DOI:
10.1039/D3RA00137G
(Paper)
RSC Adv., 2023,
13, 6747-6759
Water–SDS–[BMIm]Br composite system for one-pot multicomponent synthesis of pyrano[2,3-c]pyrazole derivatives and their structural assessment by NMR, X-ray, and DFT studies†
Received
8th January 2023
, Accepted 16th February 2023
First published on 27th February 2023
Abstract
Here, we report a simple, efficient, and green protocol for the one-pot synthesis of pyrano[2,3-c]pyrazole derivatives via a sequential three-component strategy using aromatic aldehydes, malononitrile and pyrazolin-5-one in a water–SDS–ionic liquid system. This is a base and volatile organic solvent-free approach that could be applicable to a wide substrate scope. The key advantages of the method over other established protocols are very high yield, eco-friendly conditions, chromatography-free purification and recyclability of the reaction medium. Our study revealed that the N-substituent present in pyrazolinone controls the selectivity of the process. N-unsubstituted pyrazolinone favours the formation of 2,4-dihydro pyrano[2,3-c]pyrazoles whereas under identical conditions N-phenyl substituent pyrazolinone favours the formation 1,4-dihydro pyrano[2,3-c]pyrazoles. Structures of the synthesized products were established by NMR and X-ray diffraction techniques. Energy optimized structures and energy gaps between the HOMO-LUMO of some selected compounds were estimated using density functional theory to explain the extra stability of the 2,4-dihydro pyrano[2,3-c]pyrazoles over 1,4-dihydro pyrano[2,3-c]pyrazoles.
Introduction
In the present day context, organic synthesis via multicomponent reactions (MCRs) becomes a very important method of choice to save time, solvent, energy and for executing the process in a cost effective manner. MCRs provide easy access to wide range of novel functional molecules with high efficiency in terms of yield and selectivity.1 The heterocyclic compounds are widely spread in nature and play an important role in life processes and therefore they are well recognized in biological and therapeutic arenas.2 Consequently, synthesis of novel functionally diversified compounds specially heterocyclic compounds by MCR approach has attracted tremendous attention not only to organic synthetic chemists but also pharmaceutical and medicinal chemistry researchers across the globe. Amongst them, pyrano[2,3-c]pyrazoles (Fig. 1, 1 or 1′) is a fused heterocycle comprised of pyrazole and pyran rings which are known as the sub-structural units of several biologically active compounds3 and widely used as medicinal intermediates due to their biological and pharmacological properties such as analgesic and anti-inflammatory,4a,b anticancer,5a,b anti-oxidant,6a–c anticholine-sterase,7 anti-HIV,8 anti-microbial activity,9a–d human checkpoint kinase 1 (Chk1) inhibitor,10 fungicidal,11 bactericidal12 and vasodilatory13 activity. They are also used as biodegradable agro-chemicals due to their fungicidal, bactericidal, and herbicidal properties14 and pharmaceutical ingredients.15 Some 2-amino-4H-pyrans can be used as photoactive materials16 as they can undergoes dimerization upon UV irradiation, therefore they can act as UV absorbers.17 Furthermore, these compounds are used as cosmetics and pigments.18 Fig. 2 displayed structures of some of the important pyranopyrazoles with their possible applications.4–13,19a–f
 |
| | Fig. 1 Structure of 2,4-dihydropyrano-[2,3-c]pyrazole (1) and 1,4-dihydropyrano[2,3-c]pyrazole (1′). | |
 |
| | Fig. 2 Structures and applications of some biologically active pyranopyrazoles | |
Thus, the synthesis of the heterocyclic compounds containing the pyranopyrazole moiety is of great importance and fascinated by research community as an interesting synthetic target since 19th century after its first report from Otto et al.20 Several procedures for the synthesis of pyranopyrazoles (1 or 1′) were reported,21a–c and documented by Mamaghani and co-workers in the recent review.21d Literature survey indicated that most of the synthesis of pyranopyrazoles are revolve around – (i) four component assembly of aldehydes (2), malononitrile (3) and β-ketoester (4) with hydrazine or its derivatives (5) using catalytic systems such as imidazole,22a [DSim]AlCl4,22b poly(4-vinylpyridine),22c Fe3O4 nanoparticles,22d DABCO,22e thiamine hydrochloride,22f thiourea dioxide,22g ZrO2 nanoparticles,22h ball milling process,22i electro-chemical procedures,22j zwitterionic sulfamic acid functionalized nanoclay,22k Nano-SiO2 catalyst,22l NMO/Ag2O,22m Fe3O4@chitosan-tannic acid nanocatalyst,22n BF3/MNPs,22o (ii) pseudo three-component condensation23a–d of aromatic aldehydes (2), malononitrile (3) and pyrazolinone derivatives (6) and (iii) pseudo two component assembly,24a–d by synthesizing some of its pre-combined constituents like benzylidenemalononitriles (7) and pyrazolone derivatives (6) (Scheme 1). Four component assembly involving aromatic aldehydes, Meldrum's acid, hydrazine hydrate or its derivatives, and ethyl acetoacetate are also known for pyranopyrazole synthesis.25 Rapid four-component assembly in the presence of base in aqueous medium for the synthesis of 2,4-dihydropyranopyrazole analogues were also reported.26 Although the reported methods are effective with their own advantages, however many of the existing methodologies suffer from several drawbacks such as use of toxic bases, tedious catalyst preparation and use of organic solvents rather than aqueous medium, environmental compatibility using toxic and expensive catalysts, lack of recyclability, and co-occurrence of side products. In addition, the ultrasound or microwave assisted synthesis are very fast but requires additional use of sonicator or microwave reactor and may not be suitable for large-scale synthesis. So despite of the available literature for the synthesis of pyranopyrazoles, simple, efficient, and environmentally benign approaches are still demanding. Interestingly, all the four components method started with similar starting materials and worthy to mention that they produced varying product just by changing the catalyst. As example, synthesis of 6-amino-4-substituted-3-methyl-2,4-dihydropyrano-[2,3-c]pyrazole-5-carbonitriles (1) were achieved27–29 by using ionic liquid, Amberlyst A21 or L-cysteine in water under refluxing condition whereas alternative catalysts such as starch solution, silica-coated cobalt oxide nanostructure-catalyst or taurine generates 6-amino-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile derivatives (1′).30–32 Till the date no report on such a selectivity with respect to a specific catalyst are available and details of establishment of the structure was yet to report.
 |
| | Scheme 1 Various strategies for the synthesis of pyranopyrazoles | |
With the growing concern over environment pollution and related societal health problems, green chemistry concept is emerging as one of the important tools in the development of environmentally benign chemical processes and clean technologies including generation of solvent-free protocols along with replacement of volatile organic solvents by water as a green reaction medium. Water is non-hazardous, inexpensive, abundant, and eco-friendly in nature having high boiling point. Reactions in aqueous medium not only possess negative activation volume but also help in controlling exothermic reactions. Hence organic synthesis in aqueous medium is preferred from environmental as well as economical point of view.33
However; poor solubility of organic compounds in water remains a major issue of the success of any organic reaction in water. One of the possible ways to improve the solubility of organic substrates is the use of surface-active agents that can form micelles or vesicular structures. The micellar and vesicle-forming surfactants form hydrophobic cavity in where organic substrates and reagents assembled and undergoes reaction.34
Catalysis has been playing a pivotal role in pollution prevention from fine chemical processes, pharmaceutical industries by activation of functional groups thus by lowering the reaction temperature, reaction time and increased the chemical yield. While choosing the catalyst it is important to think about its water compatibility because water shows some commendable chemical and physical properties which are unrealizable with any other organic solvents. In this respect, ionic liquids plays a larger and more diversified role in catalysis,35 which is reflected by the fact that the number of literature reports on this subject increases annually with many other advantages. To overcome the poor solubility issue of organic compounds in water and enhances the reactivity of functional group, various strategy have been reported in different occasion. In recent studies it was found that imidazolium based ionic liquids with long hydrophobic chains can reduce the CMCs of SDS greatly in water by ionic interaction as well as hydrophobic–hydrophobic interactions between alkyl chains of IL and SDS surfactant.36 Motivated with the recent report on developing the multicomponent reaction for the synthesis of pyranopyrazoles and in continuation of our on-going efforts towards the development of environmentally benign protocols for organic transformations,37a–d we reported herein a methodology for the synthesis of pyranopyrazole by sequential addition of aromatic aldehyde and active methylene compound (Knoevenagel condensation) followed by the addition of pyrazolone (6) derivatives in a one pot fashion at 80 °C in an essentially neutral condition comprising water–SDS–ionic liquid combo system (Scheme 2).
 |
| | Scheme 2 One pot sequential MCR strategy for the synthesis of pyranopyrazoles in aqueous medium. | |
Our method has distinct advantages in comparison to other existing methodology for the same synthesis and these includes – (i) volatile organic solvent and chromatography free procedure (ii) high yield and short reaction time (iii) avoidance of undesirable side reactions, (iv) complete recyclability of the reaction medium which can be used for subsequent cycles without loss of its activity (v) scale up of the protocol with same level of efficiency and (vi) selectivity. Structures of the synthesised compounds were established by 1H, 13C NMR and 1H–1H COSY and XRD studies. In addition, to find out the energetically favourable optimized structure between 2,4-dihydropyranopyrazoles and 1,4-dihydropyaranopyrazoles and to explain the reason for preferential formation of former from un-substituted pyrazolinone we have performed the density functional theory (DFT) calculations of the tautomers at B3LYP/6311G** level in the gas phase by using the computational program Gaussian 09W.
Results and discussions
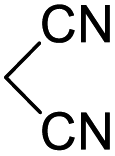
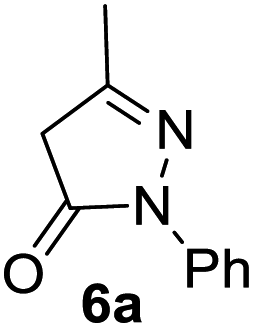
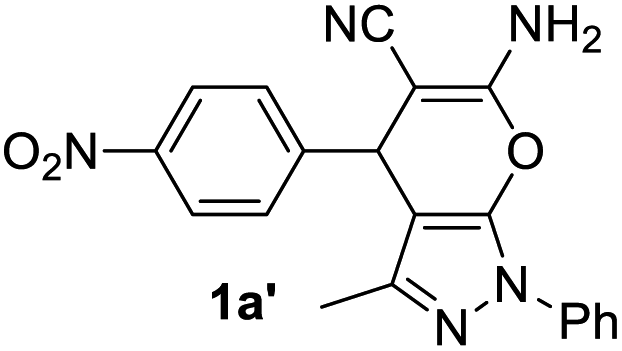
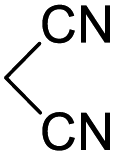
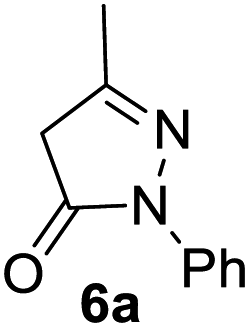
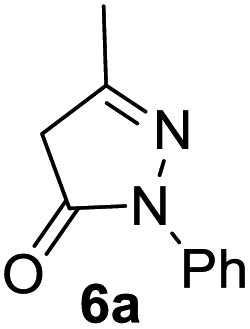
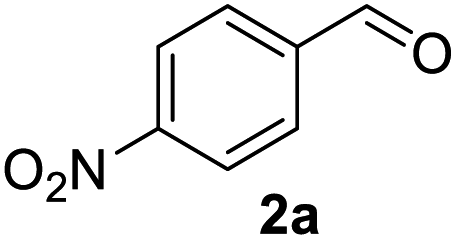
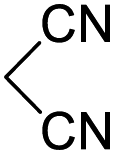
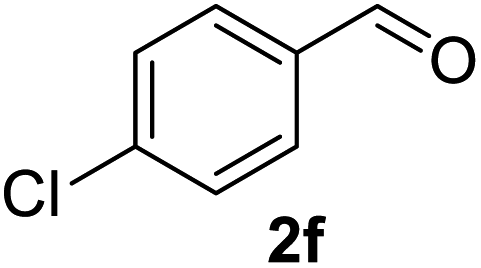
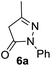
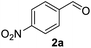
In spite of some success of MCR involving four component assembly of pyranopyrazole synthesis it is matter of concern to subside competitive side reaction between reactive aldehydes with hydrazine/substituted hydrazine to form corresponding hydrazone derivatives (8) of the aldehydes or the formation bispyrazolone (9) from the condensation between aldehyde (2) and pyrazolone (6) formed as intermediates. Three component strategy is also suffering from some drawbacks such as formation of bis-pyrazoles (9) side product (Scheme 1). Based on our recent report37a on a versatile “base free” true “on water” strategy by mobilizing SDS as surfactant and neutral ionic liquid ([BMIm]Br) as activator for Knoevenagel condensation to synthesize arylidenemalononitrile derivatives (7) which is also an important piece of orchestra for the pyranopyrazoles synthesis, initially we began with 4-nitrobenzaldehyde (2a) as a model aromatic aldehyde, malononitrile (3), ethylacetoacetate (4a) and phenylhydrazine (5a). All these components were allowed to react at 80 °C in water–SDS–[BMIm]Br system under the condition established previously for arylidenemalonitrile 7a. Unfortunately, such attempt resulted pyranopyrazole (1a′: R1 = p-NO2, R2 = Me and R3 = Ph) in 43% after 20 min along with the formation of phenylhydrazone derivative (8a, 20%) and bis-pyrazolol derivative (9a, 32%). Lessoned from these results and to suppress the cross reaction possibilities we drop down the approach to a three component variant by synthesizing intermediate 3-methyl-1-phenyl-5-pyrazolone (6a) separately from ethyl acetoacetate (4a) and phenylhydrazine (5a) and then assembling it with 4-nitrobenzaldehyde (2a) and malononitrile (3). This attempt definitely shows improvements in yields of 1a′ (65%) but still bis-pyrazolol (9a, 26%) was formed due to a tandem Knoevenagel–Michael addition between aromatic aldehyde (2a) and pyrazolone (6a). To improve further the yield of pyranopyrazole, we then decided to study the reaction in a sequential manner. In this approach initially only aldehyde (2a) and malononitrile (3) were allowed to react to form benzylidenemalononitrile (4a) in the presence of 10 mol% of [BMIm]Br in SDS-water (8.7 mM) and then pyrazolone derivative (5a) was added. We were happy to notice that this sequential approach works well and expected 6-amino-4-nitrophenyl-1, 4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1a′) was produced in 96% yield without any side reactions (Table 1). Among the two possible pyranopyrazoles 1a and 1a′ only later one is expected and formed as the substituent's R2 and R3 located in 1,3-position. This preliminary result prompted us to investigate the scope and generality of the new protocol for various aldehydes under optimized conditions. Various structurally diverse aromatic aldehydes were considered to establish the generality and functional group tolerance capability of the present methodology towards the synthesis of highly functionalized pyranopyrazole derivatives. Our studies revealed that aromatic aldehydes bearing –NO2, –OH, –OMe, –Cl etc. All underwent facile condensation (with malononitrile) – addition – cyclization with 1-phenyl-3-methyl-5-pyrazolone (5a) or 3-methyl-5-pyrazolone (5b) to give pyranopyrazole derivatives.
Table 1 Optimization of strategy employed for the synthesis of 1a′
| Entry |
Strategya |
Time (min) |
1a′b |
8ab |
9ab |
| Reactions were carried out with SDS (3.5 mol%, 10 mg) and [BMIm]Br (10 mol%, 20 mg) in H2O (4 mL) at 80 °C. Isolated yields. |
| 1 |
Four component (2a +3a + 4a +5a) |
20 |
43 |
20 |
32 |
| 2 |
Three component, non-sequential (2a +3a +6a) |
20 |
65 |
— |
26 |
| 3 |
Three component, sequential (2a+ 3a → 7a then 6a) |
20 + 20 |
96 |
— |
— |
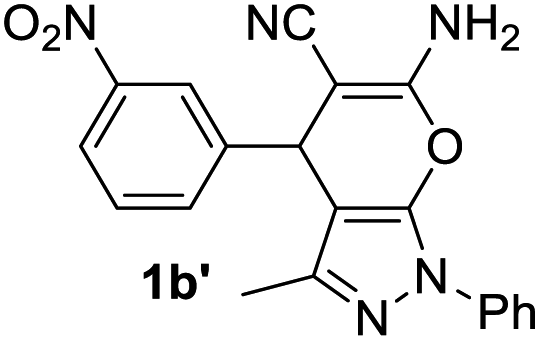
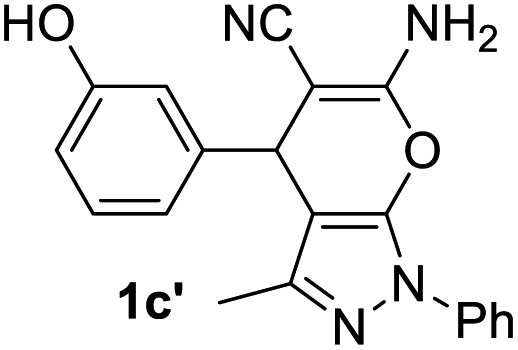
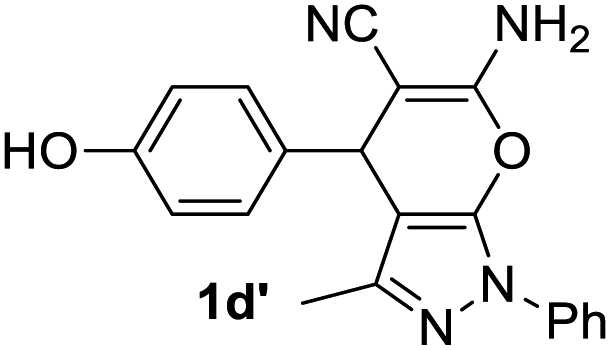
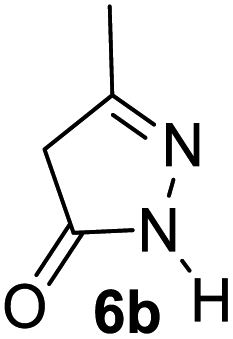
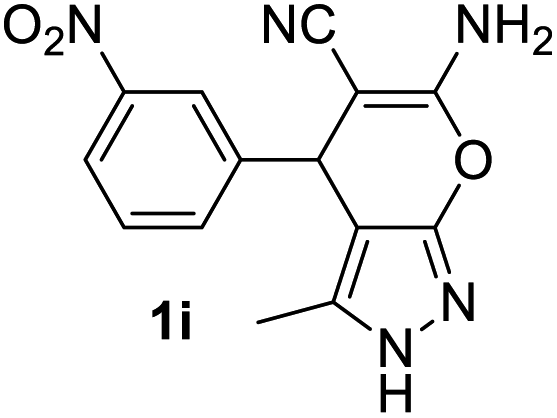
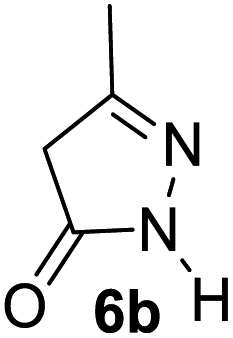
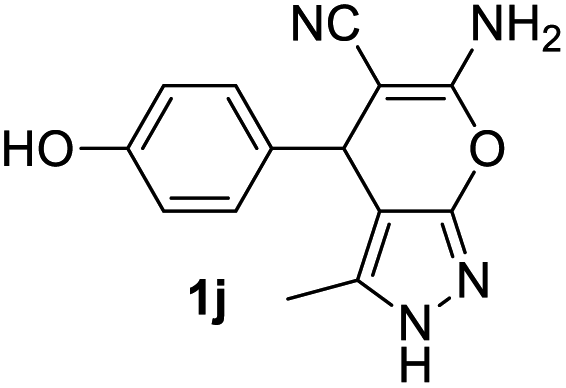
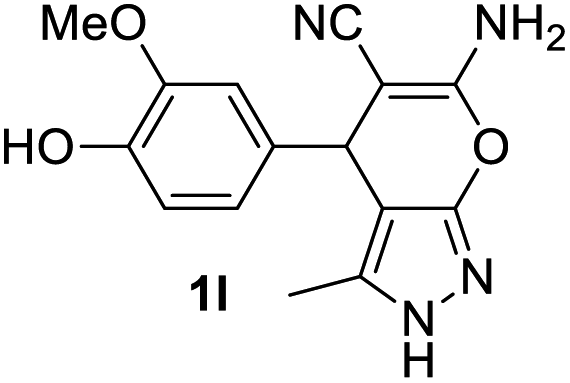
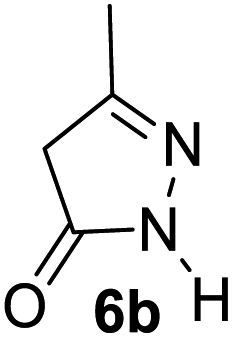
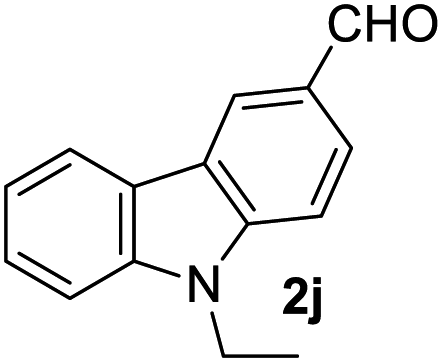
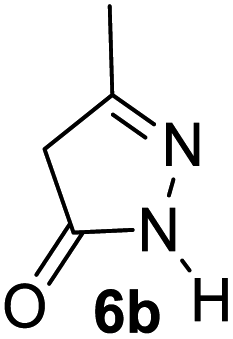
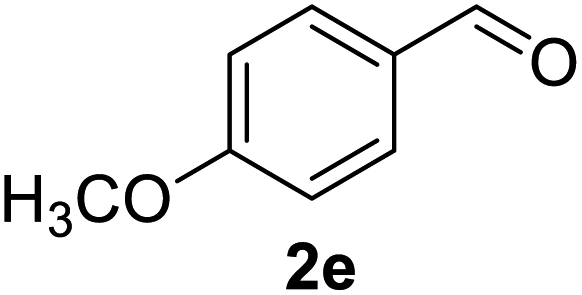
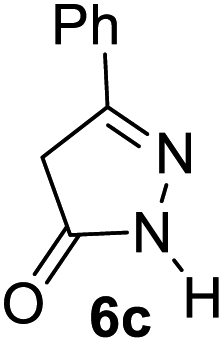
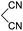
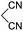
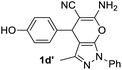
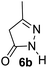
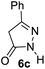
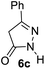
Aldehydes such as m-nitro, m-hydroxy, p-hydroxy and 4-methoxy benzaldehydes, all underwent smooth transformation under the same condition to 6-amino-4-substituted-1-phenyl-1, 4-dihydropyrano-[2,3-c]pyrazole-5-carbonitrile derivatives (1b′–e′) in excellent yield (Table 2, entries 2–5) as 4-nitrobenzaldehyde does (Table 2, entry 1). Here again, no 6-amino-4-substituted 2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile derivatives (1b–e) were detected in any case. However, benzaldehyde and other substituted benzaldehydes upon similar treatment with malononitrile followed by the addition of 3-methyl-5-pyrazolone (6b: R3 = H) produced 6-amino-4-substituted 2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile derivatives (1f–l) as sole product (78–96% isolated yield) as revealed by NMR (Table 2, entries 6–12). Other aldehydes, thiophene-2-carboxaldehyde (2h), anthracene-9-carboxaldehyde (2i) or carbazole-3-carboxaldehyde (2j) also underwent similar transformation to 2,4-dihydro pyranopyrazoles 1m–o in good yields but took unexceptionally longer reaction time (up to 12 h, Table 2, entries 13–15). The 3-phenyl-5-pyrazolone (6c: R3 = H) which was synthesised from ethyl benzoylacetate also produced pyranopyrazoles 1p–r in excellent yield (Table 2, entries 16–18). Unfortunately when we used salicylaldehyde as aldehyde for the synthesis of concerned pyranopyrazole derivatives it produced a complex mixture of products due to the presence of additional –OH group possibly occurring of cyclization product from intermediate 7 (ref. 42) as well as two more cyclization products after nucleophilic addition of pyrazolinone (6b) to the intermediate 7.26b,43 We also tested our methodology for aliphatic aldehydes such as n-butanal, n-hexanal or n-heptanal, however such attempts did not produced any expected products. Perhaps this is may be due to the presence of enolizable hydrogen in the intermediate active dicyano alkene that reduces its nucleophilicity. To check the selectivity, we have studied a control reaction using butanal (1 mmol), 4-hydroxy benzaldehyde (1 mmol) and malononitrile (2 mmol) under water–SDS–ionic liquid system at 80 °C. After consumption of aldehydes, 1 mmol of pyrazolone (5b) was added and heating was continued for 60 min. Only pyranopyrazole (1j) was isolated in 90% yield, no pyranopyrazole having aliphatic residue was detected. This experiment indicated that our protocol is highly selective for aromatic aldehydes. All the synthesized products were fully characterized by their IR, NMR and mass spectral data and are strongly resembled with the data those are reported earlier except two new compounds 1n and 1o.
Table 2 One pot multicomponent synthesis of pyrano[2,3-c]pyrazoles by sequential addition strategy using aldehydes, malononitrile and pyarazolinones in water–SDS–[BMIm]Br system
| Entry |
Aldehyde |
Active methylene |
Pyrazolone |
Product |
Time (mins) |
Yield (%) |
| 1 |
 |
 |
 |
 |
40 |
96 (ref. 22c) |
| 2 |
 |
 |
 |
 |
50 |
95 (ref. 22c) |
| 3 |
 |
 |
 |
 |
50 |
94 (ref. 38) |
| 4 |
 |
 |
 |
 |
70 |
94 (ref. 22c) |
| 5 |
 |
 |
 |
 |
70 |
95 (ref. 22c) |
| 6 |
 |
 |
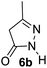 |
 |
60 |
78 (ref. 27) |
| 7 |
 |
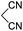 |
 |
 |
40 |
96 (ref. 27) |
| 8 |
 |
 |
 |
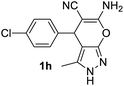 |
50 |
94 (ref. 27) |
| 9 |
 |
 |
 |
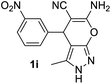 |
50 |
94 (ref. 39) |
| 10 |
 |
 |
 |
 |
60 |
94 (ref. 27) |
| 11 |
 |
 |
 |
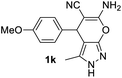 |
70 |
94 (ref. 27) |
| 12 |
 |
 |
 |
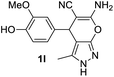 |
60 |
93 (ref. 27) |
| 13 |
 |
 |
 |
 |
120 |
92 (ref. 39) |
| 14 |
 |
 |
 |
 |
720 |
87new |
| 15 |
 |
 |
 |
 |
720 |
85new |
| 16 |
 |
 |
 |
 |
60 |
95 (ref. 40) |
| 17 |
 |
 |
 |
 |
60 |
94 (ref. 41) |
| 18 |
 |
 |
 |
 |
80 |
93 (ref. 40) |
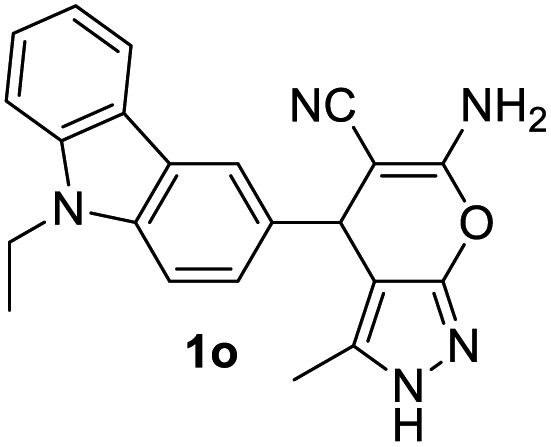
The structure of 2,4-dihydropyrano[2,3-c]pyrazoles were confirmed by 1H–1H COSY in where correlation was found between H–N and protons of methyl group suggesting that they are located at the adjacent position (see ESI†). Aiming to get single crystal of the newly synthesised compounds 1n and 1o, suitable crystals for XRD studies were grown from ethanol-water system. The crystal structure of both 1n and 1o are shown in Fig. 3. The crystallographic data were deposited to Cambridge Crystallographic Data Centre (CCDC no. for 1n and 1o are 2203408 and 2203466 respectively). Single crystal X-ray diffraction study of the these synthesized compounds confirms the identity of 1n or 1o as 2,4-dihydropyranopyrazoles. The crystal packing indicated presence of intermolecular hydrogen bonding interactions (blue dashed lines) between NH2 group of one with either nitrogen of pyrazole or –CN group of second molecule (Fig. 4). Crystal data, data collection and structure refinement details are summarized in Table 3. The structures of 1p–r (R2 = phenyl) were also confirmed as 2,4-dihydropyranopyrrazole by comparing with reported data as well NOESY spectra of 1r. In NOESY spectrum of 1r very nice interaction was seen between –NH and one of the ortho proton of phenyl group at the position 3 of pyrazole ring (ESI†). Based on the experimental results, spectroscopic and crystallographic data we concluded that the present pseudo three component strategy favours 1,4-dihydropyranopyrazoles when N-substituted (R3 = Ph) pyrazolone (6a) is used whereas N-un-substituted (R3 = H) pyrazolone produced 2,4-dihydropyrano-pyrazoles. To establish the higher stability of 2,4-dihydro pyranopyrazoles than the 1,4-dihydropyranopyrazoles, quantum mechanical calculations using Density Functional Theory (DFT)44 with B3LYP/6311G (d,p) in Gauss 09w software45 were performed initially for 1f and 1f′ as model. The optimized structures shown in Fig. 5 were verified by performing the frequency calculation at the same level of theory and found no imaginary frequency. Based on the calculated energy, 1f was found to be more stable than 1f′ by 3.47 kcal mol−1.
 |
| | Fig. 3 Single crystal structures of 1n (A) and 1o (B). | |
 |
| | Fig. 4 Crystal packing of (A) 1n and (B) 1o showing the intermolecular hydrogen bonding involving pryazole N/NH and of pyran NH2. | |
Table 3 Crystallographic experimental details of 1n and 1o
| Crystallographic parameters |
Compound 1n |
Compound 1o |
| Crystal data deposition no. |
CCDC no. 2203408 |
CCDC no. 2203466 |
| Chemical formula |
C22H16N4O |
C22H19N5O |
| Molecular weight |
352.39 g mol−1 |
369.42 g mol−1 |
| Crystal system, space group |
Tetragonal, I41/a |
Tetragonal, I41/a |
| Temperature (K) |
100 |
101 |
| a, b, c (Å) |
28.496(2), 28.496(2), 10.1953(2) |
22.861(2), 22.861(2), 14.185(2) |
| α, β, γ (°) |
90, 90, 90 |
90, 90, 90 |
| V (Å3) |
8279.0 (17) |
7413.4(17) |
| Z |
16 |
16 |
| Radiation type |
Cu Kα (λ = 1.54178) |
Cu Kα (λ = 1.54178) |
| μ (mm−1) |
0.577 |
0.682 |
| Crystal size (mm) |
0.45 × 0.35 × 0.2 |
0.18 × 0.12 × 0.12 |
| Dcalc (g cm−3) |
1.131 |
1.324 |
| Diffractometer |
Bruker Apex III CCD |
Bruker Apex III CCD |
| Absorption correction |
Multiscan (SADABS; Bruker) |
Multiscan (SADABS; Bruker) |
| Tmin, Tmax |
0.630, 0.753 |
0.607,0.753 |
| 2θ range for data collection/° |
9.212 to 136.642 |
7.334 to 136.68 |
| Reflections collected |
126![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 607 607 |
76619 |
| Independent reflections |
3782 |
3383 |
| Rint, Rsigma |
0.0660, 0.0196 |
0.0823, 0.0235 |
| R[F2 > 2σ(F2), wR(F2)] |
0.0639, 0.1999 |
0.0466, 0.1229 |
| Data completeness |
0.994 |
0.994 |
| θ (theta)max |
68.321 |
68.340 |
| Goodness-of-fit on F2 (S) |
1.075 |
1.084 |
 |
| | Fig. 5 DFT (B3LYP/6-311G**) computed optimized structure of 1f, 1f′, 1n and 1n′ | |
To assess a possible reason of the stability of 1f over tautomer 1f′, the HOMO-LUMO analysis of these structure were performed at the B3LYP/6-311G (d,p) level. Observed HOMO-LUMO energy gap of 1f (5.1230 eV) and 1f′ (5.3509 eV) is demonstrative of greater delocalization of the pi electrons in 1f and so it's extra stability. Similar quantum mechanical calculations were also done for 1n (2,4-dihydro) and its 1,4-dihydro form (1n′) using the same basis set. In this case HOMO-LUMO gap estimated as 3.548 eV (1n) and 3.558 eV (1n′) respectively (Fig. 6). Thus, the outcome of the DFT studies reliably in favour of the structure 1f and 1n as established by X-ray and NMR studies. The optimized bond lengths and bond angles of all the tautomers are collected and given in Table 4. The DFT study further indicated that 6-amino-4-(p-chlorophenyl)-3-phenyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1q) is more stable than 6-amino-4-(p-chlorophenyl)-3-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbo-nitrile (1q′) by 4.60 kcal mole−1 (details not given).
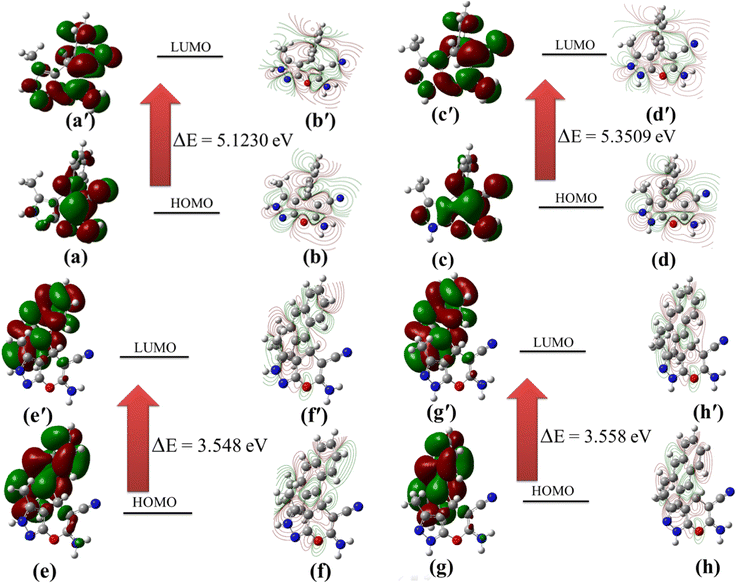 |
| | Fig. 6 (a) Contour plot of HOMO of 1f; (b) transparent surface view of HOMO of 1f; (a′) contour plot of LUMO of 1f; (b′) transparent surface view of LUMO of 1f; (c) contour plot of HOMO of 1f′; (b) transparent surface view of HOMO of 1f′; (c′) contour plot of LUMO of 1f′; (d′) transparent surface view of LUMO of 1f′; (e) contour plot of HOMO of 1n; (f) transparent surface view of HOMO of 1n; (e′) contour plot of LUMO of 1n; (f′) transparent surface view of LUMO of 1n; (g) contour plot of HOMO of 1n′; (h) transparent surface view of HOMO of 1n′; (g′) contour plot of LUMO of 1n′; (h′) transparent surface view of LUMO of 1n′ | |
Table 4 Calculated bond angles and bond distances of optimized structure of 1f/1f′ and 1n/1n′ (only fused ring part is given)
| Bond angles (°) about various centres of optimized structure |
Bond lengths (Å) about various centres of optimized structure |
Bond angles (°) about various centres of optimized structure |
Bond lengths (Å) about various centres of optimized structure |
| Centres (C) |
1f |
1f′ |
Centres(C) |
1f |
1f′ |
Centres (C) |
1n |
1n′ |
Centres(C) |
1n |
1n′ |
| C2–C1–C27 |
114.6734 |
109.0376 |
C1–C2 |
1.4029 |
1.3664 |
C2–C1–C5 |
122.6790 |
121.7823 |
C1–C2 |
1.4075 |
1.3706 |
| C1–C2–C3 |
123.0121 |
121.6485 |
C1–C7 |
1.3692 |
1.3609 |
C1–C2–C7 |
124.6190 |
127.5686 |
C1–C5 |
1.5097 |
1.5091 |
| C1–C2–C6 |
103.5541 |
103.5163 |
C2–C6 |
1.3872 |
1.4251 |
C7–C3–C4 |
123.8036 |
123.1996 |
C1–C6 |
1.3905 |
1.4277 |
| C2–C3–C4 |
106.9077 |
106.8861 |
C2–C3 |
1.5020 |
1.5014 |
C3–C4–C5 |
124.2446 |
124.5625 |
C2–C7 |
1.3914 |
1.3866 |
| C4–C3–C11 |
112.3937 |
111.9773 |
C3–C4 |
1.5325 |
1.5376 |
C3–C4–C13 |
117.5447 |
117.1809 |
C2–C9 |
1.3298 |
1.3540 |
| C3–C4–C5 |
124.3957 |
124.7251 |
C3–C11 |
1.5319 |
1.5316 |
C1–C5–C4 |
108.4460 |
108.3026 |
C3–C7 |
1.3948 |
1.4071 |
| C5–C4–C9 |
117.1141 |
116.7170 |
C4–C5 |
1.3688 |
1.3651 |
C1–C5–C26 |
115.0176 |
115.1381 |
C3–C4 |
1.3698 |
1.3673 |
| C4–C5–C7 |
124.4341 |
123.6470 |
C4–C9 |
1.4140 |
1.4156 |
C4–C5-26 |
113.5236 |
113.6170 |
C3–C8 |
1.3599 |
1.3602 |
| C4–C5–C8 |
125.5914 |
126.4661 |
C5–C7 |
1.3602 |
1.3725 |
C19–C5–C26 |
106.6745 |
106.6107 |
C4–C5 |
1.5430 |
1.5471 |
| C2–C6–C26 |
105.4214 |
111.3308 |
C5–C8 |
1.3631 |
1.3644 |
C1–C6–C10 |
105.5625 |
111.0949 |
C4–C13 |
1.4143 |
1.4157 |
| C2–C6–C22 |
131.6480 |
128.1029 |
C6–C26 |
1.3611 |
1.3298 |
C1–C6–C15 |
131.9322 |
128.8578 |
C5–C26 |
1.5434 |
1.5430 |
| C1–C7–C5 |
115.7246 |
114.1018 |
C6–C22 |
1.4932 |
1.4949 |
C10–C6–C15 |
122.5053 |
120.0452 |
C6–C10 |
1.3748 |
1.3508 |
| C6–C26–C27 |
114.2294 |
105.3770 |
C9–C10 |
1.1599 |
1.1593 |
C6–C10–C9 |
113.6359 |
104.9480 |
C6–C15 |
1.4941 |
1.4928 |
| C26–C27–C1 |
102.1212 |
110.7381 |
C26–C27 |
1.3566 |
1.3566 |
C2–C9–C10 |
101.8039 |
110.2306 |
C9–C10 |
1.3862 |
1.3929 |
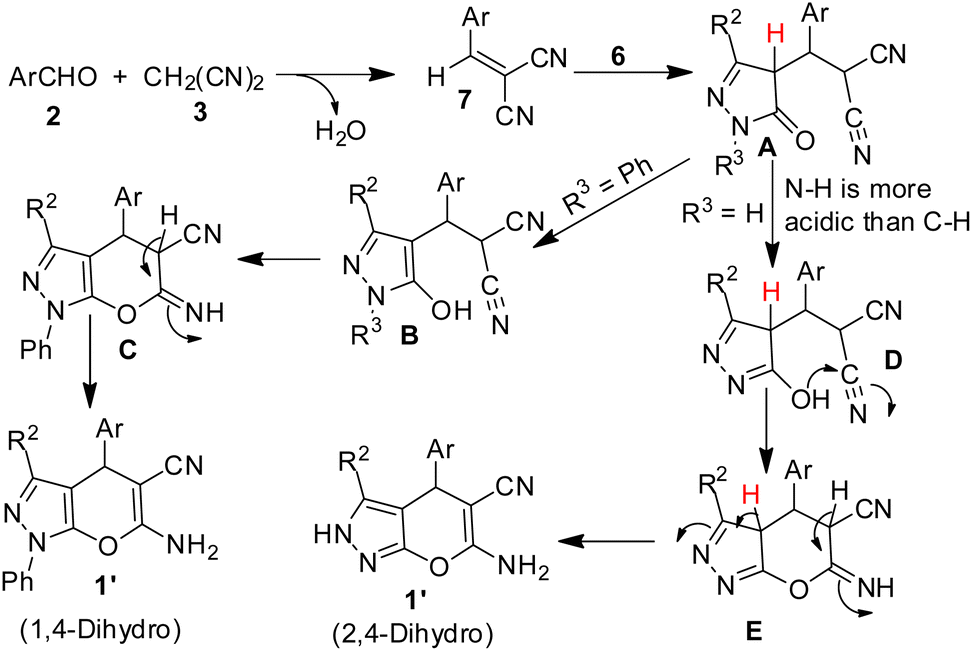
The experimental observations and our previous experiences on SDS–IL–water combo catalytic system, the following mechanistic rationale is proposed. It is perceived that SDS form hydrophobic vesicle in water of size about 190 nm which was established by DLS study and the size of this vesicle is enhanced further with the interionic attraction between imidazolium cation which forms a stern layer, resulting in an increase in mean effective diameter of the cavity.46 The resultant hydrophobic cavity eventually helps the potent transport of all reactants into the nano sized reactor for reaction and efficient formation of highly reactive benzylidenemalononitrile derivative 7. After the sequential addition of pyrazolone (6, 3rd component) a Michael addition reaction took place to form a dicyano intermediate (A) which quickly converted the enols depending upon the nature of R3. When R3 is phenyl, enol B is form which undergoes Pinner type cyclisation to give another intermediate C followed by proton shift afforded 1′ (1,4-dihydro). If R3 is H, enol D is formed due to the relatively weaker N–H bond than C–H bond, which than followed similar type of cyclisation and proton shift due to imine-enamine tautomerization gave produced 1 (Scheme 3). Pinner type cyclisation reaction involving enol –OH to the nitrile group which is extensively influenced and activated by the imidazolium cation resulted in the formation of pyran moiety.
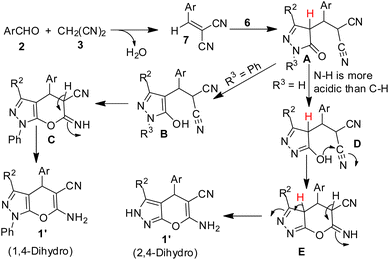 |
| | Scheme 3 Probable mechanism for pyranopyrazole synthesis. | |
To assess the recycling credibility of reaction “waste” (filtrate containing SDS, ionic liquid and water), 4-nitro benzaldehyde and malononitrile were taken in water (4 mL) containing SDS (10 mg) and butyl methyl imidazolium bromide (20 mg). The mixture was stirred vigorously at 80 °C for 15 min, then 1-phenyl-3-methyl-5-pyrazolone was added to the reaction mixture. After completion of reaction (as revealed by TLC) the precipitate so formed was filtered and washed with small amount of water. The combined filtrate was concentrated to total volume of 4 mL, if exceed and used for next cycle. The recovered medium containing water, SDS and ionic liquid (BMImBr) was used six times without notable loss of yield of 1a′ (Fig. 7). The yields of the initial and subsequent four runs were 96% and last two runs were 95% (Fig. 6). Thus, the SDS–IL–water catalytic media is proved to be recyclable without noticeable alteration of its catalytic activity.
 |
| | Fig. 7 Recyclability test of the reaction waste containing water–SDS–BMImBr. | |
Conclusion
In conclusion, we have developed a base and metal free protocol for the selective synthesis of pyrano[2,3-c]pyrazole derivatives via sequential pseudo three component strategy using aromatic aldehydes, malononitrile and pyrazolin-5-one in water–SDS–ionic liquid system. The reaction was conducted under an eco-friendly medium which is completely recyclable including the catalyst. The key advantages of the method over other established protocols are very high yield, eco-friendly condition, chromatography free purification and selectivity. Our study revealed that N-phenyl pyarazolinone favours the formation 1,4-dihydro pyrano[2,3-c]pyrazoles and whereas under identical condition 2,4-dihydro pyrano[2,3-c]pyrazoles were formed when nitrogen is un-substituted. Structures of the synthesized products were established by NMR and X-ray diffraction techniques. The preferential formation of 2,4-dihydro pyrano[2,3-c]pyrazoles was ascertained based on the energy optimization and estimation of HOMO-LUMO energy gap using density functional theory which explain the extra stability of 2,4-dihydro pyrano[2,3-c]pyrazoles over 1,4-dihydro pyrano[2,3-c]pyrazoles.
Experimental section
The starting aldehydes (aromatic, aliphatic, heterocyclic etc.), malononitrile, ethyl acetoacetate, hydrazine, phenyl hydrazine, SDS were purchased either from Sigma Aldrich chemical Co., USA or Acros Organics or SRL India and were used as received. Ionic liquid butyl methyl imidazolium bromide ([BMIm]Br) was prepared in the laboratory using standard protocol. 1H NMR and 13C NMR spectra were recorded at ambient temperature using Bruker Ascend 400 MHz spectrometers (400 MHz for 1H and 100 MHz for 13C). Chemical shifts were reported in parts per million from the tetramethylsilane internal reference, and coupling constants were reported in Hertz. Proton multiplicities were represented as s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), and m (multiplet). FTIR spectra were recorded on Bruker Alpha FTIR spectrometer on Neat or KBr pellets. Mass spectra (HRMS) were obtained from XEVO G2-XS QTOF (Waters) using 70 eV in positive ion mode. The single-crystal X-ray diffraction (XRD) data were collected on a Bruker D8 Venture system with a microfocus optics using Cu Kα radiation. The data were analysed and processed with Bruker Apex III software suite 61 incorporated with multiple tools such as cell_now and RLATT for the determination of the unit cell, SAINT-plus for data reduction, and SADABS for absorption correction. The structure solutions were performed with SHELXT and the full-matrix least-squares refinements were performed with SHELXL suite of programs incorporated in Olex 2.6. For the optimization of geometry of represented compounds the structures were drawn in Gauss view with Gaussian-09W-Gaussview-6 program to obtain the energy minimized structure. The density functional theory (DFT) with the B3LYP correlation function as basis set of calculations have been employed imposing 6-311G(d,p) as additional constraints. Frequency calculations were carried out with the DFT optimized structures using the same basis set to confirm the correctness of optimization and found no imaginary frequency.
Experimental procedure for the synthesis of 1a′–e′ and 1f–o
To a mixture of SDS (10 mg, 3.5 mol%), ionic liquid [BMIm]Br (20 mg, 10 mol%), aromatic aldehydes (2, 1 mmol), malononitrile (73 mg, 1.1 mmol) in H2O (4 mL) were stirred at 80 °C until the disappearance of aldehydes (TLC, usually 20–40 min). Then pyrazolones (6, 1 mmol) was added in the reaction mixture and continued heating at the same temperature till the complete consumption of pyrazolones (Table 2). After the completion of reaction as indicated by TLC, the reaction mixture is cooled to 40 °C, the solid obtained was filtered, washed with water and recrystallized from ethanol–water (∼1:1) mixture. Synthesized products were characterized by melting point, IR data, NMR, mass spectral analysis and compared with the reported one.
Physical and spectral data of new compounds
6-Amino-4-(anthracen-9-yl)-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1n). Yield: 87%, yellowish solid, mp 190–192 °C; IR (KBr) νmax 3394, 3297, 3171, 2888, 2188, 1719, 1636, 1541, 1391, 1155, 1046, 1012, 878, 832, 775, 716 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.71 (d, J = 8.0 Hz, 1H), 8.70 (s, 1H), 8.15–8.05 (m, 3H), 7.61–7.54 (m, 2H), 7.44 (t, J = 8.0 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 6.96 (s, 2H), 6.40 (s, 1H), 1.19 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 161.1, 155.1, 135.7, 133.4, 132.1, 131.2, 131.1, 130.1, 129.8, 129.5, 128.4, 127.1, 125.5, 125.1, 124.5, 124.0, 120.8, 99.1, 58.1, 29.8, 9.8; HRMS calcd for (C22H16N4O + H+) 353.1402 found 353.1402.
6-Amino-4-(9-ethyl-9H-carbazol-3-yl)-3-methyl-2,4-dihydro-pyrano[2,3-c]pyrazole-5-carbonitrile (1o). Yield: 85%, yellowish white solid, mp 200–202 °C; IR (KBr) νmax 3352, 3177, 2970, 2192, 1652, 1600, 1482, 1392, 1332, 1223, 1150, 1103, 1039, 752 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.98 (s, 1H), 7.59–7.53 (m, 2H), 7.44 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.17 (t, J = 8.0 Hz, 1H), 6.86 (s, 2H), 4.77 (s, 1H), 4.41 (d, J = 8.0 Hz, 2H), 1.76 (s, 3H), 1.32 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) d 161.1, 155.2, 140.3, 139.0, 136.1, 135.6, 126.1, 126.0, 122.5, 122.3, 121.5, 120.8, 119.5, 119.1, 109.6, 109.5, 98.9, 58.7, 37.5, 36.9, 14.3, 10.3. HRMS calcd (C22H19N5O + H+) 370.1668 found 370.1668.
Author contributions
S. M. designed the work. S. C. performed all experiments. U. C. D. performed DFT study. B. P. and R. N. performed XRD studies and processing of X-ray data. S. M. and S. C. analyzed the spectral data and wrote the manuscript with input from other authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. C. is thankful to University Grant commission (UGC), Govt. of India for providing UGC-Senior Research Fellowship [No. 16–6(DEC.2018)/2019(NET/CSIR)]. The authors also grateful to Department of Science and Technology (DST), Govt. of India for providing 400 MHz NMR facility under DST-FIST programme (No. SR/FST/CSI-263/2015) and Central Instrumentation Centre (CIC) Tripura University for Instrumental facilities. U. C. D. is thankful to Dr S. K. Sahoo, SVNIT, Gujarat, INDIA for providing software assistance in DFT calculations.
References
-
(a) J. Zhu, Q. Wang and M.-X. Wang, Multicomponent Reactions in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015 Search PubMed;
(b) A. C. Boukis, K. Reiter, M. Frölich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439 CrossRef PubMed;
(c) R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
-
(a) N. Elders, D. van der Born, L. J. D. Hendrickx, B. J. J. Timmer, A. Krause, E. Janssen, F. J. J. de Kanter, E. Ruijter and R. V. A. Orru, Angew. Chem., Int. Ed., 2009, 48, 5856–5859 CrossRef CAS PubMed;
(b) E. M. M. Abdelraheem, S. Shaabani and A. Dömling, Drug Discov. Today Technol., 2018, 29, 11–17 CrossRef PubMed;
(c) R. Sunke, P. V. Babu, S. Yellanki, R. Medishetti, P. Kulkarni and M. Pal, Org. Biomol. Chem., 2014, 12, 6800–6805 RSC.
-
(a) D. Das, R. Banerjee and A. Mitra, J. Chem. Pharm. Res., 2014, 6, 108–116 Search PubMed;
(b) G. M. Reddy, J. R. Garcia, V. H. Reddy, A. K. Kumari, G. V. Zyryanov and G. Yuvaraja, J. Saudi Chem. Soc., 2019, 23, 263–273 CrossRef CAS.
-
(a) S. C. Kuo, L. J. Huang and H. Nakamura, J. Med. Chem., 1984, 27, 539–544 CrossRef CAS PubMed;
(b) A. Kumar, P. Lohan, D. K.Aneja, G. K. Gupta, D. Kaushik and O. Prakash, Eur. J. Med. Chem., 2012, 50, 81–89 CrossRef CAS PubMed.
-
(a) X. Sun, L. Zhang, M. Gao, X. Que, C. Zhou, D. Zhu and Y. Cai, Molecules, 2019, 24, 624 CrossRef PubMed;
(b) S. K. Salama, M. F. Mohamed, A. F. Darweesh, A. H. Elwahy and I. A. Abdelhamid, Bioorg. Chem., 2017, 71, 19–29 CrossRef CAS PubMed.
-
(a) S. P. Khare, T. R. Deshmukh, J. N. Sangshetti, V. M. Khedkar and B. B. Shingate, Synth. Commun., 2019, 49, 2521–2537 CrossRef CAS;
(b) G. M. Reddy, D. Y. Chen, M. V. Subbaiah, L. Jianyou and J. C. Wen, J. Heterocycl. Chem., 2019, 56, 1806–1811 CrossRef CAS;
(c) R. S. Aliabadi and N. O. Mahmoodi, RSC Adv., 2016, 6, 85877–85884 RSC.
- C. Derabli, I. Boualia, A. B. Abdelwahab, R. Boulcina, C. Bensouici, G. Kirsch and A. Debache, Bioorg. Med. Chem. Lett., 2018, 28, 2481–2484 CrossRef CAS PubMed.
- M. Rueping, E. Sugiono and E. Merino, Chem. Eur. – J., 2008, 14, 6329–6332 CrossRef CAS PubMed.
-
(a) B. M. Labana, G. C. Brahmbhatt, T. R. Sutariya, N. J. Parmar, J. M. Padrón, R. Kant and V. K. Gupta, Mol. Divers., 2017, 21, 339–354 CrossRef CAS PubMed;
(b) M. S. Vasava, M. N. Bhoi, S. K. Rathwa, S. S. Shetty, R. D. Patel, D. P. Rajani, S. D. Rajani, A. Patel, H. A. Pandya and H. D. Patel, J. Mol. Struct., 2019, 1181, 383–402 CrossRef CAS;
(c) B. M. Chougala, S. Samundeeswari, M. Holiyachi, L. A. Shastri, S. Dodamani, S. Jalalpure, S. R. Dixit, S. D. Joshi and V. A. Sunagar, Eur. J. Med. Chem., 2017, 125, 101–116 CrossRef CAS PubMed;
(d) G. M. Reddy, J. R. Garcia, G. V. Zyryanov, G. Sravya and N. B. Reddy, Bioorg. Chem., 2019, 82, 324–331 CrossRef CAS PubMed.
- R. Ramtekkar, K. Kumarvel, G. Vasuki, K. Sekar and R. Krishna, Lett. Drug Des. Discovery, 2009, 6, 579–584 CrossRef CAS.
- R. Nagamallu and A. K. Kariyappa, Bioorg. Med. Chem. Lett., 2013, 23, 6406–6409 CrossRef PubMed.
- M. N. Nasr and M. M. Gineinah, Arch. Pharm., 2002, 335, 289–295 CrossRef CAS PubMed.
- V. K. Ahluwalia, A. Dahiya and V. K. Garg, Indian J. Chem., 1997, 36B, 88–90 CAS.
- S. U. Tekale, S. S. Kauthale, K. M. Jadhav and R. P. Pawar, J. Chem., 2013, 1–9 Search PubMed.
- A. M. El-Agrody, A. M. Fouda and A. A. Al-Dies, Med. Chem. Res., 2014, 23, 3187–3199 CrossRef CAS.
- W. M. Armesto, N. Horspool, A. Martin, A. Ramos and C. Seoane, J. Org. Chem., 1989, 54, 3069–3072 CrossRef.
-
(a) R. Ghorbani-Vaghei and V. Izadkhah, Appl. Organomet. Chem., 2018, 32, e4025 Search PubMed;
(b) J. W. Pavlik, V. Ervithayasuporn, J. C. MacDonald and S. Tantayanon, Arkivoc, 2008, 8, 57–68 Search PubMed.
-
(a) E. A. Hafez, M. H. Elnagdi, A. G. A. Elagemey and F. M. A. A. El-Taweel, Heterocycles, 1987, 26, 903–907 CrossRef CAS;
(b) M. A. Sofan, F. M. A. A. El-Taweel and M. H. LiebigsElnagdi, Ann. Chem., 1989, 935–936 CAS;
(c) A. Galil, F. B. Riad, S. Sherif and M. Elnagdi, Chem. Lett., 1982, 1123–1126 CrossRef;
(d) M. Kidwai, S. Saxena, M. Khan and S. Thukral, Bioorg. Med. Chem. Lett., 2005, 15, 4295–4298 CrossRef CAS PubMed.
-
(a) H. Adibi, L. Hosseinzadeh, S. Farhadi and F. Ahmadi, J. Rep. Pharm. Sci., 2013, 2, 116–124 Search PubMed;
(b) N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Front. Chem., 2020, 7, 917 CrossRef PubMed;
(c) H. T. Nguyen, M.-N. H. Truong, T. V. Le, N. T. Vo, H. D. Nguyen and P. H. Tran, ACS Omega, 2022, 7(20), 17432–17443 CrossRef CAS PubMed;
(d) M. A. Abdelgawad, M. B. Labib and M. Abdel-Latif, Bioorg. Chem., 2017, 74, 212–220 CrossRef CAS PubMed;
(e) T. E. Ali, M. A. Assiri, H. M. El-Shaaer, S. M. Abdel-Kariem, W. R. Abdel-Monem, S. M. El-Edfawy, N. M. Hassanin, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Synth. Commun., 2021, 51, 2478–2497 CrossRef CAS;
(f) O. N. Gein, T. M. Zamaraeva and V. L. Gein, Pharm. Chem. J., 2019, 53, 40–42 CrossRef CAS.
- H. H. Otto, Arch. Pharm., 1974, 307, 444–447 CrossRef CAS PubMed 19.
-
(a) M. Koohshari, M. Dabiri and P. Salehi, RSC Adv., 2014, 4, 10669–10671 RSC;
(b) N. Aslam, J. M. White, A. M. Zafar, M. Jabeen, A. Ghafoor, N. Sajid, S. Noreen and M. A. Khand, Arkivoc, 2018,(part vi), 139–203 Search PubMed;
(c) R. K. Ganta, N. Kerru, S. Maddila and S. B. Jonnalagadda, Molecules, 2021, 26, 3270 Search PubMed;
(d) M. Mamaghani and R. H. Nia, Polycyclic Aromat. Compd., 2021, 41, 223–291 Search PubMed.
-
(a) A. Siddekha, A. Nizam and M. A. Pasha, Spectrochim. Acta, Part A, 2011, 81, 431–440 CrossRef CAS PubMed;
(b) A. R. Moosavi-Zare, M. A. Zolfigol, E. Noroozizadeh, M. Tavasoli, V. Khakyzadehb and A. Zare, New J. Chem., 2013, 37, 4089–4094 RSC;
(c) J. Albadi and A. Mansournezhad, Curr. Chem. Lett., 2014, 3, 221–227 CrossRef CAS;
(d) M. A. A. El-Remaily, Tetrahedron, 2014, 70, 2971–2975 CrossRef CAS;
(e) A. S. Waghmare and S. S. Pandit, J. Saudi Chem. Soc., 2017, 21, 286–290 CrossRef CAS;
(f) M. D. Nikam, P. S. Mahajan, A. V. Chate, S. K. Dabhade and C. H. Gill, J. Chil. Chem. Soc., 2015, 60, 2847–2850 CrossRef CAS;
(g) R. H. Vekariya, K. D. Patel and H. D. Patel, Res. Chem. Intermed., 2016, 42, 4683–4696 CrossRef CAS;
(h) A. Saha, S. Payra and S. Banerjee, Green Chem., 2015, 17, 2859–2866 RSC;
(i) M. G. Dekamin, M. Alikhani, A. Emami, H. Ghafuri and S. Javanshir, J. Iran Chem. Soc., 2016, 13, 591–596 CrossRef CAS;
(j) A. Upadhyay, L. K. Sharma, V. K. Singh, R. Dubey, N. Kumar and R. K. Pal Singh, Tetrahedron Lett., 2017, 58, 1245–1249 CrossRef CAS;
(k) J. Safari and M. Ahmadzadeh, J. Taiwan Inst. Chem. Eng., 2017, 74, 14–24 CrossRef CAS;
(l) K. G. Patel, N. M. Misra, R. H. Vekariya and R. R. Shettigar, Res. Chem. Intermed., 2018, 44, 289–304 CrossRef CAS;
(m) M. Beerappa and K. Shivashankar, Synth. Commun., 2018, 48, 146–154 CrossRef CAS;
(n) M. Kamalzare, M. R. Ahghari, M. Bayat and A. Maleki, Sci. Rep., 2021, 11, 20021 CrossRef CAS PubMed;
(o) M. Abdollahi-Alibeik, A. Moaddelia and K. Masoomi, RSC Adv., 2015, 5, 74932–74939 RSC.
-
(a) S. P. Prajapati, D. P. Patel and P. S. Patel, J. Chem. Pharm. Res., 2012, 4, 2652–2655 CAS;
(b) N. R. Mohamed, N. Y. Khaireldin, A. F. Fahmy and A. A. El-Sayed, Der Pharma Chem., 2010, 2, 400–417 CAS;
(c) S.-B. Guo, S.-X. Wang and J.-T. Li, Synth. Commun., 2007, 37, 2111–2120 CrossRef CAS;
(d) B. Y. Riad, A. O. Abdelhamid, F. A. Khalifa and Y. E Saleh, Arch. Pharm. Res., 1989, 12, 201–206 CrossRef CAS.
-
(a) G. Tacconi, G. Gatti, G. Desimoni and V. Messori, J. Prakt. Chem., 1980, 322, 831–834 CrossRef CAS;
(b) Y. Peng, G. Song and R. Dou, Green Chem., 2006, 8, 573–575 RSC;
(c) A. M. Shestopalov, A. P. Yakubov, D. V Tsyganov, Y. M. Emel'yanova and V. N. Nesterov, Chem. Heterocycl. Compd., 2002, 38, 1180–1189 CrossRef CAS;
(d) V. D. Dyachenko and E. B. Rusanov, Chem. Heterocycl. Compd., 2004, 40, 231–240 CrossRef CAS.
- S. H. S. Azzam and M. A. Pasha, Tetrahedron Lett., 2012, 53, 6834–6837 CrossRef CAS.
-
(a) G. Vasuki and K. Kumaraveln, Tetrahedron Lett., 2008, 49, 5636–5638 CrossRef CAS;
(b) K. Kumaravel, B. Rajarathinam and G. Vasuki, RSC Adv., 2020, 10, 29109–29113 RSC.
- U. D. Nimbalkar, J. A. Seijas, M. P. Vazquez-Tato, M. G. Damale, J. N. Sangshetti and A. P. Nikalje, Molecules, 2017, 22, 1628 CrossRef PubMed 25.
- M. Bihani, P. P. Bora, G. Bez and H. Askari, ACS Sustain. Chem. Eng., 2013, 1, 440–447 CrossRef CAS.
- S. Sikandar, A. F. Zahoor, S. Ahmad, M. N. Anjum, M. N. Ahmad and M. S. U. Shah, Curr. Org. Synth., 2020, 17, 457–463 CrossRef CAS PubMed.
- K. G. Patel, N. M. Misra, R. H. Vekariya and R. R. Shettigar, Res. Chem. Intermed., 2018, 44, 289–304 CrossRef CAS.
- S. Shahbazi, M. A. Ghasemzadeh, P. Shakib, M. R. Zolfaghari and M. Bahmani, Polyhedron, 2019, 170, 172–179 CrossRef CAS.
- G. Mali, B. A. Shaikh, S. Garg, A. Kumar, S. Bhattacharyya, R. D. Erande and A. V. Chate, ACS Omega, 2021, 6, 30734–30742 CrossRef CAS PubMed.
-
(a) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC;
(b) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed;
(c) N. A. Harry, S. Radhika, M. Neetha and G. Anilkumar, ChemistrySelect, 2018, 4, 12337–12355 CrossRef.
-
(a) M. Shiri and M. A. Zolfigol, Tetrahedron, 2009, 65, 587–598 CrossRef CAS;
(b) A. Chakraborty, T. Chakraborty, M. I. Menendez and T. Chattopadhyay, ACS Omega, 2019, 4, 11558–11565 CrossRef CAS PubMed;
(c) N. Krause, Curr. Opin. Green Sust. Chem., 2017, 7, 18–22 Search PubMed;
(d) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
-
(a) Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed;
(b) T. Welton, Biophys. Rev., 2018, 10, 691–706 CrossRef CAS PubMed.
-
(a) K. Behera and S. Pandey, J. Colloid Interface Sci., 2007, 316, 803–814 CrossRef CAS PubMed;
(b) A. Żywociński, P. Bernatowicz, D. Pociecha, E. Górecka and J. Gregorowicz, J. Mol. Liq., 2021, 343, 117610 CrossRef;
(c) H. Kumar and A. Katal, J. Phys. Org. Chem., 2021, e4199 CAS.
-
(a) S. Chakraborty, A. Rudra Paul and S. Majumdar, Results Chem., 2022, 4, 100294 CrossRef CAS;
(b) B. Deb, S. Debnath, A. Chakraborty and S. Majumdar, RSC Adv., 2021, 11, 30827–30839 RSC;
(c) K. Das and S. Majumdar, RSC Adv., 2022, 12, 21493–21502 RSC;
(d) A. Chakraborty, S. Debnath, T. Ghosh, D. K. Maiti and S. Majumdar, Tetrahedron, 2018, 74, 5932–5941 CrossRef CAS.
- V. N. Bhosale, J. A. Angulwar, G. S. Khansole and G. S. Waghmare, J. Chem. Pharm. Res., 2014, 6, 733–737 Search PubMed.
- G. S. Kumar, C. Kurumurthy, B. Veeraswamy, P. S. Rao, P. S. Rao and B. Narsaiah, Org. Prep. Proced. Int., 2013, 45, 429–436 CrossRef CAS.
- Y. Zou, Y. Hu, H. Liu and D. Q. Shi, J. Heterocycl. Chem., 2013, 50, 1174–1179 CAS.
- M. G. Dehbalaei, N. Foroughifar, H. Pasdar, A. Khajeh-Amiri, N. Foroughifar and M. Alikarami, Res. Chem. Intermed., 2017, 43, 3035–3051 CrossRef CAS.
- A. Rabahi, M. Makhloufi-Chebli, S. M. Hamdi, A. M. Silva, D. Kheffache, B. Boutemeur-Kheddis and M. Hamdi, J. Mol. Liq., 2014, 195, 240–247 CrossRef CAS.
- K. Kumaravel and G. Vasuki, Green Chem., 2009, 11, 1945–1947 RSC.
- P. Pulay, G. Fogarasi, J. E. Boggs and A. Vargha, J. Am. Chem. Soc., 1983, 105, 7037–7047 CrossRef CAS.
- M. J. Frisch et alGaussian 09, revision A. 02, Gaussian,Inc., Wallingford CT, 2009. http://www.gaussian.com/g_tech/g_ur/m_citation.htm Search PubMed.
- A. Chakraborty, T. Ghosh, D. K. Maiti and S. Majumdar, ChemistrySelect., 2019, 4, 1841–1845 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Bhaswati Paulb,
Utpal Chandra De
a,
Bhaswati Paulb,
Utpal Chandra De