
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anticancer evaluation, and in silico ADMET analysis of novel thalidomide analogs as promising immunomodulatory agents†
Anas Ramadan Kotba,
Abdallah E. Abdallah *a,
Hazem Elkady*a,
Ibrahim H. Eissaa,
Mohammed S. Taghoura,
Dina Abed Bakhotmahb,
Tamer M. Abdelghanycd and
Mohamed Ayman El-Zahabi*a
*a,
Hazem Elkady*a,
Ibrahim H. Eissaa,
Mohammed S. Taghoura,
Dina Abed Bakhotmahb,
Tamer M. Abdelghanycd and
Mohamed Ayman El-Zahabi*a
aPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: Abdulla_emara@azhar.edu.eg; Hazemelkady@azhar.edu.eg; malzahaby@azhar.edu.eg
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
cDepartment of Pharmacology& Toxicology, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt
dDepartment of Pharmacology& Toxicology, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
First published on 3rd April 2023
Abstract
Immunomodulatory medications like thalidomide and its analogs prevent the production of some proinflammatory cytokines linked to cancer. A new series of thalidomide analogs were designed and synthesized in order to develop potential antitumor immunomodulatory agents. The antiproliferative activities of the new candidates against a panel of three human cancer cell lines (HepG-2, PC3 and MCF-7) were assessed in comparison to thalidomide as a positive control. The obtained results showed the relative significant potency of 18f (IC50 = 11.91 ± 0.9, 9.27 ± 0.7, and 18.62 ± 1.5 μM) and 21b (IC50 = 10.48 ± 0.8, 22.56 ± 1.6, and 16.39 ± 1.4 μM) against the mentioned cell lines, respectively. These results were comparable to thalidomide (IC50 = 11.26 ± 0.54, 14.58 ± 0.57, and 16.87 ± 0.7 μM, respectively). To see to what extent the biological properties of the new candidates are relative to those of thalidomide, the effects of 18f and 21b on the expression levels of TNF-α, CASP8, VEGF, and NF-κB P65 were evaluated. Significant reductions in the proinflammatory TNF-α, VEGF, and NF-κB P65 levels in HepG-2 cells were observed after exposure to compounds 18f and 21b. Furthermore, a sharp increase in CASP8 levels was detected. The obtained results revealed that 21b is of greater significance than thalidomide in TNF-α and NF-κB P65 inhibition. The in silico ADMET and toxicity studies showed that most of tested candidates have a good profile of drug-likeness and low toxicity potential.
1. Introduction
Immunomodulatory drugs modify the response of the immune system by increasing (immunostimulants) or decreasing (immunosuppressives) the production of serum antibodies.1 Immunostimulant drugs are prescribed to enhance the immune response against infectious diseases, tumors, primary or secondary immunodeficiency, and alterations in antibody transfer, among others.2 Immunosuppressive drugs are used to reduce the immune response against transplanted organs and to treat autoimmune diseases such as pemphigus, lupus, or allergies.3 In contrast to immunosuppressive agents that inhibit the immune response in transplant rejection and autoimmunity, a few immunostimulatory drugs have been developed with applicability to infection, immunodeficiency, and cancer. Problems with such drugs include systemic (generalized) effects at one extreme or limited efficacy at the other.3Thalidomide I, lenalidomide II, and pomalidomide III are a class of immunomodulatory drugs that contain imide groups targeting cereblon (CRBN). These drugs work through various mechanisms of actions that promote malignant cell death and enhance host immunity.4,5 Thalidomide is considered as a prototype of the glutarimide-containing immunomodulatory agents.6 It inhibits the production of many proinflammatory mediators such as tumor necrosis factor-alpha (TNF-α) and can affect the production of others, such as interleukin-1b (IL-1b), IL-2, IL-4, IL-5, IL-6, IL-10, and interferon-γ (IFN-γ).7 Moreover, thalidomide and its analogs inhibit the secretion of both beta fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) from cancer cells and bone marrow stromal cells, leading to reduction of endothelial cell migration and proliferation and induction of apoptosis.8
Second-generation thalidomide analogs, lenalidomide II, is a potent immunomodulator that is 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more potent than thalidomide as an inhibitor of TNF-α.9 Clinical studies have revealed that lenalidomide demonstrates fewer side effects and almost neither neurological toxicity nor teratogenicity, compared to thalidomide.10 Structural modification of thalidomide via the addition of an amino group at the 4-position of the phthaloyl ring forming pomalidomide III which was 10-fold more potent than lenalidomide as a TNF-α inhibitor and interleukin-2 (IL-2) stimulator.11 It also showed better anti-angiogenic results than thalidomide and lenalidomide.12
000 times more potent than thalidomide as an inhibitor of TNF-α.9 Clinical studies have revealed that lenalidomide demonstrates fewer side effects and almost neither neurological toxicity nor teratogenicity, compared to thalidomide.10 Structural modification of thalidomide via the addition of an amino group at the 4-position of the phthaloyl ring forming pomalidomide III which was 10-fold more potent than lenalidomide as a TNF-α inhibitor and interleukin-2 (IL-2) stimulator.11 It also showed better anti-angiogenic results than thalidomide and lenalidomide.12
Great attention has focused on the possible use of thalidomide as an anti-angiogenic agent – a property that might account for its teratogenicity. The ability of a tumor to induce new blood vessel formation is crucial for the growth of solid tumors and for metastasis. The similarities between this process in the promotion of tumor growth and in chronic inflammation supports a possible role for thalidomide in the treatment of cancers. There are reports of efficacy in patients with multiple myeloma13 and thalidomide has been reported to possess anti-angiogenic properties.14
Other CRBN targeting thalidomide analogs were developed in an attempt to overcome the toxicity of thalidomide. These compounds include CC-122 (avadomide) IV, CC-220 (iberdomide) V, and CC-885 VI.15 Avadomide is effective for the treatment of multiple myeloma and diffuse large B-cell lymphoma (DLBCL), whereas iberdomide is effective for systemic lupus erythematosus (SLE)16 (Fig. 1). CC-885 was recently identified as CRBN modulator with potent activities against solid tumors.17
 | ||
| Fig. 1 Reported thalidomide analogs having the same pharmacophoric features. | ||
Three common pharmacophoric features of thalidomide and its analogs can be identified, as is presented in Fig. 1. These features include: (i) aromatic domain, (ii) spacer, and (iii) glutarimide moiety. In this work, we designed and synthesized a new series of anticancer agents having the same essential pharmacophoric features of thalidomide.
1.1. Rationale of molecular design
Forcing by all mentioned facts and the promising findings obtained in our former work,18 we decided to start the present work. The rational of molecular design depended on ligand-based drug design approach to develop potent thalidomide analogs. In line with the pharmacophoric features of thalidomide and on the basis of the structural features of potent thalidomide analogs, we made modifications on thalidomide structure (Fig. 2). It can be noticed that the modifications were at three different sites including the aromatic ring system, the linker, and the glutarimide moiety. | ||
| Fig. 2 Rationale of the work using thalidomide as lead compound. | ||
Initially, different aromatic systems extracted from potent thalidomide analogs of significant clinical characteristics have been designed. Similar to avadomide, compounds 17a–g and 20a–c were based on quinazolinone. Meanwhile, substituted phenylthiourea moiety was constructed in compound 12 as a bio-isostere to phenylurea moiety presented in CC-885. Furthermore, anilide group was introduced in compounds 6a,b and 9 as can be seen in Fig. 2.
With respect to the linker, we can see amide group in compounds 6a,b, 9, and 12 and thioacetamide in compounds 17a–g and 20a–c. The two linkers were of different lengths to study the effect of the distance between the aromatic system and the glutarimide moiety on the activity. At the same time, the new linkers had two or three bonds of free rotation, which would contribute to the flexibility of the new molecules relative to thalidomide which contains a one bond linker with restricted rotation.
Regarding the glutarimide moiety, it was kept as it is in all the designed members, whereas it was only replaced with 3-chloro-4-fluorophenyl moiety in compounds 20a–c. Variability of the substitutions enabled us to study the structure–activity relationships of the final compounds as a main objective of this work (Fig. 2).
The antiproliferative activities of synthesized compounds were assessed against three cancer cell lines namely; hepatocellular carcinoma (HepG-2), prostate carcinoma (PC3), and breast cancer (MCF-7). The immunomodulatory activities of the synthesized compounds were evaluated against different enzymes including caspase-8, VEGF, NFκB P65, and TNF-α. Furthermore, the kinetic and toxicity profile of the synthesized compounds were assessed in silico.
2. Results and discussion
2.1. Chemistry
According to the above rationale, Schemes 1–4 were carried out for furnishing the target compounds 6a,b, 9, 12, 18a–g, and 21a–c.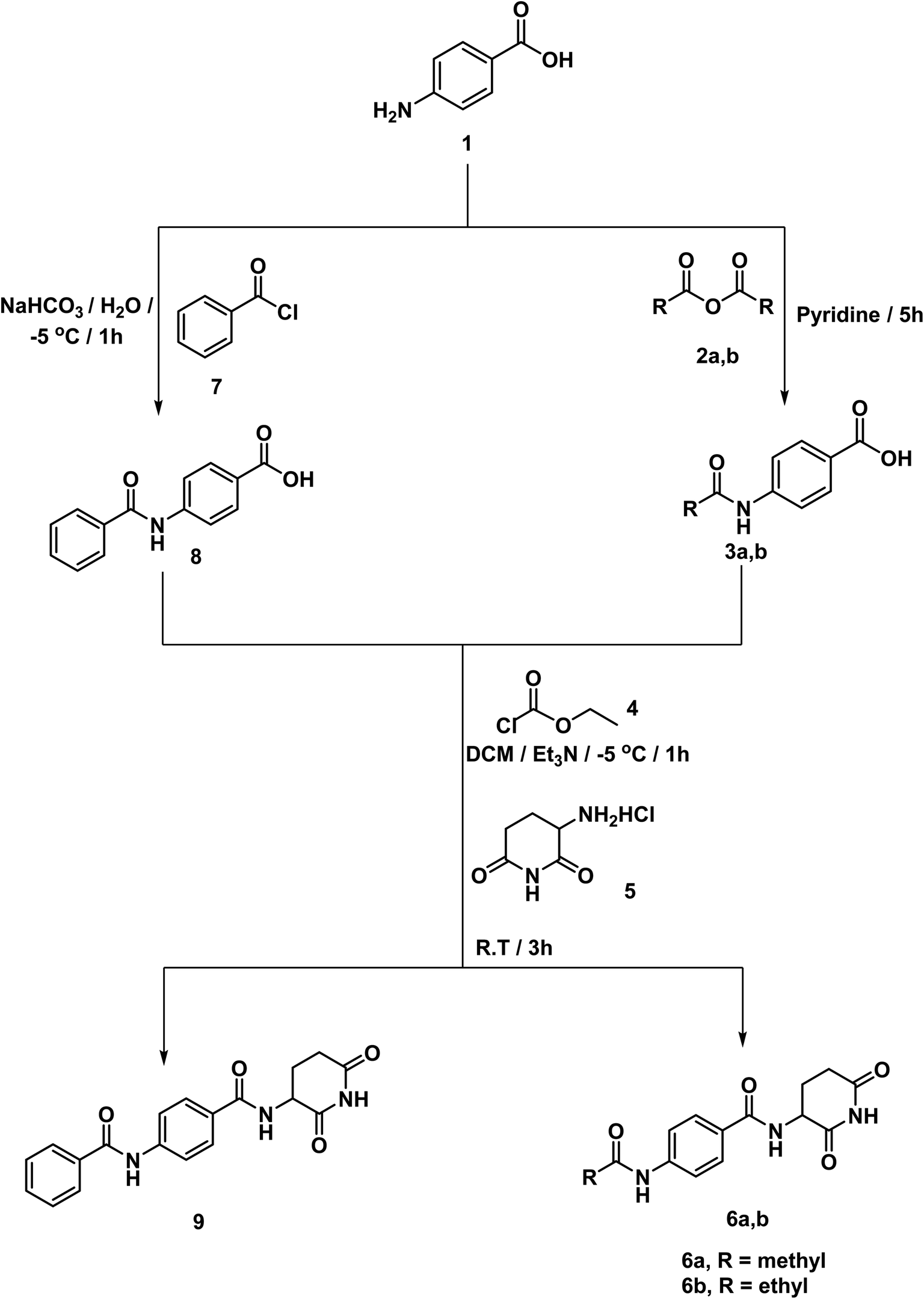 | ||
| Scheme 1 Synthesis of compounds 6a,b, and 9. | ||
Compounds 3a,b were synthesized according to the reported method which involves addition of the appropriate anhydride to 4-aminobenzoic acid in pyridine at r.t.19 Pyridine was a suitable solvent because it is of pKa 5.2. It is a stronger base than the amino group of 4-aminobenzoic acid which has pKa of 2.7. Hence, pyridinium carboxylate salt will be formed instead of zwitter ion of 4-aminobenzoic acid increasing the availability of the lone pair of the amino group. At the same time, compound 8 was formed via drop wise addition of benzoyl chloride to an aqueous solution of sodium 4-aminobenzoate with stirring. Next, the target compound was furnished using mixed anhydride method that involved reaction of ethyl chloroformate with the carboxylic group of 3a,b, and 8 to give the corresponding mixed anhydride in DCM. Then 3-aminopiperidine-2,6-dione was allowed to react with the formed anhydrides to furnish the target compounds 6a,b, and 9, respectively (Scheme 1). The structures of compounds 6a,b, and 9 were verified by their elemental and spectral data. The IR spectra of this series revealed the presence of imide carbonyl absorption bands at a range of 1690 to 1730 cm−1, amide carbonyl absorption bands at a range of 1645 to 1680 cm−1 and presence of NH bands at a range of 3176–3368 cm−1. On the other hand, the 1H NMR spectra showed peaks at about 10.85, 10.3 and 8.6 ppm for imidic NH and the two amidic NH, respectively. Moreover, their 1H NMR spectra revealed the signals of the aliphatic NCH and CH2CH2CO proton at a range of 4.79–4.72 and 2.61–1.92 ppm, respectively for the introduced glutarimide ring.
The intermediate compounds 11a,b were synthesized by refluxing 4-aminobenzoic acid with the proper isothiocyanates in ethanol in the presence of Et3N. In this reaction, the nucleophilic amino group of 4-aminobenzoic acid attacked the electron deficient carbon of isothiocyanates. Then compound 12 was synthesized in line with a reported mixed anhydride method20,21 by reaction of 4-(3-ethylthioureido)benzoic acid 11 with ethyl chloroformate in the presence of Et3N followed by 3-aminopiperidine-2,6-dione (Scheme 2). The structure of compound 12 was verified by its elemental and spectral data. The IR spectrum revealed the presence of imide carbonyl and amide carbonyl absorption band at 1726. On the other hand, the 1H NMR spectrum of compound 12 showed peaks at about 10.85, 10.3 and 8.3 ppm for imidic NH and the two amidic NH, respectively. Moreover, the 1H NMR spectrum revealed the signals of the aliphatic NCH and CH2CH2CO proton at a range of 4.77 and 2.80–2.56 ppm, respectively for the introduced glutarimide ring.
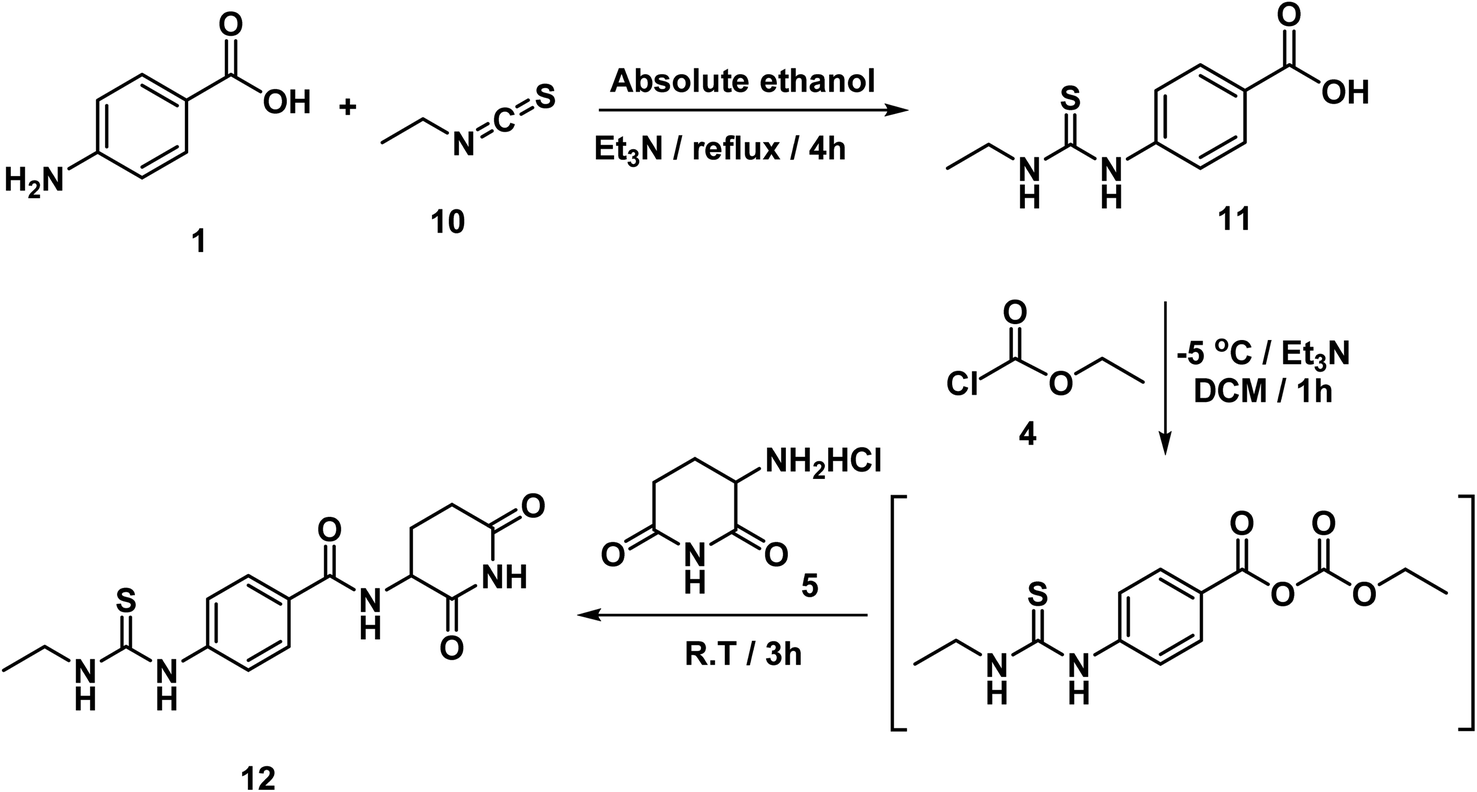 | ||
| Scheme 2 Synthesis of compounds 12. | ||
In accordance with the reported method, we prepared compounds 14a–g. This method involves refluxing an appropriate anthranilic acid derivative namely; 6-chloroanthranilic acid 13a, 3-chloroanthranilic acid 13b, 6-fluoroanthranilic acid 13c, 5-fluoroanthranilic acid 13d, and 5-chloroanthranilic acid 13e with the appropriate isothiocyanates namely; ethyl isothiocyanate 10a or allyl isothiocyanate 10c in absolute ethanol and in the presence of Et3N.22,23 The potassium salts 15a–g were furnished via heating the appropriate 2-mercaptoquinazolin-4-one derivatives 14a–g with equimolar amount of KOH in ethanol.23 On the other hand, compound 16 was produced in two phase system (DCM and water) in the presence of two molar equivalent of NaHCO3 in ice-salt bath as reported. Then, we obtained the final compounds 18a–g in good yields by stirring the appropriate mercapto salt of quinazoline derivatives 15a–g with the chloroacetamide derivative 16 and KI at r.t. in acetonitrile (Scheme 3). The structures of compounds 15a–g were verified by their elemental analyses and spectral data. The IR charts of compounds 15a–g showed bands for imide CO of glutarimide ring at about 1708 to 1720 cm−1. While amide CO showed bands from 1623 to 1684 cm−1. On the other hand, the 1H NMR spectra of compounds 15a–g showed peaks at about 10.8 and 8.6 ppm for imide NH and amide NH, respectively. Methylene protons flanked between sulfur and carbonyl appeared as a singlet peak at about 4.1 ppm.
 | ||
| Scheme 3 Synthesis of compounds 18a–g. | ||
The acetamide derivative 20 was obtained by addition of chloroacetyl chloride 16 to a mixture of 3-chloro-4-fluoroaniline 19 and NaHCO3 in DMF in ice salt bath. Stirring the appropriate mercapto salt of quinazoline derivatives 15a–g with the acetamide derivative 20 and KI at r.t. in acetonitrile afforded the final compounds 21a–c in good yields (Scheme 4). The 1H NMR spectra showed peaks at about 10.69 and 10.74 ppm for amide NH. Methylene protons flanked between sulfur and carbonyl appeared as a singlet peak at about 4.2 ppm.
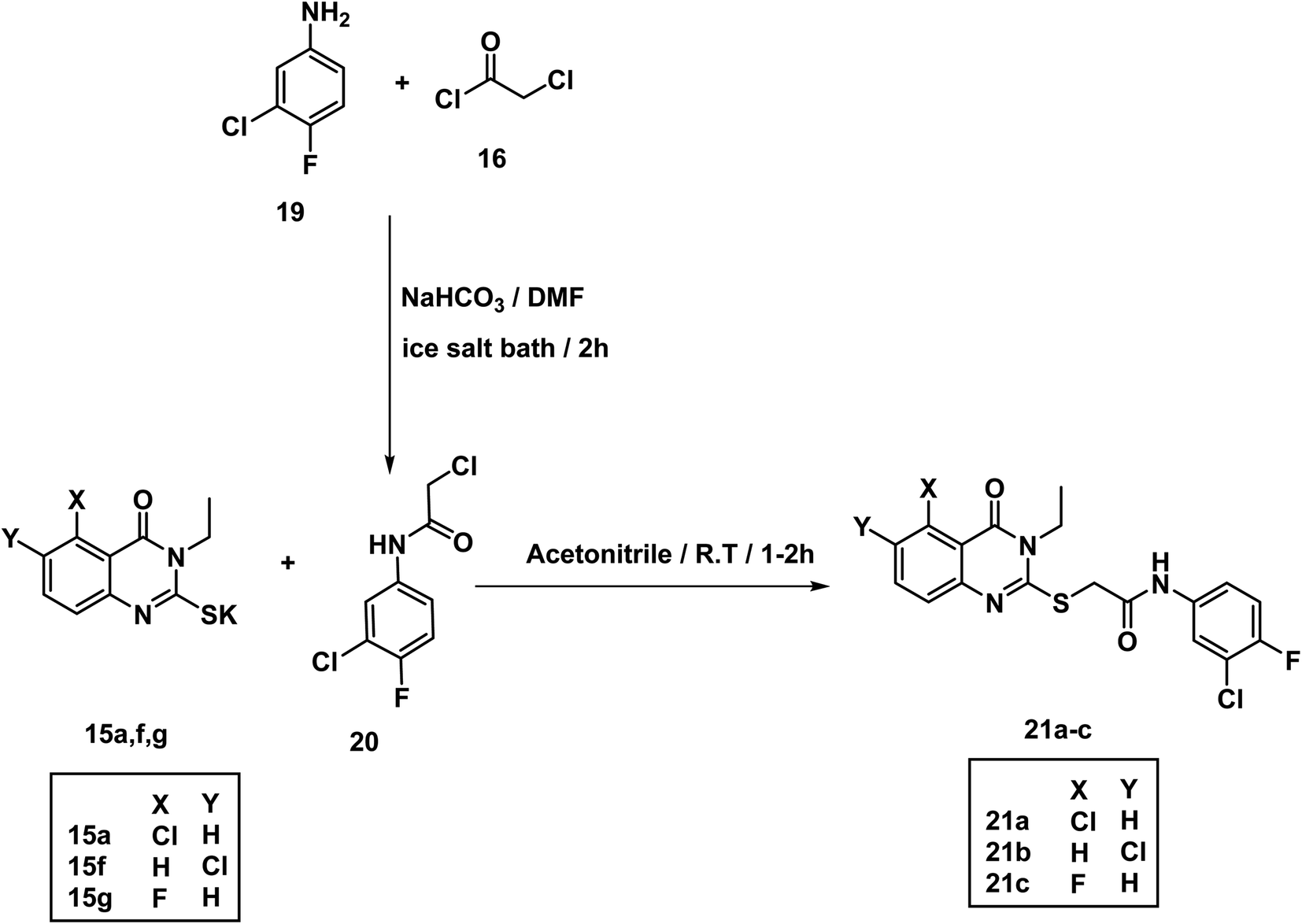 | ||
| Scheme 4 Synthesis of compounds 21a–c. | ||
2.2. Biological testing
| Comp. | In vitro cytotoxicity IC50 (μM) a | ||
|---|---|---|---|
| HepG-2 | PC3 | MCF-7 | |
| a Three independent experiments were performed for each concentration. | |||
| 6a | 49.34 ± 3.4 | 39.27 ± 2.7 | 65.27 ± 3.6 |
| 6b | 62.78 ± 3.8 | 48.02 ± 3.1 | 56.39 ± 3.3 |
| 9 | 53.39 ± 3.6 | 57.48 ± 3.4 | 78.25 ± 4.4 |
| 12 | 37.22 ± 2.6 | 25.91 ± 1.9 | 34.81 ± 2.5 |
| 18a | 18.90 ± 1.4 | 32.86 ± 2.3 | 37.95 ± 2.5 |
| 18b | 39.76 ± 2.6 | 60.29 ± 3.6 | 49.70 ± 2.9 |
| 18c | 33.82 ± 2.2 | 12.13 ± 1.1 | 26.89 ± 2.1 |
| 18d | 72.31 ± 3.9 | 82.38 ± 4.3 | 93.16 ± 4.8 |
| 18e | 42.65 ± 3.1 | 31.36 ± 2.4 | 64.03 ± 3.4 |
| 18f | 11.91 ± 0.9 | 9.27 ± 0.7 | 18.62 ± 1.5 |
| 18g | 30.23 ± 2.3 | 23.04 ± 1.9 | 36.18 ± 2.7 |
| 21a | 52.47 ± 3.1 | 39.94 ± 2.7 | 61.51 ± 3.3 |
| 21b | 10.48 ± 0.8 | 22.56 ± 1.6 | 16.39 ± 1.4 |
| 21c | 55.02 ± 3.2 | 67.72 ± 3.9 | 79.05 ± 4.0 |
| Thalidomide | 11.26 ± 0.54 | 14.58 ± 0.57 | 16.87 ± 0.7 |
 | ||
| Fig. 3 SAR of the synthesized members. | ||
Regarding 3-ethyl-2-substitutedquinazolin-4-one containing derivatives; 5-fluoro candidate 18c (IC50 = 33.82 ± 2.2, 12.13 ± 1.1, and 26.89 ± 2.1 μM against HepG-2, PC3, and MCF-7, respectively) was more potent than the corresponding 6-fluoro one 18d (IC50 = 72.31 ± 3.9, 82.38 ± 4.3, and 93.16 ± 4.8 μM against HepG-2, PC3, and MCF-7, respectively) against all cell lines. Moreover, 5-chloro derivative 18a (IC50 = 18.90 ± 1.4, 32.86 ± 2.3, and 37.95 ± 2.5 μM against HepG-2, PC3, and MCF-7, respectively) was more potent than the corresponding 8-chloro one 18b (IC50 = 39.76 ± 2.6, 60.29 ± 3.6, and 49.70 ± 2.9 μM against HepG-2, PC3, and MCF-7, respectively) against all cell lines. For 3-ally-2-substitutedquinazolin-4-derivatives, it was noticed that 6-chloro counterpart 18f (IC50 = 11.91 ± 0.9, 9.27 ± 0.7, and 18.62 ± 1.5 μM against HepG-2, PC3, and MCF-7, respectively) was more advantageous than 5-fluoro analog 18g (IC50 = 30.23 ± 2.3, 23.04 ± 1.9, and 36.18 ± 2.7 μM against HepG-2, PC3, and MCF-7, respectively). However, the 5-chloro containing derivative 18e (IC50 = 42.65 ± 3.1, 31.36 ± 2.4, and 64.03 ± 3.4 μM against HepG-2, PC3, and MCF-7, respectively) displayed less potent inhibitory activity against the tested cell lines.
Comparing the cytotoxic activity of compounds 21a–c (quinazoline derivatives containing 3-chloro-4-fluorophenyl moieties) indicated that 6-chloro 21b is more preferred biologically than 5-chloro 21a and 5-fluoro 21c members against all cell lines.
2.2.3.1. The effect on the expression level of caspase-8. As presented in Table 2, thalidomide and compounds 18f and 21b showed a statistically significant increase in caspase-8 levels compared with control cells. Also, it was found that compounds 18f and 21b were able to increase caspase-8 level in HepG-2 cells by 6.7 and 6.5 ng mL−1, respectively, compared to 8.3 ng mL−1 for thalidomide.
| Comp. no. | CASPASE-8 (ng mL−1) |
|---|---|
| 18f | 6.7 |
| 21b | 6.5 |
| Control | 1.08 |
| Thalidomide | 8.3 |
2.2.3.2. The effect on the expression level of vascular endothelial growth factor (VEGF). The data presented in Table 3 showed that thalidomide and compounds 18f and 21b showed a significant decrease in VEGF level comparing with control cells. HepG-2 cells treated with 18f and 21b revealed VEGF level of 169.6 and 162.3 pg mL−1, respectively, compared to 153.2 pg mL−1 calculated for thalidomide.
| Comp. no. | VEGF (pg mL−1) |
|---|---|
| 18f | 169.6 |
| 21b | 162.3 |
| Control | 432.5 |
| Thalidomide | 153.2 |
2.2.3.3. The effect on NF-κB P65 expression level. The obtained data indicated that the impact of 18f and 21b on NF-κB P65 were much better than thalidomide. It can be seen that NF-κB P65 level in HepG-2 cells treated with 18f and 21b were 89.4 and 85.1 pg mL−1 compared to 110.5 pg mL−1 measured for thalidomide treated HepG-2 cells. At the same time, the two tested compounds showed a significant decrease in NF-κB P65 levels when compared to control treated cells (Table 4).
| Compound | NFκB P65 (pg mL−1) |
|---|---|
| 18f | 89.4 |
| 21b | 85.1 |
| Control | 278.1 |
| Thalidomide | 110.5 |
2.2.3.4. The effect on TNF-α expression level. The data presented in Table 5 shows that both 18f and 21b significantly reduced TNF-α levels in HepG-2 cells. TNF-α levels were reduced from 162.5 pg mL−1 in control cells to 73.2 and 50.6 pg mL−1 under the effects of 18f and 21b respectively. Meanwhile, the level in cells treated with thalidomide was 53.1 pg mL−1. These findings highlight the importance of 21b as TNF-α inhibitor in comparison with thalidomide.
| Compound | TNF-α (pg mL−1) |
|---|---|
| 18f | 73.2 |
| 21b | 50.6 |
| Control | 162.5 |
| Thalidomide | 53.1 |
2.2.3.5. HepG-2 cell cycle analysis. The most promising candidate, compound 21b, was further evaluated by analyzing its effect on cycle phases of the HepG-2 cells. From the results presented in Table 6, it can be noticed that a large percentage (54.05%) of cells treated with 21b was accumulated at G0/G1 phase compared to 8.07 and 10.05% reported for thalidomide and control cells, respectively. This clearly indicates the ability of compound 21b to inhibit HepG-2 cell proliferation efficiently at G0/G1 phase (Fig. 4).
| Sample | G0/G1 | S | G2/M |
|---|---|---|---|
| Control | 10.05 | 59.55 | 29.73 |
| Thalidomide | 8.07 | 66.38 | 25.54 |
| 21b | 54.05 | 37.04 | 8.96 |
 | ||
| Fig. 4 The effect of 21b on different phases of HepG-2 cell cycle. | ||
2.2.3.6. The effect of 21b on apoptosis and necrosis rates of HepG-2. As presented in Table 7 and Fig. 5, the death rate of HepG-2 cells treated with compound 21b dramatically increased from 1.19% to 25.78% at the early stage of apoptosis. Meanwhile, there is no significant increase in the late apoptosis and necrosis. This suggests that apoptosis was the main mechanism of HepG-2 cell death that was caused by compound 21b.
| Sample | Normal cells | Early apoptosis | Late apoptosis | Necrosis |
|---|---|---|---|---|
| Control | 98.61 | 1.19 | 0.18 | 0.02 |
| Thalidomide | 95.98 | 3.76 | 0.26 | 0.00 |
| 21b | 74.00 | 25.78 | 0.19 | 0.03 |
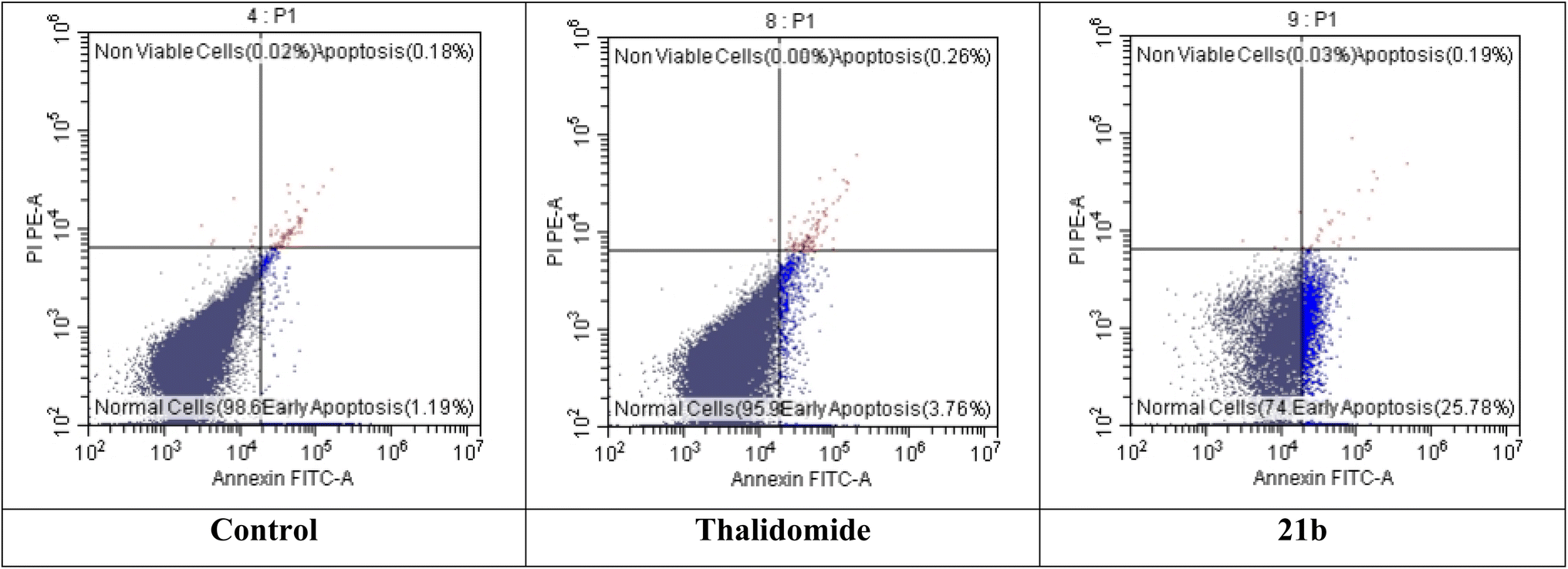 | ||
| Fig. 5 Apoptosis and necrosis rates of HepG2 cells treated with 21b compared to control and thalidomide. | ||
The most promising candidates 18f and 21b show acceptable range of the expected ADMET profile. Compound 18f was predicted to have low blood–brain barrier (BBB) penetration level. While compound 21b was expected to have high penetration level. For aqueous solubility, compounds 18f and 21b showed good and low levels, respectively. It is noteworthy that compounds 18f and 21b have good absorption levels and do not inhibit CYP2D6. Regarding plasma protein binding parameter, compounds 18f was anticipated to bind plasma protein less than 90% while 21b was expected to bind it with more than 90% (Table 8 and Fig. 6).
| Comp. | BBB levela | Solubilityb | Absorptionc | CYP2D6 inhibitiond | Plasma protein bindinge |
|---|---|---|---|---|---|
| a 1 = high, 3 = low, and 4 = very low.b 1 = very low, 2 = low, 3 = good, and 4 = optimal.c 0 = good.d False = non inhibitor.e False means less than 90% & true means more than 90%. | |||||
| 6a | 4 | 4 | 0 | False | False |
| 6b | 3 | 4 | 0 | False | False |
| 9 | 3 | 3 | 0 | False | False |
| 12 | 3 | 3 | 0 | False | False |
| 18a | 3 | 3 | 0 | False | False |
| 18b | 3 | 3 | 0 | False | False |
| 18c | 3 | 3 | 0 | False | False |
| 18d | 3 | 3 | 0 | False | False |
| 18e | 4 | 3 | 0 | False | False |
| 18f | 4 | 3 | 0 | False | False |
| 18g | 3 | 3 | 0 | False | False |
| 21a | 1 | 1 | 0 | False | True |
| 21b | 1 | 2 | 0 | False | True |
| 21c | 1 | 1 | 0 | False | True |
| Thalidomide | 3 | 3 | 0 | False | False |
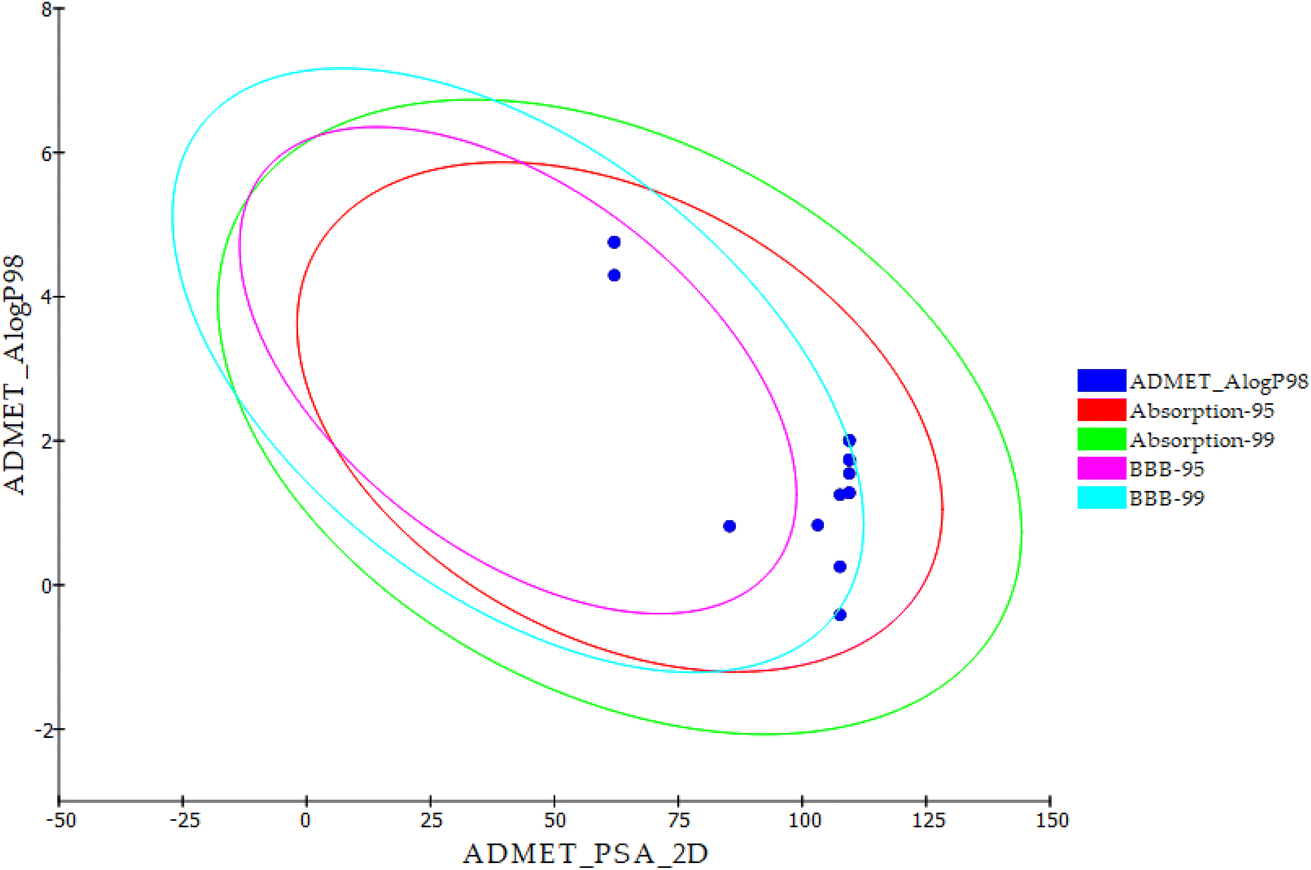 | ||
| Fig. 6 ADMET properties of the new derivatives and thalidomide. | ||
| Comp. | C-P-TD50 (rat) a | A-M | R-M-T-D (feed) b | R-O-LD50 b | R-C-LOAELb | S-I | O-I | DTP |
|---|---|---|---|---|---|---|---|---|
| a Unit: mg per kg body weight per day.b Unit: g per kg body weight. | ||||||||
| 6a | 2.890 | N-M | 0.107 | 5.079 | 0.171 | N-I | M | T |
| 6b | 2.827 | N-M | 0.119 | 5.002 | 0.107 | N-I | M | T |
| 9 | 8.040 | N-M | 0.115 | 2.375 | 0.134 | N-I | M | T |
| 12 | 10.821 | N-M | 0.220 | 3.375 | 0.071 | N-I | M | T |
| 18a | 2.240 | N-M | 0.055 | 1.287 | 0.023 | N-I | M | N-T |
| 18b | 1.665 | N-M | 0.055 | 2.222 | 0.029 | N-I | M | N-T |
| 18c | 2.397 | N-M | 0.059 | 0.673 | 0.024 | N-I | M | N-T |
| 18d | 0.908 | N-M | 0.059 | 1.205 | 0.023 | N-I | M | N-T |
| 18e | 5.760 | N-M | 0.055 | 0.564 | 0.007 | N-I | M | N-T |
| 18f | 2.182 | N-M | 0.055 | 0.692 | 0.007 | N-I | M | N-T |
| 18g | 6.173 | N-M | 0.059 | 0.295 | 0.007 | N-I | M | N-T |
| 21a | 67.057 | N-M | 0.058 | 0.660 | 0.029 | N-I | M | N-T |
| 21b | 71.894 | N-M | 0.062 | 0.693 | 0.029 | N-I | M | N-T |
| 21c | 25.404 | N-M | 0.058 | 0.484 | 0.025 | N-I | M | N-T |
| Thalidomide | 26.375 | N-M | 0.047 | 0.835 | 0.133 | N-I | None | N-T |
For ames mutagenicity (A-M), skin irritancy (S-I), ocular irritancy (O-I) models, and developmental toxicity potential model (DTP), the most promising candidates 18f and 21b were predicted as non-mutagen (N-M), non-irritant (N-I), mild irritant (M), and non-toxic (N-T), respectively.
For carcinogenic potency TD50 rat model (C-P-TD50), compound 21b showed TD50 values of 71.894 g per kg body weight, which is higher than thalidomide (26.375). For rat maximum tolerated dose (R-M-T-D) model, compound 18f and 21b demonstrated values higher than that of thalidomide. Additionally, compounds 18f and 21b revealed oral LD50 (R-O-LD50) and LOAEL (R-C-LOAEL) values lower than thalidomide.
3. Conclusion
Fourteen thalidomide analogs have been designed and synthesized for anticancer and immunomodulatory evaluation. The in vitro cytotoxicity results revealed that compounds 18f (IC50 = 11.91 ± 0.9, 9.27 ± 0.7, and 18.62 ± 1.5 μM against HepG-2, PC3, and MCF-7, respectively) and 21b (IC50 = 10.48 ± 0.8, 22.56 ± 1.6, and 16.39 ± 1.4 μM against HepG-2, PC3, and MCF-7, respectively) were the most potent members. Also, compounds as 12, 18a, 18c, and 18g displayed good anti-proliferative activities against the tested cell lines with IC50 values ranging from 12.13 to 37.95 μM. Moreover, the most promising compounds 18f and 21b were further evaluated for their immunomodulatory effects. Compounds 18f and 21b showed a significant reduction in levels of TNF-α (from 162.5 pg mL−1 to 73.2 and 50.6 pg mL−1), VEGF (from 432.5 pg mL−1 to 169.6 and 162.3 pg mL−1), and NF-κB p65 (from 278.1 pg mL−1 to 89.4 and 85.1 pg mL−1). Furthermore, compounds 18f and 21b exhibited significant elevation in CASP8 levels (from 1.08 ng mL−1 to 6.7 and 6.5 ng mL−1). These results were compared to thalidomide as a reference standard which reports 53.1, 153.2, 110.5 pg mL−1 and 8 ng mL−1 against TNF-α, VEGF, NF-κB p65, and CASP8, respectively. The obtained findings suggest that the designed members have the potential to function as powerful immunomodulators and anticancer agents. Further optimization of these derivatives may lead to the discovery of more promising immunomodulatory agents.4. Experimental
4.1. Chemistry
4.1.1.1. 4-Acetamido-N-(2,6-dioxopiperidin-3-yl)benzamide (6a).
White crystal (yield, 75%); m.p. = 245–247 °C. IR (KBr, cm−1): 3329, 3176 (NH), 3090 (CH aromatic), 2975 (CH aliphatic), 1693 (C
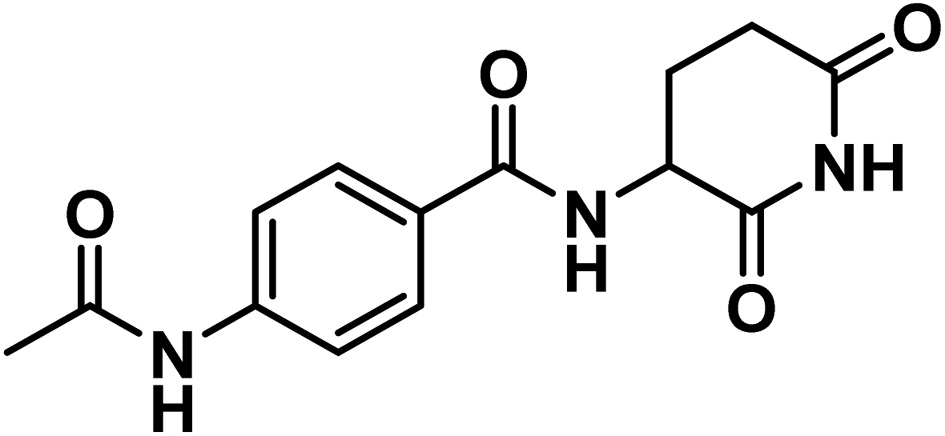
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O imide), 1645 (CO amide).; 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 10.18 (s, 1H, CH3CONH), 8.64 (d, J = 8 Hz, 1H, PhCONH), 7.84 (d, J = 8.2 Hz, 2H, Ar–H), 7.68 (d, J = 8.2 Hz, 2H, Ar–H), 4.78 (m, 1H, CH-piperidine), 2.80 (m, 1H, CHCONH-piperidine), 2.55 (m, 1H, CHCONH-piperidine), 2.15 (m, 1H, CHCH2CONH-piperidine), 2.08 (s, 3H, CH3), 1.97 (m, 1H, CHCH2CONH-piperidine); mass (m/z): 289 (M+, 31%), and 75 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.55, 172.82, 169.23, 166.07, 142.62, 128.68, 128.60, 118.56, 49.95, 31.48, 24.73, 24.59; Anal. Calcd. for C14H15N3O4 (289.29): C, 58.13; H, 5.23; N, 14.53. Found: C, 58.41; 5.40; N, 14.61%.
O imide), 1645 (CO amide).; 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 10.18 (s, 1H, CH3CONH), 8.64 (d, J = 8 Hz, 1H, PhCONH), 7.84 (d, J = 8.2 Hz, 2H, Ar–H), 7.68 (d, J = 8.2 Hz, 2H, Ar–H), 4.78 (m, 1H, CH-piperidine), 2.80 (m, 1H, CHCONH-piperidine), 2.55 (m, 1H, CHCONH-piperidine), 2.15 (m, 1H, CHCH2CONH-piperidine), 2.08 (s, 3H, CH3), 1.97 (m, 1H, CHCH2CONH-piperidine); mass (m/z): 289 (M+, 31%), and 75 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.55, 172.82, 169.23, 166.07, 142.62, 128.68, 128.60, 118.56, 49.95, 31.48, 24.73, 24.59; Anal. Calcd. for C14H15N3O4 (289.29): C, 58.13; H, 5.23; N, 14.53. Found: C, 58.41; 5.40; N, 14.61%.
4.1.1.2. N-(2,6-Dioxopiperidin-3-yl)-4-propionamidobenzamide (6b).
Grayish white crystal (yield, 67%); m.p. = 260–262 °C. IR (KBr, cm−1): 3263, 3194 (NH), 3106 (CH aromatic), 2980 (CH aliphatic), 1728 (C
 O imide), 1680 (CO amide); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 10.11 (s, 1H, CH2CONH), 8.63 (d, J = 8.2 Hz, 1H, PhCONH), 7.83 (d, J = 8.4 Hz, 2H, Ar–H), 7.69 (d, J = 8.4 Hz, 2H, Ar–H), 4.77 (m, 1H, CH-piperidine), 2.80 (m, 1H, CH2CH2CONH-piperidine), 2.57 (m, 1H, CH2CH2CONH-piperidine), 2.36 (q, J = 7.5 Hz, 2H, CH3CH2CO), 2.12 (m, 1H, CH2CH2CONH-piperidine), 1.98 (m, 1H, CH2CH2CONH-piperidine), 1.09 (t, J = 7.5 Hz, 3H, CH3); mass (m/z): 303 (M+, 100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.53, 172.86, 172.81, 166.02, 142.67, 128.66, 128.48, 118.56, 49.91, 31.48, 30.04, 24.73, 9.97; Anal. Calcd. for C15H17N3O4 (303.32): C, 59.40; H, 5.65; N, 13.85. Found: C, 59.63; H, 5.81; N, 13.97%.
O imide), 1680 (CO amide); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 10.11 (s, 1H, CH2CONH), 8.63 (d, J = 8.2 Hz, 1H, PhCONH), 7.83 (d, J = 8.4 Hz, 2H, Ar–H), 7.69 (d, J = 8.4 Hz, 2H, Ar–H), 4.77 (m, 1H, CH-piperidine), 2.80 (m, 1H, CH2CH2CONH-piperidine), 2.57 (m, 1H, CH2CH2CONH-piperidine), 2.36 (q, J = 7.5 Hz, 2H, CH3CH2CO), 2.12 (m, 1H, CH2CH2CONH-piperidine), 1.98 (m, 1H, CH2CH2CONH-piperidine), 1.09 (t, J = 7.5 Hz, 3H, CH3); mass (m/z): 303 (M+, 100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.53, 172.86, 172.81, 166.02, 142.67, 128.66, 128.48, 118.56, 49.91, 31.48, 30.04, 24.73, 9.97; Anal. Calcd. for C15H17N3O4 (303.32): C, 59.40; H, 5.65; N, 13.85. Found: C, 59.63; H, 5.81; N, 13.97%.
4.1.1.3. 4-Benzamido-N-(2,6-dioxopiperidin-3-yl)benzamide (9).
White crystal (yield, 75%); m.p. = 295–298 °C. IR (KBr, cm−1): 3626, 3298 (2NH), 3098 (CH aromatic), 2923 (CH aliphatic), 1724 (C
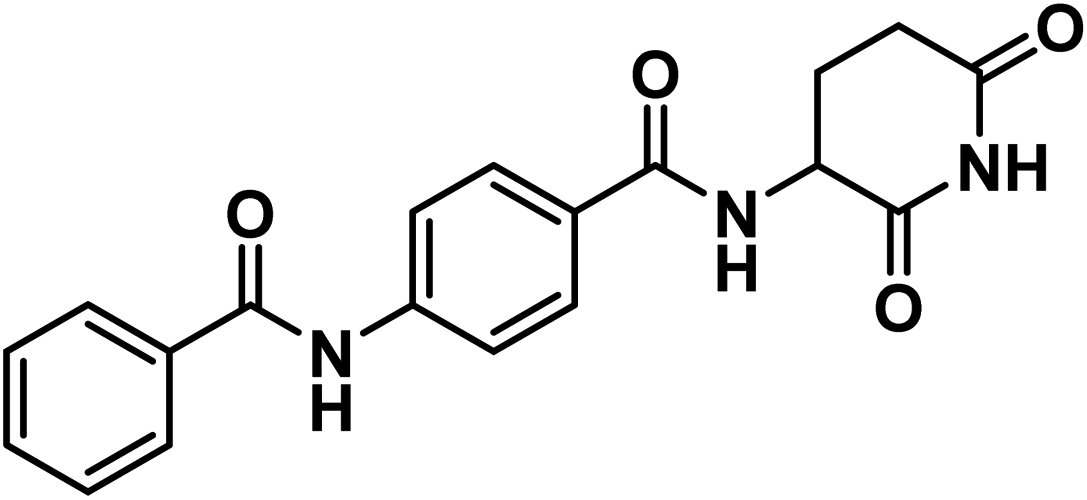 O imide), 1647 (CO amide); 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 10.49 (s, 1H, PhCONHPh), 8.70 (d, J = 8.2 Hz, 1H, PhCONH), 7.98 (m, 2H, Ar–H), 7.91 (d, J = 8.2 Hz, 4H, Ar–H), 7.62 (m, 1H, Ar–H), 7.55 (m, 2H, Ar–H), 4.79 (m, 1H, CH-piperidine), 2.81 (m, 1H, CH2CH2CONH-piperidine), 2.56 (m, 1H, CH2CH2CONH-piperidine),2.14 (m, 1H, CH2CH2CONH-piperidine), 2.01 (m, 1H, CH2CH2CONH-piperidine); 13C NMR (101 MHz, DMSO-d6) δ 173.54, 172.81, 166.32, 166.06, 142.55, 135.15, 132.27, 129.20, 128.91, 128.55, 128.23, 119.91, 49.96, 31.49, 24.75; Anal. Calcd. for C19H17N3O4 (351.36): C, 64.95; H, 4.88; N, 11.96. Found: C, 65.18; H, 4.96; N, 12.19%.
O imide), 1647 (CO amide); 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 10.49 (s, 1H, PhCONHPh), 8.70 (d, J = 8.2 Hz, 1H, PhCONH), 7.98 (m, 2H, Ar–H), 7.91 (d, J = 8.2 Hz, 4H, Ar–H), 7.62 (m, 1H, Ar–H), 7.55 (m, 2H, Ar–H), 4.79 (m, 1H, CH-piperidine), 2.81 (m, 1H, CH2CH2CONH-piperidine), 2.56 (m, 1H, CH2CH2CONH-piperidine),2.14 (m, 1H, CH2CH2CONH-piperidine), 2.01 (m, 1H, CH2CH2CONH-piperidine); 13C NMR (101 MHz, DMSO-d6) δ 173.54, 172.81, 166.32, 166.06, 142.55, 135.15, 132.27, 129.20, 128.91, 128.55, 128.23, 119.91, 49.96, 31.49, 24.75; Anal. Calcd. for C19H17N3O4 (351.36): C, 64.95; H, 4.88; N, 11.96. Found: C, 65.18; H, 4.96; N, 12.19%.
4.1.2.1. N-(2,6-Dioxopiperidin-3-yl)-4-(3-ethylthioureido)benzamide (12).
Yellow crystal (yield, 69%); m.p. = 220–222 °C. IR (KBr, cm−1): 3752, 3652, 3314 (3NH), 3100 (CH aromatic), 2972 (CH aliphatic), 1726 (C
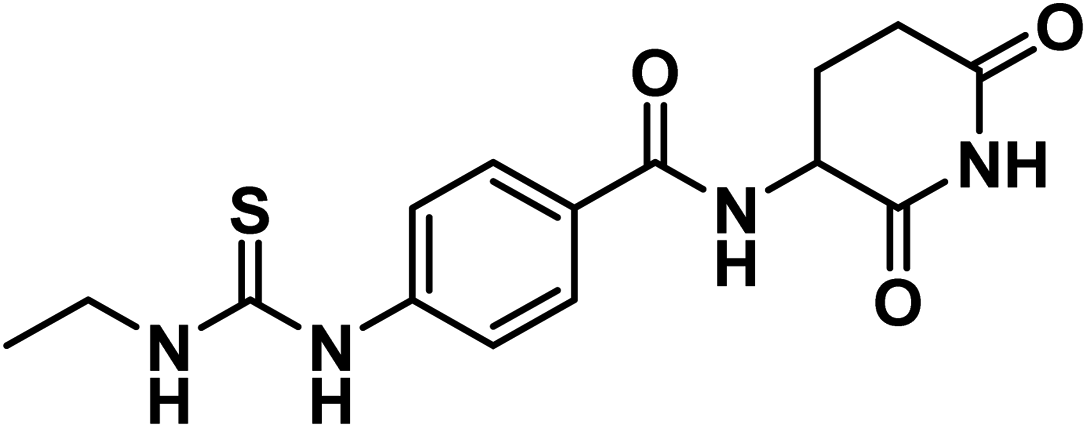 O imide), 1644 (CO amide); 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 9.68 (s, 1H, CSNHPh), 8.66 (d, J = 8.2 Hz, 1H, PhCONH), 7.95 (s, 1H, CH2NHCS), 7.82 (d, J = 8.4 Hz, 2H, Ar–H), 7.58 (d, J = 8.4 Hz, 2H, Ar–H), 4.77 (m, 1H, CH-piperidine), 3.49 (q, J = 6.8 Hz, 2H, CH3CH2NH), 2.80 (m, 1H, CH2CH2CONH-piperidine), 2.57 (m, 1H, CH2CH2CONH-piperidine), 2.13 (m, 1H, CH2CH2CONH-piperidine), 1.99 (m, 1H, CH2CH2CONH-piperidine), 1.14 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 180.40, 173.53, 172.77, 166.04, 142.97, 129.05, 128.33, 49.94, 39.15, 31.47, 24.73, 14.49; Anal. Calcd. for C15H18N4O3S (334.11): C, 53.88; H, 5.43; N, 16.76. Found: C, 53.72; H, 5.49; N, 16.98%.
O imide), 1644 (CO amide); 1H NMR (DMSO-d6) δppm: 10.85 (s, 1H, CONHCO), 9.68 (s, 1H, CSNHPh), 8.66 (d, J = 8.2 Hz, 1H, PhCONH), 7.95 (s, 1H, CH2NHCS), 7.82 (d, J = 8.4 Hz, 2H, Ar–H), 7.58 (d, J = 8.4 Hz, 2H, Ar–H), 4.77 (m, 1H, CH-piperidine), 3.49 (q, J = 6.8 Hz, 2H, CH3CH2NH), 2.80 (m, 1H, CH2CH2CONH-piperidine), 2.57 (m, 1H, CH2CH2CONH-piperidine), 2.13 (m, 1H, CH2CH2CONH-piperidine), 1.99 (m, 1H, CH2CH2CONH-piperidine), 1.14 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 180.40, 173.53, 172.77, 166.04, 142.97, 129.05, 128.33, 49.94, 39.15, 31.47, 24.73, 14.49; Anal. Calcd. for C15H18N4O3S (334.11): C, 53.88; H, 5.43; N, 16.76. Found: C, 53.72; H, 5.49; N, 16.98%.
4.1.3.1. 2-((5-Chloro-3-ethyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(2,6-dioxopiperidin-3-yl)acetamide (18a).
White crystal (yield, 74%); m.p. = 218–220 °C. IR (KBr, cm−1): 3530, 3271 (2NH), 3085 (C–H aromatic), 2981 (C–H aliphatic), 1711 (C
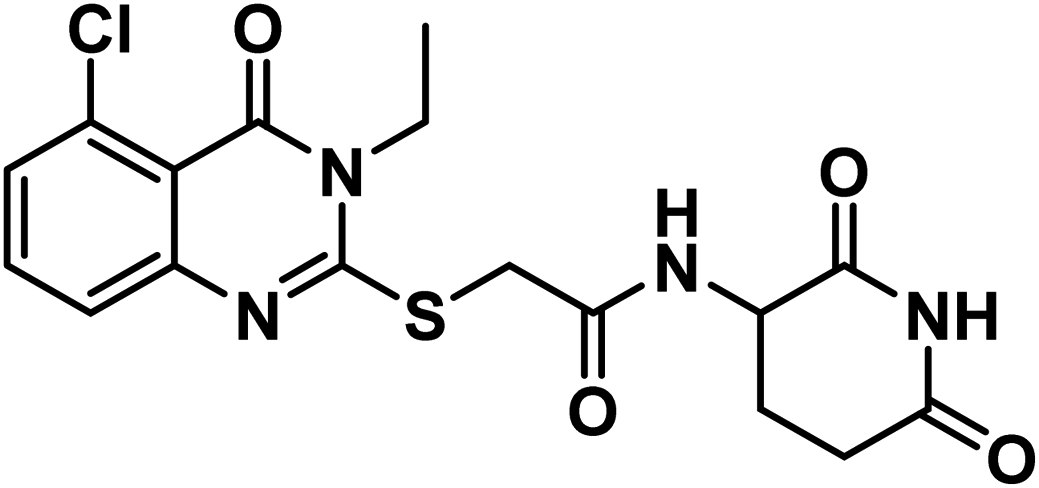 O imide), 1680 (CO amide) and 11650 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 7.68 (t, J = 8.0 Hz, 1H, Ar–H), 7.51 (d, J = 8.2 Hz, 1H, Ar–H), 7.45 (d, J = 7.8 Hz, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.09 (q, J = 7.2 Hz, 2H, CH2CH3), 4.07 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine), 1.30 (t, J = 7 Hz, 3H, CH3); mass (m/z): 408 (M+, 16%), and 241 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.33, 172.42, 167.20, 158.59, 157.25, 149.56, 134.78, 133.03, 128.74, 126.12, 116.15, 50.06, 35.87, 31.26, 24.75, 13.21; Anal. Calcd. for C17H17ClN4O4S (408.86): C, 49.94; H, 4.19; N, 13.70. Found: C, 49.71; H, 4.27; N, 13.96%.
O imide), 1680 (CO amide) and 11650 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 7.68 (t, J = 8.0 Hz, 1H, Ar–H), 7.51 (d, J = 8.2 Hz, 1H, Ar–H), 7.45 (d, J = 7.8 Hz, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.09 (q, J = 7.2 Hz, 2H, CH2CH3), 4.07 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine), 1.30 (t, J = 7 Hz, 3H, CH3); mass (m/z): 408 (M+, 16%), and 241 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.33, 172.42, 167.20, 158.59, 157.25, 149.56, 134.78, 133.03, 128.74, 126.12, 116.15, 50.06, 35.87, 31.26, 24.75, 13.21; Anal. Calcd. for C17H17ClN4O4S (408.86): C, 49.94; H, 4.19; N, 13.70. Found: C, 49.71; H, 4.27; N, 13.96%.
4.1.3.2. 2-((8-Chloro-3-ethyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(2,6-dioxopiperidin-3-yl)acetamide (18b).
White crystal (yield, 74%); m.p. = 232–234 °C. IR (KBr, cm−1): 3393, 3170 (2NH), 3083 (C–H aromatic), 2939 (C–H aliphatic), 1708 (C
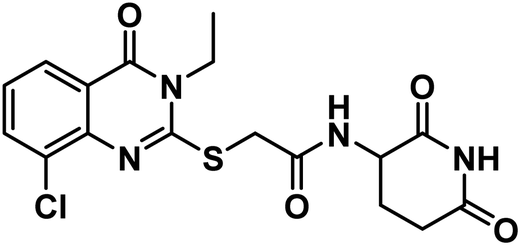 O imide), 1679 (CO amide) and 1646 (amide II band); 1H NMR (DMSO-d6) δppm: 10.82 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 8.03 (dd, J = 8.0,1.5 Hz, 1H, Ar–H), 7.92 (dd, J = 7.8, 1.4 Hz, 1H, Ar–H), 7.42 (t, J = 7.9 Hz, 1H, Ar–H), 4.58 (m, 1H, CH-piperidine), 4.18 (q, J = 7.8 Hz, 2H, CH2CH3), 4.13 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.94 (m, 2H, CH2CH-piperidine), 1.31 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 173.35, 172.33, 166.78, 160.22, 157.76, 143.51, 134.93, 129.82, 126.61, 125.89, 120.83, 50.15, 36.10, 31.26, 24.62, 13.25; Anal. Calcd. for C17H17ClN4O4S (408.86): C, 49.94; H, 4.19; N, 13.70. Found: C, 49.86; H, 4.31; N, 13.98%.
O imide), 1679 (CO amide) and 1646 (amide II band); 1H NMR (DMSO-d6) δppm: 10.82 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 8.03 (dd, J = 8.0,1.5 Hz, 1H, Ar–H), 7.92 (dd, J = 7.8, 1.4 Hz, 1H, Ar–H), 7.42 (t, J = 7.9 Hz, 1H, Ar–H), 4.58 (m, 1H, CH-piperidine), 4.18 (q, J = 7.8 Hz, 2H, CH2CH3), 4.13 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.94 (m, 2H, CH2CH-piperidine), 1.31 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 173.35, 172.33, 166.78, 160.22, 157.76, 143.51, 134.93, 129.82, 126.61, 125.89, 120.83, 50.15, 36.10, 31.26, 24.62, 13.25; Anal. Calcd. for C17H17ClN4O4S (408.86): C, 49.94; H, 4.19; N, 13.70. Found: C, 49.86; H, 4.31; N, 13.98%.
4.1.3.3. N-(2,6-dioxopiperidin-3-yl)-2-((3-ethyl-5-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (18c).
Shine yellow crystal (yield, 72%); m.p. = 252–250 °C. IR (KBr, cm−1): 3273, 3210 (2NH), 3114 (C–H aromatic), 2978 (C–H aliphatic), 1720 (C
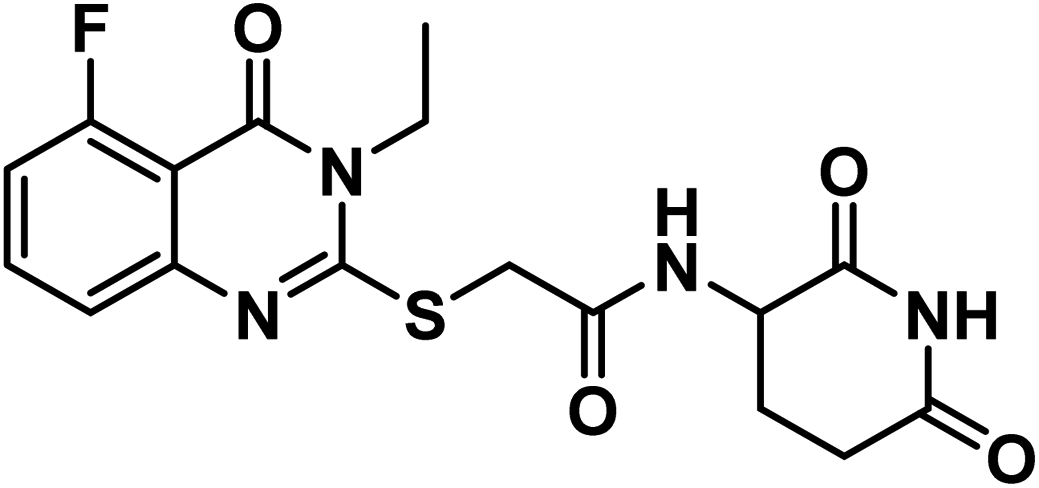 O imide), 1681 (CO amide) and 1653 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 7.74 (t, J = 8.0 Hz, 1H, Ar–H), 7.38 (d, J = 8.2 Hz, 1H, Ar–H), 7.18 (d, J = 7.8 Hz, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.09 (q, J = 7.2 Hz, 2H, CH2CH3), 4.06 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine), 1.29 (t, J = 7.0 Hz, 3H, CH3); mass (m/z): 392 (M+, 3%), and 42 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.34, 172.42, 167.19, 162.17, 159.55, 157.65, 157.61, 149.10, 135.71, 135.61, 122.60, 122.56, 112.76, 112.56, 108.85, 50.05, 35.99, 31.26, 24.75, 13.27; Anal. Calcd. for C17H17FN4O4S (392.41): C, 52.03; H, 4.37; N, 14.28. Found: C, 52.29; H, 4.50; N, 14.47%.
O imide), 1681 (CO amide) and 1653 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.63 (d, J = 8.0 Hz, 1H, CONHCH), 7.74 (t, J = 8.0 Hz, 1H, Ar–H), 7.38 (d, J = 8.2 Hz, 1H, Ar–H), 7.18 (d, J = 7.8 Hz, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.09 (q, J = 7.2 Hz, 2H, CH2CH3), 4.06 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine), 1.29 (t, J = 7.0 Hz, 3H, CH3); mass (m/z): 392 (M+, 3%), and 42 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 173.34, 172.42, 167.19, 162.17, 159.55, 157.65, 157.61, 149.10, 135.71, 135.61, 122.60, 122.56, 112.76, 112.56, 108.85, 50.05, 35.99, 31.26, 24.75, 13.27; Anal. Calcd. for C17H17FN4O4S (392.41): C, 52.03; H, 4.37; N, 14.28. Found: C, 52.29; H, 4.50; N, 14.47%.
4.1.3.4. N-(2,6-Dioxopiperidin-3-yl)-2-((3-ethyl-6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (18d).
Shine yellow crystal (yield, 72%); m.p. = 227–229 °C. IR (KBr, cm−1): 3301, 3169 (2NH), 3108 (C–H aromatic), 2954 (C–H aliphatic), 1713 (C
 O imide), 1647 (CO amide) and 1649 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.69 (d, J = 7.9 Hz, 1H, CONHCH), 7.72 (d, J = 8.8 Hz, 1H, Ar–H), 7.66 (d, J = 7.9 Hz, 1H, Ar–H), 7.64 (s, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.11 (q, J = 7.2 Hz, 2H, CH2CH3), 4.07 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.94 (m, 2H, CH2CH-piperidine), 1.30 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 173.35, 172.42, 167.25, 161.05, 160.19, 158.63, 155.77, 144.21, 129.30, 129.22, 123.64, 123.40, 120.38, 120.30, 111.41, 111.17, 50.04, 35.95, 31.26, 24.74, 13.37; Anal. Calcd. for C17H17FN4O4S (392.41): C, 52.03; H, 4.37; N, 14.28. Found: C, 52.79; H, 3.68; N, 14.04%.
O imide), 1647 (CO amide) and 1649 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.69 (d, J = 7.9 Hz, 1H, CONHCH), 7.72 (d, J = 8.8 Hz, 1H, Ar–H), 7.66 (d, J = 7.9 Hz, 1H, Ar–H), 7.64 (s, 1H, Ar–H), 4.60 (m, 1H, CH-piperidine), 4.11 (q, J = 7.2 Hz, 2H, CH2CH3), 4.07 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.94 (m, 2H, CH2CH-piperidine), 1.30 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 173.35, 172.42, 167.25, 161.05, 160.19, 158.63, 155.77, 144.21, 129.30, 129.22, 123.64, 123.40, 120.38, 120.30, 111.41, 111.17, 50.04, 35.95, 31.26, 24.74, 13.37; Anal. Calcd. for C17H17FN4O4S (392.41): C, 52.03; H, 4.37; N, 14.28. Found: C, 52.79; H, 3.68; N, 14.04%.
4.1.3.5. 2-((3-Allyl-5-chloro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(2,6-dioxo piperidin-3-yl)acetamide (18e).
White crystal (yield, 71%); m.p. = 289–291 °C. IR (KBr, cm−1): 3304, 3201 (2NH), 3093 (C–H aromatic), 2987 (C–H aliphatic), 1713 (C
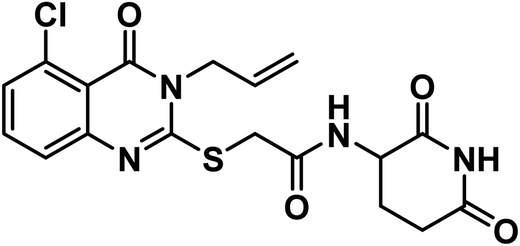 O imide), 1684 (CO amide) and 1645 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.61 (d, J = 8.0 Hz, 1H, CONHCH), 7.70 (t, J = 8.0 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.47 (d, J = 7.8 Hz, 1H, Ar–H), 5.93 (m, 1H, CH2CHCH2), 5.21 (m, 2H, CH2CHCH2), 4.68 (d, J = 5.1 Hz, 2H, NCH2), 4.59 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.92 (m, 2H, CH2CH-piperidine); mass (m/z): 420 (M+, 23%), and 364 (100%, base peak);. 13C NMR (101 MHz, DMSO-d6) δ 173.33, 172.42, 167.16, 158.59, 157.69, 149.57, 134.94, 133.14, 131.62, 128.84, 126.18, 118.21, 116.09, 50.05, 46.58, 36.01, 31.26, 24.75; Anal. Calcd. for C18H17ClN4O4S (420.87): C, 54.35; H, 4.32; N, 10.01. Found: C, 54.60; H, 4.41; N, 10.28%.
O imide), 1684 (CO amide) and 1645 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.61 (d, J = 8.0 Hz, 1H, CONHCH), 7.70 (t, J = 8.0 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.47 (d, J = 7.8 Hz, 1H, Ar–H), 5.93 (m, 1H, CH2CHCH2), 5.21 (m, 2H, CH2CHCH2), 4.68 (d, J = 5.1 Hz, 2H, NCH2), 4.59 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.92 (m, 2H, CH2CH-piperidine); mass (m/z): 420 (M+, 23%), and 364 (100%, base peak);. 13C NMR (101 MHz, DMSO-d6) δ 173.33, 172.42, 167.16, 158.59, 157.69, 149.57, 134.94, 133.14, 131.62, 128.84, 126.18, 118.21, 116.09, 50.05, 46.58, 36.01, 31.26, 24.75; Anal. Calcd. for C18H17ClN4O4S (420.87): C, 54.35; H, 4.32; N, 10.01. Found: C, 54.60; H, 4.41; N, 10.28%.
White crystal (yield, 71%); m.p. = 289–291 °C. IR (KBr, cm−1): 3305, 3189 (2NH), 3086 (C–H aromatic), 2927 (C–H aliphatic), 1712 (C
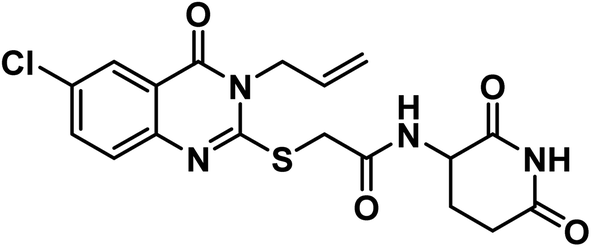 O imide), 1678 (CO amide) and 1628 (amide II band); 1H NMR (DMSO-d6) δppm: 10.83 (s, 1H, CONHCO), 8.62 (s, 1H, CONHCH), 7.99 (s, 1H, Ar–H), 7.81 (s, 1H, Ar–H), 7.60 (s, 1H, Ar–H), 5.92 (m, 1H, CH2CHCH2), 5.22 (m, 2H, CH2CHCH2), 4.72 (d, J = 5.2 Hz, 2H, NCH2), 4.59 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.70 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.92 (m, 2H, CH2CH-piperidine); 13C NMR (126 MHz, DMSO-d6) δ 172.71, 171.79, 166.59, 159.31, 156.93, 145.42, 134.72, 131.02, 129.94, 128.21, 125.28, 119.85, 117.71, 49.56, 35.59, 30.71, 24.19; Anal. Calcd. for C18H17ClN4O4S (420.87): C, 54.35; H, 4.32; N, 10.01. Found: C, 54.56; H, 4.49; N, 10.02%.
O imide), 1678 (CO amide) and 1628 (amide II band); 1H NMR (DMSO-d6) δppm: 10.83 (s, 1H, CONHCO), 8.62 (s, 1H, CONHCH), 7.99 (s, 1H, Ar–H), 7.81 (s, 1H, Ar–H), 7.60 (s, 1H, Ar–H), 5.92 (m, 1H, CH2CHCH2), 5.22 (m, 2H, CH2CHCH2), 4.72 (d, J = 5.2 Hz, 2H, NCH2), 4.59 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.70 (m, 1H, CH2CO-piperidine), 2.46 (m, 1H, CH2CO-piperidine), 1.92 (m, 2H, CH2CH-piperidine); 13C NMR (126 MHz, DMSO-d6) δ 172.71, 171.79, 166.59, 159.31, 156.93, 145.42, 134.72, 131.02, 129.94, 128.21, 125.28, 119.85, 117.71, 49.56, 35.59, 30.71, 24.19; Anal. Calcd. for C18H17ClN4O4S (420.87): C, 54.35; H, 4.32; N, 10.01. Found: C, 54.56; H, 4.49; N, 10.02%.White crystal (yield, 71%); m.p. = 289–291 °C. IR (KBr, cm−1): 3308, 3191 (2NH), 3092 (C–H aromatic), 2987 (C–H aliphatic), 1690 (C
 O imide), 1623 (CO amide) and 1640 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.62 (d, J = 8.0 Hz, 1H, CONHCH), 7.76 (t, J = 8.0 Hz, 1H, Ar–H), 7.40 (d, J = 8.2 Hz, 1H, Ar–H), 7.19 (d, J = 8.1 Hz, 1H, Ar–H), 5.92 (m, 1H, CH2CHCH2), 5.21 (m, 2H, CH2CHCH2), 4.69 (d, J = 5.1 Hz, 2H, NCH2), 4.60 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine); 13C NMR (126 MHz, DMSO-d6) δ 172.72, 171.78, 166.58, 161.41, 159.32, 157.48, 148.55, 135.30, 131.12, 122.11, 117.62, 112.29, 108.21, 49.53, 45.58, 35.60, 30.70, 24.18; Anal. Calcd. for C18H17FN4O4S (404.42): C, 56.57; H, 4.50; N, 10.42. Found: C, 56.78; H, 4.62; N, 10.68%.
O imide), 1623 (CO amide) and 1640 (amide II band); 1H NMR (DMSO-d6) δppm: 10.84 (s, 1H, CONHCO), 8.62 (d, J = 8.0 Hz, 1H, CONHCH), 7.76 (t, J = 8.0 Hz, 1H, Ar–H), 7.40 (d, J = 8.2 Hz, 1H, Ar–H), 7.19 (d, J = 8.1 Hz, 1H, Ar–H), 5.92 (m, 1H, CH2CHCH2), 5.21 (m, 2H, CH2CHCH2), 4.69 (d, J = 5.1 Hz, 2H, NCH2), 4.60 (m, 1H, CH-piperidine), 4.05 (s, 2H, SCH2), 2.71 (m, 1H, CH2CO-piperidine), 2.47 (m, 1H, CH2CO-piperidine), 1.93 (m, 2H, CH2CH-piperidine); 13C NMR (126 MHz, DMSO-d6) δ 172.72, 171.78, 166.58, 161.41, 159.32, 157.48, 148.55, 135.30, 131.12, 122.11, 117.62, 112.29, 108.21, 49.53, 45.58, 35.60, 30.70, 24.18; Anal. Calcd. for C18H17FN4O4S (404.42): C, 56.57; H, 4.50; N, 10.42. Found: C, 56.78; H, 4.62; N, 10.68%.4.1.6.1. 2-((5-Chloro-3-ethyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(3-chloro-4-fluorophenyl)acetamide (21a).
Off white crystal (yield, 85%); m.p. = 240–244 °C. IR (KBr, cm−1): 3257 (NH), 3044 (C–H aromatic), 2987 (C–H aliphatic), 1669 (C
 O amide) and 1643 (amide II band); 1H NMR (DMSO-d6) δppm: 10.69 (s, 1H, NH), 7.92 (s, 1H, Ar–H), 7.67 (t, J = 8.0 Hz, 1H, Ar–H), 7.52 (s, 1H, Ar–H), 7.44 (m, 1H, Ar–H), 7.37 (m, 2H, Ar–H), 4.21 (s, 2H, SCH2), 4.08 (q, J = 7.4 Hz, 2H, NCH2CH3), 1.31 (t, J = 7.0 Hz, 3H, CH3); MS (m/z): 428 (M++1, 34%), 426 (M+, 52%), 147 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 166.53, 158.53, 157.30, 154.80, 152.39, 149.48, 136.82, 134.88, 133.12, 128.79, 125.86, 120.94, 119.91, 119.51, 117.37, 116.16, 36.98, 13.22; Anal. Calcd. for C18H14Cl2FN3O2S (426.29): C, 50.72; H, 3.31; N, 9.86. Found: C, 50.97; H, 3.45; N, 9.98%.
O amide) and 1643 (amide II band); 1H NMR (DMSO-d6) δppm: 10.69 (s, 1H, NH), 7.92 (s, 1H, Ar–H), 7.67 (t, J = 8.0 Hz, 1H, Ar–H), 7.52 (s, 1H, Ar–H), 7.44 (m, 1H, Ar–H), 7.37 (m, 2H, Ar–H), 4.21 (s, 2H, SCH2), 4.08 (q, J = 7.4 Hz, 2H, NCH2CH3), 1.31 (t, J = 7.0 Hz, 3H, CH3); MS (m/z): 428 (M++1, 34%), 426 (M+, 52%), 147 (100%, base peak); 13C NMR (101 MHz, DMSO-d6) δ 166.53, 158.53, 157.30, 154.80, 152.39, 149.48, 136.82, 134.88, 133.12, 128.79, 125.86, 120.94, 119.91, 119.51, 117.37, 116.16, 36.98, 13.22; Anal. Calcd. for C18H14Cl2FN3O2S (426.29): C, 50.72; H, 3.31; N, 9.86. Found: C, 50.97; H, 3.45; N, 9.98%.
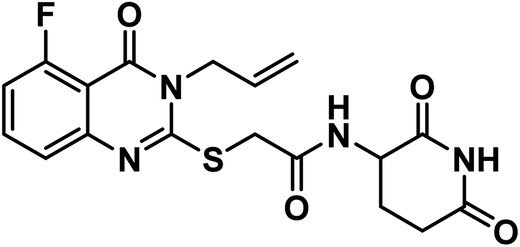
4.1.6.2. 2-((6-Chloro-3-ethyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(3-chloro-4-fluorophenyl)acetamide (21b).
Grayish white crystal (yield, 85%); m.p. = 209–211 °C. IR (KBr, cm−1): 3251 (NH), 3049 (C–H aromatic), 2985 (C–H aliphatic), 1675 (C
 O amide) and 1619 (amide II band); 1H NMR (DMSO-d6) δppm: 10.61 (s, 1H, NH), 7.91 (s, 1H, Ar–H), 7.71 (s, Hz, 1H, Ar–H), 7.50(m, 1H, Ar–H), 7.38 (d, J = 7.6 Hz, 1H, Ar–H), 7.19 (s, 2H, Ar–H), 4.20 (s, 2H, SCH2), 4.08 (q, J = 7.1 Hz, 2H, NCH2CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 166.51, 159.75, 157.10, 154.81, 152.40, 145.86, 136.81, 135.25, 130.41, 128.45, 125.77, 120.95, 120.48, 119.85, 119.52, 117.37, 37.09, 13.33; Anal. Calcd. for C18H14ClF2N3O2S (409.84): C, 52.75; H, 3.44; N, 10.25. Found: C, 52.91; H, 3.63; N, 10.12%.
O amide) and 1619 (amide II band); 1H NMR (DMSO-d6) δppm: 10.61 (s, 1H, NH), 7.91 (s, 1H, Ar–H), 7.71 (s, Hz, 1H, Ar–H), 7.50(m, 1H, Ar–H), 7.38 (d, J = 7.6 Hz, 1H, Ar–H), 7.19 (s, 2H, Ar–H), 4.20 (s, 2H, SCH2), 4.08 (q, J = 7.1 Hz, 2H, NCH2CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 166.51, 159.75, 157.10, 154.81, 152.40, 145.86, 136.81, 135.25, 130.41, 128.45, 125.77, 120.95, 120.48, 119.85, 119.52, 117.37, 37.09, 13.33; Anal. Calcd. for C18H14ClF2N3O2S (409.84): C, 52.75; H, 3.44; N, 10.25. Found: C, 52.91; H, 3.63; N, 10.12%.
4.1.6.3. N-(3-chloro-4-fluorophenyl)-2-((3-ethyl-5-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (21c).
White crystal (yield, 82%); m.p. = 279–281 °C. IR (KBr, cm−1): 3262 (NH), 3063 (C–H aromatic), 2987 (C–H aliphatic), 1686 (C
 O amide) and 1652 (amide II band); 1H NMR (DMSO-d6) δppm: 10.74 (s, 1H, NH), 8.0 (s, 1H, Ar–H), 7.92 (s, Hz, 1H, Ar–H), 7.80 (dd, J = 8.7, 2.6 Hz, 1H, Ar–H), 7.52 (d, J = 7.6 Hz, 1H, Ar–H), 7.46 (d, J = 8.7 Hz, 1H, Ar–H), 7.39 (t, J = 9.1 Hz, 1H, Ar–H), 4.23 (s, 2H, SCH2), 4.13 (q, J = 7.1 Hz, 2H, NCH2CH3), 1.31 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 166.06, 161.60, 159.51, 157.29, 154.27, 152.34, 148.66, 136.31, 135.42, 121.89, 120.68, 119.54, 117.22, 112.27, 108.49, 39.44, 36.75, 12.90. Anal. Calcd. for C18H14Cl2FN3O2S (426.29): C, 50.72; H, 3.31; N, 9.86. Found: C, 50.99; H, 3.48; N, 9.93%.
O amide) and 1652 (amide II band); 1H NMR (DMSO-d6) δppm: 10.74 (s, 1H, NH), 8.0 (s, 1H, Ar–H), 7.92 (s, Hz, 1H, Ar–H), 7.80 (dd, J = 8.7, 2.6 Hz, 1H, Ar–H), 7.52 (d, J = 7.6 Hz, 1H, Ar–H), 7.46 (d, J = 8.7 Hz, 1H, Ar–H), 7.39 (t, J = 9.1 Hz, 1H, Ar–H), 4.23 (s, 2H, SCH2), 4.13 (q, J = 7.1 Hz, 2H, NCH2CH3), 1.31 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 166.06, 161.60, 159.51, 157.29, 154.27, 152.34, 148.66, 136.31, 135.42, 121.89, 120.68, 119.54, 117.22, 112.27, 108.49, 39.44, 36.75, 12.90. Anal. Calcd. for C18H14Cl2FN3O2S (426.29): C, 50.72; H, 3.31; N, 9.86. Found: C, 50.99; H, 3.48; N, 9.93%.
4.2. Biological testing
4.2.6.1. ADMET studies. ADMET descriptors were determined using Discovery studio 4.0 as according to the reported method42–45 (ESI†).
4.2.6.2. Toxicity studies. The toxicity parameters of the synthesized compounds were calculated using Discovery studio 4.0 as described46–49 in ESI.†
Conflicts of interest
There is no conflict of interest.Acknowledgements
This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STIFA) under grant number 43040.References
- A. A. Gaber, A. H. Bayoumi, A. M. El-Morsy, F. F. Sherbiny, A. B. Mehany and I. H. Eissa, Bioorg. Chem., 2018, 80, 375–395 CrossRef CAS PubMed.
- R. A. Asherson, K. Gunter, D. Daya and Y. Shoenfeld, J. Rheumatol., 2008, 35, 1224–1227 Search PubMed.
- L. S. Chan, Clin. Dermatol., 2012, 30, 34–37 CrossRef PubMed.
- A. Mercurio, G. Adriani, A. Catalano, A. Carocci, L. Rao, G. Lentini, M. M. Cavalluzzi, C. Franchini, A. Vacca and F. Corbo, Curr. Med. Chem., 2017, 24, 2736–2744 CrossRef CAS PubMed.
- S. Joglekar and M. Levin, Drugs Today, 2004, 40, 197–204 CrossRef CAS PubMed.
- R. Knight, Seminars in Oncology, Elsevier, 2005, pp. 24–30 Search PubMed.
- J. Folkman, Seminars in Oncology, Elsevier, 2001, pp. 536–542 Search PubMed.
- M. Melchert and A. List, Int. J. Biochem. Cell Biol., 2007, 39, 1489–1499 CrossRef CAS PubMed.
- A. L. Ruchelman, H.-W. Man, W. Zhang, R. Chen, L. Capone, J. Kang, A. Parton, L. Corral, P. H. Schafer and D. Babusis, Bioorg. Med. Chem. Lett., 2013, 23, 360–365 CrossRef CAS PubMed.
- S. K. Teo, AAPS J., 2005, 7, E14–E19 CrossRef CAS PubMed.
- R. Suppiah, J. G. Srkalovic and M. A. Hussein, Clin. Lymphoma Myeloma, 2006, 6, 301–305 CrossRef CAS PubMed.
- E. Terpos, N. Kanellias, D. Christoulas, E. Kastritis and M. A. Dimopoulos, OncoTargets Ther., 2013, 6, 531 CrossRef CAS PubMed.
- S. Singhal and J. Mehta, Biomed. Pharmacother., 2002, 56, 4–12 CrossRef CAS PubMed.
- J. B. Marriott, G. Muller and A. G. Dalgleish, Immunol. Today, 1999, 20, 538–540 CrossRef CAS PubMed.
- T. Ito and H. Handa, Proc. Jpn. Acad., Ser. B, 2020, 96, 189–203 CrossRef CAS PubMed.
- C. C. Bjorklund, J. Kang, M. Amatangelo, A. Polonskaia, M. Katz, H. Chiu, S. Couto, M. Wang, Y. Ren and M. Ortiz, Leukemia, 2020, 34, 1197–1201 CrossRef PubMed.
- L. Li, W. Xue, Z. Shen, J. Liu, M. Hu, Z. Cheng, Y. Wang, Y. Chen, H. Chang and Y. Liu, Mol. Ther.-Oncolytics, 2020, 18, 215–225 CrossRef CAS PubMed.
- A. R. Kotb, D. A. Bakhotmah, A. E. Abdallah, H. Elkady, M. S. Taghour, I. H. Eissa and M. A. El-Zahabi, RSC Adv., 2022, 12, 33525–33539 RSC.
- J. Qiu, B. Xu, Z. Huang, W. Pan, P. Cao, C. Liu, X. Hao, B. Song and G. Liang, Bioorg. Med. Chem., 2011, 19, 5352–5360 CrossRef CAS PubMed.
- A. E. Abdallah, M. S. Alesawy, S. I. Eissa, E. M. El-Fakharany, M. H. Kalaba, M. H. Sharaf, N. M. A. Shama, S. H. Mahmoud, A. Mostafa and A. A. Al-Karmalawy, New J. Chem., 2021, 45, 16557–16571 RSC.
- A. E. Abdallah, R. R. Mabrouk, M. M. S. Al Ward, S. I. Eissa, E. B. Elkaeed, A. B. Mehany, M. A. Abo-Saif, O. A. El-Feky, M. S. Alesawy and M. A. El-Zahabi, J. Enzyme Inhib. Med. Chem., 2022, 37, 573–591 CrossRef CAS PubMed.
- A. S. El-Azab, A. A.-M. Abdel-Aziz, H. A. Ghabbour and M. A. Al-Gendy, J. Enzyme Inhib. Med. Chem., 2017, 32, 1229–1239 CrossRef CAS PubMed.
- A. E. Abdallah, S. I. Eissa, M. M. S. Al Ward, R. R. Mabrouk, A. B. Mehany and M. A. El-Zahabi, Bioorg. Chem., 2021, 109, 104695 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. B. Mehany, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dyn., 2022, 1–16 CrossRef PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, E. B. Elkaeed, B. A. Alsfouk, M. S. Alesawy, D. Z. Husein and A. M. Metwaly, PLoS One, 2022, 17, e0272362 CrossRef CAS PubMed.
- R. G. Yousef, H. Elkady, E. B. Elkaeed, I. M. Gobaara, H. A. Al-ghulikah, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 7719 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, M. Alswah, H. S. Elzahabi, A. M. Metwaly and I. H. Eissa, Processes, 2022, 10, 2290 CrossRef CAS.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS PubMed.
- S. A. El-Metwally, M. M. Abou-El-Regal, I. H. Eissa, A. B. Mehany, H. A. Mahdy, H. Elkady, A. Elwan and E. B. Elkaeed, Bioorg. Chem., 2021, 112, 104947 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, A. Elwan and H. Elkady, J. Enzyme Inhib. Med. Chem., 2021, 36, 1093–1114 CrossRef CAS PubMed.
- D. Gerlier and N. Thomasset, J. Immunol. Methods, 1986, 94, 57–63 CrossRef CAS PubMed.
- A. I. Abd-Elhamid, H. El-Gendi, A. E. Abdallah and E. M. El-Fakharany, Pharmaceutics, 2021, 13, 1595 CrossRef CAS PubMed.
- N. T. Dawoud, E. M. El-Fakharany, A. E. Abdallah, H. El-Gendi and D. R. Lotfy, Sci. Rep., 2022, 12, 1–17 CrossRef PubMed.
- R. M. Talaat, Viral Immunol., 2010, 23, 151–157 CrossRef CAS PubMed.
- MyBioSource ELISA Test Kits, https://www.mybiosource.com/human-elisa-kits/casp8/2513072#QLAPP_MBS2513072_TD, (accessed May 2018) Search PubMed.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Bioorg. Chem., 2020, 104, 104218 CrossRef CAS PubMed.
- J. Wang and M. J. Lenardo, J. Cell Sci., 2000, 113, 753–757 CrossRef CAS PubMed.
- W. M. Eldehna, G. S. Hassan, S. T. Al-Rashood, T. Al-Warhi, A. E. Altyar, H. M. Alkahtani, A. A. Almehizia and H. A. Abdel-Aziz, Med. Chem., 2019, 34, 322–332 CAS.
- K. K.-W. Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef CAS PubMed.
- A. Sabt, O. M. Abdelhafez, R. S. El-Haggar, H. M. Madkour, W. M. Eldehna, E. E.-D. A. El-Khrisy, M. A. Abdel-Rahman and L. A. Rashed, Med. Chem., 2018, 33, 1095–1107 CAS.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Processes, 2022, 10, 1391 CrossRef CAS.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 4606 CrossRef CAS PubMed.
- E. B. Elkaeed, I. H. Eissa, H. Elkady, A. Abdelalim, A. M. Alqaisi, A. A. Alsfouk, A. Elwan and A. M. Metwaly, Int. J. Mol. Sci., 2022, 23, 8407 CrossRef CAS PubMed.
- E. B. Elkaeed, F. S. Youssef, I. H. Eissa, H. Elkady, A. A. Alsfouk, M. L. Ashour, M. A. El Hassab, S. M. Abou-Seri and A. M. Metwaly, Int. J. Mol. Sci., 2022, 23, 6912 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy, E. B. Elkaeed, A. A. Alsfouk, M. S. Alesawy, A. M. Metwaly and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 1903–1917 CrossRef CAS PubMed.
- A. Belal, N. M. Abdel Gawad, A. B. Mehany, M. A. Abourehab, H. Elkady, A. A. Al-Karmalawy and A. S. Ismael, J. Enzyme Inhib. Med. Chem., 2022, 37, 1884–1902 CrossRef CAS PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, M. M. Alanazi, M. A. Alharbi, I. H. Eissa and M. A. Dahab, RSC Adv., 2021, 11, 30315–30328 RSC.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, J. Mol. Struct., 2021, 132220 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00066d |
| This journal is © The Royal Society of Chemistry 2023 |