DOI:
10.1039/D3RA00514C
(Paper)
RSC Adv., 2023,
13, 9539-9554
Non-covalent interaction, adsorption characteristics and solvent effect of procainamide anti-arrhythmias drug on silver and gold loaded silica surfaces: SERS spectroscopy, density functional theory and molecular docking investigations†
Received
24th January 2023
, Accepted 1st March 2023
First published on 23rd March 2023
Abstract
First-principle calculations were systematically carried out to explore the structural and electronic properties of the non-covalent interaction of procainamide (PA) anti-arrhythmias drug molecules on silver-loaded and gold-loaded silica nanostructures. Computed adsorption energies presented a higher affinity of PA towards the Ag–SiO2 as compared with Au–SiO2 surfaces. The non-covalent interaction analysis revealed a weak van der Waals type of forces and hydrogen bonding, associated with a noticeable repulsive steric interaction. It was conceived that silver and gold decorated silica can be used for drug administration in biological systems due to the fact that their frontier molecular orbital energy levels were considerably altered upon absorption, decreasing the pertinent energy gaps. Moreover, the electronic spectra of PA⋯Ag–SiO2 and PA⋯Au–SiO2 structures investigated in different solvents display a notable blue shift, suggesting that noble metal-loaded silica can be effective in the context of drug delivery systems. Therefore, silver- and gold-decorated silica of three possible drug adsorption scenarios was fully analyzed to realize the associated bioactivity and drug likeness. Theoretical findings suggest that Ag- and Au–SiO2 nanocomposites can be considered potential drug delivery platforms for procainamide in medication protocols.
1. Introduction
Monitoring the impact of pharmaceutics used for pre- and post-operative medications to prevent adverse effects on an individual's health is a crucial task. If not administered properly, the drugs used in treatment may adversely disturb the physiology of organs and thus lead to organ failure. As a result of overdosing, toxicity and the long residence time in the body, therapeutic drugs have been carefully and strictly observed since the 70's.1 To better understand pharmacokinetics and pharmacodynamics (PK-PD) factors, clinical assessments were conducted according to the risk associated with the medicinal therapy.2 In human therapy studies involving drugs acting on the central nervous system (CNS), it is essential to focus on identifying the difference between their PK/PD relationships. The use of these methods could result in a more efficient selection of doses for CNS agents.
In both of its pre and post-operative treatments, procainamide (PA) is a widely used type I antiarrhythmic, which combats various atrial and ventricular dysrhythmias.3,4 The pharmaceutical and clinical impacts of PA have been extensively investigated. For example, it is likely that PA could be used as an alternative strategy to prevent the return of ventricular tachyarrhythmias and ICD discharge if radiofrequency ablation or beta-blockers are ineffective. Recurrent ventricular tachyarrhythmias after catheter ablation remain a challenging and poorly defined problem, including treatment with amiodarone or beta-blockers when those agents are ineffective or contraindicated. Especially for patients with implantable cardioverter-defibrillators (ICDs), electrical storm (ES) incidents can cause repeated interventions to the ICD system. There is a strong association between frequent ICD discharges and worse clinical outcomes, while recurrent ES can result in life-threatening conditions.4,5 Antiarrhythmic drugs, including PA, are usually treated first, followed by catheter ablation to prevent future ES possibilities.6,7 Medication with PA can cause a myocardial depression, which is associated with a drop in blood pressure and also associated with nausea, diarrhea and other symptoms. Furthermore, there is an association between PA and drug-induced lupus erythematosus in up to 30% of patients taking it for six months or longer and also for cancer prevention by hindering DNA methyltransferase activity.8 Nonetheless, several studies have provided evidence that PA may cause adverse effects, such as developing lupus in about 25–30% of users, decreasing their bone marrow cells and causing renal failure in a small number of users.9,10 PA can also cause cardiotoxicity, myocardial depression and polymorphic ventricular tachycardia.11,12 Long term use has also been reported to result in lethargy, confusion, seizures, antimuscarinic effects, agranulocytosis and thrombocytopenia.13
The objective from this study is to explore new means that help to lessen the negative impact of the chemotherapeutic medication PA, while not affecting its therapeutic efficiency. Such an approach could be achieved with through nano-medication delivery technologies, which promote therapeutic indices and decrease side effects. As a result of the proper incorporation of nanotechnology, including clinical applications involving the use of nanoscale drug carrier, cancer diagnosis has been remarkably revolutionized.14 Another innovative use of nanoscience in that direction is relevant to the surface enhanced Raman scattering (SERS) spectroscopic techniques. SERS is commonly used to improve Raman signals of small concentrations of biomolecules by employing noble metal containing nanoparticles as SERS substrates. In the past few years, increasing interest has been shown in predicting the spectral intensities resulting from SERS when organic analytes bind to such nanosize metal-containing substrates, as a result of this approach spectral intensities can be dramatically enhanced up to thousands of millions of times.15–17 Possible molecule–nanoparticle interaction profiles leading to the SERS effect can be further explored at the atomic level by first principle calculations.18,19 Moreover, the orientations of molecular anchoring on a noble metal surface are investigated. Molecule-nanoparticle vibrational frequencies explaining the Raman spectral fingerprints can be readily assigned based on theoretical models, leading to better understanding of the nature of molecule-metal interactions.20–23 It should be noted that for the synthesis of silver oxide complexes, either silver chloride or silver nitrate may be treated with sodium hydroxide, and that will result in silver(I) oxide (Ag2O) or otherwise the oxidation of metallic silver could result in silver(II) oxide. For silicon the Na2Si3O7 form of silica can be treated with concentrated acid to yield SiO2 according to: Na2Si3O7 + H2SO4 → 3SiO2 + Na2SO4 + H2O. In this work, however, we are focusing on theoretical aspects for the formation of Ag–O and Si–O bond. The literature survey reveals that the model of Ag–SiO2 is handily used in the computation of pharmaceutical as well as catalytic activities.24–43 Recently, the synthesis and characterization of silver/silica nanocomposites (Ag–SiO2) were reported, and based on that Ag/Au–SiO2 nanocomposites structure and complexes with PA in three different sites were explored by computational tools.44,45 Metallic silver tends not to react with oxygen. Silver and gold generally don't react with oxygen even at a very high temperature, being noble or inert metals as they are less reactive and placed at the bottom of the reactivity series.
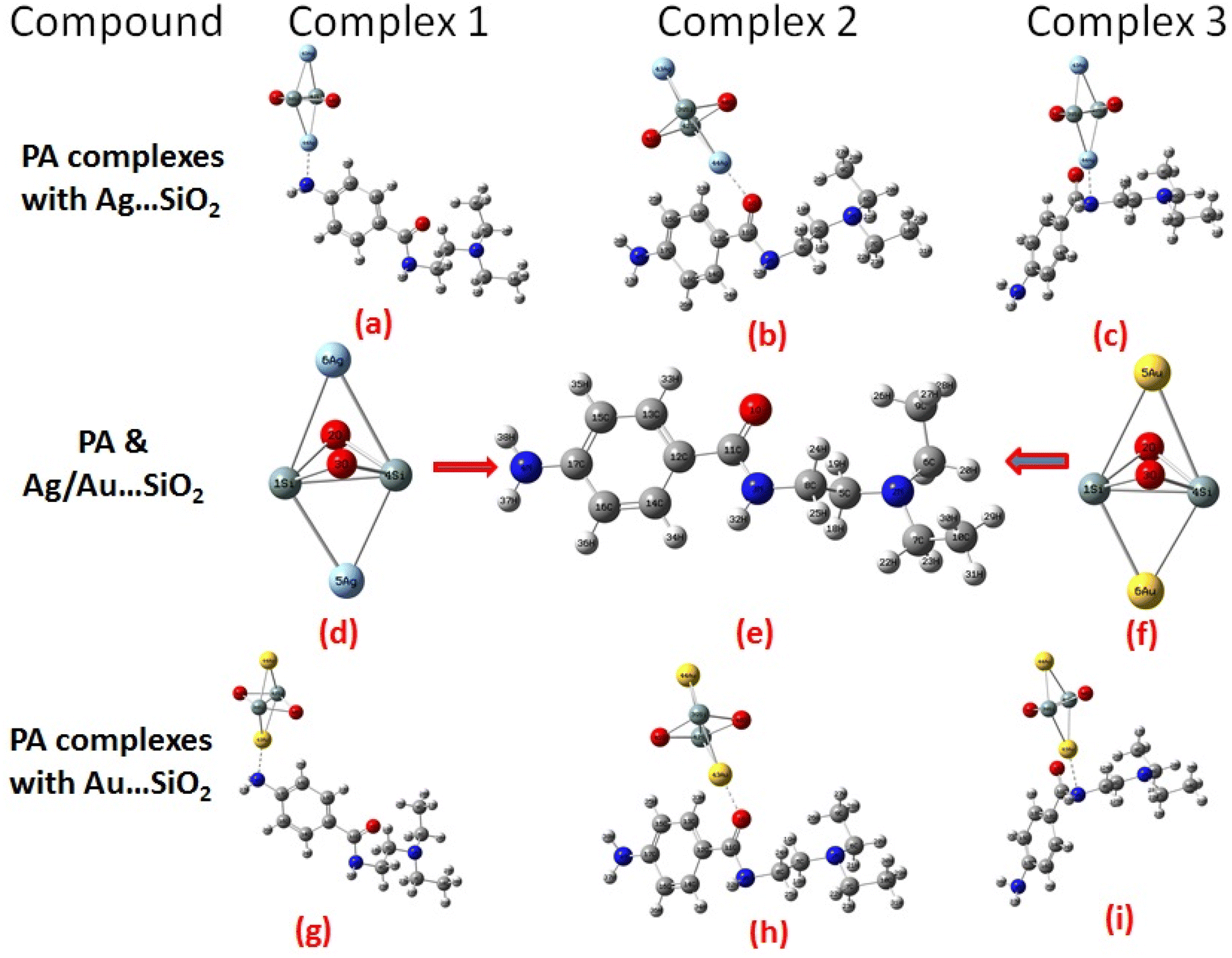
According to our knowledge, density functional theory (DFT) approach has not been applied to study PA adsorption on self-organized silver- and gold-loaded silicon substrates, aiming at revealing changes in the biological behaviors. As part of this work, the adsorption orientations and binding sites of PA on Ag– and Au–SiO2 nanocomposites are determined through Raman and SERS spectroscopies, and these results are interpreted using quantum chemical calculations.46 The adsorption spectra in different solvents have been also theoretically assessed. Owing to its structural characteristics and geometry, PA has the ability to meaningfully interact through different hot spots with the noble metal loaded silica. The possible formation of three different complexes, as depicted in Fig. 1, has been thoroughly studied. Moreover, quantitative analysis of PA was conducted with silver/gold nanoparticles decorated on SiO2 core shell, which was a key outcome of this study.
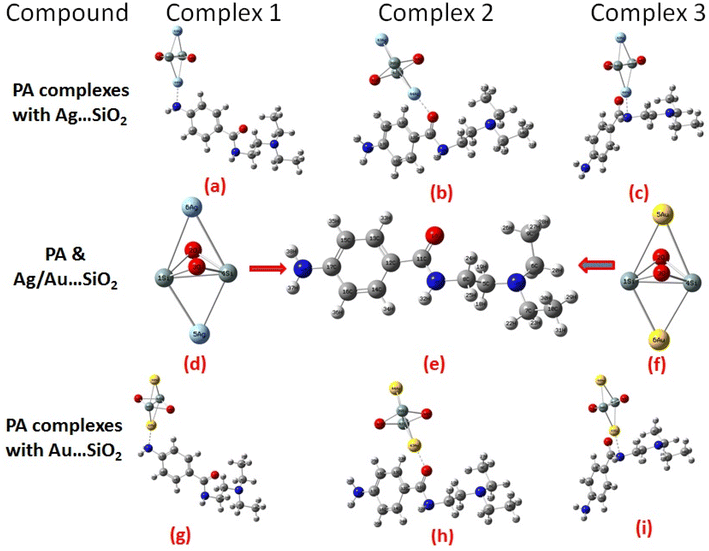 |
| | Fig. 1 Optimized structure and possible orientations of the PA drug with the Ag–SiO2 (a–c) and Au–SiO2 (g–i) complexes. The optimized structures of PA, Ag- and Au–SiO2 substrates are also shown (d–f). | |
2. Methodology
A first-principle calculation approach with the DFT method was implemented in this study. DFT calculations were shown to produce an adequate accuracy rate and found to be reliable in predicting structural and electronic features of medium–size complexes, both features are accompanied with a low computational cost.47–49 A feature like this is especially useful in studying large size molecular systems that may be difficult or impossible to study accurately using ab-inito methods. DFT calculations can be applied to individual specific problems due to its availability and reliable results that could be obtained. Based on the LANL2DZ basis set, Gaussian 0945 was used for all theoretical calculations incorporating the hybrid B3LYP functional. The LANL2DZ basis set is considered cost-effective and was shown to achieve good performance for metal–ligand interactions.50,51 A hypothetical UV analysis was conducted using the TD-DFT, and the polarizable continuum model using the integral equation formalism variant (IEFPCM) was applied to predict the solvent properties. For understanding the interaction between molecule and spherical shell clusters, as well as to determine the vibrational modes, extensive optimizations were performed to identify the global minima. The thermodynamic stability of PA, PA⋯Ag–SiO2 and PA⋯Au–SiO2 systems was investigated by assessing key parameters, namely the enthalpy, entropy and Gibbs free energy. The model of silver loaded silica was set to be with two silver atoms containing cluster. This shall better represent the silica core nature in the nanomaterial bulk system. Structural parameters of the systems, adsorption energies, MEP, HOMO–LUMO and chemical reactivity descriptors both in gas and aqueous phases were evaluated. Using Multiwfn,52 non-covalent interaction (NCI) was calculated, and electron distributions were analyzed on the basis of electron localization function (ELF), localized orbital locator (LOL), and reduced density gradient (RDG) properties. For the purpose of determining the drug properties of silica complexes of PA, a drug likeness analysis was performed. Swiss ADME53 was utilized to assess drug comparisons and lipophilicity parameters, while Pass analysis,54 Patch dock55 and Auto dock 4.2.6 56software were used to study the PA complexation with Ag and Au–SiO2 nanocomposites at the molecular level, identify binding sites of protein-ligand interactions, and predict the biological activity. It is established that the coinage metals of group XI, in particular silver and gold, are useful reagents due to their coordination chemistry and localized surface plasmonic resonance (LSPR) features.57
3. Result and discussion
3.1 Optimized structures of PA adsorbed on Ag- and Au-loaded silica nanocomposites
An overview of PA optimized structural parameters before and after the interaction with Ag–SiO2 and Au–SiO2 nanocomposites is presented in Table 1.
Table 1 Geometric parameters of PA before and after adsorption with Ag–SiO2 and PA⋯Au–SiO2 clusters, as optimized at the B3LYP/LANL2DZ level of theory. Refer to Fig. 1 for the possible optimized structures of Complexes 1–3 for each type of metals
| Parameters |
PA |
PA⋯Ag–SiO2 |
PA⋯Au–SiO2 |
| Complex 1 |
Complex 2 |
Complex 3 |
Complex 1 |
Complex 2 |
Complex 3 |
| Bond length (Å) |
| C11–O1 |
1.267 |
1.264 |
1.293 |
1.286 |
1.262 |
1.296 |
1.246 |
| N3–C8 |
1.472 |
1.476 |
1.477 |
1.476 |
1.475 |
1.481 |
1.529 |
| N3–C11 |
1.380 |
1.374 |
1.361 |
1.363 |
1.374 |
1.355 |
1.491 |
| N2–C5 |
1.478 |
1.478 |
1.475 |
1.471 |
1.476 |
1.475 |
1.478 |
| N2–C6 |
1.483 |
1.483 |
1.483 |
1.484 |
1.483 |
1.484 |
1.485 |
| N2–C7 |
1.481 |
1.482 |
1.483 |
1.483 |
1.482 |
1.483 |
1.487 |
| N4–C17 |
1.391 |
1.461 |
1.383 |
1.384 |
1.471 |
1.383 |
1.381 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Bond angles (°) |
| N4–C17–C15 |
120.7 |
119.9 |
120.2 |
120.7 |
119.7 |
120.6 |
120.7 |
| N4–C17–C16 |
120.7 |
119.7 |
121.0 |
120.7 |
119.5 |
120.7 |
120.6 |
| C12–C11–O1 |
121.6 |
120.7 |
123.0 |
119.5 |
120.7 |
123.8 |
124.3 |
| O1–C11–N3 |
120.8 |
121.9 |
118.5 |
121.5 |
121.9 |
117.3 |
118.2 |
| C11–N3–C8 |
121.4 |
121.2 |
123.0 |
124.5 |
121.3 |
123.1 |
113.6 |
| C5–N2–C6 |
114.1 |
114.0 |
114.2 |
114.6 |
114.2 |
114.2 |
113.0 |
| C5–N2–C7 |
114.9 |
114.9 |
113.5 |
113.9 |
113.6 |
114.8 |
114.1 |
There are different types of bonding present in the optimized PA molecules, such as N–C (5), N–H (3), O–C (1), C–H (18) and C–C (10). It was found that 2N–6C, 2N–7C, 2N–5C and 3N–8C bonds in PA complexes had the longest bond length in the range of 1.472–1.529 Å while 11C–1O had the shortest bond distance of 1.246 Å towards the range of double-bond. It is obvious that geometrical parameters of PA molecules were altered upon the adsorption with Ag/Au–SiO2 nanocomposites and that bond distances and bond angles values have increased. There were no changes observed in structural parameters of the Ag/Au–SiO2 nanocomposites, regardless its small cluster model, as listed in Table 1. This hints to the stable configurations of the silica-based core–shell upon the interaction with the PA drug molecules.
3.2 Electronic, chemical and non-covalent interaction analysis
A DFT analysis of PA with Ag/Au–SiO2 nanocomposites was conducted to determine the most energetically favorable conjugated configuration based on thermodynamic properties, structural, adsorption energy and other characteristics. Our studied adsorbents interact with the PA via the free amine N4, amide carbonyl O1 and amide nitrogen N3 findings are presented in Table 2. The adsorption energy (Eads) was computed for all configurations using the formula;| | |
Eads = EPA⋯Ag/Au–SiO2 − EAg/Au⋯SiO2 − EPA
| (1) |
where EPA⋯Ag/Au–SiO2 is the total energy calculated for the PA drug adsorbed on the Ag- or Au–SiO2 nanostructures, EAg/Au⋯SiO2 is the total energy calculated for the Ag- or Au–SiO2 nanostructures, and EPA is the total energy of the free drug molecule.58
Table 2 Calculated adsorption characteristics of the possible positions of Ag and Au–SiO2 nanocomplexes with the PA drug. Refer to Table 1 and Fig. 1 for the structural details
| Compound |
Type |
Energy (kcal mol−1) |
Adsorption energy (kcal mol−1) |
Bond length (Å) |
Bond angles (°) |
| PA⋯Ag–SiO2 |
Complex 1 |
216.35 |
−0.43 |
N4–Ag44 |
2.216 |
N4–C17–Ag44 |
25.4 |
| Complex 2 |
215.69 |
−1.09 |
O1–Ag44 |
2.188 |
C11–O1–Ag44 |
19.4 |
| Complex 3 |
215.48 |
−1.3 |
N3–Ag44 |
2.776 |
N3–C11–Ag44 |
101.6 |
| PA⋯Au–SiO2 |
Complex 1 |
217.03 |
−0.5 |
N4–Au44 |
2.117 |
N4–C17–Au44 |
25.5 |
| Complex 2 |
216.34 |
−0.19 |
O1–Au44 |
2.089 |
C11–O1–Au44 |
130.8 |
| Complex 3 |
216.47 |
−0.06 |
N3–Au44 |
2.135 |
N3–C11–Au44 |
110.4 |
The amine, amide carbonyl and amide nitrogen moieties in PA are considered as possible hotspots, and hence PA molecule can adsorb on the surface of Ag/Au–SiO2 via N4, O1 or N3 atoms (Fig. 1). In the adsorption facilitation step, the reactive regions of PA and Ag/Au–SiO2 approach to each other. It has been determined that in the gas and water phases, atoms N4, O1 and N3 of the drug system interacting with silica have charges of −0.7633, 0.6876, −0.688/−0.8267, −0.6878, −0.0.6835 (N4-gas) as well as −0.7575, −0.6970, −0.695 (N4-water); −0.3142, −0.4181, −0.420/-0.3104, −0.4225, −0.2336 (O1-gas) as well as −0.3765, −0.4353, −0.441/0.3766, −0.4447, −0.2807 (O1-water); −0.3852, 0.3757, −0.369/−0.3912, −0.3769, −0.6121 (N3-gas) as well as −0.3853, −0.3756,-0.373/−0.3903, −0.3747, −0.6201 (N3-water), while atoms N4, O1 and N3 in PA possess charges of −0.7103, −0.2874 and −0.4138 (gas)/−0.7217, −0.3521 and −0.4136 (water), respectively (Table S1†). These results indicate that the electronic characteristics of PA have remarkable altered upon the adsorption on silver and gold silica.
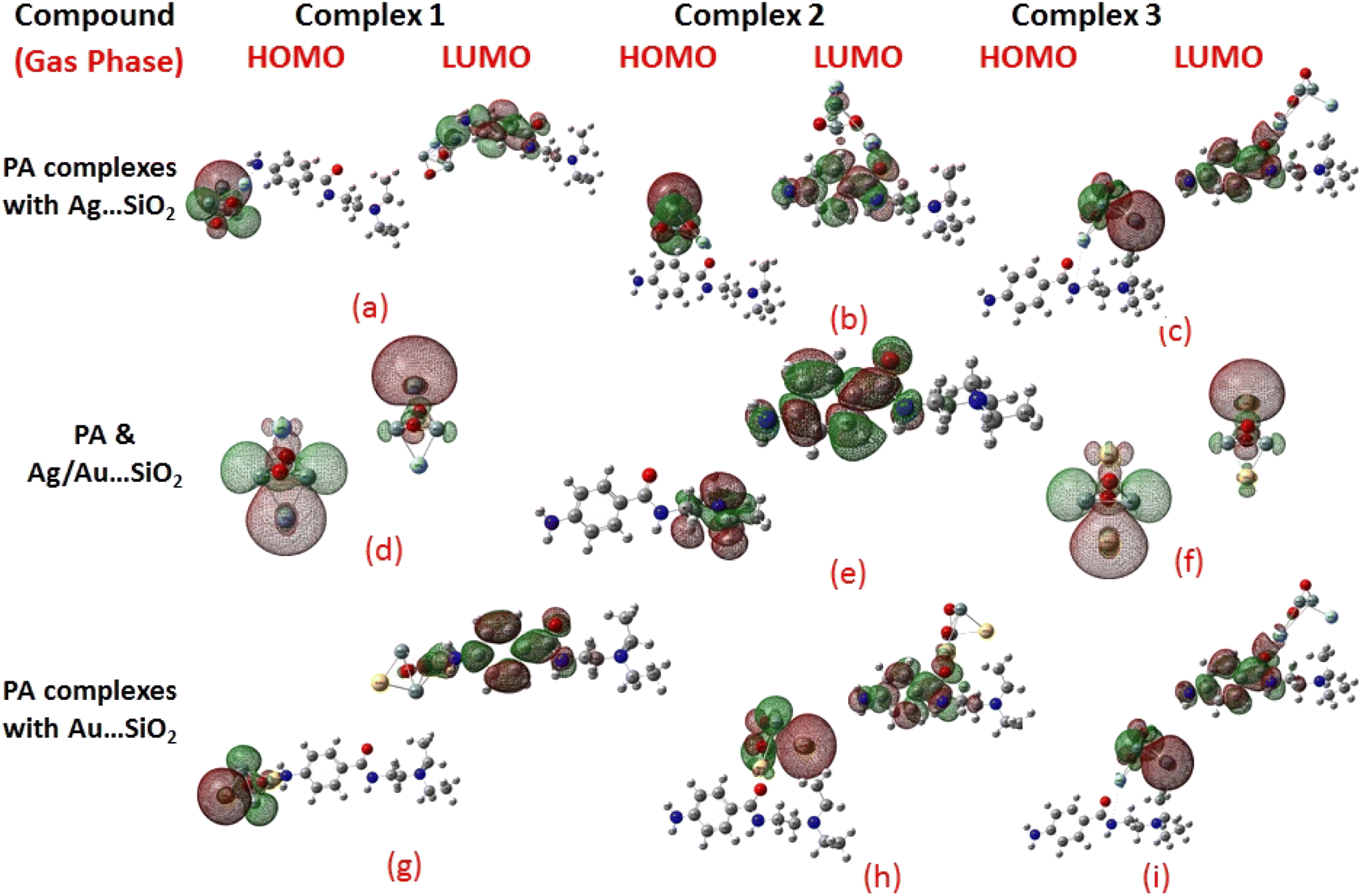
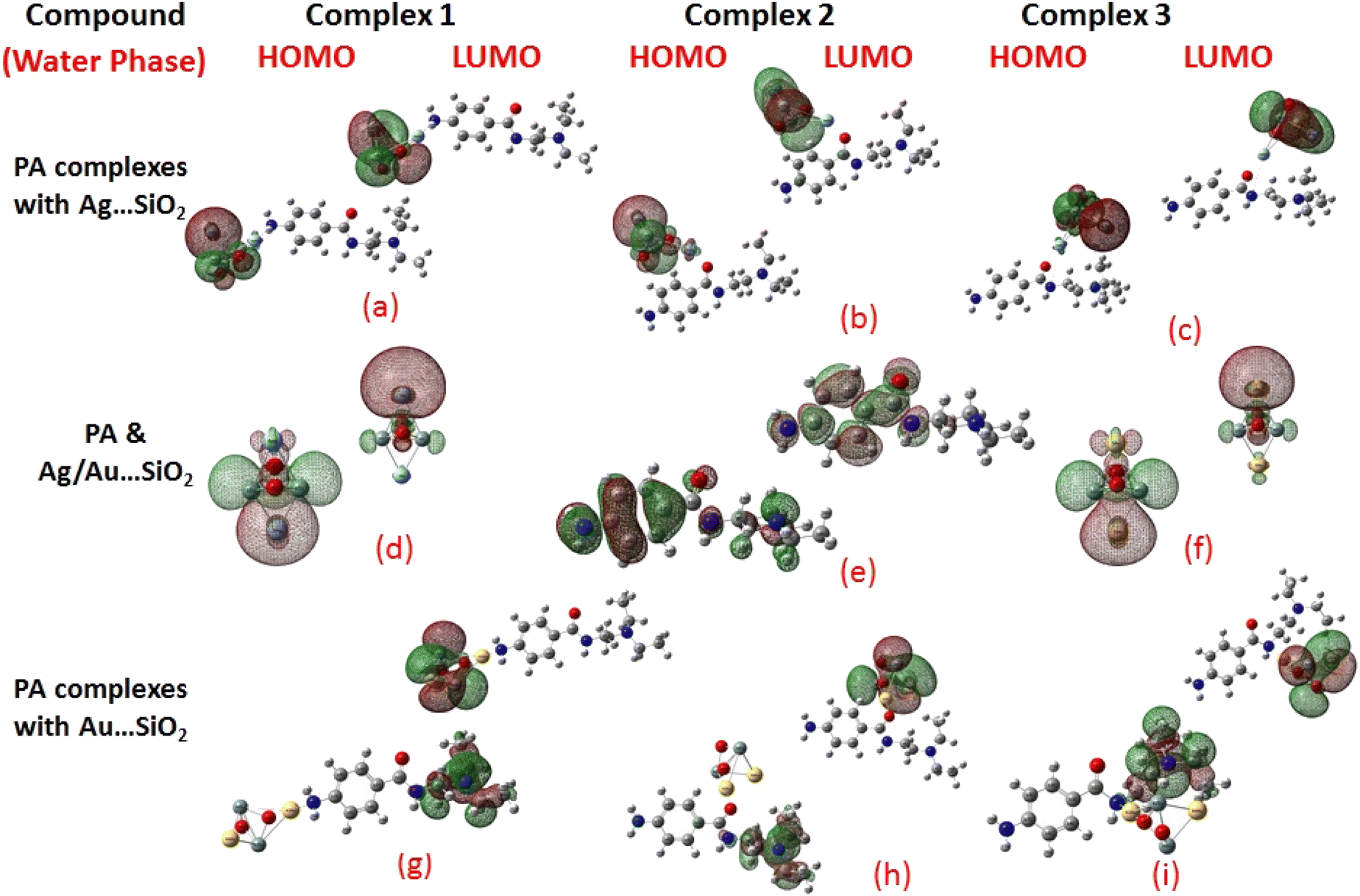
Frontier molecular orbitals (FMOs) and the molecular electrostatic potential (MEP) maps derived for PA and PA adsorbed on Ag/Au–SiO2 nanocomposites in gas and water phases are depicted in Fig. 2, 3, S1 and S2,† respectively. An investigation of procainamide electronic properties, including its HOMO, LUMO and dipole moment was carried out (Table S2†). There appear to be potential sites in the vicinity of the N4, O1, N3 and C11 atoms in PA, with a dipole moment of 5.67 (gas)/7.88 (water) Debye, which is supported by MEP diagrams (Fig. S1 and S2†). As shown in Table S2,† fermi levels are predicted as −2.98 eV and −3.20 eV, while EH and EL values are found as −5.53 eV, −5.73 eV and −0.43 eV, −0.68 eV, in gas and water media respectively. The dipole moment values of Ag/Au–SiO2 is 4.11 (in gas) and 3.77 (in water) Debye. In the PA, Ag–SiO2, Au–SiO2, PA⋯Ag–SiO2 PA⋯Au–SiO2 systems, the global reactivity values were critical to understand the reactivity and molecular stability. The frontier molecular orbitals can help in predicting the stability and reactivity of a given chemical system. The energy values of HOMO and LUMO for Ag/Au–SiO2 nanocomposite were −5.23/−6.37 eV (gas) and −3.69/−4.83 eV (aqueous), while the corresponding energy band gap values in Ag/Au–SiO2 nanocomposite were computed as 1.54/1.54 eV, respectively (Table S2†).
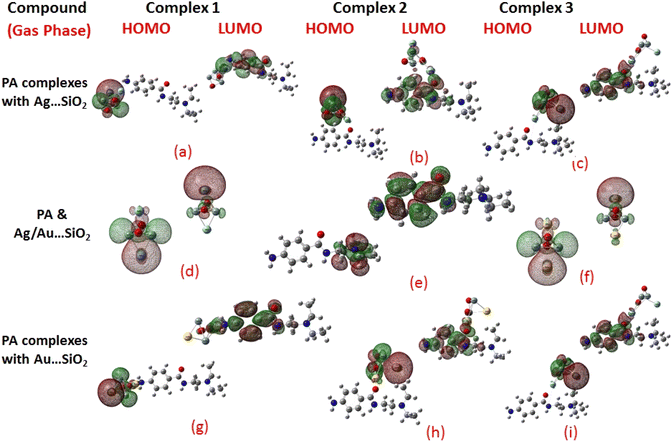 |
| | Fig. 2 HOMO and LUMO plots of PA drug with the Ag–SiO2 (a–c) and Au–SiO2 (g–i) complexes calculated in the gas medium. The corresponding frontier orbitals of PA, Ag- and Au–SiO2 substrates are presented (d–f) for sake of comparison. | |
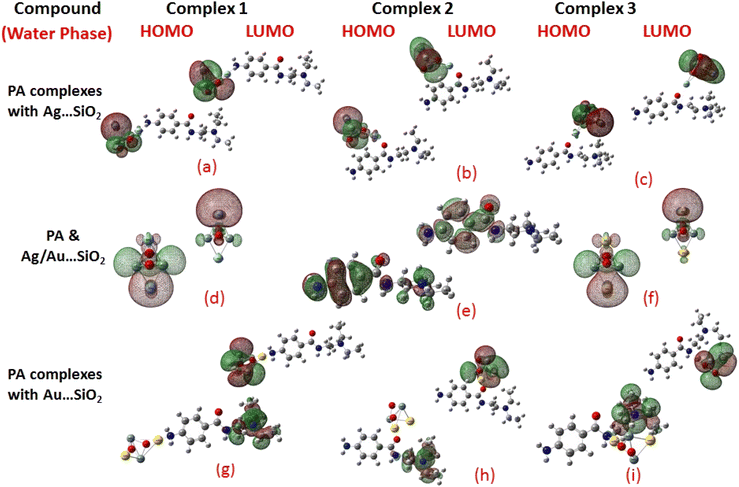 |
| | Fig. 3 HOMO and LUMO plots of PA drug with the Ag–SiO2 (a–c) and Au–SiO2 (g–i) complexes calculated in the aqueous medium. The corresponding frontier orbitals of PA, Ag- and Au–SiO2 substrates are presented (d–f) for sake of comparison. | |
When the values of dipole moment of Ag/Au–SiO2 nanocomposites and their complexes with PA were evaluated, a significant change in chemical and electronic properties as a result of the adsorption process can be noticed. The dipole moment in PA⋯Ag and PA⋯Au–SiO2 is varying from 10.66 to 18.91 Debye in gas and 11.21 to 22.63 Debye in water solvent. The dipole moment in PA is brought on by the non-uniform charge dispersion, and it is evident that after the interaction with Ag/Au–SiO2 all complex produced an appreciable dipole change (Table S2†). The charge redistribution between PA and Ag/Au–SiO2 in Complex 1, Complex 2 and Complex 3, respectively, causes improvement in dipole moments, from 10.66/12.02, 18.91/12.71 and 16.19/13.37 Debye for Ag/Au loaded clusters in gas phase to 11.21/12.41, 22.65/14.99 and 19.19/15.65 Debye for Ag/Au loaded clusters in water solvent. It is noticed that a drift in the development of dipole moment is due to interaction in the order of Au–SiO2 > Ag–SiO2. It is suggested that the PA⋯Ag- and PA⋯Au–SiO2 charge flow for Au is significantly higher than that for other adsorbents, explaining why PA⋯Au–SiO2 retains a stronger affinity. The electrons rich to electron-deficient zones of the MEP surfaces indicated by red to blue, respectively, signifies that PA⋯Ag and PA⋯Au–SiO2 nanocomposites exercise critical interactions and that charge transfer has taken from the metals on the silica to PA molecule.
PA, Ag–SiO2, Au–SiO2, PA⋯Ag–SiO2 PA⋯Au–SiO2 systems were highly dependent on the global reactivity values. As PA⋯Ag/Au–SiO2 nanocomposites reacted more efficiently than Ag/Au–SiO2 nanocomposites, the hardness of PA⋯Ag/Au–SiO2 nanocomposites decreased, allowing more PA molecules to be adsorbed (Table S2†). The outcome was a decrease in hardness, followed by an increase in softness and chemical potential. When complexing with the drug molecule, chemical potential values associated with metal-loaded silica changed from −4.46 (before complexation) to −3.23, −2.97 and −3.03 eV (gas)/−3.25, −3.22 and −3.24 eV (water) for PA⋯Ag–SiO2 system, and from −5.60 (before complexation) to −3.81, −3.59 and −3.91 eV (gas)/−3.81, −3.82 and −3.89 eV(water) for PA⋯Au–SiO2 for Complexes 1–3, respectively. A high tendency to accept electrons was observed with PA adsorption at all Ag- and Au–SiO2 complexes. A small variation to molecular orbital energies was caused by PA adsorption onto nanoclusters. HOMO, LUMO and energy gap (Eg) values of PA, Ag/Au–SiO2, and PA⋯Ag/Au–SiO2 are listed in Table S2.† A decrease in Eg was noticed in all adsorbed systems following adsorption, resulting in an increase in conductivity. As indicated by Mulliken charge values and chemical descriptors, the most reactive sites in PA and PA⋯Ag- and PA⋯Au–SiO2 presented a favorable charge transfer leading to more pronounced interactions. In addition, the thermodynamic properties of the investigated structures were analyzed to determine whether they were thermodynamically stable.59 The computed Gibbs energies indicated that PA is spontaneously adsorbed on the metal-SiO2 shell. Moreover, different Ag/Au–SiO2 showed distinct thermodynamic properties. Generally, as depicted in Table 3, negative values indicate exothermic, spontaneous and thermodynamically ordered interactions.
Table 3 Thermodynamic parameters of PA, PA⋯Ag–SiO2 and PA⋯Au–SiO2 clusters as computed in gaseous and water phases at the B3LYP/LANL2DZ level of calculations
| Atom |
PA |
Ag–SiO2 |
Au–SiO2 |
PA complexes with Ag–SiO2 |
PA complexes with Au–SiO2 |
| Complex 1 |
Complex 2 |
Complex 3 |
Complex 1 |
Complex 2 |
Complex 3 |
| Gas |
Water |
Gas |
Water |
Gas |
Water |
Gas |
Water |
Gas |
Water |
Gas |
Water |
Gas |
Water |
| Enthalpy (H) (Hartree) |
−747.0 |
−747.1 |
−449.7 |
−429.1 |
−1196.8 |
−1196.9 |
−1196.8 |
−1196.9 |
−1196.8 |
−1196.9 |
−1176.2 |
−1176.3 |
−1176.2 |
−1176.3 |
−1176.2 |
−1176.3 |
| Gibbs free energy (G) (Hartree) |
−747.1 |
−747.2 |
−449.8 |
−429.1 |
−1196.8 |
−1197.0 |
−1196.9 |
−1197.0 |
−1196.9 |
−1197.0 |
−1176.3 |
−1176.4 |
−1176.3 |
−1176.4 |
−1176.3 |
−1176.4 |
| Energy (E) (kcal mol−1) |
219.3 |
219.1 |
10.9 |
10.3 |
234.0 |
231.3 |
233.5 |
231.8 |
233.4 |
232.4 |
234.5 |
233.8 |
233.9 |
232.4 |
233.9 |
232.9 |
| Specific heat capacity (Cv)(Cal mol−1 Kelvin−1) |
56.01 |
56.2 |
24.2 |
22.3 |
96.9 |
89.3 |
98.3 |
94.9 |
98.4 |
96.9 |
96.1 |
94.1 |
97.7 |
94.2 |
97.1 |
95.6 |
| Entropy (S) (Cal mol−1 Kelvin−1) |
114.0 |
115.1 |
96.1 |
95.5 |
209.0 |
181.2 |
203.2 |
193.8 |
206.4 |
204.0 |
208.1 |
198.3 |
205.7 |
193.2 |
201.5 |
197.4 |
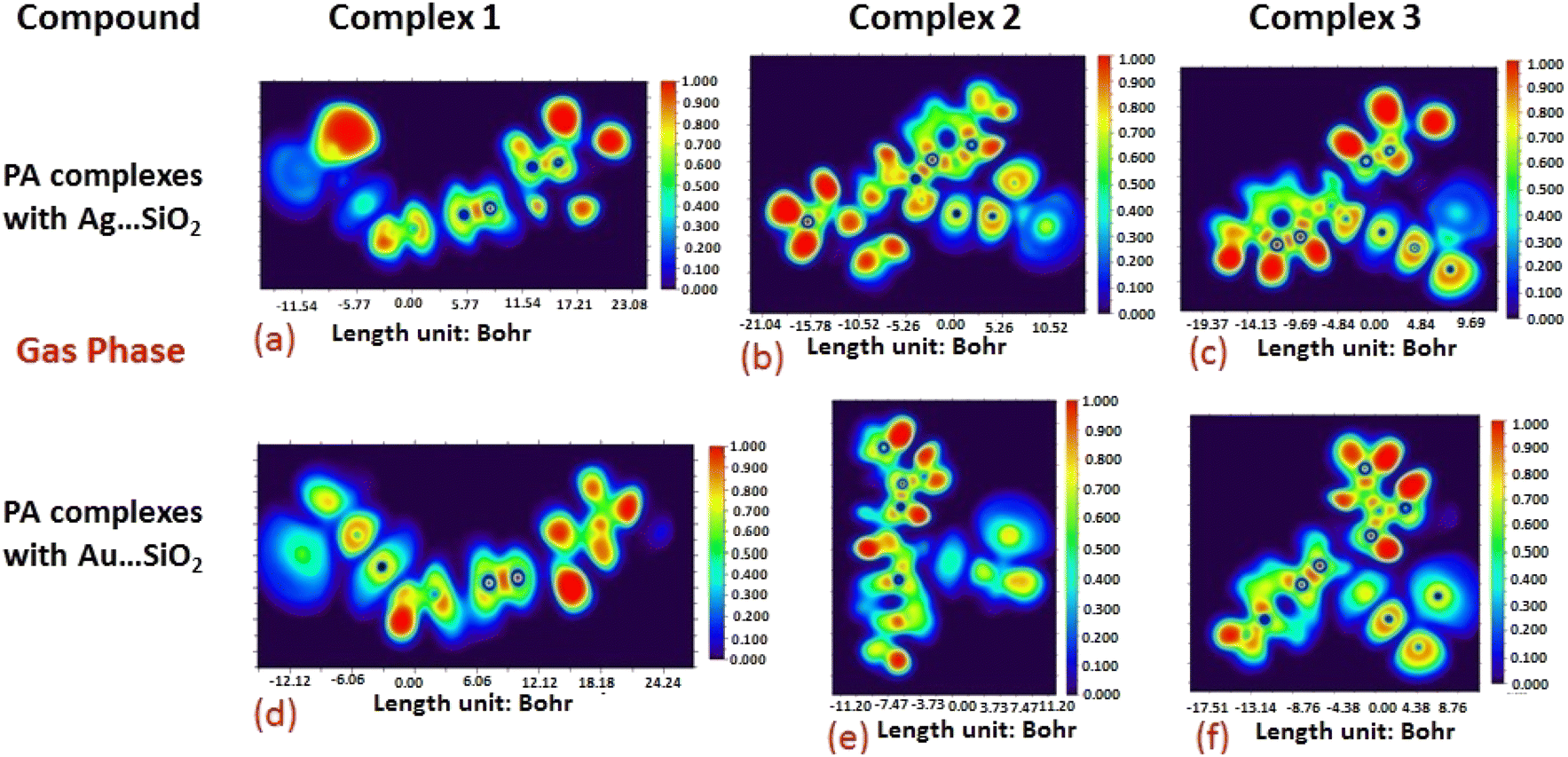
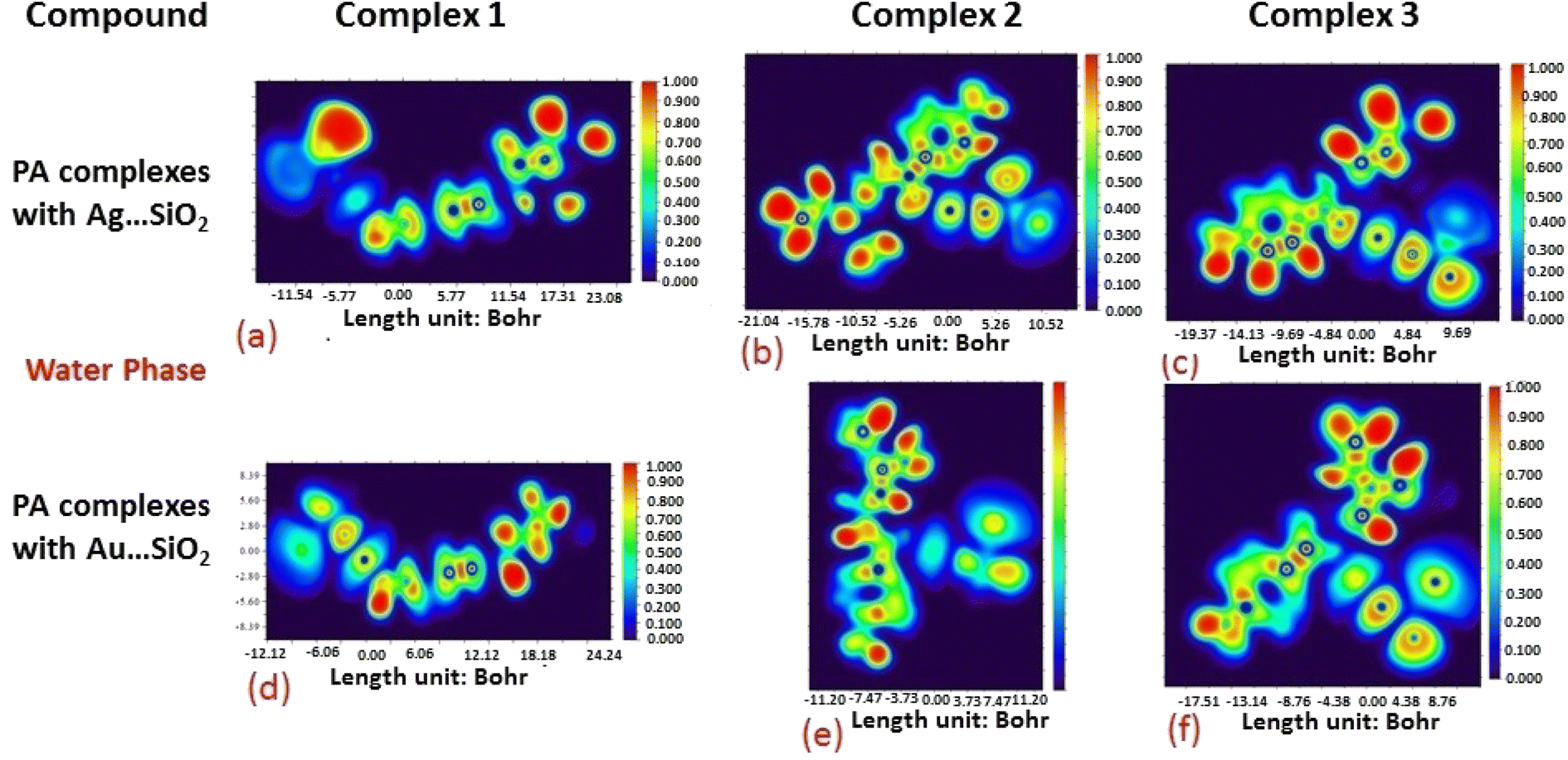
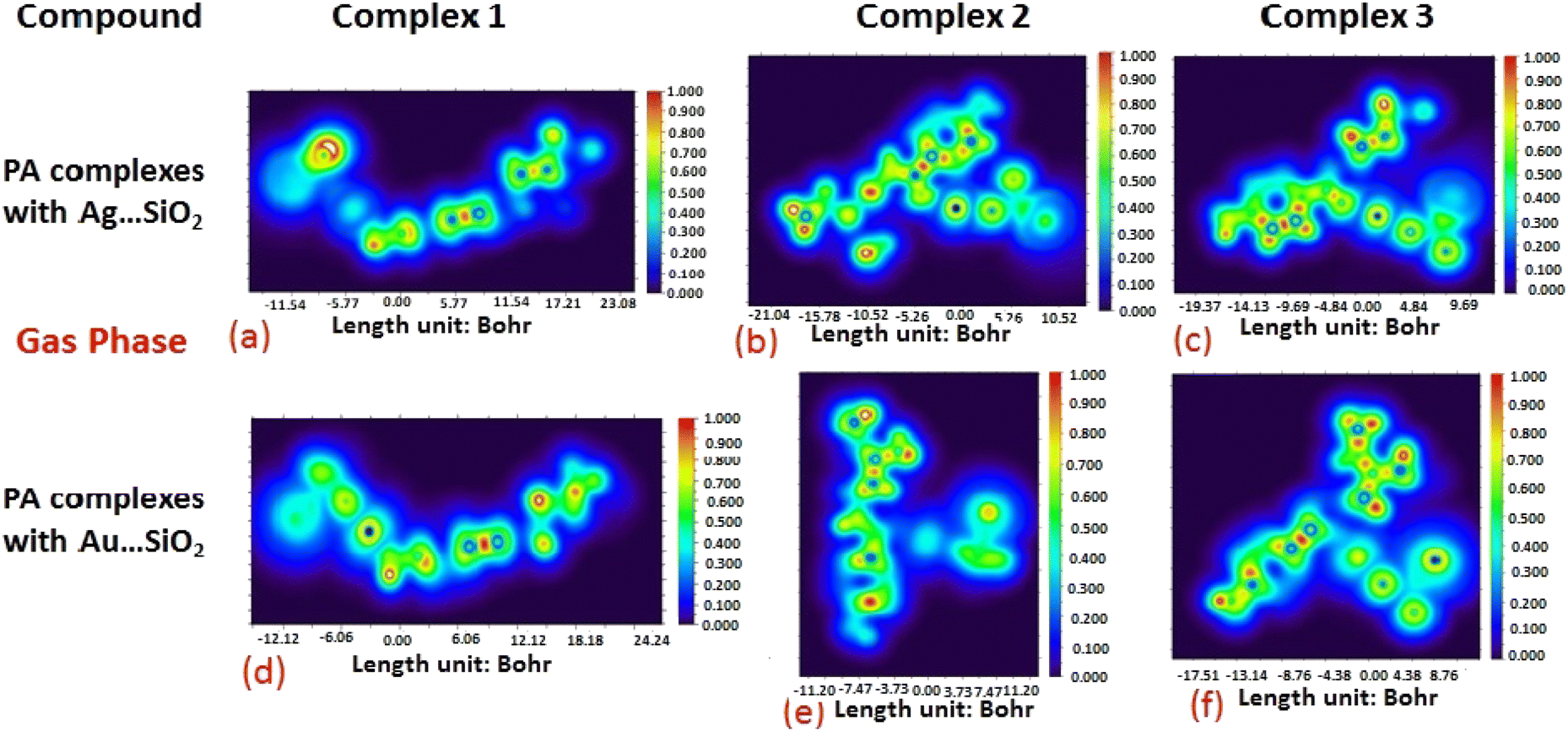
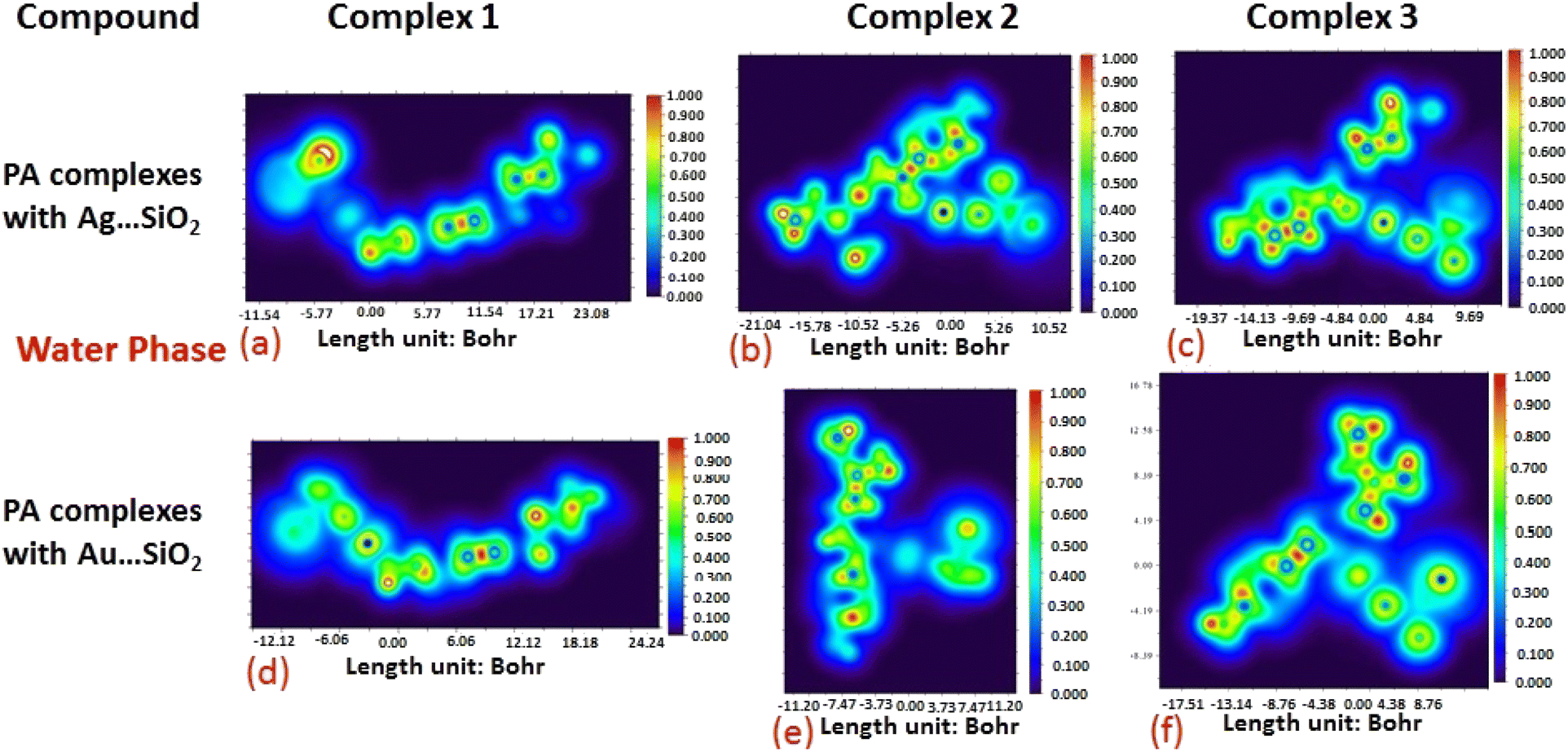
The electron density associated with the surface can be conveniently determined by electron pair density and maximum localized orbital overlap functions (see ELF and LOL diagrams depicted in Fig. 4–7).60,61 Multiwfn was used52 to generate color contours of PA⋯Ag- and PA⋯Au–SiO2 complexes in gas and water phases using electro-optical wave function analysis, as shown in Fig. 4–7. The colour code of the ELF model is based on the density of electron pairs of the drug molecule, while the LOL model is centred on the confined electron cloud. According to the computed ELF, blue to red colours correspond to parameters that range from 0.00 to 1.00 within the molecule in the range of −11.54 to 23.08, −21.02 to 10.52, and −19.37 to 9.69 Bohr3 for Complexes 1–3 of the PA⋯Ag–SiO2 cluster, respectively, from gas to water phases. For Complexes 1–3 of the gold-loaded silica, these values were calculated to be −12.12 to 24.24, −11.20 to 11.20, and −17.51 to 8.76 Bohr3 from gas and water phases. It should be noted that an electron density below 0.5 Bohr3 indicates localized bonds with non-bonded electrons, while values above 0.5 Bohr3 demonstrate a delocalized electron condition. While a high density due to delocalized electrons can be clearly viewed in the red region of the ELF contour around hydrogen atoms (H37, H27, H28, H22 and H23), low density electron clouds are located in the vicinity of carbon atoms (C7, C9, C10, C11 and C12). The value of 0.5 Bohr3 describes in what way an electron localization outweighs the density of electrons in the LOL profile, corresponding to ranges of values of (PA⋯Ag–SiO2 from gas to water phases) −11.54 to 23.08, −21.04 to 10.52, and −19.37 to 9.69 Bohr3 for Complexes 1–3, and to ranges of values of (PA⋯Au–SiO2 from gas to water phases) −12.12 to 24.24, −11.20 to 11.20, and −17.51 to 8.76 Bohr3. Here, 0.5 Bohr3 reflects that the electron localization triumphs over an electron density. A high level of covalent bonding is hence demonstrated by a green region (with high LOL values) in the diagram. These contours provide an idea of bonding and non-bonding types of electrons, and hence understand the most probable interactions involved in the process.
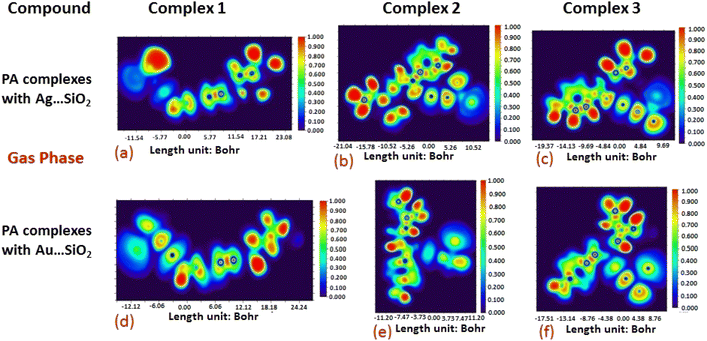 |
| | Fig. 4 Gas-phase electron localization function (ELF) contours associated with PA⋯Ag–SiO2 (a–c) and PA⋯Au–SiO2 (d–f). ELF represents the relative degree of electron localization in the periodic structures. Red contours correspond to higher ELF values, dark-blue contours correspond to lower ELF values, while green contours indicate intermediate localization of electron density. | |
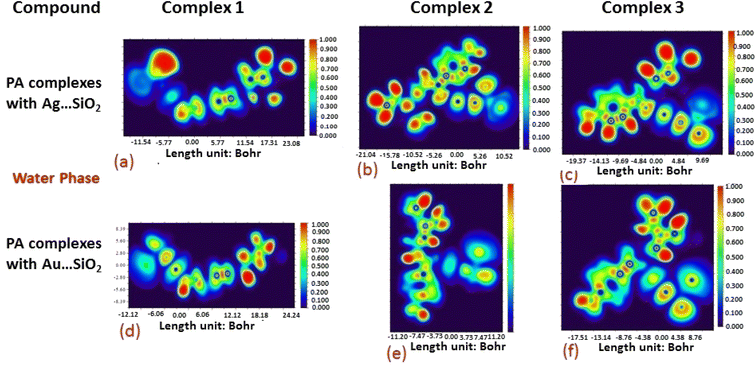 |
| | Fig. 5 Aqueous phase electron localization function (ELF) contours associated with PA⋯Ag–SiO2 (a–c) and PA⋯Au–SiO2 (d–f). Refer to caption of Fig. 4 for further descriptions. | |
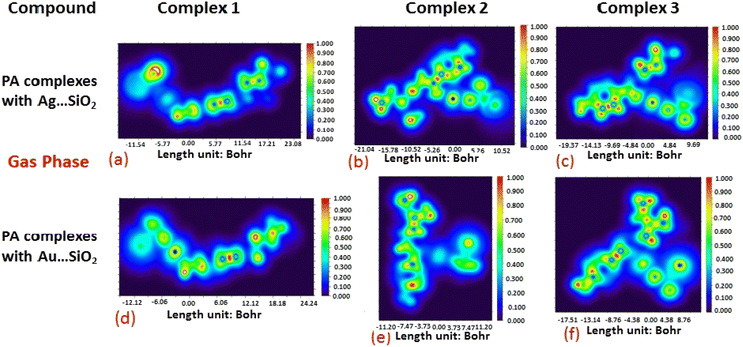 |
| | Fig. 6 Illustrations of maximum localized orbital overlap (LOL) functions depicted for PA⋯Ag–SiO2 (a–c) and PA⋯Au–SiO2 (d–f) complexes computed in the gas phase. LOL values help to distinguish the nature of atomic interaction by analyzing the localization of electronic density. Covalent regions of high LOL values are depicted by red contours, while electron depletion regions (can be indicative of non-covalent interaction) are shown by blue contours. | |
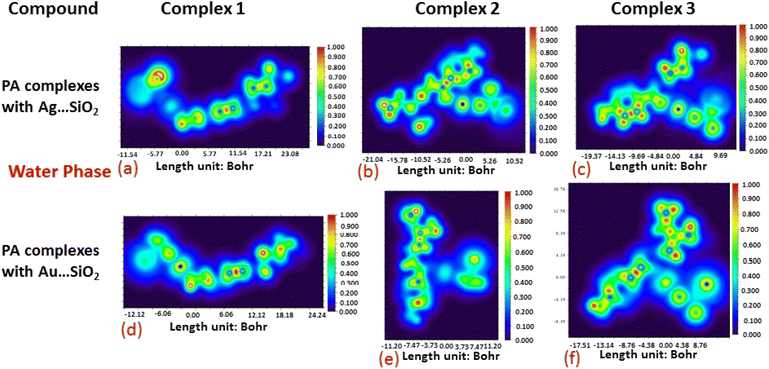 |
| | Fig. 7 Illustrations of maximum localized orbital overlap (LOL) functions depicted for PA⋯Ag–SiO2 (a–c) and PA⋯Au–SiO2 (d–f) complexes computed in water. Refer to caption of Fig. 6 for further descriptions. | |
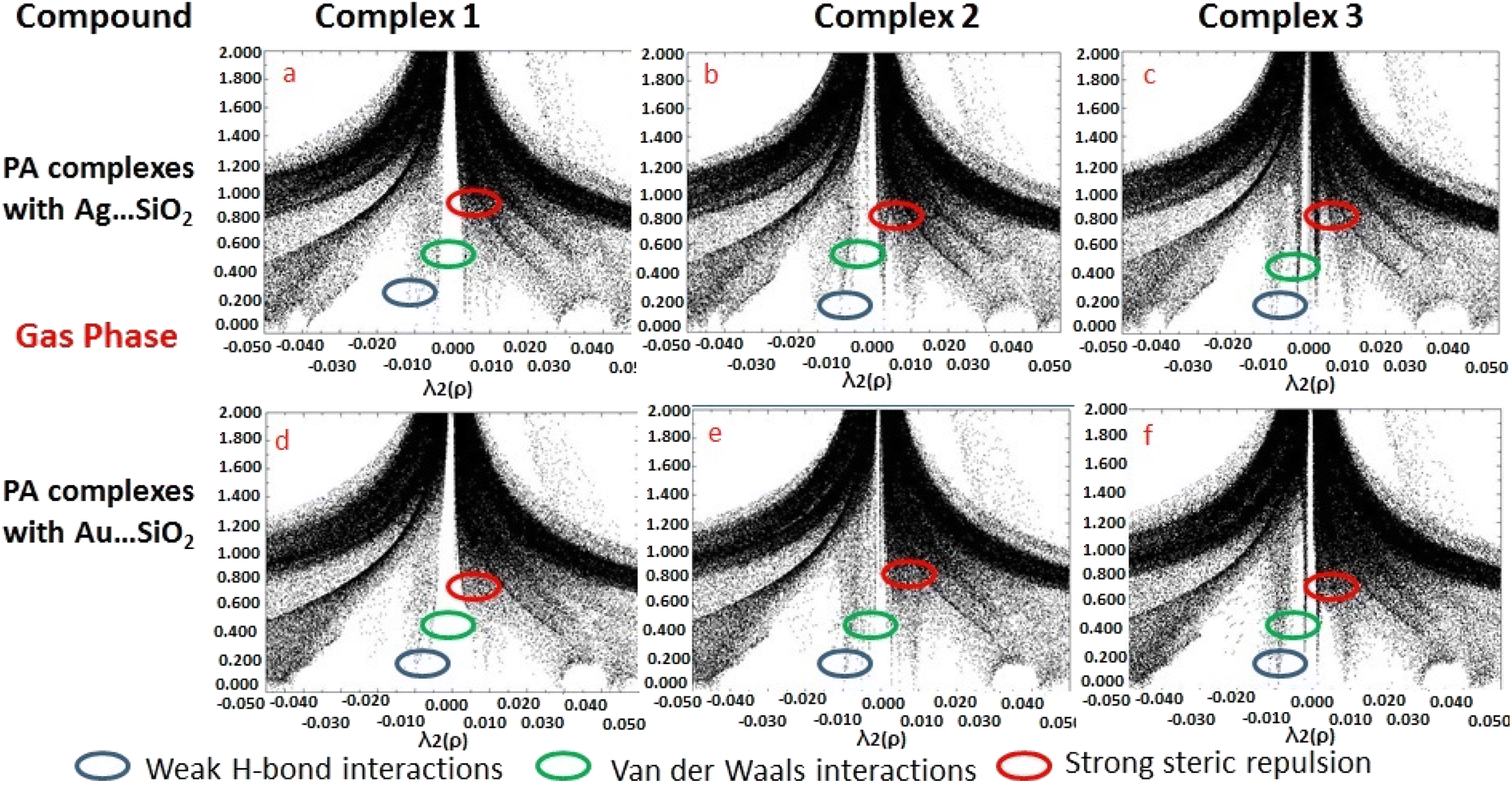
Based on the topological analysis provided by the reduced density gradient (RDG) model, non-covalent interactions are conveniently depicted visually as shown in Fig. 8. Generally, noncovalent interactions play a crucial role since they are notably weak in comparisons to covalent bonding. A low RDG value and a low electron density value isolate the weak interactions. RDG analyses were carried out to understand the nature of interactions between PA molecules and noble metal complexes. In the depicted graphs, the green, blue and red regions represent weak attractive forces (λ2 = 0) namely van der Waals interactions, strong attractive forces, and steric repulsion forces, respectively. The negative value of λ2(ρ) indicates a strong attractive interaction, while the positive values reflect a repulsive interaction force. PA adsorbing to Ag/Au–SiO2 appears to significantly interact with the metal atom.62,63 According to the RDG plot depicted in Fig. 8 and S3, the PA⋯Ag–SiO2 complexes present a stronger interaction than PA⋯Au–SiO2 analogues.
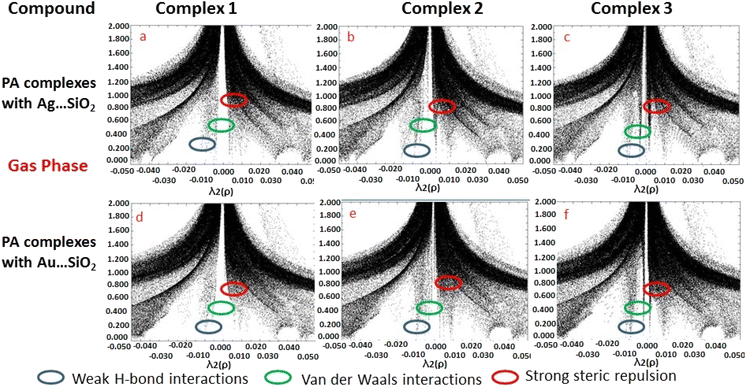 |
| | Fig. 8 Reduced density gradient (RDG) scattered graphs PA⋯Ag–SiO2 (a–c) and PA⋯Au–SiO2 (d–f) complexes modeled in the gas medium. Different possible interaction modes are labeled. | |
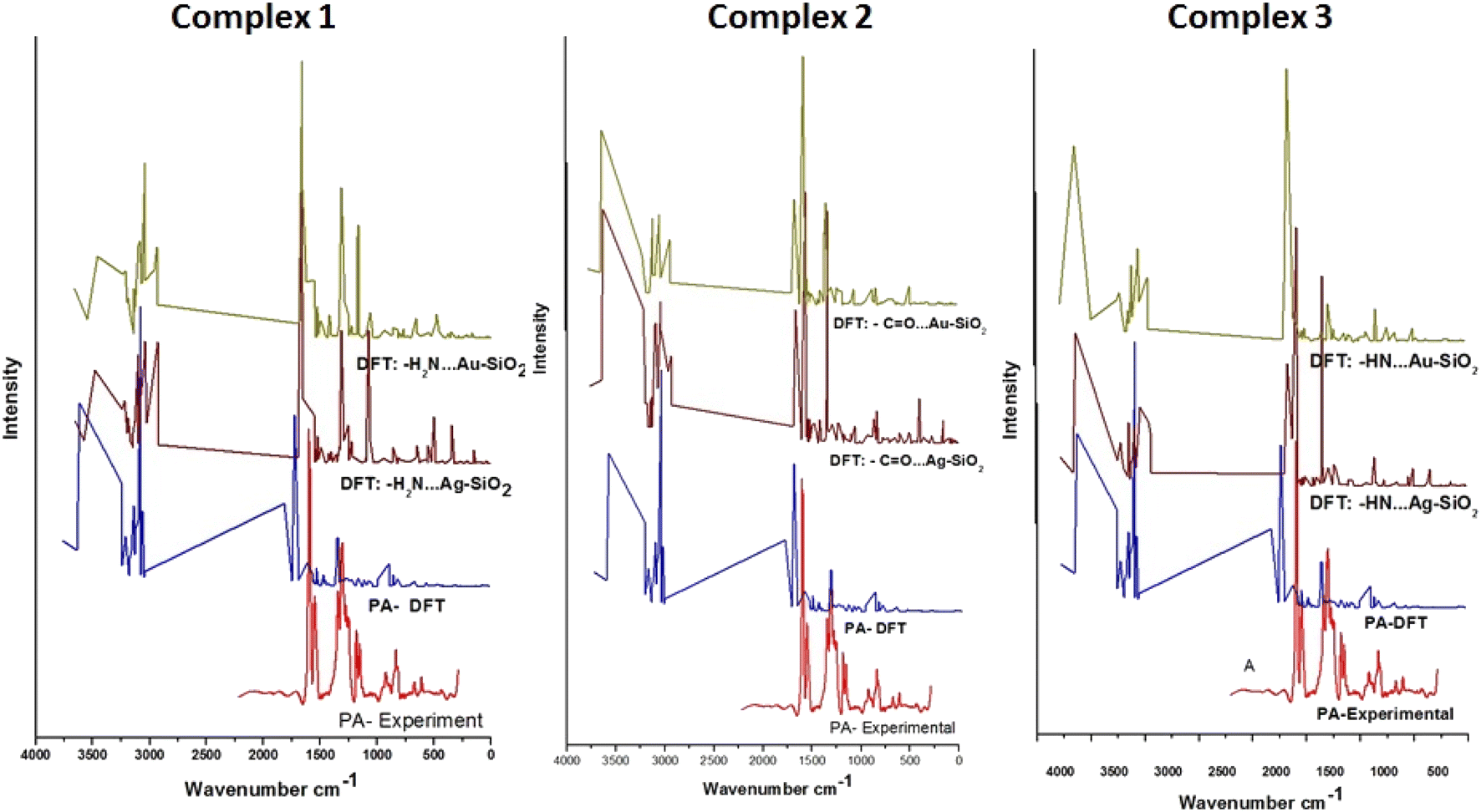
3.3 SERS and absorption spectroscopic analysis
To evaluate the SERS enhancement properties of metal-loaded silica nanomaterials, DFT calculations for each possible complexation scenario were carried out. According to Fig. 9, the enhancement of Complexes 1–3 seems significant for all fundamentals of PA⋯M–SiO2 as compared to non-interaction PA (Table 4). The Raman line at 3603 cm−1 belongs to a stretching N–H mode, which is shifted, for Complexes 1–3 respectively, to 3484, 3630 and 3633 cm−1 in the PA⋯Ag–SiO2; and to 3464, 3633 and 3630 cm−1 in the PA⋯Au–SiO2 spectra. As PA drug molecules are expected to respond in the SERS spectrum differently from their normal Raman spectrum, the line observed at 1317 cm−1 in the Raman spectrum shifted to 1556, 1575 and 1584 cm−1 (PA⋯Ag–SiO2) and to 1557, 1580 and 1417 cm−1 (PA⋯Au–SiO2) for Complexes 1–3, respectively. It is predicted that Au-Complex 3 configuration encountered the least amount of shift in band positions. According to the PED analysis, this band is attributed to N–H bending and shifts slightly towards higher frequencies. The SERS phenomenon was noticed to as well cause other sets of low-frequency bending modes corresponding to the HNH, OCN and CNH vibrational modes to enhance. The medium intensity signature peak appearing at 1802 cm−1 and assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching was drastically shifted to lower wavenumbers in the range of 1664–1623 cm−1 for the case of silver-loaded silica and of 1667–1658 cm−1 for gold-loaded silica nano-shells. Maximum theoretical Raman enhancement factor (EF) values of 2.33, 0.46 and 0.14 (Ag-loaded), and 0.68, 0.86 and 4.07 (Au-loaded) were detected for Complexes 1–3, respectively. Various modes exhibited blue or red shifts when considering PA interactions with Ag/Au–SiO2. Thus, it can be established that Ag/Au–SiO2 nanocomposites can be used as drug sensing devices using SERS techniques.64
O stretching was drastically shifted to lower wavenumbers in the range of 1664–1623 cm−1 for the case of silver-loaded silica and of 1667–1658 cm−1 for gold-loaded silica nano-shells. Maximum theoretical Raman enhancement factor (EF) values of 2.33, 0.46 and 0.14 (Ag-loaded), and 0.68, 0.86 and 4.07 (Au-loaded) were detected for Complexes 1–3, respectively. Various modes exhibited blue or red shifts when considering PA interactions with Ag/Au–SiO2. Thus, it can be established that Ag/Au–SiO2 nanocomposites can be used as drug sensing devices using SERS techniques.64
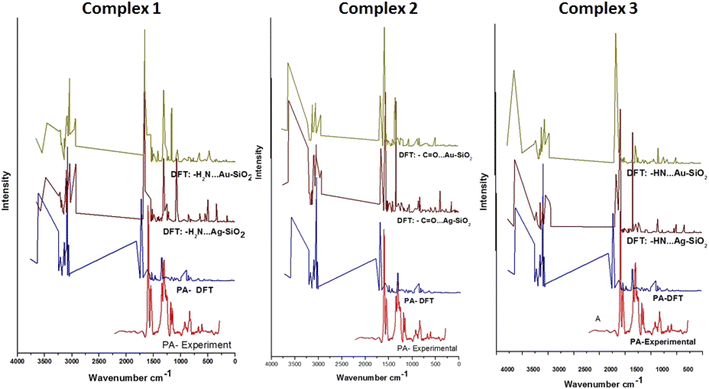 |
| | Fig. 9 Raman spectra of PA compared to the theoretical Raman and SERS spectra of PA, PA⋯Ag–SiO2 and PA⋯Au–SiO2 clusters of the three possible complex formations. Tentative vibrational mode assignments are presented in Table | |
Table 4 Experimental and calculated Raman parametersa of PA drug, and computed SERS vibrational parameters of the modeled PA⋯Ag–SiO2 and PA⋯Au–SiO2 complexes as predicted with the B3LYP/LANL2DZ level
| PA Raman |
Calculated SERS |
Assignments (PED) |
| PA⋯Ag–SiO2 |
PA⋯Au–SiO2 |
| Experimental |
Calculated |
Complex 1 |
Complex 2 |
Complex 3 |
Complex 1 |
Complex 2 |
Complex 3 |
| Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
Wave number (cm−1) |
Intensity |
| Descriptions of labels used: γ-stretching, β-in-plane bending, vs-very strong, s-strong, m-medium, w-weak, vw-very weak. |
| |
|
3603 |
250 s |
3484 |
128 m |
3630 |
292 s |
3633 |
416 vs |
3464 |
106 m |
3633 |
349 vs |
3630 |
414 vs |
γ NH |
| |
|
3235 |
142 s |
3103 |
149 s |
3220 |
169 s |
3209 |
119 m |
3101 |
119 m |
3227 |
106 m |
3216 |
103 m |
γ CH |
| |
|
3124 |
110 m |
3088 |
76 m |
3095 |
151 s |
3084 |
161 s |
3088 |
125 m |
3107 |
173 s |
3101 |
161 s |
γ CH |
| |
|
3063 |
382 vs |
3044 |
155 s |
3044 |
176 s |
3045 |
172 s |
3043 |
226 s |
3044 |
181 s |
3043 |
170 s |
γ CH |
| |
|
3048 |
103 s |
2930 |
166 s |
2939 |
109 m |
2935 |
146 s |
2937 |
117 m |
2940 |
131 m |
2950 |
136 m |
γ CH |
| 1598 |
4375 vs |
1802 |
114 s |
1664 |
380 vs |
1665 |
167 s |
1623 |
130 s |
1659 |
192 s |
1667 |
212 s |
1658 |
578 vs |
γ OC |
| 1589 |
4104 vs |
1317 |
6 vw |
1556 |
67 m |
1575 |
314 vs |
1584 |
705 vs |
1557 |
75 m |
1580 |
496 vs |
1417 |
11 vw |
γ NC |
| 1343 |
1818 vs |
1347 |
25 vw |
1316 |
182 s |
1348 |
290 vs |
1343 |
573 vs |
1317 |
195 s |
1348 |
203 vs |
1342 |
21 vw |
β HNC |
| 1181 |
1041 vs |
|
|
1073 |
182 s |
1063 |
21 vw |
|
|
1176 |
130 m |
1059 |
32 vw |
|
|
β HNH |
| 475 |
229 s |
|
|
498 |
65 w |
488 |
1 vw |
494 |
1 vw |
493 |
19 vw |
494 |
37 vw |
494 |
29 vw |
γ SiO |
| 356 |
196 s |
|
|
345 |
53 w |
407 |
56 w |
347 |
47 w |
356 |
9 vw |
407 |
2 vw |
356 |
3 vw |
β OSiSi |
Using four different solvents, water (polar protic), methanol (polar protic), DMSO (polar aprotic) and chloroform (non-polar), TD-DFT in IEFPCM simulates the electronic spectrum of PA.65 Fig S4† depicts the absorption spectra of free PA and compared with PA⋯Ag- and PA⋯Au–SiO2 complexes. Based on the trend shown, it can be realized that the adsorption bands for PA molecules associated with Ag/Au–SiO2 dissolved in water, chloroform, DMSO and methanol are estimated, respectively, at 481, 482, 481 and 488 nm (Complex 1)/468, 469, 467 and 464 nm (Complex 2)/482, 483, 481 and 483 nm (Complex 3) for PA⋯Ag–SiO2; while for PA⋯Au–SiO2 solvents bands are predicted, respectively, at 482, 483, 481 and 483 nm (Complex 1)/332, 333, 332 and 334 nm (Complex 2)/332, 333, 332 and 335 nm (Complex 3). The prominent absorption bands for non-interacting PA are estimated around 244, 244, 244, 245 nm in DFT compared to 260 nm for the experimental aqueous medium. The UV-vis red shift can be noticed in Fig. S4 and Table S3.† The strongest absorption is linked with silver silica surfaces. By virtue of their lower and higher bands gaps, the valence electrons of PA⋯Ag- and PA⋯Au–SiO2 are tightly bound in polar protic solvents and loosely bound in polar aprotic solvents. As a result of aggregation of nanomaterials, the charge transfer is observed as a band shift after interaction.
3.4 Evaluation of drug-likeness and lipophilicity properties
As a result of its bioavailability, a molecule can be considered a drug if it can be administered orally. To design drugs, scientists look for a substance that can penetrate into the body, binds to proteins and interferes with toxic effects. It is important to consider both physical and chemical properties when calculating drug-related properties.66–68 Modeled PA⋯Ag- and PA⋯Au–SiO2 nanostructures exhibited interesting bioactivity scores based on physicochemical, hydrogen bonding (HB), rotatable bonding, bio-availability scores, log-kp, BBB and water solubility results and values (Table 5). In addition, a lipophilicity of a drug molecule determines its drug-likeness based on its physicochemical properties. Table 6 shows that PA⋯Ag- and PA⋯Au–SiO2 nanoclusters transport well inside the human lipid system on basis of five free models provided by Swiss ADME, XLOGP3; WLOGP; MLOGP; SILICOS-IT LOGP, and finally ilogp.69–71
Table 5 Fundamental bioactivity scores calculated for the PA drug molecule in a free environment, and after being complexed in the forms of PA⋯Ag–SiO2 and PA⋯Au–SiO2 clusters
| Property |
PA |
PA⋯Ag–SiO2 |
PA⋯Au–SiO2 |
| Complex 1 |
Complex 2 |
Complex 3 |
Complex 1 |
Complex 2 |
Complex 3 |
| Rotatable bonds |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
| Hydrogen bond acceptors |
2 |
4 |
4 |
4 |
4 |
4 |
4 |
| Hydrogen bond donors |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
| Molar refractivity |
70.77 |
84.45 |
84.45 |
84.45 |
84.45 |
84.45 |
84.45 |
| Topological polar surface area |
58.36 |
83.42 |
83.42 |
83.42 |
83.42 |
83.42 |
83.42 |
| Bioavailability score |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
| Pure water solubility (mg ml−1) |
5.24 |
0.08 |
0.089 |
0.093 |
0.093 |
0.093 |
0.093 |
| log Kp (cm s−1) |
−7.11 |
−8.61 |
−8.61 |
−9.70 |
−9.70 |
−9.70 |
−9.70 |
| Lipinski rule violations |
0 |
1 |
1 |
1 |
1 |
1 |
1 |
Table 6 Computed lipophilicity parameters of PA drug molecule and the six PA⋯Ag–SiO2 and PA⋯Au–SiO2 complexes
| Model |
PA |
PA⋯Ag–SiO2 |
PA⋯Au–SiO2 |
| Complex 1 |
Complex 2 |
Complex 3 |
Complex 1 |
Complex 2 |
Complex 3 |
| ilogp |
2.39 |
0 |
0 |
0 |
0 |
0 |
0 |
| XLOGP3 |
0.88 |
1.38 |
1.38 |
1.38 |
1.38 |
1.38 |
1.38 |
| WLOGP |
1.35 |
0.45 |
0.45 |
0.44 |
0.44 |
0.44 |
0.44 |
| MLOGP |
1.53 |
−0.51 |
−0.51 |
−0.51 |
−0.51 |
−0.51 |
−0.51 |
| Silicos-IT logp |
1.29 |
1.29 |
1.29 |
1.29 |
1.29 |
1.29 |
1.29 |
| Consensus logp |
1.49 |
0.52 |
0.52 |
0.52 |
0.52 |
0.52 |
0.52 |
3.5 Docking analysis
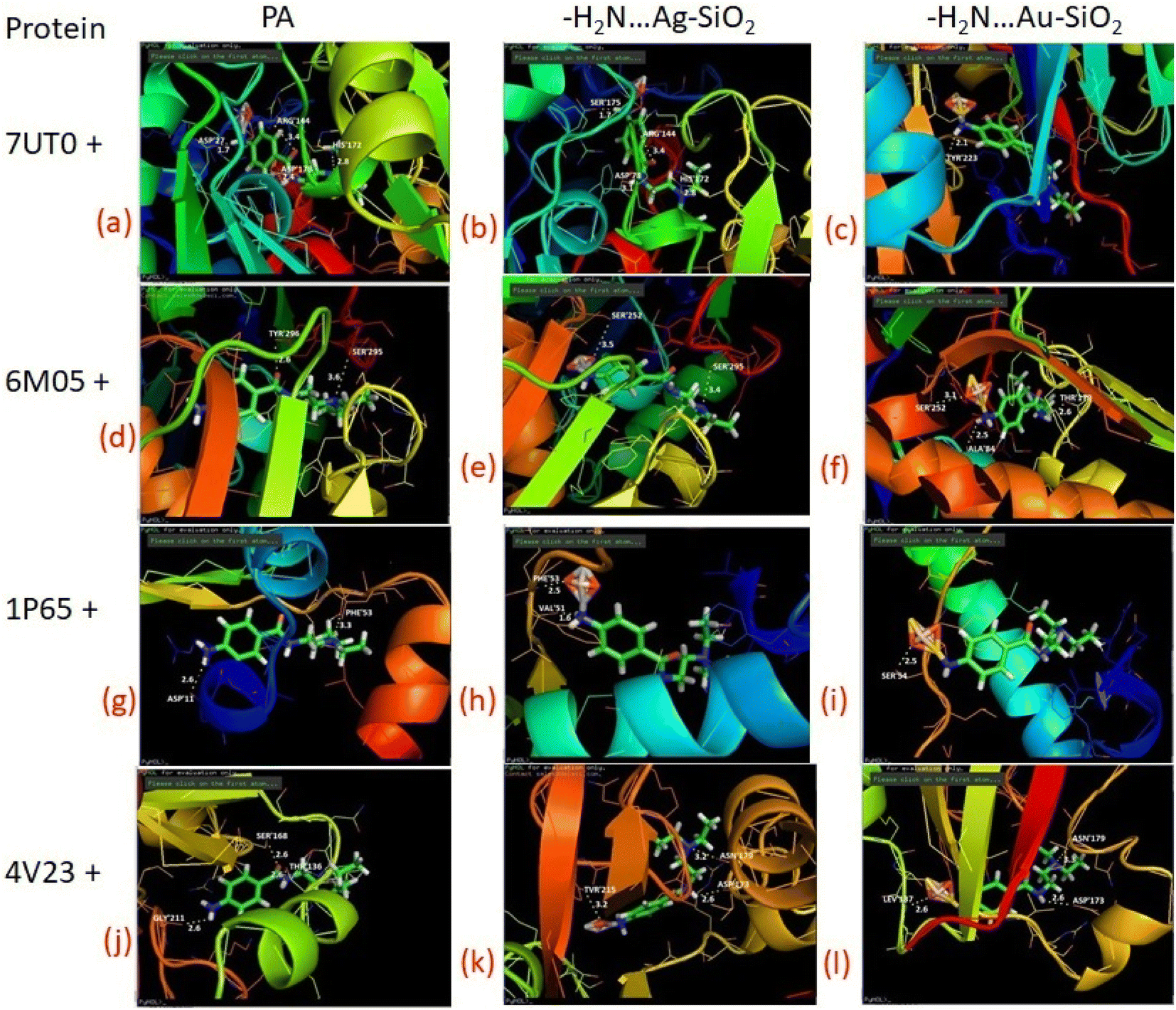
As predicted by PASS analysis (Table S4†), cardiovascular analeptic and respiratory analeptic inhibitors may be the most likely to interact with PA, thus causing free and complexes of PA⋯Ag- and PA⋯Au–SiO2 nano-formations to dock into the active site of the 7UTO, 6MO5, 1P65 and 4V23 proteins. From the above investigation, Complex 1 structure was docked with these proteins by means of Patch dock, and it was concluded that a pronounced interaction with the five types of proteins could be achieved. Both silver and gold-based systems possess inhibitory effects against receptor as presented in Fig. 10 and Table 7. These results indicate that the drugs-noble metal-silica formations well-inhibit the receptors.
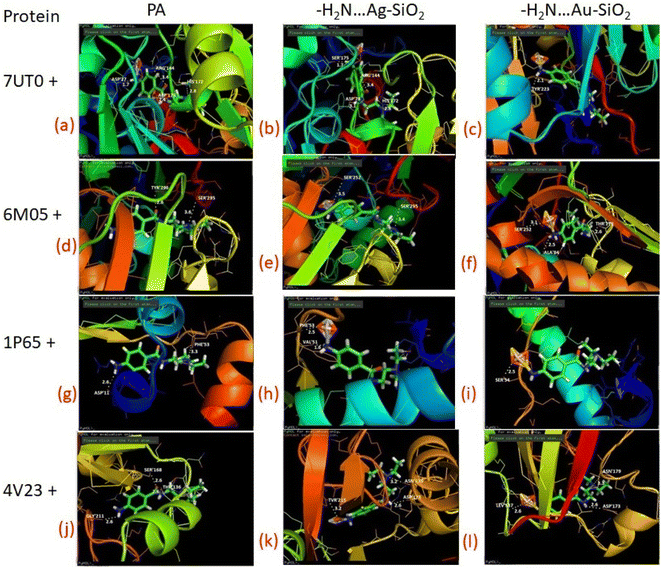 |
| | Fig. 10 Depicted possible biological interactions of PA drug molecule, PA⋯Ag–SiO2 and PA⋯Au–SiO2 (in their adsorptive modes with the amine moiety of PA) with 7UTO, 6MO5, 1P65 and 4V23 proteins. Associated docking outcomes are listed in Table 7. | |
Table 7 Docking results of PA molecule, PA⋯Ag–SiO2 and PA⋯Au–SiO2 (in their adsorptive modes with the amine moiety of PA) with 7UTO, 6MO5, 1P65 and 4V23 proteins
| Protein |
PA |
PA⋯Ag–SiO2 |
PA⋯Au–SiO2 |
| No. of H-bonds |
Residues |
No. of H-bonds |
Residues |
No. of H-bonds |
Residues |
| 7UTO |
4 |
ASP 27 |
4 |
SER 175 |
1 |
TYR 223 |
| ARG 144 |
ARG 144 |
|
| ASP 178 |
ASP 78 |
|
| HIS 172 |
HIS 172 |
|
| 6MO5 |
2 |
TYR 296 |
2 |
SER 252 |
3 |
SER 252 |
| SER 295 |
SER 295 |
ALA 84 |
| |
|
THR 179 |
| |
|
|
| 1P65 |
2 |
ASP 11 |
2 |
PHE 53 |
1 |
SER 54 |
| PHE 53 |
VAL 51 |
|
| |
|
|
| |
|
|
| 4V23 |
3 |
GLY 211 |
3 |
TVR 215 |
3 |
LEV 137 |
| SER 168 |
ASN 179 |
ASN 179 |
| THR 136 |
ASP 173 |
ASP 173 |
4. Conclusion
The adsorption process of procainamide (PA) drug over the Ag- and Au–SiO2 nanostructure was herein comprehensively investigated using different computational models. The charge distributions of the three modeled configurations indicated a considerable enhancement emanated from a non-covalent type of interaction. The thermodynamic characteristics of the silver- and gold-based nanocomposites differed in terms of exothermic, spontaneous interactions and thermodynamic orders. It was concluded that charge transfer, changes in band gap and other chemical descriptors are more significant for PA⋯Ag/Au–SiO2 clusters as compared to a non-interacting form of PA. The Raman peak intensity enhancement of PA when adsorbed to silica nanoclusters could be observed in the simulated SERS spectra. Careful examination of the ELF, LOL and NCI function parameters showed that both Ag- and Au–SiO2 core shells shall exhibit a meaningful interaction with PA. The obtained outcome from the docking analysis affirmed that the Ag/Au–SiO2 nanocomposites are promising candidates for developing a procainamide-based anti-arrhythmias drug delivery system. This will provide information on achieving a unique molecular design with an excellent pharmacological profile.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors thank King Fahd University of Petroleum & Minerals (KFUPM) for its support through project no. DF191043.
References
- I. J. Hidi, M. Jahn, M. W. Pletz, K. Weber, D. Cialla-May and J. Popp, Toward levofloxacin monitoring in human urin samples by employing the LoC-SERS technique, J. Phys. Chem. C, 2016, 120, 20613–20623, DOI:10.1021/acs.jpcc.6b01005.
- N. Srinivas, K. Maffuid and A. D. M. Kashuba, Clinicl pharmokinetics and pharmacodynamics of drugs in the central nervous system, Clin. Pharmacokinet., 2018, 5, 1059–1074, DOI:10.1007/S40262-018-0632-Y.
- Conclusion on the peer review of the pesticide risk assessment of the active substance Pepino mosaic virus strain CH2 isolate 1906, https://www.caddirect.org/caddirect/abstrct/20153086631.
- M. Uren Webster, M. H. Perry and E. M. Santos, The herbicide linuron inhibits cholesterol biosynthesis and induces celluar stress responses in brown trout, Environ. Sci. Technol., 2015, 49(5), 3110–3118, DOI:10.1021/es505498u.
- Invesigation of a Mechanism for Leydig Cell Tumorigenesis by Linuron in Rats, E-mail: https://www. sciencedirect.com/science/article/pii/Soo41008X83710604/.
- B. Horemans, J. Hofkens, E. Smolders and D. Springael, Biofilm formation of a bacterial consortium on linuron at micropollutant concentrations in continuous flow chambers and the impact of dissolved organic matter, FEMS, J. Microb. Ecol., 2014, 88(1), 184–194, DOI:10.1111/1574-6941.12280.
- E. Engelhardt, P. R. Wallnofer and R. Plapp, Degradtion of linuron and some other herbicides and fungicides by linuron-inducible enzyme obtained from Bacillus sphericus, Appl. Environ. Microbiol., 1071, 22(3), 284–288, DOI:10.1128/am.22.3.284-288.1971.
- A. Wlker and J. A. Thompson, The degradation of simazine, linuron and propyzamide in different soils, Weed Res., 1977, 17(6), 399–405, DOI:10.1111/j.1365-3180.1977.tb00500.x.
- H. Qian, M. Lin and P. Zhou, Measurement of forchlorfenuron in graphs by surface enhanced spectroscopy coupled with gold nano substrates, J. Food Sci., Nutr. Diet., 2016, 5, 258–264 CAS . https://scidoc.org/IJFS.php.
- L. Vrielynck, C. Lapouge, S. Marquis, J. Kister and N. Dupuy, Theoreticlal and experimental vibrational study of phenylurea: structure, solvent effect and inclusion process with the b-cyclodextrin in the solid state, Spectrochim. Acta A, 2004, 60(11), 2553–2559, DOI:10.1016/j.saa.2003.12.035.
- A. A. Al-saadi, M. Haroon, S. A. Popoola and T. A. Saleh, Sensitive SERS detection and charcterisation of procaine in aqueous media by reduced gold nanoparticles, Sensors, Sens. Actuators, B, 2020, 304, 127057, DOI:10.1016/j.snb.2019.127057.
- X. Chen, K. Yan, X. Xiao and G. Li, Analysis of forchlorfenuron and thidiazuron in fruits and vegetables by surfaced – enhanced Raman spectroscopy after selective solid-phase extraction with modified b-cyclodextrin, J. Sep. Sci., 2016, 39(12), 2340–2346 CrossRef CAS PubMed.
- K. Paranjpe, V. Gowariker and V. Krishnamurthy, The pesticide encyclopedia, 2014, https://books.Google.com/books?hl=en%26Ir=%26id=cnDHBgAAQBAJ%26oi=fnd%26pg=PR5%26ots=w5i0tTwU3K%26sig=3qhMUGtBskh-H_gpv907sIpIzPw Search PubMed.
- M. J. Gonzalez da la Huebra, P. Hernandez, Y. Ballesteros and L. Hernandez, Determintion of linuron in soil by stripping voltammetry with a carbon fiber microelectrode, Talnta, 2001, 54, 1077–1085, DOI:10.1016/S0039-9140(01)00372-1.
- M. Buleandra, D. E. Popa, I. G. David, E. Bacalum, V. David and A. Ciucu, Electrochemical behavior study of some selected phenylurea herbicides at activated pencil graphite electrode, Electro oxidation Linuron Monolinuron, Microchem, 2019, 147, 1109–1116 CrossRef CAS.
- M. E. A. Kribeche, T. Sehili, G. Lesage, J. Mendret and S. Brosillon, Insight into photochemical oxidation of Fenuron in water using iron oxide and oxalate: the roles of the dissolved oxygen, J. Photochem. Photobiol., A, 2016, 329, 120–129, DOI:10.1016/j.jphotochem.2016.06.021.
- Y. F. Rao and W. Chu, Linuron decomposition in aqueous semiconductor suspension under visible light irradiation with and without H2O2, Chem. Eng. J., 2010, 158(2), 181–187, DOI:10.1016/j.cej.200912.038.
- M. Takenaka, Y. Hashimoto, T. Iwasa, T. Taketsugu, G. Seniutinas, A. Balcytis, S. Juodkazis and Y. Nishijima, First Principles Calculations Toward Understanding SERS of 2,2’-Bipyridyl Adsorbed on Au, Ag, and Au-Ag Nanoalloy, J. Comput. Chem., 2019, 40, 925–932, DOI:10.1002/jcc.25603.
- T.-T. You, N. Yang, Y.-Q. Shu and P. G. Yin, A DFT study on graphene-based surface enhanced Raman spectroscopy on benzendithiol adsorbed on gold/graphene, J. Raman Spectrosc., 2019, 50, 1510–1518, DOI:10.1002/jrs.5673.
- L. B. Zhao, R. Huang, M. X. Bai, D. Y. Wu and Z. Q. Tian, Effect of aromatic amine-metal interaction on surface vibrational Rman spectroscopy of adsorbed molecules investigated by density functional theory, J. Phys. Chem. C, 2011, 115, 4174–4183, DOI:10.1021/jp1117135.
- Y. Umebayashi, T. Fujimori, T. Sukizaki, M. Asada, K. Fujji, R. Kanazaki and S. Ishiguro, Evidence of conformational equilibrium of 1- ethyl-3-methylimidazolium in its ionic liquid salts: Raman spectroscopic study and quantum chemical calculations, J. Phys. Chem. A, 2005, 109, 8976–8982, DOI:10.1021/jp053476j.
- J. P. Ceron-Carrasco, A. Reequena, C. Michaux, E. A. Perpete and D. Jacquemin, Effects of hydration on the proton transfer mechanism in the adenine-thymine base pair, J. Phys. Chem., 2009, 113, 7892–7898, DOI:10.1021/jp900782h.
- X. F. Lang, P. G. Yin, T. T. You, L. Jiang and L. Guo, ChemPhysChem, 2011, 12, 2468–2475, DOI:10.1021/jp900782h.
- J. Alimunnisa, K. Ravichandran and K. S. Meena, Synthesis and characterization of Ag@SiO2 core-shell nanoparticles for antibacterial and environmental applications, J. Mol. Liq., 2017, 231, 281–287, DOI:10.1016/j.molliq.2017.01.103.
- Y. H. Kim, Don Keun Lee, Hyun Gil Cha, Chang Woo Kim and Young Soo Kang, Synthesis and Characterization of Antibacterial Ag−SiO2 Nanocomposite, J. Phys. Chem. C, 2007, 111(9), 3629–3635, DOI:10.1021/jp068302w.
- N. Subbiah and T. Palanisamy, Collagen-Supported Amino-Functionalized Ag@SiO2 Core–Shell Nanoparticles for Visible-Light-Driven Water Remediation, ACS Appl. Nano Mater., 2022, 5(10), 14408–14424, DOI:10.1021/acsanm.2c02751.
- C.-C. Chang, K.-H. Yang, Yu-C. Liu, T.-C. Hsu and F.-D. Mai, Surface-Enhanced Raman Scattering-Active Au/SiO2 Nanocomposites Prepared Using Sonoelectrochemical Pulse Deposition Methods, ACS Appl. Mater. Interfaces, 2012, 4(9), 4700–4707, DOI:10.1021/am3017366.
- T. Munir, A. Mahmood, N. Peter, N. Rafaqat, M. Imran and H. Elhosiny Ali, Structural, morphological and optical properties at various concentration of Ag doped SiO2-NPs via sol gel method for Antibacterial and Anticancer Activities, Surf. Interfaces, 2023, 102759, DOI:10.1016/j.surfin.2023.102759.
- X. Zhang, Yu Guo, T. Wang, Z. Wu and Z. Wang, Antibiofouling performance and mechanisms of a modified polyvinylidene fluoride membrane in an MBR for wastewater treatment: Role of silver@silica nanopollens, Water Res., 2020, 176(1), 115749, DOI:10.1016/j.watres.2020.115749.
- Y. Wang, Y. Wang, L. Su, L. Yi, X. Du and X. Zhang, Effect of surface topology morphologies of silica nanocarriers on the loading of Ag nanoparticles and antibacterial performance, J. Alloys Compd., 2019, 783, 136–144, DOI:10.1016/j.jallcom.2018.12.284.
- Y. H. Kim, D. K. Lee and Y. S. Kang, Synthesis and characterization of Ag and Ag–SiO2 nanoparticles, Colloids Surf., A, 2005, 257–258, 273–276, DOI:10.1016/j.colsurfa.2004.07.035.
- H.-J. Jeon, S.-C. Yi and S.-G. Oh, Preparation and antibacterial effects of Ag–SiO2 thin films by sol–gel method, Biomaterials, 2003, 24(27), 4921–4928, DOI:10.1016/S0142-9612(03)00415-0.
- K. Aslan, M. Wu and R. Joseph, Lakowicz and Chris D. Geddes, Fluorescent Core−Shell Ag@SiO2 Nanocomposites for Metal-Enhanced Fluorescence and Single Nanoparticle Sensing Platforms, J. Am. Chem. Soc., 2007, 129(6), 1524–1525, DOI:10.1021/ja0680820.
- S. Lambert, C. Cellier, P. Grange, J.-P. Pirard and B. Heinrichs, Synthesis of Pd/SiO2, Ag/SiO2, and Cu/SiO2 cogelled xerogel catalysts: study of metal dispersion and catalytic activity, J. Catal., 2004, 221(2), 335–346, DOI:10.1016/j.jcat.2003.07.014.
- Ke Xu, J.-X. Wang, Xu-L. Kang and J.-F. Chen, Fabrication of antibacterial monodispersed Ag–SiO2 core–shell nanoparticles with high concentration, Mater. Lett., 2009, 63(1), 31–33, DOI:10.1016/j.matlet.2008.08.039.
- M. Shanthil, R. Thomas, R. S. Swathi and K. George Thomas, Ag@SiO2 Core–Shell Nanostructures: Distance-Dependent Plasmon Coupling and SERS Investigation, J. Phys. Chem. Lett., 2012, 3(11), 1459–1464, DOI:10.1021/jz3004014.
- A. Lombardi, M. P. Grzelczak, A. Crut, P. Maioli, I. Pastoriza-Santos and M. Luis, Liz-Marzan, Natalia Del Fatti and Fabrice Vallee , Optical Response of Individual Au–Ag@SiO2 Heterodimers, ACS Nano, 2013, 7(3), 2522–2531, DOI:10.1021/nn305865h.
- H. Baida, P. Billaud, S. Marhaba, D. Christofilos, E. Cottancin, A. Crut, J. Lermé, P. Maioli, M. Pellarin, M. Broyer, N. Del Fatti, F. Vallee, A. Sánchez-Iglesias, I. Pastoriza-Santos and L. M. Liz-Marzan, Quantitative Determination of the Size Dependence of Surface Plasmon Resonance Damping in Single Ag@SiO2 Nanoparticles, Nano Lett., 2009, 9(10), 3463–3469, DOI:10.1021/nl901672b.
- W. Wang, Z. Li, B. Gu, Z. Zhang and H. Xu, Ag@SiO2 Core−Shell Nanoparticles for Probing Spatial Distribution of Electromagnetic Field Enhancement via Surface-Enhanced Raman Scattering, ACS Nano, 2009, 3(11), 3493–3496, DOI:10.1021/nn9009533.
- M. Lismont and A. Carlos, Paez and Laurent Dreesen, A one-step short-time synthesis of Ag@SiO2 core–shell nanoparticles, J. Colloid Interface Sci., 2015, 447, 40–49, DOI:10.1016/j.jcis.2015.01.065.
- S.-D. Oh, Seungho Lee, Seong-Ho Choi, In-Soo Lee, Yun-Mi Lee, Jong-Han Chun and Hae-Jun Park, Synthesis of Ag and Ag–SiO2 nanoparticles by γ-irradiation and their antibacterial and antifungal efficiency against Salmonella enterica serovar Typhimurium and Botrytis cinerea, Colloids Surf., A, 2006, 228–233, DOI:10.1016/j.colsurfa.2005.11.039.
- I. Tanahashi, M. Yoshida, Y. Manabe and T. Tohda, Effects of heat treatment on Ag particle growth and optical properties in Ag/SiO2 glass composite thin films, J. Mater. Res., 1995, 10(2), 362–365, DOI:10.1557/JMR.1995.0362.
- S. Lambert, C. Cellier and M. Eric, Gaigneaux, Jean-Paul Pirard and Benoit Heinrichs, Ag/SiO2, Cu/SiO2 and Pd/SiO2 cogelled xerogel catalysts for benzene combustion: Relationships between operating synthesis variables and catalytic activity, Catal. Commun., 2007, 8(8), 1244–1248, DOI:10.1016/j.catcom.2006.11.018.
- M. Haroon, W. Iali, T. A. Saleh and A. A. Al-Saadi, NMR evidence for hydrogen bonding stabilized anti conformation of 1-methoxy-1-methyl-3-phenylurea and the concentration detection by SERS, J. Mol. Liq., 2022, 357, 119096, DOI:10.1016/j.molliq.2022.119096.
- M. J. Frisch, Gaussian 09, Revision D.01, Gaussian Inc, Wallingford CT, 2013 Search PubMed.
- K. Mahboobeh and T. L. Elham, B12Y12 (Y:N:, P) fullerene like cages for exemestane delivery; molecular modeling investigation, J. Mol. Struct., 2020, 1217, 128455, DOI:10.1016/j.molstruc.2020.128455.128455.
- Y. Zhao and D. G. Truhlar, The MO6 suite of density functional for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states and transition elements: two new functional and systematic testing of our M06-class functional and 12 other functional, Theor. Chem. Acc., 2008, 120, 215–241, DOI:10.1007/s00214-007-0310-x.
- J. S. Al-Otaibi, Y. S. Mary, S. Armakovic and R. Thomas, Hybrid and bioactive cocrystals of pyrazinamide with hydroxybenzoic acids: Detailed study of structure, spectroscopic characteristics, other potential applications and noncovalent interactions using SAPT, J. Mol. Struct., 2020, 1202, 127316, DOI:10.1016/j.molstruc.2019.127316.
- E. Catherine, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, Addition of polarization and diffuse functions to the LANL2DZ Basis set for P-Block elements, J. Phys. Chem. A, 2001, 105(34), 8111–8116, DOI:10.1021/jp0119451.
- A. A. Al-Saadi, Computational study of SERS effects in some aliphatic and cyclic carboxylic acids with silver nanomaterials, J. Phys.: Conf. Ser., 2020, 1564, 012008, DOI:10.1088/1742-6596/1564/1/012008.
- N. Mahar, V. Vetrivelan, S. Muthu, S. Javed and A. A. Al-Saadi, Surface enhanced Raman spectra(SERS), Computational study of Gemcitabine drug adsorption on to Au/Ag clusters with different complexes: Adsorption behavior and solvent effect (IEFPCM) – Anticancer agent, J. Comput. Theor. Chem., 2022, 1217, 113914, DOI:10.1016/j.comptc.2022.113914.
- J. T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885.
- https://way2drug.com/passonline/.
- Preadme, https://preadmet.bmdrc.kr/.
- https://bioinfo3d.cs.tau.ac.il/PatchDock/.
- G. M. Morris, R. Heuy, W. Lindstrom, M. F. Scanner, R. K. Belew, D. S. Goodsell and A. J. Olson, Autodock 4 and AutoDock Tools 4: automated docking with selected receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- G. M. Morris, R. Heuy, W. Lindstrom, M. F. Scanner, R. K. Belew, D. S. Goodsell and A. J. Olson, Autodock 4 and AutoDock Tools 4: automated docking with selected receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- M. Haroon, I. Abdulazeez, T. A. Saleh and A. A. Al-Saadi, SERS-based trace-level quantification of sulindac: Spectroscopic and molecular modeling evaluation, J. Mol. Liq., 2020, 313, 113402, DOI:10.1016/j.molliq.2020.113402.
- J. S. Al-Otaibi, Y. S. Mary and Y. S. Mary, DFT analysis of valproic acid adsorption onto Al12/B12-N12/P12 nanocages with solvent effects, J. Mol. Model., 2022, 28, 98 CrossRef CAS PubMed.
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371, 683–686, DOI:10.1038/371683a0.
- H. Jacobsen, Localized – orbital locator (LOL) profiles of chemical bonding, Can, J. Chem., 2008, 86, 695–702, DOI:10.1139/v08-052.
- S. Aayisha, T. S. Renuga Devi, S. Janani, S. Muthu, M. Raja, S. Sevvanthi and B. R. Raajaraman, Vibrational and computational analysis for molecular structure properties of N-(2-(trifluoromethyl))Phenyl)acetamide: density functional theory approach, Spectrosc. Lett., 2019, 52, 563–576, DOI:10.1080/00387010.2019.1678175.
- J. S. Al-Otaibi, A. H. Almuqrin, Y. S. Mary and Y. S. Mary, Comprehensive quantum mechanical studies on three bioactive anastrozole based triazole analogous and their SERS active graphene complex, J. Mol. Struct., 2020, 1217, 128388, DOI:10.1016/j.molstruc.2020.128388.
- J. S. Al-Otaibi, A. H. Almuqrin, Y. S. Mary and Y. S. Mary, Comprehensive quantum mechanical studies on three bioactive anastrozole based triazole analogues and their SERS active graphene complex, J. Mol. Struct., 2020, 1217, 128388, DOI:10.1016/j.molstruc.2020.128388.128388.
- A. Jeelani, S. Muthu and B. Narayana, Molecular structure determination, bioactivity score, spectroscopic and quantum computational studies on (E)-n’-(4-chlorobenzylidene)-2-(napthalen-2-yloxy)acetohydrazide, J. Mol. Struct., 2021, 124, 130558, DOI:10.1016/j.molstruc.2021.130558.
- V. Vetrivelan, Spectra, electronic properties, biological activities and molecular docking investigation on sulfonamide derivative compound: an experimental and computational approach, J. Nanosci. Nanotechnol., 2018, 4(2), 348–352, DOI:10.30799/jnst.sp203.18040203.
- S. Chitra, G. Mani, M. Kumar, V. Vetrivelan, S. Muthu, K. Arulaabaranam, A. Jeelani, I. Ahmed and H. Umamaheesvari, Theoretical electron excitation study in liquid phase (protic, aprotic, non-polar) and inter and intra molecular reactivity of 2-hydroxy-5-[1-hydroxy-2-(4-phenylbutan-2-ylamino)ethyl] benzamide, J. Indian Chem. Soc., 2022, 99, 100372, DOI:10.1016/jics.2022.100372.
- M. Habib Rahuman, S. Muthu, B. R. Raajaraman and M. Raja, Quantum computational, spectroscopic and molecular docking studies on 2-acetylthiophene and its bromination derivative, J. Mol. Struct., 2020, 1212, 128129, DOI:10.1016/j.molstruc.2020.128129.
- B. Arjun Kumar, V. Vetrivelan, G. Ramalingam, A. Manikandan, S. Viswanathan, P. Boomi and G. Ravi, Computational studies and experimental fabrication of DSSC device assembly on 2D-layered TiO2 and MoS2@TiO2 nanomaterials, Phys. B, 2022, 633, 413770, DOI:10.1016/j.physb.2022.413770.
- P. Chakkaravarthy, V. Vetrivelan, S. Syed Shafi and S. Muthu, Spectroscopic (FT-IR & FT-Raman), Fukui function and molecular docking analysis of 6-amino-7,9-dihydropurine-8-thione by DFT approach, J. BCC, 2020, 52(4), 440–447, DOI:10.34049/bcc.52.4.5234.
- I. Moriguchi, H. Shuichi, I. Nakagome and H. Hirano, Comparison of reliability of log P values for drugs calculated by several methods, Chem. Pharm. Bull., 1994, 42, 976–978, DOI:10.1248/cpb.42.976.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Abdulaziz A. Al-Saadi*d
*c and
Abdulaziz A. Al-Saadi*d