DOI:
10.1039/D3RA00299C
(Paper)
RSC Adv., 2023,
13, 9678-9685
Promising transition metal decorated borophene catalyst for water splitting†
Received
15th January 2023
, Accepted 2nd March 2023
First published on 24th March 2023
Abstract
Borophene has been recently reported to be a promising catalyst for water splitting. However, as a newly synthesized two-dimensional material, there are several issues that remain to be explored. In the present study, we investigate the catalytic performance of three kinds of pristine and decorated borophenes using first-principles calculations. Our calculations show that Ni-doped α borophene can be a highly active catalyst for water splitting. Doping or decorating with different transition metals such as Co or Ni at different sites shows a strong effect on the catalytic performance of α, β12 and χ3 borophenes. Ni-doped α borophene shows low Gibbs free energy of hydrogen adsorption (ΔGH ∼ 0.055 eV) for the hydrogen evolution reaction (HER) and promising overpotential (0.455 V) for the oxygen evolution reaction (OER). This study provides some critical insights into the catalytic activity of borophene for water splitting by selecting suitable decorated metal.
Introduction
The serious environmental and energy crises all over the world have greatly stimulated research interests in the exploration of sustainable energy sources.1–10 Water splitting is one of the clean and renewable energy technology, and the efficiency of this mainly depends on the performance of the catalysts. To date, the most effective catalysts are Pt (for HER) and IrO2/RuO2 (for OER).11–14 However, the scarcity and high prices prevent their large-scale usage. Therefore, developing cheaper and more effective catalysts for water splitting is extremely needed.
On account of the special physical and chemical properties and the large specific area, two-dimensional (2D) materials have been highly attractive in the past few years. Besides MoS2, which is a highly effective 2D catalyst comparable with Pt for HER, a new 2D material, borophene, has been reported to be a highly intrinsic catalyst recently.15 A 2D boron sheet, borophene, which was theoretically predicted and first prepared on Ag (111) in 2015,16 has many excellent physical properties, such as high strength comparable to graphene17,18 and super-conductivity.19,20 In addition, borophene has a substantial number of allotropes such as α, β12 and χ3 phases.21 According to recent studies,15,22 borophene also has the possibility of being used as a novel HER catalyst. As borophene is the lightest 2D material, it may become the lightest catalyst for HER. Liu et al.23 reported that boron nanosheets were excellent conductors, metal-free, and could be synthesized readily; they are promising candidates for HER. Shi et al.24 proposed that borophene is a high-performance catalyst for HER with near zero free energy for hydrogen adsorption, metallic conductivity, and plenty of active sites in the basal plane. Tai et al.25 prepared borophene nanosheets on a carbon cloth surface by chemical vapor deposition and found that borophene showed good electrocatalytic HER ability with a 69 mV dec−1 Tafel slope and good cycling stability in a 0.5 M H2SO4 solution. Zhang et al.26 reported that Au and Ni two-dimensional boron sheets might serve as HER catalysts that were competitive with Pt. Moreover, the HER activity of borophene can be further improved by some methodologies, such as doping some metal elements and strain engineering. Mir et al.27 investigated the influence of the dopants on the catalytic activity of the α phase of borophene sheets and reported that the intrinsic HER catalytic activity of borophene was improved by the doped atoms. Wang et al.28 found that strain engineering was demonstrated to be an effective way to improve the HER catalytic performance of armchair borophene nanoribbons, and free energy of almost zero was achieved at a critical compressive strain of −2%. Subsequently, borophene showed vast application prospects in OER. Singh et al.29 proposed that borophene-supported single transition metal atoms showed high catalytic performance in OER and oxygen reduction reaction (ORR). Xu et al.30 studied the borophene-supported single transition metal atoms as potential OER electrocatalysts using density functional theory calculations and revealed that the Ni atom was useful to enhance the catalytic behavior of borophenes for water splitting. Xu et al.31 designed single-atom bifunctional electrocatalysts for overall water splitting that involved 3d transition metal atom-doped borophene nanosheets. Besides, Erakulan et al.32 studied that doping nonmetal carbon atoms into borophene could reduce the σ electrons of the adjacent boron atoms making them less reactive towards O* intermediate, and dual carbon doping made β12 borophene more efficient towards OER. Moreover, a few groups have demonstrated the introduction of an additional transition metal atom on the δ3 sheet borophene monolayer,33 and the β12 sheet borophene monolayer34 can display good electrocatalytic performance. Although there exist several studies on the application of borophene in water splitting illustrated above, the systematic studies on HER/OER catalytic activity of various borophenes are inadequate and the main factors that may affect their HER/OER performance are still unclear, calling for more theoretical studies at the atomic level.
In this work, we rationally adopted two different doping methods, BCo-doping (Co atom substitutes inert B catalytic site) and Ni-doping (Ni atom adsorbs on vacancy site), to study α, β12 and χ3 borophenes for their HER and OER performance by first-principles calculations. With good structural stability and metallic conductivity, Ni-doped borophenes can be used as bifunctional HER/OER catalysts. In particular, Ni-doped α borophene shows the optimal catalytic performance for water splitting.
Computational methods
The first-principles calculations were performed based on the density functional theory, implemented in the Vienna Ab initio Simulation Package (VASP).35 The projector-augmented wave (PAW) pseudopotential36 was used and the generalized gradient approximation of the Perdew–Burke–Ernzerhof (PBE-GGA) method37 was employed in the calculations. A 380 eV cut-off energy was set for the plane-wave basis set. The convergence accuracy for the energy was set to 10−4 eV and the force converged to 0.01 eV Å−1. A 3 × 3 × 1 Monkhorst–Pack k-mesh in the Brillouin zone was used for the borophene structure relaxation. The calculations were completed on the 2 × 2 × 1, 3 × 2 × 1, and 3 × 2 × 1 supercells consisting of 32, 30, and 30 boron atoms in α-sheet, β12-sheet and χ3-sheet, respectively. The doping ratios of the transition metal in these sheets were 1/32, 1/30, and 1/30, respectively. Both the directions perpendicular to the periodic direction have a vacuum of 15 Å to avoid the interaction between neighboring units.
The Gibbs free energy of hydrogen adsorption (ΔGH) was calculated to evaluate the catalytic activity of borophenes for HER. The free energy ΔGH is defined as ΔGH = ΔEH + ΔEZPE − TΔSH, where ΔEH, ΔEZPE, and ΔSH are the hydrogen adsorption energy, the zero point energy (ZPE), and the entropy differences between the adsorbed state and the gas phase, respectively. The calculation method of ΔGH used in the present work has been validated in a previous study.38 Thus, the formula can be rewritten as ΔGH = ΔEH + 0.24. In addition, the adsorption energy per H atom (ΔEH) is defined as ΔEH = Eslab+H − Eslab − 1/2EH2, where Eslab+H and Eslab are the total energies of borophenes with and without the adsorbed H atom, and EH2 is the total energy of H2 gas.
The pathway of OER has been summarized in detail as shown below. In the acid electrolyte, OER can be decomposed into the following elementary steps:
| | |
H2O(l) +* → OH* + H+ + e−
| (1) |
| | |
O* + H2O(l) → OOH* + H+ + e−
| (3) |
| | |
OOH* → * + O2(g) + H+ + e−
| (4) |
where * represents the active site on the surfaces of the catalysts, l, and g represent the liquid phase and the gas phase, respectively.
For each elementary step of OER, the free energy has been calculated according to the method proposed by Nørskov et al.12 The free energy change from the initial state to the final state of the reaction is defined as:
| | |
ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH
| (5) |
where Δ
E, Δ
EZPE, and Δ
S are the differences in the total energy obtained from DFT calculations, zero point energy, and entropy between the reactants and the products, respectively.
T represents the temperature (298.15 K). Δ
GU = −
neU, where
U is the applied electrode potential,
e is the charge transferred and
n is the number of proton-electron transferred pairs. In addition, Δ
GpH = −
kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
ln[H
+] = pH ×
kBTln
10, where
kB is the Boltzmann constant.
The free energy change for the four elementary OER steps can be obtained as ΔG1 = ΔGOH*, ΔG2 = ΔGO* − ΔGOH*, ΔG3 = ΔGOOH* − ΔGO*, and ΔG4 = 4.92 − ΔGOOH*. Therefore, the overpotential that evaluates the performance of OER is applied according to the following equation:
| | |
ηOER = max{ΔG1, ΔG2, ΔG3, ΔG4}/e − 1.23
| (6) |
where 1.23 V represents the equilibrium potential.
11
The adsorption energies of intermediates OH*, O*, and OOH* are defined as follows:
| | |
ΔEOH = E(OH*) − E(*) − [E(H2O) − 1/2 E(H2)]
| (7) |
| | |
ΔEO = E(O*) − E(*) − [E(H2O) − E(H2)]
| (8) |
| | |
ΔEOOH = E(OOH*) − E(*) − [2E(H2O) − 3/2E(H2)]
| (9) |
E(OH*),
E(O*), and
E(OOH*) are the absolute energies computed for slabs with OH, O, and OOH adsorbed, respectively.
E(*),
E(H
2O), and
E(H
2) are the energies of the bare slab, H
2O, and H
2 molecules.
The charge density difference (ρdiff) on the borophene before and after the deposition of Ni is calculated:
| | |
ρdiff = ρ(Ni@borophene) − ρ(borophene) − ρ(Ni)
| (10) |
where
ρ(Ni@borophene),
ρ(borophene) and
ρ(Ni) are the charge densities of Ni@borophene, borophene and Ni, respectively.
The adsorption energy (Ea), which is the required energy to dissociate the Ni adatom from the boron sheet and move it away from the sheet, was determined to assess the stability of the Ni-doped system with eqn (8).
| | |
Ea = EB(Ni) − EB − ENi
| (11) |
where
EB(Ni) and
EB refer to the total energy of Ni-doped borophene and borophene, and
ENi is the energy of the isolated Ni atom.
In our simulation, the formation energy of the Co-substituted B was calculated by the following formula:
| | |
EF = E(BCo) − EB + μB − μCo
| (12) |
where
E(B
Co) is the total energy of B
Co-doped borophene,
EB is the total energy of borophene,
μB, and
μCo are the chemical potentials of isolated B and Co atoms, respectively.
The climbing image nudge elastic band (CI-NEB) method39 was performed to investigate the water dissociation on the different surfaces. To be specific, we first deposited an H2O molecule on the catalyst surface and allowed it to relax to the most stable site. After that, we started with multiple initial configurations and selected the one with the minimum energy. Then, we found the corresponding potential final locations of H+ and OH− on the surface and allowed each of these configurations to relax. Finally, we performed CI-NEB calculations on the most stable final H + OH configuration with the most stable initial H2O configuration.
Results and discussion
The active sites and HER activity of the pristine borophenes
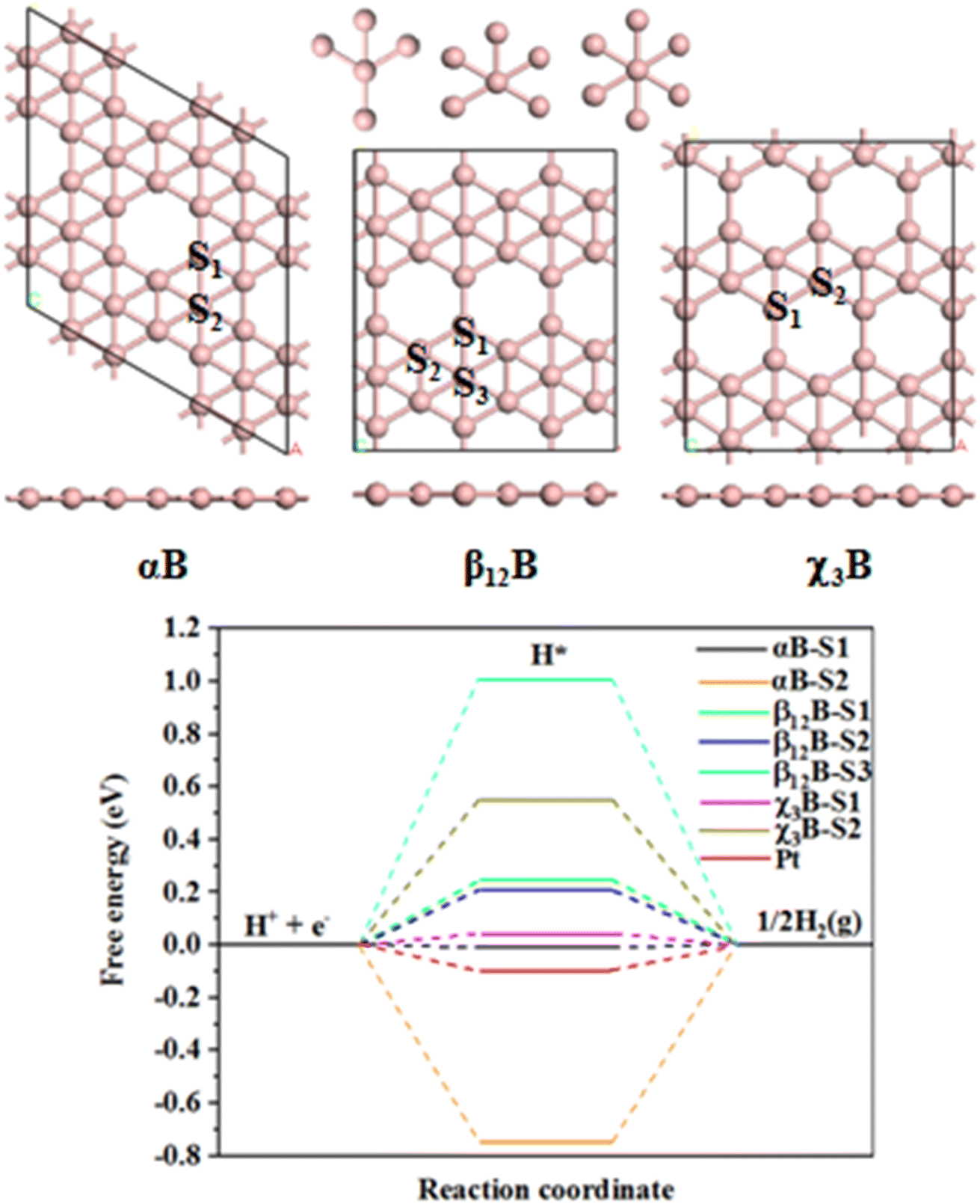
It is well-known that the HER catalytic activity of electrocatalysts highly depends on the H adsorption-free energy (ΔGH).40 Generally, the HER catalytic activity is considered to be high if |ΔGH| < 0.2 eV.41,42 The closer value of |ΔGH| is to 0, the HER catalytic activity is higher.43 We first studied the HER performance of three typical pristine borophene allotropes, namely, αB, β12B, and χ3B. Among many predicted structures, αB was confirmed to be the most stable structure,44 β12B and χ3B can be synthesized experimentally.45 These three allotropes have a typical porous characteristic structure that can provide sufficient surface space and active atoms, which are beneficial to be used in the field of catalysis. As shown in Fig. 1, For αB, there are two H adsorption sites namely, αB-S1 and αB-S2. The calculated free energies of the H adsorption at these sites are −0.005 and −0.75 eV, respectively. Thus, HER at αB-S1 site presents high activity as the H adsorption free energy at this site is better than that of Pt (−0.09 eV (ref. 46)), and the very negative free energy of the αB-S2 site shows that the strong H adsorption, which will poison the active site. For β12B, three distinct sites for H adsorption are obtained, β12B-S1, β12B-S2, and β12B-S3. However, the free energies of these sites are very positive which indicates weak H adsorption and HER performance, and the H adsorption free energies of these corresponding sites are 0.253, 0.218, and 0.99 eV, respectively. As for χ3B, we find two no-equivalent kinds of sites, χ3B-S1 and χ3B-S2. The free energy of χ3B-S1 is 0.031 eV and so this site performs good HER activity than χ3B-S2 whose free energy is 0.544 eV. In addition, all H atoms adsorb on the top sites of B atoms (see Fig. S1†), and interestingly, all these sites can be classified into three kinds of coordination numbers of B sites, which are 4, 5, and 6 coordinations, as shown in Fig. 1.
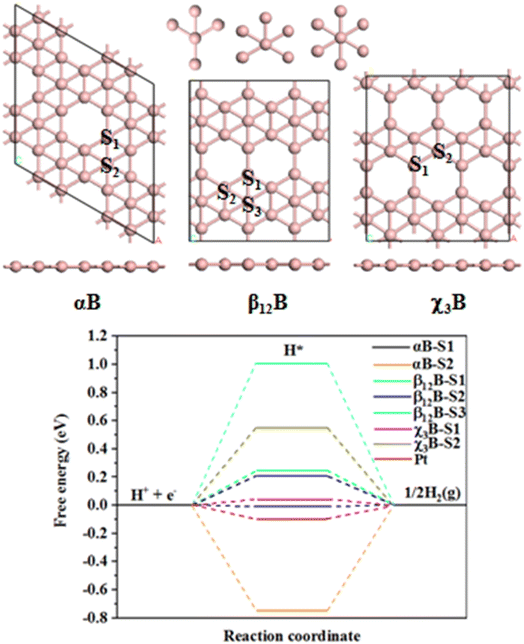 |
| | Fig. 1 Models and calculated HER performance: top and side views of atomic structures of αB, β12B, and χ3B, and calculated the free energy diagram for HER of different sites in various borophenes and cited data. | |
The stability of BCo- and Ni-doped borophenes
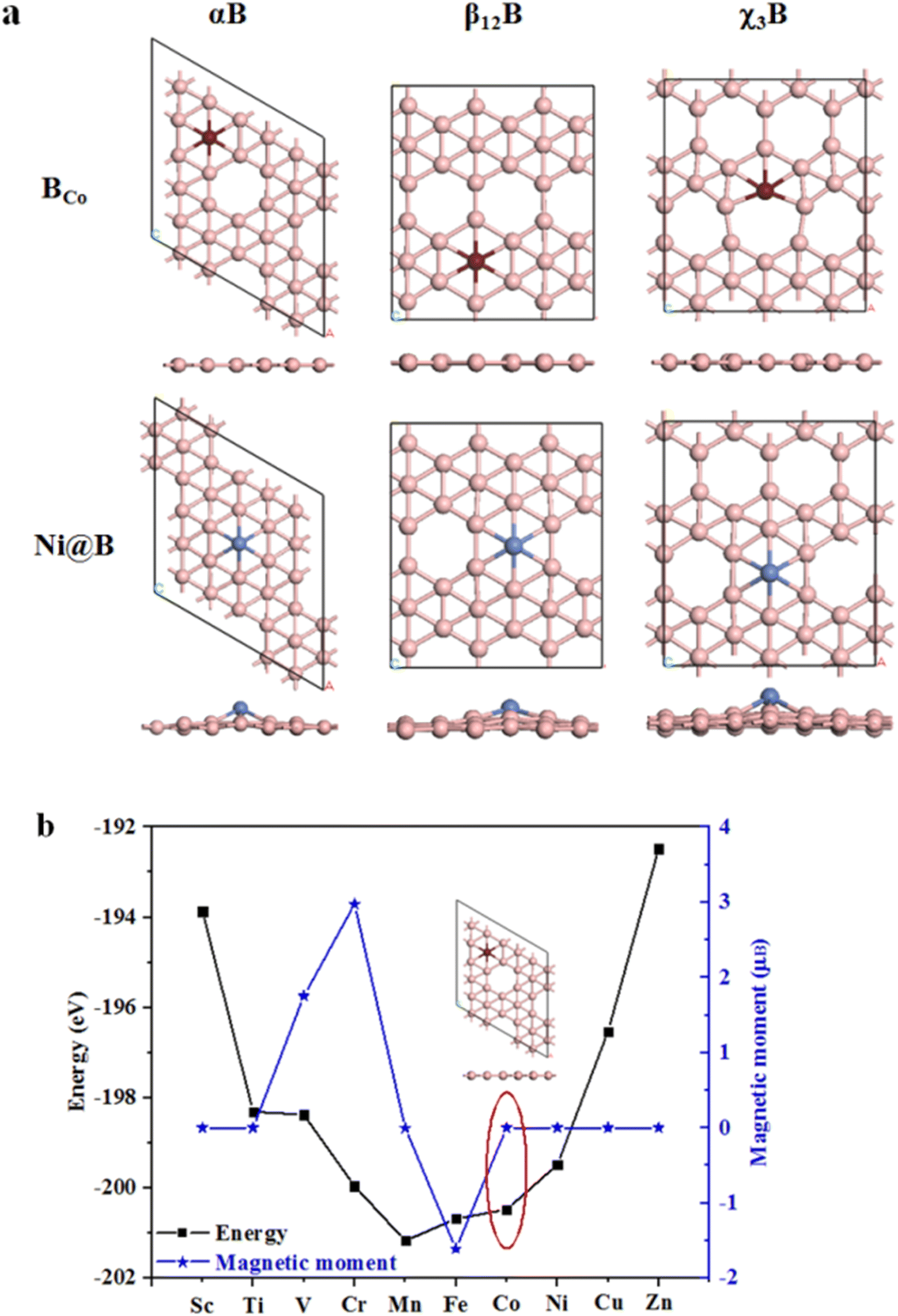
Next, we adopt two different doping methods, BCo- and Ni-doping, to the aforementioned three kinds of borophenes, i.e., αB, β12B and χ3B, then to investigate their atomic structures, electronic properties, HER and OER performance. Fig. 2a shows the BCo- and Ni-doped borophene structures. Obviously, BCo-doping barely causes structural distortions to different phases of borophenes. Here the calculated formation energies of BCo-doping into αB, β12B and χ3B are only 0.663, 0.897, and 0.974 eV, respectively, indicating that the dopant should be relatively easily doped on the surface. On the other hand, we use the Co atom to substitute the inert B catalytic site for HER in different borophene surfaces. For Ni-doping, it can show various degrees of structural distortions to borophenes and distortion degrees obey the sequence of αB < β12B < χ3B. Ni tends to adsorb on the vacancy site for αB, as shown in Fig. S2,† there are three Ni adsorption sites and the Ni adsorption energy is the highest (−5.218 eV) when it stays on the vacancy site. According to the preceding result, we study the Ni adsorption energies for corresponding vacancy sites on β12B and χ3B, which are −5.404 eV and −5.149 eV, respectively. These large negative values of Ni adsorption energies on the vacancy sites manifest the high stability of Ni-doped structures. Interestingly, the reason we chose the Co atom to substitute the B atom in our studied structures is shown in Fig. 2b, besides V (1.752μB), Cr (2.975μB), Mn (−0.003μB), and Fe (−1.611μB) dopants, other transition metal atoms can keep αB with zero magnetic moments, reflecting the nonmagnetic intrinsic property of αB. Based on zero magnetic moments, BCo-doped αB has the lowest energy. Therefore, Co-substituted B was adopted in our research.
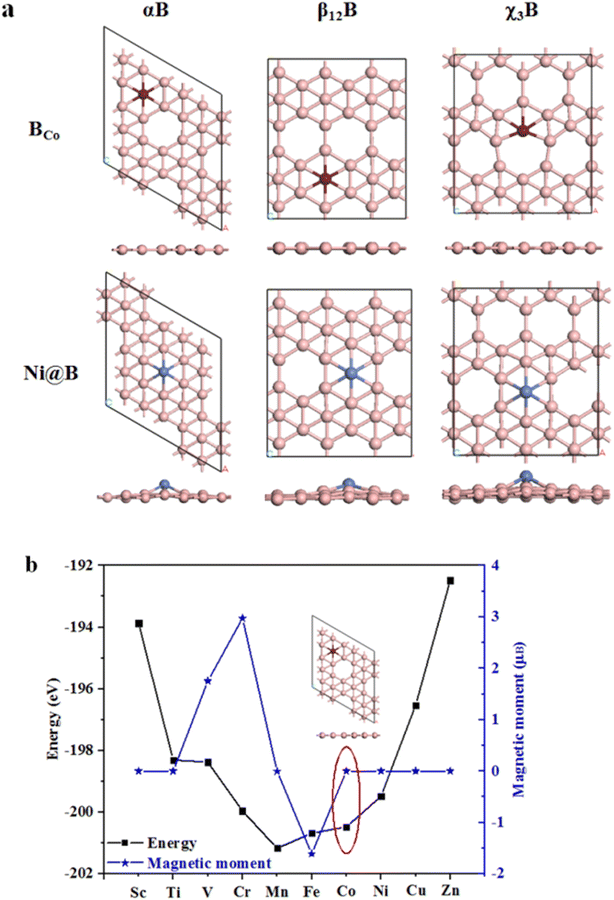 |
| | Fig. 2 (a) Top and side views of BCo- and Ni-doped structures; (b) summarized energies and magnetic moments of different metal dopant substitute B in αB. The pink, brown, and blue balls represent B, Co, and Ni atoms, respectively. | |
The HER activity and electronic properties of BCo- and Ni-doped borophenes
From the above discussion, it is concluded that the BCo- and Ni-doped borophene structures showcase high stability. The following step is that we explore the HER catalytic abilities of these modified borophene surfaces in detail (see Fig. S3 and S4†). Firstly, it is clear that BCo-doping could bring Co and Co–B bridge new sites for H adsorption toward αB, β12B, and χ3B. Ni-doping could introduce new Ni sites for H adsorption toward αB and β12B, especially, Ni-doped χ3B possesses Ni and Ni–B bridge new sites. Secondly, Co new sites perform excellent HER catalytic activities on BCo-doped β12B and χ3B as a result of near-zero hydrogen adsorption-free energies. Therefore, BCo-doped β12B and χ3B may be used as highly active single-atom catalysts for HER. Thirdly, new Ni sites of different Ni-doped borophenes showed inferior HER performance along with very positive hydrogen adsorption-free energies.
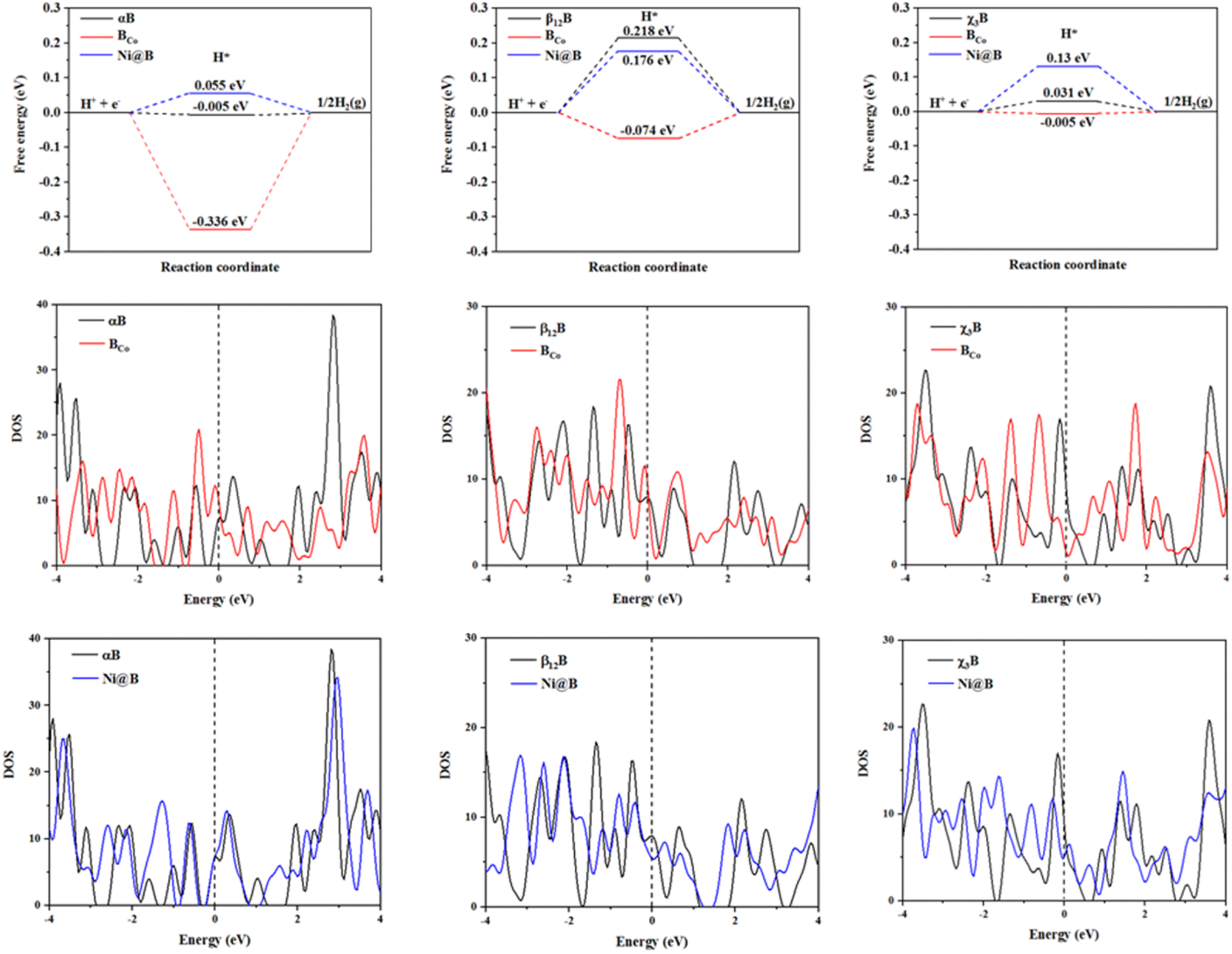
Then, as far as the best active sites of BCo- and Ni-doped surfaces for HER are concerned, as shown in Fig. 3, from three free energy diagrams for hydrogen evolution, it is clear that BCo- and Ni-doping are effective to modulate the HER performance of αB, β12B, and χ3B. Specifically, for BCo-doping, the hydrogen adsorption-free energies of three kinds of borophenes decrease to −0.336, −0.074, and −0.005 eV, respectively. Thus, BCo-doped β12B, and χ3B behave excellent HER activity as a result of zero approached free energies. Although BCo-doping is not beneficial to improve the HER catalytic activity of αB when pH = 0, BCo-doped αB possesses rich and excellent HER active sites when pH = 7 (see Fig. S5†) and all the active sites on this surface have near zero hydrogen adsorption free energies. Besides, we studied the water dissociation on this surface and find that the energy barrier (0.674 eV) for water dissociation was extremely low (see Fig. S6†); H2O molecules adsorb on the top site of Co atom and the dissociated H* adsorbs on the bridge site between the Co atom and the nearest neighbor B atom. Considering a relatively high energy barrier of 0.89 eV for the water dissociation on the Pt surface,38,46 BCo-doped αB shows superior neutral HER catalytic performance, which increases the feasibility of the splitting seawater, the most abundant water source, in H2 production. The reason is that electrolysers operating in neutral media can potentially enable the direct use of saltwater without the requirement of desalination for pH maintenance.47 For Ni-doping, the hydrogen adsorption free energies of different modified surfaces are 0.055, 0.176, and 0.13 eV, respectively, implying that Ni-doped αB, β12B, and χ3B have good HER activity.
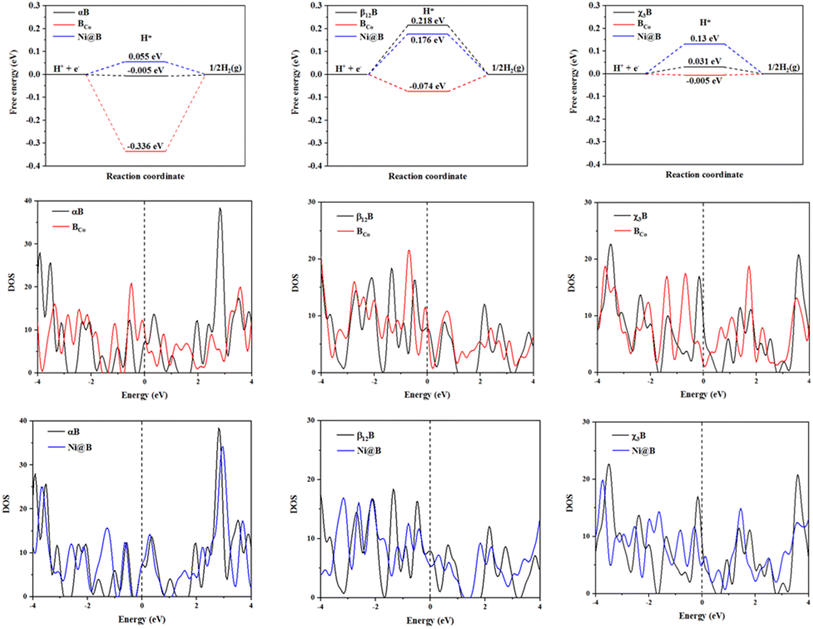 |
| | Fig. 3 The hydrogen adsorption free energies of best active sites on pristine, BCo- and Ni-doped borophene (αB, β12B and χ3B) surfaces; and the DOS of these surfaces, where the Fermi level is set to 0 eV. | |
We will employ the electronic properties of these pristine and decorated borophenes to enrich the physics insight. Compared to the pristine surfaces, the free energies of the modified surfaces work in small increments or decrements. Furthermore, by analyzing the density of states (DOS) of the pristine, BCo- and Ni-doped borophene surfaces, firstly, these surfaces are metallic as a result of DOS profiles crossing the Fermi level with no gap. Next, we noticed that BCo-doping leads to the peak values of αB and β12B near the Fermi level becoming bigger, however, the peak value of χ3B near the Fermi level declines drastically. Ni-doping makes the peak values of β12B and χ3B near the Fermi level become smaller, and the peak value of αB remains unchanged with the same DOS profile near the Fermi level. All in all, BCo-doping and Ni-doping could arouse the change in electronic properties of αB, β12B, and χ3B except Ni-doped αB. We all know that the changed electronic properties may directly affect the electrical conductivity and catalytic ability of materials, then we get different hydrogen adsorption-free energies of the surfaces mentioned above. Additionally, the phenomenon of the unchanged electronic property of Ni-doped αB explains that Ni-doping could keep the stability of αB in the electronic structure, on the other hand, Ni-doped αB maintains the outstanding HER performance from αB.
The OER activity and electronic properties of BCo- and Ni-doped borophenes
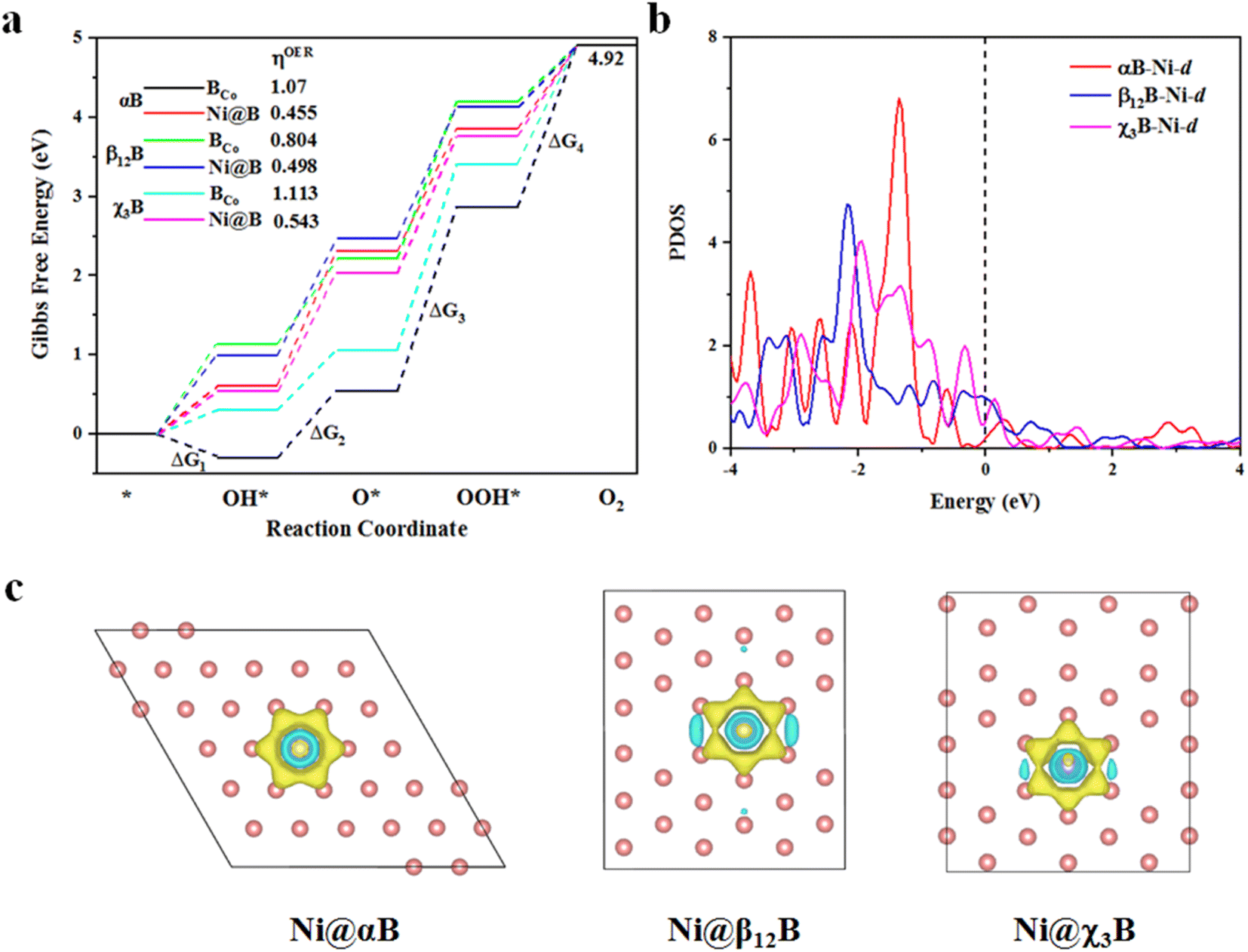
After investigating the stability and HER activity of BCo- and Ni-doped borophenes, we study the catalytic activity of these structures for OER. As depicted in Fig. 4a, four elementary steps of OER, Co, and Ni sites are chosen as the active sites for OER, and the structures of the adsorbed intermediates (OH*, O*, and OOH*) on BCo- and Ni-doped borophenes are shown in Fig. S7 and S8.† From the free energy diagram, we can see that for Ni-doped αB, the second step of OH* reacting to O* is the rate-determining step of the entire OER reaction, but for other BCo- and Ni-doped borophenes, the third step of O* reacting with H2O to form OOH* is the rate-determining step. The reaction Gibbs free energy (ΔG1, ΔG2, ΔG3, and ΔG4) of the four elementary steps are listed in Table S1.† In addition, we calculated the overpotentials for the corresponding modified borophenes and the overpotential is the most important indicator to determine the OER performance, an ideal OER catalyst is accompanied by zero overpotential.48 The overpotentials of Ni-doped αB, β12B, and χ3B are 0.445, 0.498, and 0.543 V, respectively, even lower than that of the state-of-the-art IrO2 (ηOER = 0.56 V (ref. 49)), indicating that the OER catalytic activities of Ni-doped borophenes are better than those of IrO2. Moreover, BCo-doped borophenes have much bigger overpotentials (ηOER = 1.07, 0.804, and 1.113 V, respectively) and lower OER catalytic activities. It is worth noticing that the overpotential for Ni-doped αB is the lowest in this work, implying the best OER performance. Additionally, we plotted the partial density of states (PDOS) analyses of Ni-d orbitals, as shown in Fig. 4b, and find that the lower peak value of Ni-d near the Fermi level means smaller overpotential. Thus, Ni-d of Ni-doped αB manifests the lowest peak value. Apart from Ni-doped αB with different rate-determining steps for OER, we compared Ni-doped β12B with Ni-doped χ3B combining the PDOS analyses of Ni-d orbitals. The Ni-d peak of Ni-doped χ3B near the Fermi level within the energy range (−0.5–0 eV) is higher than that of Ni-doped β12B, indicating that Ni atom of Ni-doped χ3B has more bonding electrons and then stronger adsorption energy of O*. We obtained the adsorption energies of O* of Ni-doped χ3B, and β12B as 1.97 eV and 2.45 eV (see Table. S2†), respectively. The stronger adsorption energies of O* will poison the catalytic site and need more potential to form OOH*. Thus, Ni-doped χ3B has the bigger overpotential.
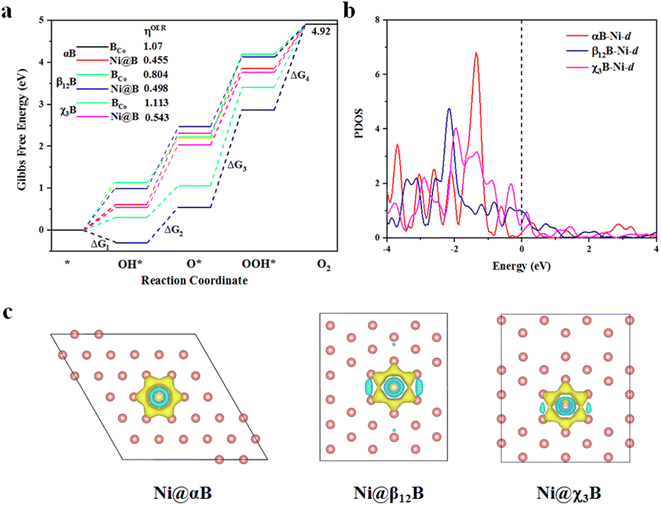 |
| | Fig. 4 (a) The Gibbs free energy diagram for the OER pathway of BCo- and Ni-doped borophenes (αB, β12B, and χ3B); (b) PDOS of Ni-d orbitals in Ni-doped borophenes; (c) charge density differences (isosurface level: 0.008 e Å−3) of Ni-doped borophenes. The yellow and cyan contours represent electron accumulation and depletion, respectively. | |
In general, Ni-doped borophenes show splendid OER catalytic activities and good HER performance from the perspective of extremely low OER overpotentials and Pt-compared hydrogen adsorption-free energies. Also, the charge density differences of Ni-doped borophenes are shown in Fig. 4c, it is visualized to discover that borophenes obtain electrons from Ni atoms and then the electron increase of the borophene systems is beneficial to improve the catalytic activity of catalysts, which may be the inner reason of high water splitting activity of Ni-doped borophenes.
Conclusions
In summary, the catalytic performance of pristine, BCo-and Ni-doped borophenes (αB, β12B, and χ3B) for HER and OER was systematically investigated using first-principle calculations. Firstly, we examine the HER activities of αB, β12B, and χ3B, and there exist active sites with hydrogen adsorption-free energies close to zero in αB and χ3B, indicating that they own excellent HER performance. Then, with good structural stability and metallic conductivity, BCo- and Ni-doping are two effective ways to modulate the HER catalytic activities of different borophenes. BCo-doped αB shows superior neutral HER catalytic performance and BCo-doped β12B and χ3B could be used as highly active single-atom catalysts for HER. Meanwhile, we studied the OER catalytic activities of BCo- and Ni-doped borophenes in great detail. Ni-doped borophenes showed excellent catalytic abilities for HER and OER; in particular, Ni-doped αB is a promising HER/OER bifunctional electrocatalyst for water splitting with a low hydrogen adsorption free energy (0.055 eV) for HER and overpotential (0.455 V) for OER. Our results indicate that Ni-doped borophenes are expected to be economical and efficient candidates for HER/OER bifunctional electrocatalysts applied to water splitting.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by the Ministry of Higher Education of Malaysia through the Fundamental Research Grant Scheme (FRGS/1/2021/STG05/XMU/01/1), and the National Natural Science Foundation of China (NSFC) (No. 12274355).
References
- T. Burger, C. Sempere, B. Roy-Layinde and A. Lenert, Joule, 2020, 4, 1660–1680 CrossRef
 .
. - I. M. Peters, C. Breyer, S. A. Jaffer, S. Kurtz, T. Reindl, R. Sinton and M. Vetter, Joule, 2021, 5, 1353–1370 CrossRef .
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef .
- P. Veers, K. Dykes and E. Lantz, et al., Science, 2019, 366, 443 CrossRef PubMed .
- T. Sun, D. Wang, M. V. Mirkin, H. Cheng, J.-C. Zheng, R. M. Richards, F. Lin and H. L. Xin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11618 CrossRef CAS PubMed .
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed .
- J. C. Zheng, Front. Phys., 2008, 3, 269–279 CrossRef .
- J. C. Zheng, Research, 2022, 2022, 9867639 CAS .
- L. Han, H. Cheng and W. Liu, et al., Nat. Mater., 2022, 21, 681 CrossRef CAS PubMed .
- A. Alagumalai, L. Yang and Y. Ding, et al., Cell Rep. Phys. Sci., 2022, 3, 101007 CrossRef CAS .
- H. Xu, D. Cheng, D. Cao and X. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS .
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef .
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS .
- J. Wang, Y. Fan, S. Qi, W. Li and M. Zhao, J. Phys. Chem. C, 2020, 124, 9350–9359 CrossRef CAS .
- Z. Wang, T. Lü, H. Wang, Y. Feng and J. C. Zheng, Front. Phys., 2019, 14, 33403 CrossRef .
- A. J. Mannix, X. Zhou and B. Kiraly, et al., Science, 2015, 350, 1513–1516 CrossRef CAS PubMed .
- B. Mortazavi, O. Rahaman, A. Dianat and T. Rabczuk, Phys. Chem. Chem. Phys., 2016, 18, 27405–27413 RSC .
- G. I. Giannopoulos, Comput. Mater. Sci., 2017, 129, 304–310 CrossRef CAS .
- X. Zhou, X. Dong, A. R. Oganov, Q. Zhu, Y. Tian and H. Wang, Phys. Rev. Lett., 2014, 112, 085502 CrossRef .
- H. Zhong, K. Huang, G. Yu and S. Yuan, Phys. Rev. B, 2018, 98, 054104 CrossRef CAS .
- X. Sun, X. Liu, J. Yin, J. Yu, Y. Li, Y. Hang, X. Zhou, M. Yu, J. Li, G. Tai and W. Guo, Adv. Funct. Mater., 2017, 27, 1603300 CrossRef .
- Y. Chen, G. Yu, W. Chen, Y. Liu, G. Li, P. Zhu, Q. Tao, Q. Li, J. Liu, X. Shen, H. Li, X. Huang, D. Wang, T. Asefa and X. Zou, J. Am. Chem. Soc., 2017, 139, 12370–12373 CrossRef CAS PubMed .
- C. Liu, Z. Dai, J. Zhang, Y. Jin, D. Li and C. Sun, J. Phys. Chem. C, 2018, 122, 19051 CrossRef CAS .
- L. Shi, C. Ling, Y. Ouyang and J. Wang, Nanoscale, 2017, 9, 533 RSC .
- G. Tai, M. Xu, C. Hou, R. Liu, X. Liang and Z. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 60987–60994 CrossRef CAS PubMed .
- Z. Zhang, Y. Yang, G. Gao and B. I. Yakobson, Angew. Chem., 2015, 127, 13214–13218 CrossRef .
- S. H. Mir, S. Chakraborty, P. C. Jha, J. Wärna, H. Soni, P. K. Jha and R. Ahuja, Appl. Phys. Lett., 2016, 109, 053903 CrossRef .
- X. Wang, R. Wu, P. Tian, Y. Yan, Y. Gao and F. Xuan, J. Phys. Chem. C, 2021, 125, 16955–16962 CrossRef CAS .
- Y. Singh, S. Back and Y. Jung, Phys. Chem. Chem. Phys., 2018, 20, 21095 RSC .
- X. Xu, R. Si, Y. Dong, L. Li, M. Zhang, X. Wu, J. Zhang, K. Fu, Y. Guo and Y. He, J. Mol. Model., 2021, 27, 67 CrossRef CAS PubMed .
- M. Xu, X. Zhang, Y. Liu, X. Zhao, Y. Liu, R. Wu and J. Wang, ChemPhysChem, 2020, 21, 2651–2659 CrossRef CAS PubMed .
- E. S. Erakulan and T. Ranjit, Appl. Surf. Sci., 2022, 574, 151613 CrossRef .
- A. Banerjee, S. Chakraborty, N. K. Jena and R. Ahuja, ACS Appl. Energy Mater., 2018, 1, 3571–3576 CrossRef CAS .
- C. Ling, L. Shi, Y. Ouyang, X. Zeng and J. Wang, Nano Lett., 2017, 17, 5133–5139 CrossRef CAS PubMed .
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed .
- G. Kresse and D. Joubert, Phys. Rev., 1999, 59, 1758 CAS .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed .
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef .
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS .
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed .
- Q. Peng, J. Zhou, J. Chen, T. Zhang and Z. Sun, J. Mater. Chem. A, 2019, 7, 26062–26070 RSC .
- G. Gao, A. P. O'Mullane and A. J. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS .
- X. Zhang, A. Chen, Z. Zhang, M. Jiao and Z. Zhou, J. Mater. Chem. A, 2018, 6, 11446–11452 RSC .
- A. R. Oganov, J. Chen, C. Gatti, Y. Ma, Y. Ma, C. W. Glass, Z. Liu, T. Yu, O. O. Kurakevych and V. L. Solozhenko, Nature, 2009, 457, 863–867 CrossRef CAS PubMed .
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed .
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed .
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. D. Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS .
- Y. Yang, J. Liu, F. Liu, Z. Wang and D. Wu, J. Mater. Chem. A, 2021, 9, 2438–2447 RSC .
- T. Zhang, B. Zhang, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 433–441 RSC .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Jin-Cheng Zheng
a and
Jin-Cheng Zheng