
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general method to predict optical rotations of chiral molecules from their structures
Hai-Feng Ji
Department of Chemistry, Drexel University, Philadelphia, PA 19104, USA. E-mail: hj56@drexel.edu; Fax: +1-215-895-1265; Tel: +1-215-895-2562
First published on 6th February 2023
Abstract
The relationship of the chiroptical response of a molecule to its absolution configuration does not exist now. In this letter, I intend to report a general rule with exceptions to predict the sign of optical rotation of chiral molecules with a RCHXY structure from their absolute configurations using the Hammett constant, σp, which is based on the electron withdrawing/donating power of functional groups. In this rule, a priority list of functional groups based on the electron withdrawing powers of the groups are used. When the lowest priority group is in the back of the molecule, a clockwise arrangement of the other three priorities from the most electron withdrawing to the least withdrawing (1-2-3) is predicted to be dextrorotatory, the counterclockwise arrangement is predicted to be levorotatory.
Introduction
2022 marks the 200th birthday of Louis Pasteur, who separated the first pair of chiral molecules in the 19th century.1 Since then, scientists have strived to make a connection between the optical rotation of a chiral molecule and its molecular structure. Unfortunately, the relationship of the chiroptical response of a molecule to its absolution configuration has still not been achieved or does not exist now. The famed Cahn–Ingold–Prelog (CIP) rules2 have been used to assign the absolute R and S configurations around a stereocenter, but every textbook of organic chemistry specifically addresses that there is no correlation between the R/S configuration and the optical rotation of chiral molecules. Experiments must be conducted in order to determine the optical properties of chiral molecules. Currently, much of the effort in this field has focused on computational chemistry to improve the accuracy in predicting the optical rotation from the structures of molecules.3–6 It is doubtful that such a correlation would ever exist. However, there is likely to be a method that is close to a general rule to predict the optical rotations of chiral molecules with some exceptions.Chiral molecules are those that have no plane of symmetry and can rotate linearly polarized light. In a simple chiral molecule with only one chiral carbon, four different groups are bonded to carbon so it can be considered a spiral object. A linearly polarized light beam can be regarded as a superposition of two circularly polarized electromagnetic waves.7 They proceed in the same frequency and direction but with the opposite rotation. The spiral shape of chiral molecules enables them to have an opposite effect on the two circularly polarized electromagnetic waves, so the two waves travel at slightly different speeds through the chiral medium,8 i.e., they no longer have the same phase on the original plane. This phase shift results in the cancellation of the two waves at a slightly different plane, causing an optical rotation (or deviation) of the linearly polarized light beam.
Since the rotation of the linearly polarized light beam is caused by the spatial arrangement of the four groups, it is expected that parameters related to the electron characteristics of the four groups could be used to correlate the optical rotation of chiral molecules with their absolute configurations. In this letter, I intend to report a general rule with exceptions to predict the optical rotation sign of chiral molecules from their absolute configurations using the order of the electron-withdrawing/donating powers of the four groups.
The electron withdrawing/donating properties of substituents have been used to compare the reactivities of various organic reactions, such as hydrolysis,9 ionization,10 esterifications,11 brominations,12 electrophilic aromatic substitutions,13 etc. The electron-withdrawing power of functional groups follows the following order in general, but some of them shift slightly depending on reactions:
NR2 < NH2 < OH < OR < NHCOR < OCOR < R < Ph < CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2 <H < X < CHO < COR < COOR < COOH < COCl < CF3 < CN < SO3H < NO2 <NH3+ < NR3+ CR2 <H < X < CHO < COR < COOR < COOH < COCl < CF3 < CN < SO3H < NO2 <NH3+ < NR3+ |
This order can be quantitively described using various substituent constants, such as the Hammett constant,8 and constants used in the Swain–Lupton equation,14 the Taft equation,15 the Grunwald–Winstein equation,16 and the Yukawa–Tsuno equation,17 etc. Among these, the Hammett equation was the first developed and has been widely used, and the Hammett constants are a set of the most complete list of substituent constants.
The Hammett constants, σp and σm, contain both inductive and resonance effects of substituents on the hydrolysis of benzoic acid esters. σp has less effect from the steric hindrance because the substituents are on the para position than that of σm at the meta position. The applications of these constants are far beyond the reactions only.18 Both σp and σm have been used in this study to predict the optical rotation from the structures of molecules. σp constants are used in this report because they have a better performance, suggesting the steric effect is not critical for the correlation between the optical rotation and the absolute configuration. σp constants of some major organic groups are listed in Table 1. Stronger electron-withdrawing groups have a greater positive number, and electron donating groups have negative numbers. All the numbers are obtained from two ref. 18 and 19.
| NMe2 | NH2 | OH | OR(OMe) | NHCOCH(Me)2 | OCOR(Me) | R(Me) | H | |
|---|---|---|---|---|---|---|---|---|
| σp | −0.83 | −0.66 | −0.37 | −0.27 | −0.1 | −0.19 | −0.17 | 0 |
| Ph | X (Cl, Br) | COOR(H) | COCl | CF3 | CN | NO2 | NR3+ | |
|---|---|---|---|---|---|---|---|---|
| σp | 0.01 | 0.23 | 0.45 | 0.61 | 0.54 | 0.66 | 0.78 | 0.82 |
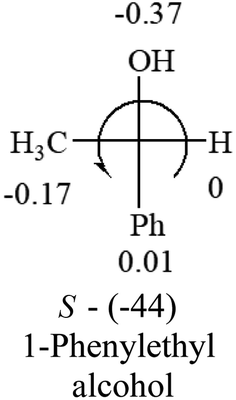
A general rule on predicting the chiroptical sign of chiral molecules can be described as: first, assign the priorities to each bonded group surrounding the stereocenter based on their electron-withdrawing power (1, most to 4, least). Second, move the lowest priority (4) group to the back of the molecule. Count the other three priorities from the most electron-withdrawing to the least withdrawing (1-2-3). A clockwise is predicted to be dextrorotatory, the counterclockwise is predicted to be levorotatory. In this rule, instead of a priority list of functional groups based on the atomic number of the elements, electron-withdrawing powers of the groups are used. The structure can be written either as a 3D structure or a fisher projection (Fig. 1).
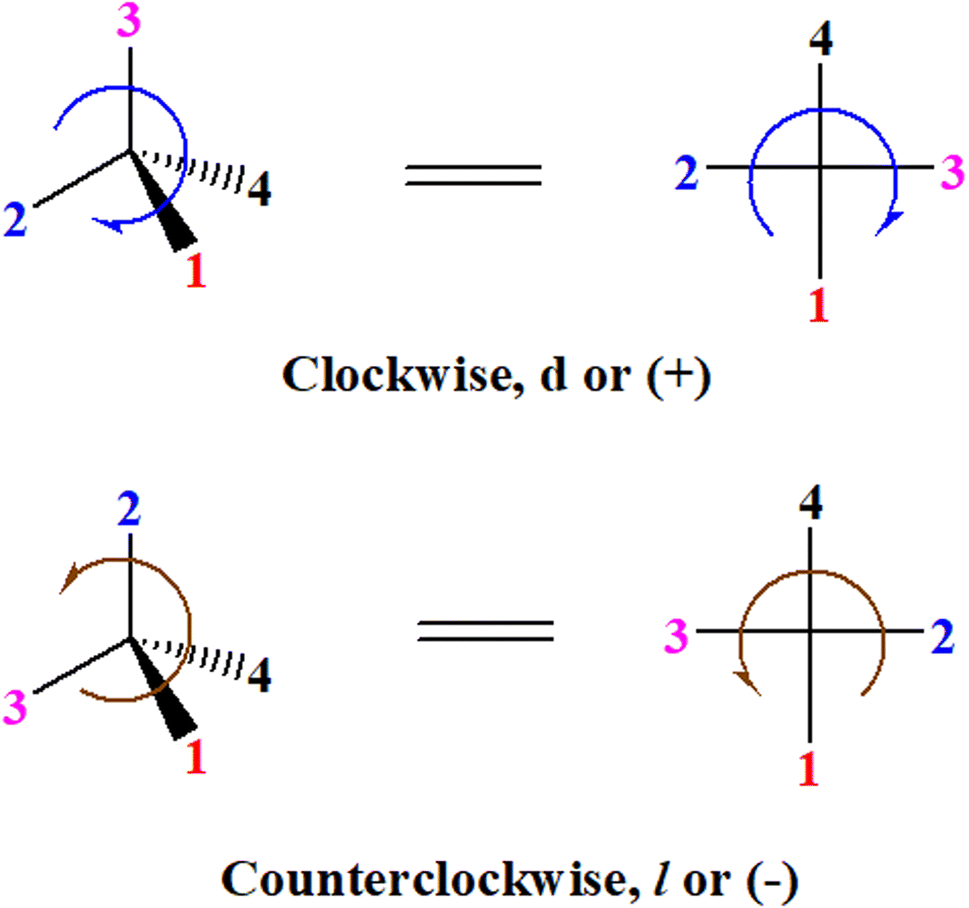 | ||
| Fig. 1 The 3D structure and fisher projection of the general rule. Electron withdrawing powers 1 > 2> 3 > 4. | ||
Two model systems were studied in this work to confirm how the above general rule applies to the prediction of the chiroptical response (sign) of chiral molecules from their molecular structures and the accuracy rate. The first is an RCHXY system. The second is the 20 L-amino acids in their acidic forms.
Discussion
Table 2 shows the prediction result of RCHXY molecules, where R is CH3 as those listed in the table, but other alkyl groups gave the same results on the sign, X and Y are two other popular organic functional groups. Several common functional groups, such as –OCOR, –NO2 were not included in the table since there are no chiroptical data on these chemicals. The group NH3+ will be specifically discussed in the amino acid section later. The rotation signs of these compounds were obtained from literature or available chemical catalogs. Those signs that could not be found were listed as —.| X | Y | |||||
|---|---|---|---|---|---|---|
| –NH2 | –OH | –OR | –OCOR | –Ph | –X | |
| –NH2 or –NR2 | n/a | 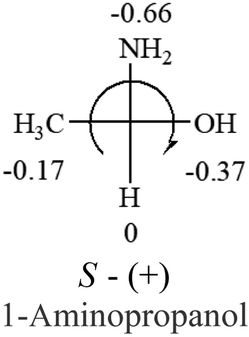 |
— | — | 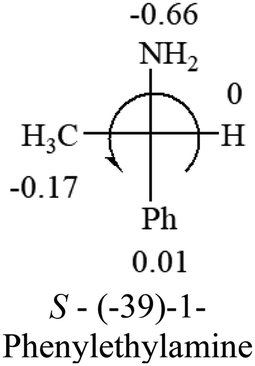 |
— |
| –OH or –OR | See row 2-column 3 | — | — | — | See row 4-column 3 | — |
| –Ph | 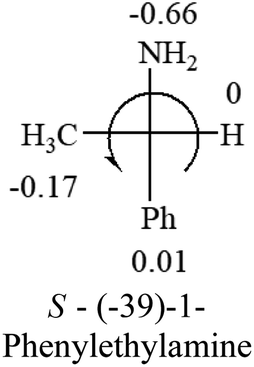 |
 |
— | 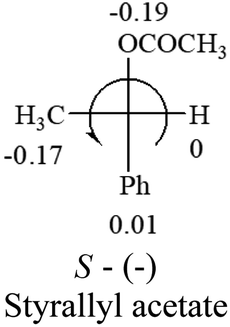 |
— | — |
| –COOR or –COOH | 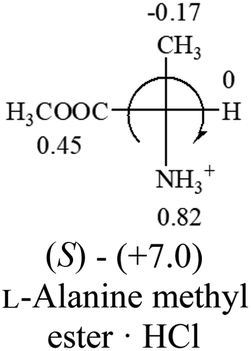 |
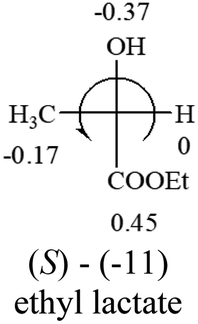 |
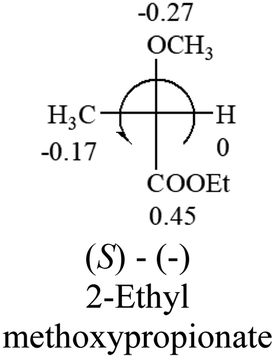 |
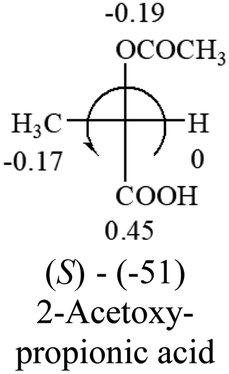 |
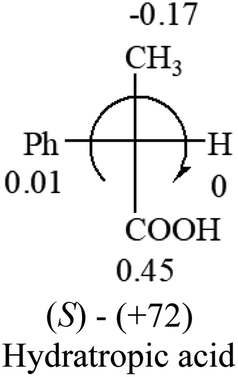 |
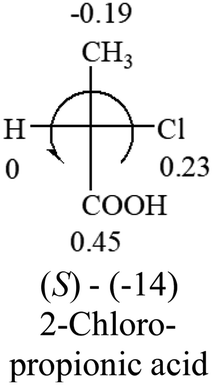 |
| –CF3 | — | 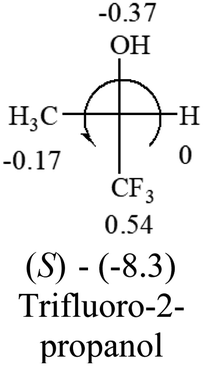 |
— | — | — | — |
| –CN | — | 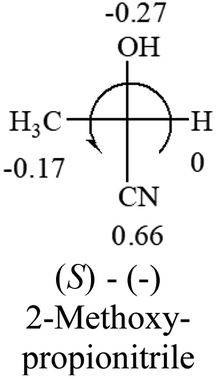 |
— | — | — | |
The result shows that all the molecules listed in Table 2, except for lactic acid (S, +2.6), obey the electron-withdrawing rule above, indicating a high accuracy in predicting the chiroptical sign of chiral molecules from their molecular structures. It is not unexpected that lactic acid disobeys the rule since the dominating species in water is its dimmer and larger self-aggregation via hydrogen bonding over its monomer form,20 which will change the electron-withdrawing powers of both –OH and –COOH groups. As a comparison, lactic acetate ethyl ester doesn't form such aggregation in water and its monomer form does obey the rule. The chiroptical rotation of lactate is included in Table 2 to show the general trend since the –COOEt in lactate has the same Hammett constant as that of –COOH in lactic acid.
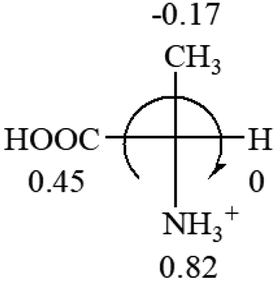
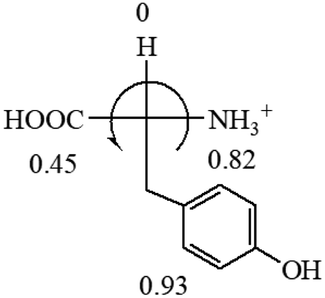
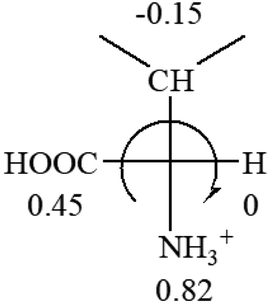
All the 20 natural L-amino acids, except for glycine and cysteine, have the S absolute configuration, but the signs of specific rotation can be both positive and negative. In the early 20th century, Lutz and Jirgensons21 reported that when a strong acid was added to an aqueous solution of natural L-amino acids, the specific rotation of the protonated amino acids became more positive than their zwitterionic forms, which is called the CLJ rule.22 This phenomenon has been used in industry due to its reliability, but no explanation has been proposed. The above prediction of the optical rotations of chiral chemicals can well explain this phenomenon. Taking L-(S)-alanine as one example, in pure water, it is in equilibrium with its zwitterionic form (eqn (1)), and this mixture of alanine has a negative sign, i.e., levorotary. According to the prediction method set in this work, the original form of alanine (CH3CHNH2COOH) is expected to have a levorotary sign since the order of the electron-withdrawing powers is counterclockwise from –COOH to –H and to –CH3 (Fig. 2 left). The zwitterionic form may not contribute to the optical rotation of alanine since the constants of both –COO− and H are zero. The addition of HCl protonates the carboxylate group (eqn (2)),23 as a result, the most electron withdrawing group in alanine changes from COOH to NH3+, and the sign of this protonated form is positive since the order of the electron-withdrawing powers is now clockwise from –NH3+ to –COOH and to –H (Fig. 2 right).
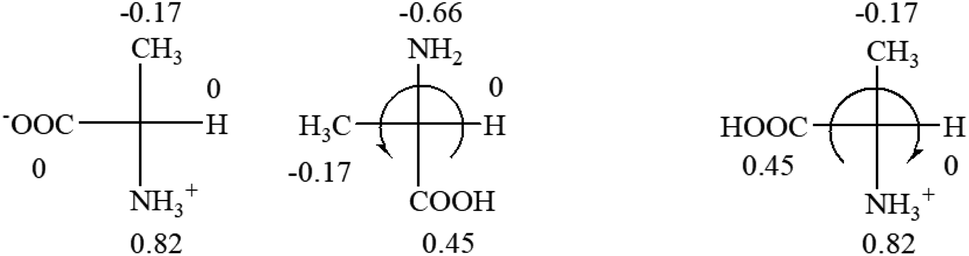 | ||
| Fig. 2 Left: optical rotation sign predicted from L-(S)-alanine (CH3CHNH2COOH). Right: sign predicted from the protonated form of L-(S)-alanine (CH3CHNH3+COOH). | ||
It is noteworthy that although the Hammett constant σp of –COO− is zero, this constant specifically for the purpose of predicting the chiroptical response of chiral molecules can be estimated from the optical rotations of amino acids in alkaline solutions. It is known that the addition of an access amount of NaOH in the aqueous solution of alanine decreases the rotation to a more negative number, i.e. rotate to the further left.20 In the alkaline solution, alanine only exists in its anionic form, CH3CHNH2COO−. According to the prediction rule in this work, in order for CH3CHNH2COO− to be levorotatory, the σp of COO− should be slightly less than 0 of –H, but greater than −0.17 of –CH3.
| CH3CHNH2COOH → CH3CHNH3+COO− | (1) |
| CH3CHNH3+COO− + HCl → CH3CHNH3+COOH | (2) |
Since the specific rotations of amino acids in pure water were recorded from a mixture of the two forms, the protonated form is used in this work to analyze how the general rule applies to the prediction of chiroptical response (sign) of L-amino acids from their molecular structures and the accuracy rate.
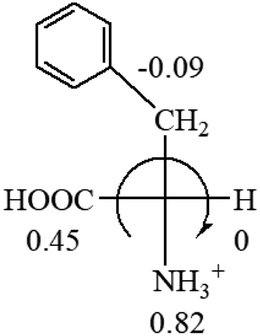
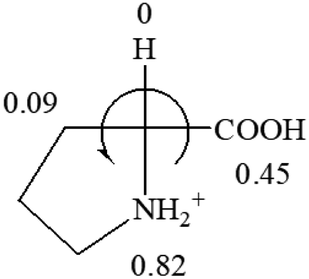
The result in Table 3 shows that all the protonated natural L-amino acids, except for phenylalanine, obey the general rule introduced in this work. The exception for phenylalanine may be caused by the π-cation interaction of the benzene ring with –NH3+, which decreases the Hammett constant of –NH3+.
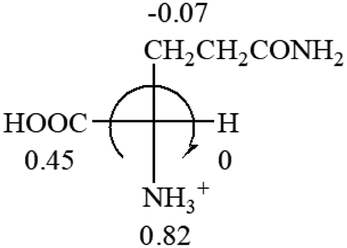
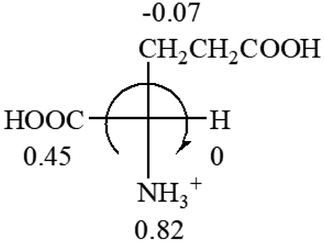
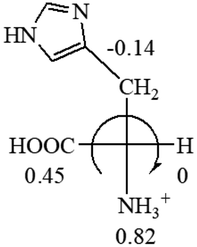
CH2 (−0.14); glutamine: no σp for CH2CH2CONH2. It will be close to CH2CH2COOH (−0.07); histidine: no σp on –CH2-ring. It will be close to –CH2CHCH2 (−0.14)
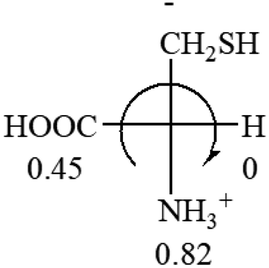
| L-Alanine (+13) | L-Arginine (+21.6) | L-Asparagine (+34) | L-Aspartic acid (+24.4) | L-Cysteine (+7.9) |
 |
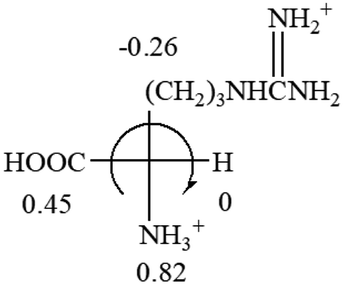 |
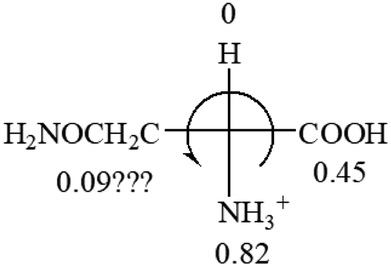 |
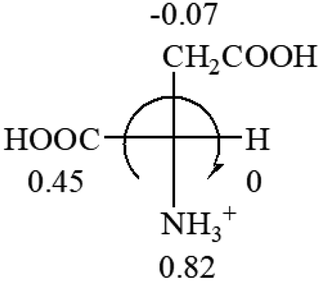 |
 |
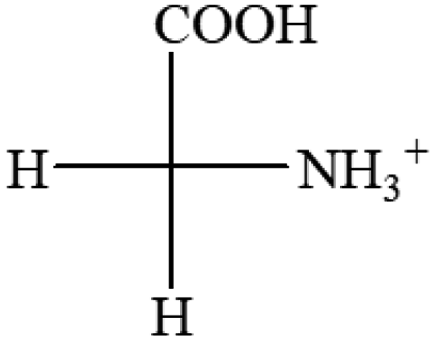
| L-Glutamine (+6.5) | L-Glutamic acid (+31.5) | Glycine (not chiral) | L-Histidine (+13) | L-Isoleucine (+51.8) two chiral centers |
 |
 |
 |
 |
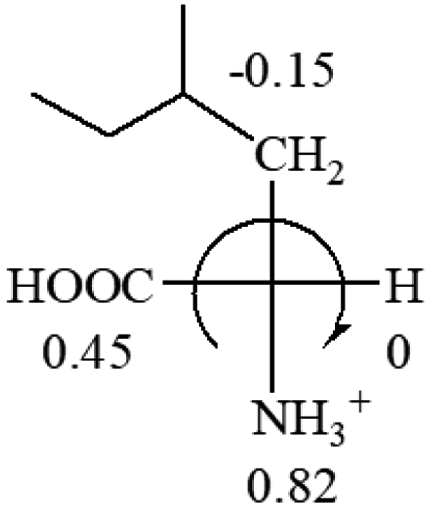 |
| L-Leucine (+14.9) | L-Lysine (+20.5) | L-Methionine (22.8) | L-Phenylalanine (−7.4) | L-Proline (−69) |
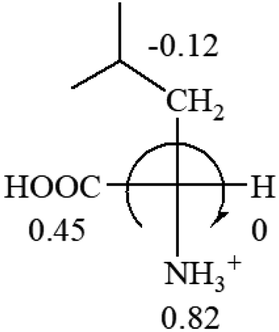 |
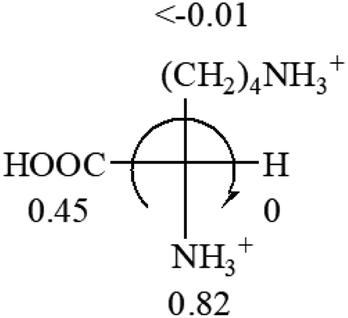 |
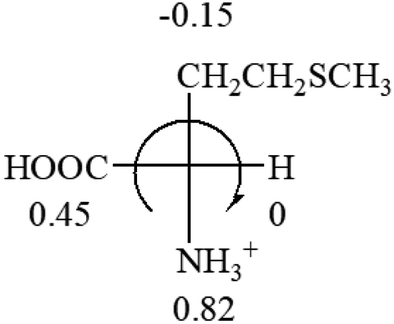 |
 |
 |
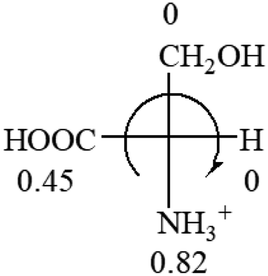
| L-Serine (+15.9) | L-Threonine (−17.9) | L-Tryptophan (+13) | L-Tyrosine (−10.1) | L-Valine (+26.9) |
 |
Two chiral centers | 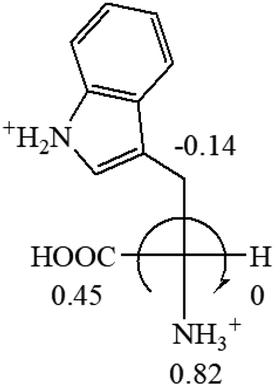 |
 |
 |
As observed in all these cases, the result showed high accuracy in predicting the chiroptical sign of chiral molecules from their molecular structures, but there are chiral molecules that do not obey the above general prediction rule. One feature in common is the existence of inter and intra- molecular interactions, such as those in lactic acid and phenylalanine. Another feature that is common, but not specifically discussed in the above examples, is that the Hammett constants of at least two groups are close to each other, such as two alkyl groups. For instance, the constants of several alkyl groups are: Me (−0.17), Et (−0.15), butyl (−0.15), isobutyl (−0.15), etc. The prediction of these molecules is difficult and their disagreement with experimental measurements of the optical rotation is expected. For accurate prediction of the optical rotation, the difference in σp between any two groups, Δσp, should be greater than a threshold number, such as 0.05. If Δσp < 0.05, even the weak interactions of the R groups with other functional groups on the asymmetric carbon become a crucial factor to determine the optical rotation of the molecule since a minor change in the electron donating/withdrawing power of R will change the optical rotation of the molecule. Other than interactions with adjacent groups, additional structural information related to the conformation of alkyl groups must also be considered, which is known to affect the magnitude and sign of optical rotations of chiral molecules.24 If both features, interactions and Δσp, exist in one molecule, the prediction will become more challenging. For example, in CH3CHNH2CH2OH, the σp of –CH2OH is 0, which is the same as –H (0), and it forms internal hydrogen bonding with –NH2. For molecules like all of these, the quantum theoretical calculation is again needed to aid in more accurate prediction of their chiroptical responses, which might explain why such prediction did not exist so far.
In conclusion, prediction of the sign of optical rotations of chiral molecules from their absolute configurations can generally be achieved using the Hammett constants, σp, based on the electron-withdrawing/donating power of functional groups. The Hammett constants, σp, may further be fine-tuned specifically for the purpose of such prediction. The modification will be based on a large number of available data on chiral molecules. Re-measurement of some chemicals may be necessary to obtain accurate optical rotations, such as a change of pH to measure the optical rotation of a single form of amine or carboxylic acid species. Once this set of data is built, it is also likely to deduce a general empirical equation to calculate the amplitude of specific rotations using a data fitting method. More importantly, the general rule based on the withdrawing/donating powers of functional groups may be applied in quantum theoretical calculations to accurately calculate the sign and amplitude of specific rotations of chiral molecules in the future, especially for molecules with small Δσp of two groups, or more than two chiral carbons and chiral molecules without chiral carbons.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks the Louis and Bessie Stein Family Fellowship for the support of the work.References
- L. Pasteur, Lecons de Chlmie en 1860, Alembic Club Reprint, 1848, vol. 14 Search PubMed.
- R. S. Cahn, C. K. Ingold and V. Prelog, Specification of Molecular Chirality, Angew. Chem., Int. Ed., 1966, 5, 385–415 CrossRef CAS.
- P. L. Polavarapu, Renaissance in chiroptical spectroscopic methods for molecular structure determination, Chem. Rec., 2007, 7, 125–136 CrossRef CAS PubMed.
- A. R. Turbomole, M. Bar, M. Haser, H. Horn and C. Kolmel, Electronic structure calculations on workstation computers: The program system turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef.
- T. D. Crawford, Ab initio calculation of molecular chiroptical properties, Theor. Chem. Acc., 2006, 115, 227–245 Search PubMed.
- P. J. Stephens, F. J. Devlin, J. R. Cheeseman and M. J. Frisch, Calculation of Optical Rotation Using Density Functional Theory, J. Phys. Chem. A, 2001, 105, 5356–5371 CrossRef CAS.
- F. A. Jenkins and H. E. White, Fundamentals of Optics, McGraw-Hill, New York, 4th edn, 1976 Search PubMed.
- A. Fresnel, Mémoire sur la double réfraction que les rayons lumineux éprouvent en traversant les aiguilles de cristal de roche suivant les directions parallèles à l'axe, in Oeuvres complètes d'Augustin Fresnel, ed. H. de Senarmont, E. Verdet and L. Fresnel, 1866, vol. 1, pp. 731–51, translated as Memoir on the double refraction that light rays undergo in traversing the needles of quartz in the directions parallel to the axis, Zenodo, 4745976, 2021 Search PubMed.
- L. P. Hammett, The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives, J. Am. Chem. Soc., 1937, 59(1), 96–103 CrossRef CAS.
- H. C. Ko, W. F. O'Hara, T. Hu and L. G. Hepler, Ionization of Substituted Phenols in Aqueous Solution, J. Am. Chem. Soc., 1964, 86, 1003–1004 CrossRef CAS.
- R. W. Taft, Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters, J. Am. Chem. Soc., 1952, 74, 3120–3128 CrossRef CAS.
- J. Toullec and M. El-Alaoui, Ring substituent effects on acetophenone dimethyl acetal formation. 2 Dual-parameter treatment of kinetic data for acid-catalyzed acetal formation and hydrolysis in methanol containing small amounts of water, J. Org. Chem., 1985, 50, 4928–4933 CrossRef CAS.
- R. O. C. Norman and J. M. Coxon, Principles of Organic Synthesis, CRC Press, 3rd edn, 1993, pp. 353–354, ISBN 9780748761623 Search PubMed.
- C. G. Swain and E. C. Lupton, Field and resonance components of substituent effects, J. Am. Chem. Soc., 1968, 90, 4328–4337 CrossRef CAS.
- R. W. Taft, Linear Free Energy Relationships from Rates of Esterification and Hydrolysis of Aliphatic and Ortho-substituted Benzoate Esters, J. Am. Chem. Soc., 1952, 74, 2729–2732 CrossRef CAS.
- E. Grunwald and S. Winstein, The Correlation of Solvolysis Rates, J. Am. Chem. Soc., 1948, 70, 846–854 CrossRef CAS.
- Y. Yasuhide and T. Yuho, Resonance Effect in Hammett Relationship. II. Sigma Constants in Electrophilic Reactions and their Intercorrelation, Bull. Chem. Soc. Jpn., 1959, 32, 965–971 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, Dordrecht, 1981, ISBN: 978-94-009-5883-8 Search PubMed.
- M. D. Kundrat and J. Autschbach, Time dependent density functional theory modeling of chiroptical properties of small amino acids in solution, J. Phys. Chem. A, 2006, 110, 4115–4123 CrossRef CAS PubMed.
- I. O. Lutz and B. Jirgensons, Über eine neue Methode der Zuteilung optisch-aktiver α-Aminosäuren zur Rechts- oder Linksreihe I Mitteil, Ber. Dtsch. Chem. Ges., 1930, 63B, 448–460 CrossRef CAS.
- J. P. Greenstein and M. Winitz, Chemistry of the Amino Acids, John Wiley & Sons, New York, 1961 Search PubMed.
- M. D. Kundrat and J. Autschbach, Time Dependent Density Functional Theory Modeling of Specific Rotation and Optical Rotatory Dispersion of the Aromatic Amino Acids in Solution, J. Phys. Chem. A, 2006, 110, 12908–12917 CrossRef CAS PubMed.
- M. Pecul, K. Ruud, A. Rizzo and T. Helgaker, Conformational Effects on the Optical Rotation of Alanine and Proline, J. Phys. Chem. A, 2004, 108, 4269–4276 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |