
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of polysubstituted imidazoles using AC-SO3H/[Urea]7[ZnCl2]2 as an efficient catalyst system: a novel method, and α-glucosidase inhibitor activity†
Hai Truong Nguyen ab,
Vy Anh Truongab and
Phuong Hoang Tran*ab
ab,
Vy Anh Truongab and
Phuong Hoang Tran*ab
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Vietnam National University, Ho Chi Minh City 700000, Viet Nam. E-mail: thphuong@hcmus.edu.vn
bVietnam National University, Ho Chi Minh City 7000000, Viet Nam
First published on 20th April 2023
Abstract
Deep eutectic solvents (DESs) act as both an organic solvent and a useful catalyst for organic synthesis reactions, especially the synthesis of heterocyclic compounds containing the element nitrogen. DESs exhibit many important properties namely large liquid fields, biodegradability, outstanding thermal stability, and moderate vapor pressure. Amorphous carbon-bearing sulfonic acid groups (AC-SO3H) are one of the new-generation solid acids showing strong acid activity. Based on the simultaneous presence of acidic functional groups such as carboxylic acid, phenolic, and sulfonic acid groups, they exhibit many important activities namely strong Brønsted acid, high surface area, high stability, reusability, and recyclability. In this study, AC-SO3H was made from rice husk via the carbonization and sulfonation processes, and the surface properties and structure were examined using contemporary methods such as FT-IR, P-XRD, TGA, SEM, and EDS. And, [Urea]7[ZnCl2]2 was synthesized from urea and ZnCl2 with a mole ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2; the structure is defined using FT-IR and TGA. By combining AC-SO3H and [Urea]7[ZnCl2]2 we aim to form an effective catalyst/solvent system for the preparation of polysubstituted imidazole derivatives through the multi-component cyclization reaction from nitrobenzenes, benzil, aldehydes, and ammonium acetate. The major products are obtained with high isolation yields above 60%. To assess the catalyst system's activity, the recovery and reusability of the AC-SO3H/[Urea]7[ZnCl2]2 system were examined with hardly any performance modification. In an effort to create potential enzyme α-glucosidase inhibitors, several novel polysubstituted imidazoles were created. Five of these compounds showed good enzyme α-glucosidase inhibitor activity. The most effective substances were IMI-13, IMI-15, and IMI-20, with IC50 values that were greater than the acarbose at 16.5, 15.8, and 11.6 μM, respectively – the acarbose (IC50, 214.5 μM) as the positive control.
:2; the structure is defined using FT-IR and TGA. By combining AC-SO3H and [Urea]7[ZnCl2]2 we aim to form an effective catalyst/solvent system for the preparation of polysubstituted imidazole derivatives through the multi-component cyclization reaction from nitrobenzenes, benzil, aldehydes, and ammonium acetate. The major products are obtained with high isolation yields above 60%. To assess the catalyst system's activity, the recovery and reusability of the AC-SO3H/[Urea]7[ZnCl2]2 system were examined with hardly any performance modification. In an effort to create potential enzyme α-glucosidase inhibitors, several novel polysubstituted imidazoles were created. Five of these compounds showed good enzyme α-glucosidase inhibitor activity. The most effective substances were IMI-13, IMI-15, and IMI-20, with IC50 values that were greater than the acarbose at 16.5, 15.8, and 11.6 μM, respectively – the acarbose (IC50, 214.5 μM) as the positive control.
Introduction
Imidazoles, five-member rings containing two nitrogen atoms, play a vital role in organic synthesis. Polysubstituted imidazoles were majorly used as a host for bioactive compounds for the fabrication of pharmaceutical drugs such as antihypertensive,1 platelet aggregation inhibitor,2 angiotensins II receptor blocker (ARB),3 potential angiotensin II receptor blockers to inhibit the COVID-19 main protease,4 anti-inflammatories,5 anti-Alzheimer's disease,6 and anticancer7 (Scheme 1). Angyal et al. reported the method for synthesis of polysubstituted imidazoles from 2H-azirines and nitrones through 1,3-dipolar cycloaddition using trifluoroacetic acid (TFA) in anhydrous MeCN.8 X. Guo et al. studied the one-pot three-component reaction for the synthesis of 2-aminoimidazoles via ring-opening of α-nitro epoxides from nitro-epoxide, aniline, and cyanamide in the presence of n-propanol.9 B. Hu et al. applied the domino reaction to synthesize polysubstituted imidazoles between 2-azido acrylates and nitrones in 1,2-dichloroethane and without a catalyst.10 Zang et al. introduced the method for synthesis of 4-aminoimidazoles from 1,2,4-oxadiazoles and ynamides through selective [3 + 2] with gold as a catalyst in the presence of PhCF3.11 J. O. Strelnikova et al. reported the procedure for the synthesis of 5-sulfonamidoimidazoles under 1,2,4-oxadiazole and N-sulfonyl-1,2,3-triazole derivatives transannulation with rhodium(II) carboxylates as the catalyst and 1,2-dichloroethane as the solvent.12 H. Mehrabi et al. have synthesized polysubstituted imidazoles from arylamidines, malononitrile, and arylglyoxals or aryl aldehydes using Et3N in CH3CN.13 The most common method used to synthesize polysubstituted imidazoles was the reaction between aniline, benzil, aldehyde, and ammonium acetate using various catalysts such as Fe3O4–PEG–Cu,14 K5CoW12O40·3H2O,15 PEG-400,16 DABCO,17 InCl3·3H2O,18 BF3/SiO2,19 silica-bonded propylpiperazine-N-sulfamic acid (SBPPSA),20 TiCl4–SiO2,21 Zr(acac)2,22 polyphosphoric acid impregnated on silica gel (PPA–SiO2),23 [Hbim]BF4,24 Yb(OTF)3,25 L-proline,26 MCM-41-SO3H,27 Fe3O4/SO3H@zeolite-Y,28 nano-Fe3O4,29 Brønsted acidic deep eutectic solvent,30 organocatalyst,31 dendrimer-PWAn nanoparticles,32 and another modern method.32–34 However, the use of acid Lewis as the catalysts and organic solvents used were an environmental impact. Thus, it was necessary to create a new and efficient method of synthesizing polysubstituted imidazoles, one of those methods was the application of new catalysts and/or solvents to research.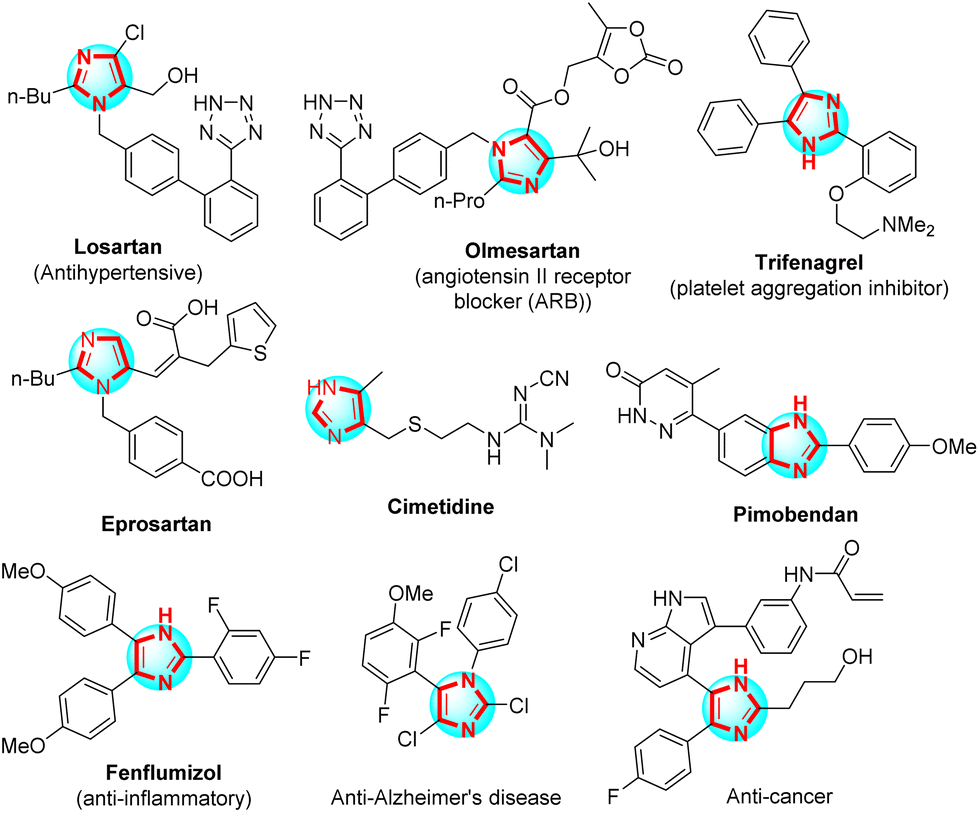 | ||
| Scheme 1 Several important bioactive compounds contain the imidazole framework. | ||
Amorphous carbon functionalized sulfonic acid was considered a new-generation solid catalyst with many characteristic features, namely strong Brønsted acid, high surface area, high stability, reusability, and recyclability.35–37 Sulfopolycyclic aromatic compounds can be incompletely carbonized in concentrated H2SO4, or natural organic materials can be sulfonated after being incompletely carbonized to produce carbon-based solid acids, which were sulfonic acid –SO3H groups, carboxylic acid –COOH groups, and phenolic –OH groups present in the carbon material. Three functional groups were unique from one another. The typical solid acids were examined; nevertheless, each only possesses a single functional group. It's possible that this was the reason why the carbon material has so powerful catalytic activity.38–40
Deep eutectic solvents (DESs) have received a great deal of interest in the last ten years in both starting materials and catalysts because of their distinctive qualities, including their large liquid range, biodegradability, outstanding thermal stability, and moderate vapor pressure.41,42 Since the first one was made by Abbott's group in the year 2001, a number of other DESs have indeed been discovered as solvents and catalysts for organic transformations such as the creation of carbon–carbon43–45 and carbon–heteroatom46 bonding, as well as multicomponent reactions. DESs have been employed in a wide range of applications, including the extraction of cellulose,47 chitin,48 polysaccharides,49 lipids,50 and other polarity chemicals,51 as well as the electrolyte in dye-sensitized solar cells,52–54 lithium-ion batteries,55–57 and electrochemical double-layer capacitors.58 As an environmentally friendly catalyst for organic transformation, deep eutectic solvent [Urea]7[ZnCl2]2 made of urea and zinc chloride has recently come to light (Scheme 2).
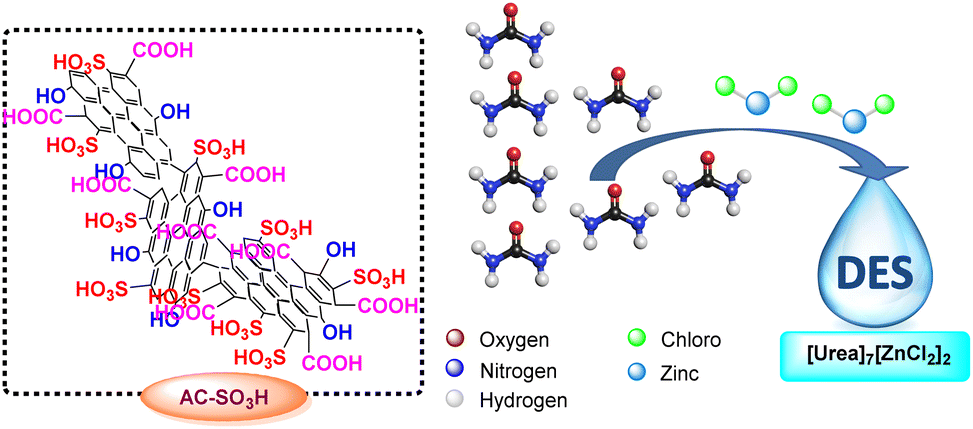 | ||
| Scheme 2 Structure of AC-SO3H and [Urea]7[ZnCl2]2. | ||
Herein, we developed a new pathway to synthesize polysubstituted imidazoles from nitro compounds via the four-component condensation reaction. By using AC-SO3H/[Urea]7[ZnCl2]2 system as the catalyst or solvent for the reaction between nitro compound, benzil, aldehyde, and ammonium acetate. The α-glucosidase inhibitor activity of the major products was tested.
Results and discussion
Chemistry
Amorphous carbon-bearing sulfonic acid groups (AC-SO3H) were introduced in the previous study, and carbonization and sulfonation were used to create them from the rice husk.59,60 Modern techniques, including FT-IR, P-XRD, TGA, SEM, and EDS were used to analyze the surface characteristics and structure of AC-SO3H materials (ESI†). Fig. S1† depicts the ATR-FTIR spectra of AC-SO3H and AC. The results indicated multiple distinguishing signals at 3212 cm−1 and 1699 cm−1, matching to O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, respectively, which were characterized as phenolic, carboxylic acid, and sulfonic acid group indicators. The bending vibrations peak at 1606 cm−1 represented CC signals within the polyaromatic structure. The AC-SO3H line exhibits significant characteristic bands at approximately 1073 cm−1, which correspond to the SO stretching vibrations of the sulfonic acid groups. This study's outcomes were compared to those of previously published research.61 Fig. S2† illustrates representative TGA graphs under N2 atmosphere. The primary mass loss events of GO may be characterized predicated upon their operating temperature: under 500 °C for the displacement of water, 500–600 °C for the removal of organic functional groups, and 600–800 °C for the oxidative decomposition of the carbon network. The results of SEM image analysis of AC and AC-SO3H showed that the material after being sulfonation with concentrated H2SO4 has an agglomerated structure (Fig. S3†). EDX analysis also shows that in the structure of AC and AC-SO3H materials, both contain elements such as carbon, oxygen, silicon, and sulfur with the corresponding element content in AC-SO3H recorded as 56.73%, 35.99%, 6.78%, and 0.5% (Fig. S4†). Fig. S5† shows the diffraction grating of AC-SO3H. The broad peaks within the P-XRD pattern around 2θ = 15–35° can be ascribed to the carbon materials. Also observed has been the wide peak C(002). The moderate and broad C(101) diffraction pattern that was clearly detected within 2θ = 40–50° was due, in part, to the fact that the a axis of the graphene sheets was involved in the phenomenon.62 Fig. S7† shows the Raman spectra of AC and AC-SO3H, which demonstrated the two distinct signals assignable to the D (∼1350 cm−1, A1g D breathing mode) and G bands (∼1580 cm−1, E2g G mode), the presence of polycyclic aromatic carbon sheets in the carbon bulk.63 The element S content in the AC-SO3H catalyst was 10243 ppm (0.320 mmol g−1), which was recorded by Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
O groups, respectively, which were characterized as phenolic, carboxylic acid, and sulfonic acid group indicators. The bending vibrations peak at 1606 cm−1 represented CC signals within the polyaromatic structure. The AC-SO3H line exhibits significant characteristic bands at approximately 1073 cm−1, which correspond to the SO stretching vibrations of the sulfonic acid groups. This study's outcomes were compared to those of previously published research.61 Fig. S2† illustrates representative TGA graphs under N2 atmosphere. The primary mass loss events of GO may be characterized predicated upon their operating temperature: under 500 °C for the displacement of water, 500–600 °C for the removal of organic functional groups, and 600–800 °C for the oxidative decomposition of the carbon network. The results of SEM image analysis of AC and AC-SO3H showed that the material after being sulfonation with concentrated H2SO4 has an agglomerated structure (Fig. S3†). EDX analysis also shows that in the structure of AC and AC-SO3H materials, both contain elements such as carbon, oxygen, silicon, and sulfur with the corresponding element content in AC-SO3H recorded as 56.73%, 35.99%, 6.78%, and 0.5% (Fig. S4†). Fig. S5† shows the diffraction grating of AC-SO3H. The broad peaks within the P-XRD pattern around 2θ = 15–35° can be ascribed to the carbon materials. Also observed has been the wide peak C(002). The moderate and broad C(101) diffraction pattern that was clearly detected within 2θ = 40–50° was due, in part, to the fact that the a axis of the graphene sheets was involved in the phenomenon.62 Fig. S7† shows the Raman spectra of AC and AC-SO3H, which demonstrated the two distinct signals assignable to the D (∼1350 cm−1, A1g D breathing mode) and G bands (∼1580 cm−1, E2g G mode), the presence of polycyclic aromatic carbon sheets in the carbon bulk.63 The element S content in the AC-SO3H catalyst was 10243 ppm (0.320 mmol g−1), which was recorded by Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
[Urea]7[ZnCl2]2-deep eutectic solvent was prepared from urea and zinc chloride with the ratio of the mole was 7:2, which was carried out according to the procedure previously published research.64–66 The structure and purity of [Urea]7[ZnCl2]2 were evaluated by FT-IR and TGA. Fig. S8† displayed the FTIR spectrum of the original urea and the deep eutectic solvent [Urea]7[ZnCl2]2. The [Urea]7[ZnCl2]2 spectrum overlaps with the urea spectrum. The outcome showed that [Urea]7[ZnCl2]2 did not degrade the structure of urea. Particularly, the twin peaks of a broad band from N–H stretching vibrations might be attributed to the urea absorption bands at 3375 cm−1 and 3300 cm−1. The CO carbonyl group's stretching vibration absorption band is located at 1700 cm−1. Other N–H vibrations have a peak absorption band at 1500 cm−1. C–N vibrations are responsible for the highest absorptions at 1250 cm−1 and 1200 cm−1, respectively. The O–H of urea participates in the creation of the hydrogen bond with the anion of zinc chloride, as seen by the shift in the N–H stretching vibration of [Urea]7[ZnCl2]2. In Fig. S9,† a thermal gravimetric analysis (TGA) of [Urea]7[ZnCl2]2 was completed. The [Urea]7[ZnCl2]2 is suited for high-temperature reaction conditions because the majority of weight loss takes place between 200 °C and 400 °C.
The four-component condensation reaction approach contains the reduction of deep eutectic solvent in the presence of AC-SO3H was developed. In screening for reaction conditions, the condensation reaction to form IMI-01 was carried out in two-step, firstly, the mixture of nitrobenzene (1.0 mmol), ammonium chloride (1.5 mmol), and iron powder (3.0 mmol) under H2O/EtOH (v/v = 1:1) was heated. Then, benzil (1.0 mmol), benzaldehyde (1.0 mmol), and ammonium acetate (1.0 mmol) in the presence of AC-SO3H (10 mg), and [Urea]7[ZnCl2]2 (1.0 mmol), which was heated at different time intervals and temperatures. In the first stage, the procedure for conversion of nitrobenzene to form aniline investigated the influence of temperature reaction, and time reaction (Table S1†). In the presence of iron powder and ammonium chloride under deionized water and ethanol (v/v = 1:1), the conversion of nitrobenzene was carried out at room temperature, and 60 °C within a variety of time ranges namely 15, 30, 45, and 60 min. The best result was shown at 60 °C for 60 min with the conversion of nitrobenzene up to 100%. For synthesis to be successful, the reaction media must be chosen carefully.49,67,68 As a simple model substrate, nitrobenzene, benzil, benzaldehyde, and ammonium acetate were used in a four-component reaction with a catalytic amount of AC-SO3H and [Urea]7[ZnCl2]2, which was then investigated to determine the viability of the technique and to optimize the process parameters at various temperatures, times, and with various amounts of catalyst (Table S2†). The effect of the temperature reaction was obtained by heating at 80 °C, 100 °C, 110 °C, and 130 °C; the best results were shown at 110 °C, which yielded product IMI-01 in high yield. The time reaction was examined from 30 min to 90 min to form the major product IMI-01, when the reaction was carried out for 60 min, the major product was recorded as the best yield with over 73%. The yield was reduced by loading the AC-SO3H catalyst from 0 mg to 15 mg in the presence of [Urea]7[ZnCl2]2, and reached the highest by loading the catalyst to 10 mg on the product of the reaction, it was seen that there was an effect. As can be seen from Table 1, the process parameter optimal was established by heating at 110 °C for 60 min using AC-SO3H (10 mg) and [Urea]7[ZnCl2]2 in the mixture of reaction, the solvent role of [Urea]7[ZnCl2]2 was also evaluated by replacing some types of deep eutectic solvents (namely [CholineCl][Urea]2, [CholineCl][Glycerin], [CholineCl][ZnCl2]) and organic solvents (namely dichloromethane, ethanol, 1,4-dioxane). The survey results indicated that the best-isolated yield was achieved when the reaction took place in [Urea]7[ZnCl2]2 as the solvent was 73%. Next, the activity of AC-SO3H was measured and compared to that of other catalysts of a similar kind, such as TsOH, SiO2–H2SO4, and in the case of without any catalyst, that of [Urea]7[ZnCl2]2 as the solvent. Based on the concentration of acid functional groups present in AC-SO3H, which possessed greater catalytic activity compared to that of other catalysts. According to the findings of the research, the AC-SO3H/[Urea]7[ZnCl2]2 system was the most essential method for the synthesis of IMI-01. However, the reaction was carried out in the absence of a catalyst and a solvent, which was a challenging reaction, and the desired product yield was less than 10%. In particular, the efficiency of the AC-SO3H/[Urea]7[ZnCl2]2 system was carried out on a large scale, 10 mmol of the reactants were carried out under optimized conditions, and the survey results showed that the yield of IMI-01-product about 70% (Table 1, entry 4). Compared with other studies, the starting materials for synthesis polysubstituted imidazole frames were used as aniline, benzil, ammonium acetate, and benzaldehyde and showed similar yields.33,69–71 This method improved the yield of the main product while producing pure polysubstituted imidazole frames in a short amount of time. It was also clean and ecologically friendly.
|
|||
|---|---|---|---|
| Entry | Catalysts | Solvents | Yieldsb (%) |
| a Condition reaction: nitrobenzene (1.0 mmol, 123 mg), ammonium chloride (1.5 mmol, 79.5 mg), and iron powder (3.0 mmol, 168 mg) under H2O/EtOH (v/v = 1:1) at 60 °C for 60 min. Then, benzil (1.0 mmol, 210 mg), benzaldehyde (1.0 mmol, 106 mg), ammonium acetate (1.0 mmol, 77 mg), catalysts (10 mg), and solvents (1.0 mmol) at 110 °C for 60 min.b Yields were recorded by isolated yield in hot ethanol (10–15 mL).c mp = 10 mmol.d mp = 2.0 mL. |
|||
| 1 | AC-SO3H | [CholineCl][Urea]2 | 40 |
| 2 | AC-SO3H | [CholineCl][Glycerin] | 44 |
| 3 | AC-SO3H | [CholineCl][ZnCl2] | 43 |
| 4 | AC-SO3H | [Urea]7[ZnCl2]2 | 73 (70)c |
| 5 | AC-SO3H | Dichloromethaned | 29 |
| 6 | AC-SO3H | Ethanold | 38 |
| 7 | AC-SO3H | 1,4-Dioxaned | 35 |
| 8 | AC-SO3H | None | 60 |
| 9 | None | [Urea]7[ZnCl2]2 | 43 |
| 10 | TsOH | [Urea]7[ZnCl2]2 | 48 |
| 11 | SiO2–H2SO4 | [Urea]7[ZnCl2]2 | 57 |
| 12 | None | None | <10 |
The impact of the AC-SO3H/[Urea]7[ZnCl2]2 system was intended to investigate the applicability of the current methodology for the preparation of polysubstituted imidazole frames under the aforementioned optimal conditions. The results were outlined in Scheme 3. Notably, under AC-SO3H (10 mg) and [Urea]7[ZnCl2]2 (1.0 mmol) system conditions, the best results were obtained at a temperature of 110 °C using different aldehydes (1.0 mmol), benzil (1.0 mmol), various nitro compound (1.0 mmol), ammonium acetate (1.0 mmol), for 60 min. The process of converting nitrobenzene to aniline and further applying it to the synthesis of polysubstituted imidazoles with benzaldehyde derivatives carrying different substituents. For benzaldehyde derivatives carrying a hydroxy substituent (–OH) at the ortho position, the 2-hydroxybenzaldehyde compound contains an intramolecular hydrogen bond, which reduces the activity of the aldehyde group, resulting in a reduced yield of the IMI-02 only 61%. The desired product formation efficiency gradually increased when the –OH group was changed at the meta and para positions, 70% and 79%, respectively. Under benign parameters, a variety of aromatic aldehydes with either electron-donating or electron-withdrawing groups were suitable for the four-component reaction catalyzed by the AC-SO3H/[Urea]7[ZnCl2]2 system. The current procedure proved very effective for producing polysubstituted imidazoles in high yields (65–86%) without any byproducts. The reaction was carried out for 45 minutes with cyclohexanecarbaldehyde and furfural, yielding a result of 67% and 70%, respectively. 4-Nitrophenol was used as starting material for the polysubstituted imidazoles synthesis. The transformation of 4-nitrophenol was carried out under similar conditions to nitrobenzene. After that, the influence of aldehyde derivatives continued to be evaluated, the aromatic aldehydes were preferred to carry the electron-donating or electron-withdrawing groups at the para position groups on the benzene-ring compared to the aldehyde group, and the product formation efficiency was over 70%.
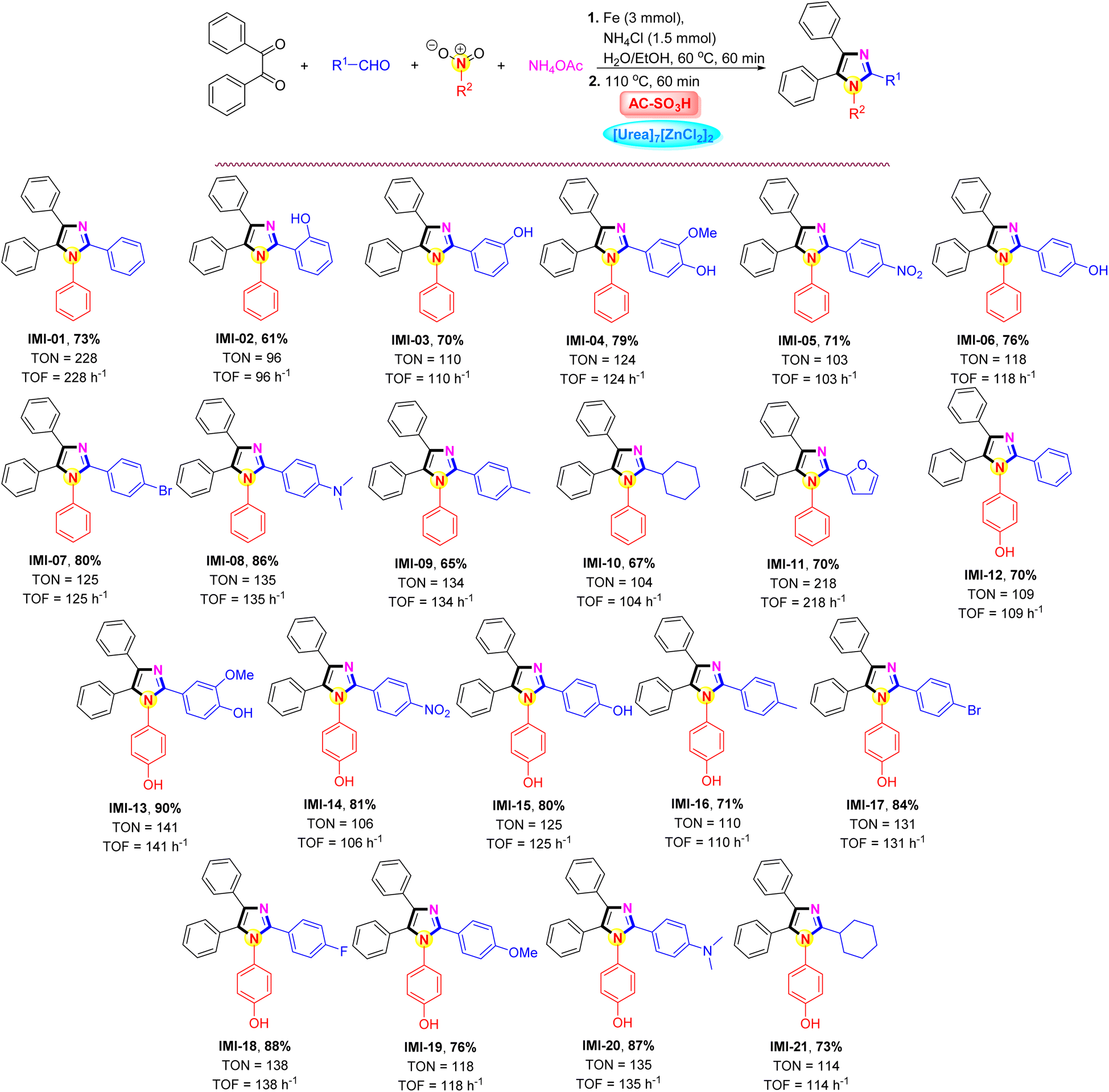 | ||
| Scheme 3 AC-SO3H-catalyzed for the preparation of polysubstituted imidazoles. Reaction condition: nitrobenzene (1.0 mmol, 123 mg), ammonium chloride (1.5 mmol, 79.5 mg), and iron powder (3.0 mmol, 168 mg) under H2O/EtOH (v/v = 1:1) at 60 °C for 60 min. Then, benzil (1.0 mmol, 210 mg), aldehyde (1.0 mmol, 106 mg), ammonium acetate (1.0 mmol, 77 mg), AC-SO3H (10 mg), and [Urea]7[ZnCl2]2 (1.0 mmol) at 110 °C for 60 min. Yields were recorded by isolated yields in hot ethanol (20 mL); TON = (moles of products)/(moles of catalyst); TOF = TON/hour (h−1). | ||
In Scheme 4, the plausible mechanism was revealed. In the first stage, the reduction of nitrobenzene to aniline was accomplished via diphenyldiazene (3) and diphenylhydrazine (4) in the presence of Fe, NH4Cl, and H2O/EtOH.72,73 Next, the mechanisms for the four-component condensation reaction were in complete agreement with previous studies.74–76 Regarding the reaction's mechanism, we proposed that the AC-SO3H catalyst first protonates the aromatic aldehyde's carbonyl group before it condenses with ammonium acetate and a substituted aromatic amine to yield the adduct intermediate (6). Intermediate (6) nucleophilically reacts with protonated benzil to produce an intermediate (7). Polysubstituted imidazoles were produced via ring closure followed by dehydration using AC-SO3H as the catalyst. 1H-NMR, 13C-NMR, and LC-MS all supported the product structure.
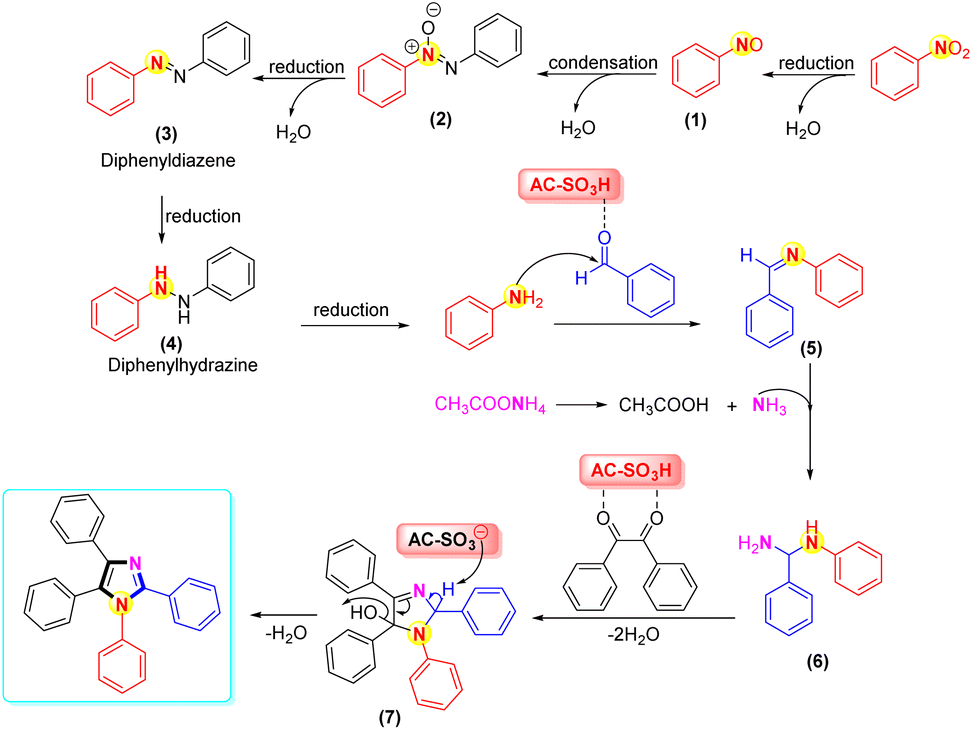 | ||
| Scheme 4 Proposed mechanism for synthesis of polysubstituted imidazole frames. | ||
The ability to recover and reuse the AC-SO3H/[Urea]7[ZnCl2]2 system was investigated to determine the level of performance of the catalyst system. Based on the optimized conditions, the catalyst after use was washed several times with ethyl acetate to remove the substrate from the catalyst system. This AC-SO3H/[Urea]7[ZnCl2]2 system continues to be used as a catalyst for the four-component cyclization reaction to form IMI-01. The performance was recorded after four times recovers and reused (Fig. 1). From that result, it can be seen that the catalytic efficiency of system AC-SO3H/[Urea]7[ZnCl2]2 was not significantly reduced. The structure and properties of the catalyst were also recorded by FT-IR, EDX, and SEM. Based on SEM images, the recovered catalyst has the agglomeration form as the original AC-SO3H catalyst (Fig. S3c†). From the results of the EDX analysis, the amount of silicon in the catalyst was significantly reduced (Fig. S4c†). The functional groups on the surface of the recovered catalyst were still maintained through the FT-IR spectrum (Fig. S6†).
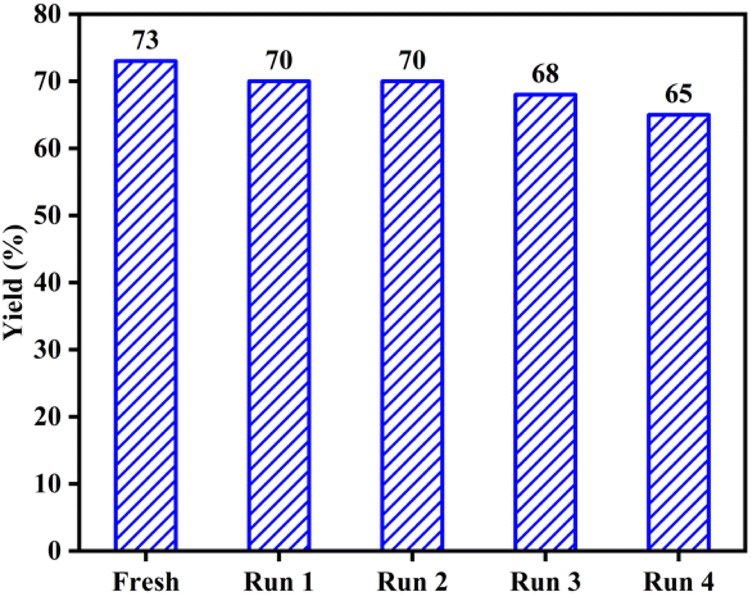 | ||
| Fig. 1 Recovery and reusing of AC-SO3H/[Urea]7[ZnCl2]2 system in the preparation of polysubstituted imidazole frames. | ||
α-Glucosidase inhibitors
The ability of each produced substance to inhibit α-glucosidase was evaluated. Acarbose, which is now employed therapeutically in combination with either diet or antidiabetic drugs to maintain the blood glucose level of patients, served as the positive control in this investigation. Table 2 displays the IC50 values for various substances. The majority of the tested drugs exhibited α-glucosidase inhibitory activity, and some even had an encouraging attribute. The experiment was run at a range of concentrations between 10 and 50 μM. IMI-01, IMI-11, IMI-13, IMI-15, and IMI-20 were selected as survey compounds because they each exhibited significant α-glucosidase inhibition effect in acarbose (IC50, 214.5 μM), which was being used in therapeutically in combination with either diet or anti-diabetic drugs to maintain blood glucose levels in patients, in a concentration-dependent manner and with more robust inhibitory effect.77 The most active substance was found to be compound IMI-20, which was 18.5 times stronger than acarbose with an IC50 of 11.6 μM (the acarbose, which had an IC50 value of 214.5 μM). The enzyme α-glucosidase was likewise very effectively inhibited by compounds IMI-13 and IMI-15, with IC50 values of 16.5 and 15.8 μM, respectively. IMI-01 and IMI-11 compounds made from nitrobenzene gave low α-glucosidase inhibitory activity, IC50 values of 119.5 and 182.9 μM, respectively (Table 2, entries 1 and 2). However, for products made up of 4-nitrophenol, namely IMI-13, IMI-15, and IMI-20, the α-glucosidase inhibitory activity was much better than that of IMI-01 and IMI-11 (Table 2, entries 3–5).| Entry | Compound | Inhibition (I%) | IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| 10 (μM) | 20 (μM) | 30 (μM) | 40 (μM) | 50 (μM) | |||
| a (*) inhibition percentage greater than 100% (I > 100%). (—) inhibition percentage less than 1% (I < 1%).b Positive control. | |||||||
| 1 | IMI-01 | 87.1 ± 1.0 | 44.4 ± 1.2 | 28.3 ± 1.6 | 15.81 ± 0.99 | — | 119.5 |
| 2 | IMI-11 | 70.8 ± 1.2 | 24.2 ± 2.3 | 8.2 ± 2.3 | — | — | 182.9 |
| 3 | IMI-13 | * | 99.28 ± 0.51 | 95.2 ± 1.0 | 81.9 ± 1.3 | 26.0 ± 1.2 | 16.5 |
| 4 | IMI-15 | * | 97.91 ± 0.98 | 91.57 ± 0.85 | 84.55 ± 0.87 | 28.4 ± 2.0 | 15.8 |
| 5 | IMI-20 | * | * | 99.42 ± 0.49 | 87.9 ± 1.0 | 44.3 ± 2.1 | 11.9 |
| 6 | Acarboseb | 214.15 | |||||
Experimental
General procedure for preparation of amorphous carbon-bearing sulfonic acid groups (AC-SO3H)
Rice husks were partially carbonized at 400 °C for 10 hours in a N2 environment. The mixture was then heated for 10 hours at 150 °C while being filled with N2, after which strong sulfuric acid was added. The black substance was washed many times with hot deionized water at 80 °C until sulfate anions were no longer detectable in the filtrate. A two-hour drying process at 100 °C was then performed on the sulfonated amorphous carbon catalyst. The characterization and structural integrity of AC-SO3H were examined by FT-IR, XRD, TGA, EDS, and SEM.Typical processes for the preparation of [Urea]7[ZnCl2]2
Urea (35.0 mmol, 2.100 g) and zinc chloride (10.0 mmol, 1.360 g) were heated at 100 °C until a homogenous mixture was produced to form [Urea]2[ZnCl2]2. By using FT-IR, and TGA to confirm the characterization of [Urea]2[ZnCl2]2.General procedure for the one-pot multi-component synthesis of polysubstituted imidazoles
A 10 mL round-bottom flask fitted with a condenser was filled with nitrobenzene (1.0 mmol, 123 mg), ammonium chloride (1.5 mmol, 80.3 mg), and iron powder (3.0 mmol, 168 mg), and the mixture was heated to 60 °C using a standard oil bath for 60 minutes. Then, benzil (1.0 mmol, 210 mg), aldehyde (1.0 mmol), ammonium acetate (1.0 mmol, 77 mg), AC-SO3H (10 mg), and [Urea]7[ZnCl2]2 (1.0 mmol, 0.692 g) was continuously heated to 110 °C for 60 min. After cooling, the reaction mixture was dissolved in ethyl acetate (2.0 mL × 5). The solvent was recovered from the ethyl acetate layer, which was then recrystallized with hot ethanol (15 mL) to produce the main product in its pure form. All of the products were verified by using 1H-NMR, 13C-NMR, and LC-MS.General procedure gram-scale for the synthesis of IMI-01
Nitrobenzene (10.0 mmol, 1.23 g), ammonium chloride (15.0 mmol, 0.803 g), and iron powder (30.0 mmol, 1.68 g) were heated to 60 °C for 60 minutes. Then, benzil (10.0 mmol, 2.10 g), benzaldehyde (10.0 mmol, 1.06 g), ammonium acetate (10.0 mmol, 0.77 g), AC-SO3H (100 mg), and [Urea]7[ZnCl2]2 (10.0 mmol, 6.92 g) was continuously heated to 110 °C for 60 min. After cooling, the reaction mixture was dissolved in ethyl acetate (10.0 mL × 5). The solvent was recovered from the ethyl acetate layer, which was then recrystallized with hot ethanol (15 mL) to form IMI-01 with a high yield of 70% (2.605 g).General procedure for recycling AC-SO3H
After the reaction was completed, the mixture was washed with ethyl acetate (2.0 mL × 5) to obtain the product, the catalyst system continued to be washed with ethyl acetate (2.0 mL × 5) followed by TLC. Then, the catalyst system was dried over 80 °C prolonged 2 h, aniline (from nitrobenzene), benzil, benzaldehyde, and ammonium acetate was added to the catalyst system, and the reaction was carried out under optimized conditions.General procedure for α-glucosidase inhibitory
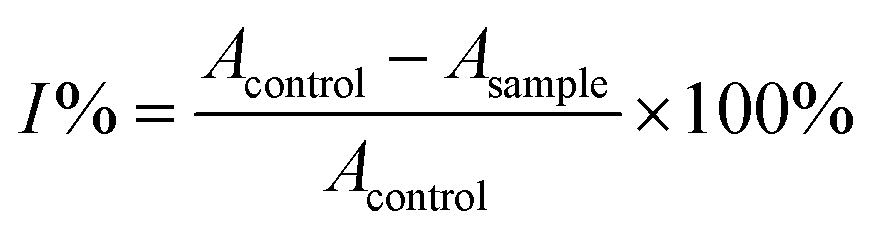
The revised strategy of Kim et al. was used to determine the inhibitory activity of α-glucosidase.78 In 0.01 M phosphate buffer (pH = 7.0), 25 μL of 3 mM p-nitrophenyl-α-D-glucopyranoside and 25 μL of 0.2 U mL−1 α-glucosidase were added to start the reaction. Each reaction took place for 30 minutes at 37 °C before being halted by the addition of 0.1 M Na2CO3 (375 μL). The quantification of enzyme activity involved measuring absorbance at 401 nm. The amount of p-nitrophenol (1.0 M) that was released by the enzyme per minute was used one of α-glucosidase activity must be defined (Scheme 5). The quantity of an inhibitor of α-glucosidase that the concentration at which α-glucosidase activity was reduced by 50% was referred to as the IC50 value. As a positive control, acarbose, a well-known inhibitor of α-glucosidase, was utilized.In which: Acontrol: value of the solution's optical density without the survey sample. Asample: value of the survey sample's solution's optical density.
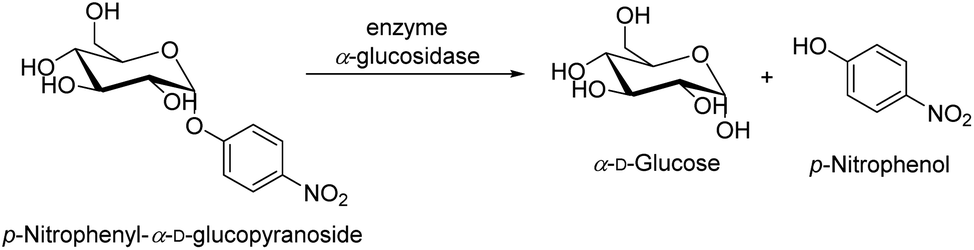 | ||
| Scheme 5 The enzymatic activity of the enzyme α-glucosidase in the hydrolysis of p-nitrophenyl-α-D-glucopyranoside. | ||
Conclusions
In conclusion, we have successfully synthesized AC-SO3H material from rice husks through carbonization at 400 °C for 10 hours under N2 conditions. This was followed by sulfonation in the same medium with concentrated sulfonic acid at 150 °C a prolonged 10 h. The chemical properties of AC-SO3H were evaluated by FT-IR, P-XRD, TGA, SEM, and EDS. From that, it was concluded that AC-SO3H material has an agglomerated structure with the content of elements carbon (C = 56.73%), oxygen (O = 35.99%), silicon (Si = 6.78%), and sulfur (S = 0.50%) from EDX analysis. In addition, the combination of [Urea]7[ZnCl2]2 with AC-SO3H created an effective catalyst system for the preparation of polysubstituted imidazoles from nitrobenzenes, benzil, aldehydes, and ammonium acetate at 110 °C for 60 minutes. The highlight of this study is the application of the reduction of the nitro group of the nitro compound to aniline, as a starting material for the synthesis of polysubstituted imidazoles, with the yield over 60% with the TON value is recorded 96–141, and TOF is recorded 96–141 h−1. This catalyst system is also evaluated for efficiency through gram-scale reaction and recovery and reuse. In addition, the formed product was evaluated for its ability to inhibit enzyme α-glucosidase with IC50 values obtained from 11.9–182.9 μM.Author contributions
Hai Truong Nguyen: investigation, methodology, resources, formal analysis, validation, data curation, writing-original drafts. Vy Anh Truong: investigation, methodology, resources, writing-original drafts. Phuong Hoang Tran: methodology, resources, formal analysis, validation, data curation, writing-review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number 562-2022-18-03.Notes and references
- L. S. Ikeda, S. C. Harm, K. E. Arcuri, A. I. Goldberg and C. S. Sweet, Blood Pressure, 1997, 6, 35–43 CrossRef CAS PubMed.
- S. L. Abrahams, R. J. Hazen, A. G. Batson and A. P. Phillips, J. Pharmacol. Exp. Ther., 1989, 249, 359 CAS.
- J. Agata, N. Ura, H. Yoshida, Y. Shinshi, H. Sasaki, M. Hyakkoku, S. Taniguchi and K. Shimamoto, Hypertens. Res., 2006, 29, 865–874 CrossRef CAS PubMed.
- R. Alnajjar, A. Mostafa, A. Kandeil and A. A. Al-Karmalawy, Heliyon, 2020, 6, e05641 CrossRef CAS PubMed.
- J. Aono, H. Nabata and K. Sakai, Jpn. J. Pharmacol., 1986, 42, 575–578 CrossRef CAS PubMed.
- A.-S. Cornec, L. Monti, J. Kovalevich, V. Makani, M. J. James, K. G. Vijayendran, K. Oukoloff, Y. Yao, V. M. Y. Lee, J. Q. Trojanowski, A. B. Smith, K. R. Brunden and C. Ballatore, J. Med. Chem., 2017, 60, 5120–5145 CrossRef CAS PubMed.
- I. Ali, M. N. Lone and H. Y. Aboul-Enein, MedChemComm, 2017, 8, 1742–1773 RSC.
- A. Angyal, A. Demjén, J. Wölfling, L. G. Puskás and I. Kanizsai, J. Org. Chem., 2020, 85, 3587–3595 CrossRef CAS PubMed.
- X. Guo, W. Chen, B. Chen, W. Huang, W. Qi, G. Zhang and Y. Yu, Org. Lett., 2015, 17, 1157–1159 CrossRef CAS PubMed.
- B. Hu, Z. Wang, N. Ai, J. Zheng, X.-H. Liu, S. Shan and Z. Wang, Org. Lett., 2011, 13, 6362–6365 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, J. Xie, B. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2017, 19, 1020–1023 CrossRef CAS PubMed.
- J. O. Strelnikova, N. V. Rostovskii, G. L. Starova, A. F. Khlebnikov and M. S. Novikov, J. Org. Chem., 2018, 83, 11232–11244 CrossRef CAS PubMed.
- H. Mehrabi, M. Hajipour, F. Rezazadeh-Jabalbarezi and F. Alizadeh-Bami, J. Heterocycl. Chem., 2020, 57, 3361–3368 CAS.
- Z. Zarnegar and J. Safari, New J. Chem., 2014, 38, 4555–4565 RSC.
- L. Nagarapu, S. Apuri and S. Kantevari, J. Mol. Catal. A: Chem., 2007, 266, 104–108 CrossRef CAS.
- X. C. Wang, H. P. Gong, Z. J. Quan, L. Li and H. L. Ye, Chin. Chem. Lett., 2009, 20, 44–47 CrossRef CAS.
- S. N. Murthy, B. Madhav and Y. V. D. Nageswar, Tetrahedron Lett., 2010, 51, 5252–5257 CrossRef CAS.
- S. Das Sharma, P. Hazarika and D. Konwar, Tetrahedron Lett., 2008, 49, 2216–2220 CrossRef CAS.
- B. Sadeghi, B. B. F. Mirjalili and M. M. Hashemi, Tetrahedron Lett., 2008, 49, 2575–2577 CrossRef CAS.
- K. Niknam, A. Deris, F. Naeimi and F. Majleci, Tetrahedron Lett., 2011, 52, 4642–4645 CrossRef CAS.
- B. F. Mirjalili, A. H. Bamoniri and L. Zamani, Sci. Iran., Trans. C, 2012, 19, 565–568 CrossRef CAS.
- A. R. Khosropour, Ultrason. Sonochem., 2008, 15, 659–664 CrossRef CAS PubMed.
- N. Montazeri, K. Pourshamsian, M. Khoddadi and K. Khoddadi, Orient. J. Chem., 2011, 27, 1023–1027 CAS.
- S. A. Siddiqui, U. C. Narkhede, S. S. Palimkar, T. Daniel, R. J. Lahoti and K. V. Srinivasan, Tetrahedron, 2005, 61, 3539–3546 CrossRef CAS.
- L.-M. Wang, Y.-H. Wang, H. Tian, Y.-F. Yao, J.-H. Shao and B. Liu, J. Fluorine Chem., 2006, 127, 1570–1573 CrossRef CAS.
- J. Li, S. Lin, J. Dai and W. Su, J. Chem. Res., 2010, 34, 196–199 CrossRef CAS.
- G. H. Mahdavinia, A. M. Amani and H. Sepehrian, Chin. J. Chem., 2012, 30, 703–708 CrossRef CAS.
- M. Kalhor and Z. Zarnegar, RSC Adv., 2019, 9, 19333–19346 RSC.
- N. Montazeri, K. Pourshamsian, H. Rezaei, M. Fouladi and S. Rahbar, Asian J. Chem., 2013, 25, 3463–3466 CrossRef CAS.
- L. Wang, X. Zhong, M. Zhou, W.-y. Zhou, Q. Chen and M.-Y. He, J. Chem. Res., 2013, 37, 236–238 CrossRef CAS.
- M. Salimi, M. A. Nasseri, T. D. Chapesshloo and B. Zakerinasab, RSC Adv., 2015, 5, 33974–33980 RSC.
- M. Esmaeilpour, J. Javidi, F. Dehghani and S. Zahmatkesh, Res. Chem. Intermed., 2017, 43, 163–185 CrossRef CAS.
- G. Patel, A. R. Patel and S. Banerjee, New J. Chem., 2020, 44, 13295–13300 RSC.
- O. Merino, G. Fundora-Galano, R. Luque and R. Martinez-Palou, ACS Sustainable Chem. Eng., 2018, 6, 4122–4129 CrossRef CAS.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, K. Domen, T. Tatsumi, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039–3045 CrossRef CAS.
- M. Hara, Top. Catal., 2010, 53, 805–810 CrossRef CAS.
- K. Fukuhara, K. Nakajima, M. Kitano, S. Hayashi and M. Hara, ChemCatChem, 2015, 7, 3945–3950 CrossRef CAS.
- S. H. Y. S. Abdullah, N. H. M. Hanapi, A. Azid, R. Umar, H. Juahir, H. Khatoon and A. Endut, Renewable Sustainable Energy Rev., 2017, 70, 1040–1051 CrossRef CAS.
- L. J. Konwar, P. Mäki-Arvela and J.-P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS PubMed.
- Q. Xie, X. Yang, K. Xu, Z. Chen, B. Sarkar and X. Dou, Environ. Res., 2020, 188, 109887 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- T. El Achkar, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS.
- M. Tiecco, R. Germani and F. Cardellini, RSC Adv., 2016, 6, 43740–43747 RSC.
- G. Carrasco-Huertas, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Commun., 2020, 56, 3592–3604 RSC.
- B. Saavedra and D. J. Ramón, ACS Sustainable Chem. Eng., 2021, 9, 7941–7947 CrossRef CAS.
- D. A. Alonso, S.-J. Burlingham, R. Chinchilla, G. Guillena, D. J. Ramón and M. Tiecco, Eur. J. Org. Chem., 2021, 4065–4071 CrossRef CAS.
- Y. Liu, W. Chen, Q. Xia, B. Guo, Q. Wang, S. Liu, Y. Liu, J. Li and H. Yu, ChemSusChem, 2017, 10, 1692–1700 CrossRef CAS PubMed.
- S. Hong, Y. Yuan, Q. Yang, P. Zhu and H. Lian, Carbohydr. Polym., 2018, 201, 211–217 CrossRef CAS PubMed.
- W. Zhang, S. Cheng, X. Zhai, J. Sun, X. Hu, H. Pei and G. Chen, Nat. Prod. Commun., 2020, 15, 1934578X19900708 CAS.
- E. Tommasi, G. Cravotto, P. Galletti, G. Grillo, M. Mazzotti, G. Sacchetti, C. Samorì, S. Tabasso, M. Tacchini and E. Tagliavini, ACS Sustainable Chem. Eng., 2017, 5, 8316–8322 CrossRef CAS.
- O. A. O. Alshammari, G. A. A. Almulgabsagher, K. S. Ryder and A. P. Abbott, Green Chem., 2021, 23, 5097–5105 RSC.
- R. Ahmadi, M. Heydari Dokoohaki, J. Tashkhourian, A. R. Zolghadr and A. Safavi, Int. J. Energy Res., 2022, 46, 14546–14557 CrossRef CAS.
- D. Nguyen, T. Van Huynh, V. S. Nguyen, P.-L. Doan Cao, H. T. Nguyen, T.-C. Wei, P. H. Tran and P. T. Nguyen, RSC Adv., 2021, 11, 21560–21566 RSC.
- D. J. Boogaart, J. B. Essner and G. A. Baker, Green Chem. Lett. Rev., 2022, 15, 615–626 CrossRef CAS.
- D. L. Thompson, I. M. Pateli, C. Lei, A. Jarvis, A. P. Abbott and J. M. Hartley, Green Chem., 2022, 24, 4877–4886 RSC.
- N. Peeters, K. Binnemans and S. Riaño, Green Chem., 2020, 22, 4210–4221 RSC.
- Y. Chen, Y. Lu, Z. Liu, L. Zhou, Z. Li, J. Jiang, L. Wei, P. Ren and T. Mu, ACS Sustainable Chem. Eng., 2020, 8, 11713–11720 CrossRef CAS.
- K. T. T. Tran, L. T. M. Le, A. L. B. Phan, P. H. Tran, T. D. Vo, T. T. T. Truong, N. T. B. Nguyen, A. Garg, P. M. L. Le and M. V. Tran, J. Mol. Liq., 2020, 320, 114495 CrossRef CAS.
- H. T. Nguyen, T. V. Le and P. H. Tran, J. Environ. Chem. Eng., 2021, 9, 105228 CrossRef CAS.
- H. T. Nguyen, M.-N. H. Truong, T. V. Le, N. T. Vo, H. D. Nguyen and P. H. Tran, ACS Omega, 2022, 7, 17432–17443 CrossRef CAS PubMed.
- N. Nagasundaram, M. Kokila, P. Sivaguru, R. Santhosh and A. Lalitha, Adv. Powder Technol., 2020, 31, 1516–1528 CrossRef CAS.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed., 2004, 43, 2955–2958 CrossRef CAS PubMed.
- K. Fukuhara, K. Nakajima, M. Kitano, H. Kato, S. Hayashi and M. Hara, ChemSusChem, 2011, 4, 778–784 CrossRef CAS PubMed.
- H. T. Nguyen, D.-K. Nguyen Chau and P. H. Tran, New J. Chem., 2017, 41, 12481–12489 RSC.
- H. T. Nguyen and P. H. Tran, RSC Adv., 2016, 6, 98365–98368 RSC.
- P. H. Tran, H. T. Nguyen, P. E. Hansen and T. N. Le, RSC Adv., 2016, 6, 37031–37038 RSC.
- S. Yamabe and S. Yamazaki, J. Phys. Org. Chem., 2016, 29, 361–367 CrossRef CAS.
- N. Daems, J. Wouters, C. Van Goethem, K. Baert, C. Poleunis, A. Delcorte, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2018, 226, 509–522 CrossRef CAS.
- S. S. Dipake, V. D. Ingale, S. A. Korde, M. K. Lande, A. S. Rajbhoj and S. T. Gaikwad, RSC Adv., 2022, 12, 4358–4369 RSC.
- S. Samai, G. C. Nandi, P. Singh and M. S. Singh, Tetrahedron, 2009, 65, 10155–10161 CrossRef CAS.
- K. Sanchooli Tazeh, R. Heydari and M. Fatahpour, Bull. Korean Chem. Soc., 2021, 42, 1464–1472 CrossRef CAS.
- A. Mahata, R. K. Rai, I. Choudhuri, S. K. Singh and B. Pathak, Phys. Chem. Chem. Phys., 2014, 16, 26365–26374 RSC.
- Q. Liu, S. Tadrent, C. Proust, F. Gomez, A. Khelfa, D. Luart and C. Len, Chem. Eng. Sci., 2020, 211, 115275 CrossRef CAS.
- J. J. Gabla, S. R. Mistry and K. C. Maheria, Catal. Sci. Technol., 2017, 7, 5154–5167 RSC.
- M. Nejatianfar, B. Akhlaghinia and R. Jahanshahi, Appl. Organomet. Chem., 2018, 32, e4095 CrossRef.
- A. R. Moosavi-Zare, M. A. Zolfigol, V. Khakyzadeh, C. Böttcher, M. H. Beyzavi, A. Zare, A. Hasaninejad and R. Luque, J. Mater. Chem. A, 2014, 2, 770–777 RSC.
- F. A. Van de Laar, P. L. Lucassen, R. P. Akkermans, E. H. Van de Lisdonk, G. E. Rutten and C. Van Weel, Cochrane Database Syst. Rev., 2005, 2005, Cd003639 Search PubMed.
- K. Y. Kim, K. A. Nam, H. Kurihara and S. M. Kim, Phytochemistry, 2008, 69, 2820–2825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00755c |
| This journal is © The Royal Society of Chemistry 2023 |