DOI:
10.1039/D3RA00259D
(Paper)
RSC Adv., 2023,
13, 4924-4931
Unraveling the effect of solvents on the excited state dynamics of C540A by experimental and theoretical study†
Received
13th January 2023
, Accepted 23rd January 2023
First published on 8th February 2023
Abstract
In this work, the excited-state dynamics including intramolecular charge transfer (ICT) and the redshift of C540A have been investigated in a series of solvents on the basis of the Kamlet–Taft solvatochromic parameters (π*, α, β) using femtosecond transient absorption spectra and systematic theoretical calculation. We demonstrate that the redshift of the emission peak has a linear relationship with the α and π* scales and the effect of the π* scale is slightly stronger than that of the α scale. Meanwhile, the ICT rates can be suggested as relevant to not only the α scale but also the π* scale. Additionally, C540A-AN has proved that the excited state molecules have a unique inactivation mechanism because of the dark feature of the S1 (CT) state. The valuable mechanistic information gleaned from the excited-state dynamics by the experimental and theoretical study would facilitate the design of organic materials for prospective applications in photochemistry and photobiology.
1 Introduction
Excited-state intramolecular charge transfer (ICT) has attracted extensive attention for in-depth understanding of the fundamental deactivation processes and key mechanisms in photochemistry and photobiology.1–5 The ICT processes are sensitive to the intermolecular hydrogen bond interactions between solute and solvent molecules as an important external factor.6–9
To explore the influence of intermolecular hydrogen bonds on the excited-state dynamics,10–15 the Kamlet–Taft (K–T) solvatochromic parameters (π*, α, β) have been developed to depict the hydrogen bond interaction between solute and solvent molecules and to depict the spectral redshift in the steady-state absorption and fluorescence spectroscopy induced by solvents.16–19 For instance, Horng et al. studied the vertical excitation energies and fluorescence emission energies of coumarin 153 in about 40 solvents.20 The observed results reveal that both vertical excitation energies and fluorescence emission energies are associated with the polarity of simple non-aromatic aprotic solvents. Additional transition energy redshifts are produced in hydrogen bond-donating solvents (α > 0). Li et al. proposed that the intermolecular hydrogen bonding length, as well as the redshifts of the steady-state absorption peak and fluorescence emission peak, has a linear relationship with the solvent polarity function by studying the influence of solvent polarity on C500 and C500 hydrogen-bonded complexes.21 Li et al. confirmed that as the solvent's hydrogen-donating capacity (α) increases, the intermolecular hydrogen bonds in the excited state are strengthened, thereby promoting the ICT process.17 Moreover, tremendous efforts have been made to elucidate the influence of hydrogen bonds on the inactivation mechanism of the excited states. Venkatraman et al. demonstrated that the hydrogen-bonding interaction of protic solvent molecules can modify the rate of intersystem crossing (ISC) of benzophenone from its S1(nπ*) state to its triplet manifold of states using steady-state resonance Raman, femtosecond time-resolved electronic and vibrational absorption, and ultrafast Raman loss (URL) spectroscopic experiments combined with theoretical calculations.22 Han et al. proved that the excited state hydrogen bonds will be strengthened to varying degrees in many hydrogen-bonded systems through the femtosecond time-resolved spectroscopy23–27 and high-precision quantitative calculations. The hydrogen bond strengthening is crucial to explaining the fluorescence quenching phenomenon. Regarding the experimental study of fluorescence quenching of coumarin 102 by Barman et al.,28 they demonstrated that the fluorescence of coumarin 102 drastically quenches upon hydrogen bonding (H-bonding) with phenol in non-polar solvent cyclohexane due to H-bond driven photoinduced electron transfer (PET).29,30 Nevertheless, the excited-state dynamics influenced by the K–T parameters α, β, and π* of the solvents is yet to be developed in detail.
Herein, C540A has been chosen as a model system to study the excited-state dynamics in a series of solvents and it can be anticipated to occur ICT from the electron donor amino group to the electron acceptor carbonyl group. Also, C540A is a typical hydrogen-bond acceptor (HBA) molecule and the intermolecular hydrogen bond can be formed between the C540A molecule and the donor solvent such as Aniline (AN), methanol (MeOH), and formamide (FA). We look into the impact of the solvents on the excited-state dynamics through femtosecond transient absorption (fs-TA) spectra and the theoretical calculation by taking C540A-AN, C540A-MeOH, and C540A-FA as the contrasting models. The distinct spectral redshift and ICT process are observed as solvents vary. Therefore, the excited-state dynamics can be suggested as relevant to the K–T parameters. The hydrogen bond dynamics is investigated by the density functional theory (DFT) and time-dependent density functional theory (TDDFT). Firstly, the optimized geometric configurations of C540A in the three solvents in ground state (S0) and excited state are accomplished. Secondly, the electrostatic potential (ESP) has been analyzed on C540A, MeOH, FA, and AN molecules to determine the hydrogen bond formation sites. Based on the optimized geometric configurations, the hydrogen-bond binding energies of different complexes are calculated by atoms in molecules (AIM) theory. Besides, the reduced density gradient (RDG) analysis,31 infrared (IR) spectroscopy analysis, and molecular orbital analysis have been carefully performed to indicate the effects of solvent parameters α on the hydrogen bond interactions. Finally, the absorption and emission spectroscopy are calculated to determine the impact of the K–T parameters α and π* on the redshift of the electron spectra. ICT process has been demonstrated that it is related to the α and π* scale. In addition, a unique inactivation mechanism is proposed in C540A-AN complex.
2 Experiments
2.1 Experimental methods
The C540A sample is purchased from Exciton Inc (USA) and has not been further purified. The FA, MeOH, and AN solvents are spectrum pure quality reagents. The C540A was dissolved in FA, AN, and MeOH solvents with a concentration of 80 μM.
The femtosecond laser (Astrella, Coherent) is an integrated oscillator and regenerative amplifier laser system.24 The seed laser of the system generates 5 mJ pulses with a repetition rate of 1000 Hz, a spectral center of 800 nm, and a pulse duration of 35 fs. The 800 nm fundamental beam of the Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sapphire laser is split into two beams of 7:3. The stronger beam generates a 400 nm pumping pulse through an optical parametric amplifier (TOPAS-Prime, Coherent). The pump pulse is passed through a mechanical chopper to excite the sample at a repetition rate half that of the fundamental laser. The other beam, acting as a probe laser, is focused into a rotating CaF2 window with a thickness of 2 mm to produce a super-continuum white light of 350 to 750 nm. The optical delay line is used to vary the delay time between the pump pulse and the probe pulse from 1 fs to 4 ns. The pump and probe beams are incident at a small angle to the sample in a quartz cuvette with an optical path length of 1 mm. After passing through the sample, the probe beam is collimated and focused on the calibrated spectrometer (AvaSpec-ULS2048CL-EVO, Avantes) to record the temporal and spectrum of the transient absorption (TA) signal.25–27,32 All the above experiments were measured at room temperature.
:sapphire laser is split into two beams of 7:3. The stronger beam generates a 400 nm pumping pulse through an optical parametric amplifier (TOPAS-Prime, Coherent). The pump pulse is passed through a mechanical chopper to excite the sample at a repetition rate half that of the fundamental laser. The other beam, acting as a probe laser, is focused into a rotating CaF2 window with a thickness of 2 mm to produce a super-continuum white light of 350 to 750 nm. The optical delay line is used to vary the delay time between the pump pulse and the probe pulse from 1 fs to 4 ns. The pump and probe beams are incident at a small angle to the sample in a quartz cuvette with an optical path length of 1 mm. After passing through the sample, the probe beam is collimated and focused on the calibrated spectrometer (AvaSpec-ULS2048CL-EVO, Avantes) to record the temporal and spectrum of the transient absorption (TA) signal.25–27,32 All the above experiments were measured at room temperature.
2.2 Computational methods
The geometries of the C540A monomer and hydrogen-bonded complexes in ground and excited states are optimized by DFT and TDDFT methods.33–35 Through vibrational frequencies calculation and analysis, all vibrational frequencies have no imaginary frequency, so as to ensure that all optimized geometric configurations are in the local minima. Based on the optimized structures, we investigate electrostatic potential analysis,36–38 frontier molecular orbital, hydrogen bond binding energy, vertical excitation energy, fluorescence emission energy, and corresponding oscillator strength. The popular Becke's three-parameter hybrid exchange functional with Lee–Yang–Parr gradient-corrected correlation (B3LYP) in conjunction with the triple-ζ valence quality with one set of polarisation functions (TZVP) basis set are used throughout all the calculations.7,39 Considering the solvent effect, the integral equation formalism version of polarizable continuum model (IEFPCM) is suitable for hydrogen-bonded systems.40 The non-covalent bond interactions are analyzed by reducing the gradient density using the Multiwfn program.41 The non-covalent bond interactions are visually analyzed by the VMD program.42 In addition to the analysis above, all calculations in the present work are performed by Gaussian 09 package.
3 Results and discussion
3.1 The excited-state dynamics by transient absorption spectroscopy
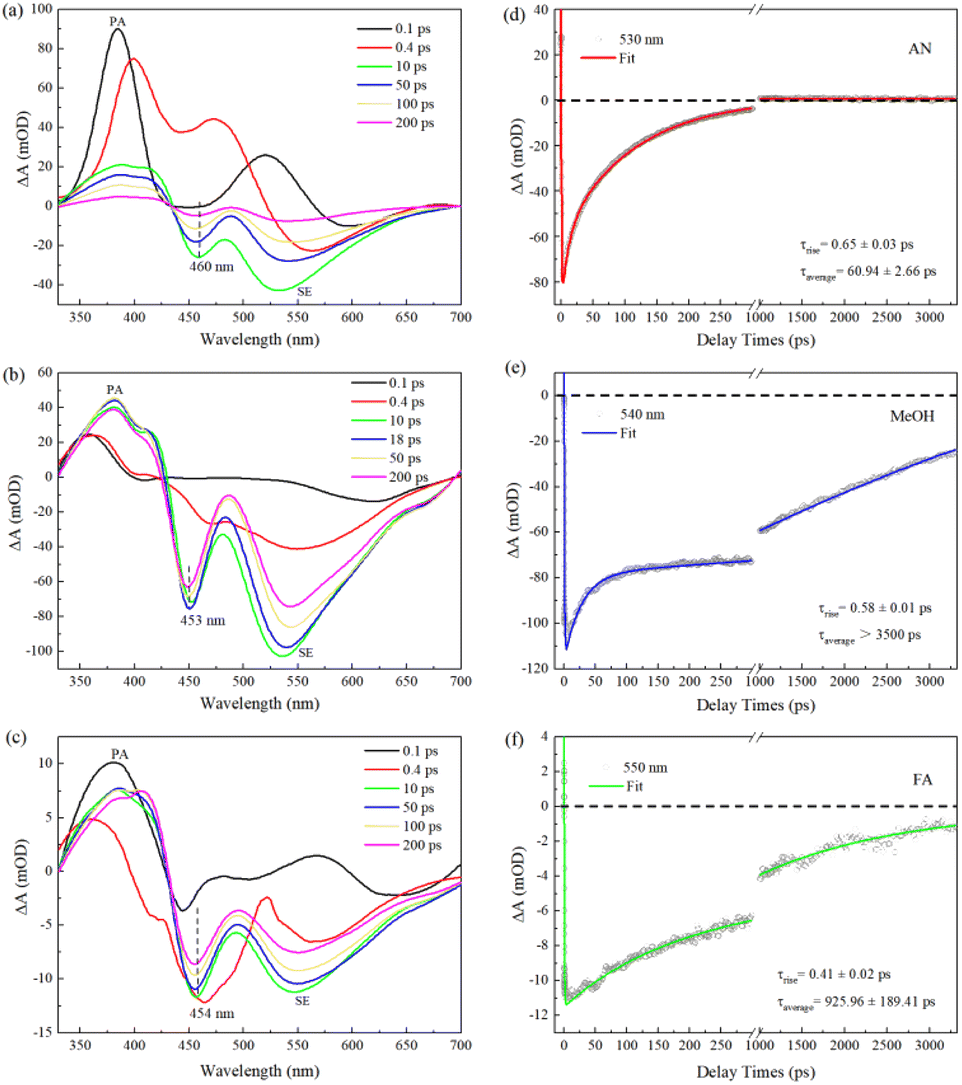
We carry out femtosecond transient absorption (fs-TA) spectroscopic characterizations to investigate the excited-state dynamics of C540A. The left panels a, b, and c of Fig. 1 display the TA spectra of C540A in AN, MeOH, and FA solvents with different polarities (π*) and different hydrogen-bond-donating capacity (α) at the specified delay times, respectively. The positive TA signals of 330–430 nm region can be assigned to the photoinduced absorption (PA), while the negative signals of lower energy bands peaking at 460, 453, and 454 nm marked by dashed black lines can be attributed to the ground state bleaching signals for C540A-AN, C540A-MeOH, and C540A-FA, respectively. And the negative TA signals of higher energy bands can be attributed to the stimulated emission (SE). As time delays, the feature peaks of SE gradually shift to 530 nm, 540 nm, and 550 nm within 200 ps for C540A in AN, MeOH, and FA, respectively, indicating the occurrence of ICT process. In addition, it is worth nothing that a discrepancy between the absorption peak displayed in Fig. S1† and the bleach peak can be seen by comparison, which is due to the coincidence of the PA signal. To gain more insights into the impact of hydrogen bonds on excited state dynamics, the decay kinetics monitored at maximum emission peaks are presented in the right panels d, e, and f of Fig. 1 for C540A-AN, C540A-MeOH, and C540A-FA complexes, respectively. For better presentation of all the relaxation process, the kinetics traces are shown with a break in the X-axis in Fig. 1d–f. An exponential rise time can be ascribed to the formation of intramolecular charge transfer (ICT) states and the lifetimes of ICT are ∼0.65, ∼0.58, and ∼0.41 ps, respectively, for C540A-AN, C540A-MeOH, and C540A-FA complexes. The decay processes are well fitted by a biexponential function which can be assigned to two different channels of radiative recombination and all the fitting parameters are displayed in Table S1.† The average lifetimes of radiative recombination are shown in Fig. 1: for C540A-AN complexes, τaverage = 60.94 ± 2.66 ps; for C540A-MeOH complexes, τaverage > 3500 ps; for C540A-FA complexes, τaverage = 925.96 ± 189.41 ps. Thus, according to the average lifetimes, we can deduce that the C540A-AN complexes have a unique inactivation mechanism. Moreover, the redshift wavelength increase gradually while the lifetime of ICT slow down in turn for C540A-AN, C540A-MeOH, and C540A-FA, respectively. This order doesn't correspond to neither the order of solvent's polarity nor the hydrogen-donating capacity which are shown in Table 1. Thus, it is obvious that either the redshift or the ICT rate is not just related to parameters α or π*.
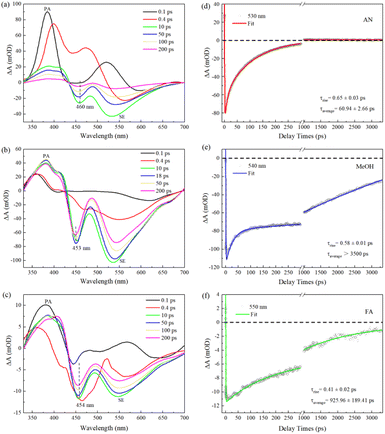 |
| | Fig. 1 The transient absorption spectra of C540A in AN (a), MeOH (b), and FA (c) solvents after excitation at 400 nm. The TA kinetic curves and fitting curves of C540A (d)–(f). | |
Table 1 The Kamlet–Taft (K–T) solvatochromic parameters (π*, α, β) of FA, MeOH, and AN
| Solvent |
π* |
α |
β |
| AN |
0.73 |
0.26 |
0.50 |
| MeOH |
0.60 |
0.98 |
0.66 |
| FA |
0.97 |
0.71 |
0.48 |
3.2 The effect of solvents on the ICT process and the spectral red shift in the complex systems by theoretical calculation
In order to further elucidate how the K–T parameters α and π* influence the excited state dynamics containing the ICT process and the spectral redshift in C540A-solvent hydrogen-bonded complexes, we systemically performed theoretical analysis based on DFT and TDDFT methods.
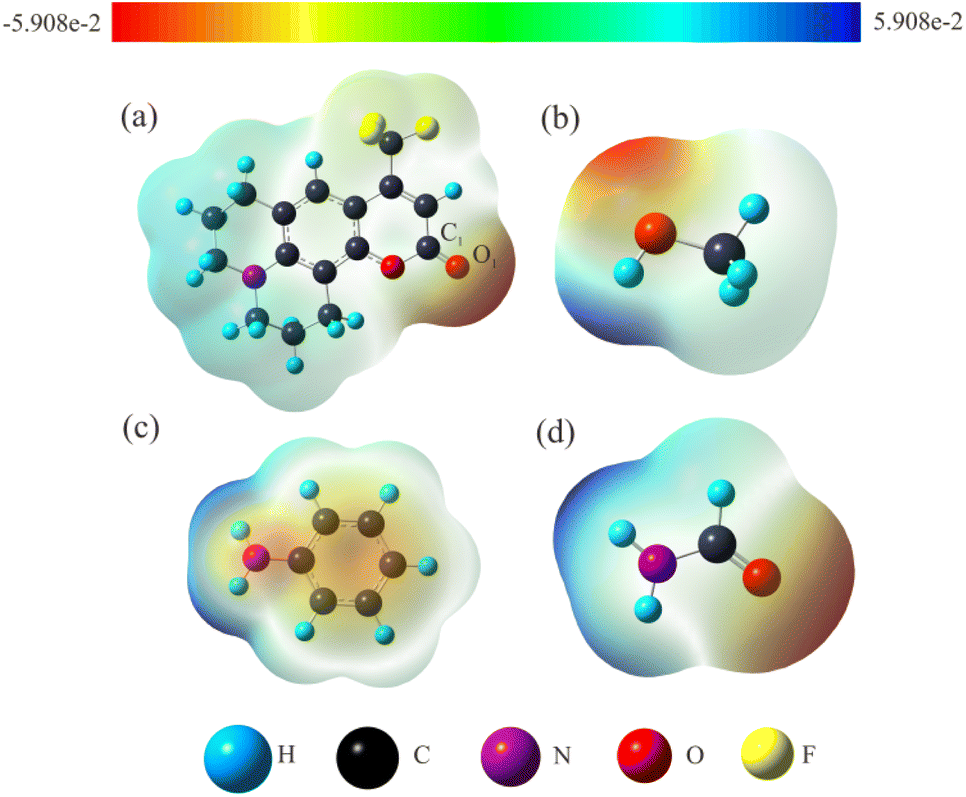
3.2.1 Electrostatic potential. The intermolecular hydrogen bonding interactions between solute and solvent molecules which can be as an external factor have noticeable effects on the ICT process. The electrostatic potential (ESP) can visually and intuitively display the charge distribution and electronegativity properties of molecular functional groups, and can also accurately predict the formation sites of hydrogen bonds. The ESP maps of C540A and solvent molecules were plotted in Fig. 2, respectively. The color bar in the ESP maps from red to blue represents the high electronegative area (nucleophilic) to the low electronegative area (electrophilic). The C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O1 in the ESP map of C540A appears bright red, which corresponds to the region with strong electronegativity. This result indicates that the C1O1 group of the C540A can interact with the electrophilic region. While, the O–H group of MeOH and the N–H groups of FA and AN are overlaid in blue, indicating that these groups are located in the electrophilic region. In summary, the H atom of the electrophilic group in the solvent can be functioned as a hydrogen-bond donor to form an intermolecular hydrogen bond with the hydrogen-bond acceptor CO group in C540A. This phenomenon further implies that intermolecular hydrogen bonding can form in MeOH, FA, and AN solvent, consistent with the α scale of the corresponding hydrogen-bond donor solvents.
O1 in the ESP map of C540A appears bright red, which corresponds to the region with strong electronegativity. This result indicates that the C1O1 group of the C540A can interact with the electrophilic region. While, the O–H group of MeOH and the N–H groups of FA and AN are overlaid in blue, indicating that these groups are located in the electrophilic region. In summary, the H atom of the electrophilic group in the solvent can be functioned as a hydrogen-bond donor to form an intermolecular hydrogen bond with the hydrogen-bond acceptor CO group in C540A. This phenomenon further implies that intermolecular hydrogen bonding can form in MeOH, FA, and AN solvent, consistent with the α scale of the corresponding hydrogen-bond donor solvents.
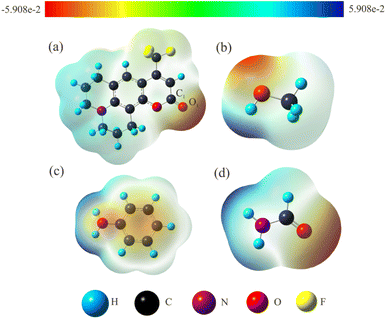 |
| | Fig. 2 The ESP maps of C540A (a), MeOH (b), AN (c), and FA (d) molecules. | |
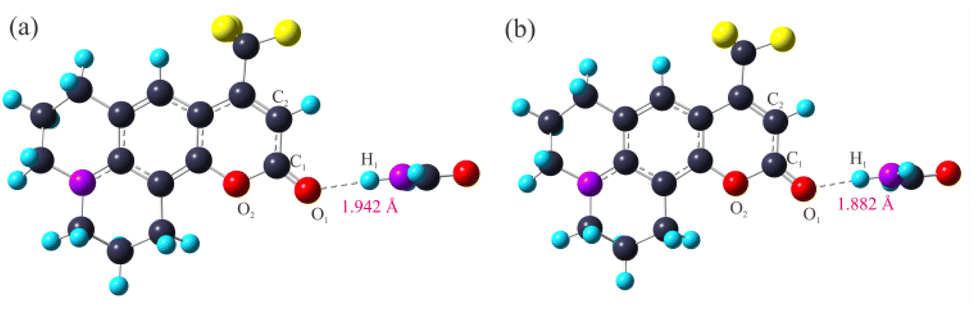
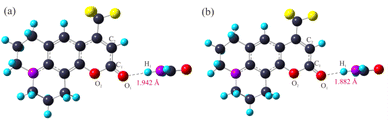
3.2.2 Optimized geometric configurations. The ground-state and excited-states geometrical configurations of the C540A monomer and the hydrogen bonding complexes have been optimized at the level of B3LYP/TZVP. The optimized geometric configurations of the C540A-FA complex as a representative in the ground state (S0) and the first excited states (S1) are presented in Fig. 3. The geometric configurations of the other two hydrogen-bonded complexes are also shown in Fig. S2 and S3.† And the critical geometric parameters are summarized in Table 2. Notably, as compared to the C1O1 bond length of the C540A monomer in the S0 state which is 1.203 Å, the C1O1 bond length in the C540A-MeOH, C540A-FA, and C540A-AN increased by 0.020, 0.019, and 0.015 Å, respectively, according to the formation of the intermolecular hydrogen bond C1O1⋯H1. Additionally, the C1–O2 and C1–C2 bonds of C540A molecules appeared to be a certain degree of change. Thus, the above results demonstrate that the formation of intermolecular hydrogen bonds affects the structure of the C540A molecule.
 |
| | Fig. 3 Optimized geometric structures for the C540A-FA complex in S0 (a) and S1 (b) at the B3LYP/TZVP/IEF-PCM (FA) level. | |
Table 2 Calculated important bond lengths (Å) of the isolated C540A and hydrogen-bonded complexes in three solvents
| Molecule |
C540A |
C540A-MeOH |
C540A-FA |
C540A-AN |
| S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
S0 |
S2 |
| C1–O1 |
1.203 |
1.213 |
1.223 |
1.240 |
1.222 |
1.239 |
1.218 |
1.233 |
| C1–O2 |
1.391 |
1.453 |
1.370 |
1.400 |
1.372 |
1.402 |
1.376 |
1.409 |
| C1–C2 |
1.450 |
1.419 |
1.435 |
1.414 |
1.435 |
1.414 |
1.439 |
1.417 |
| C1–O1···H2 |
— |
— |
1.859 |
1.805 |
1.942 |
1.882 |
2.063 |
2.002 |
Based on the optimized molecular structure in the ground state, the geometric configurations in the electron excited state of the C540A monomer and the hydrogen bonding complexes were further optimized at the level of B3LYP/TZVP. The relevant bond lengths are also listed in Table 2. Apparently, the hydrogen bonding lengths in the excited state are shortened to 1.805, 1.882, and 2.002 Å for C540A-MeOH, C540A-FA, and C540A-AN, respectively, compared with the lengths of the hydrogen bonds C1O1⋯H1 in the ground state which are 1.852, 1.942, and 2.063 Å, respectively. In addition, it is worth noting that the maximum oscillator strength of the C540A-AN corresponds to the transition to the second excited state (S2) state. Therefore, we focus on the S2 state of C540A-AN. Based on the analysis above, it can be demonstrated that the intermolecular hydrogen bonds were strengthened in the excited state.
Additionally, aiming to accurately study the change of intermolecular hydrogen bond strength, the hydrogen bonds were analyzed by AIM theory. The electron density (ρ) and its Laplacian (∇2ρ) at the critical point and the binding energy of the hydrogen bond are listed in Table 3. The electron density (ρ) can be used to describe the bond strength, and the greater the value of ρ is, the stronger the bond is. In the S0 state, the hydrogen-bond binding energies of the C540A-MeOH, C540A-FA, and C540A-AN are −24.9479, −19.4259, and −14.1056 kJ mol−1, respectively. However, upon photo-excitation to the first excited state, the hydrogen-bond binding energies increased to −29.1762, −23.3553, and −16.9593 kJ mol−1, respectively. The increase of the intermolecular hydrogen-bond binding energies in the excited state compared with the S0 state again intuitively confirms the fact that the intermolecular hydrogen bonds are strengthened in the excited state.
Table 3 The electron density (ρ), Laplacian of the electron density (∇2ρ) at the hydrogen bond critical point and the energy of the hydrogen bond (EHB)
| Molecule |
C540A-MeOH |
C540A-FA |
C540A-AN |
| S0 |
S1 |
S0 |
S1 |
S0 |
S2 |
| ρ(r) |
0.0301 |
0.0346 |
0.0241 |
0.0284 |
0.0184 |
0.0215 |
| ∇2ρ(r) |
0.1025 |
0.1106 |
0.0920 |
0.1016 |
0.0737 |
0.0830 |
| E/kJ mol−1 |
−24.95 |
−29.19 |
−19.43 |
−23.36 |
−14.11 |
−16.96 |
Based on the analysis above, a negative (positive) relationship between the α scales and hydrogen-bond lengths (hydrogen-bond binding energies) can be proposed for the C540A-MeOH, C540A-FA, and C540A-AN complexes. Thus, the α scales increase followed by the increase of the intermolecular hydrogen-bond strength, and the results are consistent with the study on LD490 by Li et al.17
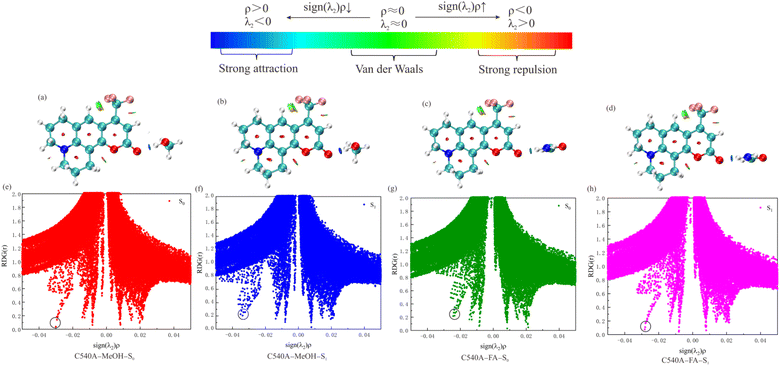
3.2.3 Non-covalent interaction (NCI) analysis. The analysis of the non-covalent bond interaction proposed by Johnson et al.31 can effectively help researchers to identify intermolecular hydrogen bond interactions. The analysis is based on normalized and dimensionless reduced density gradients:| |
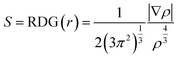 | (1) |
The ρ represents the total electron density, which has the following relationship with the eigenvalue λ2 in the Hessian matrix of electron density:
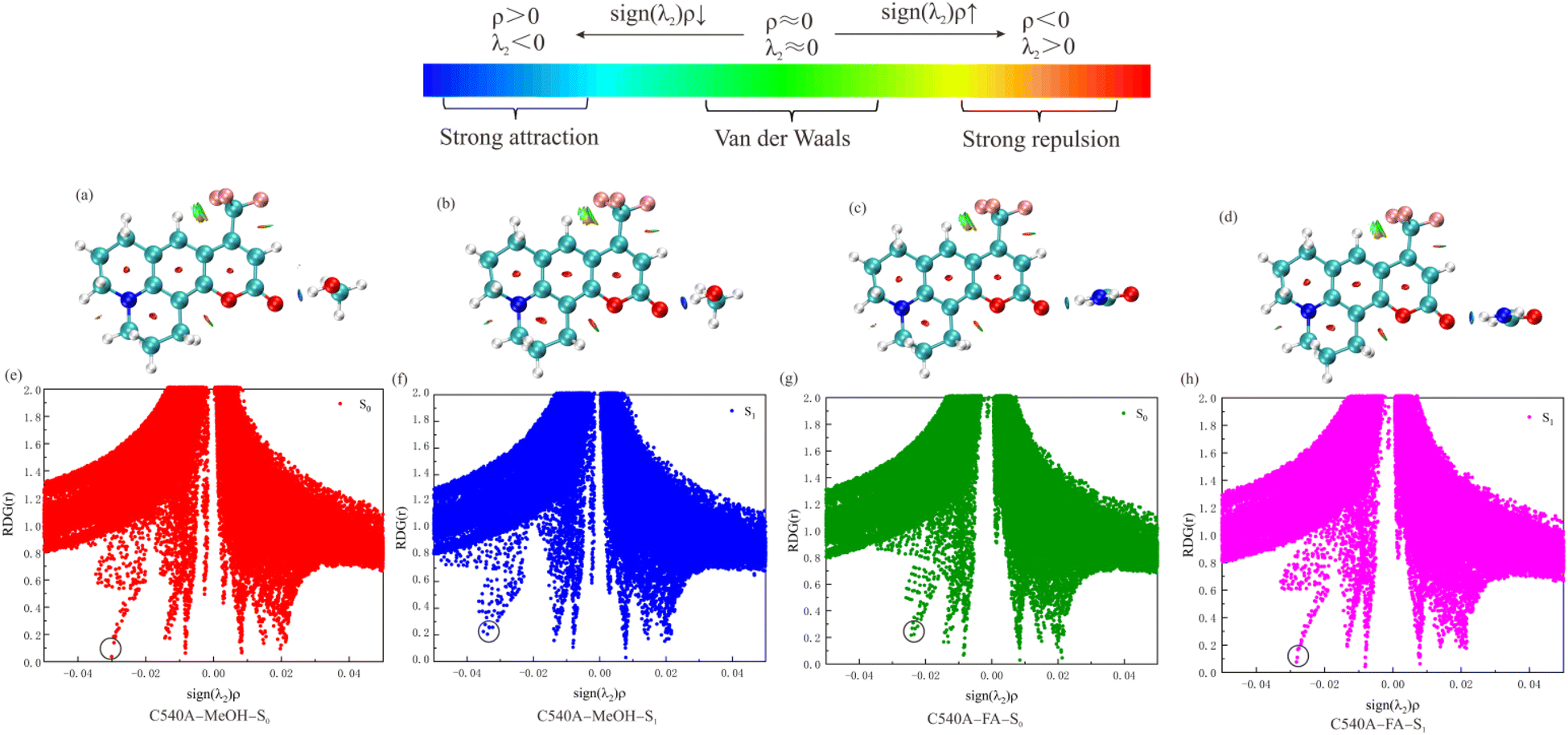
Different forms of the non-covalent bond interaction can be distinguished by the above formula sign(λ2)ρ. The non-covalent bond interaction analysis diagrams were drawn in Fig. 4. The upper panel shows the iso-surfaces and different colors represent different forms of interactions in this portion. The continuous color-coding scheme represents the value of sign(λ2)ρ. When sign(λ2)ρ < 0, it indicates that there is a mutually attractive interaction, the larger the negative values of sign(λ2)ρ, the deeper the blue in the iso-surface, the greater the interaction force, the most common interaction force is hydrogen bond. When sign(λ2)ρ ≈ 0, the green part represents weak interaction such as van der Waals interaction. When sign(λ2)ρ > 0, the red part generally corresponds to antibonding interactions, such as steric hindrance effects in rings. The blue interactions can be observed clearly from Fig. 4(a)–(d) between C540A monomer and MeOH or FA, so the interaction between them is the hydrogen bond interaction. As is evident from Fig. 4(a) and (b), the blue color of the hydrogen bond interaction points of the C540A-MeOH and the C540A-FA complexes in S1 state are both darker than that in S0 states, so it can be concluded that the hydrogen bonds are strengthened in S1 states. Moreover, the blue interaction region of the C540A-MeOH complex is deeper than that of the C540A-FA complex in both S0 and S1 states. Hence, the hydrogen bond interaction of the C540A-MeOH is stronger than that of the C540A-FA in both S0 and S1 states.
 |
| | Fig. 4 NCI isosurface of the C540A-MeOH complex in S0 (a), S1 state (b), and the C540A-FA complex in S0 (c), S1 state (d). Plots of sign(λ2)ρ versus RDG(r) for hydrogen-bonded complexes C540A-MeOH and C540A-FA in the S0 (e), (g), and S1 state (f) and (h), respectively. | |
Additionally, the bottom panel of Fig. 4 shows the scatter diagrams of the RDG(r) value versus the sign(λ2)ρ value for the C540A-MeOH and C540A-FA in S0 and S1 states. In Fig. 4(e) and (h), the circled position is the hydrogen bond C1O1⋯H1 spike. The C1O1⋯H1 spike value of the C540A-MeOH is −0.030 in the S0 state, and the C1O1⋯H1 spike value shifted to −0.035 upon photoexcitation. It indicates that the hydrogen bond is strengthened in the excited state. The same trend is observed in the C540A-FA complexes. Besides, the hydrogen bond spike value of the C540A-MeOH complexes in S0 and S1 states is more negative than that of the C540A-FA complexes. It turns out that the hydrogen bond of the C540A-MeOH complexes is stronger than that of the C540A-FA complexes in both S0 and S1 states. In addition, the non-covalent bond analysis of C540A-AN was also performed as shown in Fig. S4.† The non-covalent bond analysis is consistent with the configuration analysis result. It also proves the strength of the intermolecular hydrogen bonds increases gradually as the increase of α scales.
3.2.4 Infrared spectra (IR) analysis. Since the C1O1 group is a considerable group for forming hydrogen bonds in the complex systems, the stretching vibrational information of the C1O1 was calculated as shown in Table S2.† The vibrational frequencies of C1O1 for the C540A monomer, C540A-MeOH, C540A-FA, and C540A-AN complexes in S0 state are 1799, 1706, 1708, and 1726 cm−1, respectively. The vibrational frequencies of the C1O1 are red-shifted to 1791, 1702, 1704, and 1721 cm−1 after the photoexcitation. Compared with the C540A monomer, due to the formation of hydrogen bonds, the stretching vibrational frequencies of the C1O1 group for the complexes all have a significant red-shift phenomenon in the ground and the excited states. Thus, we propose that the hydrogen bonds are strengthened in the excited states according to the results above. The vibrational frequencies of the C1O1 group in the ground states of C540A-MeOH, C540A-FA, and C540A-AN are red-shifted by 93, 91, and 70 cm−1 compared to the C540A monomer. The red-shift order of the vibrational frequencies is consistent with the order of the solvent's α scales, which is also consistent with the configuration analysis, the non-covalent bond analysis results, and with the results of previous studies.17
3.2.5 Electronic excitation energy and electronic spectrum. The vertical excitation energy, fluorescence emission energy, and the corresponding oscillator strength are calculated and listed in Table 4 and 5. It demonstrates that the maximum oscillator strengths of C540A-MeOH and C540A-FA correspond to the transition to the S1 state. The S1 state is derived from the transition between HOMO and LUMO, and the transition contribution is greater than 95%. Especially, the maximum oscillator strength of the C540A-AN corresponds to the transition to the S2 state, and the S2 state of C540A-AN corresponds to the transition from the HOMO-1 to the LUMO. The vertical excitation energies and fluorescence emission energies of the complexes have a different degree of red-shift compared with the C540A monomer. This phenomenon demonstrates that the formation of the intermolecular hydrogen bond can effectively reduce the energy gap between the ground state and the excited state, which is in accordance with the research of Li et al.21 Interestingly, it is found that the S1 state of C540A-AN is a dark state, and its unique fluorescence quenching mechanism will be investigated below. The red-shifts of vertical excitation energy and fluorescence emission energy compared to the C540A are C540A-FA, C540A-MeOH, and C540A-AN in descending order, which coincide with the above experimental results.
Table 4 Calculated electronic excitation energies (nm) and corresponding oscillator strengths of the singlet excited states for the isolated C540A and hydrogen-bonded complexes by IEFPCM
| |
C540A |
C540A-FA |
C540A-MeOH |
C540A-AN |
| S1 |
379 (0.336) H → L 97.1% |
424 (0.469) H → L 98.0% |
422 (0.441) H → L 97.7% |
450 (0.005) H → L 98.7% |
| S2 |
323 (0.024) |
334 (0.050) |
334 (0.050) |
419 (0.504) H-1 → L 97.2% |
| S3 |
282 (0.000) |
285 (0.076) |
285 (0.072) |
332 (0.049) |
| S4 |
279 (0.040) |
274 (0.000) |
283 (0.002) |
299 (0.000) |
| S5 |
258 (0.068) |
263 (0.147) |
263 (0.139) |
284 (0.075) |
| S6 |
240 (0.046) |
262 (0.000) |
260 (0.000) |
236 (0.062) |
Table 5 Calculated fluorescence emission energy (nm) and corresponding oscillator strengths of the isolated C540A and hydrogen-bonded complexes by IEFPCM
| |
C540A |
C540A-FA |
C540A-MeOH |
C540A-AN |
| S1 |
425 (0.192) |
514 (0.508) |
512 (0.491) |
534 (0.003) |
| S2 |
379 (0.106) |
381 (0.098) |
381 (0.101) |
497 (0.442) |
| S3 |
297 (0.000) |
301 (0.000) |
314 (0.001) |
380 (0.101) |
Regarding the influence of the solvent parameters on electron transition energy, Taft et al.19 proposed that solutes such as coumarin, which accept hydrogen bonds in protic solvents but cannot provide hydrogen bonds. The observed electron transition energies for certain solutes are expected to follow this relationship:
where,
v represents the electronic transition energy in solvent and
v0 represents the electronic transition energy without considering any solvent effect (gas phase). The
α scale represents the ability of the solvent to provide hydrogen atoms to form intermolecular hydrogen bonds with the solute, and the
π* scale represents the polarity of the solvent. In
eqn (3),
a and
s are the constants properties of the solute, and their magnitude (sign) reflects the relative influence of the corresponding solvent–solute interactions on the electron transition energy. Previously, Li
et al. studied the effect of solvent
α scale on spectral redshift, but did not exclude the effect of solvent
π* scale.
17 Therefore, to obtain the relationship between electron transition energy and solvent based on the above discussion, the vertical excitation energies and fluorescence emission energies of the C540A in five solvents were calculated and the results are listed in
Table 6. In order to make the fitting results more accurate, the calculations of vertical excitation energies and fluorescence emission energies for the additional C540A-EtOH and C540A-DCM complexes are also provided. The vertical excitation and fluorescence emission energies of the complexes all have redshifts in the five solvents, and the red-shift can be expressed as (
v −
v0). It is that the red-shifts of vertical excitation energy and fluorescence emission energy had the following relationship with
α and
π* scales of the solvent (cm
−1):
| | |
v − v0 = −1635 − 621.4α − 679.8π*
| (4) |
Table 6 Redshift data (v − v0) of absorption spectrum (ABS) and emission spectrum (EM) were fitting by Gaussian and eqn (4)a
| Molecule |
Gaussian |
Fitting |
| ABS |
EM |
ABS/EM |
| Redshift data (v − v0) is as described in eqn (4) and are in units of 103 cm−1. The uncertainty in each value is estimated to be 154 cm−1. |
| C540A-MeOH |
−2.65 |
−2.65 |
−2.65 |
| C540A-FA |
−2.81 |
−2.70 |
−2.74 |
| C540A-AN |
−2.52 |
−2.03 |
−2.30 |
| C540A-EtOH |
−2.67 |
−2.60 |
−2.54 |
| C540A-DCM |
−2.33 |
−2.05 |
−2.27 |
In conclusion, the redshifts of the absorption spectra and the fluorescence emission spectra have a linear relationship with the α and π* scale of the solvents. Moreover, the effect of the polarity on the red-shift of the electron spectrum is slightly stronger than the hydrogen-donating ability.
3.2.6 Frontier molecular orbitals. The frontier molecular orbitals (MOs) analyses can visualize the ICT process and obtain the relationship between the ICT process and the solvents. The MOs analyses of C540A-MeOH, C540A-FA, and C540A-AN are depicted in Fig. 5. The S1 states of C540A-FA and C540A-MeOH correspond mainly to the transition from the highest occupied orbital (HOMO) to the lowest unoccupied orbital (LUMO). The S2 state of C540A-AN corresponds to the transition from the HOMO-1 to LUMO. It should be noted that the S1 states of C540A-FA and C540A-MeOH and the S2 state of C540A-AN show a typical ππ* feature. Furthermore, the electron densities are distributed on the C540A molecule, there is no charge distribution on the solvent molecules. Hence, the S1 states of C540A-FA and C540A-MeOH and the S2 state of the C540A-AN are locally excited (LE) states. For the HOMO orbital of C540A-FA and C540A-MeOH, as well as the HOMO-1 orbital of C540A-AN, the electron densities are mainly distributed near the N atom and the benzene ring. When the complexes are photoexcited, the electron densities of the LUMO orbital are redistributed, and most of the charge is transferred to the C1O1 group and trifluoromethyl group. The transfer and rearrangement of charge densities prove that the S1 states of C540A-FA and C540A-MeOH and the S2 state of the C540A-AN show significant ICT characteristics. Another significant feature to note here is that the electronegativity of the O1 atom in the C540A-MeOH increased by 0.037 C from −0.4289 C (S0) to −0.4659 C (S1). The same trend also exists in the other two complexes, and the electronegativity of the O1 atom is distinctively enhanced with an increase in the solvent's α acidity. It indicates that the strength of the intermolecular hydrogen bond increase with the increase of the α scales. However, it is worth noting that the relationship between the ICT process and α scales seems different with the previous study by Li et al., in which they suggest that the strengthened intermolecular hydrogen bond facilitates the ICT process as α increases. Herein, we demonstrate that the ICT processes accelerate gradually accompanying the increase of redshift for C540A-AN, C540A-MeOH, and C540A-FA in turn. Consequently, we can safely infer that the effect of the polarity of solvent on ICT process cannot be ignored, and efforts about the influence of the π* scale on ICT process will be made in our future study.
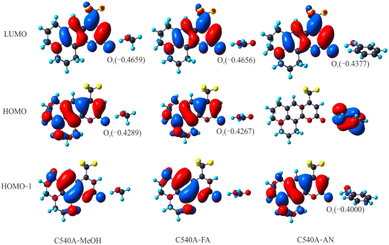 |
| | Fig. 5 Frontier molecular orbitals and electronegativity changes (Coulomb: C) in the hydrogen-bonded complexes C540A-MeOH, C540A-FA, and C540A-AN. | |
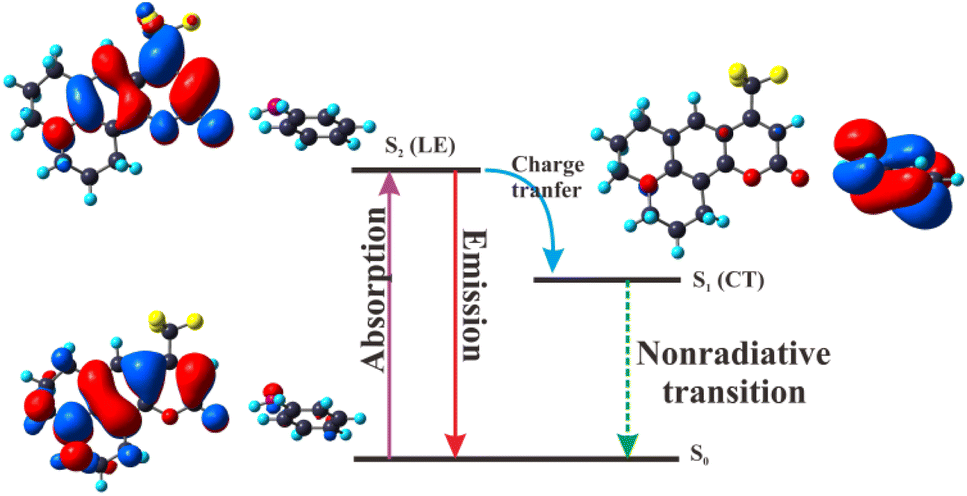
3.2.7 The mechanism of deactivation for C540A-AN complex. Based on the calculation of vertical excitation energy for the C540A-AN complex, it can be drawn that the oscillator strength of C540A-AN is the largest in S2 state. Thus, the C540A-AN complex can be directly excited to S2 state. Since the S1 state of the C540A-AN is an intramolecular charge transfer (CT) state which is a dark state and possesses extremely weak oscillator strength, indicating that the C540A has a unique excited state inactivation mechanism in Aniline solvent, and the mechanism diagram is shown in Fig. 6. The C540A-AN is excited to the S2 (LE) state and then relaxes to the S0 state radiatively or undergoes internal conversion (IC) to the S1 (CT) state. The nonradiative CT channel accelerates the radiative recombination of S2 state, accounting for the fastest recombination rate in these three complex systems. Moreover, the strengthened hydrogen bond of C540A-AN in S1 state can effectively promote the intramolecular charge transfer (CT), which induces the unique inactivation mechanism for the C540A-AN complex in accordance with the research on C500 by Li et al.21
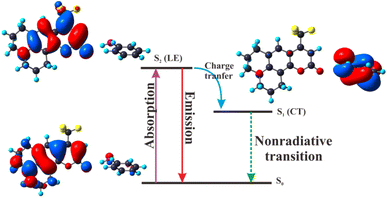 |
| | Fig. 6 Deactivation mechanism of the C540A-AN complex in the excited states. | |
4 Conclusions
We have investigated the excited state dynamics of C540A in AN, MeOH, and FA solvents by transient absorption spectroscopy and systematically performed a set of theoretical calculations to interrogate the effect of K–T parameters on excited state dynamics in the hydrogen-bonded complex systems. The different solvents in complex systems turn out to have a pronounced impact on the spectral redshift and ICT process. The redshift of the emission peak has a linear relationship with the α and π* scale of the solvents. And the effect of polarity on the redshift of the electron spectrum is slightly stronger than that of hydrogen donating ability. Meanwhile, the ICT rates can be suggested as relevant to not only the α scale but also the π* scale, which seems different from the previous study. In addition, a unique inactivation mechanism has been found in the C540A-AN complex due to the dark feature of the S1 (CT) state. This work provides in-depth insights into the photophysical properties of C540A in different solvents.
Author contributions
Jing Ge: conceptualization, formal analysis, supervision, writing – original draft, review & editing. Xue-Dong Zhang: methodology, data curation, software, visualization, writing – original draft. Yue Peng: methodology, investigation, visualization. Xi-Lin Bai: methodology, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Project supported by the National Natural Science Foundation of China (NSFC) (No. 21903050), Applied Basic Research Program in Shanxi Province (No. 20210302124124), Research Project Supported by Shanxi Scholarship Council of China Scientific (No. 2021-092), Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20220024), and Technological Innovation Programs of Higher Education Institutions in Shanxi (No. 2021L256).
Notes and references
- Y. A. Salawu, J. H. Yun, J. S. Rhyee, M. Sasaki and H. J. Kim, Sci. Rep., 2022, 12, 1–10 CrossRef PubMed.
- K. Sugimoto, T. Okubo, M. Maekawa and T. Kuroda-Sowa, Cryst. Growth Des., 2021, 21, 4178–4183 CrossRef CAS.
- Z. M. Wang, B. Cai, Y. J. Ren, W. H. Wang, L. K. Feng, S. L. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 19674–19681 CrossRef CAS PubMed.
- R. Zhang, S. H. Ma, Q. B. Wei, Q. Ye, B. Yu, J. Van Der Gucht and F. Zhou, Macromolecules, 2015, 48, 6186–6196 CrossRef CAS.
- H. Yamane and N. Kosugi, J. Phys. Chem. C, 2018, 122, 26472–26479 CrossRef CAS.
- P. Bajaj, J. O. Richardson and F. Paesani, Nat. Chem., 2019, 11, 367–374 CrossRef CAS PubMed.
- H. Wang, Y. F. Zhao, F. T. Zhang, Z. G. Ke, B. X. Han, J. F. Xiang, Z. P. Wang and Z. M. Liu, Sci. Adv., 2017, 7, 0396 Search PubMed.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami and H. Mori, Nat. Commun., 2013, 4, 1344–1346 CrossRef PubMed.
- R. K. Venkatraman and A. J. Orr-Ewing, J. Am. Chem. Soc., 2019, 141, 15222–15229 CrossRef CAS PubMed.
- H. Li, J. H. Han, H. F. Zhao, X. C. Liu, Y. Luo, Y. Shi, C. L. Liu, M. X. Jin and D. J. Ding, J. Phys. Chem. Lett., 2019, 10, 748–753 CrossRef CAS PubMed.
- C. Wang, Q. L. Qiao, W. J. Chi, J. Chen, W. J. Liu, D. Tan, S. McKechnie, D. Lyu, X. F. Jiang, W. Zhou, N. Xu, Q. S. Zhang, Z. C. Xu and X. G. Liu, Angew. Chem., Int. Ed., 2020, 59, 10160–10172 CrossRef CAS PubMed.
- F. A. S. Chipem, A. Mishra and G. Krishnamoorthy, Phys. Chem. Chem. Phys., 2012, 14, 8775–8790 RSC.
- Y. L. Cui, H. Zhao, J. F. Zhao, P. Y. Li, P. Song and L. X. Xia, New J. Chem., 2015, 39, 9910–9917 RSC.
- Y. Wang, G. Yang, M. Jia, X. Y. Song, Q. L. Zhang and D. P. Yang, J. Phys. Org. Chem., 2019, 32, 1–8 Search PubMed.
- X. Y. Liu, J. F. Zhao and Y. J. Zheng, RSC Adv., 2017, 7, 51318–51323 RSC.
- M. Kohantorabi, M. Fakhraee, H. Salari and M. R. Gholami, RSC Adv., 2016, 6, 18515–18524 RSC.
- H. Li, H. Yin, X. C. Liu, Y. Shi, M. X. Jin and D. J. Ding, Spectrochim. Acta, Part A, 2017, 184, 270–276 CrossRef CAS PubMed.
- R. S. Moog, W. W. Davis, S. G. Ostrowski and G. L. Wilson, Chem. Phys. Lett., 1999, 299, 265–271 CrossRef CAS.
- Y. Marcus, M. J. Kamlet and R. W. Taft, J. Phys. Chem., 1988, 92, 3613–3622 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- D. L. Li, C. Z. Li and Y. F. Liu, J. Mol. Liq., 2016, 218, 670–675 CrossRef CAS.
- R. K. Venkatraman, S. Kayal, A. Barak, A. J. Orr-Ewing and S. Umapathy, J. Phys. Chem. Lett., 2018, 9, 1642–1648 CrossRef CAS PubMed.
- D. P. Morgan and D. F. Kelley, J. Phys. Chem. C, 2020, 124, 8448–8455 CrossRef CAS.
- X. Z. Hua, L. Q. Hua and X. J. Liu, J. Phys. Chem. A, 2015, 119, 12985–12989 CrossRef CAS PubMed.
- L. Wang, S. Zhang, Y. Wang and B. Zhang, RSC Adv., 2020, 10, 18093–18098 RSC.
- J. Ge, Q. Zhang, J. Jiang, Z. Geng, S. Jiang, K. Fan, Z. Guo, J. Hu, Z. Chen, Y. Chen, X. Wang and Y. Luo, Phys. Chem. Chem. Phys., 2015, 17, 13129–13136 RSC.
- B. Gu and S. Mukamel, J. Phys. Chem. Lett., 2020, 11, 5555–5562 CrossRef CAS PubMed.
- N. Barman and K. Sahu, RSC Adv., 2014, 4, 58299–58306 RSC.
- G. J. Zhao and K. L. Han, J. Phys. Chem. A, 2007, 111, 9218–9223 CrossRef CAS PubMed.
- G. J. Zhao, J. Y. Liu, L. C. Zhou and K. L. Han, J. Phys. Chem. B, 2007, 111, 8940–8945 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. V. Lockard, A. B. Ricks, D. T. Co and M. R. Wasielewski, J. Phys. Chem. Lett., 2010, 1, 215–218 CrossRef CAS.
- D. P. Yang, Y. G. Yang and Y. F. Liu, Comput. Theor. Chem., 2012, 997, 42–48 CrossRef CAS.
- K. Bhattacharyya, Chem. Phys. Lett., 2021, 779, 138827 CrossRef CAS.
- X. H. Zhao, Y. F. Liu, L. C. Zhou, Y. Z. Li and M. D. Chen, J. Lumin., 2010, 130, 1431–1436 CrossRef CAS.
- P. A. Hunt, B. Kirchner and T. Welton, Chem.–Eur. J., 2006, 12, 6762–6775 CrossRef CAS PubMed.
- G. E. Jara, C. A. Solis, N. S. Gsponer, J. J. Torres, C. A. Glusko, C. M. Previtali, A. B. Pierini, D. M. A. Vera, C. A. Chesta and H. A. Montejano, Dyes Pigm., 2015, 112, 341–351 CrossRef CAS.
- D. P. Yang, Y. G. Yang and Y. F. Liu, Cent. Eur. J. Chem., 2013, 11, 171–179 CAS.
- L. Cong, H. Yin, Y. Shi, M. X. Jin and D. J. Ding, RSC Adv., 2015, 5, 1205–1212 RSC.
- H. Li, J. H. Han, H. F. Zhao, X. C. Liu, L. N. Ma, C. F. Sun, H. Yin and Y. Shi, J. Cluster Sci., 2018, 29, 585–592 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence