
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step fabrication of three-dimensional macropore copolymer-modified polycarbonate array by photo-crosslinking for protein immunoassay†
Kaimei Peng ab,
Runping Wanga and
Jianhua Zhou*b
ab,
Runping Wanga and
Jianhua Zhou*b
aSchool of Chemistry and Chemical Engineering, Qiannan Normal University for Nationalities, Duyun 558000, China
bKey Laboratory of Sensing Technology and Biomedical Instruments of Guangdong Province School of Biomedical Engineering, Sun Yat-sen University, Guangzhou 510275, China. E-mail: zhoujh33@mail.sysu.edu.cn
First published on 28th February 2023
Abstract
A photocross-linked copolymer was prepared, and could rapidly form a macropore structure in phosphate buffer solution (PBS) without the addition of porogen. The photo-crosslinking process contained the crosslinking of the copolymer itself and that with the polycarbonate substrate. The three-dimensional (3D) surface was achieved through one-step photo-crosslinking of the macropore structure. The macropore structure can be finely regulated by multiple dimensions, including monomer structure of the copolymer, PBS and copolymer concentration. Compared with the two-dimensional (2D) surface, the 3D surface has a controllable structure, a high loading capacity (59 μg cm−2) and immobilization efficiency (92%), and the effect of inhibiting the coffee ring for protein immobilization. Immunoassay results show that a 3D surface immobilized by IgG has high sensitivity (LOD value of 5 ng mL−1) and broader dynamic range (0.005–50 μg mL−1). This simple and structure-controllable method for preparing 3D surfaces modified by macropore polymer has great potential applications in the fields of biochips and biosensing.
1. Introduction
Biochips, which consist of a protein/nuclein array on a substrate, are widely used in biodetection and disease diagnosis1–4 due to the merits of cost-efficiency, portability and scalability.5–8 Compared to inorganic glass substrates, polymer substrates are potential valuable alternatives. For example, polydimethylsiloxane (PDMS), cyclic olefin copolymer (COC), polymethyl methacrylate (PMMA), and polycarbonate (PC) are widely used in biochips,9–12 with the advantages of solvent compatibility, optical clarity, and low-cost fabrication. PC is regarded as a good candidate for application in biochip substrates.13 However, although a lot of work have been done,14–16 the preparation of protein arrays on these polymer substrates still suffers from two problems: the difficulty of modifying the polymer substrate surface and the cumbersome steps of preparing three-dimensional (3D) structures with immobilizing high capacity of antibody for biodetection with broad dynamic range.In view of the inert nature of the polymer surfaces, it is necessary to transform the inert surface into a bioanalytical surface with active sites capable of binding capture molecules. A common strategy is to introduce reactive functional groups, for example, epoxy, carboxyl or aldehyde to the surface for protein by covalent coupling.17 However, the modifications of inert polymers require a multistep chemical and physical process that is cumbersome and time-consuming, thus increasing the cost and making it difficult to control the quality of the chip product. With the application of the photoinitiated surface grafting technology, plastic surface modification could be realized by one-step photo-grafting. The photo-grafting method has high stability to the modified surface, and is simple to operate. The functional groups used in the photoinitiated surface grafting technology can be benzophenones and diazirines. Benzophenone (BP) group has been extensively studied due to its excellent photochemical activity, good thermal and chemical stability.18 The BP is used to modify any surface with a C–H group,19 whereas polymers containing the BP groups serve as a simple and efficient photo-cross-linking tool for modifying different surfaces. These surfaces modified by BP-containing polymers are widely used in biosensing,20,21 coating,22 composite material23 and surface patterning.24–26
Photo-crosslinking technology can solve the problem of difficult and tedious problems in the modification of polymer substrate surface, while the 3D structural surface provides the basis for excellent biodetection performance of biochips. Compared with the 2D flat surfaces, the 3D matrixes can greatly increase the immobilization amount of the biological probes, provide a more natural water environment and maintain the biological activity of the immobilized biomolecules.27,28 Except for 3D structures such as papers and membranes, most 3D surfaces are obtained by modifying 2D planar substrates, and hydrogel and porous polymer monolith modification are two widely used strategies for 3D surface fabrication.17 Rigid macropore polymer monoliths are a porous polymer frit whose structure is composed of a non-swellable, highly cross-linked continuous phase containing interconnected pores.29,30 The cross-linked network structure and absorbent swelling properties of hydrogels will produce an “entropic shielding” effect on macromolecules such as proteins and prevent them entering and exiting from the matrices.31 In contrast, the porous polymer monoliths have a non-swelling open porous space, which not only ensures the unhindered penetration of large biomolecules such as proteins, but also a short time and less residue in the cleaning.30,32 Due to these properties of macropore polymers, they are widely used in thin-layer chromatography and protein/DNA microarrays.33–36
Given the advantages of photo-crosslinking technology and the macropore 3D structure, we prepared a photo-crosslinked copolymer, whose PBS solution can form 3D macropore copolymer film on a polymer substrate through just one step of photo-crosslinking. The three-dimensional (3D) array on polymer substrate surface was achieved through one-step photo-crosslinking of the macropore structure. The macropore structure can be finely regulated by multiple dimensions, including monomers structure of copolymer, PBS and copolymer concentration. The 3D array on polymer substrate a high loading capacity (59 μg cm−2) and immobilization efficiency (92%) of protein, and the effect of inhibiting the coffee ring for protein immobilization. Immunoassay results show that the 3D surface immobilized by IgG has high sensitivity (LOD value of 5 ng mL−1) and broader dynamic range (0.005–50 μg mL−1) for biodetection. This simple and structure-controllable method for preparing the 3D surfaces modified by macropore polymer has great potential applications in the fields of biochips and biosensing.
2. Materials and methods
2.1. Materials
Polycarbonate (PC) slides (75 × 25 × 0.5 mm3) were obtained from Bositai Biotechnology Co. (Chongqing, China). Methacryloxybenzophenone (MABP), N,N-dimethylacrylamide (DMAA), Na-4-styrenesulfonate (SSNa), acrylic acid (AA), butyl methacrylate (BMA), azobis(isobutyronitrile) (AIBN), diethyl ether, N,N-dimethylformamide (DMF) and dichloromethane were purchased from Titan Technology Co. (Shanghai, China). Phosphate-buffered saline (PBS, 0.1 M, pH 7.4), rabbit IgG (IgG) and Cy3-labeled goat anti-rabbit IgG H&L (Cy3-anti-IgG) were obtained from Bioss Biotechnology Co. (Beijing, China). FITC-labeled human IgG (FITC-IgG) was supplied by Sigma-Aldrich Co. (Shanghai, China). All other chemicals were used as received.2.2. Instruments
A Bruker NMR Spectrometer (Bruker Avance III HD) was used to determine the chemical composition of copolymer, and all the obtained spectra were measured in D2O. The molecular weights of copolymers were determined by GPC (Waters 1525/2414, US). N,N-Dimethylformamide (DMF) was used as eluant, with a flow rate of 0.8 mL min−1. Calibration was carried out with polymethyl methacrylate (PMMA) standards.The morphology of the copolymer-based macropore structure was studied in a Scanning Electron Microscope (SEM, GeminiSEM 300) with an accelerating voltage of 3 kV. Prior to the measurement, the samples were covered with a thin layer of gold (Au) by sputtering. Porosity and pore size distribution were obtained by ImageJ analysis of SEM images. Fluorescent microscopy images were taken to confirm protein immobilization or interaction of immunoassay tests. Images were achieved with a MF43 fluorescent microscopy (Micro-shot Technology Co., China), and fluorescence intensity was analyzed using the ImageJ software.
2.3. General synthetic procedures
For the synthesis of poly(SSNa-co-MABP-co-BMA-co-DMAA-co-AA) (PSMBDA), the monomers were dissolved in DMF to a concentration of 2 mol L−1, followed by adding 0.2 mol% α,α′-azoisobutyronitrile (AIBN). The solution was degassed with nitrogen and kept at 65 °C for 18 h, from which the copolymers were precipitated by ether. The remaining unreacted monomers and solvents were removed in water by dialysis (MWCO = 3500). After freeze-drying, the resulting copolymers were a white solid (70–80%, Mw ≅ 2.8–4.1 g mol−1).37 According to the content of AA and the absence of monomer BMA or AA, six copolymers (PSMBDA10–PSMBDA40, PSMDA and PSMBD) were prepared, with chemical compositions and molecular properties, respectively, as shown in Table 1.| Monomer unit composition (mol%) | Yield (%) | Molecular weightb | Solubilityc | ||||
|---|---|---|---|---|---|---|---|
| In feed SSNa/MABP/BMA/DMAA/AA | In copolymera SSNa/MABP/BMA/DMAA/AA | Mw × 104 | Mw/Mn | Ethanol | Water | ||
| a Determined by 1H-NMR spectrum in D2O.b Molecular weights were determined by GPC in DMF, polymethyl methacrylate standards. Mn and Mw represent number and weight average molecular weight, respectively.c Polymer solubility was measured by 25 mg mL−1 and described as (+) or insoluble (−) at room temperature. | |||||||
| PSMBDA10 | 8/5/19/58/10 | 8/7/21/58/6 | 75 | 4.1 | 1.8 | + | + |
| PSMBDA20 | 8/5/19/48/20 | 9/6/20/49/16 | 70 | 3.5 | 1.9 | + | + |
| PSMBDA30 | 8/5/19/38/30 | 9/6/21/39/25 | 72 | 3.2 | 1.9 | + | + |
| PSMBDA40 | 8/5/19/28/40 | 9/7/21/29/34 | 71 | 4.1 | 1.8 | − | + |
| PSMDA | 8/5/—/57/30 | 9/6/—/62/23 | 77 | 3.1 | 1.6 | − | + |
| PSMBD | 8/5/19/68/— | 10/7/20/63/— | 78 | 2.8 | 1.9 | + | + |
2.4. Fabrication of the macropore copolymer-modified PC (MC-PC) surface
Bulk commercially available transparent foils of polycarbonate (PC), with a thickness of 0.8 mm, were laser-cut to 75 × 25 mm slide dimensions, with the protective adhesive foil being removed. The PC slides were sonicated in deionized water for 3 min, and dried in a nitrogen flow. The copolymers in PBS were spotted on the cleaned PC films with pipette. The spots were dried in air and cross-linked for 6 min under UV irradiation at 365 nm with the intensity of 80 mW cm−2. The macropore copolymer-modified PC (MC-PC) slides were thoroughly rinsed with water, and stored in a 4 °C refrigerator for later use.To monitor the photo-crosslinking properties of the benzophenone groups in the copolymers and hydrophilicity, PC slides (75 × 25 mm) were coated with 150 μL of aqueous PSMBDA30 solution (25 mg mL−1). The variations of the copolymer solution, under different UV illumination time (365 nm, 80 mW cm−2), were measured using a TU-1901 UV-Vis spectrophotometer (Beijing Purkinje General Instrument Co., China). The water contact angles, on the copolymer-modified PC surfaces, were measured by the sessile drop method using OCA20 contact angle analyzer (Data Physics, Germany), with 2 μL water droplet at room temperature. The film thickness, on the surface modified with different concentration copolymers, was measured by Profilometer (Bruke, Dektak XT), i.e., a polymer solution of 0.7 μL was spotted on the PC surface, dried, and washed after UV illumination, with the thickness of the spot being measured.
2.5. Protein immobilization on the MC-PC surface
The prepared MC-PC slide was added to a mixed aqueous solution of EDC (40 mM) and NHS (20 mM), which was left for 30 min at room temperature to activate the carboxyl groups. The activated slide was rinsed with deionized water, exposed to protein IgG or FITC-IgG, and left for 2 h at room temperature. The immobilized IgG or FITC-IgG immobilized macropore copolymer-modified PC (IgG-MC-PC or FITC-IgG-MC-PC) slide was obtained. For the FITC-IgG-MC-PC slides, the fluorescence intensity was determined by the fluorescence microscopy. Subsequently, the proteins-modified slides were blocked by bovine serum albumin (BSA, 10 mg mL−1) in phosphate buffer solution (PBS) for 1 hour, followed by three alternating washes with deionized water and PBST (10 mM PBS, 0.05% Tween-20, pH 7.4). Their fluorescence intensity was measured again. Furthermore, the ratio of the fluorescence intensity after and before blocking by BSA was the immobilization efficiency. The immobilization density was calculated from the calibration curve (Fig. S1†).2.6. Immunoassay tests
On the IgG-MC-PC slide was spotted the Cy3-labeled goat anti-rabbit IgG H&L (Cy3-anti-IgG) in 10 mM PBS. Here, the immunoreaction was allowed to proceed for 1 h at room temperature. The slide was then thoroughly rinsed six times with PBST. After nitrogen blowing, the fluorescence signal of the Cy3 dye was measured by fluorescence microscope. The gray mean value was evaluated using ImageJ software.3. Results and discussion
3.1. Synthesis of copolymers
The copolymers (PSMBDA) with different feed ratios of the monomers were synthesized as random copolymers by free radical polymerization. The designed copolymers consist of (i) DMAA units that provide a hydrophilic matrix, (ii) AA providing active sites for covalent linking proteins, (iii) MaBP for the light-induced immobilization on the PC surface and the cross-linking of polymer segments, (iv) ionic monomers (SSNa) and butyl methacrylate (BMA) for controlling the hydrophilic–hydrophobic balance to form the macropore polymer structure.To investigate the relationship between the copolymer composition and the macropore structure, to determine the optimal composition for protein immobilization, copolymers with different monomer compositions were prepared. Key analytical data of the copolymers are summarized in Table 1. The chemical structures of the copolymers were confirmed by 1H NMR (Fig. S2†). Chemical shifts at 6.60–8.40 ppm were assigned to the aromatic protons of DMAA and SSNa moieties. The other characteristic peaks were assigned as follows:37–39 3.57–4.20 ppm (–OCH2–) for the BMA unit, 2.33–3.18 ppm ((CH3)2N–) for the DMAA unit and the protons on the backbone, 1.20–1.43 ppm (–CH2–CH2–) for the BMA unit, and 0.69–1.03 ppm (–CH3) for the MABP and BMA units. Details of the characteristic peak attribution are shown in Fig. S2,† and the copolymer compositions closely match the original feed ratios (Table 1). The average molecular mass (Mw) and polydispersity (Mw/Mn) of the copolymers are in the range of 28–41 kg mol−1 and 1.6–1.9 (Table 1), respectively. All the copolymers are soluble in water, laying a foundation for the construction of the aqueous solution-based macropore copolymer.
3.2. Fabrication of the macropore copolymer-modified PC (MC-PC) surface
Polymers of macropore structures are mainly prepared by two methods, one is free radical polymerization of monomers, cross-linking agents, initiators and porogens, and the other is HIPE polymerization technology.30,40 The two methods form porous structures through the addition of unreacted organic porogens or internal phase separation, where all processes are completed in one step. Their advantage is fewer steps, but it is difficult to control the formation process of the macropore structure. Instead, the formation of the macropore structure herein was divided into three steps composed of polymer preparation, macropore structure formation and photo-crosslinking fixation. Those can facilitate the quality control of each process, and quickly build macropores structure in the required area by coating or spotting. In brief, the step-by-step operation strategy could be beneficial to improvement in the stability of product quality and the preparation on-demand.The preparation of the PC surface decorated by macropore copolymers is shown in Scheme 1. All the prepared copolymers were hydrosoluble on account of the presence of water-soluble DMAA and SSNa monomers (Table 1). The copolymers were dissolved in PBS, and spotted on the PC plate with a pipette. As the water evaporated, the salt concentration in the solution gradually increased, and the polymer began to precipitate from the solution and formed a macropore structure. After the formation of the macropore structure, the self-crosslinking of the copolymer and fixation of the copolymer on the PC surface were realized by one step of photo-crosslinking (Fig. 1A). The photo-crosslinking mechanism of BP has been clearly studied.39,41,42 The benzophenone (BP) in MABP comonomer can generate a triplet biradical via triggering n–π* or π–π* transition of the carbonyl group under UV light (250–365 nm) irradiation. The biradical then abstracts a hydrogen atom from neighboring polymer segment, leading to the creation of a benzophenone ketyl radical and a polymer radical. These radicals produce covalent bonds between the polymer and BP, thus curing the structure and morphology of the macropore polymer frit. The photo-crosslinking capability of the copolymer was analyzed by monitoring the absorption spectra at 250–290 nm (Fig. S3†). The absorption intensity decreases with the increase in the degree of crosslinking.43 When the copolymer was exposed to UV light of 365 nm with an intensity of 80 mW cm−2, the maximum absorption peak intensity decreased significantly (Fig. 1B), indicating a successful photo-crosslinking process.
 | ||
| Scheme 1 Process for the preparation of 3D protein chip with macropore copolymer on polymer substrate for immunoassay. | ||
 | ||
| Fig. 1 (A) The schematics showing the crosslinking reaction of copolymer (PSMBDA30) at the benzophenone group on PC plastic substrate and (B) absorption spectra of PSMBDA30 during UV illumination. (C)–(E) SEM images of the modified PC film prepared with a 25 mg mL−1 of PSMBDA30 in H2O, in 10 mM PBS, or in 50 mM PBS, respectively. | ||
After UV illumination, the copolymers were cross-linked and immobilized on the surface of PC film. Some of non-crosslinked copolymers and inorganic salts were washed, forming a pore-like structure on the PC surface (Fig. S4†). In addition, the macropore structural modification layer can be immobilized on the surface of different plastic substrates (PE, PET, PMMA, PS, PVC or PP, Fig. S5†). This result can verify the universality of the photo-crosslinking immobilization method with benzophenone functional groups. The porosity of PSMBD is about 20%, and its pore size distribution range is wide (Fig. S6†). When the molar percentage of the monomer AA increased to 30 mol%, the porosity increased to 45%, with the pore size gradually becoming uniform and being concentrated in the range of from 0.1 to 0.5 μm. However, as the AA content continued to increase, the porosity decreased. These results indicate that the proportion of hydrophilic monomer AA can adjust the morphology of macropore copolymer. Furthermore, the copolymer PSMDA, without the hydrophobic monomer BMA did not form a pore-like structure on the PC surface, although its AA content is 30 mol% (Fig. S4F†). The hydrophilic and hydrophobic properties of copolymers may play a key role in the formation of macropore structures. When the copolymer has a good solubility, it has a gel-like polymer structure in aqueous solution. When the water evaporates, the gel-like polymer structure collapses to a non-porous glassy structure.44 As the solubility decreases, phase separation occurs in the aqueous solution of the copolymer and a phase boundary is formed, and upon removal of the aqueous solvent, the copolymer is able to form a porous structure.45 Therefore, the hydrophilic and hydrophobic property of copolymer has an important influence on phase separation46 and may be a key factor in the formation of macropore structures.
To clarify the effect of PBS on the pore formation process, the SEM images of the PSMBDA30 at different PBS concentrations are shown in Fig. 1. The PC surface has no pore structure when the phosphate is absent in the copolymer solution (Fig. 1C). The macropore structure formed after the addition of PBS, and some long cracks will appear when the concentration of PBS is too high (Fig. 1D and E). As reported in the literature,47,48 salt promoted the self-hydration caused by the interaction between ion and water; simultaneously, it destroys the clathrate structure formed by water molecules around the hydrophobic moieties of the polymer, resulting in the reduction of polymer solubility (salt-out effect) and the forming macropore structures. These findings suggest that the main role of phosphate may be to promote the orderly deposition of copolymers in water, thereby creating the conditions for the formation of macropore structures, while the role of PBS is equivalent to that of a porogen.
In addition to hydrophilicity and hydrophobicity of copolymers and PBS, the concentration of the copolymer also affects the thickness of the modified layer. In the range of concentration from 5 to 50 mg mL−1, the thickness of the layer modified with the macropore copolymer increases linearly with the copolymer concentration (Fig. S7†). When the concentration of the copolymer is too low, there is not enough copolymer to build up the complex pore structures, resulting in most of the PC surface being not covered by the pore structures of polymer. With the increase in the copolymer concentration, on the PC surface increases the coverage of the pore structures. When the polymer concentration is above 40 mg mL−1, the pores are partially blocked by the copolymer, resulting in a reduced porosity. In conclusion, the hydrophilicity and hydrophobicity of the copolymer molecular chain and PBS are crucial for the formation and regulation of the macropore copolymer structure. The adjustment of the copolymer concentration can finely control the thickness of the layer modified by macropore copolymer.
3.3. Immobilization of protein on the PC surface modified by macropore copolymer
Due to the porous structure of the MC-PC surface, the hydrophilicity and hydrophobicity of the structure have an important influence on the penetration efficiency of the aqueous protein solution.49 The water contact angle (CA) of the pristine PC surface is 78°, and the CA value of the PC surface modified by copolymer decreases (Fig. S8†). The concentration of the copolymers is greater than 50 mg mL−1 in water (Table 1) because it contains hydrophilic monomers DMAA and SSNa, facilitating the immobilization of proteins on the copolymer-modified PC surface.Since the monomer AA contains carboxyl groups, the PSMBDA-modified PC surface can immobilize the FITC-IgG proteins very well after the carboxyl groups are activated by EDC/NHS.50 The immobilization amount on the surface significantly increases with the increase in the carboxyl group content, and almost reaches the maximum when the AA content reaches 30 mol% (Fig. 2A and D). Those should be attributed to two changes during the increase of AA content, namely the increase of the carboxyl group number and the porosity of the macropore structure. Furthermore, at the 30 mol% of AA content, in terms of the amount of the protein immobilized on the surface, the amount on that with macropore structures modified by PSMBDA30 is 12 times higher than that on the surface without macropore structure modified with PSMDA. The results suggest that both the number of carboxyl groups and the macropore structure are crucial for the immobilization of proteins.
 | ||
| Fig. 2 Fluorescence images (A) and intensity (D) of the immobilized FITC-IgG (1 mg mL−1) spots prepared by using different copolymers (25 mg mL−1) in PBS (10 mM). Fluorescence images (B) and intensity (E) of immobilized FITC-IgG (1 mg mL−1) spots prepared by using PSMBDA30 (25 mg mL−1) in different PBS concentrations, respectively. Fluorescence images (C) and intensity (F) of immobilized FITC-IgG (1 mg mL−1) spots prepared by using different PSMBDA30 concentrations in PBS (10 mM), respectively. | ||
The above results imply that the content of monomer AA affects both the active linked sites and the porosity of macropore structure, thus influencing the load of protein, simultaneously, the structure and thickness of the macropore-modified layer can also been adjusted by changing the concentration of PBS and copolymer, respectively. To clarify the relationship between these structural adjustments and the sample loading, under the condition of 1 mg mL−1 of the FITC-IgG protein, it was investigated whether the concentrations of PBS (from 0 to 50 mM) and the copolymer PSMBDA30 (from 5 to 50 mg mL−1) influenced the fluorescence intensity of the FITC-IgG-MC-PC. As shown in Fig. 2B and E, when the medium dissolving the copolymer was deionized water, where the concentration of PBS is 0 mol L−1, the fluorescence intensity of the spots was only 3.7 au in the absence of macropore structures (Fig. 1C). A pore structure was formed and the fluorescence intensity of the spots reached a maximum at 10 mM of PBS. With the increase in the PBS concentration, the fluorescence intensity of the spots decreased instead, probably due to some cracks in the macropore structures (Fig. 1E).
The fluorescence intensity increases with the copolymer concentration and reaches a maximum at 40 mg mL−1 (Fig. 2C and F). Those may be due to the increase of polymer concentration resulting in the increased coverage and thickness of the modified layer possessing macropore structures on the PC surface. However, too high a polymer concentration will reduce the porosity of the modified layer (Fig. S7†). Similarly, the pore size is too small, to facilitate the late cleaning of the proteins. In conclusion, the concentration variation of PBS and copolymer can regulate the hierarchy, coverage and thickness of the macropore structure, ultimately changing the surface area of the macropore structure, thus affecting the immobilization amount of the protein.
When PSMBDA30 was mixed within 10 mM PBS, macropore structures were formed, and activated by EDC/NHS. FITC-IgG protein in the range of concentration from 0.01 to 5 mg mL−1 was spotted and immobilized on the modified PC surface. The fluorescence intensity in the spotted area increased by 18 times as the protein concentration increased from 0.01 to 5 mg mL−1 (Fig. 3A and S9†). When the concentration of FITC-IgG reach 2 mg mL−1, the increase of fluorescence intensity is not significant, and the loading capacity of the MC-PC surface is near saturation. After blocking with BSA, the fluorescence intensity of the spots decreases slightly, with the immobilization efficiency and density of protein being as high as 83–92% and 1–59 μg cm−2, respectively (Fig. 3B and C). Those indicate that the surface modified by macropore structure still maintains a high immobilization efficiency at high protein concentration (2 mg mL−1). According to the reported results, the immobilization efficiency of the surface modified by polymer brush is 63–88% at the protein concentration of 1–13 μg mL−1,51 while that of the hydrogel-modified surface is 60% at the protein concentration of about 1 mg mL−1.37 The immobilization efficiency of the aldehyde-derivatized agarose surface is 34–57% at protein concentrations from 50 to 360 μg mL−1.52 Therefore, the immobilization amount on the MC-PC surface is comparable to that on the hydrogel 3D surface, and the immobilization efficiency can be improved due to its macropore structure.
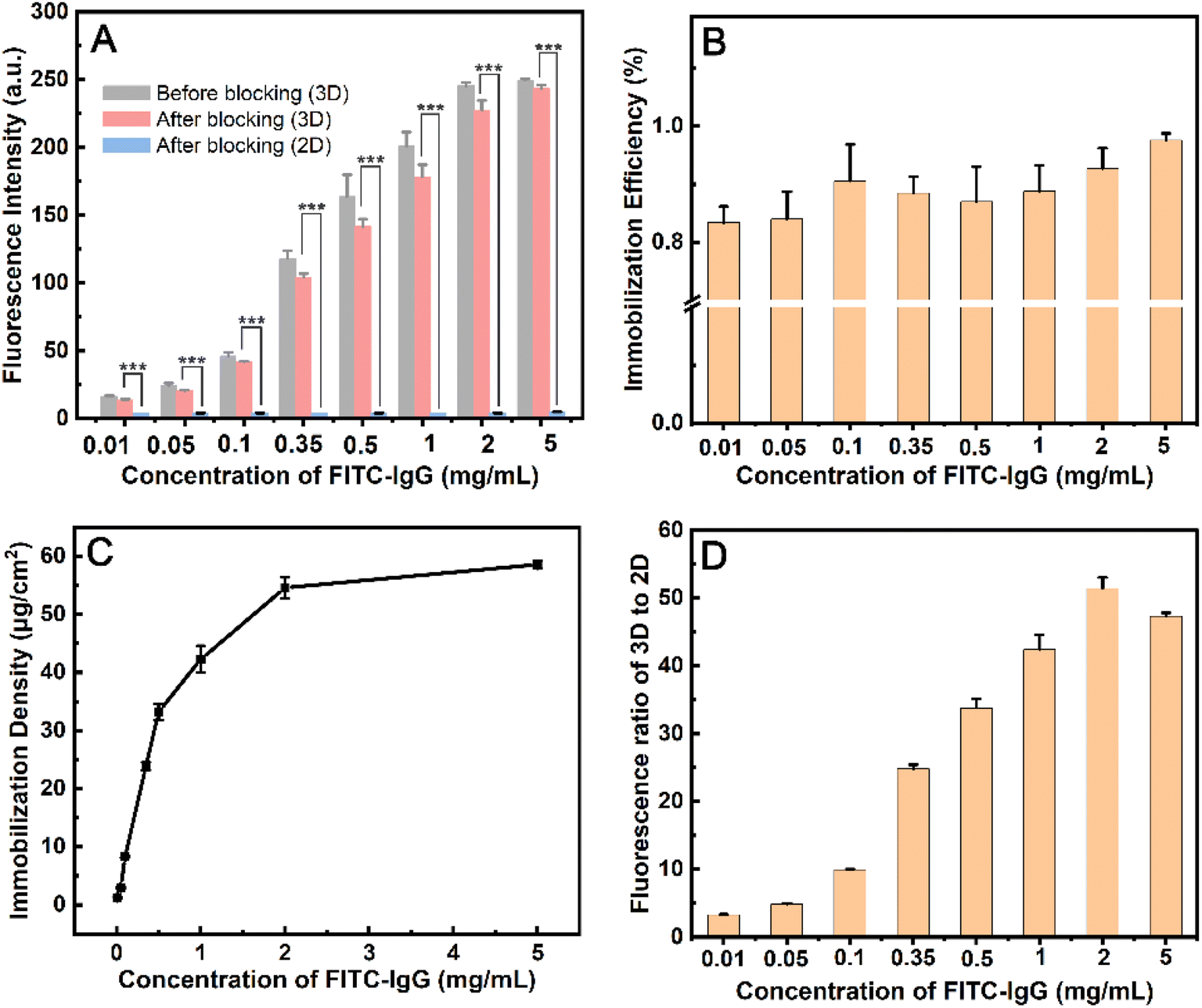 | ||
| Fig. 3 (A) Fluorescence intensity of PSMBDA30 PBS solution modified PC film (3D surface) before and after blocking, and the fluorescence intensity of PSMBDA30 aqueous solution modified PC film (2D surface). (B) Immobilization efficiency and (C) immobilization density of 3D surface, and (D) fluorescence ratio of 3D to 2D surface under different FITC-IgG concentrations. Asterisks (***) indicates statistically significant difference (p < 0.0001, n = 4). | ||
The modified PC surface was a non-porous structure when the medium for dissolving PSMBDA30 was deionized water, resulting in the fluorescence intensity of the spot region being only about 4 au (Fig. 3D and S9†). The fluorescence intensity of the surface modified with macropore structure is 51 times higher than that of the surface modified without pore structure at the FITC-IgG concentration of 2 mg mL−1. The reported results show that the protein loading capacity of nanotextured PMMA, PS/PSMA fibers or Ag@SiO2-PCL 3D surface is 5–51 times higher than that of 2D flat.15,53,54 Those may be attributed to the large surface area to volume ratio of 3D surfaces.15
The coffee ring phenomenon is a problem in the field of protein chips, resulting in a large number of protein molecules aggregated in a small region, whose the active sites are insufficient to immobilize so many protein molecules, while other regions have only a small number of immobilized protein molecules (Fig. 4A and B). Therefore, the inhibition of the coffee ring is beneficial to improve the uniformity of the protein distribution in the spot region, which can greatly improve the immobilization amount and immobilization efficiency of the protein. There are many methods to inhibit the coffee ring, including two commonly used methods, namely changing the surface structure55,56 and adding inhibitors.57,58 The surface modified by the macropore structure can also significantly inhibit the formation of coffee rings (Fig. 4C and D). The distribution of the fluorescence intensity is uniform at different FITC-IgG concentrations, indicating that the macropore structure could inhibit the coffee-ring distribution of the spot solution. The macropore structure has high immobilization amount and efficiency, inhibiting the coffee ring effect, and has potential applications in the field of microarray biosensing.
 | ||
| Fig. 4 (A) Fluorescence images and (B) the diametrical distributions of fluorescence intensity of the different concentration of FITC-IgG spots on PC surface, and (C) fluorescence images and (D) distributions of fluorescence intensity of the different concentration of FITC-IgG spots on PSMBDA30-modified PC surface. | ||
3.4. Immunoassay of the protein-immobilized macropore polymer structure
The macropore copolymer-based modification offers a simple and efficient method to obtain uniform protein immobilization spots, and provides a solid foundation for the preparation protein chips with high sensitivity and reproducibility. The immunoassay performance of the IgG-MC-PC was evaluated using different concentrations (0.005–80 μg mL−1) of the analyte Cy3-anti-IgG. The spots specifically bound to the analyte (Cy3-anti-IgG) show a uniform red fluorescence, and the fluorescence intensity gradually increases with the analyte concentration (Fig. 5A and B). The protein chip had satisfactory responses to the analyte in the concentration range of 0.005–50 μg mL−1 because of the high loading capacity of the macropore structure (Fig. 5B). The detection range of macropore structure is wider than that of 2D surfaces,51,59 which is consistent with the broader dynamic range of 3D surfaces.60,61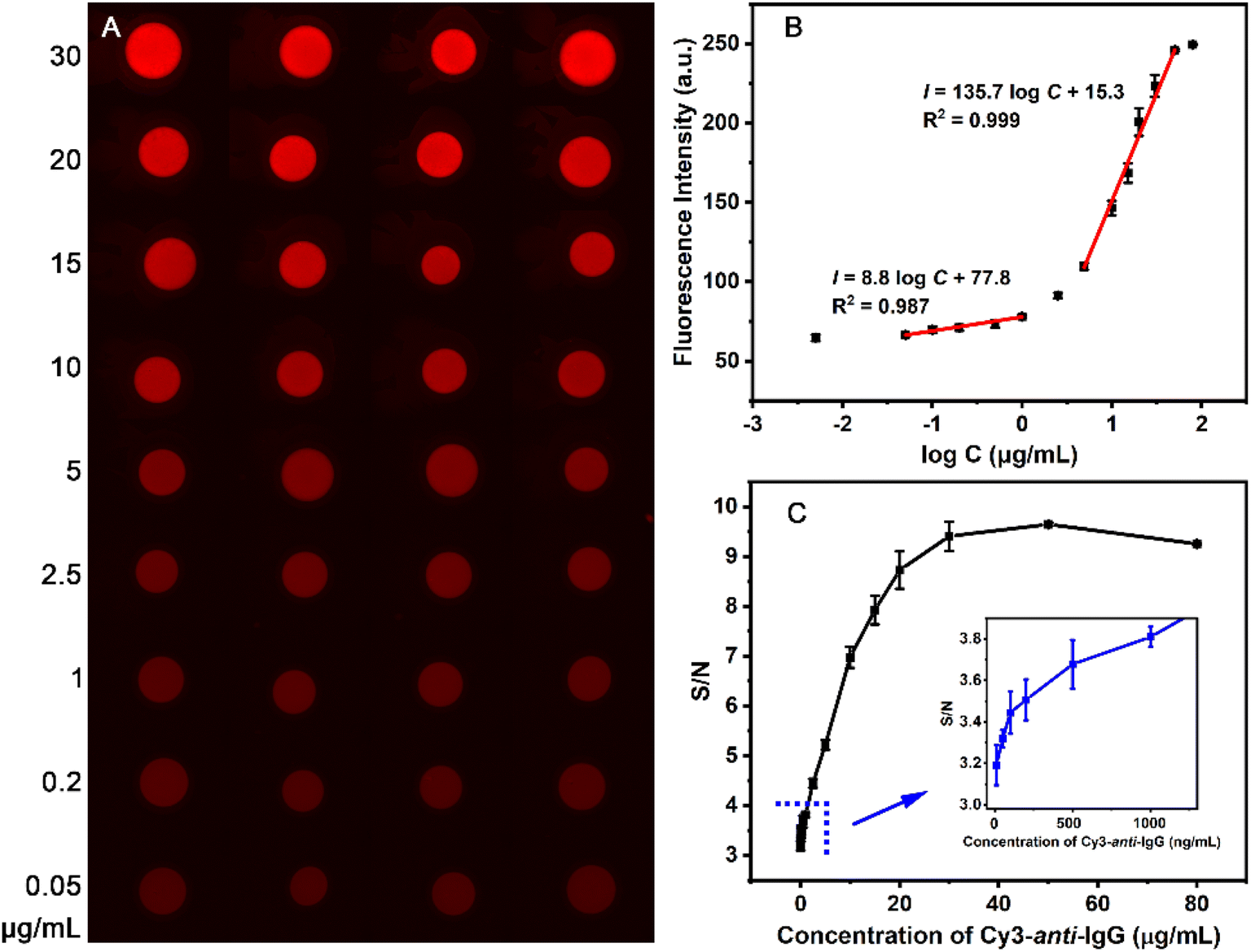 | ||
| Fig. 5 Immunoassay images (A), immunofluorescence intensity (B) and S/N ratio (C) of IgG-MC-PC at different concentrations of Cy3-anti-IgG. | ||
The fluorescence intensity increases linearly with the logarithm of the Cy3-anti-IgG concentration in the range of 0.05–1 μg mL−1 and 5–50 μg mL−1. The linear response range of the 3D surfaces constructed by hollow silica nanoparticles14 and nanobranched silica structure62 is 1–100 ng mL−1 and 10−6 to 1 μg mL−1, respectively, which is characterized by a linear response at low analyte concentration (Fig. 5B). Compared with them, the macroporous modified 3D surface has a linear response range when the analyte concentration is low or high. The limit of detection (LOD) is determined by a signal-to-noise (S/N) ratio equal to or greater than 3.63 Accordingly, the LOD of the IgG-MC-PC surface is 5 ng mL−1 (S/N = 3.1, Fig. 5C), which is comparable to that of 3D surfaces modified by various structures (1.0–27 ng mL−1).14,64–66 In summary, the high density and uniform distribution of protein immobilization on the MC-PC surface not only expands the detection range of the analyte, but also improves the repeatability of the protein chip.
4. Conclusion
A photocross-linked copolymer was synthesized and could rapidly form a macropore structure on PC surface in the PBS environment without the addition of porogen. The macropore copolymer was immobilized on PC surface by UV irradiation to form the MC-PC structure. Those macropore structures can be finely tuned through multiple dimensions including monomer structure, PBS and copolymer concentration. As a comparison, the characteristics of traditional free radical polymerization and high internal phase emulsion (HIPE) polymerization to form macropore structures are that, monomer polymerization and surface modification are carried out simultaneously and require the participation of porogen. This novel one-step photo-crosslinking, that separates the polymer preparation from the formation of the porous polymer surface, contributes to the regulation of polymer structure and function. In addition, the process is simple and environmentally friendly, since the macropore structure was formed in PBS without the addition of porogen and immobilized by one-step UV illumination. Furthermore, the MC-PC surface has high loading capacity (59 μg cm−2) and immobilization efficiency (92%) for protein immobilization, and the effect of inhibiting the coffee ring for protein immobilization. Immunoassay results show that the IgG-MC-PC surface has high sensitivity (LOD value of 5 ng mL−1) and broader dynamic range (0.005–50 μg mL−1) for biodetection of protein. The simplicity of the process, the high loading capacity and immobilization efficiency of the macropore structures provide a new way to prepare high-density 3D protein biochips based on polymer substrates.Author contributions
J. H. Z. and K. M. P. designed research; K. M. P. and R. P. W. performed research; J. H. Z., K. M. P., and R. P. W. performed data analyses; J. H. Z. and K. M. P. wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study is funded by the Guizhou Provincial Science and Technology Foundation (No. [2020]1Y043), Guizhou Provincial Department of Education Foundation (No. Qian-Cai-Jiao [2021]78), Special Fund of Qiannan Normal University for Nationalities (No. QNSY2018BS013), Shenzhen Science and Technology Program (No. JCYJ20190807160401657 and JCYJ20190807160415074), the National Natural Science Foundation of China (No. 22174167).References
- Y. Wang, Q. Li, H. Shi, K. Tang, L. Qiao, G. Yu, C. Ding and S. Yu, Lab Chip, 2020, 20, 4632–4637 RSC
.
- Y. Chu, Y. Gao, W. Tang, L. Qiang, Y. Han, J. Gao, Y. Zhang, H. Liu and L. Han, Anal. Chem., 2021, 93, 5129–5136 CrossRef CAS PubMed
- Y. Wang, K. Li, G. Xu, C. Chen, G. Song, Z. Dong, L. Lin, Y. Wang, Z. Xu, M. Yu, X. Yu, B. Ying, Y. Fan, L. Chang and J. Geng, Research, 2021, 2021, 1–9 CAS
- S. Han, Y. Xu, J. Sun, Y. Liu, Y. Zhao, W. Tao and R. Chai, Biosens. Bioelectron., 2020, 154, 112073 CrossRef CAS PubMed
- S. Liu, Z. Shen, L. Deng and G. Liu, Biosens. Bioelectron., 2022, 209, 114251 CrossRef CAS PubMed
- M. Park, B.-H. Kang and K.-H. Jeong, BioChip J., 2018, 12, 1–10 CrossRef CAS
- A. Hassibi, N. Wood and A. Manickam, presented in part at the 2018 IEEE Custom Integrated Circuits Conference (CICC), 8-11 April 2018, 2018 Search PubMed
- P. Howladar, P. Roy and H. Rahaman, IETE J. Res., 2020, 66, 662–676 CrossRef
- J. Grant, A. Özkan, C. Oh, G. Mahajan, R. Prantil-Baun and D. E. Ingber, Lab Chip, 2021, 21, 3509–3519 RSC
- S. A. Aghvami, A. Opathalage, Z. K. Zhang, M. Ludwig, M. Heymann, M. Norton, N. Wilkins and S. Fraden, Sens. Actuators, B, 2017, 247, 940–949 CrossRef CAS
- K. T. L. Trinh, D. A. Thai, W. R. Chae and N. Y. Lee, ACS Omega, 2020, 5, 17396–17404 CrossRef CAS PubMed
- A. M. Kaba, H. Jeon, A. Park, K. Yi, S. Baek, A. Park and D. Kim, Sens. Actuators, B, 2021, 346, 130511 CrossRef CAS
- M.-J. Bañuls, S. B. Morais, L. A. Tortajada-Genaro and Á. Maquieira, in Microarray Technology: Methods and Applications, ed. P. C. H. Li, A. Sedighi and L. Wang, Springer New York, New York, NY, 2016, pp. 37–51, DOI: DOI:10.1007/978-1-4939-3136-1_4
- J. Yan, C. Zhao, Y. Ma and W. Yang, Biomacromolecules, 2022, 23, 2614–2623 CrossRef CAS PubMed
- Y. Lee, H. J. Lee, K. J. Son and W.-G. Koh, J. Mater. Chem., 2011, 21, 4476–4483 RSC
- M. Drobota, S. Ursache and M. Aflori, Polymers, 2022, 14, 2307 CrossRef CAS PubMed
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7, 041501 CrossRef PubMed
- J. Gao, E. M. White, Q. Liu and J. Locklin, ACS Appl. Mater. Interfaces, 2017, 9, 7745–7751 CrossRef CAS PubMed
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS
- T. Brandstetter, S. Bohmer, O. Prucker, E. Bisse, A. zur Hausen, J. Alt-Morbe and J. Ruhe, J. Virol. Methods, 2010, 163, 40–48 CrossRef CAS PubMed
- B. Shrestha, C. Pipatpanukul, N. Houngkamhang, T. Brandstetter, J. Rühe and T. Srikhirin, Sens. Actuators, B, 2020, 320, 128358 CrossRef CAS
- D. Pospiech, D. Jehnichen, S. Starke, F. Müller, T. Bünker, A. Wollenberg, L. Häußler, F. Simon, K. Grundke, U. Oertel, M. Opitz and R. Kruspe, Appl. Surf. Sci., 2017, 399, 205–214 CrossRef CAS
- Z. M. Abdul Hamid, M. Florea, S. Fliegener, M. Schober, J. Hohe and J. Rühe, Adv. Eng. Mater., 2018, 21, 1800590 CrossRef
- V. Dhyani and N. Singh, ACS Appl. Bio. Mater., 2018, 1, 1355–1361 CrossRef CAS PubMed
- V. T. Widyaya, E. K. Riga, C. Müller and K. Lienkamp, Macromolecules, 2018, 51, 1409–1417 CrossRef CAS PubMed
- K. Wang, H. Dong, D. Zhou, Y. Ito, L. Hu, Z. Zhang and X. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 8722–8729 CrossRef CAS PubMed
- P. Jonkheijm, D. Weinrich, H. Schröder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS PubMed
- S. Zhang, Nat. Mater., 2004, 3, 7–8 CrossRef CAS PubMed
- X.-J. Li, M. Jia, Y.-X. Zhao, Z.-S. Liu and H. Akber Aisa, J. Chromatogr. A, 2016, 1438, 171–178 CrossRef CAS PubMed
- E. Korzhikova-Vlakh, M. Antipchik and T. Tennikova, Polymers, 2021, 13, 1059 CrossRef CAS PubMed
- A. Wörz, B. Berchtold, K. Moosmann, O. Prucker and J. Rühe, J. Mater. Chem., 2012, 22, 19547–19561 RSC
- K. B. Lynch, J. Ren, M. A. Beckner, C. He and S. Liu, Anal. Chim. Acta, 2019, 1046, 48–68 CrossRef CAS PubMed
- J. A. Kraai, G. L. Rorrer and A. X. Wang, J. Chromatogr. A, 2019, 1591, 162–170 CrossRef CAS PubMed
- Y. Cao, M. Lv, H. Xu, F. Svec, T. Tan and Y. Lv, Anal. Chim. Acta, 2015, 896, 111–119 CrossRef CAS PubMed
- M. Volokitina, M. Krutyakova, V. Sirotov, M. Larionov, T. Tennikova and E. Korzhikova-Vlakh, J. Pharmaceut. Biomed., 2019, 165, 242–250 CrossRef CAS PubMed
- A. S. Glotov, E. S. Sinitsyna, M. M. Danilova, E. S. Vashukova, J. G. Walter, F. Stahl, V. S. Baranov, E. G. Vlakh and T. B. Tennikova, Talanta, 2016, 147, 537–546 CrossRef CAS PubMed
- M. Rendl, A. Bonisch, A. Mader, K. Schuh, O. Prucker, T. Brandstetter and J. Ruhe, Langmuir, 2011, 27, 6116–6123 CrossRef CAS PubMed
- H. Schenderlein, A. Voss, R. W. Stark and M. Biesalski, Langmuir, 2013, 29, 4525–4534 CrossRef CAS PubMed
- X. Lin, K. Fukazawa and K. Ishihara, ACS Appl. Mater. Interfaces, 2015, 7, 17489–17498 CrossRef CAS PubMed
- P. A. Levkin, F. Svec and J. M. J. Fréchet, Adv. Funct. Mater., 2009, 19, 1993–1998 CrossRef CAS PubMed
- G. Dorman, H. Nakamura, A. Pulsipher and G. D. Prestwich, Chem. Rev., 2016, 116, 15284–15398 CrossRef CAS PubMed
- Z. Lin and J. M. Goddard, Colloids Surf., B, 2018, 172, 143–151 CrossRef CAS PubMed
- V. P. Dhende, S. Samanta, D. M. Jones, I. R. Hardin and J. Locklin, ACS Appl. Mater. Interfaces, 2011, 3, 2830–2837 CrossRef CAS PubMed
- O. Okay, Prog. Polym. Sci., 2000, 25, 711–779 CrossRef CAS
- M. Merhar, A. Podgornik, M. Barut, M. Žigon and A. Štrancar, J. Sep. Sci., 2003, 26, 322–330 CrossRef CAS
- N. Toshikj, J.-J. Robin, M. Ramonda, S. Catrouillet and S. Blanquer, ACS Appl. Polym. Mater., 2021, 3, 4966–4976 CrossRef CAS
- F. J. Hua, J. D. Nam and D. S. Lee, Macromol. Rapid Commun., 2001, 22, 1053–1057 CrossRef CAS
- M. Annaka, K. Motokawa, S. Sasaki, T. Nakahira, H. Kawasaki, H. Maeda, Y. Amo and Y. Tominaga, J. Chem. Phys., 2000, 113, 5980–5985 CrossRef CAS
- Y. Qi, Y. Wang, C. Zhao, Y. Ma and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 28690–28698 CrossRef CAS PubMed
- H. Lisalova, E. Brynda, M. Houska, I. Visova, K. Mrkvova, X. C. Song, E. Gedeonova, F. Surman, T. Riedel, O. Pop-Georgievski and J. Homola, Anal. Chem., 2017, 89, 3524–3531 CrossRef CAS PubMed
- Y. Qi, Y. Wang, C. Chen, C. Zhao, Y. Ma and W. Yang, ACS Appl. Bio. Mater., 2020, 3, 3203–3209 CrossRef CAS PubMed
- S. A. Rodriguez-Segui, J. I. Pons Ximenez, L. Sevilla, A. Ruiz, P. Colpo, F. Rossi, E. Martinez and J. Samitier, J. Biomed. Mater. Res., Part A, 2011, 98, 245–256 CrossRef PubMed
- K. Tsougeni, A. Tserepi, V. Constantoudis, E. Gogolides, P. S. Petrou and S. E. Kakabakos, Langmuir, 2010, 26, 13883–13891 CrossRef CAS PubMed
- B. J. Yun, J. E. Kwon, K. Lee and W.-G. Koh, Sens. Actuators, B, 2019, 284, 140–147 CrossRef CAS
- L. Cui, J. Zhang, X. Zhang, Y. Li, Z. Wang, H. Gao, T. Wang, S. Zhu, H. Yu and B. Yang, Soft Matter, 2012, 8, 10448–10456 RSC
- M. Dicuangco, S. Dash, J. A. Weibel and S. V. Garimella, Appl. Phys. Lett., 2014, 104, 201604 CrossRef
- L. Cui, J. Zhang, X. Zhang, L. Huang, Z. Wang, Y. Li, H. Gao, S. Zhu, T. Wang and B. Yang, ACS Appl. Mater. Interfaces, 2012, 4, 2775–2780 CrossRef CAS PubMed
- X. Shao, F. Duan, Y. Hou and X. Zhong, Adv. Colloid Interface, 2020, 275, 102049 CrossRef CAS PubMed
- S. Li, M. Dong, R. Li, L. Zhang, Y. Qiao, Y. Jiang, W. Qi and H. Wang, Nanoscale, 2015, 7, 18453–18458 RSC
- K. Tsougeni, K. Ellinas, G. Koukouvinos, P. S. Petrou, A. Tserepi, S. E. Kakabakos and E. Gogolides, Colloids Surf., B, 2018, 165, 270–277 CrossRef CAS PubMed
- Y. Qi, K. Li, C. Zhao, Y. Ma and W. Yang, J. Chem. Technol. Biotechnol., 2021, 96, 1902–1908 CrossRef CAS
- Y. Chen, L. P. Xu, J. Meng, S. Deng, L. Ma, S. Zhang, X. Zhang and S. Wang, Biosens. Bioelectron., 2018, 102, 418–424 CrossRef CAS PubMed
- M. Hu, J. Yan, Y. He, H. Lu, L. Weng, S. Song, C. Fan and L. Wang, ACS Nano, 2010, 4, 488–494 CrossRef CAS PubMed
- Y. Zhao, X. Zhao, C. Sun, J. Li, R. Zhu and Z. Gu, Anal. Chem., 2008, 80, 1598–1605 CrossRef CAS PubMed
- V. de Lange and J. Vörös, Anal. Chem., 2014, 86, 4209–4216 CrossRef CAS PubMed
- S. W. Han, S. Lee, J. Hong, E. Jang, T. Lee and W.-G. Koh, Biosens. Bioelectron., 2013, 45, 129–135 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00696d |
| This journal is © The Royal Society of Chemistry 2023 |