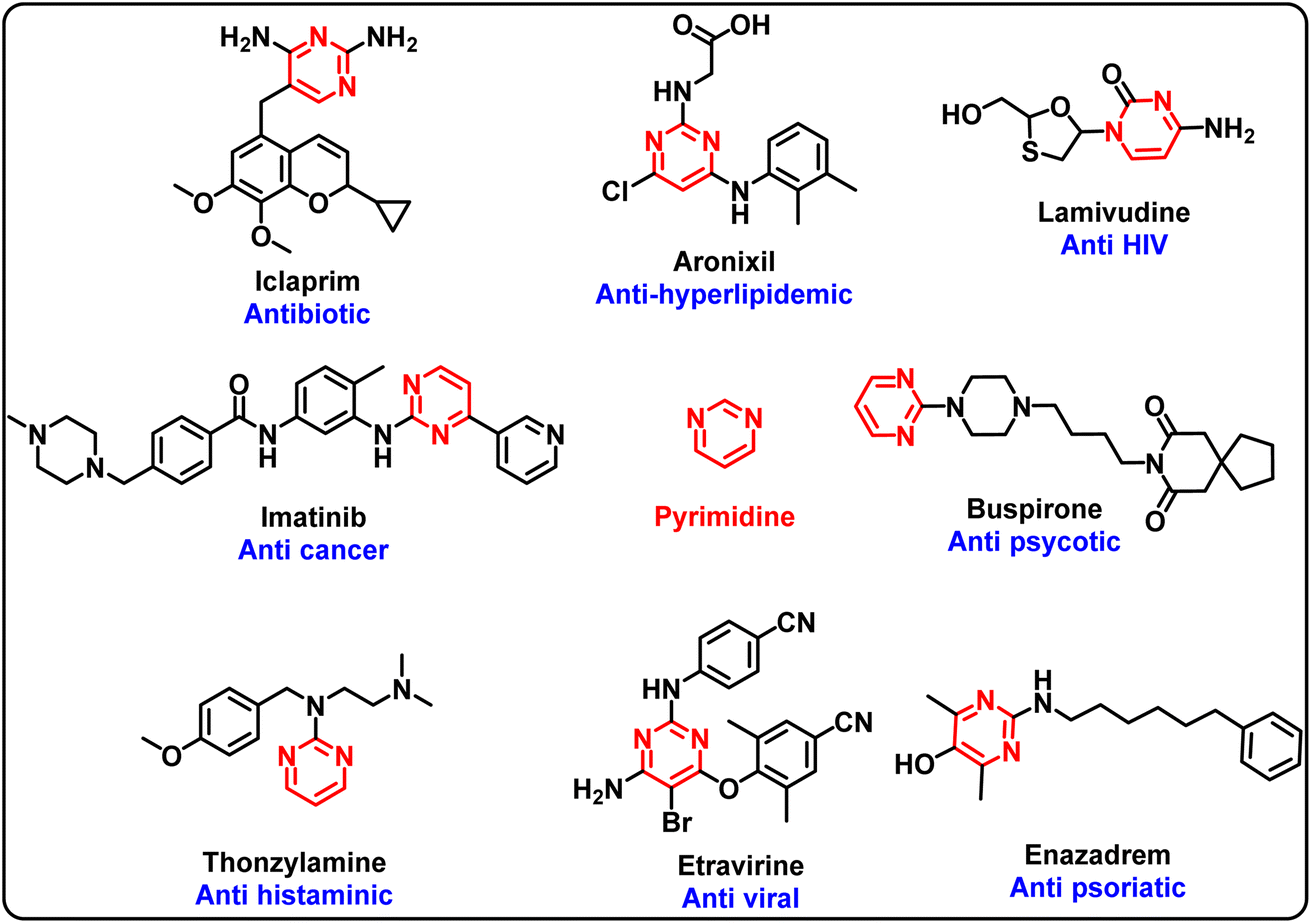 | ||
| Fig. 1 Biologically active compounds with a pyrimidine nucleus. | ||
Alkylation of pyrimidin-2(1H)-ones with alkyl halides predominantly produced N-alkylated products.8,17 Hopkins et al. studied the ambident anion chemistry of 2-hydroxypyrimidines for their alkylation towards various alkyl halides and alkali salts. The alkylation reactions were found to favour the nitrogen site and N to O ratio for the substitution was found to be insensitive to the nature of alkali metal. A slight improvement in the O-alkylated product was observed when silver salts were used.18 Later, Gacek and Undheim also showed the same characteristics for the alkylation of 5-halopyrimidin-2-ones and established that yield for O-isomer can be increased by using silver salts. Exclusive O-isomer was synthesized through a multistep protocol.14 Similarly, Meščić et al. synthesised 5-hydroxyalkyl substituted N-acyclic and O-acyclic pyrimidines in almost equimolar ratio using K2CO3 and DMF.19 There is hardly any report for the selective O-alkylation of pyrimidinones in a single step. In an attempt to regio-selectively synthesize O-alkylated product in a single step, B. S Møller and co-workers reported selective synthesis of O-alkyloxy products using Cs2CO3 in anhydrous DMF at 0 °C.20 However, α-haloethers were used to carry out the selective O-alkylation and α-oxygen atom was an indispensable requirement in the form of ether or ester to functionalise the alkyl chlorides. It was observed that in the absence of the α-oxygen atom, generally N-alkylated regioisomer was obtained as a major product. Recently, Mittersteiner et al. reported regioselective O-alkylation of 4-(trifluoromethyl)pyrimidin-2(1H)-ones but scope of electrophile used for these transformations was only limited to 4-(iodomethyl)pyrimidines.21 Therefore, a synthetic methodology is desired for the regioselective O-alkylation of pyrimidinones/quinazolinones under mild conditions. Herein, we report a method for the regioselective O-alkylation of pyrimidinones under benign conditions using Cs2CO3 in dimethyl formamide as solvent. The products were obtained regioselectivity in high to excellent yields.
2. Experimental section
2.1 General
All the chemicals were purchased from Sigma-Aldrich, Avra synthesis, Qualikems Fine Chem, Spectrochem and other chemical companies and used without further purification. 4,6-Bis(4-methoxyphenyl)pyrimidin-2(1H)-ones were synthesised via Biginelli type reaction.22 Progress of the reaction was monitored through thin layer chromatography (TLC) and mass-spectroscopy. Ethyl acetate and petroleum ether solutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:3 & 1:2) were used as the mobile phase for the purification of the compounds through column chromatography. Mass spectra were recorded on GC-MS (ESI), Department of Pharmaceutical Sciences and Natural Products (PSNP) & Central Instruments Laboratory (CIL), Central University of Punjab, Bathinda. 1H and 13C spectra were recorded on JEOL JNM ECS400 or Bruker Avance HD III 500 instruments at 400 MHz and 500 MHz in CDCl3 as solvent at IIT Ropar and GNDU Amritsar, respectively. Chemical shifts were recorded in parts per million downfield to internal standard tetramethyl silane.
:5, 1:3 & 1:2) were used as the mobile phase for the purification of the compounds through column chromatography. Mass spectra were recorded on GC-MS (ESI), Department of Pharmaceutical Sciences and Natural Products (PSNP) & Central Instruments Laboratory (CIL), Central University of Punjab, Bathinda. 1H and 13C spectra were recorded on JEOL JNM ECS400 or Bruker Avance HD III 500 instruments at 400 MHz and 500 MHz in CDCl3 as solvent at IIT Ropar and GNDU Amritsar, respectively. Chemical shifts were recorded in parts per million downfield to internal standard tetramethyl silane.
2.2 Typical procedure for alkylation of 4,6-diphenylpyrimidin-2(1H)-one
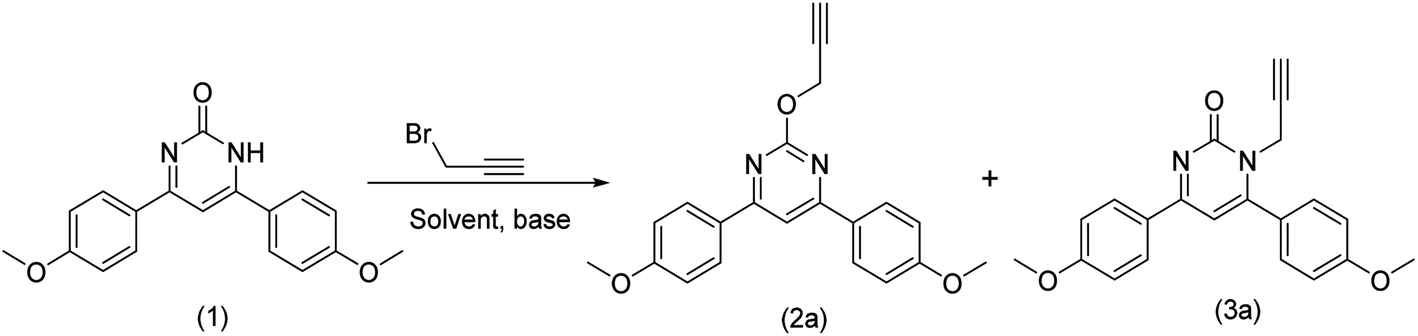
To a 25 mL round bottom flask (RBF), 4,6-diphenylpyrimidin-2(1H)-one (0.16 mmol), caesium carbonate (0.16 mmol) was added and solubilized in DMF. The RBF was kept inside an ice bath and organic halide (0.19 mmol) was carefully added to it. Thereafter, the reaction mixture was kept for stirring at room temperature for 8 hours. Progress of the reaction was monitored through TLC and GC-MS. To the reaction mixture (15 mL) water was added and aqueous phase was extracted with ethyl acetate (20 mL × 3), washed with brine, dried over sodium sulphate and the organic solvent was evaporated under vacuum using rotary evaporator to obtain 2a and 3a (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Yieldb (%) 2ac |
| a Unless mentioned, 4,6-bis(4-methoxyphenyl)-pyrimidin-2(1H)-one (0.16 mmol), base (0.16 mmol), and solvents (5 mL) were used in the reaction; addition of propargyl bromide (0.19 mmol) was done at 0 °C and thereafter stirring at room temperature for 8 h.b Isolated yield.c Yield of 3a is given in parenthesis wherever applicable. | ||||
| 1 | DMF | K2CO3 | 8 | 58(42) |
| 2 | DMF | Na2CO3 | 8 | 36(30) |
| 3 | DMF | NaHCO3 | 8 | 29(8) |
| 4 | DMF | KOC(CH3)3 | 8 | 52(38) |
| 5 | DMF | Cs2CO3 | 8 | 89(11) |
| 6 | NMP | Cs2CO3 | 24 | 70 |
| 7 | THF | Cs2CO3 | 24 | 61 |
| 8 | DMSO | Cs2CO3 | 24 | 79 |
| 9 | can | Cs2CO3 | 24 | 69 |
| 10 | Dioxane | Cs2CO3 | 48 | — |
| 11 | DMF | NaH | 8 | 2 |
| 12 | DMF | Cs2CO3 | 8(80 °C) | 62(38) |
| 13 | DMF | Cs2CO3 | 8(120 °C) | 57(43) |
2.3 Computational details
All the calculations were performed using DFT method in the Gaussian 16 suite.23 The intermediates and the transition states were optimized in the gas phase without any symmetry constraints using B3LYP exchange–correlation functional for all the structures24 with Grimme empirical dispersion correction D3 (ref. 25) in conjunction with Karlsruhe def2-SVP basis set for all the atoms.26 Frequency analysis was conducted at the same level of theory on the optimized geometries to determine whether the stationary points are minima. Using the Linear Synchronous Transit (LST)27 method, transition state guesses were searched on the potential energy surface, and the subsequent optimization was performed using the Berny algorithm. The transition states and the adjacent intermediates were connected by performing Intrinsic reaction coordinates (IRC) calculations.28 Furthermore, to refine the energetics of the system single-point energy calculations were carried out on the optimized structures using the same functional B3LYP-D3 for all the structures employing a higher valence triple-zeta polarization valence basis set def2-TZVP. Solvation energies in N,N-dimethylformamide (DMF) solvent was evaluated by a self-consistent reaction field (SCRF) approach using the SMD continuum solvation model29 as conducted in the experimental setup. Tight wave function convergence criteria were used during single-point energy calculations. The figures and 3D images of the optimized geometries were drawn using ChemDraw Professional 16.0 and CYLview visualization software, respectively.303. Results and discussion
Since last few years, we are working on the design and synthesis of pyrimidine and quinazoline based heterocycles. The synthesized compounds are being screened for multi-potent activity against different targets for the treatment of Alzheimer's disease.10,11,31–37 In continuation of this, we got interested in the functionalization of pyrimidine and quinazoline with propargyl bromide. Initially, propargyl bromide mediated alkylation of 6-bis(4-methoxyphenyl)pyrimidin-2(1H)-one (1) was attempted using K2CO3 and dimethyl formamide (DMF) as solvent. The reaction completes after 8 h of reaction time and O-alkylated and N-alkylated products were obtained in 58% and 42% yield, respectively. The literature search revealed that N-alkylated product is common under basic conditions however, single step O-alkylation is not much explored. The regioselective O-alkylation of 2,4-diphenylpyrimidinones was achieved by Wan et al.38 using BOP as reagent. Thus, we got interested in the regioselective O-alkylation of pyrimidinones and quinazolinones under milder conditions and without using any catalyst or coupling reagent.4,6-Bis(4-methoxyphenyl)pyrimidin-2(1H)-one (1) was taken as a model substrate and different set of conditions including different bases and solvents were explored for the regioselective O-alkylation. Different combination of bases like Na2CO3, Cs2CO3, NaHCO3, NaH, KOC(CH3)3 and solvents like CH3CN, dioxane, DMSO, NMP, THF were explored for the optimization of product yield (Table 1). The combination of Na2CO3/DMF, NaHCO3/DMF and KOC(CH3)3/DMF provided mixture of N- and O-alkylated products in varying yield. Cs2CO3 provided O-alkylated product regioselectively with CH3CN, THF, DMSO and NMP in 69%, 61%, 79% and 70% yield, respectively. Surprisingly, no alkylated product could be obtained with Cs2CO3/dioxane even after 48 h and NaH/DMF combination provided O-alkylated product in 2% yield only. When the reaction was performed at the elevated temperatures at 80 °C and 120 °C, regioselectivity lost and both N- & O-alkylated products were obtained. The optimum yield of 89% of the O-alkylated product was obtained with Cs2CO3/DMF in 8 h with high regioselectivity at room temperature.
NMR spectrum was used to confirm the functionalization of O- and N-alkylated regioisomers. In the O-alkylated regioisomer (2a) the methylene group would directly attach to more electronegative atom as compared to the N-regioisomer (3a) and expected to be in the downfield region of 1H-NMR. As shown in Fig. 2, methylene proton for 2a was observed at δ value of 5.19 (d, J = 2.0 Hz, 2H) whereas for 3a it was observed at δ value of 4.64 (d, J = 2.4 Hz, 2H) in 1H NMR. In addition, aromatic proton of the aromatized pyrimidine ring appeared downfield (δ = 7.70) as compared to aromatic proton of the pyrimidinone ring (δ = 6.69). Similarly, in the 13C NMR spectrum, O-alkylated product (2a) showed no peak for the carbonyl carbon while N-alkylated product displayed peak for carbonyl carbon at δ 202.88 (Fig. 2). By gradually evaporating an ethanol solution, single crystals of compounds 2a were produced and subsequently analysed using single crystal X-ray. The ORTEP projections of compounds 2a (Fig. 3) confirms O-alkylation of 4,6-bis(4-methoxyphenyl)pyrimidin-2(1H)-one.
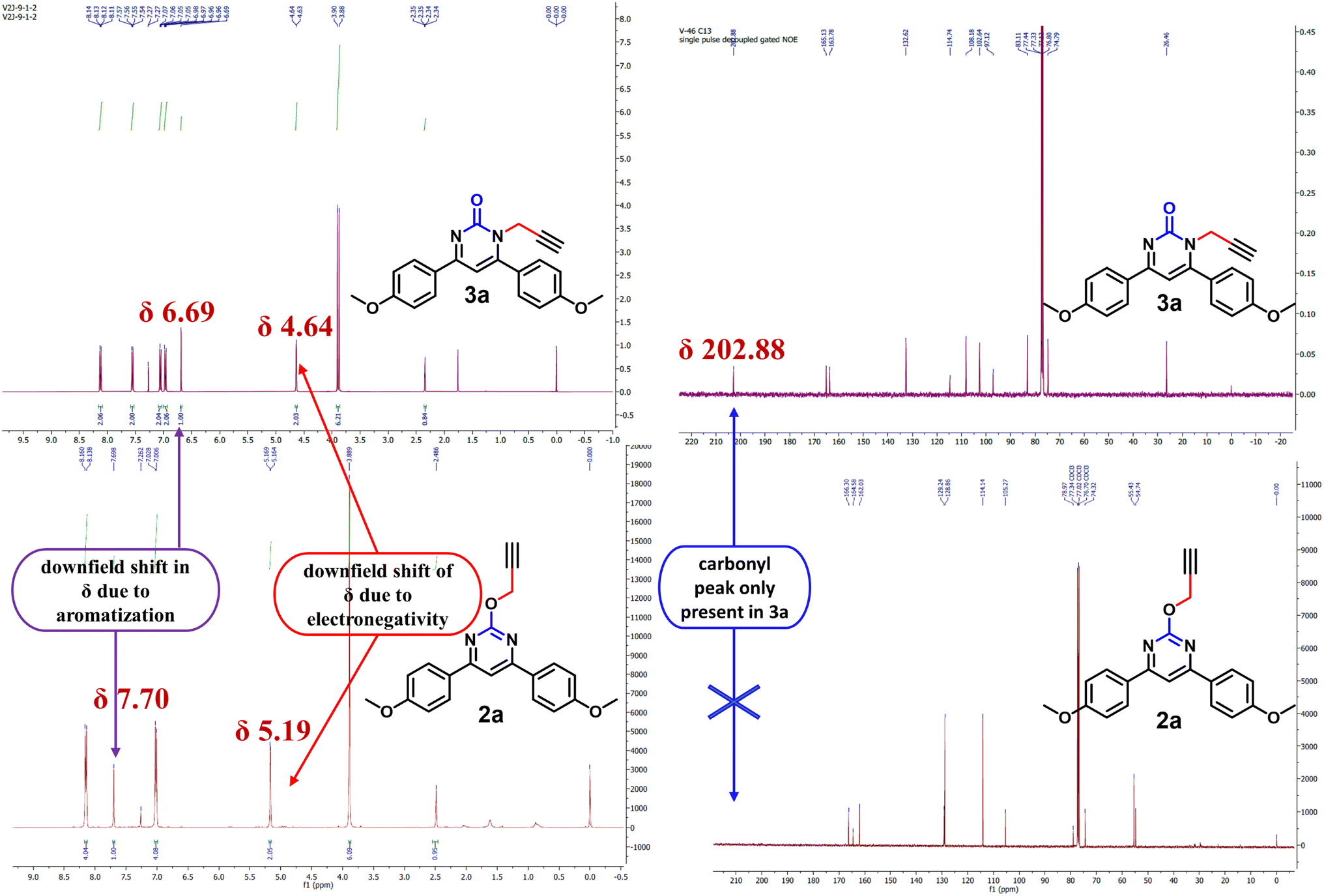 | ||
| Fig. 2 Comparison of chemical shift values for 2a and 3a. | ||
 | ||
| Fig. 3 ORTEP of compound 2a. | ||
It was envisaged that the regioselective O-alkylation was achieved through the in situ formation of a stable oxygen–caesium complex via aromatization of the pyrimidinone ring system.
Thus, several substituted biphenyl pyrimidinones were alkylated using propargyl bromide under the developed reaction conditions. Both electron releasing and electron withdrawing groups were found compatible for the regioselective O-alkylation and products were obtained in 81 to 91% yield (Scheme 1).
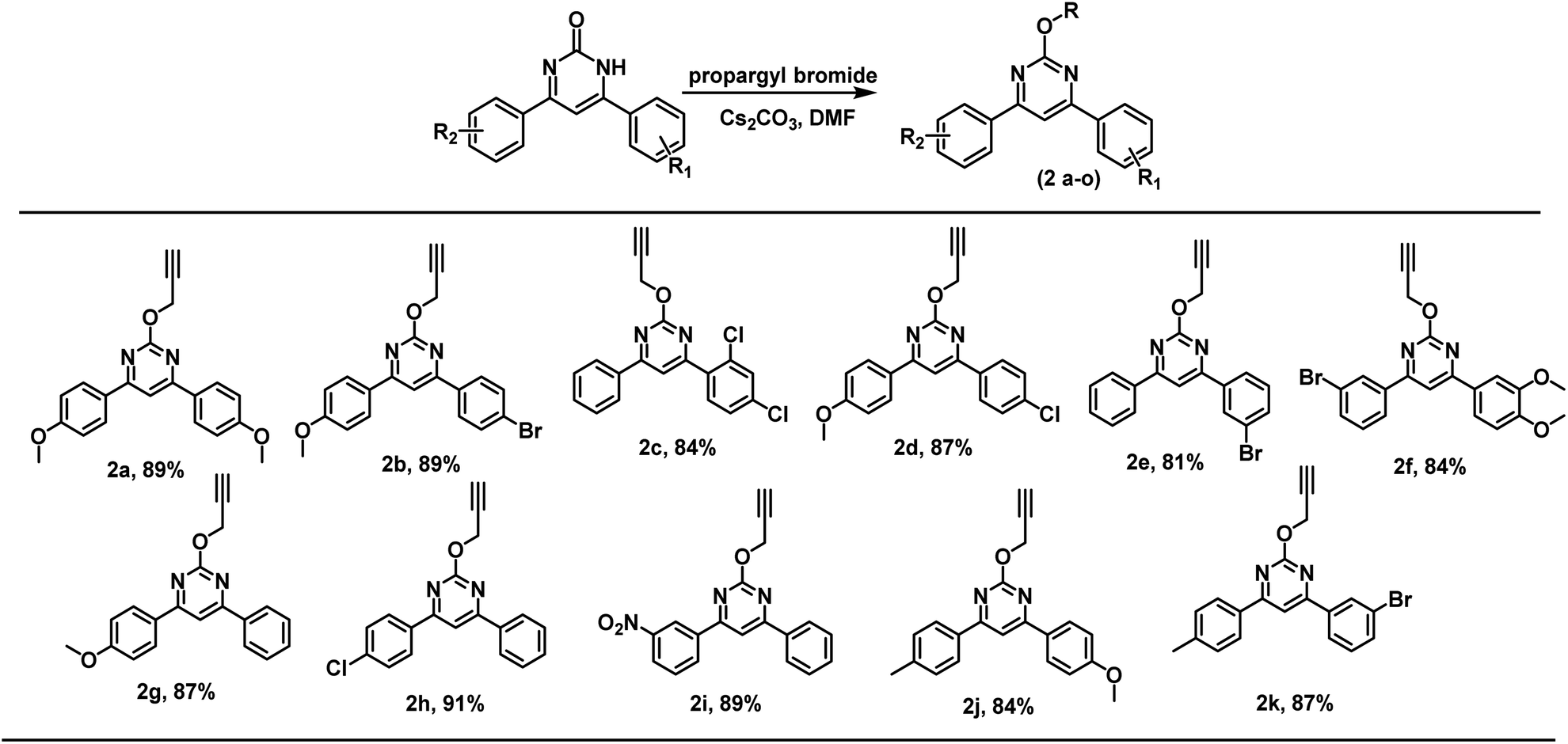 | ||
| Scheme 1 Scope of the reaction for different substrates.a Unless mentioned, 4,6-bis(4-methoxyphenyl)-pyrimidin-2(1H)-one or 2-phenylquinazolin-4(3H)-one (0.16 mmol), base (0.16 mmol), and solvents (5 mL); addition of organic halide (0.19 mmol) done at 0 °C and thereafter stirring at room temperature for 8 h. | ||
Further, the propargyl bromide was also replaced with other alkylating agents like propyl bromide, allyl bromide, benzyl bromide, etc. (Scheme 2). In all the cases, alkylation proceeded regioselectivity and corresponding O-alkylated products were obtained in excellent yields.
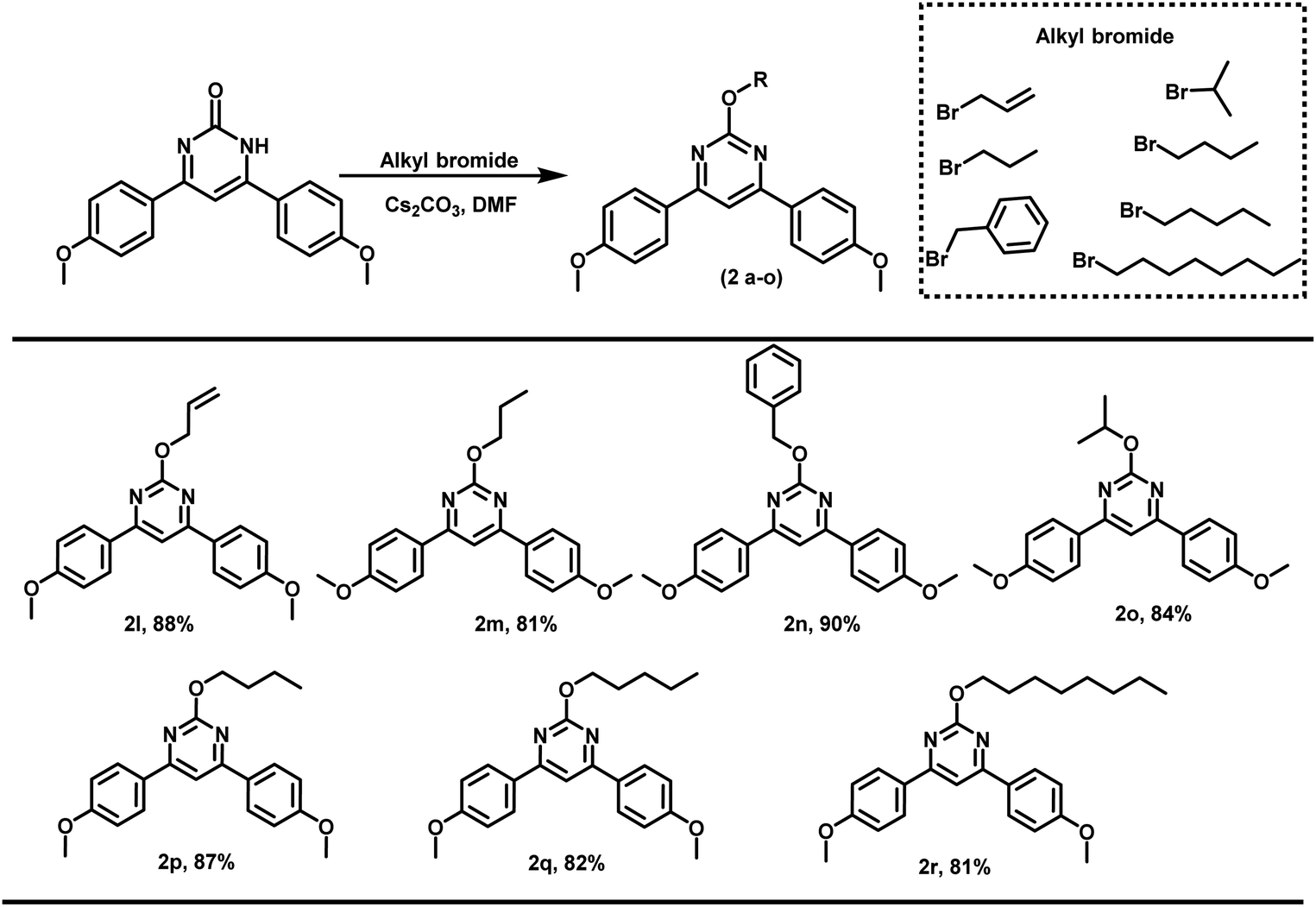 | ||
| Scheme 2 Scope of the reaction for different alkylating agents.a Unless mentioned, 4,6-bis(4-methoxyphenyl)-pyrimidin-2(1H)-one or 2-phenylquinazolin-4(3H)-one (0.16 mmol), base (0.16 mmol), and solvent (5 mL); addition of organic halide (0.19 mmol) done at 0 °C and thereafter stirring at room temperature for 8 h. | ||
3.1 Plausible reaction mechanism
To study the reaction mechanism for selectivity, DFT studies were performed using Gaussian 16 suite. DFT studies revealed that abstraction of proton from 1 proceeds with the formation of INT1 through exergonic process (ΔGLS = −8.0 kcal mol−1) (Fig. 4). Loss of proton in the form of CsHCO3 from INT1 produces INT2 through exergonic fashion (ΔGLS = −14.3 kcal mol−1). In the optimized structure of INT2, negative charge is delocalized over O–C–N atoms of the pyrimidine and bond lengths between Cs–N and Cs–O were found to be 2.3 Å and 2.4 Å, respectively. Further, electrophilic attack of propargyl bromide on INT2 result in the formation of 2a and 3a via the transition states TS1‡, and TS2‡, respectively. The free energy barrier was found to be lower for the formation of O-isomer 2a (ΔGLS = 16.6 kcal mol−1) as compared to N-isomer 3a (ΔGLS = 20.1 kcal mol−1). It justifies the regioselective formation of O-functionalized product as compared to N-functionalized product in Cs2CO3. Similarly, DFT studies were also conducted using K2CO3 as base however, in this case the transition state TS3‡ for the O-alkylated product was found to be more energy demanding (see ESI†). | ||
| Fig. 4 Free energy diagram for the O/N-functionalization of pyrimidinones using Cs2CO3. | ||
The DFT studies clearly indicate that the electrophilic attack of propargyl bromide favours the formation of 2a via TS1‡. From the optimized structure of TS1‡ and TS2‡, it was found that Cs+ ion play crucial role for the formation of 2a.
Further, the present reaction conditions were also explored for the regioselective alkylation of quinazolinones. Substituted quinazolinone derivatives were alkylated with propargyl bromide however, a mixture of O- and N-alkylated product was obtained in almost 70:30 ratio wherein O-alkylated regio-isomer was obtained as a major product in the alkylation of quinazolinones (Scheme 3).
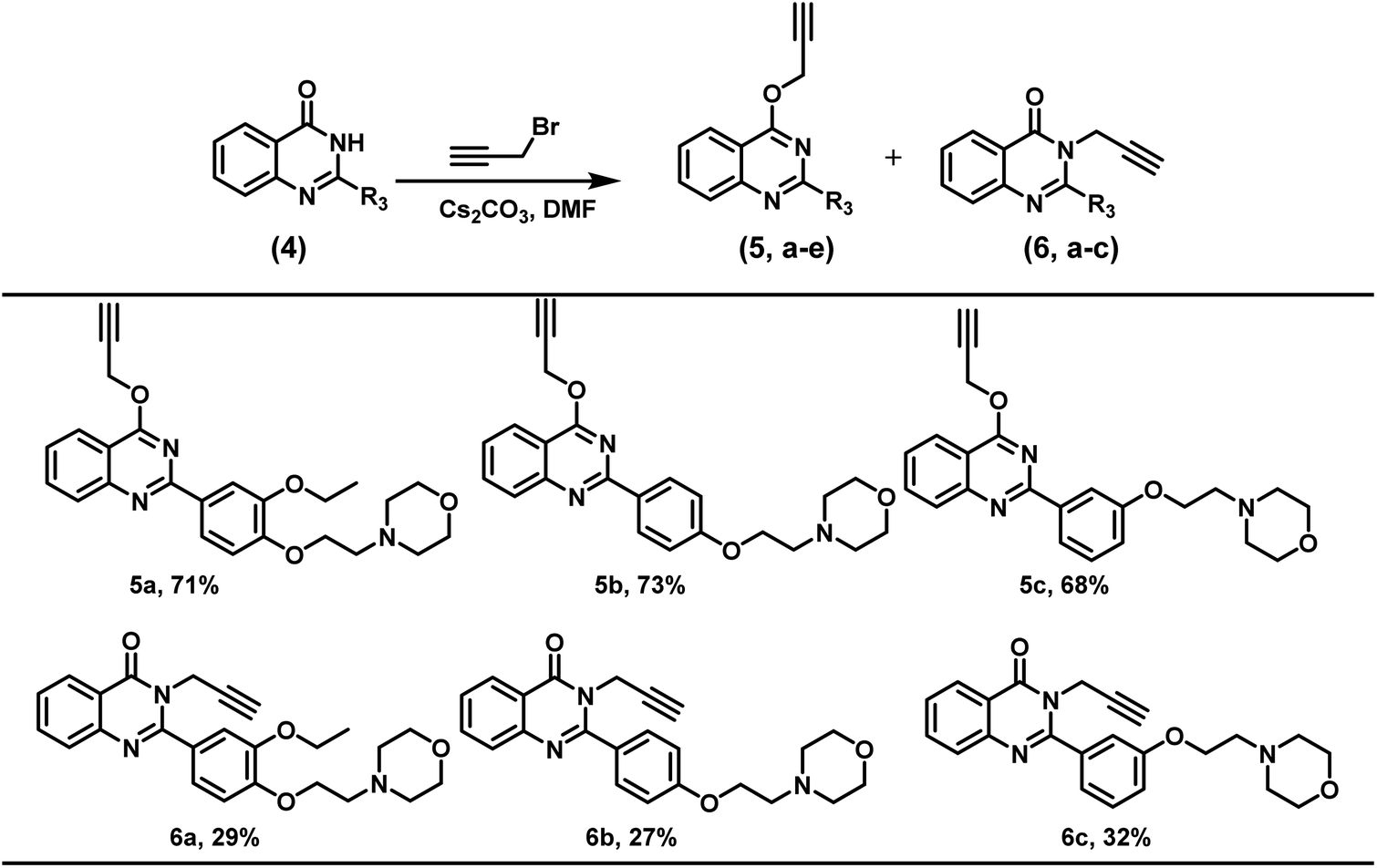 | ||
| Scheme 3 Synthesis of N & O functionalized 2-phenylquinazolines.a Unless mentioned, 2-phenylquinazolin-4(3H)-one derivatives (0.16 mmol), Cs2CO3 (0.16 mmol), and DMF (5 mL); addition of organic halide (0.19 mmol) done at 0 °C and stirring at room temperature for 8 h. | ||
4. Conclusion
Pyrimidine is a versatile scaffold and present in number of bioactive compounds. The pyrimidine scaffold is also used as an important intermediate in a number of chemical transformations. In the drug discovery process a number of pyrimidine derivatives are being synthesized and screened against various diseases. In this regard, the currently developed protocol provides easy access for the predominant O-alkylation of pyrimidinones. The reaction conditions were mild and regioselectivity was achieved under milder conditions without use of any catalyst or coupling agents. The protocol was found to be compatible with a range of substituted pyrimidinones and alkyl halides. In the DFT studies, it was found that Cs+ ion play crucial role in the regioselective O-functionalization of the pyrimidinones.Conflicts of interest
The authors declare no conflict of interest, financial or otherwise.Acknowledgements
VK is thankful to the Council of Scientific and Industrial Research, New Delhi for the financial grant no. 02/(0354)/19/EMRII. Vijay Kumar and NK are thankful to University Grant Commission for providing senior research fellowship. ARD is thankful to the DST for fellowship. PPS is thankful to CSIR for providing NET-JRF fellowship.References
- M. Löffler, L. D. Fairbanks, E. Zameitat, A. M. Marinaki and H. A. Simmonds, Trends Mol. Med., 2005, 11, 430–437 CrossRef PubMed.
- S. Kumar and B. Narasimhan, Chem. Cent. J., 2018, 12, 38 CrossRef PubMed.
- I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1–50 CrossRef CAS PubMed.
- M. Sahu and N. Siddiqui, Int. J. Pharm. Pharmaceut. Sci., 2016, 8, 8–21 CAS.
- M. L. Gatton, L. B. Martin and Q. Cheng, Antimicrob. Agents Chemother., 2004, 48, 2116–2123 CrossRef CAS PubMed.
- G. H. Hitchings, G. B. Elion, H. Vander Werff and E. A. Falco, J. Biol. Chem., 1948, 174, 765–766 CrossRef CAS PubMed.
- S. Ichikawa, Yakugaku Zasshi, 2008, 128, 1403–1430 CrossRef CAS PubMed.
- G. Joshi, H. Nayyar, J. M. Alex, G. S. Vishwakarma, S. Mittal and R. Kumar, Curr. Top. Med. Chem., 2016, 16, 3175–3210 CrossRef CAS PubMed.
- S. Prachayasittikul, R. Pingaew, A. Worachartcheewan, N. Sinthupoom, V. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Mini-Rev. Med. Chem., 2017, 17, 869–901 CAS.
- B. Kumar, A. R. Dwivedi, B. Sarkar, S. K. Gupta, S. Krishnamurthy, A. K. Mantha, J. Parkash and V. Kumar, ACS Chem. Neurosci., 2019, 10, 252–265 CrossRef CAS PubMed.
- B. Kumar, V. Kumar, V. Prashar, S. Saini, A. R. Dwivedi, B. Bajaj, D. Mehta, J. Parkash and V. Kumar, Eur. J. Med. Chem., 2019, 177, 221–234 CrossRef CAS PubMed.
- B. Feng, Y. Li, H. Li, X. Zhang, H. Xie, H. Cao, L. Yu and Q. Xu, J. Org. Chem., 2018, 83, 6769–6775 CrossRef CAS PubMed.
- C. Xu, C. Cheng, H. Liu and B. Liu, Lett. Org. Chem., 2011, 8, 545–548 CrossRef CAS.
- M. Gacek and K. Undheim, Acta Chem. Scand., 1981, 35B, 69–71 CrossRef.
- N. Zanatta, D. Faoro, L. da S. Fernandes, P. B. Brondani, D. C. Flores, A. F. C. Flores, H. G. Bonacorso and M. A. P. Martins, Eur. J. Org. Chem., 2008, 5832–5838 CrossRef CAS.
- A.-E. E. Mansouri, M. Zahouily and H. B. Lazrek, Synth. Commun., 2019, 49, 1802–1812 CrossRef CAS.
- B. Laxminarayana and L. M. Kundu, Synth. Commun., 2015, 45, 1342–1353 CrossRef CAS.
- G. C. Hopkins, J. P. Jonak, H. Tieckelmann and H. J. Minnemeyer, J. Org. Chem., 1966, 31, 3969–3973 CrossRef CAS.
- A. Meščić, D. Glavač, A. Osmanović, D. Završnik, M. Cetina, D. Makuc, J. Plavec, S. M. Ametamey and S. Raić-Malić, J. Mol. Struct., 2013, 1039, 160–166 CrossRef.
- B. S. Møller, M. L. Falck-Pedersen, T. Benneche, K. Undheim, F. Piedade, M. M. E. Kady and S. B. Christensen, Acta Chem. Scand., 1992, 46, 1219–1222 CrossRef.
- M. Mittersteiner, G. S. Pereira, L. A. Wessjohann, H. G. Bonacorso, M. A. P. Martins and N. Zanatta, ACS Omega, 2022, 7(22), 18930–18939 CrossRef CAS PubMed.
- M. B. Madhusudana Reddy and M. A. Pasha, Synth. Commun., 2011, 41, 1875–1880 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, M. J. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- E. Goldstein, B. Beno and K. N. Houk, J. Am. Chem. Soc., 1996, 118, 6036–6043 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- N. K. Nandi, R. Bhatia, S. Saini, R. Rawat, S. Sharma, K. Raj, N. Rangra and B. Kumar, J. Mol. Struct., 2023, 1276, 134671 CrossRef CAS.
- V. Kumar, B. Kumar, A. Ranjan Dwivedi, D. Mehta, N. Kumar, B. Bajaj, T. Arora, V. Prashar, J. Parkash and V. Kumar, ChemistrySelect, 2020, 5, 8021–8032 CrossRef CAS.
- B. Kumar, A. R. Dwivedi, T. Arora, K. Raj, V. Prashar, V. Kumar, S. Singh, J. Prakash and V. Kumar, ACS Chem. Neurosci., 2022, 13, 2122–2139 CrossRef CAS PubMed.
- A. R. Dwivedi, S. S. Rawat, V. Kumar, N. Kumar, V. Kumar, R. P. Yadav, S. Barnwal, A. Prasad and V. Kumar, Curr. Cancer Drug Targets, 2022, 23(4), 278–292 Search PubMed.
- A. R. Dwivedi, S. S. Rawat, V. Kumar, N. Kumar, P. Anand, R. P. Yadav, S. Baranwal, A. Prasad and V. Kumar, Bioorg. Med. Chem., 2022, 72, 116976 CrossRef CAS PubMed.
- A. R. Dwivedi, V. Kumar, R. P. Yadav, N. Kumar, K. Jangid, P. Anand, D. K. Sharma, S. Barnawal and V. Kumar, J. Mol. Struct., 2022, 1267, 133592 CrossRef CAS.
- A. R. Dwivedi, V. Kumar, V. Prashar, A. Verma, N. Kumar, J. Parkash and V. Kumar, RSC Med. Chem., 2022, 13, 599–609 RSC.
- Z.-K. Wan, S. Wacharasindhu, C. G. Levins, M. Lin, K. Tabei and T. S. Mansour, J. Org. Chem., 2007, 72, 10194–10210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2262808. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00773a |
| This journal is © The Royal Society of Chemistry 2023 |