
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in synthesis and application of furoxan
Muhammad Abu Yousef
 and
Ryosuke Matsubara
*
and
Ryosuke Matsubara
*
Department of Chemistry, Graduate School of Science, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. E-mail: matsubara.ryosuke@people.kobe-u.ac.jp
First published on 9th February 2023
Abstract
This review highlights recent developments in the synthesis and application of furoxan. The chemistry of furoxan is relatively underdeveloped compared to that of other heterocycles owing to its difficult synthesis, which is ascribed to the labile nature of this molecule under various reaction conditions. Nevertheless, recent studies have conducted a variety of bond-forming reactions on the furoxan ring via a post-ring introduction of substituents (PRIS) strategy. This strategy enables the synthesis of furoxan molecules of interest more directly than the conventional methods that rely on the pre-installation of substituents on the furoxan ring precursors. In this review, the PRIS strategy for furoxan synthesis is classified and discussed according to the type of bond formed. Additionally, recent progress in the application of furoxan molecules, predominantly facilitated by the development of new synthetic methods, is covered in this review.
 Muhammad Abu Yousef | Muhammad Abu Yousef was born in Amman, Jordan. He received his BSc in chemistry from University of Jordan in 2016. In 2019, he joined the group of Professor Ryosuke Matsubara at the department of organic chemistry in Kobe University, Japan, where he obtained his MSc in chemistry in 2021. His research focused on design and synthesis of novel antitubercular drugs. |
 Ryosuke Matsubara | Ryosuke Matsubara was born in 1978 in Chiba, Japan. He received his PhD degree from the pharmaceutical department, the University of Tokyo in 2007 under supervision of Prof. Shu Kobayashi. He worked as an assistant professor in the pharmaceutical and science departments, the University of Tokyo from 2005 to 2009. He studied at the Massachusetts Institute of Technology as a visiting researcher from 2009 to 2011 under supervision of Prof. Timothy F. Jamison. In 2011, he was appointed as an associate professor at Kobe University and started his independent laboratory. His research interests include furoxan chemistry, photochemistry, and energy science. |
1. Introduction
Furoxan is the conventional name of 1,2,5-oxadiazole-2-oxide (Scheme 1a). It is characterized by an out-of-ring oxygen atom comprising N-oxide, which makes the furoxan ring asymmetric. Therefore, two regioisomers exist when the two substituents, R1 and R2, are not identical. Conventionally, the oxygen atom on the ring is numbered 1, the nitrogen atom with the out-of-ring oxygen atom is numbered 2, and other atoms are numbered 3,4, and 5 in order. | ||
| Scheme 1 Chemical structures of furoxan and its relatives. (a) Structure of furoxan, furazan, and benzofuroxan. (b) Wrongly proposed structures of furoxan. | ||
The history of furoxan has been well summarized in several review articles,1–3 and it is briefly described here. The first synthesis of furoxan dates back to the synthesis of dibromofuroxan (1) by Kekulé in 1857 (Scheme 2).4 It is noteworthy that he had synthesized furoxan before 1865, when he proposed the structure of benzene (Kekulé structure). However, Kekulé wrongly reported that he had synthesized dibromonitroacetonitrile (the same molecular formula as that of dibromofuroxan). Several other chemists have also proposed incorrect structures of furoxan (Scheme 1b). The controversy over the structure of furoxan continued for approximately 100 years until the 1960s, when NMR spectroscopy5 and X-ray crystallography6 confirmed the true structure. Reasons for this misassignment were the scarcity of spectroscopic techniques in those days, and difference in thoughts among chemists about the existence of isomers. When R1 and R2 are not identical, if the C2-asymmetric structure is correct, isomers should exist, whereas there should be no isomerism if the C2-symmetric structure is correct. However, some furoxan molecules undergo rapid isomerization, which makes the differentiation of isomers difficult (a typical example of such a molecule is benzofuroxan (Scheme 1a)). Additionally, even if two isomers were observed, some chemists insisted that they had different ring frameworks and were not regioisomers.1 The correct structure of furoxan was first proposed in 1903 by Wieland (Nobel Prize in Chemistry in 1927 for his work on bile acids),7 but he later abandoned the idea and proposed a different structure.1
 | ||
| Scheme 2 First synthesis of furoxan. | ||
The general properties of the furoxans are described. The furoxan ring moiety is an aromatic, 6π-electron system, but its aromatic stabilization energy is relatively small, partly due to the presence of multiple heteroatom–heteroatom bonds. Depending on the substituents, furoxan is generally stable in oxygen and water and can be purified by silica gel chromatography. Many furoxans with relatively low molecular weights are solid at ambient temperatures, which enables reliable structure determination via single-crystal X-ray diffraction analysis. In 13C NMR characterization of furoxans with common carbon substituents, the chemical shifts of the C3 and C4 carbons are generally around 115 and 160 ppm, respectively. The resonance of C3 carbon at a higher magnetic field than the C4 carbon is owing to mesomeric electron donation from the out-of-ring oxygen atom to the C3 carbon. This large difference in the chemical shifts is useful for the determination of isomer structures.
Each regioisomer of furoxans is stable at ambient temperatures, but most furoxans undergo isomerization at temperatures above 100 °C (Scheme 3). This thermal isomerization is proposed to proceed with dinitrosoalkene 2 as an intermediate.5,8,9 It is also known that irradiation with ultraviolet light causes isomerization, although there are only a few examples.10 The mechanism for photochemical isomerization remains to be clarified.
 | ||
| Scheme 3 Isomerization of furoxan. | ||
As with other heteroaromatic compounds, furoxan molecules have been reported to have various physiological activities, such as anti-cancer, antiviral, and antinematode activities.11 However, the ability to release nitric oxide (NO) makes furoxan unique and distinctive from other heteroaromatics. Not all furoxan molecules can release NO; in other words, a specific substituent pattern is required. In 1992, furoxan 3 was reported to have the NO-releasing ability,12 and in 1994, furoxan 4 was reported as another NO donor (Fig. 1).13 Neither furoxan 5, a regioisomer of 4, nor furazan 6 released NO. NO is released from furoxans only under physiological conditions (37 °C, aqueous solution) in the presence of excess thiol. Several mechanisms of NO release have been proposed,13–15 but no conclusion has been reached. One of the proposed mechanisms is illustrated here (Scheme 4).14
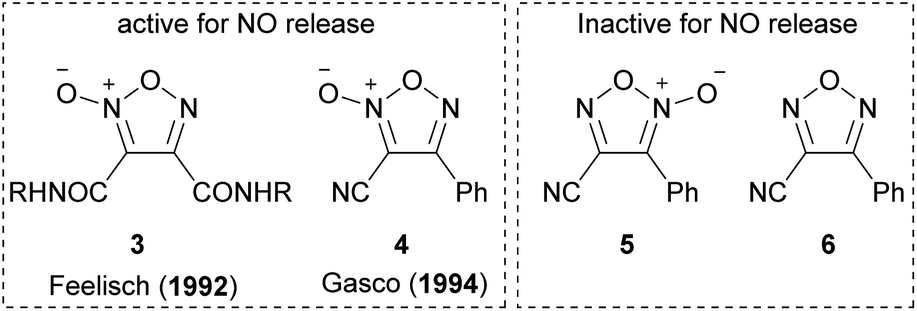 | ||
| Fig. 1 NO-releasing furoxans. | ||
 | ||
| Scheme 4 Proposed mechanism for the release of NO from furoxan. | ||
Despite more than 150 years of history, synthetic methods for furoxans are rather scarce than those for other aromatic molecules. One contributing factor to this scarcity, especially when compared to other N-oxide-containing heterocycles, is the fact that furoxans and benzofuroxans cannot be obtained through direct oxidation of parent systems, i.e., furazan and benzofurazan, respectively, and thus, require the development of independent synthetic methods. With regards to furoxan, there are five major methods for constructing furoxan rings (Scheme 5a). Methods i–iv require the preparation of elaborate precursors and lack generality. Additionally, method v, which uses readily available alkenes as starting materials, has significant restrictions on the tolerated substituents, and can only be applied to the synthesis of simple-structured furoxans. The synthesis of benzofuroxans is usually achieved through one of three oxidative routes (Scheme 5b). In method vi, benzofuroxans are obtained from intramolecular cyclization of 1-azido-2-nitrobenzenes triggered by thermo-16 or photochemical17 decomposition of the azide moiety. Method vii is the oxidative cyclization of N-hydroxy-2-nitroanilines treated with picryl chloride.18,19 This method provides a safer alternative to benzofuroxans by avoiding the use of the less safe azide substrates. The oxidative cyclization of 2-nitroanilines (method viii) is also a useful synthetic method for benzofuroxans.20,21
 | ||
| Scheme 5 Methods for the ring formation of furoxan (a) and benzofuroxan (b). | ||
There are only two positions (3- and 4-positions) on the furoxan ring which can be substituted; therefore, after a furoxan ring is constructed, if the desired substituents can be introduced at the 3- and 4-positions in sequence (this method is called post-ring introduction (PRI) of substituents or PRIS in this paper) at will, numerous furoxans can be synthesized. However, the vulnerability of the furoxan ring hampers this strategy. First, one may assume a chemical species in which a metal (such as Li or Mg) is introduced at the 3- or 4-position to make furoxan a nucleophile; however, to date, there is no example of the synthesis of this chemical species or even its application as a transient species. This is due to the rapid ring-opening reaction (Scheme 6). Even transition metals are not an exception; thus, cross-coupling reactions on the furoxan ring are currently unknown.
 | ||
| Scheme 6 Ring-opening reactions of metal-substituted furoxans. | ||
However, since furoxan has a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond in the ring and belongs to an electron-deficient aromatic ring, an aromatic nucleophilic substitution reaction (SNAr reaction) is an effective PRIS method (Scheme 7). Several PRIS methods for introducing oxygen or sulfur substituents, and a few for nitrogen substituents, have been reported. However, there are almost no reports on other nucleophiles.22
N double bond in the ring and belongs to an electron-deficient aromatic ring, an aromatic nucleophilic substitution reaction (SNAr reaction) is an effective PRIS method (Scheme 7). Several PRIS methods for introducing oxygen or sulfur substituents, and a few for nitrogen substituents, have been reported. However, there are almost no reports on other nucleophiles.22
 | ||
| Scheme 7 SNAr reactions of furoxans. | ||
This review paper discusses the recent developments in the synthesis of furoxans based on the PRIS strategy, classifying them according to the type of substituents to be introduced. Several new methods of substituent modification other than PRIS have also been developed in recent years,23 but are not included in this review. In addition to synthesis, several topics of applied research on furoxan molecules, most of which have been made possible based on the recent synthetic developments, will also be presented.
2. Furoxan synthesis based on PRIS
2.1. Leaving groups in SNAr reaction of furoxans
The leaving groups in the SNAr reaction of furoxans were almost exclusively nitro (–NO2) and arylsulfonyl (–SO2Ar) groups (Scheme 7). Halogen was also used as a leaving group, but very rarely. The primary reason for this limited number of leaving groups is the scarcity of known synthetic routes to substrates. Almost no examples of the synthesis of furoxans with halogen, carboxylate (–OCOR), and sulfonate (–OSO2R) groups, which are generally used as good leaving groups, are known. Nitrofuroxan, which can be readily synthesized by the reaction of terminal alkenes with N2O3,24,25 is the most commonly used substrate in the SNAr reaction. Arylsulfonylfuroxans can be readily synthesized by the oxidation of the corresponding arylsulfanylfuroxans.Generally, the leaving group is installed at 4-position and then subjected to the SNAr reaction, because the C4 carbon is more electron-deficient than the C3 carbon (vide supra). In most cases, furoxans with leaving groups at both the 3- and 4-positions selectively react with nucleophiles at the 4-position.
2.2. C–O bond formation
Alkoxyfuroxans or aryloxyfuroxans were obtained by the treatment of alkoxide or phenoxide nucleophiles with a furoxan having –NO2 or –SO2Ar as the leaving group (Scheme 8a). This reaction is the most commonly used method for introducing a furoxan ring into the molecular structure because the yields are generally high and reliable and starting materials are readily available. Since many review papers describe this method,1,2,11 this paper only cites seminal works,26–32 and does not discuss it in detail. When the leaving groups are at both the 3- and 4-positions, oxygen nucleophiles are often introduced to the C4 carbon with high selectivity if the reaction conditions are controlled. When a leaving group exists only at the 3-posion, the SNAr reaction at the 3-position could also proceed smoothly without ring opening. | ||
| Scheme 8 C–O bond formation on the furoxan ring. (a) C–O bond formation can occur at both the 3- and 4-positions. (b) The reaction of furoxans having leaving groups at both the 3- and 4-positions with oxygen nucleophiles is generally selective. | ||
Recently, not only NO2 and ArSO2, but also halogens (Cl, Br) have been reported to serve as leaving groups for the SNAr reaction with alkoxy nucleophiles (Scheme 9).32,33 Dichlorofuroxan and dibromofuroxan also underwent monoalkoxylation in moderate yields (Scheme 10).33 This reaction is a useful method for producing monohalofuroxans, a rare class of furoxans. Since there was an equilibrium in the alkoxylation reaction of dichlorofuroxan (Scheme 10a), adding more than a stoichiometric amount of silver salt after the completion of the substitution reaction was required to increase the yield, where silver salt traps the dissociated chloride. Depending on the type of alcohol used, 3- and 4-alkoxylated products were obtained as a mixture. Nevertheless, the formation of the 4-alkoxylated product was preferred, and its selectivity increased when an electron-deficient alcohol was used. With regard to the alkoxylation of dibromofuroxan, only the methoxide nucleophile afforded a good yield (Scheme 10b). The other alkoxides yielded adducts in poor yields (Scheme 10c), which was ascribed to a side reaction in which a dissociated bromide attacks the carbon at the α-position of the oxygen atom of the introduced alkoxy group, leading to the dissociation of the furoxanyl oxide anion (Scheme 10d).33
 | ||
| Scheme 9 C–O bond formation on the furoxan ring with halogens as a leaving group (chloride (a) and bromide (b)). | ||
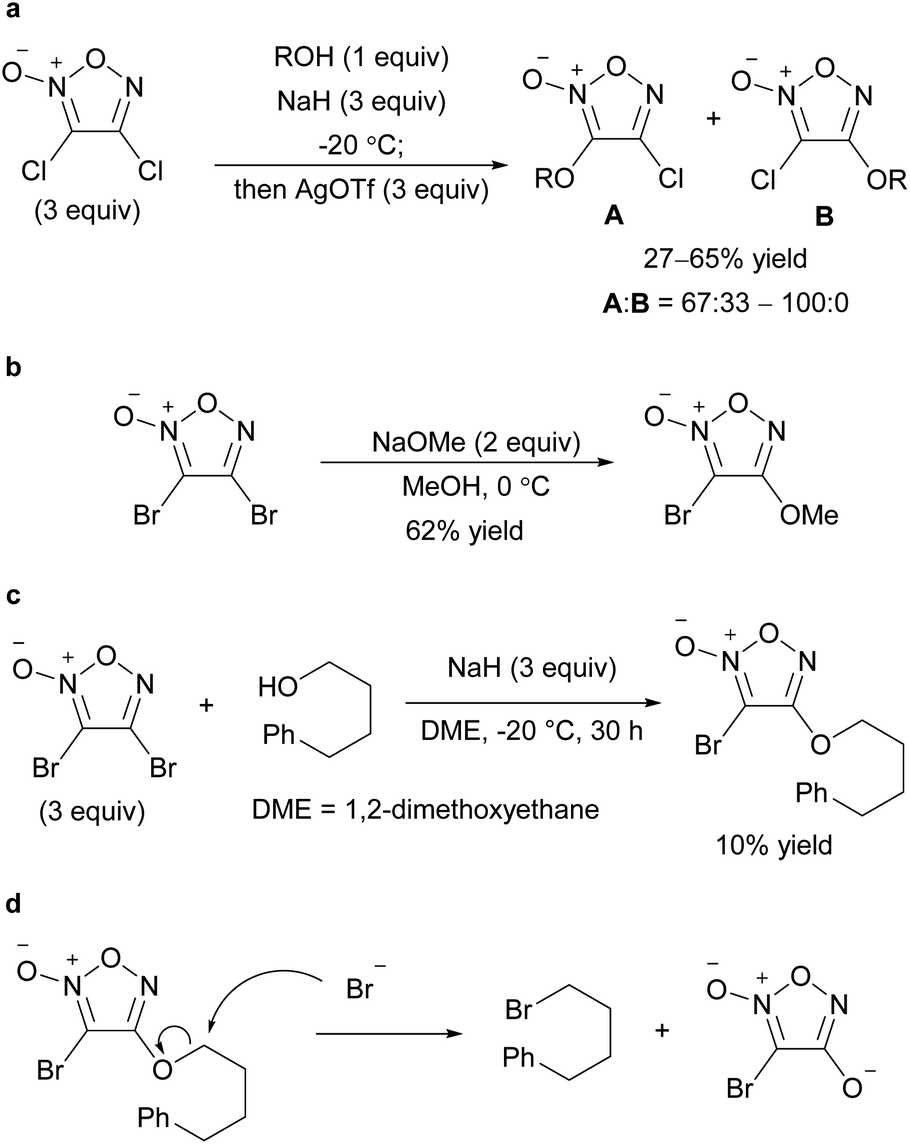 | ||
| Scheme 10 C–O bond formation on the furoxan ring with dihalofuroxans. (a–c) Alkoxylation of dichloro- and dibromofuroxan. (d) Side reaction in the alkoxylation of dibromofuroxan. | ||
Besides alcohol and phenol derivatives, hydroxy heteroarenes also reacted with 4-nitrofuroxans with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base to produce hetaryloxyfuroxans (Scheme 11).34 This class of molecules were not previously reported. Because both nitrofuroxans and hydroxy heteroarenes are readily available, this reaction is a practical method for preparing a new library of molecules.
 | ||
| Scheme 11 Reactions of 4-nitrofuroxans with hydroxy heteroarenes. | ||
The reaction of nitrofuroxan with the simplest oxygen nucleophile, OH−, yields hydroxyfuroxan. This reaction was reported by Wieland in 1903,35,36 but its generality had not been investigated, and several groups have pointed out its reproducibility issue.37,38 The reinvestigation revealed that the amount of hydroxide anion used and choice of solvent had a significant impact on the yield. Eventually, the optimal conditions to give a high yield of 4-hydroxyfuroxan were found (Scheme 12).37 The synthesis of 3-hydroxyfuroxan using the same method has not been reported.
 | ||
| Scheme 12 SNAr reaction of 4-nitrofuroxans with a hydroxide. | ||
When tetrabutylammonium hydroxide was used as an OH− nucleophile, 4-hydroxyfuroxan ammonium salts 7 were obtained in high yields (Scheme 13).38 Ammonium salt 7 was stable enough to be purified by silica gel chromatography. Alkyl and aryl groups were tolerated as substituents at the 3-position. Ammonium salts 7 were further transformed into 4-sulfonyloxy furoxan 8 by reaction with sulfonic acid anhydrides. The use of sulfonyl chlorides instead of sulfonic acid anhydrides reduced the yield, and a significant amount of nitrile compound was formed as a byproduct. This side reaction was due to the furoxan ring opening caused by a reaction of product 8 with Bu4NCl, a co-product, under neat or concentrated conditions.
 | ||
| Scheme 13 Synthesis and application of 4-hydroxyfuroxan ammonium salts. | ||
2.3. C–S bond formation
Similar to the C–O bond formation, PRI methods for sulfur substituents using a thiolate nucleophile have been commonly reported (Scheme 14a), and only seminal works are cited here.10,39–42 Most of the reported reactions for C–S bond formation also used NO2 and ArSO2 groups as leaving groups, and substitution could occur at both the 3- and 4-positions. The reaction was not selective when disulfonyl furoxan 9a was subjected to a thiolate nucleophile (Scheme 14b), in contrast to the reaction with an alkoxide nucleophile;39 this is presumably because thiolate is more nucleophilic than an alkoxide and is non-selective. | ||
| Scheme 14 C–S bond formation on the furoxan ring. (a) C–S bond formation can occur at both the 3- and 4-positions. (b) Reaction of disulfonyl furoxan 9a with sulfur nucleophile is not selective. | ||
The reaction of hetarylthiols with 4-nitrofuroxan to form C–S bonds on the furoxan ring has recently been reported (Scheme 15a).34,43 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was the base of choice. Due to the wide availability of substrate 4-nitrofuroxans,25 a compound library of hetarylsulfanyl furoxans was readily generated using this method. 4-Nitrofuroxans were exclusively employed as substrates whereas 3-mononitrofuroxan was not investigated. When dinitrofuroxan was used, the expected target 10 was obtained in low yield, and disulfide 11 was the main product (Scheme 15b). Dinitrofuroxan served as the oxidant in this reaction. Because the same byproduct was obtained when 4-nitrofuroxan with a carbonyl substituent at the 3-position was used as the substrate, an electron-deficient 4-nitrofuroxan prioritizes its function as an oxidant.
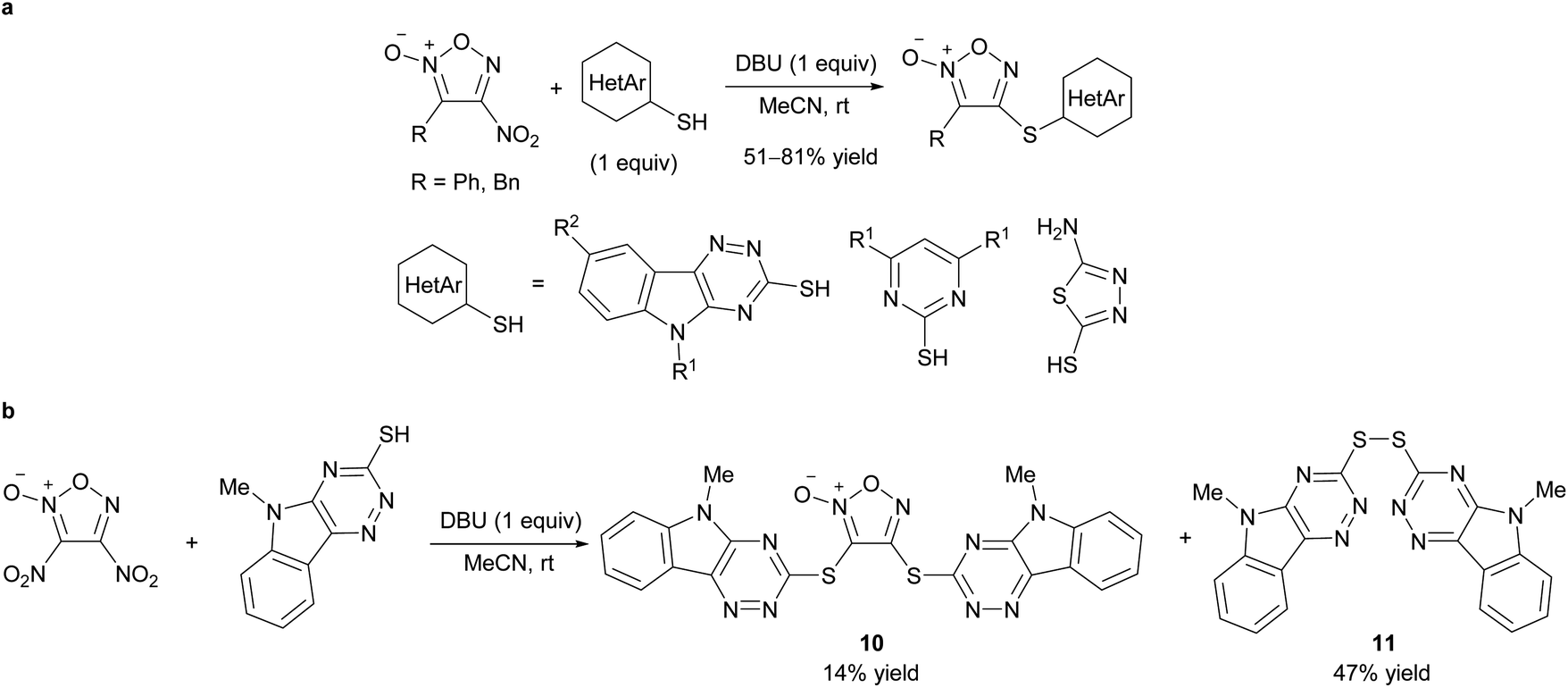 | ||
| Scheme 15 Reactions of 4-nitrofuroxans with hetarylthiols. (a) Synthetic scheme. (b) Attempt on sulfanylation of dinitrofuroxan led to oxidation of sulfur nucleophile. | ||
2.4. C–N bond formation
Before the development of a method to reduce a nitro group to an amino group using SnCl2 from readily available nitrofuroxanes,24,25 which opened the way for the facile synthesis of aminofuroxans,44 furoxans with a nitrogen substituent other than a nitro group were not well known. Compared to the C–O and C–S bond-forming reactions, a limited number of PRI methods for nitrogen substituents have been reported so far (Scheme 16).28,30,45–48 | ||
| Scheme 16 C–N bond formation on the furoxan ring. | ||
One reason for the scarcity of C–N bond-forming reactions on the furoxan ring by a nitrogen nucleophile is that ring-opening side reactions caused by a nitrogen nucleophile proceed readily. For example, 3-chloro-4-nitrofuroxan (12) underwent ring opening when primary and secondary aliphatic amines were used as nucleophiles. By using trimethylsilyl derivatives of amines, the expected adducts 13 were obtained without ring opening (Scheme 17a).49 The same strategy was applied to azidation (Scheme 17b),50 where fluoride salt was added to activate the nucleophile.
 | ||
| Scheme 17 C–N bond formation on the furoxan ring using trimethylsilyl derivatives of amine nucleophiles. (a) Amination. (b) Azidation. | ||
As a PRI method of amide substituents, the intramolecular SNAr reaction (Smiles rearrangement) was successfully applied (Scheme 18).41 Substrate 4-sulfonylfuroxan 14, synthesized by the PRI of the sulfanyl group followed by oxidation with H2O2 and CF3CO2H, underwent Smiles rearrangement upon treatment with NaOH in acetone to afford N-furoxanyl amide 15. Acid treatment of 15 with 1 M HCl aq. caused the elimination of SO2 and provided acetamide derivative 16 in 40% yield.
 | ||
| Scheme 18 Smiles rearrangement for PRI of the nitrogen group. | ||
With the same concept as the Smiles rearrangement, where a nitrogen atom attacks the furoxan ring intramolecularly, Schmidt and Curtius rearrangements involving a sextet rearrangement at the nitrogen atom have been applied to PRIS.51–55 A representative example is shown here (Scheme 19).55 When azidocarbonyl furoxan 17 was heated in aqueous dioxane, the Curtius rearrangement, in which the furoxanyl entity transferred onto the nitrogen atom, occurred to form isocyanate 19. Subsequently, 19 was hydrolyzed to 3-aminofuroxan 20. Because its isomer, 4-aminofuroxan 18, was thermodynamically more stable than 20, the reaction gave a mixture of 18 and 20, which converged to 18 by heating in toluene at 110 °C. This type of sextet rearrangement at the nitrogen atom occurred at both the 3- and 4-positions, although the reaction rates differed.53,54
 | ||
| Scheme 19 Curtius rearrangement for the PRI of the nitrogen group. | ||
2.5. C–C bond formation
 | ||
| Scheme 20 The first C–C bond-forming reactions on the furoxan ring, reported in 2005. | ||
 | ||
| Scheme 21 Alkynylation on furoxan ring. (a) General scheme. (b) Various compounds can be synthesized by derivatization of alkynylfuroxan 21. (c) Failed attempt to alkylate furoxan. Fib = fibroin, DDQ = 2,3-dichloro-5,6-dicyano-p-benzoquinone. | ||
The reaction of alkynyl lithium reagent with dichlorofuroxan provided 4-chloro-3-alkynylfuroxan 25, a rare class of chlorofuroxans (Scheme 22a).33 In contrast to most of the SNAr reactions (such as alkoxylation) of furoxans with two leaving groups, which generally undergo substitution reaction at the 4-position, this alkynylation reaction proceeded selectively at the 3-position. This selectivity was proposed to originate from the coordination of the metal atom of the nucleophile with the out-of-ring oxygen atom of the furoxan at the 2-position, which serves as a directing group. This reaction also yielded small amounts of chlorofurazans 26 and 27 as byproducts. These byproducts were generated by metal–halogen exchange followed by the [3 + 2] cycloaddition of 28 leading to 1,2-oxazole 29, which then converted to 26 and 27 (Scheme 22b).
 | ||
| Scheme 22 Alkynylation of dichlorofuroxan leading to the 4-chlorofuroxan. (a) General scheme. (b) Proposed mechanism for the formation of byproducts 26 and 27. | ||
 | ||
| Scheme 23 Cyanation of furoxans. (a) Using a stoichiometric amount of Bu4NCN afforded moderate yield of product 31 owing to the backward reaction during the aqueous workup. (b) Catalytic use of Bu4NCN successfully afforded 31 in high yield. | ||
4-Cyanofuroxan 31 has minimal NO-releasing ability, while its regioisomer 3-cyanofuroxan 33 is known to be a good NO donor.13 Therefore, the isomerization of 31 to 33 was studied (Scheme 24).56 Upon heating 31 at 110 °C in toluene, thermal equilibrium was reached within a few hours, and the ratio of the compounds at equilibrium was 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33 = 35:65. When compound 31 was exposed to ultraviolet (UV) light at room temperature in deuterated benzene, the photostationary state was reached in approximately 30 min with the ratio of 31:33 = 3:97. Given that NO donor 3-cyanofuroxans had previously been synthesized from the corresponding hydroxymethylfuroxans in a multi-step process, the PRIS method for cyano group installation is a useful alternative. The PRI of cyano groups to arylsulfonyl furoxans using 18-crown-6 as a catalyst has also been reported by different authors.57
:33 = 35:65. When compound 31 was exposed to ultraviolet (UV) light at room temperature in deuterated benzene, the photostationary state was reached in approximately 30 min with the ratio of 31:33 = 3:97. Given that NO donor 3-cyanofuroxans had previously been synthesized from the corresponding hydroxymethylfuroxans in a multi-step process, the PRIS method for cyano group installation is a useful alternative. The PRI of cyano groups to arylsulfonyl furoxans using 18-crown-6 as a catalyst has also been reported by different authors.57
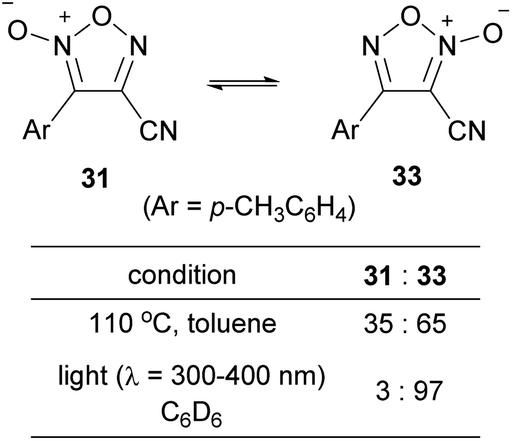 | ||
| Scheme 24 Isomerization of cyanofuroxans. | ||
 | ||
| Scheme 25 Alkylation of furoxans via a radical pathway. (a) General scheme. (b) Proposed reaction mechanism. | ||
Radical addition proceeded not only with disulfonyl furoxans but also with 3-sulfonylfuroxans bearing various substituents, such as alkoxy, sulfanyl, and carbon substituents, at the 4-position. Therefore, using this methodology, a variety of furoxans can be synthesized in a modular fashion in combination with SNAr reactions (Scheme 26).
 | ||
| Scheme 26 Modular synthesis of carbon-substituted furoxans by utilizing a radical reaction. (a) AgNO3 (20 mol%), carboxylic acid (2 equiv.), 50–90 °C, up to 78% yield; (b) toluene, 140 °C, 33% yield; (c) BuLi (1.1 equiv.), oct-1-yne (1.3 equiv.), 0 °C, 44% yield; (d) CsF (2.5 equiv.), Me3SiCF3 (2 equiv.), −20 °C, 53% yield; (e) RXH, base, SNAr reaction, up to quant. | ||
Density functional theory (DFT) calculations were performed to rationalize the 3-position selectivity of radical addition (Scheme 27). The activation energy for the addition to the 3-position was calculated to be 13.3 kcal mol−1, less than that for the 4-position (18.6 kcal mol−1), which was consistent with the experimental results. There was a large difference in the thermodynamic stability of the intermediates 35 and 37 of each reaction (−37.2 vs. −22.1 kcal mol−1, respectively), and this difference was reflected in the transition states. Intermediate 35 is in resonance with nitroxyl radical 36. The nitroxyl radicals (R1R2NO˙), represented by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), are known to be stable radicals, which were proposed to be the origin of the stability of 35.
 | ||
| Scheme 27 Rationalization of the selectivity for the radical addition at the 3-position over 4-position. | ||
Carbon radicals generated from C–H bonds instead of carboxylic acids could be added to the furoxans (Scheme 28a).60 This reaction proceeded only with persulfate salt, and a silver catalyst was not required. Simple hydrocarbon compounds, as well as molecules with C–H bonds at the benzyl position or at the α-position of the oxygen atom, could be used as coupling partners of furoxan. This reaction was proposed to proceed via hydrogen atom abstraction of C–H bonds by SO4˙– generated by the thermal decomposition of persulfate (Scheme 28b).
 | ||
| Scheme 28 Reactions of 3-sulfonylfuroxans with carbon radicals generated from abundant C–H bonds. (a) General scheme. (b) Proposed reaction mechanism. | ||
The PRI of the carbon substituents using carbon radical reagents also proceeded with bromofuroxans, where the bromo radical served as a radical-leaving group (Scheme 29a).33 In this reaction, the radical addition selectively occurred at the 3-position. In addition to 3-monoboromo furoxans, dibromofuroxan (1) could serve as a substrate that reacts with carbon radicals to provide 4-bromofuroxans, a rare class of molecules. In contrast to the previous case where the arylsulfonyl radical was the leaving group, this reaction required more than a stoichiometric amount of AgNO3 and base. The proposed reaction mechanism is illustrated in Scheme 29b. The Ag(II) species is generated from persulfate and AgNO3. Ag(II) and carboxylic acid form complex 38, which undergoes decarboxylation to provide a carbon radical (R˙) and Ag(I). The carbon radical reacts with 3-bromofuroxan 39, affording product 40 with the liberation of bromo radical. A catalytically inactive insoluble AgBr is eventually formed during this process; therefore, stoichiometric AgNO3 is necessary. The base traps HBr and HNO3, which are generated as coproducts.
 | ||
| Scheme 29 Reaction of 3-bromofuroxans with carbon radicals. (a) General scheme. (b) Proposed reaction mechanism. | ||
 | ||
| Scheme 30 Reaction of 3-sulfonylfuroxan with aryl radicals generated from potassium aryltrifluoroborates. | ||
Arylfuroxans were obtained by the polar reaction of Grignard reagents with chlorofuroxans as a substrate (Scheme 31).33 The reaction proceeded selectively at the 3-position, which was attributed to the directing effect of the out-of-ring oxygen atom, as observed in the alkynylation of furoxan (Scheme 22). Using dichlorofuroxan as the substrate, 3-aryl-4-chlorofuroxans were obtained; thus, this reaction enables easy access to chlorofuroxans, a rare class of molecules.
 | ||
| Scheme 31 Arylation of 3-chlorofuroxans. | ||
2.6. C–B bond formation
Introducing an electropositive metal element on the furoxan ring causes fast ring opening (Scheme 6). Therefore, no furoxans substituted with an element more electropositive than a hydrogen atom have been reported to date. However, it was surmised that ring decomposition might not occur in furoxans substituted with a metalloidal atom that can form a relatively strong covalent bond with carbon atoms. Indeed, 3-borylfuroxans 43 were isolated as stable compounds (Scheme 32a).62 They were synthesized by the reaction of 3-sulfonylfuroxan 41 with N-heterocyclic carbene-ligated borane 42 in the presence of 2,2-azobis(isobutyronitrile) (AIBN). Photoirradiation of 3-borylfuroxan induces isomerization to 4-borylfuroxan; thus, both regioisomers of borylfuroxan have become accessible. The proposed mechanism is shown in Scheme 32b. Boryl radical 44 is generated from 42 via hydrogen atom abstraction.63,64 Boryl radical 44 is nucleophilic, similar to the carbon radical; therefore, it can react with 3-sulfonylfuroxan 41 and provide intermediate 45. 3-Borylfuroxan product 43 is obtained after the dissociation of arylsulfonyl radical 46 from 45. Because the overall reaction proceeded with a catalytic amount of AIBN, arylsulfonyl radical 46 is proposed to abstract a hydrogen atom from the next substrate 42. | ||
| Scheme 32 Synthesis of borylfuroxans. (a) Representative example. (b) Proposed reaction mechanism. | ||
After the first synthesis of borylfuroxans, their reactivities were studied (Scheme 33).62 Hydrogenation of 3-borylfuroxan 47 catalyzed by Pd/C afforded boryloxime 48 and its derivative 49. This is the first report of the synthesis of boryldioxime. The hydrogenation of 3-borylfuroxan 43 with the alkoxide group on the furoxan ring also provided the boryldioxime derivative 50, though it required harsher conditions due to the electron-donating effect of ethoxy group. Currently, the cross-coupling reaction of the synthesized borylfuroxans has not been successful.
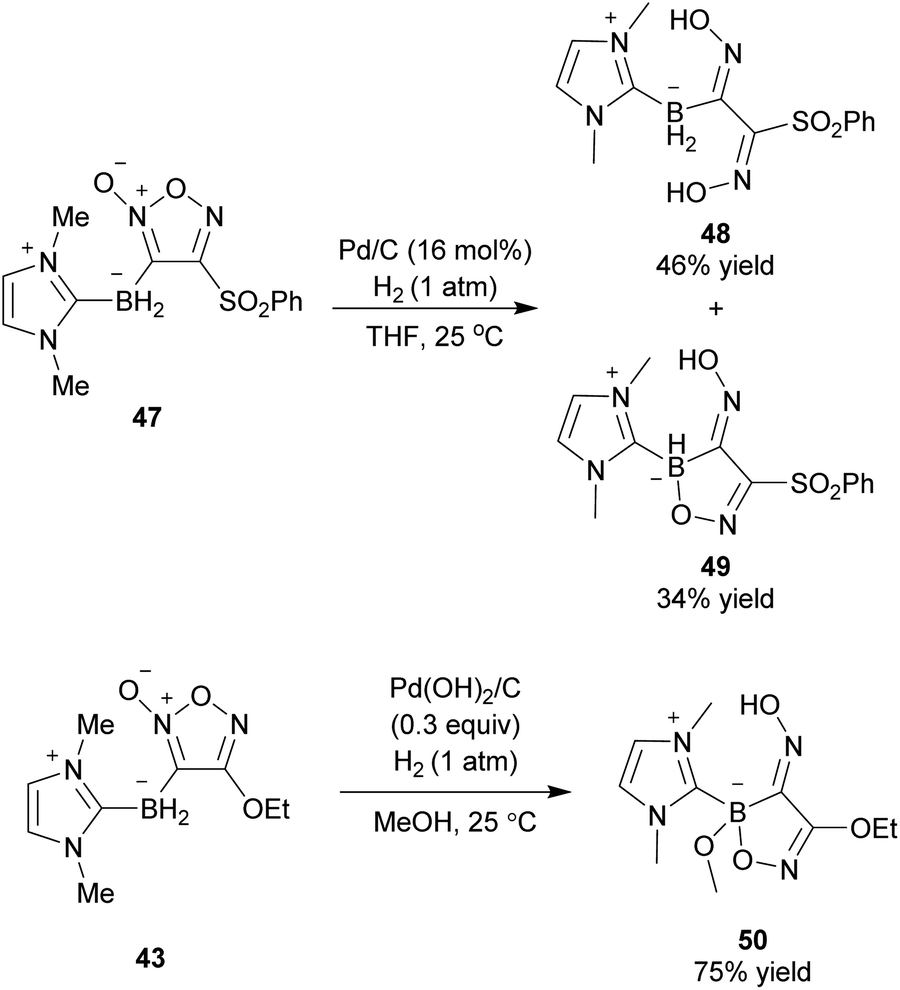 | ||
| Scheme 33 Derivatization of borylfuroxans. | ||
2.7. Carbon–halogen bond formation
There are few synthetic examples of halofuroxans, and research on their properties and bioactivity is scarce. As with other haloarenes, various physiological activities are expected, and from the viewpoint of synthetic chemistry, halogens can be expected to be used as a stepping stone for substituent introduction via cross-coupling reactions. In the previous sections, a few examples of the synthesis of halofuroxans based on the PRIS strategy were reported (Schemes 10, 17, 22, 29 and 31); however, none of them involved direct introduction of the carbon–halogen bond on the furoxan ring.4-Fluorofuroxan 51 was synthesized from 4-nitrofuroxan with Bu4NF (Scheme 34a),65 which is the first example of fluorofuroxan and of the PRI of any halogen atom. Fluorofuroxans were sufficiently stable to be purified and isolated using the usual procedure. Fluorination hardly proceeded when CsF was used instead of Bu4NF. Both alkyl and aryl groups were tolerated in this fluorination reaction as untouched substituents at the 3-position. Under the same conditions, 3-nitro-4-phenylfuroxan, the regioisomer of 4-nitrofuroxan, did not react with Bu4NF. This is probably because the C3 carbon of 3-nitrofuroxan is less electron-deficient than the C4 carbon of 4-nitrofuroxan (vide supra), and thus, nucleophilic attack of the fluoride anion cannot occur at the 3-position. The photoisomerization of 51 to 52 proceeded under UV-light irradiation, and the photostationary state was reached with a product ratio of 51:52 = 14:86. Because 52 was completely converted to 51 when heated at 110 °C in toluene, 51 is thermodynamically stable. This is a good example to demonstrate that the photoreactions of furoxans can synthesize one regioisomer of the two in defiance of their thermodynamic stabilities.
 | ||
| Scheme 34 Synthesis of fluorofuroxans via C–F bond formation on the furoxan ring. (a) General scheme. (b) Photochemical isomerization of fluorofuroxans. | ||
An attempt was made to synthesize chlorofuroxan in the same manner as fluorofuroxan, but the reaction was slow under the same conditions and required polar solvents and heating (Scheme 35a).65 The expected 4-chlorofuroxan 53 was obtained as the major product. However, regioisomer 54 and arylnitrile 55 were also obtained. It was proposed that ring opening from the Meisenheimer intermediate 56 proceeded and that byproducts 54 and 55 were formed via intermediate 57 (Scheme 35b). Analogously, the synthesis of bromofuroxans was attempted using Bu4NBr, but no target product was obtained. Side reaction giving nitrile 55 proceeded.
 | ||
| Scheme 35 C–Cl bond formation on the furoxan ring. (a) Representative example. (b) Proposed mechanism for the formation of product 53 and byproducts 54 and 55. | ||
3. Applied research of furoxan molecules
3.1. As a synthetic intermediate
As stated earlier (Section 2.5.4), carbon radicals can react with 3-sulfonylfuroxan to introduce alkyl substituents onto the furoxan ring. In this reaction, carboxylic acids or C–H bonds, which are prevalent functional groups, can serve as radical sources. From another perspective, this reaction can be described as a one-step “furoxanization” of prevalent functional groups. Furthermore, if we take the property that furoxans are prone to ring-opening decomposition, it can be interpreted that furoxans can be easily converted to other functional groups. Therefore, one can propose that a short-step functional group transformation can be achieved by the furoxanization of prevalent functionalities (“build”) followed by ring-opening degradation of furoxan (“scrap”). An example of this “build-and-scrap” method is shown in Scheme 36.59 The radical addition reactions to the furoxan ring could form furoxans 59–61 in a short step. Thereafter, these furoxans could be converted into various nitrogen-containing functional groups under various reaction conditions. This “build-and-scrap” methodology should find its application in the drug discovery research requiring structural diversity. | ||
| Scheme 36 “Build-and-scrap” of furoxans leading to a variety of functional groups. (a) P(OEt)3 (3 equiv.), 100 °C, 74% yield. (b) Pd/C (10 mol%), H2 (1 atm), MeOH, rt, 100% (62), 82% yield (63), 81% yield (64, dr = 3:1). (c) (1) hν (λ = 300–400 nm), C6D6, rt, 55% yield. (2) Pd/C (10 mol%), H2 (1 atm), MeOH, rt, 100% yield. (d) LiAlH4 (5 equiv.), THF, 0 °C, 39% yield. (e) (1) procedure (b). (2) LiAlH4 (5 equiv.), THF, 0 °C, 57% yield. (f) Bu3SnH (2 equiv.), benzene, 40 °C, 41% yield. (g) Bu3SnH (5 equiv.), benzene, 40 °C, 43% yield. (h) KOH (2.3 equiv.), THF, rt, 46% yield. (i) KOH (5.0 equiv.), THF, rt, 18% yield. (j) (1) Bu4NOH (2 equiv.), THF, rt. (2) (CF3SO2)2O (1.1 equiv.), CH2Cl2, 0 °C, 21% yield (2 step). (3) Pd(PPh3)4 (1 equiv.), benzene, rt, 52% yield. (k) 1,3-dipolarophile (3 equiv.), DMF, 130–150 °C, 33% yield (65, using phenylacetylene), 12% yield (66, using styrene), 18% yield (67, using oct-1-yne). (l) phenylacetylene (3 equiv.), toluene, 130 °C, 8% yield. | ||
3.2. As a component of hybrid drugs
Molecular hybridization is a versatile drug design and discovery approach that has emerged as a useful tool in medicinal chemistry. A hybrid molecule consists mainly of two covalently bonded parent molecules that retain their structural features and original biological activity within the hybrid.66 The additive and synergistic biological action of the parent molecules within the hybrid can introduce favorable therapeutic effects in multiple ways. The most evident, although sometimes elusive, is obtaining multitargeted therapeutic agents that target two different pathologies at the same time. More prevalently, however, hybridization of two biologically active molecules often acts as a tool to optimize the activity of one component through the synergistic action of the other component(s). Regarding the topic of this review, furoxan-based hybrids, the latter outcome is usually sought after, in which the presence of the furoxan moiety introduces higher potencies and enhanced selectivities than the parent molecule alone (Fig. 2). It is of note that most of the hybrid molecules mentioned in this section were synthesized based on the PRIS strategy to combine furoxan and drug moieties. | ||
| Fig. 2 Recently reported hybrids of furoxan and drug molecules. | ||
β-Elemene, a natural product extracted from the rhizome of Curcuma wenyujin, is a molecular scaffold that exhibits broad anti-tumor activities. This has led to its adoption as a broad-spectrum drug by the China Food and Drug Administration (CFDA) for the treatment of lung, liver, esophageal, and brain cancers, and bone metastasis in 1994.67 However, the lipophilicity and low bioavailability of β-elemene limited its applications, which led researchers to conduct various lead optimization studies to enhance its anti-tumoral activities. Hybridization of β-elemene with furoxan as the NO-donating moiety was reported, and showed improvements in the anti-cancer properties in relation to those of the parent β-elemene molecule.68,69 Six series of hybrids were synthesized and, when evaluated biologically, displayed significant anti-proliferative effects against malignant brain glioma cells. Hybrid 68 significantly and continuously suppressed the growth of gliomas in mice, with the brain glioma of the treatment group inhibited by more than 90%.
Introducing the sulfonyl furoxan moiety to phenstatin, a microtubule interfering agent (MIA), showed moderate to potent anti-tumor activities against several human cancer cell lines.70 Among the synthesized compounds, hybrid 69 showed the most potent anti-proliferative activities against both chemo-sensitive and resistant cancer cell lines with IC50 values ranging from 0.008 to 0.021 μM.
Photodynamic therapy (PDT) is an emerging minimally invasive cancer treatment approach that selectively generates reactive oxygen species (ROS) through the excitation of photosensitizers (PS) by a specific-wave-length light. In turn, ROS causes cancerous cells to perish directly or indirectly via induction of tumor vascular stasis.71 Thus, the efficacy of any PDT protocol is dependent on the concentration of oxygen surrounding the tumor tissues, a longstanding limiting factor in the therapeutic adoption of PDT. Chlorin E6 is a photosensitizer employed as a photodynamic anti-cancer agent. To enhance the PD therapeutic effect of chlorin E6, various NO donors were introduced into the chlorin E6 skeleton to mitigate the issue of low oxygen concentrations in cancerous tissues. A synergistic anti-tumor action between NO and ROS was expected to enhance the PDT performance of the parent chlorin E6.72 Hybrid 70, a furoxan-hybridized chlorin E6, was synthesized. The biological evaluation results revealed that the furoxan-containing photosensitizers exhibited synergistic ROS and NO effects, increasing the intracellular levels of both ROS and NO. Elevated ROS levels exhibited enhanced anti-tumor effects during PDT by disturbing the intracellular redox balance and inducing oxidative stress in tumor cells.
Another anti-tumor natural product that shows enhanced bioactivity upon hybridization with furoxan is chelidonine. A study synthesized and examined a series of furoxan-based, NO-donating derivatives of chelidonine, one of which is hybrid 71.73 Compared to chelidonine, 71 exhibited lower IC50 values against human hepatoma cells HepG2, breast cancer cells MCF-7, colon cancer cells HCT-116, and leukemia cells K562. In addition to exhibiting the strongest anti-proliferative activity against the aforementioned cancer cell types, 71 also demonstrated a high selectivity between normal and cancer blood cells, as indicated by its IC50 value of >40 μM against human peripheral blood mononuclear cells (PBMCs).
A recent study reported the synthesis and in vitro anti-proliferative evaluation of 15 novel furoxan-based NO-releasing hybrids of estradiol derivatives.74 The endogenous estrogen metabolite 2-methoxyestradiol, formed from estradiol, has been researched for several decades as a potential anti-cancer agent. Based on a previous work on the synthesis of various anti-cancer agents by introducing several furoxan-based NO-releasing moieties to coumarin cores, it was hypothesized that substituent modification on the phenyl ring of estradiol might enhance its anti-cancer activity. Most of the synthesized molecules exhibited stronger anti-proliferative effects than phenylsulfonylfuroxan and 2-methoxyestradiol in the MDA-MB-231, A2780, and HeLa cell and HUVEC lines. Hybrid 72 showed the highest activity against MDA-MB-231 cells, with an IC50 value of 0.00083 μM. This activity was related to the compound's higher capacity to release NO, as demonstrated by the diminished activity when the NO scavenger, hemoglobin, was introduced into the system.
Coumarin is a natural product scaffold with anti-tumor properties. A recent study reported the synthesis of a series of novel furoxan-based coumarin derivatives and evaluation of their in vitro anti-proliferative activities in human cervical cancer HeLa cells.75 All the compounds displayed more potent inhibition than the reference compound coumarin-3-carboxylic acid. Among the synthesized compounds, hybrid 73 exerted the highest anti-proliferative activity (IC50 = 0.60 μM) against human breast cancer MCF-7 cells, while hybrid 74 exhibited a wider spectrum of activity against five cancer cells.
To combat multidrug chemo-resistant leukemia cancer cells, a series of NO-releasing bifendate derivatives were synthesized.76 In vitro and in vivo biological evaluations demonstrated that hybrid 75 produced relatively high levels of NO and significantly inhibited the proliferation of drug-resistant K562/A02 cells. A substantial decrease in the anti-tumor activity of 75 was observed when an NO scavenger was introduced, establishing, at least partially, the dependence of 75 cytotoxicity against drug-resistant K562/A02 cells on its NO-releasing capabilities.
Augmenting the anti-tumoral properties of tocopherol through hybridization with furoxan has been reported.77 A series of tocopherol-furoxan analogues were synthesized and evaluated for their anti-proliferative and NO-releasing activity. Hybrid 76 emerged as a promising anti-cancer agent owing to its good anti-proliferative activity against bladder cancer cell lines, T24 and 253 J, and low toxicity against normal HaCaT cells. The study demonstrated that the NO-releasing ability of 76 is necessary for its anti-proliferative properties, as demonstrated by the diminished activity of 76 in the presence of NO scavengers in cancerous cell media.
Recently, it was reported that the synthesis of a set of mitochondria-targeting compounds with varying NO-releasing capacities was achieved through the hybridization of rhodamine B with various 3-R-substituted furoxan moieties.78 Models equipped with weaker NO-releasing furoxan moieties bearing –CH3 and –CONH2 groups at the 3-position on the furoxan exhibited cytotoxicity only at the highest concentrations. On the contrary, models equipped with stronger NO-releasing furoxan moieties bearing –CN and –SO2C6H5 (hybrid 77) at the 3-position exhibited much stronger anti-cancer activity, probably because of their higher NO-releasing ability and their capability to inhibit cellular proteins by covalent binding.
β-Carboline alkaloid scaffolds have been well-researched as anti-cancer agents because of their cytotoxic effects and ability to reverse resistance to some anti-cancer drugs. To enhance the anti-tumoral performance of β-carboline alkaloids, a recent study reported the synthesis of 30 β-carboline-(phenylsulfonyl)furoxan hybrids.79 The inhibitory activities of the synthesized compounds against the human breast cancer cell lines MCF-7 and MDA-MB-231 were evaluated, which revealed that hybrid 78 stood out as a promising antimetastatic agent for breast cancer, as it significantly inhibited the migration and invasion of MDA-MB-231 cells.
Scopoletin is another natural product scaffold that shows therapeutic promise against human breast cancer upon hybridization with furoxan. The synthesis of 11 scopoletin-furoxan derivatives has been previously reported. They exhibited improved anti-proliferative activity against MCF-7 and MDA-MB-231 cells and weaker cytotoxicity against the human breast epithelial cell line MCF-10A than parent scopoletin.80 Of these, hybrid 79 produced the highest levels of NO intracellularly and exhibited the best anti-cancer activity and low toxicity.
Many classes of furoxan hybrids have been developed for therapeutic purposes other than anti-cancer activities. A prime example of this is the development of furoxan-based antileishmanial agents. Leishmaniasis is a neglected tropical disease (NTD) caused by infection with the protozoan parasite Leishmania and occurs mainly in the tropical regions of Africa, Asia, and North and South Americas. It was previously reported that furoxan derivatives containing the N-acylhydrazone subunit showed remarkable leishmanicidal activity (hybrid 80).81 To further enhance the antileishmanial potency of 80, a novel series of N-oxide compounds containing the N-acylhydrazone subunit was designed, represented by phenyl-furoxan, amide-furoxan, and benzofuroxan.82 In vitro and in vivo evaluations showed that hybrid 81 demonstrates remarkable antileishmanial potency.
3.3. As a photo-induced NO donor
4-Fluorofuroxan 82, which was recently synthesized for the first time by PRIS, was isomerized to 3-fluorofuxoan 83 upon UV irradiation at a high conversion rate (Scheme 34). After the isolation of 82 and 83, the NO-releasing ability of each in the presence of L-cysteine was investigated, revealing that 3-fluorofuroxan 83 had a much higher NO-releasing ability than 4-fluorofuroxan 82 (Scheme 37).65 These results suggest that 4-fluorofuroxan 82 can function as a photo-induced NO donor, a compound that can release NO upon UV irradiation. | ||
| Scheme 37 Fluorofuroxans as a photo-induced NO donor. | ||
UV irradiation of an aqueous solution of 4-fluorofuroxan 82 in the presence of L-cysteine resulted in an increase in NO release with increasing irradiation time (Scheme 38).65 In comparison with the molecular design of conventional photo-induced NO donors that have a weak X–NO bond in their structure and release NO by breaking the bond upon light irradiation,83,84 4-fluorofuroxan is unique in that isomerization is the mechanism for photo-responsiveness.
 | ||
| Scheme 38 NO-releasing profile of 82 as a function of irradiation time. A buffered aqueous solution of 82 (100 μM) containing L-cysteine (5 mM) was irradiated in a Pyrex vial (λ = 300–400 nm) at 37 °C (oil bath) for the indicated time. | ||
The high photoisomerization ratio of fluorofuroxan in the photostationary state (Scheme 34) inspired the development of a photo-induced NO donor. However, if isomerization occurs at equilibrium on light irradiation, the isomerization ratio in the photostationary state needs not necessarily be high. In other words, if light is irradiated continuously in the presence of a thiol additive necessary for NO release, even if the isomerization ratio in the photostationary state is low, NO should be sequentially released from a small portion of the NO-releasing active regioisomer, after which it is continually supplied from the inactive regioisomer by photoisomerization. Therefore, theoretically, both furoxan regioisomers are involved in NO release. This hypothesis was verified using alkoxyfuroxans (Scheme 39).85 The NO-releasing ability of 4-alkoxyfuroxan 84 was as low as 2%, while that of its regioisomer, 3-alkoxyfuroxan 85, was 81%. However, the isomerization ratio in the photostationary state was not high, that is, 84:85 = 81:19. UV irradiation of an aqueous solution of 84 in the presence of L-cysteine resulted in an increase in NO release over the duration of irradiation, and the NO release reached up to 50% yield (Fig. 3); the hypothesis was thus proved.
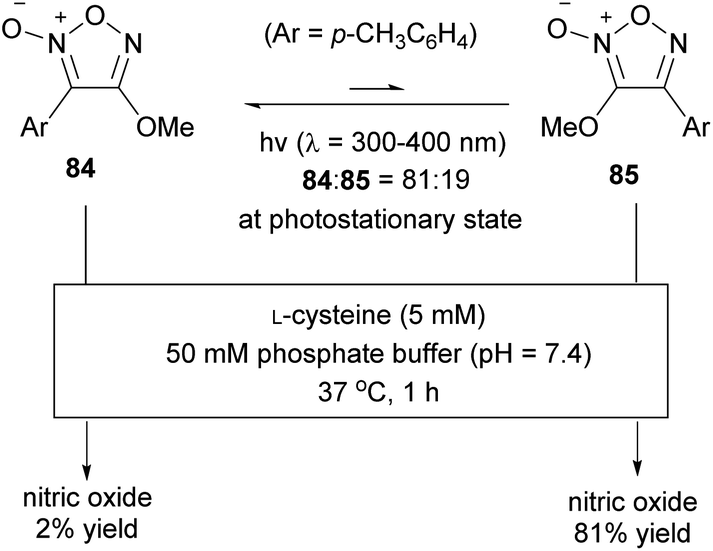 | ||
| Scheme 39 NO-releasing abilities of alkoxyfuroxans. | ||
 | ||
| Fig. 3 NO-releasing profile of 4-alkoxyfuroxan 84 as a function of irradiation time. A buffered aqueous solution of 84 (100 μM) that contained L-cysteine (5 mM) was irradiated (λ = 300–400 nm) at 37 °C. | ||
Thiol additives are required for the release of NO from furoxans. The required structure of the thiol additives depends on the type of furoxan used. For the release of NO from alkoxyfuroxans and fluorofuroxans, thiols having an amino group at the β-position, such as L-cysteine and cysteamine, acted as mediators, but simple thiols such as ethanethiol did not.85 However, 3-cyano-4-phenylfuroxan (4) released NO even with benzenethiol as a mediator.13 The proposed mechanism for the release of NO from alkoxyfuroxan in the presence of a thiol mediator is delineated in Scheme 40. Upon addition of a thiol to 3-alkoxyfuroxan at the 3-position, ring degradation occurs to form nitronate 86 with the liberation of aryl nitrile. This species undergoes cyclization to afford the C-nitroso compound 87. NO is released via homolytic cleavage of the weakened C–N bond along with the formation of radical species 88. Although experimental evidence is lacking and the mechanism remains elusive, it was experimentally confirmed that aryl nitrile was formed quantitatively as a co-product, which supports this mechanism.
 | ||
| Scheme 40 Proposed mechanism for the release of NO from alkoxyfuroxan in the presence of a thiol mediator. | ||
The photoresponsive NO-donor furoxans mentioned so far had short absorption wavelengths and required the use of UV light.65,85 To use them in vivo, visible light irradiation is more desirable because it is less harmful for cells and can penetrate deeper into the tissue than UV light. The investigation of the photochemical properties of 4-fluorofuroxan revealed that the phosphorescence of 4-fluorofuroxan 51 (R = p-CH3C6H4) (Scheme 34) was observed at 550 nm and indicated the possibility of the sensitized excitation of 4-fluorofuroxan by visible light with a suitable triplet photosensitizer. Various photosensitizers could excite 4-fluorofuroxan to isomerize it to 3-fluorofuroxan 52 by visible light irradiation (λex = 400–500 nm).86 Not only 4-fluorofuroxan but 4-alkoxyfuroxan could also be photoexcited using a triplet photosensitizer.87 Based on these results, two furoxan molecules 89 and 90 that come with furoxan and photosensitizer entities in one molecule have been developed as visible light-triggered NO donors (Fig. 4).86,87
 | ||
| Fig. 4 Visible light-induced NO donor furoxans. | ||
4. Conclusions
Because of their weak aromaticity and multiple heteroatom–heteroatom bonds, furoxans are vulnerable to ring degradation under various reaction conditions. Therefore, the main synthetic strategy for furoxans is to introduce the desired substituents into the precursor prior to the formation of the furoxan ring. However, recent research has led to the development of various PRIS methodologies, in which the furoxan ring is first constructed, and then substituents are introduced on the ring, enabling the synthesis of various types of furoxan molecules in a more direct manner. The formation of C–O, C–S, C–N, C–B, and carbon–halogen bonds on the furoxan ring was achieved. The types of reactions varied; in addition to the SNAr reaction, which takes advantage of the electron-deficient nature of furoxan, radical-involved methods and intramolecular rearrangement reactions were also reported. The selectivity between the 3- and 4-positions in the introduction of substituents varied from reaction to reaction, and a high selectivity was observed in several reactions.With the development of synthetic methods, research on the various applications of furoxans has also progressed. A new synthetic strategy named “build-and-scrap” method was developed, in which the ease of ring degradation of furoxans is regarded as a merit and utilized to enable the use of furoxans as intermediates for the synthesis of various nitrogen-containing functional groups. Hybrid molecules, which covalently link the NO-releasing furoxan with existing drugs to provide the molecule with two functions or to enhance each other's functions, have been frequently reported. The study of a photo-induced NO donor based on a unique switching mechanism utilizing furoxan isomerization is also being developed.
Indeed, studies on furoxan synthesis are growing, but there are still many furoxans that have not yet been synthesized, or for which there are no efficient synthetic methods. In chemistry, the development of synthetic methods is often the foundation for applied research; therefore, the development of synthetic methods for furoxans is increasingly desirable in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI Grant Number JP21K05054 to R. M. M. A. would like to thank the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan for continuous support through its Scholarship Program.Notes and references
- A. Gasco and A. J. Boulton, Furoxans and benzofuroxans, in Adv. Heterocycl. Chem., eds. A. R. Katritzky and A. J. Boulton, Academic Press, 1981, vol. 29, pp. 251–340 Search PubMed.
- A. B. Sheremetev, N. N. Makhova and W. Friedrichsen, Monocyclic furazans and furoxans, in Adv. Heterocycl. Chem., Academic Press, 2001, vol. 78, pp. 65–188 Search PubMed.
- H. Cerecetto and W. Porcal, Pharmacological properties of furoxans and benzofuroxans: recent developments, Mini-Rev. Med. Chem., 2005, 5, 57–71 CrossRef CAS PubMed.
- A. Kekulé, Ueber die Constitution des Knallquecksilbers, Ann. Chem. Pharm., 1857, 101, 200–213 CrossRef.
- F. B. Mallory and A. Cammarata, Evidence for the Transient Existence of 1,2-Dinitrosoalkenes, J. Am. Chem. Soc., 1966, 88, 61–64 CrossRef CAS.
- M. Calleri, G. Ferraris and D. Viterbo, On the isomerism of aryl-alkyl-furoxans: The molecular structure of methyl-p-bromophenyl-furoxans, Tetrahedron Lett., 1967, 8, 4549–4551 CrossRef.
- H. Wieland, Zur Kenntniss der Pseudonitrosite, Justus Liebigs Ann. Chem., 1903, 329, 225–268 CrossRef CAS.
- D. L. Hammick, W. A. M. Edwakdes and E. R. Steiner, CCCCLVIII.—The constitution of benzfurazan and benzfurazan oxide, J. Chem. Soc., 1931, 3308–3313 RSC.
- F. B. Mallory, S. L. Manatt and C. S. Wood, The rates of some degenerate rearrangements as determined by nuclear magnetic resonance spectroscopy, J. Am. Chem. Soc., 1965, 87, 5433–5438 CrossRef CAS.
- R. Calvino, V. Mortarini, A. Gasco, M. A. Bianco and M. L. Ricciardi, Furazan and furoxan sulfones - Synthesis and antimicrobial activity, Eur. J. Med. Chem., 1977, 12, 157–159 CAS.
- H. Cerecetto and M. González, Benzofuroxan and furoxan. Chemistry and biology, in Bioactive Heterocycles IV. Topics in Heterocyclic Chemistry, ed. M. T. H. Khan, Springer, Berlin, Heidelberg, 2007, ch. 64, vol. 10, pp. 265–308 Search PubMed.
- M. Feelisch, K. Schönafingeri and H. Noack, Thiol-mediated generation of nitric oxide accounts for the vasodilator action of furoxans, Biochem. Pharmacol., 1992, 44, 1149–1157 CrossRef CAS PubMed.
- C. Medana, G. Ermondi, R. Fruttero, A. Di Stilo, C. Ferretti and A. Gasco, Furoxans as nitric oxide donors. 4-Phenyl-3-furoxancarbonitrile: Thiol-mediated nitric oxide release and biological evaluation, J. Med. Chem., 1994, 37, 4412–4416 CrossRef CAS PubMed.
- G. Rai, A. A. Sayed, W. A. Lea, H. F. Luecke, H. Chakrapani, S. Prast-Nielsen, A. Jadhav, W. Leister, M. Shen, J. Inglese, C. P. Austin, L. Keefer, E. S. J. Arnér, A. Simeonov, D. J. Maloney, D. L. Williams and C. J. Thomas, Structure-mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis, J. Med. Chem., 2009, 52, 6474–6483 CrossRef CAS PubMed.
- O. N. Burov, M. E. Kletskii, N. S. Fedik, A. V. Lisovin and S. V. Kurbatov, Mechanism of thiol-induced nitrogen(II) oxide donation by furoxans: a quantum-chemical study, Chem. Heterocycl. Compd., 2015, 51, 951–960 CrossRef CAS.
- A. J. Boulton, P. B. Ghosh and A. R. Katritzky, N-oxides and related compounds. Part XXVIII. 5-Amino- and 5-hydroxy-benzofuroxans, J. Chem. Soc. C, 1966, 971–976 RSC.
- R. D. McCulla, G. Burdzinski and M. S. Platz, Ultrafast study of the photochemistry of 2-azidonitrobenzene, Org. Lett., 2006, 8, 1637–1640 CrossRef CAS PubMed.
- V. Armani, C. Dell'Erba, N. Marino, G. Petrillo and C. Tavani, Synthetic exploitation of the ring-opening of 3,4-dinitrothiophene. Part 7. Access to disubstituted 1,2,5-oxadiazole-2-oxides and 2-phenyl-2H-1,2,3-triazole-1-oxides, Tetrahedron, 1997, 53, 1751–1758 CrossRef.
- O. R. Klyuchnikov, V. I. Starovoitov, F. G. Khairutdinov and V. V. Golovin, Synthesis of condensed nitrofuroxanes using hydroxylamine, Russ. J. Appl. Chem., 2004, 77, 853–854 CrossRef CAS.
- A. G. Green and F. M. Rowe, LV.—The conversion of o-nitroamines into isooxadiazole oxides, and of o-nitrosoamines into isooxadiazoles, J. Chem. Soc., Trans., 1917, 111, 612–620 RSC.
- D. Britton, W. E. Noland, M. J. Pinnow and V. G. Young Jr., Crystal Packing: An examination of the packing of molecules approximately isosteric with 4,5-dichlorophthalic anhydride, Helv. Chim. Acta, 2003, 86, 1175–1192 CrossRef CAS.
- E. Del Grosso, D. Boschi, L. Lazzarato, C. Cena, A. Di Stilo, R. Fruttero, S. Moro and A. Gasco, The furoxan system: design of selective nitric oxide (NO) donor inhibitors of COX-2 endowed with anti-aggregatory and vasodilating activities, Chem. Biodivers., 2005, 2, 886–900 CrossRef CAS PubMed.
- L. L. Fershtat and F. E. Teslenko, Five-membered hetarene N-oxides: recent advances in synthesis and reactivity, Synthesis, 2021, 53, 3673–3682 CrossRef CAS.
- L. L. Fershtat and N. N. Makhova, 1,2,5-Oxadiazole-based high-energy-density materials: synthesis and performance, ChemPlusChem, 2020, 85, 13–42 CrossRef CAS.
- L. L. Fershtat, M. I. Struchkova, A. S. Goloveshkin, I. S. Bushmarinov and N. N. Makhova, Dinitrogen trioxide–mediated domino process for the regioselective construction of 4-nitrofuroxans from acrylic acids, Heteroat. Chem., 2014, 25, 226–237 CrossRef CAS.
- W. V. Farrar, The 3,4-bisarenesulphonylfuroxans, J. Chem. Soc., 1964, 904–906 RSC.
- G. Sorba, C. Medana, R. Fruttero, C. Cena, A. Di Stilo, U. Galli and A. Gasco, Water soluble furoxan derivatives as NO prodrugs, J. Med. Chem., 1997, 40, 463–469 CrossRef CAS PubMed.
- T. I. Godovikova, O. A. Rakitin, S. P. Golova, S. A. Vozchikova and L. I. Khmel’nitskii, 3,4-Dinitrofuroxan—the first example of a pernitro heterocycle, Mendeleev Commun., 1993, 3, 209–210 CrossRef.
- R. Fruttero, G. Sorba, G. Ermondi, M. Lolli and A. Gasco, Unsymmetrically substituted furoxans. Part 17. Structural investigations in benzenesulfonylfuroxan derivatives and related compounds, Farmaco, 1997, 52, 405–410 CAS.
- T. I. Godovikova, O. A. Rakitin, S. P. Golova, S. A. Vozchikova, M. V. Povorin and L. I. Khmel'nitskii, Synthesis and nucleophilic substitution reactions of 3,4-dinitrofuroxan, Chem. Heterocycl. Compd., 1994, 30, 465–469 CrossRef.
- R. Calvino, A. Gasco, A. Serafino and D. Viterbo, Unsymmetrically substituted furoxans. Part 6. 3-Nitro-4-phenylfuroxan: reaction with sodium methoxide and X-ray structural analysis, J. Chem. Soc., Perkin Trans., 1981, 2, 1240–1242 RSC.
- G. Ponzio, Ricerche sulle diossime-LXXXIII, Gazz. Chim. Ital., 1932, 62, 127–131 CAS.
- R. Matsubara, A. Ando, H. Hasebe, H. Kim, T. Tsuneda and M. Hayashi, Synthesis and synthetic application of chloro- and bromofuroxans, J. Org. Chem., 2020, 85, 5959–5972 CrossRef CAS PubMed.
- L. L. Fershtat, M. A. Epishina, A. S. Kulikov, M. I. Struchkova and N. N. Makhova, Synthesis of hetarylsulfanyl- and hetaryloxyfuroxans by nucleophilic substitution of nitro group in nitrofuroxans with heterocyclic thiol and hydroxy derivatives, Chem. Heterocycl. Compd., 2015, 51, 176–186 CrossRef CAS.
- H. Wieland, Ueber Additionsreactionen mit nitrosen Gasen, Justus Liebigs Ann. Chem., 1903, 328, 154–255 CrossRef CAS.
- H. Takayama, S. Shirakawa, M. Kitajima, N. Aimi, K. Yamaguchi, Y. Hanasaki, T. Ide, K. Katsuura, M. Fujiwara, K. Ijichi, K. Konno, S. Sigeta, T. Yokota and M. Baba, Utilization of Wieland furoxan synthesis for preparation of 4-aryl-1,2,5-oxadiazole-3-yl carbamate derivatives having potent anti-HIV activity, Bioorg. Med. Chem. Lett., 1996, 6, 1993–1996 CrossRef CAS.
- L. L. Fershtat, M. A. Epishina, I. V. Ovchinnikov, M. I. Struchkova, A. A. Romanova, I. V. Ananyev and N. N. Makhova, Side-chain prototropic tautomerism of 4-hydroxyfuroxans in methylation reactions, Tetrahedron Lett., 2016, 57, 5685–5689 CrossRef CAS.
- R. Matsubara, Y. Katsuragi, T. Sakaguchi, S. Eguchi, M. Hayashi and A. Ando, Synthesis of sulfonyloxy furoxans via hydroxyfuroxan ammonium salts, Tetrahedron, 2018, 74, 3642–3651 CrossRef CAS.
- G. Sorba, G. Ermondi, R. Fruttero, U. Galli and A. Gasco, Unsymmetrically substituted furoxans. Part 16. Reaction of benzenesulfonyl substituted furoxans with ethanol and ethanethiol in basic medium, J. Heterocycl. Chem., 1996, 33, 327–334 CrossRef CAS.
- R. Calvino, A. Gasco, E. Menziani and A. Serafino, Unsymmetrically substituted furoxans. VIII. Chloromethylfuroxans, J. Heterocycl. Chem., 1983, 20, 783–785 CrossRef CAS.
- D. Boschi, G. Sorba, M. Bertinaria, R. Fruttero, R. Calvino and A. Gasco, Unsymmetrically substituted furoxans. Part 18. Smiles rearrangement in furoxan systems and in related furazans, J. Chem. Soc., Perkin Trans., 2001, 1, 1751–1757 RSC.
- R. Fruttero, D. Boschi, A. Di Stilo and A. Gasco, The Furoxan system as a useful tool for balancing "hybrids" with mixed α1-antagonist and NO-like vasodilator activities, J. Med. Chem., 1995, 38, 4944–4949 CrossRef CAS PubMed.
- L. L. Fershtat, M. A. Epishina, A. S. Kulikov and N. N. Makhova, Design of hetarylthiofuroxans by nucleophilic substitution of NO2 group in nitrofuroxans, Mendeleev Commun., 2015, 25, 36–38 CrossRef CAS.
- L. L. Fershtat, D. M. Bystrov, E. S. Zhilin and N. N. Makhova, N- Oxide-controlled chemoselective reduction of nitrofuroxans, Synthesis, 2019, 51, 747–756 CrossRef CAS.
- H. Wieland, Über die einwirkung der halogene auf knallquecksilber, Ber. Dtsch. Chem. Ges., 1909, 42, 4192–4199 CrossRef.
- Y. Q. Huang, M. M. Liu, L. F. Meng, P. Feng, Y. L. Guo, M. H. Ying, X. Y. Zhu and Y. Chen, Synthesis and antitumor evaluation of novel hybrids of phenylsulfonylfuroxan and epiandrosterone/dehydroepiandrosterone derivatives, Steroids, 2015, 101, 7–14 CrossRef CAS PubMed.
- O. G. Vlasova, O. A. Rakitin and L. I. Khmelnitski, A simple synthesis of the heterocyclic S,S-diphenylsulfilimines, Org. Prep. Proced. Int., 1994, 26, 331–335 CrossRef CAS.
- A. Gasco, V. Mortarini, G. Ruà and A. Serafino, Unsymmetrically substituted furoxans. IIII. Methylnitrofuroxan: Its structure and behavior toward nucleophilie substitution, J. Heterocycl. Chem., 1973, 10, 587–590 CrossRef CAS.
- O. V. Zavarzina, O. A. Rakitin and L. I. Khmel’nitskii, Substitution of the nitro group in chloronitrofuroxan by N- and O-trimethylsilyl derivatives, Mendeleev Commun., 1994, 4, 135 CrossRef.
- L. L. Fershtat, M. A. Epishina, A. S. Kulikov, I. V. Ovchinnikov, I. V. Ananyev and N. N. Makhova, An efficient access to (1H-tetrazol-5-yl)furoxan ammonium salts via a two-step dehydration/[3+2]-cycloaddition strategy, Tetrahedron, 2015, 71, 6764–6775 CrossRef CAS.
- A. Gasco, V. Mortarini, G. Ruà and E. Menziani, Alkyl N-methylfuroxanylcarbamates. Synthesis and structure. II, J. Heterocycl. Chem., 1972, 9, 837–841 CrossRef CAS.
- I. V. Ovchinnikov, K. A. Lyssenko and N. N. Makhova, An unexpected transformation of 3,4-bis(isocyanato)furoxan into 3,3’-bi(1,2,4-oxadiazol-5-one), Mendeleev Commun., 2009, 19, 144–146 CrossRef CAS.
- N. N. Makhova, A. N. Blinnikov and L. I. Khmel’nitskii, The Schmidt Rearrangement of methyl furoxanyl ketones and furoxancarboxylic acids: a new synthetic route to aminofuroxans, Mendeleev Commun., 1995, 5, 56–58 CrossRef.
- I. V. Ovchinnikov, A. N. Blinnikov, N. N. Makhova and L. I. Khmel’nitskii, The Curtius rearrangement of azidocarbonylfuroxans: Some peculiarities and the synthesis of aminofuroxans, Mendeleev Commun., 1995, 5, 58–60 CrossRef.
- E. L. Baryshnikova, A. S. Kulikov, I. V. Ovchinnikov, V. V. Solomentsev and N. N. Makhova, The base-induced cascade rearrangement of 4-acetylamino-3-arylazo-1,2,5-oxadiazole 2-oxides (furoxans) into 4-acetylamino-2-aryl-5-nitro-2H-1,2,3-triazoles, Mendeleev Commun., 2001, 11, 230–232 CrossRef.
- R. Matsubara, S. Eguchi, A. Ando and M. Hayashi, Synthesis of alkynyl furoxans. Rare carbon-carbon bond-forming reaction on a furoxan ring, Org. Biomol. Chem., 2017, 15, 1965–1969 RSC.
- L. Annaratone, S. Guglielmo and K. Y. Chegaev, Direct introduction of cyano group on furoxan ring, Mendeleev Commun., 2017, 27, 565–566 CrossRef CAS.
- J. M. Anderson and J. K. Kochi, Silver(I)-catalyzed oxidative decarboxylation of acids by peroxydisulfate. Role of silver(II), J. Am. Chem. Soc., 1970, 92, 1651–1659 CrossRef CAS.
- R. Matsubara, H. Kim, T. Sakaguchi, W. B. Xie, X. F. Zhao, Y. Nagoshi, C. Y. Wang, M. Tateiwa, A. Ando, M. Hayashi, M. Yamanaka and T. Tsuneda, Modular synthesis of carbon-substituted furoxans via radical addition pathway. Useful tool for transformation of aliphatic carboxylic acids based on "build-and-scrap" strategy, Org. Lett., 2020, 22, 1182–1187 CrossRef CAS PubMed.
- C. Dong, X. Zhao, Y. Katsuragi, H. Kim, M. Hayashi and R. Matsubara, Furoxan incorporation into C–H bonds enabling nitrogen-containing functional group installation into the same, J. Org. Chem., 2021, 86, 15807–15817 CrossRef CAS PubMed.
- C. Dong, M. Hayashi and R. Matsubara, Direct arylation of furoxan using potassium aryltrifluoroborates, Heterocycles, 2022, 104, 1770–1781 CrossRef CAS.
- W. Xie, M. Hayashi and R. Matsubara, Borylfuroxans: synthesis and applications, Org. Lett., 2021, 23, 4317–4321 CrossRef CAS PubMed.
- S.-H. Ueng, M. Makhlouf Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, Complexes of borane and N-heterocyclic carbenes: A new class of radical hydrogen atom donor, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS PubMed.
- S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, N-Heterocyclic carbene boryl radicals: A new class of boron-centered radical, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed.
- A. Ando, R. Matsubara, S. Takazawa, T. Shimada and M. Hayashi, Fluorofuroxans: Synthesis and application as photoinduced nitric oxide donors, Asian J. Org. Chem., 2016, 5, 886–890 CrossRef CAS.
- M. Decker, Hybrid molecules incorporating natural products: Applications in cancer therapy, neurodegenerative disorders and beyond, Curr. Med. Chem., 2011, 18, 1464–1475 CrossRef CAS PubMed.
- Z. Bai, C. Yao, J. Zhu, Y. Xie, X.-Y. Ye, R. Bai and T. Xie, Anti-tumor drug discovery based on natural product β-elemene: Anti-tumor mechanisms and structural modification, Molecules, 2021, 26, 1499 CrossRef CAS PubMed.
- J. Chen, T. Wang, S. Xu, P. Zhang, A. Lin, L. Wu, H. Yao, W. Xie, Z. Zhu and J. Xu, Discovery of novel antitumor nitric oxide-donating β-elemene hybrids through inhibiting the PI3K/Akt pathway, Eur. J. Med. Chem., 2017, 135, 414–423 CrossRef CAS PubMed.
- R. Bai, J. Zhu, Z. Bai, Q. Mao, Y. Zhang, Z. Hui, X. Luo, X.-Y. Ye and T. Xie, Second generation β-elemene nitric oxide derivatives with reasonable linkers: potential hybrids against malignant brain glioma, J. Enzyme Inhib. Med. Chem., 2022, 37, 379–385 CrossRef CAS PubMed.
- X. Huang, Y.-S. Wang, D. Ma, Y.-Y. Wang, S.-D. Bian, B. Zhang, Y. Qiao, Z.-R. He, M. Lv, G.-L. Cai, Z.-X. Wang, X.-S. Liu, J.-B. Shi and M.-M. Liu, Synthesis and biological evaluation of novel hybrids of phenylsulfonyl furoxan and phenstatin derivatives as potent anti-tumor agents, Eur. J. Med. Chem., 2022, 230, 114112 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Overcoming the Achilles' heel of photodynamic therapy, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- X. Guo, H. Yu, W. Shen, R. Cai, Y. Li, G. Li, W. Zhao and S. Wang, Synthesis and biological evaluation of NO-donor containing photosensitizers to induce ferroptosis of cancer cells, Bioorg. Chem., 2021, 116, 105355 CrossRef CAS PubMed.
- X. Huang, K. Cheng, L. Liu, X. Hu, X. Gao, H. Li, F. Xu, Z. Li, H. Hua and D. Li, Design, synthesis and apoptosis-related antiproliferative activities of chelidonine derivatives, Bioorg. Med. Chem. Lett., 2020, 30, 126913 CrossRef CAS PubMed.
- Q. Wan, Y. Deng, Y. Huang, Z. Yu, C. Wang, K. Wang, J. Dong and Y. Chen, Synthesis and antitumor evaluation of novel hybrids of phenylsulfonylfuroxan and estradiol derivatives, ChemistryOpen, 2020, 9, 176–182 CrossRef CAS PubMed.
- Z. Zhang, Z.-W. Bai, Y. Ling, L.-Q. He, P. Huang, H.-X. Gu and R.-F. Hu, Design, synthesis and biological evaluation of novel furoxan-based coumarin derivatives as antitumor agents, Med. Chem. Res., 2018, 27, 1198–1205 CrossRef CAS.
- X. Gu, Z. Huang, Z. Ren, X. Tang, R. Xue, X. Luo, S. Peng, H. Peng, B. Lu, J. Tian and Y. Zhang, Potent inhibition of nitric oxide-releasing bifendate derivatives against drug-resistant K562/A02 cells in vitro and in vivo, J. Med. Chem., 2017, 60, 928–940 CrossRef CAS PubMed.
- F. Pérez, M. Varela, L. Canclini, S. Acosta, W. Martínez-López, G. V. López and P. Hernández, Furoxans and tocopherol analogs–furoxan hybrids as anticancer agents, Anticancer Drugs, 2019, 30, 330–338 CrossRef PubMed.
- F. Sodano, E. Gazzano, B. Rolando, E. Marini, L. Lazzarato, R. Fruttero, C. Riganti and A. Gasco, Tuning NO release of organelle-targeted furoxan derivatives and their cytotoxicity against lung cancer cells, Bioorg. Chem., 2021, 111, 104911 CrossRef CAS PubMed.
- X. Hu, X. Gao, G. Gao, Y. Wang, H. Cao, D. Li and H. Hua, Discovery of β-carboline-(phenylsulfonyl)furoxan hybrids as potential anti-breast cancer agents, Bioorg. Med. Chem. Lett., 2021, 40, 127952 CrossRef CAS PubMed.
- N. Yu, N. Li, K. Wang, Q. Deng, Z. Lei, J. Sun and L. Chen, Design, synthesis and biological activity evaluation of novel scopoletin-NO donor derivatives against MCF-7 human breast cancer in vitro and in vivo, Eur. J. Med. Chem., 2021, 224, 113701 CrossRef CAS PubMed.
- L. A. Dutra, L. d. Almeida, T. G. Passalacqua, J. S. Reis, F. A. E. Torres, I. Martinez, R. G. Peccinini, C. M. Chin, K. Chegaev, S. Guglielmo, R. Fruttero, M. A. S. Graminha and J. L. d. Santos, Leishmanicidal activities of novel synthetic furoxan and benzofuroxan derivatives, Antimicrob. Agents Chemother., 2014, 58, 4837–4847 CrossRef PubMed.
- L. d. C. Clementino, G. F. S. Fernandes, I. M. Prokopczyk, W. C. Laurindo, D. Toyama, B. P. Motta, A. M. Baviera, F. Henrique-Silva, J. L. d. Santos and M. A. S. Graminha, Design, synthesis and biological evaluation of N-oxide derivatives with potent in vivo antileishmanial activity, PLoS One, 2021, 16, e0259008 CrossRef CAS PubMed.
- Y. Hotta, T. Kataoka, T. Mori and K. Kimura, Review of a potential novel approach for erectile dysfunction: Light-controllable nitric oxide donors and nanoformulations, Sex. Med. Rev., 2020, 8, 297–302 CrossRef PubMed.
- S. Sortino, Light-controlled nitric oxide delivering molecular assemblies, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC.
- R. Matsubara, S. Takazawa, A. Ando, M. Hayashi, R. Tohda and M. Tsubaki, Study on the photoinduced nitric-oxide-releasing ability of 4-alkoxy furoxans, Asian J. Org. Chem., 2017, 6, 619–626 CrossRef CAS.
- C. P. Seymour, R. Tohda, M. Tsubaki, M. Hayashi and R. Matsubara, Photosensitization of fluorofuroxans and its application to the development of visible light-triggered nitric oxide donor, J. Org. Chem., 2017, 82, 9647–9654 CrossRef CAS PubMed.
- C. P. Seymour, A. Nakata, M. Tsubaki, M. Hayashi and R. Matsubara, A fluorescent naphthalenediimide-alkoxyfuroxan photoinduced nitric oxide donor, Bull. Chem. Soc. Jpn., 2019, 92, 162–169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |