
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and biological evaluation with molecular dynamics study of novel pyrazolo[3,4-d]pyrimidine derivatives as anti-cancer agents†
Rania M. Shaban a,
Nermin Samirb,
Yassin M. Nissanac and
Khaled A. M. Abouzid*b
a,
Nermin Samirb,
Yassin M. Nissanac and
Khaled A. M. Abouzid*b
aPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, October University for Modern Sciences and Arts (MSA), Giza, Egypt. E-mail: rmshaban@msa.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Ain Shams University, Abbassia, Cairo 11566, Egypt. E-mail: khaled.abouzid@pharma.asu.edu.eg
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo, Egypt
First published on 7th June 2023
Abstract
In continuation of our efforts to discover new structural chemotypes with significant chemotherapeutic activities, a novel series of pyrazolo[3,4-d]pyrimidine-based compounds linked to a piperazine ring, bearing different aromatic moieties, through different linkages was designed and synthesized as FLT3 inhibitors. All of the newly synthesized compounds were evaluated for their cytotoxicity on 60-NCI cell lines. Compounds with the piperazine acetamide linkage XIIa–f & XVI exhibited a remarkable anticancer activity among all of the tested compounds, especially against non-small cell lung cancer, melanoma, leukemia and renal cancer models. Furthermore, compound XVI (NSC no – 833644) was further screened with a 5-dose assay on nine subpanels and exhibited a GI50 between 1.17 and 18.40 μM. On the other hand, molecular docking and dynamics studies were performed to predict the binding mode of the newly synthesized compounds in the FLT3 binding domain. Finally, through a predictive kinetic study, several ADME descriptors were calculated.
Introduction
Cancer is a serious health problem and is ranked as the second leading cause of mortality worldwide.1,2 Mutation or overexpression of certain classes of signaling proteins releasing the natural cellular control leads to cancer development. RTKs (receptor tyrosine kinases) are one of the most important extracellular signaling mediators that are known to be frequently mutated in cancer. The mammalian genome contains 56 RTKs that can be subdivided into 20 different families based on their structures.3,4FLT3 (FMS-like tyrosine kinase 3) is a receptor tyrosine kinase that is essential for immune response and stem cell proliferation and present in both normal and malignant lymphohematopoietic cells and. It is one of the most frequently altered genes in acute myeloid leukemia. Recently FLT3 targeted tyrosine kinase inhibitors (TKIs) have attracted a lot of interest as targeted therapeutic agents because they exert a significant cytotoxic effect on cancer cells.5
Since RTKs overexpression or oncogenic mutation is common in cancers, several RTKs inhibitors targeting FLT3 have become an attractive approach for the treatment of AML such as midostaurin (Rydapt, also known as PKC-412),6 lestaurtinib (CEP-701),7 tandutinib (MLN518),8 dovitinib (TKI258, CHIR258),9 sunitinib (Sutent),10 ponatinib (Iclusig),11 sorafenib (Nexavar),12 and amuvatinib (MP-470)13 which were used as first-generation FLT3 inhibitors.14 Although many of these inhibitors had undergone preclinical and clinical trials and showed promising results, they lacked sensitivity and selectivity, hence, second-generation inhibitors, such as gilteritinib (ASP2215),15 quizartinib (AC220),16 and crenolanib,17 have been approved5,18 (Fig. 1).
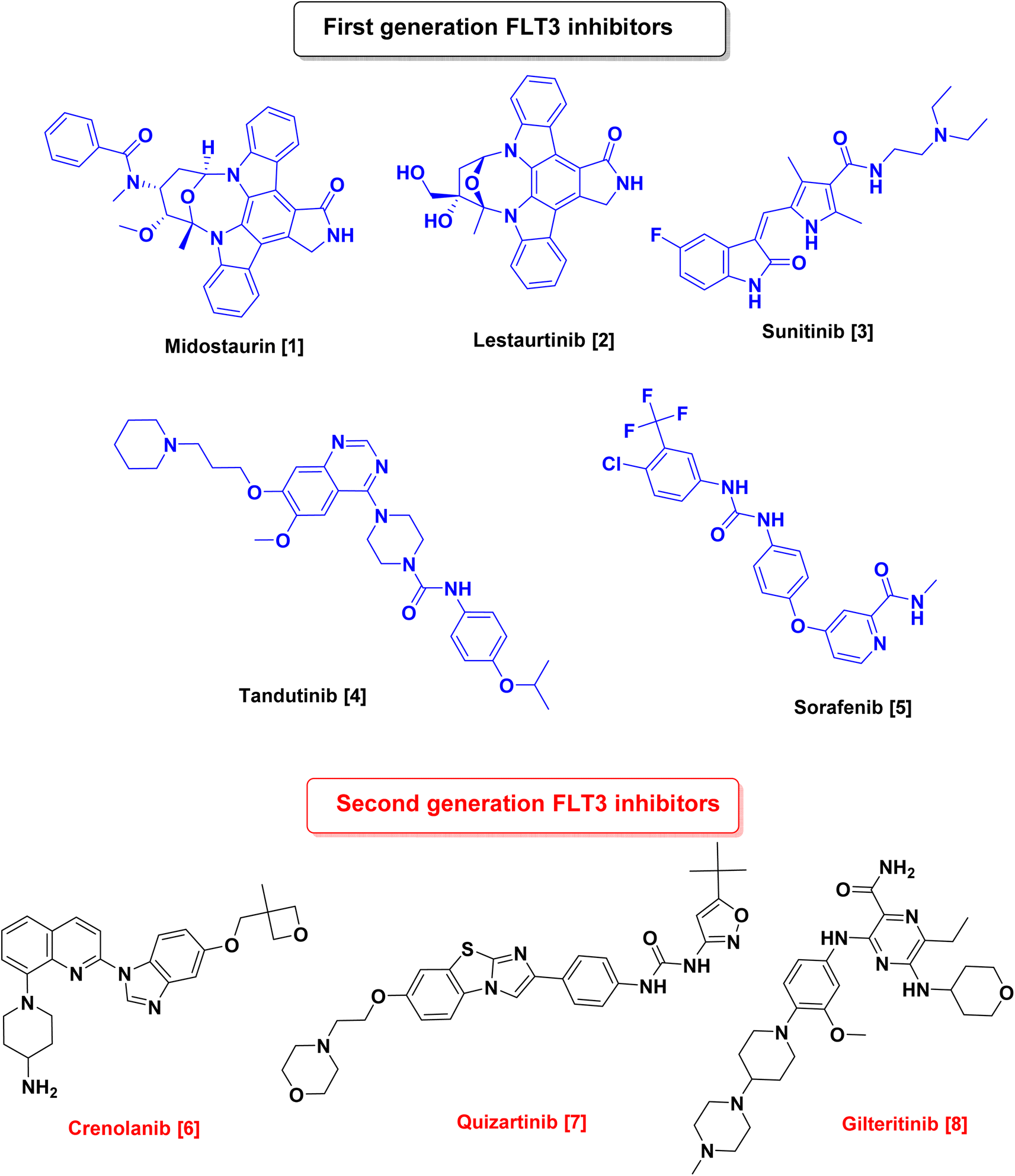 | ||
| Fig. 1 Chemical structures of first & second-generation FLT3 inhibitors. | ||
On the other hand, in recent years, there has been a lot of interest in the chemotherapeutic activities of pyrazolo-pyrimidines as antimicrobial,19 antiviral,20 anti-inflammatory,21 and potential anticancer agents.22 Also pyrazolo-pyrimidines show their anticancer activity by inhibiting a variety of enzymes such as cyclin-dependent kinase,23 Src and Abl tyrosine kinase,24,25 glycogen synthase kinase-3,26 adenosine deaminase,27 epidermal growth factor receptor protein tyrosine kinase28 and FMS-like tyrosine kinase 3 (ref. 29 and 30) (Fig. 2).
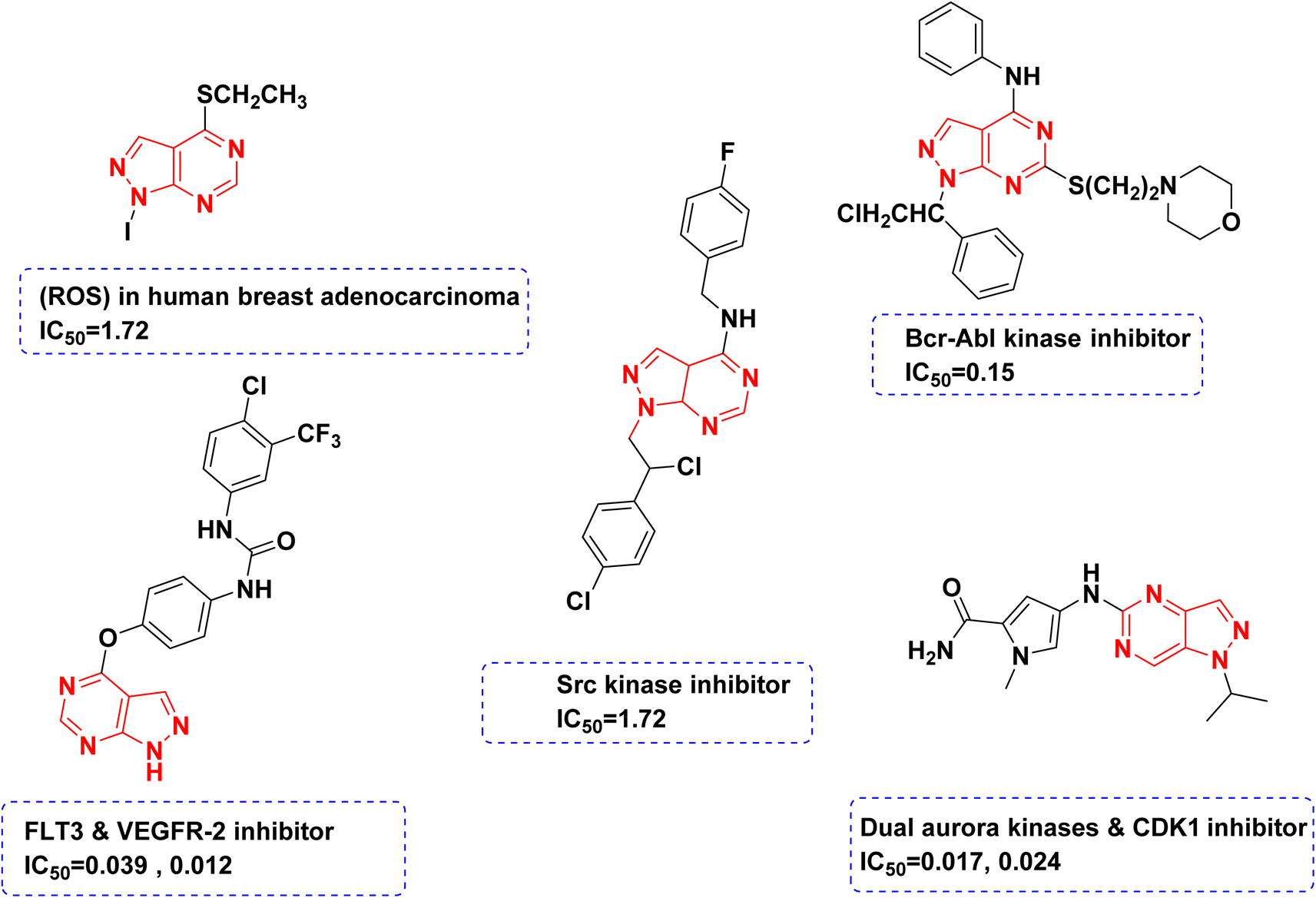 | ||
| Fig. 2 Structures of different reported compounds containing pyrazolo[3,4-d]pyrimidine scaffold as antitumor agents with their corresponding IC50 in μM. | ||
Due to these facts, and in order to find novel lead structures with potential chemotherapeutic activities, it was rationalized to design, synthesize and study the antitumor activity of some new compounds comprising the pyrazolo[3,4-d]pyrimidine as the main scaffold and to design these novel compounds, the following modifications were made to the reported lead compound (Fig. 3).
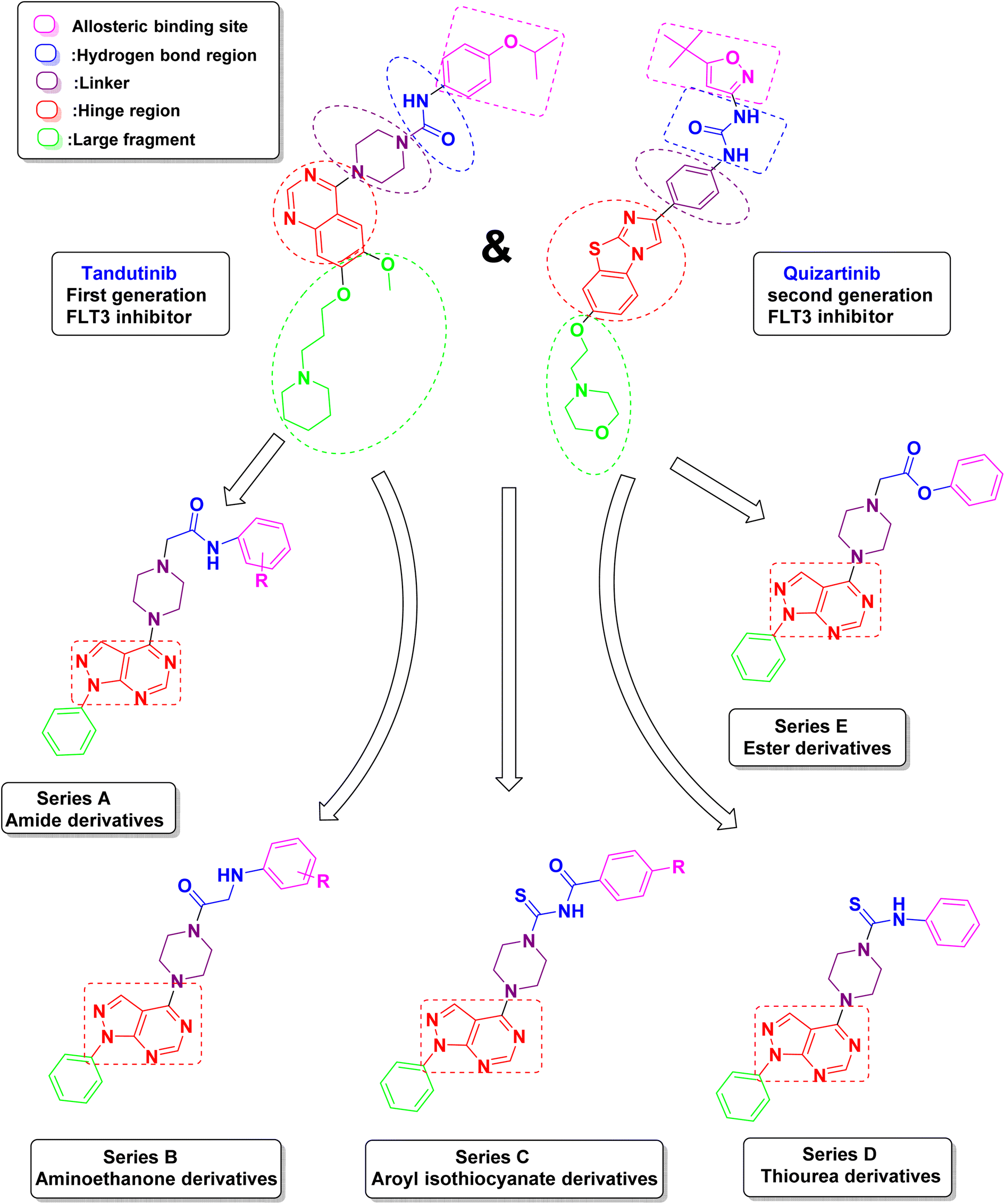 | ||
| Fig. 3 Design strategy of the proposed novel pyrazolo[3,4-d]pyrimidine as FLT3 inhibitors. | ||
(1) The red area represents the hinge region in which the quinazoline ring was replaced by 1H-pyrazolo[3,4-d]pyrimidine because it is considered to be an isostere of purine so it can achieve a promising anti-cancer activity. The main scaffold is the critical inhibitor interacting group, which is responsible for the hydrogen bond with Cys 694 in the hinge region.
(2) The purple area contains a piperazine ring as a linker which was kept the same as the lead compound.
(3) The blue area represents the hydrogen bond region where the urea carbonyl or amide carbonyl accepts a hydrogen bond from the NH found in aspartate and the NH from the urea or amide forms a hydrogen bond to the side chain of the conserved glutamate. This area could be an amide, ester, thiourea, or aroyl isothiocyanates.
(4) The pink area represents the allosteric binding site which contains different aromatic moieties either substituted by electron-donating, electron-withdrawing, or unsubstituted to study their effect on the activity.
(5) The green area represents an unnecessary large fragment which was replaced by a simple phenyl ring in the newly synthesized compounds.
Results and discussion
Chemistry
The synthetic pathways used for the preparation of the intermediates and target products are outlined in Schemes 1–3. Scheme 1, started by reacting phenylhydrazine VI with ethyl 2-cyano-3-ethoxyacrylate VII to give ethyl 5-amino-1-phenyl-1H-pyrazolo-4-carboxylate VIII.31 Cyclization of compound VIII was afforded via its reflux with formamide to yield 1-phenyl-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one IX.32 After that chlorination of compound IX was carried out by reflux in POCl3 followed by a coupling reaction with excess anhydrous piperazine (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) in isopropanol to obtain the key intermediate XI.33
:10) in isopropanol to obtain the key intermediate XI.33
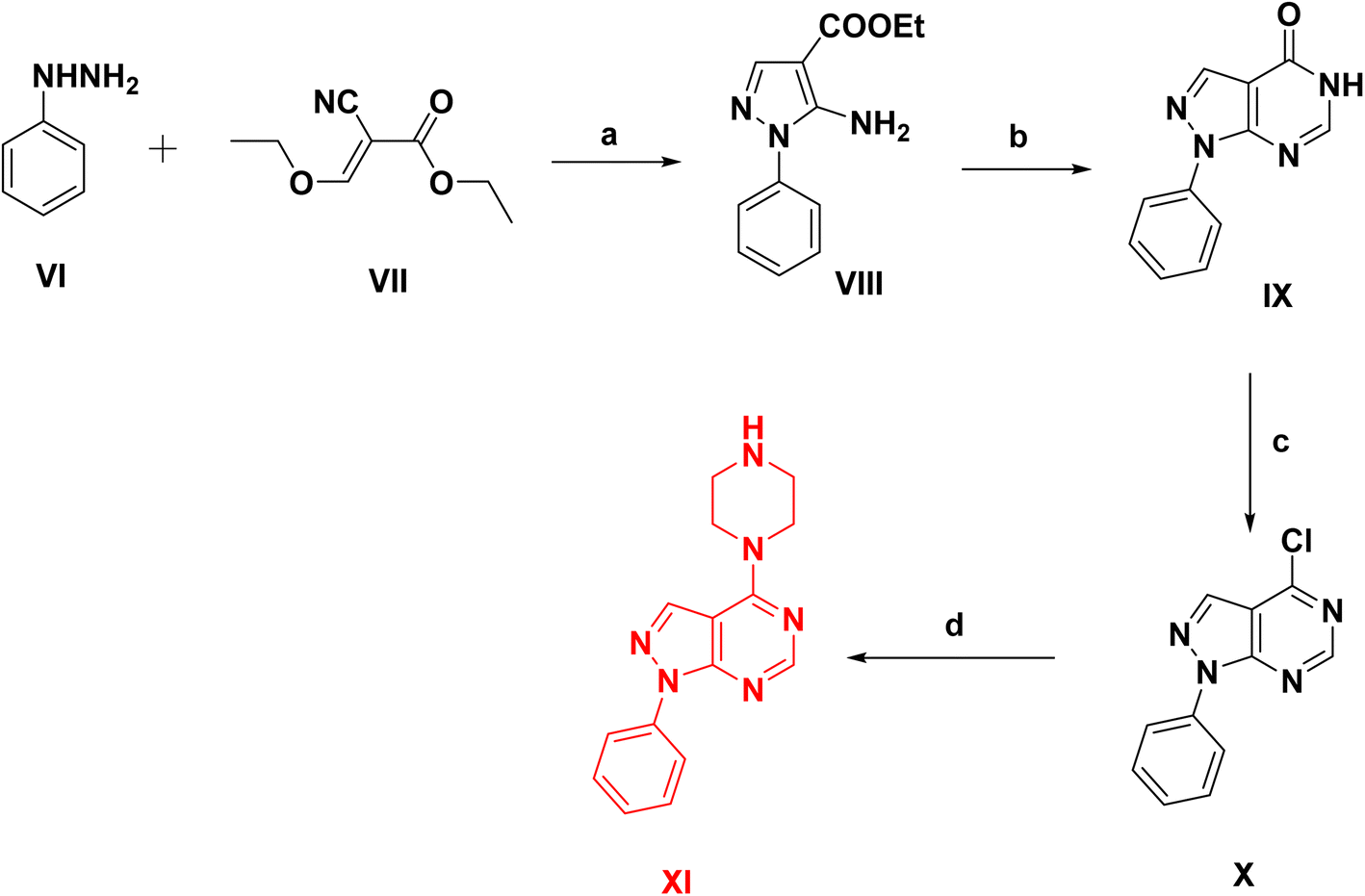 | ||
| Scheme 1 Synthesis of phenyl pyrazolo-pyrimidine intermediates, reagents and conditions: (a) ethanol, reflux 6 h, 80%, (b) formamide, reflux 10 h, 70%, (c) POCl3, reflux 18 h, 65%, (d) anhydrous piperazine, isopropanol, reflux 6 h, 70%. | ||
In Scheme 2, the starting intermediates IIa–f, III, IV & V were prepared via the reaction of chloroacetyl chloride (I) with aniline derivatives to give IIa–f.34–36 Similarly, precursor III was prepared by reacting chloroacetyl chloride with α-naphthol. Also, compound IV was prepared by reacting chloroacetyl chloride with phenol.37 Reacting chloroacetyl chloride with 1,3,4-thiadiazol-2-amine yields intermediate V (Scheme S1) (ESI†).
 | ||
| Scheme 2 Reagents and conditions: (a) NaHCO3, KI, DMF, rt, 36 h, 65–83% (b) NaHCO3, KI, DMF, rt, 24 h, 55–65%, (c) NaHCO3, KI, DMF, rt, 24 h, 50%, (d) phenyl isothiocyanate, dry DCM, reflux, 2 h, 75%, (e) NaHCO3, KI, DMF, rt, 36 h, 75%, (f) 2-chloroacetyl chloride, anhydrous sodium acetate, DMF, reflux, 16 h, 65%, (g) aromatic amine derivatives, NaHCO3, KI, DMF, rt, 48 h, 62–80%. | ||
Final compounds XIIa–f, XIII, XIV & XVI were synthesized via a nucleophilic substitution reaction of intermediates IIa–f, III, IV & V respectively with the key intermediate XI. The 1H NMR spectra for compounds XIIa–f & XVI showed one signal appeared downfield corresponding to the D2O exchangeable proton of the NH group in the range of δ (9.70–9.99 ppm). Also, appearance of a new singlet signal in the range of δ (2.71–3.25 ppm) referring to –CH2 protons. An extra singlet signal corresponding to the methyl protons for compounds XIIe, XIIf respectively appeared at δ 2.26, 2.69 ppm, and a singlet signal at δ 3.73 ppm attributed to the methoxy protons for compound XIId. Compounds XIIe, XIIf showed appearance of a singlet signal corresponding to CH3 at δ 20.92 ppm. Also, an extra signal corresponding to –OCH3 at δ 55.64 ppm for compound XIId. FT-IR spectra displayed the amidic carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the range of 1670–1695 cm−1. 1H NMR of compound XIIa & XIV showed disappearance of NH signal, and a singlet signal referring to –CH2 aliphatic at δ 4.27 ppm. Moreover, compound XV was obtained via nucleophilic addition reaction of isothiocyanate derivative with intermediate XI. 1H NMR spectra showed appearance of new signals for aromatic protons of the additional aromatic ring and a singlet signal appeared downfield corresponding to the D2O exchangeable proton of urea group at δ 9.83 ppm. On the other hand, the synthesis of compounds XVIIIa–f, initially involved reacting chloroacetyl chloride with XI, to afford the intermediate XVII which was stirred at room temperature with amine derivatives to yield XVIIIa–f. 1H NMR of compounds XVIIIa–f showed 1 singlet NH signal in the range of δ (5.51–6.79 ppm) which disappeared by D2O & a singlet peak referring to –CH2 aliphatic in the range of δ (2.71–3.25 ppm). Compound XVIIId showed an extra signal corresponding to the methoxy protons at δ 3.73 ppm. Additionally, an extra singlet signal corresponds to methyl protons for compound XVIIIe at δ 2.26 ppm.
O) in the range of 1670–1695 cm−1. 1H NMR of compound XIIa & XIV showed disappearance of NH signal, and a singlet signal referring to –CH2 aliphatic at δ 4.27 ppm. Moreover, compound XV was obtained via nucleophilic addition reaction of isothiocyanate derivative with intermediate XI. 1H NMR spectra showed appearance of new signals for aromatic protons of the additional aromatic ring and a singlet signal appeared downfield corresponding to the D2O exchangeable proton of urea group at δ 9.83 ppm. On the other hand, the synthesis of compounds XVIIIa–f, initially involved reacting chloroacetyl chloride with XI, to afford the intermediate XVII which was stirred at room temperature with amine derivatives to yield XVIIIa–f. 1H NMR of compounds XVIIIa–f showed 1 singlet NH signal in the range of δ (5.51–6.79 ppm) which disappeared by D2O & a singlet peak referring to –CH2 aliphatic in the range of δ (2.71–3.25 ppm). Compound XVIIId showed an extra signal corresponding to the methoxy protons at δ 3.73 ppm. Additionally, an extra singlet signal corresponds to methyl protons for compound XVIIIe at δ 2.26 ppm.
Finally, in Scheme 3, the targeted compounds XXIa–f were synthesized by suitable several steps starting from the chlorination of benzoic acid and its derivatives with thionyl chloride to form the corresponding acid chloride XIXa–f. These acid chlorides were treated with ammonium thiocyanate to produce 4-un/substituted benzoyl isothiocyanates XXa–f which reacted with 1-phenyl-4-(piperazin-1-yl)-1H-pyrazolo[3,4-d]pyrimidine XI to give the respective aroyl isothiocyanate derivative.38 The 1H NMR spectra for compounds XXIa–f showed one signal appeared downfield corresponding to the D2O exchangeable proton of NH group in the range of δ (10.80–11.12) ppm. Also, appearance of additional signals which refer to the newly formed aromatic protons. Compound XXIe spectrum showed a new singlet signal at δ 4.31 ppm corresponding to the methoxy protons. Compound XXIf spectrum showed an additional singlet signal at δ 2.39 ppm corresponding to the three protons of the methyl group. Compound XXIf showed an extra signal corresponding to the carbon of the methyl group at δ 20.02 ppm. Compound XXIe showed a new signal at δ 61.99 ppm attributed to the carbon of the methoxy group.
 | ||
| Scheme 3 Reagents & conditions: (a) ammonium thiocynate, dry acetone, vigorous stirring, rt, 45 min. (b) Dry acetone, rt, 36 h, 65–90%. | ||
Biological evaluation
In vitro anticancer activity against NCI-60 cell line panel
All of the designed compounds were submitted to National Cancer Institute “NCI” (https://www.dtp.nci.nih.gov), Bethesda, Maryland, USA, and all compounds were selected by the Developmental Therapeutics Program (DTP) for evaluation of their antitumor activity. The compounds were screened against 60 different human tumor cell lines, including leukemia, non-small lung cancer, colon, CNS (central nervous system), melanoma, ovarian, renal, prostate, and breast cancers at an initial single dose of 10−5 M concentration under the NCI drug discovery program. Furthermore, compound (XVI) (NSC no – 833644) showed a remarkable cell growth inhibition at 10−5 M so it was further screened for 5log dose molar range.
Primary single high dose (10−5 M) full NCI 60 cell panel in vitro assay
A primary in vitro one-dose anticancer assay was conducted in a full NCI 60 cell panel. Results for each compound were displayed as a mean graph of the percent growth of the treated cells when compared to the untreated control cells. Analysis of the NCI-60 results showed the following:At 10 μM concentration, compounds with the acetamide linkage XIIa–f & XVI showed the best anticancer activity among all of the tested compounds. Compound XVI incorporating 1,3,4-thiadiazole linked via piperazine acetamide linker to the parent scaffold demonstrated a highly potent inhibition percentage against most of the NCI cell panels. As it exhibited antiproliferative activity leukemia cell lines; MOLT-4 with inhibition percentages of 96.27%, colon cancer cell lines; KM12 with inhibition percentages of 95.45%, melanoma cell lines; M14 with inhibition percentages of 95.18% & MDA-MB-435 with inhibition percentages of 92.86%, breast cancer cell lines; HS 578T with inhibition percentages of 96.08%, BT-549 with inhibition percentages of 185.88% (Table 2).
Compounds XIIa–f incorporating a terminal un/substituted phenyl ring showed significant anticancer activity, especially against melanoma, leukemia, renal cancer & breast cancer. The broad spectrum cell inhibition was attributed to the 4-Br & 4-Cl analogs XIIb & XIIc as they exhibited antiproliferative activity against non-small cell lung cancer cell lines; NCI-H522 with inhibition percentages of 180.32, 183.09% respectively, against melanoma cell lines; MALME-3M with inhibition percentage of 154.39 & 156.24% respectively, against renal cancer cell lines; ACHN with inhibition percentage of 188.11 & 180.68% respectively, CAKI-1 with inhibition percentage of 157.01 & 139.72% respectively & UO-31 with inhibition percentage 176.96 & 160.46% respectively. Followed by 4-H & 4-CH3 analogs XIIe & XIIa which showed good anticancer activity demonstrated in (Table 1). In addition compounds XIIa–d showed good anticancer activity against leukemia cell lines (MOLT-4) with percentage inhibition of 60.21%, 85.02%, 92.94% & 68.77% respectively, and a significant anticancer activity against melanoma cell lines (SK-MEL-5) with inhibition percentage of 89.62%, 90.56%, 99.65% & 71.20% respectively. Compound XIIf (3-CH3 analog) didn't show any significant cell inhibition percentage except for NCI-H522 cell line as it showed an inhibition percentage 139.56% (Table 1).
| Cell line | Compound no. | |||||
|---|---|---|---|---|---|---|
| XIIa | XIIb | XIIc | XIId | XIIe | XIIf | |
| a NT, not tested. | ||||||
| Leukemia | ||||||
| CCRF-CEM | NT | 38.41 | 48.08 | 36.39 | 21.61 | 23.28 |
| HL-60(TB) | 35.49 | 35.69 | 41.41 | 17.31 | 4.62 | 11.66 |
| K-562 | 61.47 | 57.25 | 57.65 | 46.40 | 40.15 | 32.50 |
| MOLT-4 | 60.21 | 85.02 | 92.94 | 68.77 | 31.94 | 25.03 |
| RPMI-8226 | 33.84 | 57.43 | 55.63 | 45.31 | 24.17 | 24.81 |
| SR | 45.61 | 59.65 | 63.12 | 44.98 | 47.42 | 44.11 |
|
||||||
| Non-small cell lung cancer | ||||||
| A549/ATCC | 13.93 | 24.81 | 33.00 | 14.49 | 10.91 | 2.99 |
| EKVX | 37.47 | 53.14 | 54.99 | 45.12 | 18.97 | 14.09 |
| HOP-62 | 20.79 | 15.43 | 14.26 | 11.63 | 4.42 | 3.77 |
| HOP-92 | 27.73 | 25.37 | 30.21 | 28.33 | 9.06 | 1.79 |
| NCI-H226 | 36.03 | 37.52 | 32.46 | 24.13 | 23.53 | 15.23 |
| NCI-H23 | 49.27 | NT | NT | NT | NT | NT |
| NCI-H322M | 18.12 | 22.82 | 21.07 | 11.51 | 13.48 | 8.67 |
| NCI-H460 | 23.75 | 42.31 | 46.71 | 20.68 | 25.97 | 26.99 |
| NCI-H522 | 41.99 | 180.32 | 183.09 | 37.64 | 141.06 | 139.56 |
|
||||||
| Colon cancer | ||||||
| COLO 205 | 3.66 | 29.42 | 43.32 | 13.25 | −10.90 | −9.41 |
| HCC-2998 | 1.69 | 21.22 | 44.23 | −10.65 | −2.01 | −7.15 |
| HCT-116 | 45.12 | 47.55 | 48.15 | 25.62 | 23.71 | 41.36 |
| HCT-15 | 43.02 | 54.83 | 54.97 | 40.95 | 49.75 | 44.65 |
| HT29 | 16.07 | 26.71 | 36.56 | 12.80 | 6.16 | 23.90 |
| KM12 | 28.82 | 42.50 | 46.85 | 25.04 | 10.08 | 9.58 |
| SW-620 | 13.30 | 21.88 | 21.63 | 2.27 | 0.11 | 8.02 |
|
||||||
| CNS cancer | ||||||
| SF-268 | 17.75 | 41.16 | 55.72 | 26.14 | 19.35 | 11.56 |
| SF-295 | 45.05 | 17.02 | 19.73 | −2.88 | −15.82 | −17.04 |
| SF-539 | 13.14 | 17.69 | 23.93 | 6.04 | 15.44 | −1.83 |
| SNB-19 | 26.69 | 30.71 | 33.50 | 16.22 | 14.09 | 16.89 |
| SNB-75 | 37.81 | 19.11 | 30.34 | −0.02 | −3.25 | −0.44 |
| U251 | 16.85 | 23.83 | 33.26 | 14.59 | 13.49 | 10.99 |
|
||||||
| Melanoma | ||||||
| LOX IMVI | 32.63 | 18.24 | 60.12 | 30.94 | 76.75 | 11.85 |
| MALME-3M | 38.46 | 154.39 | 156.24 | 1.18 | 9.24 | 0.56 |
| M14 | 28.65 | 40.68 | 40.60 | 18.49 | −0.21 | 5.60 |
| MDA-MB-435 | 36.50 | 46.78 | 52.99 | 22.24 | 5.77 | 6.60 |
| SK-MEL-2 | 61.99 | 70.09 | 65.82 | 46.01 | 12.24 | 19.83 |
| SK-MEL-28 | 13.59 | 28.02 | 27.24 | 5.38 | 13.51 | 9.89 |
| SK-MEL-5 | 89.62 | 90.56 | 99.65 | 71.20 | 22.37 | 21.54 |
| UACC-257 | 32.44 | 41.35 | 50.08 | 24.51 | 7.22 | −1.73 |
| UACC-62 | 51.52 | 54.90 | 54.66 | 50.43 | 30.67 | 32.10 |
|
||||||
| Ovarian cancer | ||||||
| IGROV1 | 50.14 | 41.88 | 51.98 | 43.20 | 18.74 | 16.12 |
| OVCAR-3 | 16.99 | 39.45 | 48.21 | 30.54 | −3.49 | 1.05 |
| OVCAR-4 | 58.25 | 43.54 | 55.54 | 36.54 | 17.13 | 8.65 |
| OVCAR-5 | 6.29 | 21.80 | 14.40 | 5.17 | 2.05 | 5.28 |
| OVCAR-8 | 26.00 | 35.31 | 43.36 | 27.07 | 28.63 | 3.97 |
| NCI/ADR-RES | 36.74 | 43.85 | 51.95 | 30.40 | 20.48 | 10.59 |
| SK-OV-3 | 29.90 | 23.31 | 20.77 | 8.64 | −12.48 | 0.27 |
|
||||||
| Renal cancer | ||||||
| 786-0 | 1.86 | 161.68 | 59.55 | −1.53 | −4.76 | 1.95 |
| A498 | 5.96 | 9.14 | 21.35 | −1.31 | −4.13 | 2.01 |
| ACHN | 36.75 | 188.11 | 180.68 | 34.31 | 132.69 | 20.13 |
| CAKI-1 | 39.43 | 157.01 | 139.72 | 34.58 | 22.23 | 17.12 |
| RXF 393 | 14.49 | 24.02 | 25.56 | 5.14 | 13.26 | 9.98 |
| SN12C | 22.41 | 60.92 | 53.38 | 31.70 | 25.36 | 16.62 |
| TK-10 | −17.93 | −20.84 | 2.70 | −29.22 | −51.91 | −30.21 |
| UO-31 | 63.03 | 176.96 | 160.46 | 57.85 | 36.99 | 45.05 |
|
||||||
| Prostate cancer | ||||||
| PC-3 | 30.30 | 50.94 | 61.20 | 41.03 | 34.48 | 36.86 |
| DU-145 | 21.60 | 32.14 | 42.52 | 21.09 | 7.47 | 3.28 |
|
||||||
| Breast cancer | ||||||
| MCF7 | 60.38 | 63.24 | 72.43 | 52.81 | 25.80 | 25.18 |
| MDA-MB-231/ATCC | 26.94 | 33.31 | 36.66 | 24.57 | 19.43 | 12.65 |
| HS 578T | 22.07 | 29.25 | 35.36 | 19.42 | 22.28 | 11.82 |
| BT-549 | 46.33 | 49.95 | 60.86 | 46.27 | 14.96 | 10.12 |
| T-47D | 73.06 | 58.43 | 73.32 | 69.54 | 21.08 | 27.85 |
| MDA-MB-468 | 85.22 | 50.81 | 57.49 | 46.51 | 12.93 | 10.77 |
| Mean | 66.84 | 48.34 | 45.02 | 74.12 | 81.11 | 85.10 |
Replacement of the acetamide linkage with aminoethanone linkage XVIIIa–f resulted in a decrease in percentage inhibition. Increased anticancer activity within this series was observed in the 4-Br, 4-Cl & 3-CH3 analogues XVIIIb, XVIIIc & XVIIIf as it showed significant antiproliferative activity against non-small cell lung cancer NCI-H522 with inhibition percentage of 73.62, 50.17 & 106.93% respectively, against melanoma (SK-MEL-5) with inhibition percentage of 67.48, 74.36 & 62.50 respectively, against renal cancer UO-31 with inhibition percentage 68.99, 68.08 & 65.74% respectively and against breast cancer T-47D with inhibition percentage 72.72, 63.26 & 66.43% respectively. Followed by 4-OCH3 & 4-CH3 analogues which showed good anticancer activity against melanoma SK-MEL-5 with inhibition percentages 83.62, 64.82% respectively and against breast cancer T-47D with inhibition percentages 74.01, 62.13% respectively (Table S1) (ESI†).
Compounds XXIa–f incorporating piperazine linked to n-aroyl thioamide showed a good anticancer activity against non-small cell lung cancer, CNS cancer & renal cancer which was demonstrated in (Table S2) (ESI†) especially compound XXIf which showed a remarkable activity against leukemia SR (inhibition 80.09%), against non-small cell lung cancer HOP-62 (inhibition 102.01%), HOP-92 (inhibition 85.26%), against CNS cancer SF-539 (inhibition 88.81%), SNB-19 (inhibition 71.82%), SNB-75 (inhibition 97.65%) and against renal cancer 786-0 (inhibition 72.18%), ACHN (inhibition 75.48%), CAKI-1 (inhibition 72.33%) & SN12C (inhibition 71.70%).
On contrary, compounds XIV & XV didn't show any significant cell inhibition percentage, while compound XIII showed significant anticancer activity against non-small cell lung cancer, NCI-H322M (inhibition 69.30%), NCI-H460 (inhibition 65.00), against colon cancer COLO 205 (inhibition 97.35%), HCC-2998 (inhibition 86.37%), HCT-15 (inhibition 72.94%), HT29 (inhibition 83.52%), against melanoma MALME-3M (inhibition 72.76%), against ovarian OVCAR-3 (inhibition 94.00%), against renal cancer 786-0 (inhibition 80.79%), A498 (inhibition 107.50%), SN12C (inhibition 76.90%), TK-10 (inhibition 93.85%) (Table 2).
| Cell line | Compound no. | |||
|---|---|---|---|---|
| XIII | XIV | XV | XVI | |
| a NT, not tested. | ||||
| Leukemia | ||||
| CCRF-CEM | 4.35 | 14.88 | −3.11 | NT |
| HL-60(TB) | 3.30 | 10.45 | 1.88 | 145.54 |
| K-562 | 2.63 | 25.34 | −0.25 | 89.44 |
| MOLT-4 | 0.73 | 11.59 | −7.57 | 96.27 |
| RPMI-8226 | 3.55 | 24.02 | 0.80 | 116.64 |
| SR | 6.80 | 26.04 | 4.74 | 128.89 |
|
||||
| Non-small cell lung cancer | ||||
| A549/ATCC | −9.39 | −5.50 | −6.49 | 132.54 |
| EKVX | 12.37 | 17.80 | 5.12 | 144.79 |
| HOP-62 | −2.98 | −3.48 | 5.00 | 152.69 |
| HOP-92 | −6.68 | 4.35 | 4.93 | 170.93 |
| NCI-H226 | 11.49 | 32.35 | 17.11 | 56.09 |
| NCI-H23 | NT | NT | NT | 155.11 |
| NCI-H322M | 69.30 | 10.33 | 1.59 | 15.17 |
| NCI-H460 | 65.00 | 3.85 | −6.06 | 122.10 |
| NCI-H522 | −6.93 | 20.20 | 10.84 | 179.35 |
|
||||
| Colon cancer | ||||
| COLO 205 | 97.35 | −4.70 | −7.70 | 150.21 |
| HCC-2998 | 86.37 | −6.65 | −3.24 | 40.29 |
| HCT-116 | 58.65 | 8.74 | −9.85 | 91.77 |
| HCT-15 | 72.94 | 2.34 | −11.65 | 130.72 |
| HT29 | 83.52 | −1.02 | −4.77 | 161.53 |
| KM12 | 40.60 | 9.07 | 2.76 | 95.45 |
| SW-620 | 81.53 | 3.20 | −1.88 | 123.86 |
|
||||
| CNS cancer | ||||
| SF-268 | 73.30 | 8.83 | 6.94 | 134.37 |
| SF-295 | 46.58 | −11.73 | −9.76 | 131.40 |
| SF-539 | 59.89 | 2.29 | −3.61 | 188.20 |
| SNB-19 | 88.90 | 1.89 | −1.52 | 41.60 |
| SNB-75 | 53.50 | −8.62 | 19.30 | 174.96 |
| U251 | 87.14 | 2.45 | −0.28 | 172.52 |
|
||||
| Melanoma | ||||
| LOX IMVI | 53.82 | 21.59 | 11.67 | 187.39 |
| MALME-3M | 72.76 | 0.21 | −10.37 | 167.26 |
| M14 | 62.16 | 3.99 | −5.15 | 95.18 |
| MDA-MB-435 | 58.98 | 3.15 | −3.11 | 92.86 |
| SK-MEL-2 | 46.66 | 8.32 | −5.97 | 72.03 |
| SK-MEL-28 | 59.00 | 4.21 | −4.39 | 27.65 |
| SK-MEL-5 | 37.50 | 27.16 | 5.86 | 70.64 |
| UACC-257 | 66.91 | −3.73 | −10.30 | 45.74 |
| UACC-62 | 34.57 | 18.17 | 3.34 | 82.55 |
|
||||
| Ovarian cancer | ||||
| IGROV1 | 59.41 | 10.34 | 49.06 | 141.81 |
| OVCAR-3 | 94.00 | 12.46 | −5.47 | 155.56 |
| OVCAR-4 | 51.61 | 8.69 | 0.64 | 86.42 |
| OVCAR-5 | 83.82 | 3.43 | −1.45 | 104.09 |
| OVCAR-8 | 65.24 | 5.72 | 0.41 | 121.38 |
| NCI/ADR-RES | 60.62 | 8.96 | −0.12 | 132.63 |
| SK-OV-3 | 67.09 | 1.90 | −10.10 | 71.86 |
|
||||
| Renal cancer | ||||
| 786-0 | 80.79 | 0.36 | 5.63 | 180.94 |
| A498 | 107.50 | −4.72 | 3.13 | 51.04 |
| ACHN | 45.35 | 20.47 | 9.38 | 187.46 |
| CAKI-1 | 47.75 | 21.68 | 28.23 | 195.69 |
| RXF 393 | 76.34 | 4.44 | −10.84 | 171.34 |
| SN12C | 76.90 | 17.97 | 3.48 | 154.68 |
| TK-10 | 93.85 | −34.09 | −15.74 | 156.49 |
| UO-31 | 34.26 | 45.53 | 31.93 | 197.16 |
|
||||
| Prostate cancer | ||||
| PC-3 | 70.01 | 19.04 | 10.97 | 136.14 |
| DU-145 | 75.39 | −1.94 | −5.36 | 196.08 |
|
||||
| Breast cancer | ||||
| MCF7 | 45.57 | 27.53 | 16.49 | 83.90 |
| MDA-MB-231/ATCC | 64.96 | 6.69 | −0.80 | 70.26 |
| HS 578T | 60.40 | 12.15 | 9.50 | 96.08 |
| BT-549 | 76.87 | 13.66 | 0.30 | 185.88 |
| T-47D | 33.57 | 30.68 | 20.07 | 72.12 |
| MDA-MB-468 | 35.78 | 31.66 | 2.52 | 139.82 |
| Mean | 94.47 | 90.78 | 97.85 | −23.26 |
The investigation of the structure–activity relationship of all the newly synthesized compounds revealed that the highest anticancer activity was observed in the series with the acetamide linkage (NHCO) (XIIa–f). Increasing the density of the hetero atom of the structure of (XIIa–f) by introducing a thiazole ring seems to enhance the activity (XVI) as this compound showed promising anticancer activity against most of the cell lines. Meta substitution on the phenyl ring diminished the activity, whereas unsubstitution and para substitution on the benzene ring improved the activity. This is validated by compounds containing para or unsubstituted phenyl (XIIa–e) which demonstrated promising activity in comparison to compound (XIIf) with 3-CH3 which showed no significant activity. The introduction of a small electron withdrawing group such as chloro (XIIc, XVIIIc) or bromo (XIIb, XVIIIb) increases the activity while introducing an electron donating group (XIIe, XVIIIe) slightly decreases the activity. The presence of a phenyl ring seems to be essential for activity. It was predicted that increasing the ring size to the naphthyl group in compound (XIII) enhance the hydrophobic interaction and thus increases the activity, but, in our study, the naphthyl group has lower activity than phenyl, so we concluded that the bulkier group is not preferable.
In vitro 5 dose full NCI 60 cell panel assay
Due to the significant cell growth inhibition of compound XVI against a variety of cell lines, it was further screened for NCI full panel 5 dose assays. All 60 cell lines, representing nine tumor subpanels, were incubated at five different concentrations (0.01, 0.1, 1, 10, and 100 μM of compound XVI). In order to calculate the three response parameters (GI50, TGI & LC50) for each cell line, the assay results were used to create log concentration vs. % growth curves. GI50 (growth inhibitory activity) refers to the concentration of the compound causing a decrease in cell growth by 50%. Total growth inhibition or TGI, refers to the concentration of the compound resulting in total growth inhibition. LC50 or cytotoxic activity refers to the concentration of the compound that causes net 50% loss of initial cells at the end of 48 hours incubation period of. Compound XVI demonstrated remarkable antitumor activity against most of the cell lines representing nine subpanels with GI50 between 1.17 and 18.40 μM (Table 3).| Pannel | Cell line | GI50 | TGI | LC50 |
|---|---|---|---|---|
| Leukemia | CCRF-CEM | 2.55 | 7.17 | 100 |
| HL-60(TB) | 10.40 | 39.50 | 100 | |
| K-562 | 7.91 | 36.60 | 100 | |
| MOLT-4 | 4.08 | 31.30 | 100 | |
| RPMI-8226 | 2.83 | 8.67 | 62.8 | |
| SR | 2.11 | 5.26 | 100 | |
| Non-small cell lung cancer | A549/ATCC | 2.07 | 4.31 | 9.01 |
| EKVX | 2.90 | 11.10 | 39.90 | |
| HOP-62 | 1.62 | 3.29 | 6.68 | |
| HOP-92 | 1.57 | 3.46 | 7.62 | |
| NCI-H226 | 1.84 | 5.14 | 43.90 | |
| NCI-H23 | 1.74 | 3.57 | 7.33 | |
| NCI-H322M | 14.40 | 27.50 | 52.40 | |
| NCI-H460 | 4.25 | 14.70 | 41.30 | |
| NCI-H522 | 1.62 | 3.09 | 5.90 | |
| Colon cancer | COLO 205 | 14.04 | 27.00 | 52.00 |
| HCC-2998 | 13.60 | 26.00 | 51.40 | |
| HCT-116 | 3.75 | 14.50 | 38.10 | |
| HCT-15 | 1.79 | 3.42 | 6.55 | |
| HT29 | 6.91 | 19.30 | 43.90 | |
| KM12 | 4.46 | 15.90 | 39.90 | |
| SW-620 | 4.99 | 16.10 | 40.10 | |
| CNS cancer | SF-268 | 4.79 | 19.30 | 59.60 |
| SF-295 | 10.30 | 22.30 | 48.20 | |
| SF-539 | 1.76 | 3.32 | 6.26 | |
| SNB-19 | 12.20 | 24.60 | 49.60 | |
| SNB-75 | 15.10 | 30.50 | 16.50 | |
| U251 | 1.92 | 3.99 | 8.27 | |
| Melanoma | LOX IMVI | 1.49 | 2.81 | 5.30 |
| MALME-3M | 1.69 | 3.30 | 6.41 | |
| M14 | 4.17 | 16.20 | 40.30 | |
| MDA-MB-435 | 13.60 | 27.50 | 55.30 | |
| SK-MEL-2 | 12.60 | 25.80 | 52.80 | |
| SK-MEL-28 | 13.90 | 26.90 | 51.90 | |
| SK-MEL-5 | 10.20 | 21.90 | 46.80 | |
| UACC-257 | 14.10 | 28.10 | 56.20 | |
| UACC-62 | 4.97 | 18.70 | 43.30 | |
| Ovarian cancer | IGROV1 | 2.68 | 7.65 | 29.50 |
| OVCAR-3 | 1.93 | 3.67 | 6.98 | |
| OVCAR-4 | 10.30 | 28.10 | 76.70 | |
| OVCAR-5 | 6.41 | 19.30 | 44.00 | |
| OVCAR-8 | 1.93 | 4.02 | 8.38 | |
| NCI/ADR-RES | 1.77 | 3.69 | 7.72 | |
| SK-OV-3 | 1.25 | 25.00 | 50.00 | |
| Renal cancer | 786-0 | 1.99 | 3.56 | 6.37 |
| A498 | 18.40 | 35.00 | 6.67 | |
| ACHN | 1.79 | 3.18 | 5.64 | |
| CAKI-1 | 1.60 | 2.97 | 5.54 | |
| RXF 393 | 1.68 | 3.31 | 6.53 | |
| SN12C | 1.94 | 5.06 | 16.80 | |
| TK-10 | 3.09 | 6.14 | 16.80 | |
| UO-31 | 1.17 | 2.39 | 4.89 | |
| Prostate cancer | PC-3 | 4.33 | 17.10 | 44.40 |
| DU-145 | 1.80 | 3.24 | 5.82 | |
| Breast cancer | MCF7 | 4.32 | 18.00 | 47.70 |
| MDA-MB-231/ATCC | 1.78 | 3.87 | 8.41 | |
| HS 578T | 12.40 | 63.00 | 100.00 | |
| BT-549 | 1.55 | 3.02 | 5.88 | |
| T-47D | 7.93 | 24.10 | 62.80 | |
| MDA-MB-468 | 3.06 | 13.80 | 44.10 |
Compound (XVI) showed significant activity against non-small cell lung cancer HOP-62 with GI50 1.62, a TGI value of 3.29 and LC50 value of 6.68, HOP-92 with GI50 1.57, a TGI value of 3.46 and LC50 value of 7.62, NCI-H522 with GI50 1.62, a TGI value of 3.09 and LC50 value of 5.90, against melanoma LOX IMVI with GI50 1.49, a TGI value of 2.81 and LC50 value of 5.30, MALME-3M with GI50 1.69, a TGI value of 3.30 and LC50 value of 6.41, against ovarian cancer OVCAR-3 with GI50 1.93, a TGI value of 3.67 and LC50 value of 6.98, against renal cancer CAKI-1 with GI50 1.60, a TGI value of 2.97 and LC50 value of 5.54, RXF 393 with GI50 1.68, a TGI value of 3.31 and LC50 value of 6.53, UO-31 with GI50 1.17, a TGI value of 2.39 and LC50 value of 4.89, and against breast cancer BT-549 with GI50 1.55, a TGI value of 3.02 and LC50 value of 5.88 (Table 3).
Molecular docking
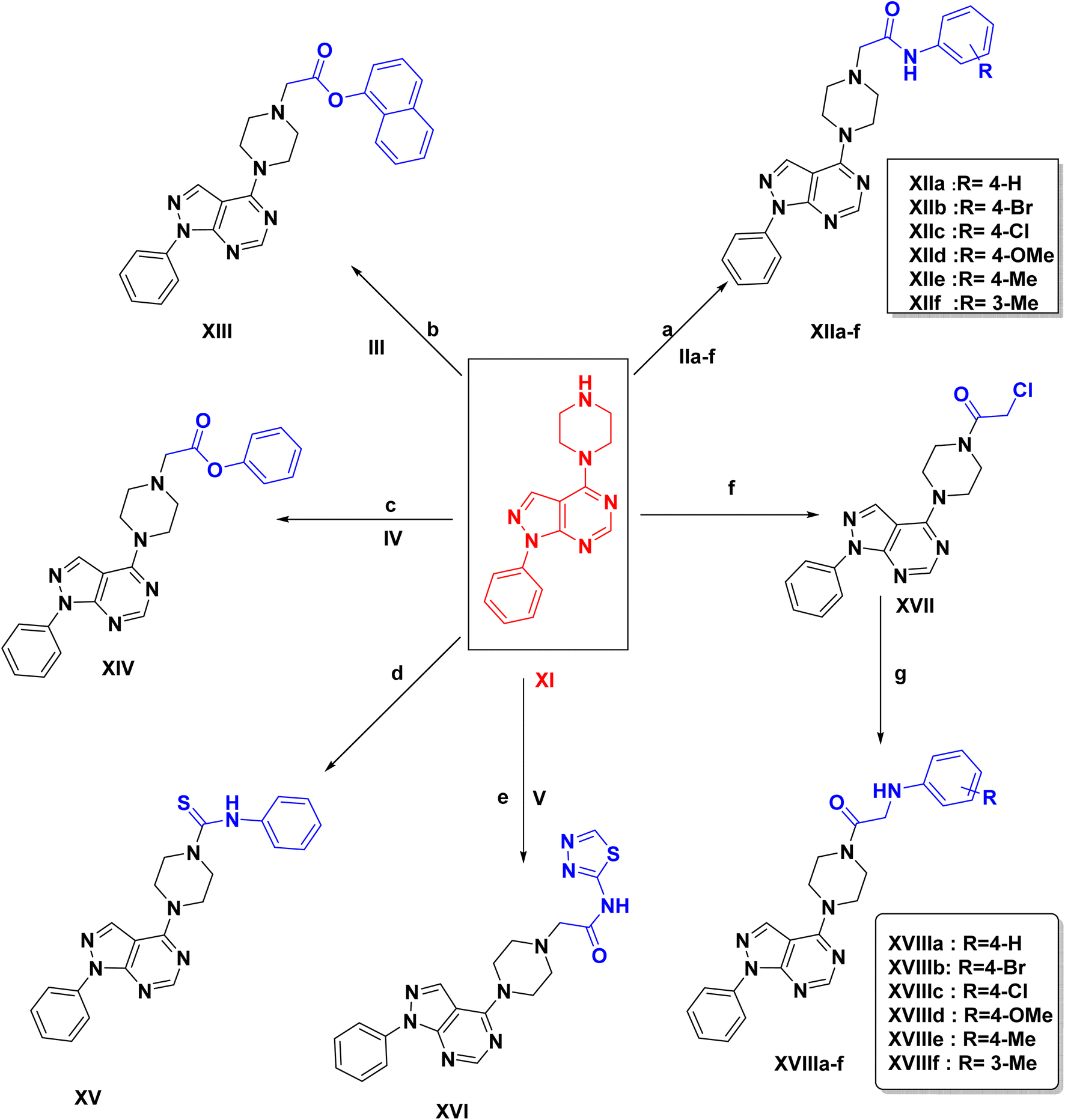
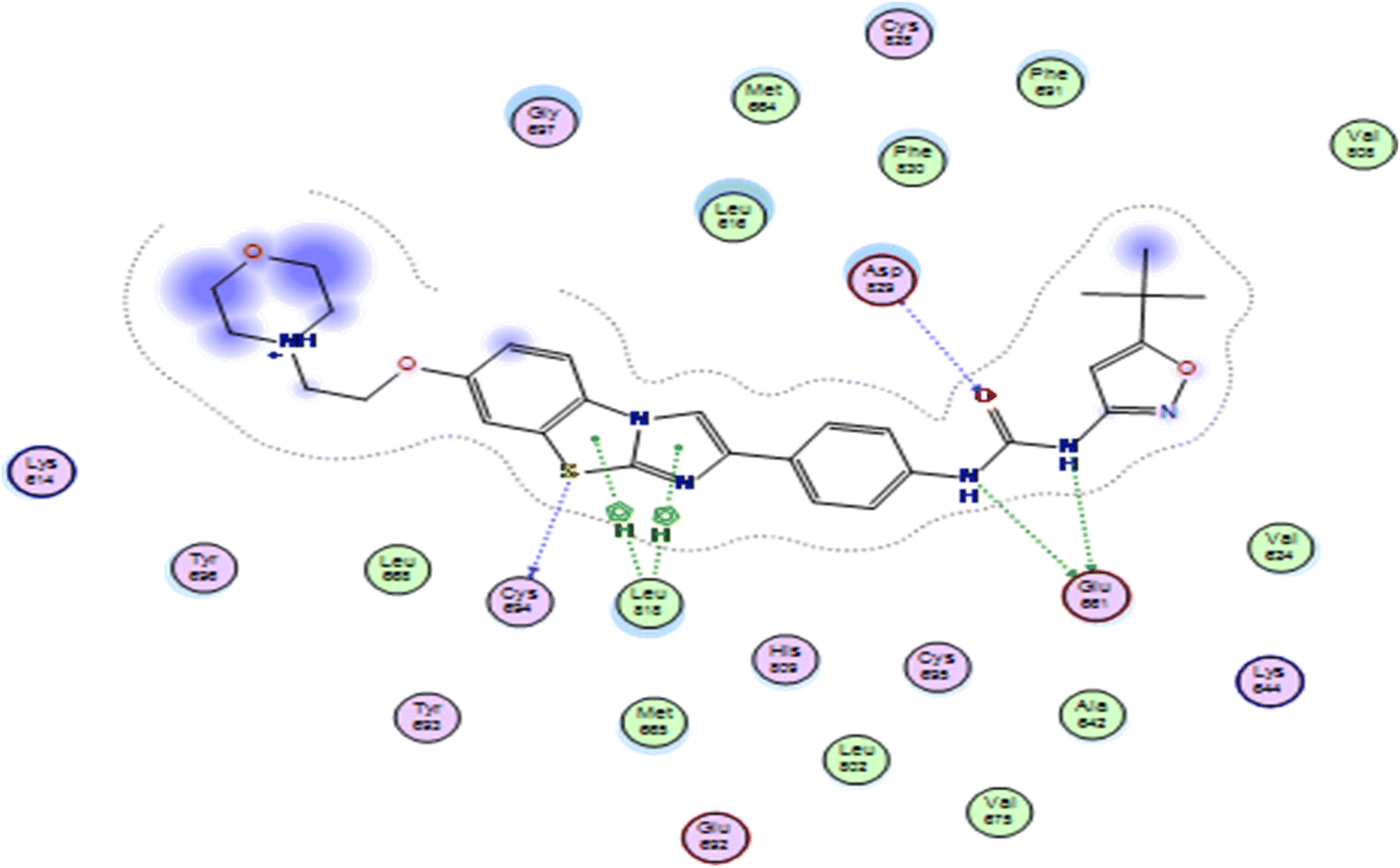
In this study, the docking was carried out using MOE software. The molecular docking study was conducted between the newly synthesized compounds and the binding site of FLT3. Each test molecule gave 10 possible docked poses. The ideal pose of each molecule was selected according to the similarity of its binding mode in the binding site to that of the lead compound.To validate our docking, we re-docked the selective inhibitor (quizartinib) in the FLT3 kinase domain. We compared its binding mode and interactions with interactions of the newly synthesized pyrazolo[3,4-d]pyrimidine derivatives. Quizartinib binds to the crystal structure of the FLT3 kinase domain (PDB code: 4RT7) through different amino acids. The benzo(d)imidazo[2,1-b]thiazole forms 1 hydrogen bond with Cys 694 & fits into the hinge region. In the allosteric site, there are two essential interaction of the urea moiety with the protein; the NH makes a hydrogen bond with Glu 661 while the carbonyl group of urea interacts with Asp 829 (Fig. 4 and 5).
 | ||
| Fig. 4 2D representation of quizartinib (re-docked) in FLT3 kinase binding domain (PDB code: 4RT7), showing the same key interactions as reported. | ||
 | ||
| Fig. 5 Alignment of the original ligand quizartinib (green) and re-docked ligand (red) at FLT3 kinase binding domain using MOE software. | ||
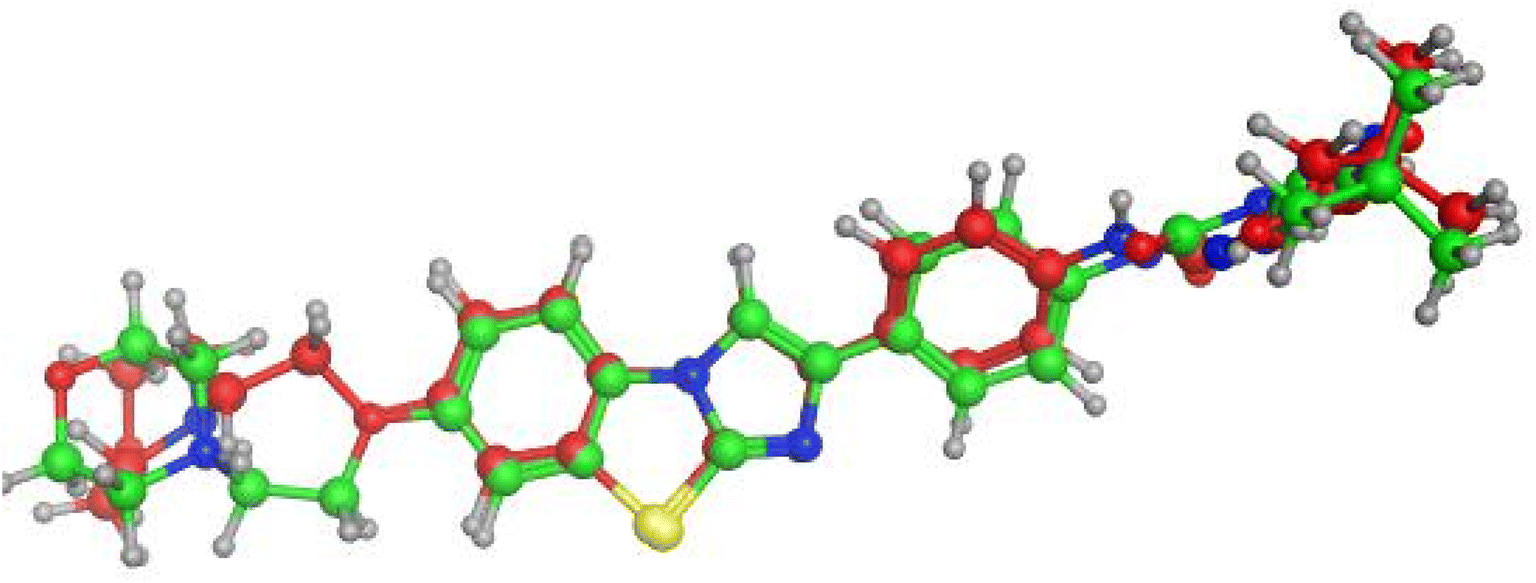
Compounds with amide linkage XIIa–f fulfilled all of the essential features as done by the lead compound in the FLT3 ATP binding site as they formed one hydrogen bond with Cys 694 in the hinge region and two hydrogen bonds with Glu 661, Asp 829 in the allosteric binding site, this could explain the high anti-cancer activity of these compounds except for compound XIIa that binds only with Cys 694 in the hinge region. One of the best-scored poses is presented in Fig. 6.
 | ||
| Fig. 6 (A–D) 2D & 3D representation of compounds XIIb (A and B) & XIIe (C and D) in FLT3 kinase binding domain (PDB code: 4RT7), they established the same key interactions as the lead compound (quizartinib); N2 pyrimidine form one hydrogen bond with Cys 694 residue; NH of amide linker makes a hydrogen bond with Glu661; CO of amide moiety makes a hydrogen bond with Asp 829. | ||
Compound XVI lost the binding interaction with Asp 829 in the allosteric binding site (Fig. 7). Compounds XVIIa–f, XIII, XIV & XV fulfilled the binding interaction in the hinge region but binds only with Asp 829 in the allosteric binding site, except for compound XVIIIC lost the binding interaction with the main amino acid (Cys 694) in the hinge region, resulting in lower docking scores which explains the lower anticancer activity in comparison with compounds with the amide linkage XIIa–f (Fig. 6). Finally, compounds XXIa–f lost the binding interaction in the allosteric binding site and formed an additional hydrogen bond with Lys 644.
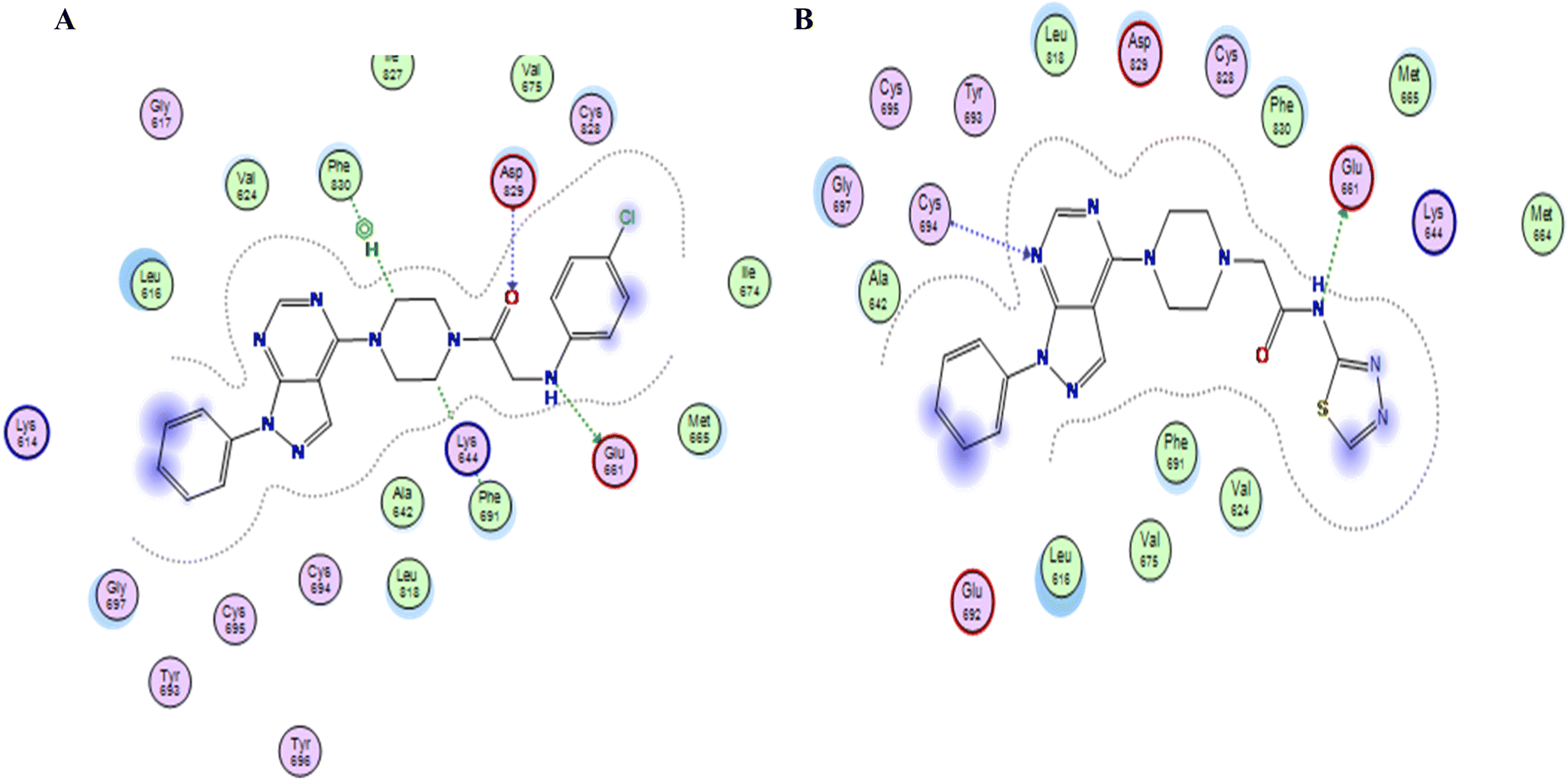 | ||
| Fig. 7 (A & B) 2D representation of compound XVIIIc & XVI in FLT3 kinase binding domain respectively (PDB code: 4RT7), missing one key interaction. | ||
In vitro enzyme inhibition assay
In an attempt to investigate the possible mechanism of action of all the synthesized compounds based on the structural similarities with tandutinib and quizartinib, all of them were evaluated for their FLT3 inhibitory activity at Thermo Fisher Scientific, USA (https://www.thermofischer.com/selectscreen). The assay was conducted using the Z′-LYTE biochemical assay which applies a fluorescence-based, coupled-enzyme method. All of the compounds were tested against FLT3 in a single-point concentration assay at 10 μM to measure their inhibition percentage. Unfortunately, the targeted compounds showed weak inhibitory activity against FLT3, where most of the compounds activity was between 5% and 29%. Because of these results, an additional investigation was performed to understand the low activity of the synthesized compounds as described next (Table 4).| No. | CPD ID | FLT3% inhibition |
|---|---|---|
| 1 | XIIa | 26 |
| 2 | XIIb | 15 |
| 3 | XIIc | 15 |
| 4 | XIId | 24 |
| 5 | XIIe | 16 |
| 6 | XIIf | 14 |
| 7 | XIII | 9 |
| 8 | XIV | 18 |
| 9 | XV | 5 |
| 10 | XVI | 29 |
| 11 | XVIIIa | 20 |
| 12 | XVIIIb | 10 |
| 13 | XVIIIc | 8 |
| 14 | XVIIId | 9 |
| 15 | XVIIIe | 13 |
| 16 | XXIa | 11 |
| 17 | XXIb | 9 |
| 18 | XXIc | 5 |
| 19 | XXId | 8 |
| 20 | XXIe | 6 |
| 21 | XXIf | 5 |
Molecular dynamics (MD)
Even though the targeted compounds had structural similarities with tandutinib and quizartinib, they fulfilled most of the key interactions as done by the lead compound in molecular docking, moreover, they showed significant antiproliferative activity against most of the cell lines in NCI sixty cell panel, they showed low enzymatic activity against FLT3 so to understand these findings, molecular dynamics was performed to gain more insight into the binding mode of 4RT7, molecular dynamics simulations were performed on the docked complexes of the highest-ranked docking compounds with the highest percentage of enzyme inhibition against FLT3 kinase domain XIIb, XIIc & XVI. Quizartinib was also subjected to MD simulations to account for its binding mode and protein dynamics as a reference, resulting in a total of four MD runs. MD was carried out for 50 ns at NPT ensemble (constant number of particles, pressure, and temperature), for minimization and equilibration purposes. Various computational techniques such as RMSD, RG, RMSF, and H-bond analysis were used to analyze the structural changes and dynamic behavior in complexes (Fig. 8).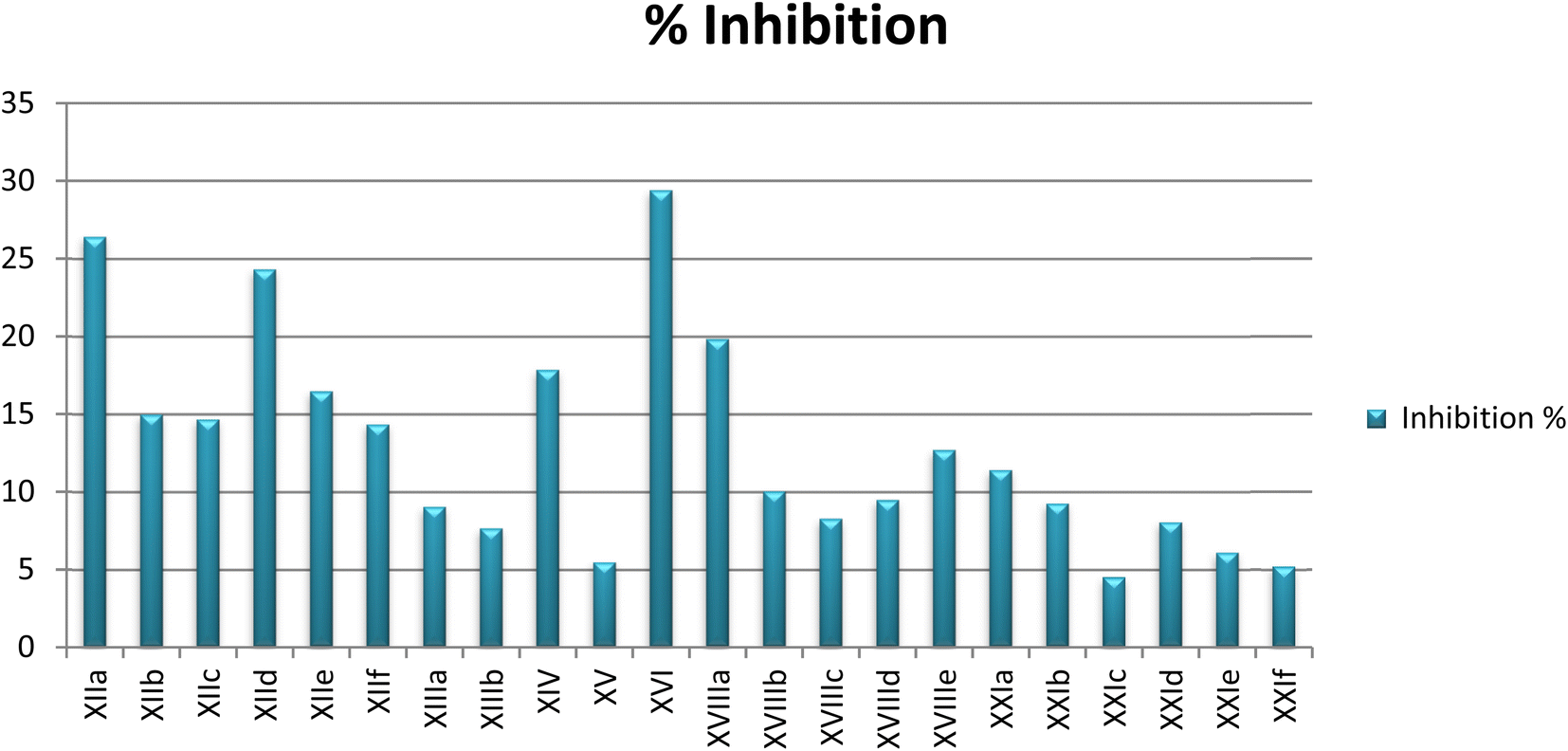 | ||
| Fig. 8 Percent inhibition at 10 μM against FLT3 enzymatic activity achieved by designed compounds. | ||
To monitor structural and conformational changes, RMSD analysis was carried out. RMSD (root mean square deviation) is used to measure the stability of the protein–ligand complex as it measures the average distance between a group of atoms. The stability for each compound was compared to that of quizartinib as a reference over the trajectory of 50 ns. The RMSD for the ligand in each trajectory is plotted against simulation time in ns and illustrated in (Fig. 9). RMSD of the protein backbone (Cα, C, and N) in the 4 dynamics runs show stability after 10 ns of the simulation with RMSD 1.24 ± 0.12, 1.08 ± 0.15, 1.18 ± 0.12 and 1.41 ± 0.13 Å (mean ± SD) for XIIb, XIIc, XVI, and quizartinib respectively. Low RMSD values (1–2 Å) indicate the stability of the protein–ligand complex. Compounds XIIc & XVI showed a slight fluctuation at 10 & 50 ns respectively, it seems that the ligand tries to explore other conformations but they remain stable for the rest of the simulation. In conclusion, the RMSD fluctuation analysis suggests that the MD trajectories were overall stable complex (Fig. 10).
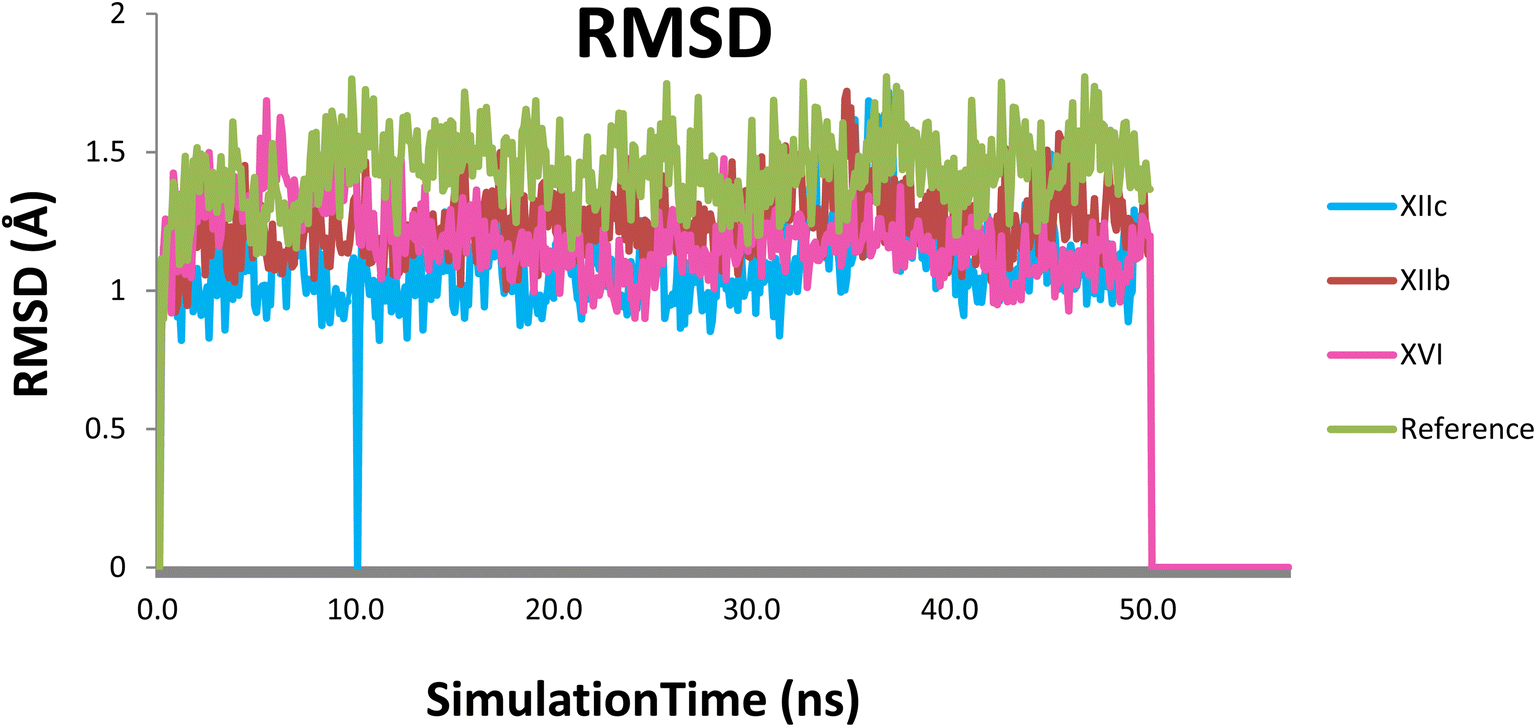 | ||
| Fig. 9 RMSD plot of each compound with the protein, (XIIb: red, XIIc: blue, XVI: pink & reference: green). | ||
 | ||
| Fig. 10 RMSF for the protein residues in the 4 dynamic runs, (XIIb: red, XIIc: blue, XVI: pink & reference: green). | ||
RMSF (root mean square fluctuation) was used to calculate the average fluctuation of each amino acid of the protein. RMSF values appear to be relatively high (> 2 Å) in the regions of amino acids 25–50, 160 & 270 for compounds XIIc & XVI which indicates that they are terminal or far away from the binding site; hence this may affect their biological activity. The rest of the binding site amino acids showed lower RMSF values (<2 Å) indicating their stability during the simulation.
The radius of gyration (Rg) is a measure of the compactness of the protein during the simulation. As shown in (Fig. 11), Rg values of XIIb, XIIc, XVI & reference were 19.25 ± 0.10, 19.27 ± 0.11, 19.29 ± 0.56, and 19.15 ± 0.77 Å (mean ± SD), respectively. Rg is stable for each protein throughout the simulation; hence, no major conformational changes occur during the simulation.
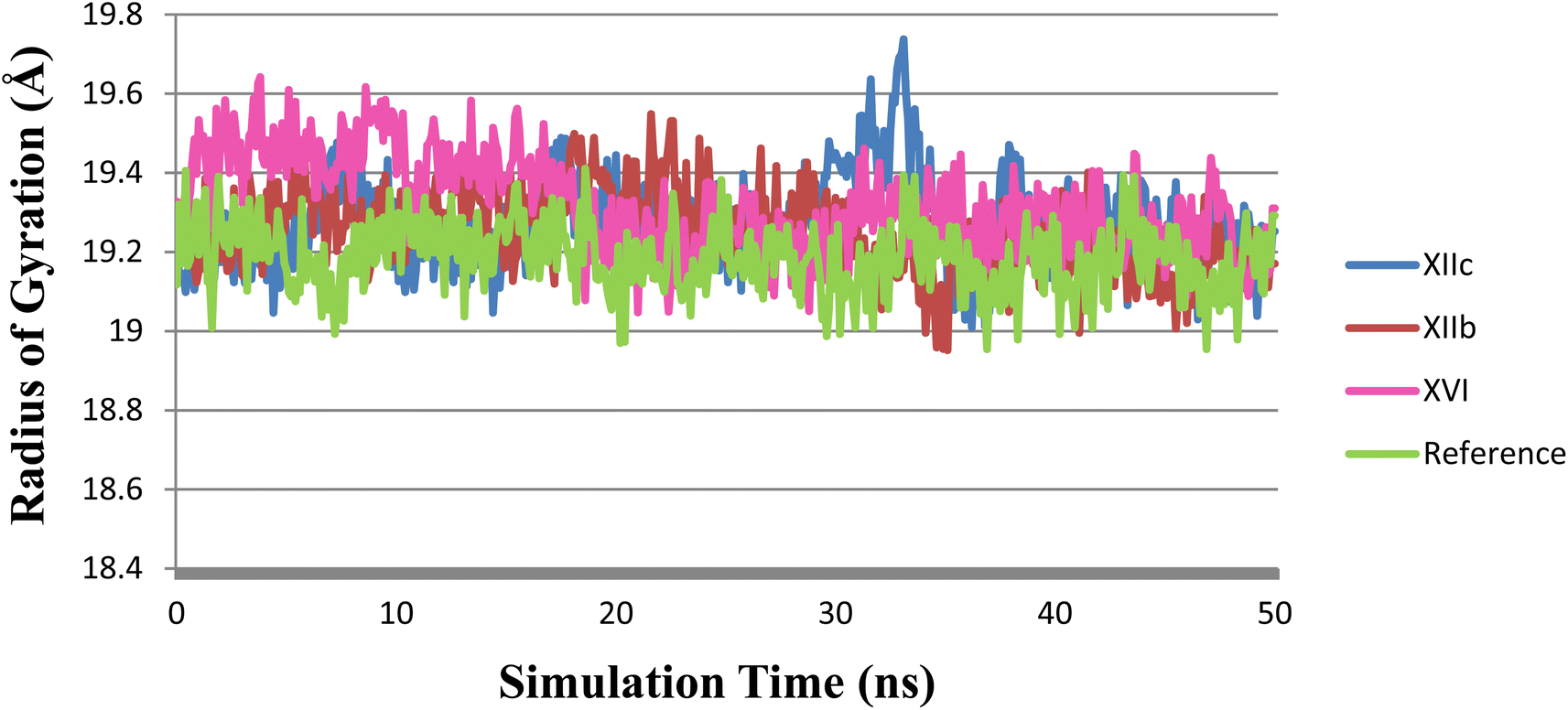 | ||
| Fig. 11 Rg plot during the simulation time of 50 ns, (XIIb: red, XIIc: blue, XVI: pink & reference: green). | ||
Hydrogen bonding analysis between each compound and the protein was performed to investigate the hydrogen bonding contribution to the overall binding interactions. (Fig. 12) describes the number of H-bonds formed during the simulation time. The reference compound (quizartinib) shows the highest number of H-bonds to the protein which indicates its strong binding. This also reflects its high docking score. On the other hand, compounds XIIb, XIIc, and XVI show a lower number of formed H-bonds when compared to the reference, also these compounds do not seem to form any stable H-bonds with the three essential amino acids as the lifetime of hydrogen bond was very low (Table 5) which indicate their weaker binding, which can reflect their low enzymatic activity in the biological evaluation.
 | ||
| Fig. 12 The number of H-bonds formed between each compound and the protein, (XIIb: red, XIIc: blue, XVI: pink & reference: green). | ||
| Contributing amino acid | Lifetime of H-bond | |||
|---|---|---|---|---|
| XIIb | XIIc | XVI | Reference | |
| Cys694 | 18.76% | — | — | 89.31% |
| Glu661 | 40.81% | 50.14% | 33.75% | 75.51% |
| Asp829 | — | — | 16.37% | 81.95% |
In silico predictive ADME study for targeted compounds
In an attempt to further examine the pharmacokinetics properties of synthesized compounds, a computer-aided ADMET study was conducted by using Accelrys Discovery Studio 2.5 software. These studies were based on various molecular descriptors and involved the calculation of some parameters as atom-based logP98 (AlogP98), aqueous solubility level (AQ SOl LEV), 2D polar surface area (ADMET 2D PSA), blood–brain barrier level (BBB LEV), cytochrome P450 2D6 (CYP2D6), hepatotoxicity level (HEPATOX LEV) and hepatotoxicity probability (HEPATOX PROB). All the calculated parameters are tabulated in (Table 6). The results of the ADMET study are presented as ADMET 2D plot drawn using calculated PSA_2D and AlogP98 properties (Fig. 13).
| CPD ID | BBB-Leva | Absorp-levb | Sol-levc | Hepatotoxd | Hepatox prob | CYP2D6e | AlogP98 |
PSA-2Df |
|---|---|---|---|---|---|---|---|---|
| a BBB_level; 4 = undefined, 2 = medium penetration, 1 = high penetration.b Absorp_level; 3 = very low absorption, 2 = low absorption, 1 = moderate absorption, 0 = good absorption.c SOl LEV; 2 = low solubility, 1 = very low but soluble, 0 = extremely low solubility.d Hepatotox_level; 1 = toxic, 0 = non-toxic.e CYP 2D6; 1 = likely to inhibit, 0 = non-inhibitor.f PSA (polar surface area); cpds must have logP value not greater than 5.0 to attain a reasonable probability of being well absorbed. |
||||||||
| XIIa | 2 | 0 | 2 | 0 | 0.231 | 1 | 2.832 | 75.947 |
| XIIb | 2 | 0 | 2 | 0 | 0.172 | 1 | 3.318 | 75.947 |
| XIIc | 2 | 0 | 2 | 0 | 0.278 | 1 | 3.496 | 75.947 |
| XIId | 3 | 0 | 2 | 0 | 0.258 | 1 | 2.815 | 84.877 |
| XIIe | 2 | 0 | 2 | 0 | 0.251 | 1 | 3.58 | 75.947 |
| XIIf | 2 | 0 | 2 | 0 | 0.192 | 1 | 3.318 | 75.947 |
| XIII | 1 | 0 | 2 | 0 | 0.298 | 1 | 4.387 | 72.067 |
| XIV | 2 | 0 | 2 | 0 | 0.205 | 1 | 3.479 | 72.067 |
| XV | 1 | 0 | 2 | 0 | 0.337 | 1 | 3.926 | 58.646 |
| XVI | 4 | 0 | 2 | 0 | 0.403 | 0 | 4.361 | 98.469 |
| XVIIIa | 2 | 0 | 2 | 0 | 0.225 | 1 | 2.779 | 75.947 |
| XVIIIb | 2 | 0 | 2 | 0 | 0.152 | 0 | 3.266 | 75.947 |
| XVIIIc | 2 | 0 | 2 | 0 | 0.271 | 0 | 3.444 | 75.947 |
| XVIIId | 3 | 0 | 2 | 0 | 0.258 | 1 | 2.763 | 84.877 |
| XVIIIe | 2 | 0 | 2 | 0 | 0.258 | 1 | 3.528 | 75.947 |
| XXIa | 2 | 0 | 2 | 0 | 0.218 | 0 | 3.798 | 75.947 |
| XXIb | 1 | 0 | 2 | 0 | 0.245 | 1 | 4.463 | 75.947 |
| XXIc | 2 | 0 | 2 | 0 | 0.225 | 0 | 3.782 | 84.877 |
| XXId | 2 | 0 | 2 | 0 | 0.192 | 1 | 4.284 | 75.947 |
| XXIe | 1 | 0 | 1 | 0 | 0.218 | 0 | 4.547 | 75.947 |
| XXIf | 2 | 0 | 2 | 0 | 0.218 | 0 | 3.926 | 75.947 |
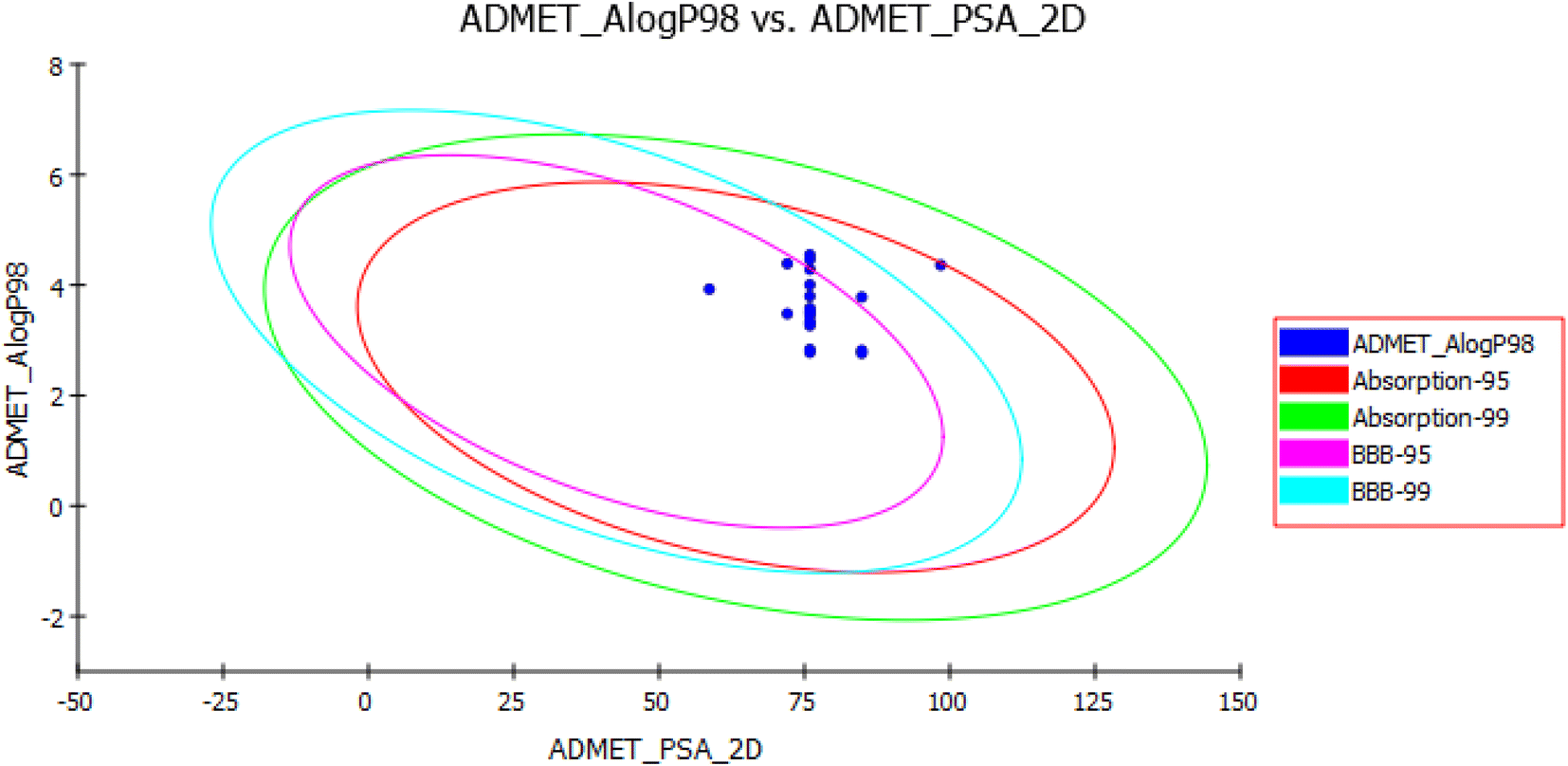 | ||
| Fig. 13 Human intestinal absorption (HIA) and blood–brain barrier plot for the newly synthesized compounds. | ||
Blood–Brain Barrier (BBB) and Human Intestinal Absorption (HIA) plots (Fig. 13) were drawn using all the compounds. In the BBB plot, compound XVI falls outside the 99% confidence ellipse (undefined), whereas the rest of the compounds fall inside the 95–99% ellipse. Hence, compound XVI may have very low penetration to the blood–brain barrier, therefore it may have a low chance for CNS side effects. On the contrary, the rest of the compounds may have a medium to high degree of penetration so these compounds may be good candidates for brain tumors. In the HIA plot, all compounds fall inside the 99% ellipse. Hence, they are predicted to possess good human intestinal absorption with an absorption level 0.
The aqueous solubility level was predicted to be one or two which indicates very low aqueous solubility. The ADME HEPATOTOX.PROB values of most of the compounds range from 0.15–0.40 with hepatotoxicity level 0 which is considered to be not toxic. The CYP2D6 value predicts the inhibitory and non-inhibitory aspects of the titled chemical structure on cytochrome P450 2D6 enzyme. Most of the compounds are foreseen as inhibitors of CYP2D6, so drug–drug interactions are expected upon administration of these compounds. Seven compounds are predicted as non-inhibitor for CYP2D6, so side effects such as liver dysfunction are not expected upon administration of these compounds.39
PSA is an important property that is associated with the drug bioavailability. Thus, molecules that are passively absorbed with PSA > 140 thought to have low bioavailability. All the synthesized compounds have PSA ranging from 58.64–98.46, thus theoretically they should show good passive oral absorption.
Conclusion
In the present study, a novel 21 compounds of pyrazolo[3,4-d]pyrimidine derivatives were designed and synthesized based on their structural similarities with tandutinib and quizartinib. Their structures were confirmed by various spectral and micro analytical data (1H NMR, 13C NMR, FT IR, mass spectrum, elemental analyses, and high resolution mass). Moreover, compounds were biologically evaluated for their antiproliferative activity against NCI sixty cell line panels as well as in vitro FLT3 inhibitory activity.Out of the titled compounds, compound XVI was further screened for NCI full panel 5 dose assays and demonstrated a remarkable anticancer activity against most of the cell lines representing nine subpanels. Additionally, compounds with the piperazine acetamide linkage bearing a terminal substituted phenyl ring XIIa–f exhibited a significant anticancer activity at 10 μM concentration, particularly against melanoma, leukemia, renal cancer, and breast cancer. Molecular docking studies were conducted to investigate the effect of our compounds on FLT3 and revealed the ability of piperazine acetamide based derivatives to fulfill all the key features as done by the lead compound. However, other compounds missed a key interaction, resulting in lower docking scores. Unfortunately, all compounds showed a weak in vitro FLT3 inhibitory activity which couldn't explain the antiproliferative activity of these compounds so molecular dynamics was performed to gain more insight into the binding mode of these compounds and the results revealed that the low lifetime of hydrogen bond affect hydrogen bond stability with the essential amino acids.
In silico ADME study was conducted and predicted these molecules to exhibit good oral bioavailability and that they can pass BBB so they may be good candidates for brain tumors. Finally and according to the in vitro and in silico studies, these compounds have a significant antiproliferative activity but their mechanism of action will be a subject under investigation in our future work.
Experimental
Chemistry
Starting materials, reagents and solvents were purchased from Sigma Aldrich, Loba Chem and Al Gomhouria and were used without further purification. Benzoyl chloride and its derivatives were purchased from Alfa Aesar Pharmaceuticals and were used without further purification. All the reactions were monitored and the purity of the compounds was checked by ascending thin layer chromatography (TLC) on silica gel-coated aluminum plates (Merck 60 F254, 0.25 mm) using mixture of DCM and methanol (9:1) and the spots were seen under ultra violet light at 254 and 366 nm. Melting points were calculated without correction in open capillaries using Stuart (Biocote) scientific melting point apparatus.
1H NMR spectra were recorded in δ scale given in ppm on a Bruker 400 MHz spectrophotometer and referred to TMS at Drug Discovery Center at Faculty of Pharmacy, Ain-Shams University. Whereas 13C NMR spectra were recorded at 100 MHz. FT-IR spectra were determined (KBr) using Shimadzu infrared spectrometer (IR-435) and FT-IR 1650 (PerkinElmer), Faculty of Pharmacy, MSA University. EI-MS spectra were recorded on Finnigan Mat SSQ 7000 (70 eV) mass spectrometer at Regional Center for Mycology and Biotechnology at El Azhar University. Elemental analyses were performed at Regional Center for Mycology and Biotechnology at El-Azhar University. High resolution mass spectra have been recorded on QTOF Firmware Version 26.723 at Natural Products Research Lab at Faculty of Pharmacy, Fayoum University. Starting intermediates in Scheme 1 was prepared according to the reported procedures.
General procedure for the preparation of compound XIIa–f
To a solution of compound XI (0.28 g, 1 mmol) in DMF (20 mL), sodium bicarbonate (0.08 g, 1 mmol) and potassium iodide (0.16 g, 1 mmol) were added. 2-Chloro-N-substituted phenyl acetamide derivatives IIa–f (1 mol) was added to the previous reaction mixture and stirred at room temperature for 36 hours. The reaction mixture was added on to ice cold water to afford a precipitate which was filtered and left to dry. The resulting solid was recrystallized from absolute ethanol to give the titled compounds XIIa–f.O, 168.2, 157.0, 155.8, 154.4, 139.1, 136.5, 135.5, 132.8 2C, 129.5 2C, 126.8, 121.7 2C, 120.0 2C, 101.5, 61.7, 52.8 2C, 40.6 2C; FT-IR (υ max, cm−1): 3316 (NH), 3034 (CH aromatic), 2948 (CH aliphatic), 1697 (CO amide).O, 156.9, 155.8, 154.3, 139.0, 138.3, 135.4, 131.9 2C, 129.5 2C, 126.9, 122.0, 121.7 2C, 115.6 2C, 101.5, 61.7, 52.7 2C, 40.4 2C; FT-IR (υ max, cm−1): 3298 (NH), 3047 (CH aromatic), 2819 (CH aliphatic), 1694 (CO amide); MS: (Mw: 492.38), m/z: 492.59 (14.59%), 474.07 (100%); anal. calcd for C23H22BrN7O, C 56.11, H 4.50, N 19.91 found C 56.38, H 4.72, N 20.04. HRMS (ESI) calcd for C23H22BrN7O [M + H]+, 492.1069; found, 492.1132, [M + 2] 494.1111.O, 156.9, 155.8, 154.3, 139.0, 138.3, 135.4, 131.9 2C, 129.5 2C, 126.9, 122.0 2C, 121.7 2C, 115.6, 101.5, 61.7, 52.7 2C, 40.4 2C; FT-IR (υ max, cm−1): 3287 (NH), 3132 (CH aromatic), 2865 (CH aliphatic), 1649 (CO amide); MS: (Mw: 447.93), m/z: 447.19 (7.02%), 329.00 (100%); anal. calcd for C23H22ClN7O, C 61.67, H 4.95, N 21.89 found C 61.85, H 5.12, N 21.72.O, 157.0, 155.8, 154.4, 139.0, 135.5, 132.4, 129.6 2C, 127.0, 121.7 2C, 121.7 2C, 114.2 2C, 101.5, 64.9, 61.9, 55.6, 52.8 2C, 45.9 2C; FT-IR (υ max, cm−1): 3326 (NH), 3134 (CH aromatic), 2922 (CH aliphatic), 1686 (CO amide); MS: (Mw: 443.21), m/z: 443.83 (19.99%), 157.53 (100%); anal. calcd for C24H25N7O2, C 65.00, H 5.68, N 22.11 found C 64.93, H 5.86, N 21.97. HRMS (ESI) calcd for C24H25N7O2 [M + H]+, 444.2070; found, 444.2144.O amide); MS: (Mw: 427.51), m/z 427.62 (9.06%), 158.89 (100%); anal. calcd for C24H25N7O, C 67.43, H 5.89, N 22.93 found C 67.59, H 6.11, N 23.16. HRMS (ESI) calcd for C24H25N7O [M + H]+, 428.2121; found, 428.2229.O amide); MS: (Mw: 428.51), m/z: 428.27 (37.36%), 204.61 (100%); anal. calcd for C24H25N7O, C 67.43, H 5.89, N 22.93 found C 67.54, H 6.13, N 22.75.General procedure for preparation of XIII & XIV
To a solution of compound XI (0.28 g, 1 mmol) in DMF (20 mL), sodium bicarbonate (0.08 g, 1 mmol) and potassium iodide (0.16 g, 1 mmol) were added. Naphthyl chloroacetate derivatives IIa,b (1 mmol) or phenyl chloroacetate IV (0.17 g, 1 mmol) were added to the previous reaction mixture and stirred at room temperature for 24 hours. The reaction mixture was added on to water (30 mL) to afford a precipitate which was filtered and left to dry. The resulting solid was recrystallized from ethyl acetate/n-hexane (3:1) to give the titled compounds.
O, 163.1, 157.0, 155.8, 154.4, 139.1, 136.5, 135.5, 132.8, 129.5 2C, 129.5 2C, 126.8 2C, 121.7 2C, 120.0 2C, 115.6, 114.2, 101.5, 61.7, 52.8 2C, 40.6 2C; FT-IR (υ max, cm−1): 3042 (CH aromatic), 2873 (CH aliphatic), 1735 (CO ester); MS: (Mw: 465.53), m/z 465.46 (15.70%), 220.24 (100%); anal. calcd for C27H24N6O2, C 69.81, H 5.21, N 18.09 found C 70.04, H 5.39, N 18.35.O, 155.8, 154.4, 139.1, 136.5, 135.5, 132.8 2C, 129.5 2C, 129.5 2C, 126.8, 121.7 2C, 120.0, 101.5, 62.7, 52.8 2C, 40.6 2C IR (υ max, cm−1): 3122 (CH aromatic), 2940 (CH aliphatic), 1759 (CO ester); MS: (Mw: 414.06), m/z: 414.06 (44.9%), 114.43 (100%); anal. calcd for C23H22N6O2 (Mw: 414.47), C 66.65, H 5.35, N 20.28 found C 66.73, H 5.19, N 20.37.The titled compound was separated as a white solid 0.31 g (yield = 75%); mp 175–177 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 9.38 (s, 1H, NH, D2O exchangeable), 8.66 (s, 1H, CH pyrimidine), 8.46 (s, 1H, CH pyrazole), 8.18 (d, J = 8, 2H, ArH), 7.57 (t, J = 8, 4, 2H, ArH), 7.39 (t, J = 8, 8, 2H, ArH), 7.37 (m, 4H, ArH), 4.13 (broad s, 4H, piperazine), 3.29 (broad s, 4H, piperazine); 13C-NMR (DMSO-d6, 100 MHz): δ (ppm): 170.3 (CS), 163.1, 157.0, 155.8, 145.3, 138.9, 135.8, 135.4, 130.3, 129.6 2C, 128.9 2C, 127.6, 127.1, 121.8 2C, 101.6, 55.6 2C, 40.1 2C; FT-IR (υ max, cm−1): 3474 (NH), 3238 (CH aromatic), 2928 (CH aliphatic); MS: (Mw: 415.52): m/z 415.96 (32.49%), 416.80 (24.82%), 84.87 (100%); anal. calcd for C22H21N7S, C 63.59, H 5.09, N 23.60 found C 63.45, H 5.17, N 23.84.
The titled compound was separated as buff crystals 0.13 g (yield = 75%); mp 190–192 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 9.44 (s, 1H, NH, D2O exchangeable), 9.43 (s, 1H, ArH), 8.66 (s, 1H, CH pyrimidine), 8.46 (s, 1H, CH pyrazole), 8.18 (d, J = 8, 2H, ArH), 7.57 (t, J = 8, 8, 2H, ArH), 7.38 (t, J = 4, 4, 1H, ArH), 4.21 (t, J = 4, 4, 4H, piperazine), 3.28 (t, J = 4, 4, 4H, piperazine), 2.72 (s, 2H, CH2); 13C-NMR (DMSO-d6, 100 MHz): δ (ppm): 168.9 CO, 156.9, 155.8, 139.0, 138.3, 135.4, 131.9, 129.5 2C, 126.9, 122.0, 121.7 2C, 101.5, 61.7, 52.7 2C, 40.2 2C; FT-IR (υ max, cm−1): 3404 (NH), 3113 (CH aromatic), 2918 (CH aliphatic), 1690 (CO amide); MS: (Mw: 421.14), m/z: 421.46 (47.71%), 57.12 (100%); anal. calcd for C19H19N9OS (Mw: 421.48), C 54.14, H 4.54, N 29.91 found C 54.31, H 4.68, N 30.15.
General procedure for preparation of compound XVIIIa–f
To a solution of compound XVII (0.35 g, 1 mmol) in DMF (25 mL), sodium bicarbonate (0.08 g, 1 mmol) and potassium iodide (0.16 g, 1 mmol) were added. Aromatic amine derivatives (1 mmol) were added to the previous reaction mixture and stirred at room temperature for 48 hours. The reaction mixture was added on to ice cold water to afford a precipitate which was filtered and left to dry. The resulting solid was recrystallized from absolute ethanol to give the titled compounds XVIIIa–f.O amide); anal. calcd for C23H23N7O (Mw: 413.49), C 66.81, H 5.61, N 23.71 found C 66.9, H 5.71, N 23.94.O amide); MS: (Mw: 492.38), m/z 492.35 (29.68%). 436.13 (100%); anal. calcd for C23H22BrN7O (Mw: 492.38), C 56.11, H 4.50, N 19.91 found C 56.34, H 4.73, N 20.15. HRMS (ESI) calcd for C23H22BrN7O [M + H]+ 492.1069; found, 492.1129, [M + 2] 494.1114.O amide); MS: (Mw: 447.93) m/z 447.08, (40.52%), 103.30 (100%); anal. calcd for C23H22ClN7O (Mw: 447.93), C 61.67, H 4.95, N 21.89 found C 61.88, H 5.11, N 21.75. HRMS (ESI) calcd for C23H22ClN7O [M + H]+ 448.1574; found, 448.1651, [M + 2] 450.1630.O amide); MS: (Mw: 443.51), m/z 443.39 (35.91%), 160.18 (100%); anal. calcd for C24H25N7O2 (Mw: 443.51), C 65.00, H 5.68, N 22.11 found C 64.79, H 5.79, N 21.98.O amide); MS: (Mw: 427.51): m/z 427.15 (30.83%), 69.12 (100%); anal. calcd for C24H25N7O (Mw: 427.51), C 67.43, H 5.89, N 22.93 found C 67.59, H 6.05, N 23.78.Aroyl isothiocyanate XXa–f
To a solution of ammonium thiocyanate (0.07 g, 1 mmol) in dry acetone (30 mL), the appropriate freshly prepared aroyl chloride derivatives (1 mmol) were added dropwise with stirring. The reaction mixture was continuously stirred vigorously for 45 minutes. The reaction was monitored by TLC until a new spot of intermediate XXa–f is formed and used directly in the following step.General procedure for the preparation of compound XXIa–f
The solution resulted from the previous reaction XXa–f was added to a solution of intermediate XI (0.28 g, 1 mmol) in dry acetone (15 mL) and stirred for 36 hours at room temperature until a precipitate is formed. The formed precipitate was washed with cold water to yield our target compounds XXIa–f.O amide); MS: (Mw: 443.53), m/z 443.54 (40.18%), 77.91 (100%); anal. calcd for C23H21N7OS, C 62.29, H 4.77, N 22.11 found C 62.41, H 4.89, N 21.97.O amide); anal. calcd for: C23H20FN7OS, C 59.86, H 4.37, N 21.24 found C 60.04, H 4.46, N 21.45. HRMS (ESI) calcd for C23H20FN7OS [M + H]+ 462.1434; found, 462.1500.O amide); MS: (Mw: 522.47), m/z 522.47 (22.33), 310.23 (100%): anal. calcd for: C23H20BrN7OS, C 52.88, H 3.86, N 18.77 found C 52.67, H 3.98, N 19.03. HRMS (ESI) calcd for C23H20BrN7OS [M + H]+ 522.0633; found, 522.0747, [M + 2] 524.0728.O amide); MS: (Mw: 477.11), m/z 477.14 (69.88%), 446.49 (100%): anal. calcd for C23H20ClN7OS, C 57.80, H 4.22, N 20.51 found C 58.04, H 4.36, N 20.78. HRMS (ESI) calcd for C23H20ClN7OS [M + H]+ 478.1139; found, 478.1204, [M + 2] 480.1184.O amide); MS: (Mw: 473.56), m/z 473.02 (14.52%), 318.12 (100%): anal. calcd for C23H20ClN7OS, C 57.80, H 4.22, N 20.51 found C 58.04, H 4.36, N 20.78.O amide); MS: (Mw: 457.17), m/z 457.46 (31.83%), 420.55 (100%): anal. calcd for C24H23N7OS, C 63.00, H 5.07, N 21.43 found C 62.78, H 5.31, N 21.278.Biological evaluation
The method used for docking was general docking. The scoring methodology was selected as GBVI/WSA dG for the refinement methodology. On the other hand, the scoring methodology was applied as London dG for the placement methodology, which was selected as a triangle matcher. The applied forcefield was Amber10:EHT. The prepared MDB file was inserted containing all newly synthesized compounds then docking calculations was performed automatically. Only one pose was selected for each compound and the selection was based on the binding scores, rmsd_refine values, and interaction modes (ESI†).
P98 (AlogP98), 2D polar surface area (ADMET 2D PSA), aqueous solubility (AQ SOl), aqueous solubility level (AQ SOl LEV), blood–brain barrier value (BBB), blood–brain barrier level (BBB LEV), cytochrome P450 2D6 (CYP2D6), cytochrome P450 2D6 probability (CYP PROB), logarithmic value of plasma protein binding (PPB LOG), and logarithmic level of plasma protein binding (PPB LEV).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Dr Reem K. Arafa, Professor of Biomedical Sciences, University of Science and Technology, Zewail City of Science and Technology, Giza, Egypt for the assistance in molecular docking study.Notes and references
- S. A. Rostom, Synthesis and in vitro antitumor evaluation of some indeno [1, 2-c] pyrazol (in) es substituted with sulfonamide, sulfonylurea (-thiourea) pharmacophores, and some derived thiazole ring systems, Bioorg. Med. Chem., 2006, 14(19), 6475–6485 CrossRef CAS PubMed.
- L. A. Torre, et al., Global cancer statistics, 2012, Ca-Cancer J. Clin., 2015, 65(2), 87–108 CrossRef PubMed.
- P. Blum-Jensen and T. Hunter, Oncogenic kinase signaling, Nature, 2001, 411(3), 355–365 CrossRef PubMed.
- N. N. Kabir and J. U. Kazi, Comparative analysis of human and bovine protein kinases reveals unique relationship and functional diversity, Genet. Mol. Biol., 2011, 34(4), 587–591 CrossRef CAS PubMed.
- J. A. Orgueira and D. A. Pérez, et al., FLT3 inhibitors in the treatment of acute myeloid leukemia: current status and future perspectives, Minerva Med., 2020, 111(5), 427–444 Search PubMed.
- E. Weisberg, et al., Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412, Cancer Cell, 2002, 1(5), 433–443 CrossRef CAS PubMed.
- M. Levis, et al., A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo, Blood, 2002, 99(11), 3885–3891 CrossRef CAS PubMed.
- L. M. Kelly, et al., CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML), Cancer Cell, 2002, 1(5), 421–432 CrossRef CAS PubMed.
- D. E. L. de Menezes, et al., CHIR-258: a potent inhibitor of FLT3 kinase in experimental tumor xenograft models of human acute myelogenous leukemia, Clin. Cancer Res., 2005, 11(14), 5281–5291 CrossRef PubMed.
- A.-M. O'Farrell, et al., SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo, Blood, 2003, 101(9), 3597–3605 CrossRef PubMed.
- J. M. Gozgit, et al., Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies, Mol. Cancer Ther., 2011, 10(6), 1028–1035 CrossRef CAS PubMed.
- E. Lierman, et al., The ability of sorafenib to inhibit oncogenic PDGFRβ and FLT3 mutants and overcome resistance to other small molecule inhibitors, Haematologica, 2007, 92(1), 27–34 CrossRef CAS PubMed.
- P. Baxter, et al., Plasma and cerebrospinal fluid pharmacokinetics of MP470 in non-human primates, Cancer Chemother. Pharmacol., 2011, 67(4), 809–812 CrossRef CAS PubMed.
- S. Scholl, et al., Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments, Cells, 2020, 9(11), 2493 CrossRef CAS PubMed.
- L. Y. Lee, et al., Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor, Blood, 2017, 129(2), 257–260 CrossRef CAS PubMed.
- P. P. Zarrinkar and et al, ., AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML), Blood, 2009, 114(14), 2984–2992 CrossRef CAS PubMed.
- E. I. Zimmerman, et al., Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia, Blood, 2013, 122(22), 3607–3615 CrossRef CAS PubMed.
- S. Bertoli, et al., Outcome of Relapsed or Refractory FLT3-Mutated Acute Myeloid Leukemia before Second-Generation FLT3 Tyrosine Kinase Inhibitors: A Toulouse–Bordeaux DATAML Registry Study, Cancers, 2020, 12(4), 773 CrossRef CAS PubMed.
- C. N. Khobragade, et al., Synthesis and antimicrobial activity of novel pyrazolo [3, 4-d] pyrimidin derivatives, Eur. J. Med. Chem., 2010, 45(4), 1635–1638 CrossRef CAS PubMed.
- A. E. Rashad, et al., Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives, Bioorg. Med. Chem., 2008, 16(15), 7102–7106 CrossRef CAS PubMed.
- S. B. Yewale, et al., Novel 3-substituted-1-aryl-5-phenyl-6-anilinopyrazolo [3, 4-d] pyrimidin-4-ones: docking, synthesis and pharmacological evaluation as a potential anti-inflammatory agents, Bioorg. Med. Chem. Lett., 2012, 22(21), 6616–6620 CrossRef CAS PubMed.
- M. Celano, et al., Cytotoxic effects of a novel pyrazolopyrimidine derivative entrapped in liposomes in anaplastic thyroid cancer cells in vitro and in xenograft tumors in vivo, Endocr.-Relat. Cancer, 2008, 15(2), 499–510 CrossRef CAS PubMed.
- J.-Y. Le Brazidec, et al., Synthesis, SAR and biological evaluation of 1, 6-disubstituted-1H-pyrazolo [3, 4-d] pyrimidines as dual inhibitors of Aurora kinases and CDK1, Bioorg. Med. Chem. Lett., 2012, 22(5), 2070–2074 CrossRef CAS PubMed.
- A. Spreafico, et al., Antiproliferative and proapoptotic activities of new pyrazolo [3, 4-d] pyrimidine derivative Src kinase inhibitors in human osteosarcoma cells, FASEB J., 2008, 22(5), 1560–1571 CrossRef CAS PubMed.
- F. Manetti, et al., Structure-based optimization of pyrazolo [3, 4-d] pyrimidines as Abl inhibitors and antiproliferative agents toward human leukemia cell lines, J. Med. Chem., 2008, 51(5), 1252–1259 CrossRef CAS PubMed.
- N. S. Ismail, et al., Pyrazolo [3, 4-d] pyrimidine based scaffold derivatives targeting kinases as anticancer agents, Future J. Pharm. Sci., 2016, 2(1), 20–30 CrossRef.
- C. La Motta, et al., Exploiting the pyrazolo [3, 4-d] pyrimidin-4-one ring system as a useful template to obtain potent adenosine deaminase inhibitors, J. Med. Chem., 2009, 52(6), 1681–1692 CrossRef CAS PubMed.
- A. A. Gaber, et al., Pharmacophore-linked pyrazolo [3, 4-d] pyrimidines as EGFR-TK inhibitors: synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies, Arch. Pharm., 2021, e2100258 Search PubMed.
- L.-L. Yang, et al., Structure–activity relationship studies of pyrazolo [3, 4-d] pyrimidine derivatives leading to the discovery of a novel multikinase inhibitor that potently inhibits FLT3 and VEGFR2 and evaluation of its activity against acute myeloid leukemia in vitro and in vivo, J. Med. Chem., 2013, 56(4), 1641–1655 CrossRef CAS PubMed.
- G.-B. Li, et al., Drug Discovery against Psoriasis: Identification of a New Potent FMS-like Tyrosine Kinase 3 (FLT3) Inhibitor, 1-(4-((1 H-Pyrazolo [3, 4-d] pyrimidin-4-yl) oxy)-3-fluorophenyl)-3-(5-(tert-butyl) isoxazol-3-yl) urea, That Showed Potent Activity in a Psoriatic Animal Model, J. Med. Chem., 2016, 59(18), 8293–8305 CrossRef CAS PubMed.
- A. Agrebi, et al., Synthesis and biological evaluation of new pyrazolo [3, 4-d] pyrimidine derivatives, Mediterr. J. Chem., 2014, 3(2), 864–876 CrossRef CAS.
- M. M. Kandeel, et al., Design, synthesis, and antitumor evaluation of novel pyrazolo [3, 4-d] pyrimidine derivatives, Sci. Pharm., 2012, 80(3), 531–546 CrossRef CAS PubMed.
- K. Matsuno, et al., Nitrogenous heterocyclic compounds, European Patent, 1999, EP1067123 Search PubMed.
- M. G. Salem, et al., Synthesis, molecular modeling, selective aldose reductase inhibition and hypoglycemic activity of novel meglitinides, Bioorg. Chem., 2021, 111, 104909 CrossRef CAS PubMed.
- H. D. Souza, et al., Synthesis, in silico study and antimicrobial evaluation of new selenoglycolicamides, J. Braz. Chem. Soc., 2019, 30, 188–197 CAS.
- R. Mishra, et al., Synthesis and Anticonvulsant Activity of Some Novel 2-Methyl Imidazole Derivatives, Lett. Drug Des. Discovery, 2012, 9(4), 402–408 CrossRef CAS.
- A. M. Qandil and L. I. Fakhouri, a-Anilinoketones, esters and amides: a chemical study, Pharmaceuticals, 2012, 5(6), 591–612 CrossRef CAS PubMed.
- K. H. Oudah, et al., Design, synthesis and molecular docking of novel pyrazolo [1, 5-a][1, 3, 5] triazine derivatives as CDK2 inhibitors, Bioorg. Chem., 2019, 92, 103239 CrossRef CAS PubMed.
- D. R. Parmar, et al., Discovery of new anticancer thiourea-azetidine hybrids: design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies, Bioorg. Chem., 2021, 115, 105206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00446e |
| This journal is © The Royal Society of Chemistry 2023 |