
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning anticancer properties and DNA-binding of Pt(II) complexes via alteration of nitrogen softness/basicity of tridentate ligands†
Kamelah S. Al-Rashdiab,
Bandar A. Babgi *a,
Ehab M. M. Alicd,
Abdesslem Jedidia,
Abdul-Hamid M. Emwase,
Bambar Davaasurene,
Mariusz Jaremkof and
Mark G. Humphreyg
*a,
Ehab M. M. Alicd,
Abdesslem Jedidia,
Abdul-Hamid M. Emwase,
Bambar Davaasurene,
Mariusz Jaremkof and
Mark G. Humphreyg
aDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia. E-mail: bbabgi@kau.edu.sa; Tel: +966 555563702
bDepartment of Chemistry, Al-Qunfudah University College, Umm Al-Qura University, Al-Qunfudah 1109, Saudi Arabia
cDepartment of Biochemistry, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
dDivision of Biochemistry, Department of Chemistry, Faculty of Science, Tanta University, Tanta 31527, Egypt
eCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia
fBiological and Environmental Science and Engineering (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia
gResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
First published on 21st March 2023
Abstract
Nine tridentate Schiff base ligands of the type (N^N^O) were synthesized from reactions of primary amines {2-picolylamine (Py), N-phenyl-1,2-diaminobenzene (PhN), and N-phenyl-1,2-diaminoethane(EtN)} and salicylaldehyde derivatives {3-ethoxy (OEt), 4-diethylamine (NEt2) and 4-hydroxy (OH)}. Complexes with the general formula Pt(N^N^O)Cl were synthesized by reacting K2PtCl4 with the ligands in DMSO/ethanol mixtures. The ligands and their complexes were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. The DNA-binding behaviours of the platinum(II) complexes were investigated by two techniques, indicating good binding affinities and a two-stage binding process for seven complexes: intercalation followed by switching to a covalent binding mode over time. The other two complexes covalently bond to ct-DNA without intercalation. Theoretical calculations were used to shed light on the electronic and steric factors that lead to the difference in DNA-binding behavior. The reactions of some platinum complexes with guanine were investigated experimentally and theoretically. The binding of the complexes with bovine serum albumin (BSA) indicated a static interaction with higher binding affinities for the ethoxy-containing complexes. The half maximal inhibitory concentration (IC50) values against MCF-7 and HepG2 cell lines suggest that platinum complexes with tridentate ligands of N-phenyl-o-phenylenediamine or pyridyl with 3-ethoxysalicylimine are good chemotherapeutic candidates. Pt-Py-OEt and Pt-PhN-OEt have IC50 values against MCF-7 of 13.27 and 10.97 μM, respectively, compared to 18.36 μM for cisplatin, while they have IC50 values against HepG2 of 6.99 and 10.15 μM, respectively, compared to 19.73 μM for cisplatin. The cell cycle interference behaviour with HepG2 of selected complexes is similar to that of cisplatin, suggesting apoptotic cell death. The current work highlights the impact of the tridentate ligand on the biological properties of platinum complexes.
1 Introduction
Cisplatin (Fig. 1) was the first approved alkylating-like chemotherapeutic drug that is known to bind DNA covalently. It is activated for binding by the hydrolysis of its labile chlorido ligands, to produce cis-[Pt(NH3)2Cl(H2O)]+.1 Inside the cell, the aqua ligand is exchanged with N7-guanine, producing a cis-[Pt(NH3)2Cl(DNA)]+ adduct.2 The second chlorido ligand undergoes a similar process to form a cross-linked DNA (either intra-strand or inter-strand).3 The drug-DNA binding causes conformational changes to DNA, promoting apoptotic cell death.4,5 Although cisplatin is clinically approved and is used to treat a range of cancer types, it has several drawbacks, including lack of selectivity toward cancer cells, acquired resistance, and severe side effects.6–10 Many platinum(II) complexes have been developed as possible alternatives, modifying the structure of cisplatin by replacing either the leaving groups (chlorido) and/or the non-leaving groups (ammine) by a range of ligands. These modifications have resulted in a few clinically approved platinum(II)-based drugs11 (Fig. 1), all of which act by a mechanism similar to that of cisplatin. However, the use of bidentate leaving groups in these complexes has been found to kinetically slow the activation process compared to cisplatin, reducing the efficacy.12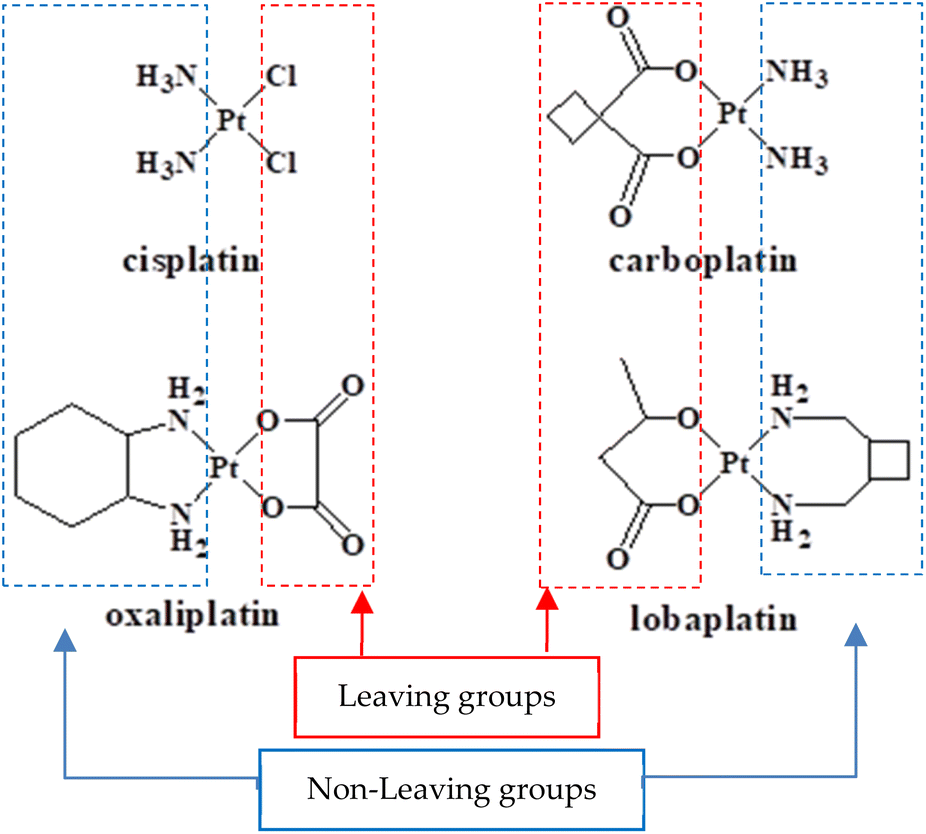 | ||
| Fig. 1 Some of the clinically approved Pt-based anticancer drugs, illustrating the ligand roles. | ||
In the search for more efficient/less toxic platinum(II) candidates, attention has focused on complexes that bind to DNA non-covalently, including via electrostatic, groove and intercalation interactions. Terpyridine and its derivatives have been employed as good intercalating ligands in designing Pt(II) complexes. For instance, binding studies of the complex [Pt(tpy)Cl]+ have suggested fast intercalation of the ligand with DNA which is followed by chlorido/Nnucleobase ligand exchange (loss of the labile chlorido ligand).13–17 In contrast, complexes with the formula [Pt(tpy)(thiol)]+ demonstrate DNA-binding via intercalation exclusively. The thiols are poorer leaving groups than chlorido, and some of these complexes have shown promising in vitro cytotoxicity.18,19 Other ligands have been used to generate platinum(II) complexes, resulting in complexes that can bind DNA covalently and through intercalation.20–22
In a recent article, we described the synthesis of Pt(N^N^O)Cl complexes (Fig. 2), employing tridentate Schiff base ligands synthesized from 2-picolylamine and salicylaldehyde derivatives {3-ethoxy (OEt), 4-diethylamino (NEt2) and 4-hydroxy (OH)}.23 The DNA-binding studies of these platinum(II) complexes suggested fast intercalation followed by a slow shift towards covalent binding. Among the platinum(II) complexes, the ethoxy-containing complex exhibited the most promising IC50 against both MCF-7 and HepG2 cell lines, and comparable to that observed for cisplatin. DFT calculations for the B-DNA trimer adduct with platinum complexes showed hydrogen bonding interactions between the ligands and nucleobases, affecting the inter-strand hydrogen bonding within the DNA, which highlights the strong ability of the complexes to induce conformational changes in the DNA that lead to the activation of apoptotic cell death, a premise supported by cell cycle arrest studies.23 The current work is an extension of this recent work; in the synthesis of the ligands, 2-picolylamine has been replaced by N-phenyl-1,2-diaminobenzene or N-phenyl-1,2-diaminoethane. Such modifications have produced tridentate ligands with variant nitrogen basicity, and hence platinum(II) complexes with different electronic properties (Fig. 2). The effect of the basicity of the nitrogen donating atoms on the DNA-binding and anticancer properties have been studied, and theoretical studies have been undertaken to rationalize the outcomes.
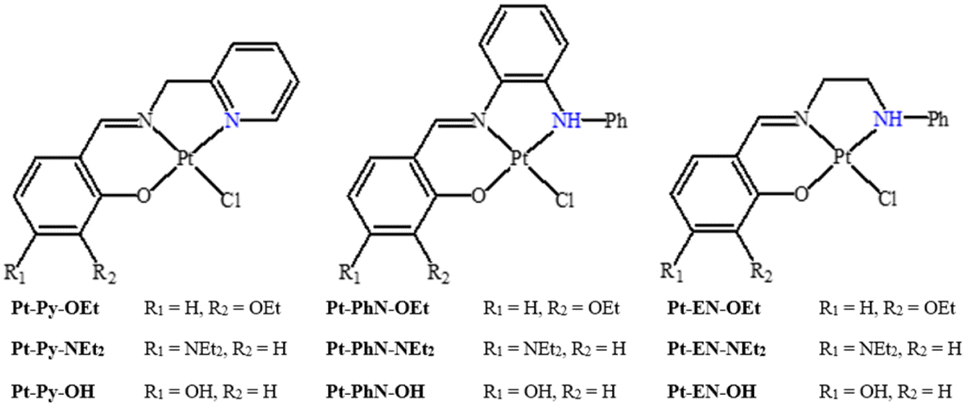 | ||
| Fig. 2 Platinum(II) compounds in this work. | ||
2 Materials and methods
2.1 Materials
All the reactions were conducted under an inert atmosphere (argon), using standard Schlenk techniques with oven-dried glassware. Molecular sieves (A4) were used to dry all solvents for at least 48 hours before use. Propidium iodide (PI) was purchased from Thermo-Fisher Scientific. All other reagents and chemicals were sourced from Sigma-Aldrich and used as received. N-(2-Picolyl)salicylimine ligands (L-Py-OEt, L-Py-NEt2 and L-Py-OH) and their platinum(II) complexes (Pt-Py-OEt, Pt-Py-NEt2 and Pt-Py-OH) were synthesized according to the previously reported procedure.232.2 Methods and instrumentation
High-resolution (HR) electrospray ionization (ESI) (positive ionization mode) mass spectra were conducted employing a Bruker Apex 4.7 FTICR-MS instrument; all mass spectrometry (MS) peaks are listed as m/z (formula). The elemental analyses were carried out at King Abdulaziz University. The infrared (IR) spectra were collected as KBr disks on a PerkinElmer Spectrum 100 instrument; the reported peaks are in cm−1. 1H NMR (600 MHz), 13C NMR (151 MHz), and 31P NMR (242 MHz) spectra were obtained, using a Bruker Avance 600 MHz spectrometer equipped with a BBO probe; the spectra were referenced to residual chloroform or DMSO (7.26 or 2.50, 1H), CDCl3 or DMSO-d6 (77.0 or 39.5 13C), or external H3PO4 (0.0 31P).24 Atom labelling in NMR follows the numbering scheme in Chart 1. UV-vis absorption spectra were obtained as chloroform solutions on a Genesys-10s UV-VIS spectrophotometer (Thermo Fischer), using 1 cm path-length quartz cells; the absorption maxima wavelengths are given in nanometres.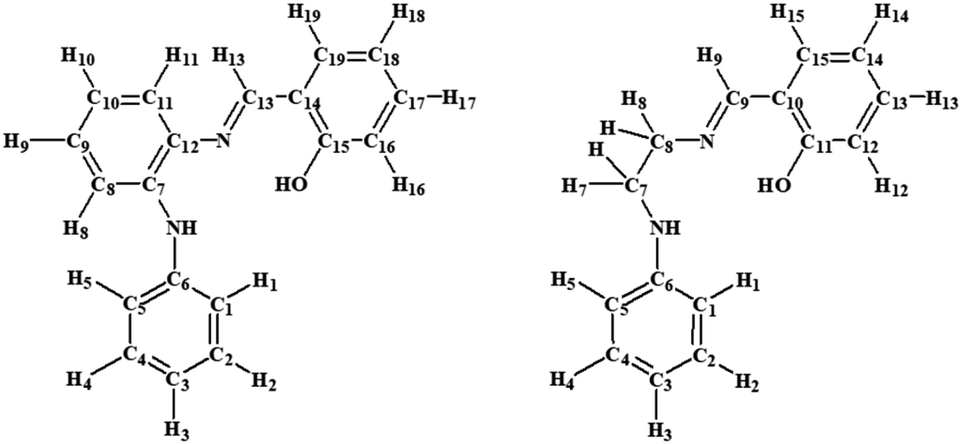 | ||
| Chart 1 NMR labelling scheme for compounds in the current work. | ||
2.3 Synthesis and characterization
2.3.1.1 Ligand 1 (L-PhN-OEt). N-Phenyl-o-phenylenediamine (0.50 g, 2.70 mmol) and 3-ethoxysalicylaldehyde (0.45 g, 2.70 mmol) were reacted to yield L1 as an orange powder (0.71 g, 82%). IR (cm−1): 1622 ν(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 2891–3053 ν(O–H), 3428 ν(aromatic NH). 1H-NMR (CDCl3) δ: 13.18 (s, 1H, OH), 8.59 (s, 1H, H13), 7.26 (d, JHH = 8.1, 2H, H1,5), 7.22 (t, JHH = 7.6, 2H, H2,4), 7.11 (d, JHH = 7.7, 2H, H11), 7.07 (d, JHH = 7.7, 2H, H17,19), 6.97 (d, JHH = 7.7, 2H, H8), 6.93 (t, JHH = 4.9, 1H, H9,10), 6.82 (q, JHH = 8.0, 1H, H3,18), 6.15 (s, 1H, NH), 4.06 (q, JHH = 7.0, 2H, CH2), 1.43 (t, JHH = 7.0, 3H, CH3). 13C-NMR (CDCl3) δ: 162.9 (C13), 151.0 (C15), 147.7 (C16), 141.9 (C12), 138.0 (C7), 136.9 (C6), 129.3 (C2,4), 127.9 (C11), 123.9 (C9,10), 122.3 (C19), 120.2 (C1,5), 119.6 (C14), 118.9 (C17), 118.7 (C3), 116.2 (C18), 115.2 (C8), 64.6 (CH2), 14.9 (CH3). HR ESI MS calcd for [C21H20N2O2]+: 332.1525, found 332.1462. Anal. calcd for C21H20N2O2: C, 75.88; H, 6.06; N, 8.43%; found: C, 76.21; H, 5.79; N, 8.06%.
N), 2891–3053 ν(O–H), 3428 ν(aromatic NH). 1H-NMR (CDCl3) δ: 13.18 (s, 1H, OH), 8.59 (s, 1H, H13), 7.26 (d, JHH = 8.1, 2H, H1,5), 7.22 (t, JHH = 7.6, 2H, H2,4), 7.11 (d, JHH = 7.7, 2H, H11), 7.07 (d, JHH = 7.7, 2H, H17,19), 6.97 (d, JHH = 7.7, 2H, H8), 6.93 (t, JHH = 4.9, 1H, H9,10), 6.82 (q, JHH = 8.0, 1H, H3,18), 6.15 (s, 1H, NH), 4.06 (q, JHH = 7.0, 2H, CH2), 1.43 (t, JHH = 7.0, 3H, CH3). 13C-NMR (CDCl3) δ: 162.9 (C13), 151.0 (C15), 147.7 (C16), 141.9 (C12), 138.0 (C7), 136.9 (C6), 129.3 (C2,4), 127.9 (C11), 123.9 (C9,10), 122.3 (C19), 120.2 (C1,5), 119.6 (C14), 118.9 (C17), 118.7 (C3), 116.2 (C18), 115.2 (C8), 64.6 (CH2), 14.9 (CH3). HR ESI MS calcd for [C21H20N2O2]+: 332.1525, found 332.1462. Anal. calcd for C21H20N2O2: C, 75.88; H, 6.06; N, 8.43%; found: C, 76.21; H, 5.79; N, 8.06%.
2.3.1.2 Ligand 2 (L-NPh-NEt2). N-Phenyl-o-phenylenediamine (0.50 g, 2.70 mmol) and 4-diethylaminosalicylaldehyde (0.52 g, 2.70 mmol) were reacted to yield L2 as a yellow powder (0.74 g, 76%). IR (cm−1):1600 ν(C
N), 2819–3140 ν(O–H), 3386 ν(aromatic NH). 1H-NMR (d6-DMSO) δ: 13.13 (s, 1H, OH), 8.59 (s, 1H, H13), 7.25 (d, JHH = 5.9, 1H, H1), 7.23 (d, JHH = 5.8, 1H, H5), 7.18 (t, JHH = 5.8, 2H, H2,4), 7.11 (t, JHH = 5.5, 1H, H9), 7.32 (d, JHH = 6.6, 1H, H8), 7.48 (s, 1H, H16), 6.98 (d, JHH = 5.8, 1H, H11), 6.77 (t, JHH = 5.5, 1H, H10), 6.30 (dd, JHH = 1.7, 1H, H3), 6.02 (d, JHH = 1.6, 1H, H18,19), 3.38 (q, JHH = 5.3, 2H, CH2), 1.12 (t, JHH = 5.2, 3H, CH3). 13C-NMR (d6-DMSO) δ: 162.9 (C13), 161.0 (C15), 151.2 (C6), 144.2 (C12), 140.5 (C7), 136.4 (C9), 133.9 (C2,4), 128.9 (C8.11), 125.8 (C10), 121.9 (C19), 119.1 (C1,5), 119.0 (C3), 109.1 (C18), 103.5 (C16), 96.8 (C14), 43.8 (CH2), 12.4 (CH3). HR ESI MS calcd for [C23H25N3O]+: 360.2076, found 360.2070. Anal. calcd for C23H26N3O: C, 76.85; H, 7.01; N, 11.69%; found: C, 76.57; H, 6.59; N, 11.35%.
2.3.1.3 Ligand 3 (L-NPh-OH). N-Phenyl-o-phenylenediamine (0.50 g, 2.70 mmol) and 2,4-dihydroxybenzaldehyde (0.37 g, 2.70 mmol) were reacted to yield L3 as an orange powder (0.73 g, 88%). IR (cm−1): 1630 ν(C
N), 2781–3289 ν(O–H), 3437 ν(aromatic NH). 1H-NMR (CDCl3) δ: 11.40 (s, 1H, OH), 9.69 (s, 1H, OH), 8.43 (s, 1H, H13), 7.40 (d, JHH = 6.4, 2H, H1,5), 7.33 (d, JHH = 5.9, 2H, H8), 7.21 (t, JHH = 4.1, 2H, H2), 7.17 (t, JHH = 5.6, 1H, H4), 7.06 (d, JHH = 5.8, 1H, H11), 6.99 (t, JHH = 5.6, 1H, H9), 6.90 (t, JHH = 5.6, 1H, H10), 6.60 (s, 1H, H16), 6.48 (dd, JHH = 1.6, 1H, H3), 6.39 (d, JHH = 1.6, 1H, H18,19). 13C-NMR (CDCl3) δ: 164.7 (C13), 163.4 (C15,17), 137.0 (C12,19), 136.1 (C6,7), 129.3 (C2,4), 127.7 (C8,11), 121.6 (C3), 118.9 (C1,5), 115.5 (C9,10), 108.7 (C14), 103.6 (C18), 103.2 (C16). HR ESI MS calcd for [C19H16N2O2]+: 305.1290, found 305.1279. Anal. calcd for C19H16N2O2: C, 74.98; H, 5.30; N, 9.20%; found: C, 74.72; H, 5.20; N, 9.39%.
2.3.2.1 Ligand 4 (L-EN-OEt). N-Phenylethylenediamine (0.50 g, 3.62 mmol) and 3-ethoxysalicylaldehyde (0.60 g, 3.62 mmol) reacted to yield L4 as a yellow powder (0.71 g, 92%). IR (cm−1): 1587 ν(C
C), 1647 ν(CN aromatic), 3402 ν(NH). 1H-NMR (d6-DMSO) δ: 13.66 (s, 1H, OH), 8.48 (s, 1H, H9), 7.07 (t, JHH = 5.8, 1H, H2), 6.99 (t, JHH = 5.5, 1H, H4), 6.76 (t, JHH = 2.3, 1H, H3), 6.60 (d, JHH = 6.0, 2H, H1,5), 6.53 (t, JHH = 5.4, 1H, H14), 5.07 (s, 1H, NH), 4.04 (q, JHH = 5.2, 2H, CH2), 3.77 (t, JHH = 6.6, 1H, H7), 3.34 (t, JHH = 4.6, 1H, H8), 1.33 (t, JHH = 5.2, 3H, CH3). 13C-NMR (d6- DMSO) δ: 166.8 (C9), 152.1 (C11), 148.4 (C12), 147.1 (C6), 128.8 (C2,4), 123.28 (C3), 123.26 (C12), 118.4 (C14), 117.4 (C13), 116.2 (C1,5), 111.9 (C10,15), 63.9 (C7), 56.8 (C8), 43.4 (CH2), 14.7 (CH3). HR ESI MS [C17H20N2O2]+: calcd 284.1525, found 284.1722. Anal. calcd for C17H20N2O2: C, 71.81; H, 7.07; N, 9.85%; found: C, 71.49; H, 6.85; N, 9.62%.
2.3.2.2 Ligand 5 (L-EN-NEt2). N-Phenylethylenediamine (0.50 g, 3.62 mmol) and 4-diethylaminosalicylaldehyde (0.70 g, 3.62 mmol) reacted to yield L5 as a yellow powder (0.71 g, 83%). IR (cm−1): 1508 ν(C
C), 1589 ν(CN aromatic), 1612 ν(CN aliph.), 3411 ν(NH). 1H-NMR (d6-DMSO) δ: 13.54 (s, 1H, OH), 8.17 (s, 1H, H9), 7.07 (t, JHH = 4.3, 2H, H2,4), 6.59 (d, JHH = 5.9, 2H, H1,5), 6.52 (t, JHH = 5.3, 1H, H3), 6.16 (dd, JHH = 1.3 and 6.5, 1H, H14), 5.92 (d, JHH = 1.1, 1H, H15), 5.67 (s, 1H, H12), 3.64 (t, JHH = 4.6, 2H, H7), 3.28 (t, JHH = 4.1, 1H, H8), 3.34 (t, JHH = 5.2, 2H, CH2), 1.10 (t, JHH = 5.3, 3H, CH3). 13C-NMR (d6-DMSO) δ: 165.3 (C9), 164.5 (C11), 150.9 (C13), 148.5 (C6), 133.0 (C15), 128.8 (C2,4), 115.6 (C3), 111.9 (C1,5), 107.8 (C10), 102.7 (C14), 97.3 (C12), 55.6 (C7,8), 43.7 (CH2), 12.5 (CH3). HR ESI MS [C19H26N3O]+: calcd 312.2075, found 312.2070. Anal. calcd for C19H25N3O: C, 73.28; H, 8.09; N, 13.49%; found: C, 72.94; H, 7.81; N, 13.19%.
2.3.2.3 Ligand 6 (L-EN-OH). N-Phenylethylenediamine (0.50 g, 3.62 mmol) and 2,4-dihydroxybenzaldehyde (0.49 g, 3.62 mmol) reacted to yield L6 as a yellow powder (0.79 g, 92%). IR (cm−1): 1595 ν(C
C), 1639 ν(CN aromatic), 3436 ν(NH). 1H-NMR (CDCl3) δ: 7.83 (s, 1H, H9), 7.14 (t, JHH = 5.8, 1H, H2,4), 6.89 (d, JHH = 6.4, 1H, H1,5), 6.70 (t, JHH = 5.5, 1H, H3), 6.59 (d, JHH = 5.9, 2H, H15), 6.23 (s, 1H, H12), 6.22 (dd, JHH = 1.50 and 6.42, 1H, H14), 3.67 (t, JHH = 4.1, 2H, H7), 3.44 (t, JHH = 4.1, 2H, H8). 13C-NMR (CDCl3) δ: 164.6 (C9), 163.6 (C11,13), 147.2 (C6), 134.3 (C15), 129.3 (C2, 4), 117.9 (C3), 111.1 (C1,5), 107.2 (C14), 104.2 (C12), 54.0 (C8), 44.0 (C9). HR ESI MS [C15H16N2O2]+: calcd 256.1212, found 256.1254. Anal. calcd for C17H20N2O2: C, 71.81; H, 7.09; N, 9.85%; found: C, 71.58; H, 6.98; N, 9.56%.
2.3.3.1 Pt-NPh-OEt. K2PtCl4 (100 mg, 0.241 mmol) and L-NPh-OEt (80 mg, 0.241 mmol) reacted to yield Pt-NPh-OEt as a green powder (0.119 g, 88%). IR (cm−1): 1517 ν(C
C), 1604 ν(CN aromatic), 3437 ν(CN aliph.). 1H-NMR (d6-DMSO) δ: 8.58 (s, 1H, H13), 7.62 (d, JHH = 5.4, 2H, H1,5), 7.47 (t, JHH = 5.4, 2H, H2,4), 7.44 (t, JHH = 7.8, 1H, H9), 7.37 (t, JHH = 7.5, 1H, H10), 7.14 (d, JHH = 8.2, 2H, H8,11), 6.82 (d, JHH = 5.7, 1H, H17), 6.33 (q, JHH = 8.9, 1H, H3,18), 3.98 (q, JHH = 6.9, 2H, CH2), 1.34 (t, JHH = 6.9, 3H, CH3). 13C-NMR (d6-DMSO) δ: 157.0 (C13), 150.9 (C15), 146.5 (C16), 138.3 (C12), 136.4 (C7), 135.3 (C6), 129.8 (C2,4), 127.6 (C11), 124.8 (C9,10), 123.9 (C19), 120.7 (C1,5), 118.6 (C14), 116.4 (C3), 116.2 (C17), 114.5 (C18), 111.1 (C8), 63.4 (CH2), 14.7 (CH3). HR ESI MS calcd for [C21H22N2O2ClPt]+: 564.1018, found 564.4267. Anal. calcd for C21H19ClN2O2Pt: C, 44.89; H, 3.41; N, 4.99%; found: C, 44.51; H, 3.14; N, 5.23%.
2.3.3.2 Pt-NPh-NEt2. K2PtCl4 (100 mg, 0.241 mmol) and L-NPh-NEt2 (86 mg, 0.241 mmol) reacted to yield Pt-NPh-NEt2 as an orange powder (0.118 g, 83%). IR (cm−1): 1610 ν(C
C), 1603 ν(CN aromatic), 3448 ν(NH). 1H-NMR (d6-DMSO) δ: 9.97 (s, 1H, NH), 8.52 (s, 1H, H13), 8.09 (d, JHH = 8.5, 1H, H11), 7.42 (d, JHH = 9.2, 2H, H1,5), 7.35 (t, JHH = 7.7, 1H, H3), 7.30 (t, JHH = 8.0, 2H, H9,10), 7.18 (d, JHH = 8.2, 1H, H19), 7.12 (t, JHH = 7.4, 2H, H2,4), 6.89 (d, JHH = 7.9, 1H, H8,11), 6.29 (d, JHH = 9.2, 1H, H18), 5.90 (s, 1H, H16), 3.36 (q, JHH = 7.3, 2H, CH2), 1.11 (t, JHH = 7.0, 3H, CH3). 13C-NMR (d6-DMSO) δ: 166.1 (C13), 154.5 (C15), 150.2 (C6), 148.8 (C12), 148.2 (C7), 142.0 (C9), 137.2 (C2,4), 129.3 (C8,11), 127.7 (C10), 127.2 (C19), 126.0 (C1,5), 116.5 (C3), 112.3 (C18), 105.1 (C16), 97.8 (C14), 44.5 (CH2), 13.2 (CH3). HR ESI MS calcd for [PtC23H25N3OCl]+: 589.1334, found 589.1239. Anal. calcd for C23H24ClN3OPt: C, 46.90; H, 4.11; N, 7.13%; found: C, 47.43; H, 4.39; N, 6.59%.
2.3.3.3 Pt-NPh-OH. K2PtCl4 (100 mg, 0.241 mmol) and L-NPh-OH (73 mg, 0.241 mmol) reacted to yield Pt-NPh-OH as a blue powder (0.096 g, 75%). IR (cm−1): 1525 ν(C
C), 1636 ν(CN aromatic), 3453 ν(NH). 1H-NMR (d6-DMSO) δ: 10.33 (s, 1H, NH), 9.36 (s, 1H, OH), 7.99 (s, 1H, H13), 7.63 (t, JHH = 7.6, 1H, H2), 7.54 (d, JHH = 7.3, 2H, H1,5), 7.48 (t, JHH = 7.3, 1H, H4), 7.14 (d, JHH = 8.3, 1H, H18), 7.02 (d, JHH = 7.2, 1H, H8), 6.98 (t, JHH = 7.1, 1H, H9), 6.77 (d, JHH = 7.4, 1H, H11), 6.74 (d, JHH = 8.6, 1H, H19), 6.67 (q, JHH = 6.4, 1H, H3), 6.46 (t, JHH = 7.0, 1H, H10), 6.15 (d, JHH = 8.3, 1H, H11).13C-NMR (CDCl3) δ: 155.9 (C13), 154.4 (C15,17), 148.7 (C11,19), 148.1 (C6.7), 129.4 (C11), 129.0 (C2,4), 127.5 (C8), 127.0 (C3), 125.8 (C1,5), 123.3 (C9,10), 120.5 (C14), 116.3 (C18), 115.4 (C16). HR ESI MS calcd for [C19H15N2O2ClPt]+: 531.0314, found 531.4069. Anal. calcd for C19H15ClN2O2Pt: C, 42.75; H, 2.83; N, 5.25%; found: C, 42.96; H, 2.56; N, 4.89%.
2.3.3.4 Pt-EN-OEt. K2PtCl4 (100 mg, 0.241 mmol) and L-EN-OEt (68 mg, 0.241 mmol) reacted to yield Pt-EN-OEt as a greenish yellow powder (0.103 g, 84%). IR (cm−1): 1577 ν(C
C), 1612 ν(CN aromatic), 3490 ν(NH). 1H-NMR (d6-DMSO) δ: 8.91 (s, 1H, NH), 8.51 (s, 1H, H9), 7.77 (d, JHH = 7.9, 2H, H15), 7.63 (d, JHH = 7.7, 1H, H13), 7.45 (t, JHH = 7.5, 1H, H2), 7.37 (t, JHH = 7.9, 1H, H4), 7.25 (t, JHH = 7.0, 1H, H3), 7.11 (d, JHH = 8.1, 1H, H1), 7.00 (d, JHH = 6.3, 1H, H5), 6.49 (t, JHH = 7.8, 1H, H14), 3.97 (q, JHH = 6.8, 2H, CH2), 2.99 (t, JHH = 6.7, 1H, H7), 2.78 (t, JHH = 5.6, 1H, H8), 1.25 (t, JHH = 7.0, 3H, CH3). 13C-NMR (d6-DMSO) δ: 153.7 (C9), 153.6 (C11), 150.5 (C12), 148.1 (C6), 129.1 (C10), 127.1 (C15), 125.9 (C12), 124.4 (C14), 122.2 (C13), 117.0 (C1,5), 115.3 (C10), 111.9 (C15), 64.5 (C8), 61.8 (C7), 60.7 (CH2), 15.4 (CH3). HR ESI MS [C17H20N2O2Pt]+: calcd 514.0861, found 514.0856. Anal. calcd for C17H19N2O2Pt: C, 39.73; H, 3.73; N, 5.45%; found: C, 39.27; H, 3.31; N, 4.99%.
2.3.3.5 Pt-EN-NEt2. K2PtCl4 (100 mg, 0.241 mmol) and L-EN-NEt2 (75 mg, 0.241 mmol) reacted to yield Pt-EN-NEt2 as an orange powder (0.103 g, 79%). IR (cm−1): 1525 ν(C
C), 1604 ν(CN aromatic), 3446 ν(NH). 1H-NMR (d6-DMSO) δ: 9.27 (s, 1H, NH), 8.69 (s, 1H, H9), 8.16 (d, JHH = 3.1, 1H, H1), 7.73 (d, JHH = 7.3, 1H, H5), 7.59 (d, JHH = 7.5, 1H, H14), 7.44 (t, JHH = 7.4, 1H, H2), 7.35 (t, JHH = 7.4, 1H, H4), 7.24 (t, JHH = 7.0, 1H, H3), 7.21 (t, JHH = 9.2, 1H, H3), 6.34 (d, JHH = 8.8, 1H, H15), 6.23 (s, 1H, H12), 6.15 (d, JHH = 7.1, 1H, H14), 3.83 (t, JHH = 7.3, 2H, H7,8), 2.73 (q, JHH = 3.9, 2H, CH2), 1.09 (t, JHH = 6.7, 3H, CH3). 13C-NMR (d6-DMSO) δ: 162.9 (C9), 156.4 (C11), 153.4 (C13), 151.7 (C6), 147.6 (C15), 146.4 (C4), 135.8 (C3), 134.5 (C2), 128.5 (C1), 123.7 (C5), 111.6 (C10), 103.3 (C14), 98.3 (C12), 60.3 (C7,8), 43.7 (CH2), 12.5 (CH3). HR ESI MS calcd for [C19H26ClN3OPt]+: 541.1391, found 541.1310. Anal. calcd for C19H24ClN3OPt: C, 42.19; H, 4.47; N, 7.77%; found: C, 41.84; H, 4.20; N, 7.34%.
2.3.3.6 Pt-EN-OH. K2PtCl4 (100 mg, 0.241 mmol) and L-EN-OH (62 mg, 0.241 mmol) reacted to yield Pt-EN-OH as a greenish yellow powder (0.096 g, 82%). IR (cm−1): 1543 ν(C
C), 1639 ν(CN aromatic), 3428 ν(NH). 1H-NMR (d6-DMSO) δ: 9.85 (s, 1H, OH), 8.07 (s, 1H, H7), 7.95 (d, JHH = 3.6, 1H, H14), 7.82 (s, 1H, H12), 7.58 (d, JHH = 5.9, 1H, H15), 7.51 (d, JHH = 5.8, 2H, H1,5), 7.34 (t, JHH = 5.7, 1H, H2). 7.20 (t, JHH = 5.3, 1H, H4), 6.12 (t, JHH = 6.5, 1H, H3), 5.60 (s, 1H, NH), 2.83 (t, JHH = 7.1, 2H, H7), 2.86 (t, JHH = 5.5, 2H, H8). 13C-NMR (d6-DMSO) δ: 164.8 (C9), 163.6 (C13), 157.7 (C11), 147.2 (C6), 135.7 (C15), 128.3 (C2,4), 126.0 (C14), 123.6 (C3), 113.5 (C1,5), 105.9 (C10), 103.4 (C12), 59.3 (C7), 44.9 (C8). HR ESI MS [C15H16ClN2O2Pt]+: calcd 486.0527, found 486.0543. Anal. calcd for C15H15ClN2O2Pt: C, 37.08; H, 3.11; N, 5.77%; found: C, 36.85; H, 2.79; N, 5.62%.
| Empirical formula | C19H25N3O |
| Formula weight | 311.42 |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a, b, c (Å) | 10.7397(8), 10.9415(9), 16.0325(12) |
| α, β, γ (°) | 88.444(3), 72.754(3), 70.469(3) |
| Volume (Å3) | 1690.3(2) |
| Z | 4 |
| ρcalc (g cm−3) | 1.224 |
| μ (mm−1) | 0.077 |
| F(000) | 672.0 |
| Crystal size (mm3) | 0.407 × 0.317 × 0.14 |
| 2θ range for data collection (°) | 3.962 to 55.032 |
| Index ranges | −13 ≤ h ≤ 13, −14 ≤ k ≤ 14, −20 ≤ l ≤ 20 |
| Reflections collected | 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 881 881 |
| Independent reflections | 7752 [Rint = 0.0477, Rsigma = 0.0256] |
| Data/restraints/parameters | 7752/0/419 |
| Goodness-of-fit on F2 | 1.046 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0465, wR2 = 0.1163 |
| Final R indexes [all data] | R1 = 0.0625, wR2 = 0.1318 |
| Largest diff. Peak/hole (e Å−3) | 0.62/−0.61 |
2.4 DNA-binding studies
| F0/F = 1 + Ksv [Pt complex] | (1) |
| KEB[EB] = Kapp [Pt complex]50% | (2) |
| Ksv = Kq × τ0 | (3) |
2.5 Assessment of covalent binding of the Pt(II) complexes against guanine
Cisplatin and some platinum(II) based drugs are known to form adducts with DNA through forming coordination bonds with the nitrogen atoms on nucleobases.2,12 To examine if our platinum(II) complexes are capable of forming nucleobase-adducts, the following procedure was applied: Pt-Py-OEt (10 mg, 0.02 mmole) was stirred with equimolar amounts of guanine nucleobase and silver(I) hexafluorophosphate in 10 mL dichloromethane for 24 h. The mixture was filtered to remove the precipitated AgCl. Removal of the solvent afforded a yellow powder. A 1H NMR was obtained and used to evaluate the binding with guanine. The same protocol was adopted for Pt-EN-OEt and Pt-NPh-OEt with guanine.2.6 Protein binding studies
Bovine serum albumin (BSA) solution (30 μM) was prepared in Tris–HCl/EDTA aqueous buffer (pH = 7.4). The solution was titrated with different concentrations of the platinum complexes in DMSO (the amount of DMSO was maintained at ca. 20% v/v). The changes in the emission intensity of BSA were tracked at 340 nm upon excitation at 280 nm after incubating the complexes for 30 min at 295 K. The Stern–Volmer quenching constants (KSV) were obtained from eqn (4), where F0 and F are the intensities of emission of the protein in the absence and the presence of the complexes, respectively. The [Pt complex] was plotted against [F0/F]; the slope of the resultant line was KSV.29 The Stern–Volmer constant is related to the quenching constant by eqn (5), where Kq is the quenching rate constant, and τ0 is the average lifetime of the fluorescence for free BSA (τ0 = 5.8 ns).34,35 The binding constant (Kb, M−1) and the number of binding sites (n) were obtained by plotting log[I0 − I/I] against log[Pt complex], where n equals the slope and log(Kb) is equivalent to the intercept according to eqn (6).36 The Gibbs free energy (ΔG0) of the binding to BSA protein can be estimated by employing eqn (7), using the binding constant (Kb, M−1).36–38| F0/F = 1 + Ksv[Pt complex] | (4) |
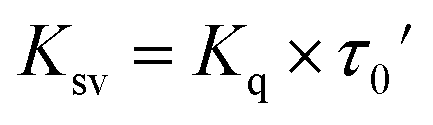 | (5) |
| log(I0 − I)/I = log(Kb) + n(log[Pt complex]) | (6) |
| ΔG0 = − RT[ln(Kb)] | (7) |
2.7 Computational studies
The covalent binding properties of the platinum complexes with guanine nucleobase were investigated computationally, employing the Gaussian09 suite.39 Gaussview software was used for visualization.40 The density functional theory (DFT) method was implemented with the B3LYP functional41 with a split-valence double-zeta basis set (6-31G) and five d-type Cartesian-Gaussian polarization functions42 for all atoms except platinum, for which the SDD basis set was employed.43 The theoretical calculations were directed to study the energies and structural features of selected [Pt]-Cl and [Pt-guanine]+ complexes. No symmetry constraints were applied in the geometry optimizations of the Pt-adducts. The final geometries (minimum potential energy structures with no imaginary frequencies) were obtained through frequency calculations. Weak interactions have been implemented in the energy evaluations using Grimme D3 corrections.44 The delocalization interactions of the proposed complexes were estimated by using the Natural Bond Orbital (NBO) program (Version 3.1).452.8 Assessments of IC50 by MTT assay
Liver (HepG2) and breast (MCF-7) cancer cell lines were provided by the Tissue Culture Unit at the Department of Biochemistry in King Abdulaziz University. Attached cancer cell lines were cultured for 1 day in a flask containing complete media. Dulbecco's Modified Eagle's Medium (DMEM) at 37 °C and 95% humidity in a sterile 5% CO2 incubator. DMEM were purchased from Life Technologies Gibco. A 0.25% trypsin solution with EDTA (4 mL) was added to the individual cells after 90% of the confluent cells were collected, and placed in a CO2 incubator for 5 min. The trypsin reaction was ceased by introducing 5 mL of complete medium. The unattached cells containing the medium were then centrifuged, followed by washing the pellets twice with sterile phosphate buffer saline (PBS).46The number of cells was assigned with a hemocytometer and counted in the four major squares after staining 20 μL of this cell-containing media with 20 μL 0.4% trypan blue. A 96-well microplate was equipped with 0.1 mL of 104 cells suspended in complete media, and the plate was incubated for 24 h. For each of the platinum coordination compounds, different amounts (3.5, 7, 14, 28, 56 μM) were added to the media when 70% of the cells in each well were confluent. Each concentration was replicated four times. The incubation time for the plate was 48 h.
In each well, the medium was exchanged with 100 μL of 0.5 mg MTT/mL free media for 4 hours in the incubator. Each well was filled with 100 μL DMSO and incubated at room temperature for 15 min before being observed using a Bio-RAD microplate reader at 595 nm. The half maximal inhibitory concentrations (IC50) of the complexes were calculated from the curve of the percentage of cell viability versus different concentration of the complex, employing GraphPad prism 9 software.47
2.9 Flow cytometry analysis of the cell cycle of HepG2 treated with platinum compounds
The DNA binder propidium iodide was applied in the DNA staining to quantify cellular aggregation in different phases of the cell cycle by flow cytometry.48 In a 6-well plate, ca. 1 × 106 HepG2 cells were seeded for 24 hours. The medium was treated with Pt-Py-OEt, Pt-PhN-OEt and Pt-EN-OEt at IC50 concentrations. After 24 h incubation, collection of the treated HepG2 cells was accomplished by the addition of 0.5 mL 0.25% trypsin and then ceasing the activity of trypsin by adding 0.5 mL complete medium to each well. The suspended cells were then centrifuged for 5 min at 1500 rpm followed by washing the cells twice with PBS. The cells were fixed with 1 mL of ethanol (70%) for 4 h at −20 °C. 100 μl of suspended cells with cold PBS with RNase A was then stained with propidium iodide solution (50 mg mL−1) and incubated in the dark for 1 h. All the stained cells were observed, employing a flow cytometer (Applied Bio-system, USA).3 Results and discussion
3.1 Synthesis and characterization
Schiff bases are normally synthesized via the condensation of primary amines and aldehydes. The reaction involves removing water from the hemiaminal intermediate. The reactions of 2-picolylamine and N-phenylethylenediamine with different salicylaldehyde derivatives in refluxing absolute ethanol were messy and provided oily mixture of materials. However, the reactions with N-phenyl-o-phenylenediamine proceeded very well in refluxing ethanol, precipitating the desired products from the reaction mixture in good yields (Fig. 3). To facilitate the Schiff base formation of 2-picolylamine and N-phenylethylenediamine, the precursors were stirred with the aldehydes in hexane, leading to mass precipitation of the desired products (Fig. 3). 1H NMR spectra of the products show the disappearance of the aldehyde signals around 10 ppm and the appearance of the imine signals in the range 7.8–8.8 ppm, confirming reaction progression. Signals in the 1H NMR spectra of the ligands can be divided into three distinctive groups: O–H in the range 10–14 ppm, aromatic protons in the range 6–9.5 ppm and aliphatic protons between 1–5 ppm. When the ligands react with the platinum centre, they produce complexes with 1H NMR data matching the signals pattern of the ligands with slight shifts from that seen in the ligands and the loss of phenolic protons due to the formation of the O–Pt bond (Fig. 3). In particular, the signals of the CHN are clearly shifted due to the coordination of the nitrogen to the platinum. Mass spectra of all the ligands and complexes exhibit [M]+ or [M + H]+ except Pt–OH which shows [M–Cl + MeCN]+ as the major peak in the spectrum.
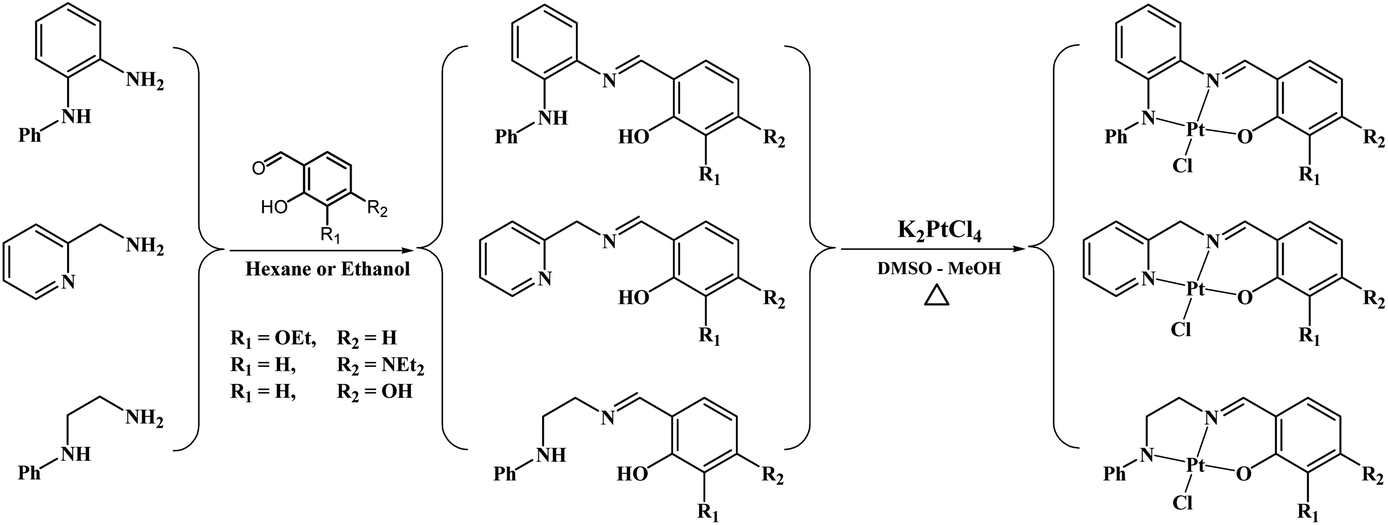 | ||
| Fig. 3 Synthesis of the ligands and their platinum(II) compounds. | ||
3.2 Crystal structure of L-EN-NEt2
The structure of L-EN-NEt2 has triclinic (P) symmetry (Fig. 4). One of the features of the ligand is the absence of delocalization through the imine bond, as the CN and N–CH2 bond lengths are 1.3036(18)–1.3110(18) Å and 1.4548(17)–1.4576(18) Å, respectively; hence, the lone pair is free for coordination. Intramolecular hydrogen bonding is observed between OH, the nitrogen of the Schiff base, and the nitrogen of the amine. Two different types of intermolecular interactions are responsible for the crystal packing: (a) C13–H⋯N5 hydrogen bonding with a distance of 0.99 Å and (b) C31–H⋯O1 hydrogen bonding with a distance of 0.99 Å. Other structural data are available in the ESI.†
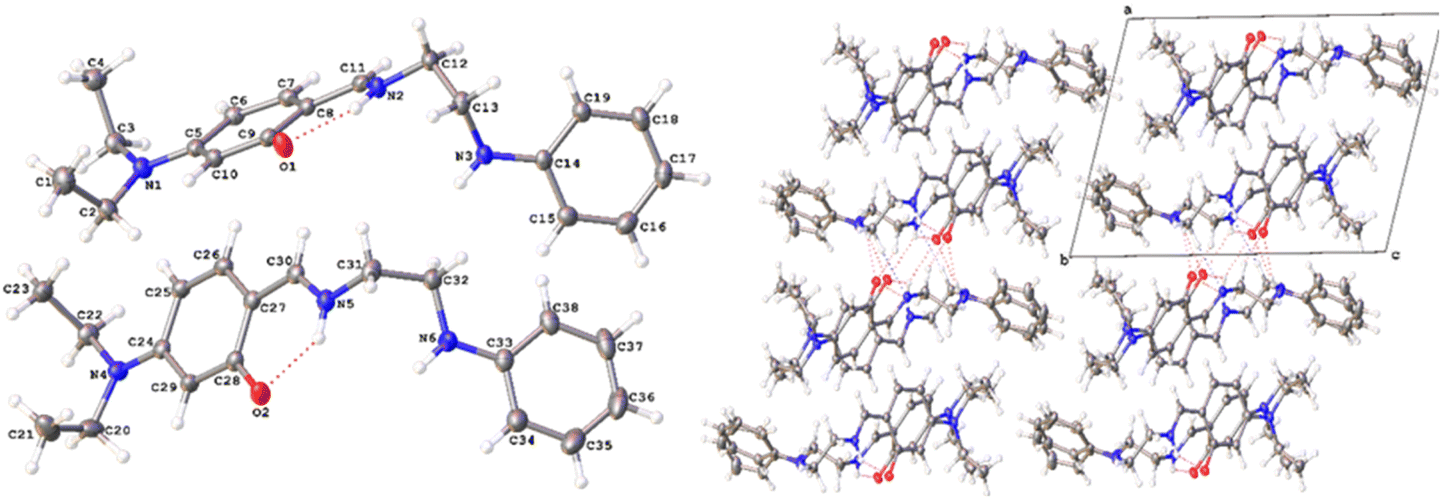 | ||
| Fig. 4 Crystal structure of L-EN-NEt2. | ||
3.3 DNA-binding studies
While cisplatin covalently binds to DNA, binding studies of [Pt(tpy)Cl]+ have suggested intercalation of the 2,2′; 6′,2′′-terpyridine with DNA, followed by covalent bonding between the metal and one of the nucleobases.49–51 In our recent work, platinum(II) complexes containing N-(2-picolyl)salicylimine ligands (Pt-Py-OEt, Pt-Py-NEt2 and Pt-Py-OH) were investigated. We concluded that the intercalation mode of binding with DNA changes over time to covalent binding, with Pt-Py-NEt2 exhibiting the largest apparent binding constant.23 We explored the binding behavior of the other six complexes with ct-DNA by studying changes in (a) the fluorescence behavior of EB-DNA upon addition of platinum(II) complexes, and (b) the relative viscosity of ct-DNA upon addition of platinum(II) complexes. Ethidium bromide (EB) binds to DNA via intercalation, resulting in an intense fluorescent adduct with a wavelength maximum at 590 nm.52 The platinum(II) complexes act as quenchers by binding with ct-DNA and destabilizing the EB-DNA adduct; hence, its binding behavior was not explored. All the other complexes in the current study can quench the EB-DNA adduct to different extents (Fig. 5). The quenching process can be a dynamic process (collisions between the excited fluorophore (EB-DNA) with the quencher) or static process (interaction between the fluorophore and the quencher at the ground state forming a non-fluorescent system). The calculated quenching constants are in the range (4.1–98.9) × 1012 M s−1, indicating a static quenching mechanism (the maximum rate for dynamic quenching is 2.00 × 1010 M s−1).53 The calculated Stern–Volmer quenching constants (Ksv) from eqn (1) are considered as the apparent binding constants (Kapp) for the complexes and they are listed in Table 2. The values are higher than the apparent binding constant for EB for all the complexes except Pt-EN-OEt; pyridine containing complexes possess the highest apparent binding constants, which indicates the importance of the ligand planarity in improving the DNA binding. The other five complexes have binding affinities within the range (0.94–9.89) × 105 M s−1. The quenching with Pt-EN-OEt causes a distinct shift of the emission maximum following addition of the complex; this phenomenon was noted previously with Pt-Py-OEt.23 We believe that steric hindrance induced by the ethoxy group causes conformational changes near the EB units and hence modify its emission maximum. The emission quenching of the EB-DNA adduct indicates the strength of the binding, but is not conclusive for the mode of binding adduct. It is worth mentioning that Pt-PhN-OEt is intensively emissive and its emission overlaps with the emission of the EB-DNA.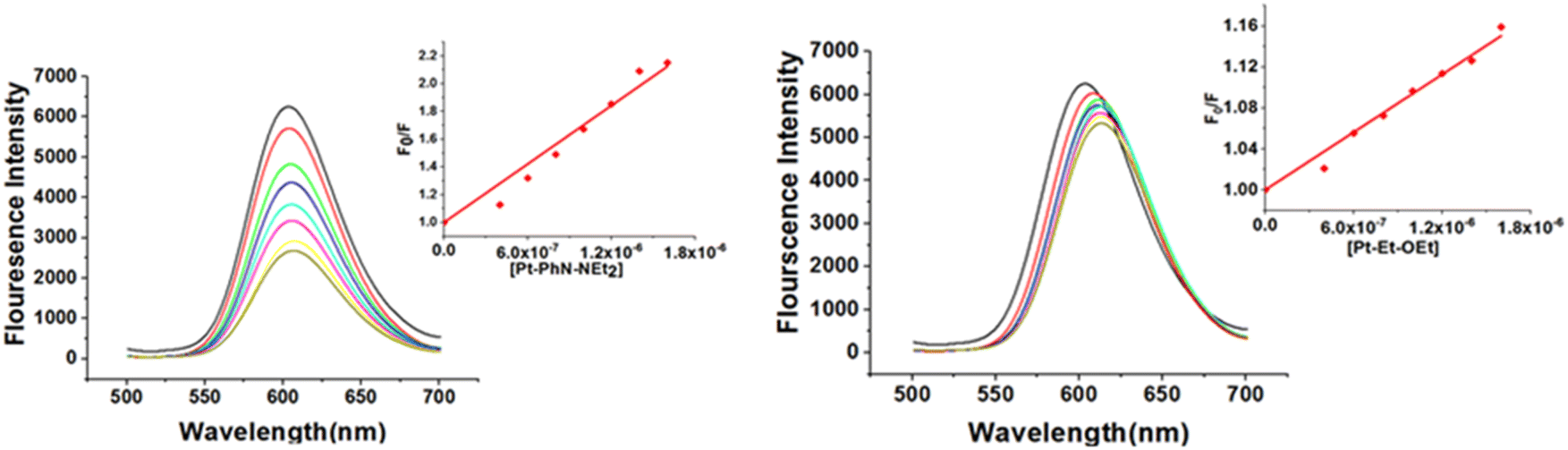 | ||
| Fig. 5 DNA binding affinities of Pt-PhN-NEt2 and Pt-EN-OEt from fluorescence competitive quenching of the EB-DNA adduct. | ||
| Complex | Pt-Py-OEt | Pt-Py-NEt2 | Pt-Py-OH | Pt-PhN-NEt2 | Pt-PhN-OH | Pt-EN-OEt | Pt-EN-NEt2 | Pt-EN-OH | |
|---|---|---|---|---|---|---|---|---|---|
| DNA-binding | Apparent binding constant (Kapp) (×105) | (12.5 ± 4.0) | (20.6 ± 0.8) | (15.4 ± 0.9) | (7.02 ± 0.3) | (5.38 ± 0.3) | (0.94 ± 0.4) | (8.38 ± 0.4) | (9.89 ± 0.6) |
| Protein binding | Stern–Volmer constant (Ksv) (×106) | 6.98 | 2.92 | 1.09 | 2.17 | 0.89 | 2.91 | 2.46 | 1.57 |
| Binding constant (Kb) (×107) | 2.35 | 0.125 | 0.98 | 0.465 | 0.062 | 4.90 | 0.048 | 0.175 | |
| Number of binding sites (n) | 1.09 | 0.94 | 1.16 | 1.06 | 0.37 | 1.2 | 0.92 | 1.01 | |
| ΔG0 (kJ mol−1) | −42 | −34.8 | −39.9 | −38.0 | −33.0 | −43.9 | −32.4 | −35.3 | |
The changes in the viscosity of ct-DNA that resulted from adding increasing concentrations of the platinum(II) complexes were monitored, so as to help identify the mode of interaction. After 10 min of mixing, Pt-PhN-OH, Pt-EN-OH, Pt-EN-NEt2 and Pt-EN-OEt induced increases in the relative viscosity of ct-DNA, as a result of an intercalation mode of interaction54 (Fig. 6). Pt-PhN-OH, Pt-EN-OH, Pt-Py-OH, Pt-EN-NEt2, Pt-Py-NEt2, Pt-EN-OEt, and Pt-Py-OEt, as well as Pt(terpy)Cl, are structurally related complexes and exhibit DNA binding that shifts gradually from the intercalation mode to the covalent binding mode with slow rates of chloride substitution (substantial enough to observe the intercalation: Fig. 6). However, Pt-PhN-NEt2 and Pt-PhN-OEt bind covalently to ct-DNA without the intermediacy of intercalation, leading to reduced relative viscosity as a result of DNA double helix unwinding.55
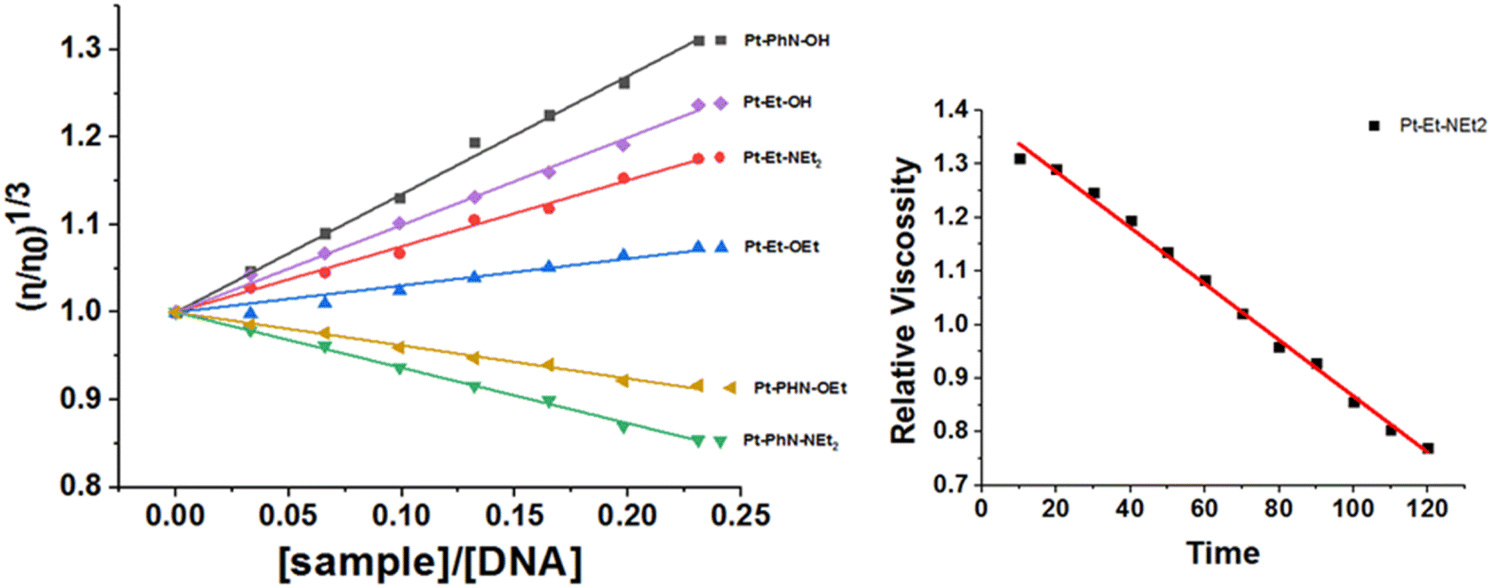 | ||
| Fig. 6 Changes in relative viscosity of ct-DNA upon treatment with platinum(II) complexes (left). Change in relative viscosity of ct-DNA upon addition of 10 μM Pt-EN-NEt2 over 120 min (right). | ||
Several platinum(II) complexes including cisplatin are activated via chlorido/aqua exchange process.56 The rate of the aquation process should not be so fast as to activate the anticancer agent before reaching the nuclear DNA.57 The faster rate of dissociation of the chlorido ligand and the coordination of the ct-DNA in Pt-PhN-NEt2 and Pt-PhN-OEt is clear from the DNA-binding study. Square planar complexes with 16 electron counts are normally inert, with their substitution proceeding slowly. Two factors could be related to the rate enhancement in complexes derived from N-phenyl-o-phenylenediamine: the ligand bulkiness (the presence of the N-phenyl group), and the higher trans effect of the nitrogen of the CHN bond (compared to the others). In the absence of crystal structures of all the complexes, theoretical studies were conducted to evaluate the structural differences between the various complexes and relate them to the dissociation rate of the chlorido ligand. Optimized structures of Pt-Py-OEt, Pt-Py-NEt2, Pt-PhN-OEt, Pt-PhN-NEt2, Pt-EN-OEt, and Pt-EN-NEt2 were obtained (Fig. 7), the important bond lengths and angles being listed in Table 3. In the optimized structures, the most significant difference in bonding is at the Pt–N1 bond (N1 being the nitrogen of the pyridine or of the amine). This bond length increases with the increase in the nitrogen basicity. In term of angles, the N1–Pt–Cl and N2–Pt–O angles are larger than 91° and differ significantly between the complexes, highlighting the role of the pyridine, phenylamine or ethylamine moieties on the molecular geometry. This difference in angles is reflected in the planarity of the molecule and the degree of distortion from square planar that is believed to be responsible for the difference in kinetic liability. Complexes containing the phenylamine fragments show the largest deviation from planarity (distortion from square planar), and exhibit the most labile DNA-covalent binding. The electronic role of the pyridine, phenylamine and ethylamine moieties was also evaluated by NBO (charge) analysis (Table 4). Note that the charge difference (polarity) for the Pt–Cl bond is 0.045 (for Pt-EN-OEt), 0.053 (for Pt-Py-OEt), and 0.07 (for Pt-PhN-OEt). These differences are mainly due to the different charge on the platinum atom of the three complexes. The Pt–Cl polarity correlates inversely with the rate of the covalent binding in the complexes.
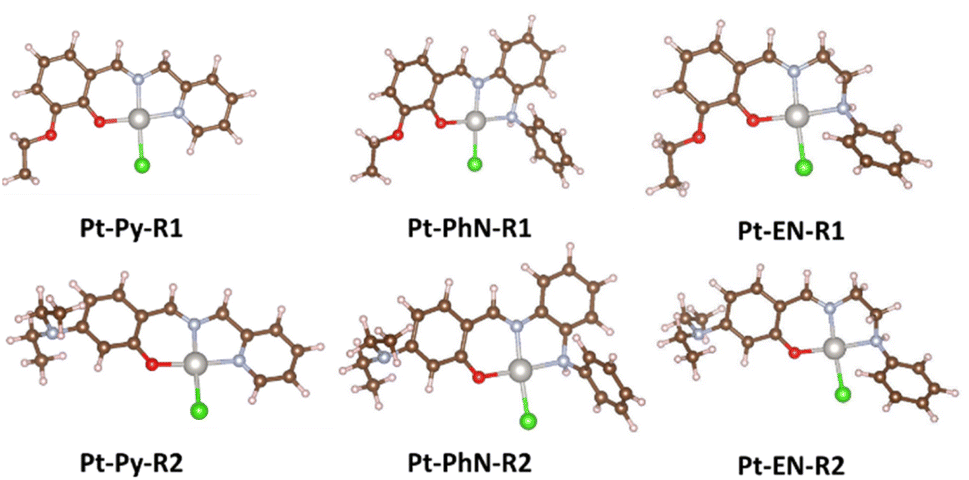 | ||
| Fig. 7 Optimized structures of the different complexes. | ||
| Complexes | Bond lengths (Å) | Angles (°) | Dihedrals (°) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Pt–N1 | Pt–N2 | Pt–O | Pt–Cl | O–Pt–Cl | N1–Pt–N2 | N1–Pt–Cl | Pt–N–C–H | O–Pt–N–C | |
| Pt-Py-OEt | 2.04 | 2.00 | 2.01 | 2.36 | 89.08 | 82.28 | 95.31 | −180.00 | 0.01 |
| Pt-Py-NEt2 | 2.04 | 2.00 | 2.01 | 2.36 | 88.89 | 82.18 | 179.99 | 0.14 | |
| Pt-PhN-OEt | 2.09 | 2.01 | 2.01 | 2.35 | 91.01 | 83.29 | 91.70 | 178.69 | 3.96 |
| Pt-PhN-NEt2 | 2.09 | 2.02 | 2.01 | 2.35 | 90.86 | 83.13 | 177.54 | 5.48 | |
| Pt-EN-OEt | 2.12 | 2.00 | 2.01 | 2.35 | 90.50 | 83.59 | 93.21 | −179.43 | 4.44 |
| Pt-EN-NEt2 | 2.12 | 2.01 | 2.01 | 2.35 | 90.18 | 83.49 | −179.43 | 4.36 | |
| Complex | Charge as obtained by NBO analysis | Δ charge | ||||
|---|---|---|---|---|---|---|
| Pt | O | CN (Schiff base) |
N (pyridine or amine) | Cl | Pt–Cl | |
| Pt-Py-OEt | +0.602 | −0.638 | −0.585 | −0.505 | −0.549 | +0.053 |
| Pt-PhN-OEt | +0.613 | −0.630 | −0.591 | −0.689 | −0.543 | +0.070 |
| Pt-EN-OEt | +0.582 | −0.640 | −0.592 | −0.694 | −0.537 | +0.045 |
3.4 Assessment of covalent binding of the Pt(II) complexes against guanine
Selected platinum complexes were reacted with guanine, using silver(I) hexafluorophosphate as chloride scavenger. All of these platinum complexes formed complexes with guanine as can be seen from 1H NMR data, but we were unable to conclude which nitrogen on the nucleobases bound to the metal due to: (1) the broadening of the signals in the 1H NMR, (b) the tautomeric nature of the guanine and the presence of only one proton with a strong signal in the 1H NMR, and (c) the low signal-to-noise ratio in the 13C NMR spectra. As a result, theoretical calculations were undertaken to evaluate the preferred nitrogen for binding. Two types of guanine adduct of Pt-Py-OEt, Pt-Py-NEt2, Pt-PhN-OEt, and Pt-PhN-NEt2 were optimized: N7-guanine and N3-guanine. Energy calculations were undertaken assuming weak (noncovalent) interactions (Fig. 8). The N7-guanine adducts of the pyridyl-containing complexes have lower energies than their N3-guanine analogues. No hydrogen bonding is observed between the guanine and the tridentate ligand. In contrast, the N3-guanine adduct is energetically dominant when the phenylamine ligands are present. This is clearly due to the significant hydrogen-bonding between the amino group of the guanine and the phenolic oxygen on the tridentate ligand. Moreover, steric hindrance is observable in the N7-guanine adduct between the N-phenyl group and the carbonyl-oxygen, which enforces a greater twist out of planarity for the guanine. The present theoretical calculations highlight the importance of the tridentate ligand design on the bonding of the guanine to the platinum. Fig. 7. Optimized structures of the different complexes.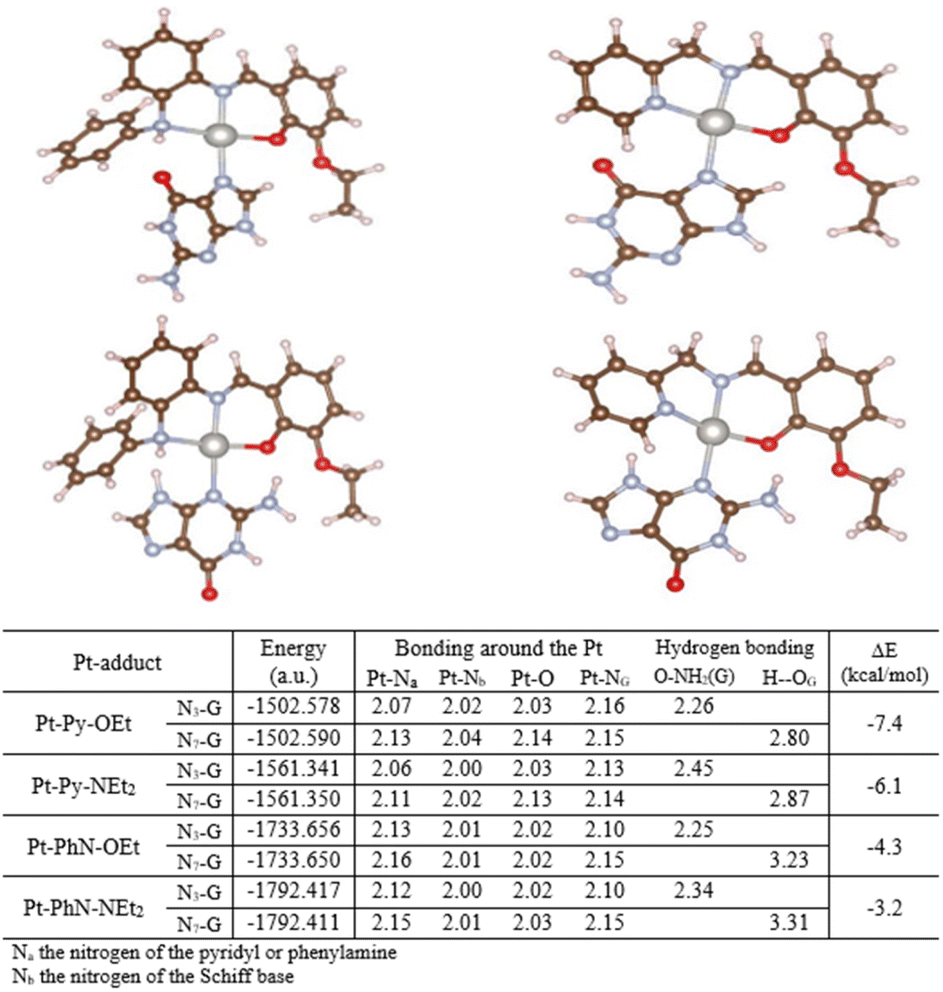 | ||
| Fig. 8 Optimized structures for guanine adducts with structural parameters and energies from theoretical calculations. | ||
3.5 BSA-binding studies
The interaction of drugs with blood plasma proteins is important in their transportation and metabolism. Human serum albumin (HSA) accounts for 55% of the protein in blood plasma.58,59 The interactions of the complexes with bovine serum albumin (BSA) (as an alternative to HSA) were evaluated by BSA fluorescence quenching (at 335–340 nm), varying the concentration of the platinum(II) complexes at 298 K (Fig. 9). Tryptophan-134 in domain I and tryptophan-212 in domain II are responsible for the fluorescence behavior of the BSA.60 The binding of our complexes changes the environment around the tryptophan units, leading to fluorescence quenching. There are several possibilities for our complexes to bind either covalently to nearby histidine units or in a noncovalent fashion. The data for eight of the platinum complexes are summarized in Table 2. The emission quenching progresses through either dynamic or static mechanisms. The quenching constants (Kq) reveal a static quenching process, because our compounds have rates much higher (in the range 1014–1015) than the maximum rate for dynamic quenching (2.00 × 1010 M s−1).53 A static quenching mechanism supports the formation of protein-complex adducts. From the Scatchard equation (eqn 5), the binding constants were extracted, showing values between 4.90 × 107 and 4.8 × 105. This highlights the importance of the ligand structural features on the binding to BSA (Table 2). The ethoxy-containing complexes have better binding constants with BSA. The average binding site “n” describes the stoichiometric ratio of the compounds to BSA;61 all of our complexes interact with BSA at one site. The formation of protein-complex adducts is a spontaneous process, with difference in Gibbs free energies ranging from −32.4 to −42.0 kJ mole−1.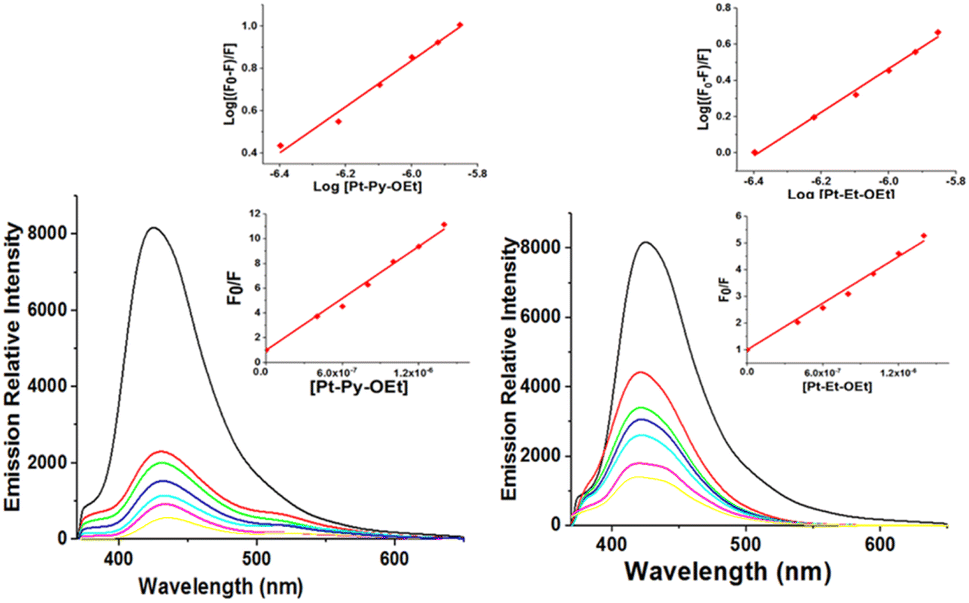 | ||
| Fig. 9 . Assessment of Protein binding affinities of Pt-Py-OEt and Pt-EN-OEt by fluorescence quenching. | ||
3.6 Preliminary in vitro antiproliferative activity
The current work extends our previous work to structurally related complexes;23 hence, we proceeded with anticancer screening against the same cancer cell lines chosen in our previous work: HepG2 and MCF-7. The IC50 values are listed in Table 5 and allow us to extract the following trends:| Compound | IC50 (μM) | |
|---|---|---|
| HepG2 | MCF-7 | |
| L-Py-OEt | 66.65 ± 6.39 | 79.57 ± 7.55 |
| L-PhN-OEt | 71.18 ± 3.04 | 97.62 ± 3.09 |
| L-EN-OEt | 116.23 ± 9.19 | 154.84 ± 11.2 |
| Cisplatin | 19.73 ± 1.53 | 18.36 ± 2.72 |
| Pt-Py-OEt | 6.99 ± 1.74 | 13.27 ± 2.10 |
| Pt-PhN-OEt | 10.15 ± 0.24 | 10.97 ± 0.96 |
| Pt-EN-OEt | 31.46 ± 0.98 | 38.96 ± 6.28 |
| Pt-PhN-NEt2 | 14.81 ± 0.18 | 13.96 ± 0.87 |
| Pt-EN-NEt2 | 27.14 ± 3.53 | 28.32 ± 1.27 |
| Pt-PhN-OH | 63.87 ± 6.44 | 66.91 ± 0.88 |
| Pt-EN-OH | 77.25 ± 6.43 | 76.55 ± 4.92 |
(1) The minimum inhibitory concentrations for the chosen ligands are relatively poor compared to those of their platinum(II) complexes, highlighting the importance of the platinum in the cytotoxic effects.
(2) Complexes and ligands that contain N-phenylethylenediamine are less cytotoxic than their N-phenyl-o-phenylenediamine and pyridyl containing complexes. The presence of planar aromatic fragments (pyridyl or phenyl) seems to be important for the activity of the complexes.
(3) 4-Hydroxysalicylimine-containing complexes show weak cytotoxic effects compared to the other salicylimine derivatives (3-ethoxy- and 4-diethylamino-containing). The hydroxy group readily dissociates to form a phenoxide moiety, resulting in an overall negative charge on the complex, so DNA binding is electrostatically unfavorable because of the strong repulsion on the backbone.62
(4) Three complexes display IC50 values significantly less (25% to 65%) than that of cisplatin: Pt-Py-OEt, Pt-PhN-OEt and Pt-PhN-NEt2. It seems that an ethoxy group at the 3-position of the salicylimine improves the anticancer properties compared to the other two substituents. Deconvoluting the electronic effect of the ethoxy from its steric effect will be addressed in a forthcoming work.
(5) All the examined compounds show slightly better anticancer effects toward the HepG2 cancer cell line compared to the MCF-7 cancer cell line.
3.7 Cell cycle phase analysis of HepG2 treated with selected compounds
The flow cytometry analysis of cell cycle phase distribution was examined for HepG2 cells using PI staining. The cell cycle interference behaviour of Pt-Py-OEt, Pt-PhN-OEt, and Pt-EN-OEt were investigated and compared to that of cisplatin at concentrations around the IC50 values of each of the complexes for 24 h. Compared with control cells, treatment of HepG2 cells with cisplatin for 24 h caused an increase in the cell population in the sub-G phase (from ca. 1% to 47.3%), with concomitant decreases in the cell population in the G1/G0, S and G2/M phases, suggesting the activation of an apoptotic cell death pathway (Fig. 10). The cell cycle assays of HepG2 treated with Pt-Py-OEt, Pt-PhN-OEt, and Pt-EN-OEt resemble that of HepG2 treated with cisplatin, but with notably different populations in the sub-G (60–65%) and G1/G0 (21–27%) phases (Fig. 10). An apoptotic cell death may be proposed by comparing the similarity in the pattern of the current data of cisplatin and our platinum complexes. Previous literature proposed that the cytotoxic effect of cisplatin is due to DNA damage, involving ATR, p53, p73, and mitogen-activated protein kinases.63 Our complexes can produce DNA damage by their DNA-binding, although they have only one leaving group (chlorido) compared to two for cisplatin. The ethoxy group causes different conformational changes and activates cell death in ways that differ from that of cisplatin.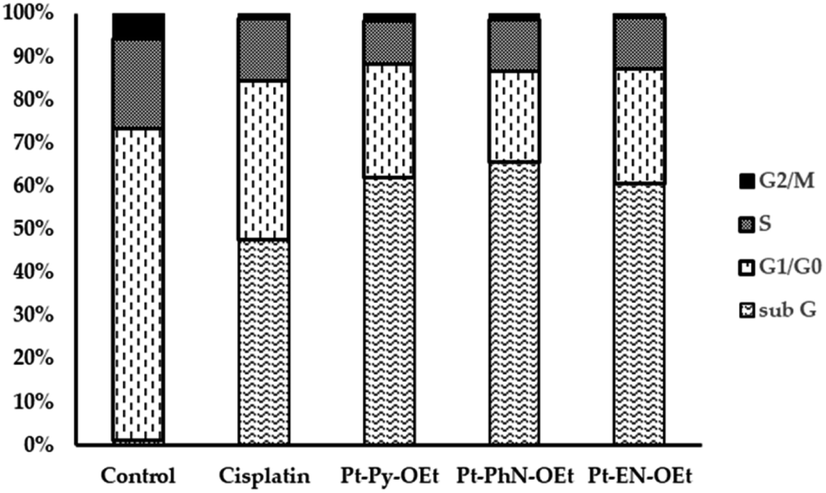 | ||
| Fig. 10 . Cell cycle assay of HepG2 upon treatment with different compounds. | ||
4 Conclusion
Our recent work on platinum(II) complexes of N-(2-picolyl)salicylimine ligands highlighted their promising anticancer activities and the high structural flexibility of the ligands which allow the study of other candidates. In the current work, new ligands were designed by replacing 2-picolylamine with N-phenyl-o-phenylenediamine and N-phenylethylenediamine in the synthesis of tridentate ligands of the type N^N^OH. The new set of ligands were employed in the synthesis of complexes with the formula Pt(N^N^O)Cl. We explored the binding behavior of the complexes with ct-DNA by (a) studying fluorescence quenching behavior of EB-DNA upon addition of the platinum(II) complexes, and (b) studying the changes in relative viscosity of ct-DNA upon addition of the platinum(II) complexes. All the complexes in the current study quench the EB-DNA adduct statically, indicating the formation of complex-DNA adducts. The calculated apparent binding constants (Kapp) for the pyridine-containing complexes are the highest apparent binding constants, which indicates the importance of the ligand planarity in improving the DNA binding. Moreover, ethoxy-containing complexes induce a distinct shift of the emission maximum of the EB-DNA adduct, presumably indicating the presence of some conformational changes in the DNA structure due to the steric hindrance induced by the ethoxy group. The changes in relative viscosity of ct-DNA indicate that most complexes interact by rapid intercalation, followed by a switch to covalent binding mode over time. However, Pt-PhN-NEt2 and Pt-PhN-OEt bind covalently without evidence for intercalation at ct-DNA; this difference in behavior, along with the theoretical modeling, stems from the electronic and steric impact of the ligand. The reaction of some platinum complexes with guanine was investigated, revealing a strong tendency to react with guanine; theoretical calculations suggest a strong impact from the tridentate ligand in determining the preferred guanine nitrogen (N3 or N7) to bind the platinum. Similarly, the binding of our complexes with BSA indicated a static interaction with higher binding affinities for the ethoxy-containing complexes. The most critical part of the current study is the anticancer properties. The current data suggest that platinum complexes with tridentate ligands containing a combination of N-phenyl-o-phenylenediamine or pyridyl with 3-ethoxysalicylimine are good chemotherapeutic candidates. Comparing the cell cycle interference behaviour of cisplatin and our platinum complexes with HepG2 suggests a similar apoptotic cell death. DNA-binding of our complexes produces DNA damage, with conformational changes that depend on the specific ligand, and differences in the activation to cell death from that seen with cisplatin. The current work illustrates the design flexibility of our tridentate ligands and the resultant platinum complexes and highlights the impact of this design flexibility on the anticancer potential. In addition to the good anticancer properties of some of the synthesized platinum complexes, the design flexibility allows more possibilities for the improvement of their physiochemical and pharmacological properties.Author contributions
Conceptualization, B. A. B.; data curation, K. S. A.; formal analysis, K. S. A., E. M. M. A., B. D., A.-H. M. E. and M. J.; funding acquisition, B. A. B.; investigation, K. S. A. and B. A. B; methodology, K. S. A, A. J., and A.-H. M. E.; project administration, B. A. B.; resources, B. A. B; software, A. J.; supervision, B. A. B.; visualization, K. S. A. and B. A. B; writing–original draft, B. A. B. and M. G. H; writing–review & editing, M. G. H.Conflicts of interest
The authors declare no conflicts of interest in this publication.Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, Saudi Arabia, under grant no. (KEP-PhD: 36-130-1443). The authors, therefore, acknowledge with thanks DSR technical and financial support. Computation for the work described in this paper was supported by King Abdulaziz University's High-Performance Computing Center (Aziz Supercomputer).References
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- D. P. Bancroft, C. A. Lepre and S. J. Lippard, Platinum-195 NMR kinetic and mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to DNA, J. Am. Chem. Soc., 1990, 112, 6860–6871 CrossRef CAS.
- A. M. Fichtinger-Schepman, J. L. van der Veer, J. H. den Hartog, P. H. Lohman and J. Reedijk, Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation, Biochemistry, 1985, 24, 707–713 CrossRef CAS PubMed.
- U.-M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins, Nature, 1999, 399, 708–712 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Direct cellular responses to platinum-induced DNA damage, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- P. J. Sadler and Z. Guo, Metal complexes in medicine: design and mechanism of action, Pure Appl. Chem., 1998, 70, 863–871 CrossRef CAS.
- M. D. Hall, M. Okabe, D.-W. Shen, X.-J. Liang and M. M. Gottesman, The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 495–535 CrossRef CAS PubMed.
- H. Timmer-Bosscha, N. H. Mulder and E. G. de Vries, Modulation of cis-diamminedichloroplatinum(II) resistance: a review, Br. J. Cancer, 1992, 66, 227–238 CrossRef CAS PubMed.
- D.-W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes, Pharmacol. Rev., 2012, 64, 706–721 CrossRef CAS PubMed.
- M. M. Gottesman, Mechanisms of cancer drug resistance, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Platinum coordination compounds with potent anticancer activity, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- S. P. Fricker, Metal based drugs: from serendipity to design, Dalton Trans., 2007, 43, 4903–4917 RSC.
- Ž. D. Bugarčić, F. W. Heinemann and R. van Eldik, Substitution reactions of [Pt(terpy)X]2+ with some biologically relevant ligands. Synthesis and crystal structure of [Pt(terpy)(cyst-S)](ClO4)2·0.5H2O and [Pt(terpy)(guo-N7)](ClO4)2·0.5guo·1.5H2O, Dalton Trans., 2004, 2, 279–286 RSC.
- D. Petrović, B. Stojimirović, B. Petrović, Z. M. Bugarčić and Ž. D. Bugarčić, Studies of interactions between platinum(II) complexes and some biologically relevant molecules, Bioorg. Med. Chem., 2007, 15, 4203–4211 CrossRef PubMed.
- C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Single-stranded nucleic acid-induced helical self-assembly of alkynylplatinum(II) terpyridyl complexes, roc. Natl. Acad. Sci., 2006, 103, 19652–19657 CrossRef CAS PubMed.
- S. D. Cummings, Platinum complexes of terpyridine: Interaction and reactivity with biomolecules, Coord. Chem. Rev., 2009, 253, 1495–1516 CrossRef CAS.
- P. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Metal complex interactions with DNA, Dalton Trans., 2015, 44, 3505–3526 RSC.
- K. Becker, C. Herold-Mende, J. J. Park, G. Lowe and R. H. Schirmer, Human thioredoxin reductase is efficiently inhibited by (2,2':6',2''-terpyridine)platinum(II) complexes. Possible implications for a novel antitumor strategy, J. Med. Chem., 2001, 44, 2784–2792 CrossRef CAS PubMed.
- G. Lowe, A. S. Droz, T. Vilaivan, G. W. Weaver, J. J. Park, J. M. Pratt, L. Tweedale and L. R. Kelland, Cytotoxicity of 2,2':6',2''-terpyridineplatinum(II) complexes against human ovarian carcinoma, J. Med. Chem., 1999, 42, 3167–3174 CrossRef CAS PubMed.
- R. J. Holmes, M. J. McKeage, V. Murray, W. A. Denny and W. D. McFadyen, cis-Dichloroplatinum(II) complexes tethered to 9-aminoacridine-4-carboxamides: synthesis and action in resistant cell lines in vitro, J. Inorg. Biochem., 2001, 85, 209–217 CrossRef CAS PubMed.
- B. E. Bowler, K. J. Ahmed, W. I. Sundquist, L. S. Hollis, E. E. Whang and S. J. Lippard, Synthesis, characterization, and DNA-binding properties of (1,2-diaminoethane)platinum(II) complexes linked to the DNA intercalator acridine orange by trimethylene and hexamethylene chains, J. Am. Chem. Soc., 1989, 111, 1299–1306 CrossRef CAS.
- Z. Ma, J. R. Choudhury, M. W. Wright, C. S. Day, G. Saluta, G. L. Kucera and U. Bierbach, A non-cross-linking platinum−acridine agent with potent activity in non-small-cell lung cancer, J. Med. Chem., 2008, 51, 7574–7580 CrossRef CAS PubMed.
- K. S. Al-Rashdi, B. A. Babgi, E. M. M. Ali, B. Davaasuren, A. Jedidi, A.-H. M. Emwas, M. A. Alrayyani, M. Jaremko, M. G. Humphrey and M. A. Hussien, Tuning Anticancer Properties of Pt(II) Complexes via Structurally Flexible N-(2-Picolyl)salicylimine Ligands, RSC Adv., 2022, 12, 27582–27595 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- C. S. Devi, B. Thulasiram, S. Satyanarayana and P. Nagababu, Analytical techniques used to detect DNA binding modes of ruthenium(II) complexes with extended phenanthroline ring, J. Fluoresc., 2017, 27, 2119–2130 CrossRef CAS PubMed.
- W. Villarreal, L. Colina-Vegas, G. Visbal, O. Corona, R. S. Corrêa, J. Ellena, M. R. Cominetti, A. A. Batista and M. Navarro, Copper(I)–phosphine polypyridyl complexes: synthesis, characterization, DNA/HSA binding study, and antiproliferative activity, Inorg. Chem., 2017, 56, 3781–3793 CrossRef CAS PubMed.
- A. Erxleben, Investigation of non-covalent interactions of metal complexes with DNA in cell-free systems, Chimia, 2017, 71, 102–111 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, GC base sequence recognition by oligoimidazolecarboxamide and C-terminus-modified analogs of distamycin deduced from circular dichroism, proton nuclear magnetic resonance, and methidiumpropylethylenediaminetetraacetate-iron(II) footprinting studies, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- D. P. Heller and C. L. Greenstock, Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye, Biophys. Chem., 1994, 50, 305–312 CrossRef CAS PubMed.
- S. Alsaedi, B. A. Babgi, M. H. Abdellattif, M. N. Arshad, A.-H. M. Emwas, M. Jaremko, M. G. Humphrey, A. M. Asiri and M. A. Hussien, DNA-Binding and cytotoxicity of copper(I) complexes containing functionalized dipyridylphenazine ligands, Pharmaceutics, 2021, 13, 764–777 CrossRef CAS PubMed.
- M. Amiri, K. Jankeje and J. R. Albani, Origin of Fluorescence Lifetimes in Human Serum Albumin. Studies on Native and Denatured Protein, J. Fluoresc., 2010, 20, 651–656 CrossRef CAS PubMed.
- S. Raut, R. Chib, S. Butler, J. Borejdo, Z. Gryczynski and I. Grycznski, Evidence of energy transfer from tryptophan to BSA/HSA protected gold nanoclusters, Methods Appl. Fluoresc., 2014, 2, 035004 CrossRef CAS PubMed.
- H. Lineweaver and D. Burk, The Determination of Enzyme Dissociation Constants, J. Am. Chem. Soc., 1934, 56, 658–666 CrossRef CAS.
- A. S. Abdelhameed, A. H. Bakheit, F. M. Almutairi, H. AlRabiah and A. A. Kadi, Biophysical and In Silico Studies of the Interaction between the Anti-Viral Agents Acyclovir and Penciclovir, and Human Serum Albumin, Molecules, 2017, 22, 1906 CrossRef PubMed.
- B. A. Babgi, J. Alsayari, H. M. Alenezi, M. H. Abdellatif, N. E. Eltayeb, A.-H. M. Emwas, M. Jaremko and M. A. Hussien, Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties, Pharmaceutics, 2021, 13, 461 CrossRef CAS PubMed.
- - M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09. revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009 Search PubMed.
- - R. Dennington, R. T. Keith, J. Millam and V. Gauss, Version 5, vol Shawnee Mission, Semichem, Inc., 2009 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichromism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- D. Moran, A. C. Simmonett, F. E. Leach III, W. D. Allen, P. v. R. Schleyer and H. F. Schaefer, Popular theoretical methods predict benzene and arenes to be nonplanar, J. Am. Chem. Soc., 2006, 128, 9342–9343 CrossRef CAS PubMed.
- M. Dolg, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the rare earth elements, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- - E. D. Glendenning, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO; version 3.1, Gaussian Inc., Pittsburg, PA, USA, 2001 Search PubMed.
- E. M. Ali, A. A. Elashkar, H. Y. El-Kassas and E. I. Salim, Methotrexate loaded on magnetite iron nanoparticles coated with chitosan: Biosynthesis, characterization, and impact on human breast cancer MCF-7 cell line, Int. J. Biol. Macromol., 2018, 120, 1170–1180 CrossRef CAS PubMed.
- J. A. Plumb Cell sensitivity assays: the MTT assay, in Cancer cell culture, Springer, 2004, pp. 165–169 Search PubMed.
- J. Fried, A. G. Perez and B. D. Clarkson, Flow cytofluorometric analysis of cell cycle distributions using propidium iodide. Properties of the method and mathematical analysis of the data, J. Cell Biol., 1976, 71, 172–181 CrossRef CAS PubMed.
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Baueri, Metallointercalation Reagents. 2-Hydroxyethanethiolato(2,2',2"-terpyridine)-platinum(I1) Monocation Binds Strongly to DNA By Intercalation, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed.
- M. Howe-Grant, K. C. Wu, W. R. Bauer and S. J. Lippard, Binding of Platinum and Palladium Metallointercalation Reagents and Antitumor Drugs to Closed and Open DNAs, Biochemistry, 1976, 15, 4339–4346 CrossRef CAS PubMed.
- P. J. Bond, R. Langridge, K. W. Jennette and S. J. Lippard, X-ray fiber diffraction evidence for neighbor exclusion binding of a platinum metallointercalation reagent to DNA, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4825–4829 CrossRef CAS PubMed.
- F. J. Meyer-Almes and D. Porschke, Mechanism of intercalation into the DNA double helix by ethidium, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS PubMed.
- W. R. Ware, Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS.
- Z. Seferoğlu, M. M. A. Mahmoud and H. Ihmels, Studies of the binding interactions of dicationic styrylimidazo[1,2-a]pyridinium dyes with duplex and quadruplex DNA, Dyes Pigm., 2016, 125, 241–248 CrossRef.
- N. Busto, M. Martínez-Alonso, J. M. Leal, A. M. Rodríguez, F. Domínguez, M. I. Acuña, G. Espino and B. García, Monomer–dimer divergent behavior toward DNA in a half-sandwich ruthenium(II) aqua complex. Antiproliferative biphasic activity, Organometallics, 2015, 34, 319–327 CrossRef CAS.
- J. A. Platts, M. Ravera, E. Gabano, M. Sardi, S. Bianco and D. Osella, Solvolysis of a series of cisplatin-like complexes – Comparison between DNA-biosensor and conductivity data, Eur. J. Inorg. Chem., 2012, 5625–5631 CrossRef CAS.
- T. Lazarevic, A. Rilak and Z. D. Bugarcic, Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef CAS PubMed.
- D. Gibellini, F. Vitone, P. Schiavone, C. Ponti, M. La Placa and M. C. Re, Quantitative detection of human immunodeficiency virus type 1 (HIV-1) proviral DNA in peripheral blood mononuclear cells by SYBR green real-time PCR technique, J. Clin. Virol., 2004, 29, 282–289 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, NY, USA, 3rd edn, 2006 Search PubMed.
- G. Vignesh, S. Arunachalam, S. Vignesh and R. A. James, BSA binding and antimicrobial studies of branched polyethyleneimine–copper(II)bipyridine/phenanthroline complexes, Spectrochim. Acta, Part A, 2012, 96, 108–116 CrossRef CAS PubMed.
- E. Lissi, C. Calderón and A. Campos, Evaluation of the Number of Binding Sites in Proteins from their Intrinsic Fluorescence: Limitations and Pitfalls, Photochem. Photobiol., 2013, 89, 1413–1416 CrossRef CAS PubMed.
- S. Alsaedi, B. A. Babgi, M. H. Abdellatif, A.-H. Emwas, M. Jaremko, M. G. Humphrey and M. A. Hussien, Effect of Net Charge on DNA-Binding, Protein-Binding and Anticancer Properties of Copper(I) Phosphine-Diimine Complexes, J. Inorg. Organomet. Polym., 2021, 31, 3943–3952 CrossRef CAS.
- S. Tanida, T. Mizoshita, K. Ozeki, H. Tsukamoto, T. Kamiya, H. Kataoka, D. Sakamuro and T. Joh, Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment, Int. J. Surg. Oncol., 2012, 2012, 862–879 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2203351. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00395g |
| This journal is © The Royal Society of Chemistry 2023 |