DOI:
10.1039/D2RA08272A
(Paper)
RSC Adv., 2023,
13, 7828-7833
1,10-Phenanthroline-based periodic mesoporous organosilica: from its synthesis to its application in the cobalt-catalyzed alkyne hydrosilylation†
Received
28th December 2022
, Accepted 1st March 2023
First published on 9th March 2023
Abstract
1,10-Phenanthroline (Phen) is a typical ligand for metal complexation and various metal/Phen complexes have been applied as a catalyst in several organic transformations. This study reports the synthesis of a Phen-based periodic mesoporous organosilica (Phen-PMO) with the Phen moieties being directly incorporated into the organosilica framework. The Phen-PMO precursor, 3,8-bis[(triisopropoxysilyl)methyl]-1,10-phenanthroline (1a), was prepared via the Kumada–Tamao–Corriu cross-coupling of 3,8-dibromo-1,10-phenanthroline and [(triisopropoxysilyl)methyl]magnesium chloride. The co-condensation of 1a and 1,2-bis(triethoxysilyl)ethane in the presence of P123 as the template surfactant afforded Phen-PMO 3 with an ordered 2-D hexagonal mesoporous structure as confirmed by nitrogen adsorption/desorption measurements, X-ray diffraction, and transition electron microscopy. Co(OAc)2 was immobilized on Phen-PMO 3, and the obtained complex showed good catalytic activity for the hydrosilylation reaction of phenylacetylene with phenylsilane.
Introduction
In 1999, periodic mesoporous organosilica (PMO) materials were developed independently by three groups, Inagaki, Ozin, and Stein.1–3 Since then, these materials have been attracting attention as organic/inorganic hybrid materials owing to their ordered mesoporous structures.4 In particular, PMOs are promising candidates in catalysis, where they are used as solid ligands to immobilize active metal complexes in the mesopores.5 While the direct incorporation of pre-formed metal complexes into organosilica frameworks during the synthesis of PMOs has been already reported,6 PMOs can act as versatile solid ligands by (a) the direct incorporation of a coordination site into the organosilica framework followed by complexation,7,8 or (b) the post-functionalization of PMOs by grafting a metal complex/a coordination site via the surface silanol groups or functional groups integrated into the organosilica framework.9
In the direct incorporation approach, 2,2′-bipyridine-based periodic mesoporous organosilica (BPy-PMO), prepared from a 5,5′-bis(triisopropoxysilyl)-2,2′-bipyridine precursor, has been the most widely developed solid ligand for an array of metal complexes.7,10 The immobilized complexes obtained by this approach have been employed as catalysts for various organic transformations and photocatalytic reactions. Furthermore, the synthesis of BPy-PMO analogs that fine-tune the catalytic activity and/or impart new reactivity have been reported,11 which can further enhance the potential of PMOs in catalysis.
1,10-Phenanthroline (Phen) has a coordination geometry similar to 2,2′-bipyridine because both have two sp2 nitrogen atoms and can form five-membered chelates upon their coordination with a metal center.12 The C2–C2′ bond of 2,2′-bipyridine can rotate freely, whereas Phen possesses a more rigid planar geometry because of the additional –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– bridge. Therefore, the two sp2 nitrogen atoms of Phen are juxtaposed to readily form a chelate. Furthermore, the additional bridge extends its π-conjugated system. Consequently, Phen and 2,2′-bipyridine sometimes exhibit different catalytic activities when applied as ligands for metal complexes.13
CH– bridge. Therefore, the two sp2 nitrogen atoms of Phen are juxtaposed to readily form a chelate. Furthermore, the additional bridge extends its π-conjugated system. Consequently, Phen and 2,2′-bipyridine sometimes exhibit different catalytic activities when applied as ligands for metal complexes.13
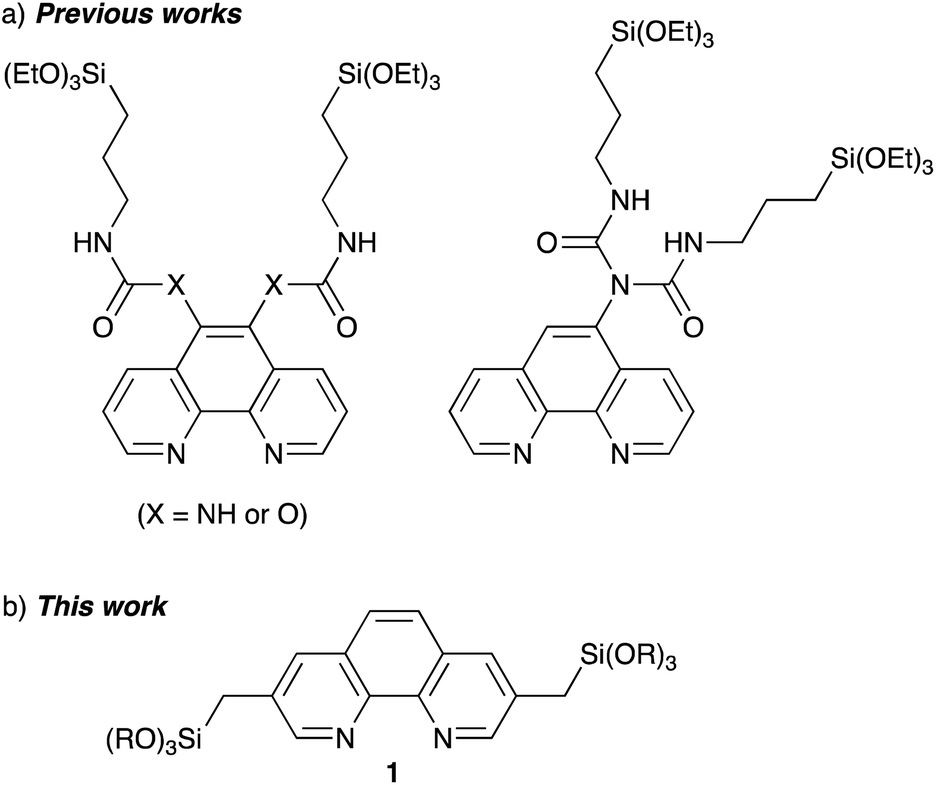
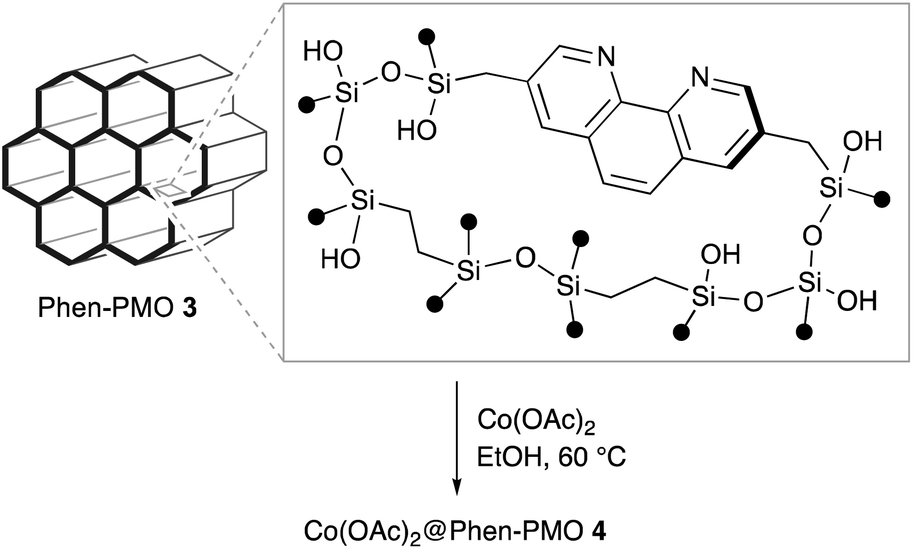
Several Phen-PMOs, in which the Phen moieties have been directly incorporated into the organosilica framework, have been previously reported (Fig. 1a).14 In these precursors, trialkoxysilyl groups are connected to the Phen moiety at the 5,6 or 5 position(s) through a conformationally flexible propylene linker with a urea or urethane function. This study reports the synthesis and characterization of a novel class of Phen-PMOs, in which the trialkoxysilyl group was linked to Phen at the 3,8 positions via a less flexible methylene linker (Fig. 1b). The immobilization of Co(OAc)2 on the as-synthesized Phen-PMO and its preliminary application as an immobilized catalyst for alkyne hydrosilylation are also investigated.
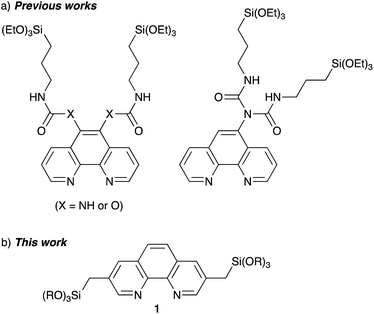 |
| | Fig. 1 Phen-PMO precursors in (a) previous works and (b) this work. | |
Results and discussion
Synthesis of the Phen-PMO precursor
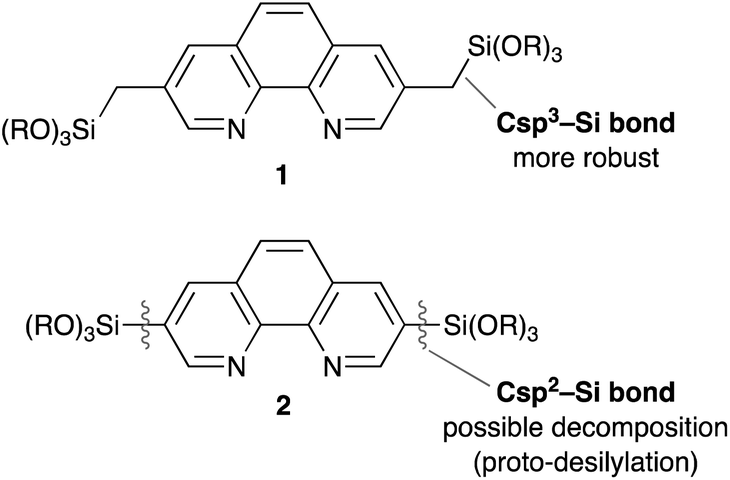
Although Phen derivative 2 bearing trialkoxysilyl groups directly at the 3,8 positions is a possible precursor for Phen-PMOs, precursor 2 might decompose by proto-desilylation during the preparation of PMOs, especially under acidic conditions (Fig. 2).15 The extended π-conjugated system of 1,10-phenanthroline would promote protonation on the ipso-carbon atom by stabilizing the corresponding σ-complex, thus resulting in proto-desilylation. Consequently, Phen derivative 1 incorporating trialkoxysilylmethyl groups at the 3,8 positions was synthesized as a Phen-PMO precursor. Since the trialkoxysilyl group is attached to the Phen moiety through an sp3 carbon atom, the proto-desilylation reaction was not expected to proceed.
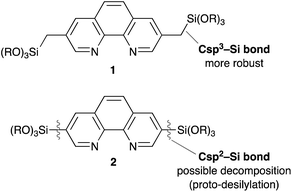 |
| | Fig. 2 Comparison of Phen-PMO precursor candidates 1 and 2. | |
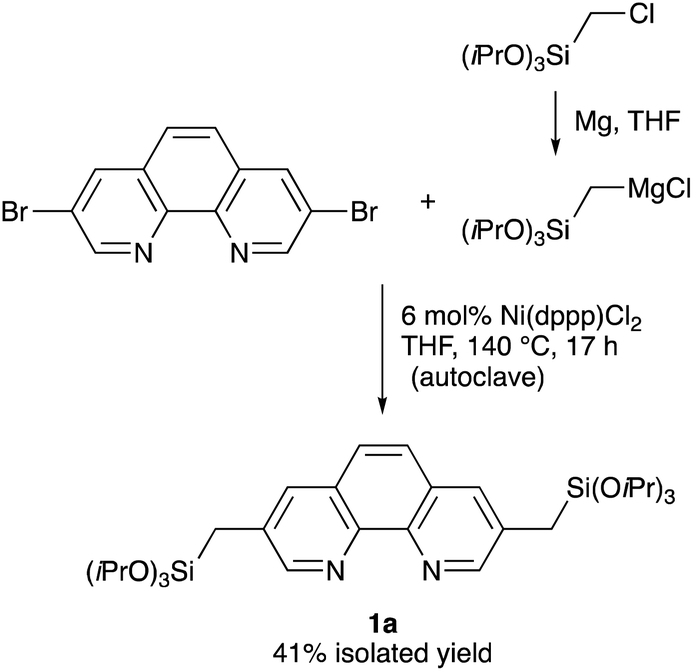
A straightforward route for the synthesis of Phen-PMO precursor 1 is via the cross-coupling of 3,8-halo-1,10-phenanthroline and a [(trialkoxysilyl)methyl]metal reagent. However, the Phen moiety exhibits a potent coordination ability with the metal center, so its coordination to transition metal catalysts for the desired cross-coupling should be avoided. Therefore, the Kumada–Tamao–Corriu cross-coupling, a robust C–C bond formation reaction, was employed for the synthesis of Phen-PMO precursor 1 (Scheme 1). The Kumada–Tamao–Corriu cross-coupling of the commercially available 3,8-dibromo-1,10-phenanthroline and [(triisopropoxysilyl)methyl]magnesium chloride,16 prepared from (chloromethyl)triisopropoxysilane and magnesium powder in THF, was carried out in the presence of Ni(dppp)Cl2 (6 mol%) at 140 °C in an autoclave with a PTFE inner vessel. Consequently, 3,8-bis[(triisopropoxysilyl)methyl]-1,10-phenanthroline (1a) was isolated in a moderate but acceptable yield (41%) (Table 1, entry 1). High temperatures were essential for the desired cross-coupling reaction to proceed. Under reflux conditions (THF: bp 66 °C) and in a standard glassware, only a trace amount of 1a was detected (entry 2). Performing the reaction in an autoclave at 90 °C afforded 1a in a slightly lower yield (entry 3). The yield decreased in the presence of dppe ligand having a narrower bite angle and dppf with a wider bite angle than dppp (entries 4 and 5).
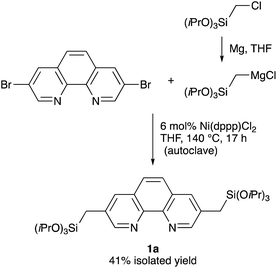 |
| | Scheme 1 Synthesis of Phen-PMO precursor 1a. | |
Table 1 Optimization of nickel/diphosphine catalysts and reaction temperature
| Entry |
Cat. |
T (°C) |
Yielda (%) |
| Determined by 1H NMR analysis using mesitylene as internal standard. Isolated yield. Reaction was carried out in a glassware. |
| 1 |
Ni(dppp)Cl2 |
140 |
56 (41)b |
| 2c |
Ni(dppp)Cl2 |
66 (reflux) |
Trace |
| 3 |
Ni(dppp)Cl2 |
90 |
53 |
| 4 |
Ni(dppe)Cl2 |
140 |
46 |
| 5 |
Ni(dppf)Cl2 |
140 |
46 |
Synthesis and characterization of Phen-PMO
The synthesis of Phen-PMO using 1a as the sole precursor did not afford any mesoporous materials. Consequently, the co-condensation of 1a and 1,2-bis(triethoxysilyl)ethane (BTEE) was investigated in the presence of three template surfactants: C22TMACl, polyoxyethylene(10) stearyl ether (Brij76), and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123). The co-condensation in the presence of C22TMACl as an ionic template surfactant under basic conditions resulted in the partial cleavage of the Si–C bonds of the precursor, as indicated by the detection of peaks corresponding to the Q units around −100 ppm in the 29Si dipolar decoupling/magic-angle spinning (DD/MAS) NMR spectroscopy (Fig. S1†). In the presence of Brij76 as a nonionic surfactant template under acidic conditions, the Si–C bond cleavage was sufficiently suppressed (Fig. S2†), and the formation of a mesoporous structure was confirmed by the nitrogen (N2) adsorption/desorption analysis which exhibited a typical type IV isotherm (Fig. S3†). However, transmission electron microscopy (TEM) images revealed that the obtained Phen-PMO had a disordered structure (Fig. S4†). An ordered 2-D hexagonal mesoporous material, Phen-PMO 3, was obtained when the co-condensation was performed in the presence of P123 and KCl under acidic conditions (Scheme 2).
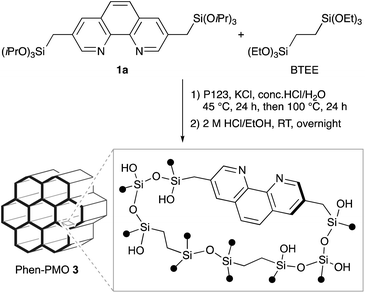 |
| | Scheme 2 Synthesis of Phen-PMO 3. | |
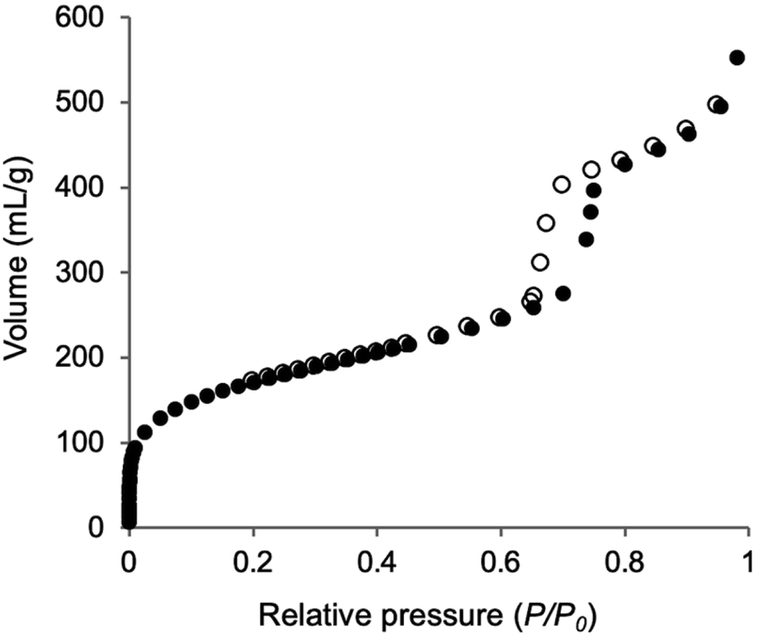
The incorporation of the Phen unit of 1a and the ethylene unit of BTEE into Phen-PMO 3 was confirmed by the 13C cross-polarization (CP)/MAS NMR spectroscopy (Fig. S5†). Furthermore, the 29Si CP/MAS NMR spectrum of 3 showed peaks corresponding to the T2 and T3 units in the −50 to −70 ppm range (Fig. S6†). Moreover, significant peaks corresponding to the Q units around −100 ppm were not observed, thus indicating that the Si–C bonds of the precursors were maintained in Phen-PMO 3. The amount of Phen units introduced into Phen-PMO 3 was 0.75 mmol g−1 as calculated from the nitrogen content (2.1%) determined by elemental analysis. The N2 adsorption/desorption analysis showed a type IV isotherm with hysteresis in the relative pressure (P/P0) range from 0.65 to 0.75, which indicated the formation of a mesoporous structure (Fig. 3). The Brunauer–Emmett–Teller surface area (SBET) and pore diameter (dBJH) were 607 m2 g−1 and 8.2 nm, respectively. The X-ray diffraction (XRD) analysis of Phen-PMO 3 showed a sharp peak at 2θ = 0.86° (d = 10.3 nm) with two diffraction peaks at 2θ = 1.48° (d = 6.00 nm) and 1.72° (d = 5.60 nm), corresponding to the 2-D hexagonal lattice (a0 = 12.0 nm) (Fig. 4). The absence of a peak at the medium scattering angles indicated that the organosilica wall constructed by the co-condensation of Phen-PMO precursor 1a and BTEE was amorphous, unlike the crystal-like BPy-PMO and PEPy-PMO in which the organic units are regularly arranged in the pore wall (PEPy = pyridinylethynylpyridine).7,17 The thickness of the pore wall was 3.8 nm calculated by subtracting the pore diameter (dBJH) from the lattice constant (a0). The TEM images of Phen-PMO 3 confirmed the formation of a 2-D hexagonal mesoscopically ordered structure (Fig. 5a) and uniform 1-D channels much longer than 100 nm (Fig. 5b). The channel size was in the 8–10 nm range, which was consistent with that obtained by N2 adsorption isotherm (8.2 nm).
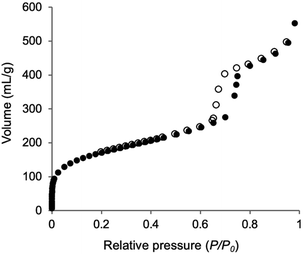 |
| | Fig. 3 N2 adsorption/desorption isotherm of Phen-PMO 3 (closed symbols: adsorption; open symbols: desorption). | |
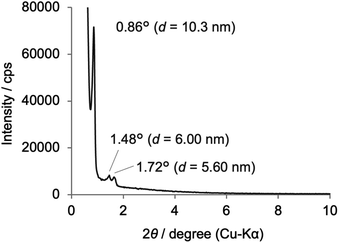 |
| | Fig. 4 XRD pattern of Phen-PMO 3. | |
 |
| | Fig. 5 TEM images (a) and (b) of Phen-PMO 3. | |
Immobilization of Co(OAc)2 on Phen-PMO and its application in alkyne hydrosilylation
In order to examine the potential of Phen-PMO 3 as a solid ligand to immobilize metal complexes, the immobilization of Co(OAc)2 onto Phen-PMO 3 was investigated (Scheme 3). The immobilization was achieved by simply mixing Co(OAc)2 and Phen-PMO 3 in EtOH at 60 °C at an initial molar ratio of 1/2 (Co/Phen units) to obtain Co(OAc)2@Phen-PMO 4. The immobilized Co content was 0.21 mmol g−1 determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES), thus indicating that approximately 30% of Phen units introduced into Phen-PMO 3 was coordinated with Co. Due to the thickness and amorphous nature of the organosilica walls, Phen-PMO 3 could contain less accessible and even inaccessible Phen moieties.
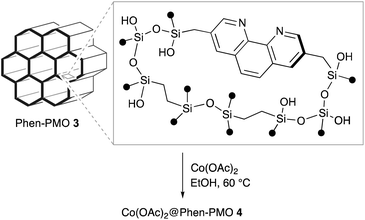 |
| | Scheme 3 Synthesis of Co(OAc)2@Phen-PMO 4. | |
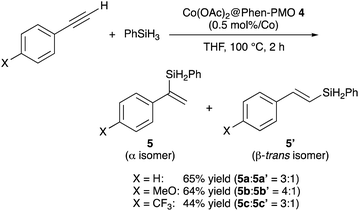
The catalytic performance of Co(OAc)2@Phen-PMO 4 was preliminary investigated in the hydrosilylation reaction of phenylacetylene with phenylsilane (Scheme 4).18 In the presence of a catalytic amount of Co(OAc)2@Phen-PMO 4 (0.5 mol%/Co), phenylacetylene underwent the hydrosilylation to give the corresponding alkenylsilanes, α-isomer 5a and β-trans-isomer 5a′, in 65% yield and at a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see also Table S1†). The introduction of an electron-donating methoxy group at the para-position did not affect the yield but slightly improved the regioselectivity. The reaction of a phenylacetylene derivative with an electron-withdrawing trifluoromethyl group resulted in a lower yield of 44%. The reaction did not proceed with Co(OAc)2 in the absence of any external ligands under the studied conditions, whereas a homogeneous counterpart, a combination of Co(OAc)2 and 1,10-phenanthroline, afforded the alkenylsilanes 5a and 5a′ with a 10:1 ratio in 53% yield. It was thus assumed that the cobalt species which coordinated to the Phen moieties was the active catalyst. Although the detailed reaction mechanism and the structure of the active species are still unclear, the mesopore wall in which the Phen moieties are embedded would affect the regioselectivity. Since ligands can control the regioselectivity in cobalt-catalyzed hydrosilylation of terminal alkynes,19–21 Co(OAc)2@Phen-PMO 4 which has the unique steric hindrance around the Phen moieties showed the different regioselectivity from the combination of Co(OAc)2 and 1,10-phenanthroline. Although we attempted catalyst recycle experiments, the Co(OAc)2@Phen-PMO 4 recovered by filtration under a N2 atmosphere after the first hydrosilylation cycle of phenylacetylene with phenylsilane showed diminished yield of 5% in the second hydrosilylation cycle.
:1 ratio (see also Table S1†). The introduction of an electron-donating methoxy group at the para-position did not affect the yield but slightly improved the regioselectivity. The reaction of a phenylacetylene derivative with an electron-withdrawing trifluoromethyl group resulted in a lower yield of 44%. The reaction did not proceed with Co(OAc)2 in the absence of any external ligands under the studied conditions, whereas a homogeneous counterpart, a combination of Co(OAc)2 and 1,10-phenanthroline, afforded the alkenylsilanes 5a and 5a′ with a 10:1 ratio in 53% yield. It was thus assumed that the cobalt species which coordinated to the Phen moieties was the active catalyst. Although the detailed reaction mechanism and the structure of the active species are still unclear, the mesopore wall in which the Phen moieties are embedded would affect the regioselectivity. Since ligands can control the regioselectivity in cobalt-catalyzed hydrosilylation of terminal alkynes,19–21 Co(OAc)2@Phen-PMO 4 which has the unique steric hindrance around the Phen moieties showed the different regioselectivity from the combination of Co(OAc)2 and 1,10-phenanthroline. Although we attempted catalyst recycle experiments, the Co(OAc)2@Phen-PMO 4 recovered by filtration under a N2 atmosphere after the first hydrosilylation cycle of phenylacetylene with phenylsilane showed diminished yield of 5% in the second hydrosilylation cycle.
 |
| | Scheme 4 Hydrosilylation of phenylacetylene derivatives catalyzed by Co(OAc)2@Phen-PMO 4. | |
Experimental
General information
All experiments were performed under nitrogen atmosphere using either the Schlenk technique or a glove box. All chemicals were purchased from Kanto Chemical Co., Inc., FUJIFILM Wako Chemicals, Sigma-Aldrich, or Tokyo Chemical Industry (TCI), and used as received without further purification. 3,8-Dibromo-1,10-phenanthroline was prepared according to a previously reported procedure.22 Silica gel column chromatography was performed on a Yamazen AI-580 Single Channel Automated Flash Chromatography System. 1H, 13C{1H} and 29Si{1H} NMR spectra were recorded on a Bruker AVANCE III HD NMR spectrometer (1H NMR at 600 MHz; 13C{1H} NMR at 151 MHz; 29Si{1H} NMR at 119 MHz). 13C CP/MAS NMR and 29Si CP/MAS NMR measurements were performed using a 4 mm diameter ZrO2 rotor at a sample spinning frequency of 8 kHz using a Bruker AVANCE II NMR spectrometer (13C CP/MAS NMR at 100.6 MHz; 29Si CP/MAS NMR at 79.49 MHz). ICP-AES was performed on a Hitachi High-Tech PS3520VDDII in the Toray Research Center, Inc. Nitrogen adsorption/desorption isotherms were measured using a MicrotracBEL BELSORP MAX-II instrument. BET surface areas were calculated from the linear sections of the BET plots (P/P0 = 0.1–0.2). Pore-size distributions were calculated from adsorption branch using the Barrett–Joyner–Halenda (BJH) method. XRD patterns were recorded on a Rigaku RINT-TTR diffractometer under Cu Kα radiation (50 kV, 300 mA). TEM images were obtained on a JEOL JEM-2100F operating at 200 kV.
Preparation of [(triisopropoxysilyl)methyl]magnesium chloride
(iPrO)3SiCH2Cl (3.18 g, 12.5 mmol) was dissolved in THF (18.1 mL). Mg powder (332 mg, 13.7 mmol) was placed in a two-necked flask (50 mL) to which the THF solution of (iPrO)3SiCH2Cl (9.4 mL) was added dropwise under continuous stirring. 3 drops of dibromoethane were then added to the mixture, and the flask was heated to 80 °C. The remaining THF solution of (iPrO)3SiCH2Cl was added dropwise over 10 min. The mixture was then stirred at room temperature for 4 h. The resulting solution of (iPrO)3SiCH2MgCl was used in the cross-coupling reaction without further purification.
Preparation of 3,8-bis[(triisopropoxysilyl)methyl]-1,10-phenanthroline (1a)
3,8-Dibromo-1,10-phenanthroline (1.49 g, 4.4 mmol) and a stirring bar were placed in an autoclave (125 mL) with a PTFE inner vessel and Ni(dppp)Cl2 (138 mg, 6 mol%) and THF (50 mL) were gradually added. A THF solution of (iPrO)3SiCH2MgCl (12.5 mmol) was added dropwise over 3 min under continuous stirring. The autoclave was sealed and stirred at room temperature for 1 h, and then heated to 140 °C. After 17 h, the mixture was cooled to room temperature and then quenched with water. The mixture was extracted with CHCl3 and the organic layer was washed with brine and dried over Na2SO4. The solvents were removed by rotary evaporator. The crude product was dissolved in n-hexane and stirred for 15 min. The mixture was filtered through a Celite pad and the solvent was removed by rotary evaporator. The crude product was purified by silica gel column chromatography (n-hexane/AcOEt) to give the Phen-PMO precursor, 3,8-bis[(triisopropoxysilyl)methyl]-1,10-phenanthroline (1a), as a pale-yellow solid (1.11 g, 41%). 1H NMR (600 MHz, CDCl3): δ 9.00 (d, J = 2.1 Hz, 2H), 8.00 (d, J = 2.1 Hz, 2H), 7.65 (s, 2H), 4.21 (sept, J = 6.1 Hz, 6H), 2.36 (s, 4H), 1.15 ppm (d, J = 6.1 Hz, 36H). 13C{1H} NMR (151 MHz, CDCl3): δ 151.9, 143.9, 134.8, 133.4, 127.9, 126.2, 65.7, 25.7, 19.6 ppm. 29Si{1H} NMR (119 MHz, CDCl3): δ −56.5 ppm. Anal. calcd for C32H52N2O6Si2: C, 62.30; H, 8.50; N, 4.54. Found: C, 62.30; H, 8.90; N, 4.50.
Preparation of Phen-PMO 3
To a screw-capped vial (110 mL) equipped with a stirring bar, P123 (0.811 g), KCl (5.17 g), distilled water (28.5 mL), and concentrated hydrochloric acid (4.05 mL) were added under ambient conditions, and the vial was sealed. The mixture was cooled to 0 °C and allowed to stand until the surfactant was completely dissolved. The mixture was then heated to 45 °C and stirred for 30 min. An ethanol solution (1.0 mL) of Phen-PMO precursor 1a (384 mg, 0.623 mmol) and 1,2-bis(triethoxysilyl)ethane (1.25 g, 3.52 mmol) was added to the mixture at 45 °C and the resulting mixture was stirred at 45 °C for 1 day. The mixture was then heated to 100 °C and allowed to stand for 1 day. After cooling to room temperature, a pale pink precipitate was collected by filtration, washed with water, and dried under vacuum at 50 °C. The precipitate was then suspended in a solution of 2 M HCl (2.8 mL)/EtOH (100 mL) and then stirred overnight at room temperature. The precipitate was collected by filtration, washed with water and ethanol, and dried under vacuum at 50 °C to give Phen-PMO 3 (667 mg).
Immobilization of Co(OAc)2 on Phen-PMO 3
Phen-PMO 3 (201 mg) was charged in a three-neck flask (50 mL) equipped with a mechanical stirring system. After drying Phen-PMO 3 in vacuum at 60 °C for 2 h, dehydrated EtOH (25 mL) was added, and the solution was stirred for 1 h. A solution of Co(OAc)2 (53 mg, 0.3 mmol) in EtOH (20 mL) was then added dropwise for 30 min with stirring. After the addition, the slurry was stirred at room temperature for 2 h and then at 60 °C for 16 h. After cooling to room temperature, the slurry was filtered through a PTFE membrane and then washed with dehydrated EtOH. The obtained solid was finally dried under vacuum to give Co(OAc)2@Phen-PMO 4 (178 mg).
Hydrosilylation of phenylacetylene
Co(OAc)2@Phen-PMO 4 (11 mg, 2.4 μmol/Co), THF (1 mL), phenylacetylene (51 mg, 0.5 mmol), and PhSiH3 (68 mg, 0.6 mmol) were gradually added in a screw-caped vial (10 mL). After the mixture was stirred at 100 °C for 2 h, mesitylene was added as an internal standard at room temperature. The product yield was determined by 1H NMR analysis.
Conclusions
This study reported the synthesis of Phen-PMO 3, in which the Phen moieties were bridged to the organosilica framework through a methylene linker, through the co-condensation of 3,8-bis[(triisopropoxysilyl)methyl]-1,10-phenanthroline (1a) and 1,2-bis(triethoxysilyl)ethane in the presence of P123 as the template surfactant under acidic conditions. Phen-PMO 3 had an ordered 2-D hexagonal mesoporous structure as confirmed by the nitrogen adsorption/desorption analysis, XRD, and TEM results. Co(OAc)2 was immobilized on Phen-PMO 3 to afford an immobilized complex, Co(OAc)2@Phen-PMO 4, which exhibited good catalytic activity for the alkyne hydrosilylation reaction. The application of Phen-PMO 3 as a solid ligand for other metal complexes is under investigation. The results of this study reveal the great potential of Phen-PMO 3 in catalysis.
Author contributions
Xiao-Tao Lin, Yusuke Ishizaka, and Yoshifumi Maegawa investigated the work. Katsuhiko Takeuchi verified the methodology. Shinji Inagaki supervised this work and helped in writing, reviewing, and editing of the manuscript. Kazuhiro Matsumoto verified the methodology, supervised the work, and wrote the original draft. Jun-Chul Choi devised the conceptual ideas and helped in the supervision, writing, reviewing, and editing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the New Energy and Industrial Technology Development Organization (NEDO) for partially supporting this research (project code: P19004).
Notes and references
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS
 .
. - T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CrossRef CAS .
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS .
-
(a) W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634 CrossRef CAS PubMed ;
(b) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC ;
(c) P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913 RSC ;
(d) J. G. Croissant, X. Cattoën, M. Wong Chi Man, J.-O. Durand and N. M. Khashab, Nanoscale, 2015, 7, 20318 RSC ;
(e) B. Karimi, N. Ganji, O. Pourshiani and W. R. Thiel, Prog. Mater. Sci., 2022, 125, 100896 CrossRef CAS .
-
(a) U. Díaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083 RSC ;
(b) Y. Liang, Nanoscale Adv., 2021, 3, 6827 RSC .
- For selected examples, see:
(a) C. Baleizãao, B. Gigante, D. Das, M. Álvaro, H. Garcia and A. Corma, J. Catal., 2004, 223, 106 CrossRef ;
(b) P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756 CrossRef CAS ;
(c) D. Zhang, J. Xu, Q. Zhao, T. Cheng and G. Liu, ChemCatChem, 2014, 6, 2998 CrossRef CAS ;
(d) H. Takeda, M. Ohashi, Y. Goto, T. Ohsuna, T. Tani and S. Inagaki, Adv. Funct. Mater., 2016, 26, 5068 CrossRef CAS .
- M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003 CrossRef CAS .
- For selected examples, see:
(a) R. Jin, K. Liu, D. Xia, Q. Qian, G. Liu and H. Li, Adv. Synth. Catal., 2012, 354, 3265 CrossRef CAS ;
(b) A. N. Prasad, B. M. Reddy, E.-Y. Jeong and S.-E. Park, RSC Adv., 2014, 4, 29772 RSC ;
(c) F. Zhou, X. Hu, M. Gao, T. Cheng and G. Liu, Green Chem., 2016, 18, 5651 RSC ;
(d) E. Gu, W. Zhong and X. Liua, RSC Adv., 2016, 6, 98406 RSC .
- For selected examples, see:
(a) S. p. Shylesh, M. Jia, A. Seifert, S. Adappa, S. Ernsta and W. R. Thiel, New J. Chem., 2009, 33, 717 RSC ;
(b) X. Shi, B. Fan, H. Li, R. Li and W. Fan, Microporous Mesoporous Mater., 2014, 196, 277 CrossRef CAS ;
(c) D. Elhamifar and E. Nazari, ChemPlusChem, 2015, 80, 820 CrossRef CAS PubMed ;
(d) A. Lazar, S. Silpa, C. P. Vinod and A. P. Singh, Mol. Catal., 2017, 440, 66 CrossRef CAS .
- For recent examples, see:
(a) M. Waki, S. Shirai, K.-i. Yamanaka, Y. Maegawa and S. Inagaki, RSC Adv., 2020, 10, 13960 RSC ;
(b) S. Kataoka and S. Inagaki, Eur. J. Inorg. Chem., 2020, 43, 4083 CrossRef ;
(c) A. M. Kaczmarek, Y. Maegawa, A. Abalymov, A. G. Skirtach, S. Inagaki and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2020, 12, 13540 CrossRef CAS .
- For selected examples, see:
(a) A. Jana, J. Mondal, P. Borah, S. Mondal, A. Bhaumik and Y. Zhao, Chem. Commun., 2015, 51, 10746 RSC ;
(b) H. Duan, M. Li, G. Zhang, J. R. Gallagher, Z. Huang, Y. Sun, Z. Luo, H. Chen, J. T. Miller, R. Zou, A. Lei and Y. Zhao, ACS Catal., 2015, 5, 3752 CrossRef CAS ;
(c) S. Zhang, H. Wang, M. Li, J. Han, X. Liu and J. Gong, Chem. Sci., 2017, 8, 4489 RSC ;
(d) S. Ishikawa, Y. Maegawa, M. Waki and S. Inagaki, ACS Catal., 2018, 8, 4160 CrossRef CAS ;
(e) Y. Naganawa, Y. Maegawa, H. Guo, S. S. Gholap, S. Tanaka, K. Sato, S. Inagaki and Y. Nakajima, Dalton Trans., 2019, 48, 5534 RSC ;
(f) X.-T. Lin, K. Matsumoto, Y. Maegawa, K. Takeuchi, N. Fukaya, K. Sato, S. Inagaki and J.-C. Choi, New J. Chem., 2021, 45, 9501 RSC ;
(g) S. Zhang, M. Li, W. Qiu, J. Han, H. Wang and X. Liu, Appl. Catal., B, 2019, 259, 118113 CrossRef CAS ;
(h) M. Shimizu, K. Michikawa, Y. Maegawa, S. Inagaki and K.-i. Fujita, ACS Appl. Nano Mater., 2020, 3, 2527 CrossRef CAS .
- For selected reviews, see:
(a) P. G. Sammes and G. Yahioglu, Chem. Soc. Rev., 1994, 23, 327 RSC ;
(b) G. Accorsi, A. Listorti, K. Yoosafa and N. Armaroli, Chem. Soc. Rev., 2009, 38, 1690 RSC ;
(c) A. Bencini and V. Lippolis, Coord. Chem. Rev., 2010, 254, 2096 CrossRef CAS .
- For selected examples, see:
(a) K. Gao and N. Yoshikai, Angew. Chem., Int. Ed., 2011, 50, 6888 CrossRef CAS ;
(b) B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS ;
(c) F. Teng, J. Cheng and C. Bolm, Org. Lett., 2015, 17, 3166 CrossRef CAS PubMed ;
(d) S. Nakatani, Y. Ito, S. Sakurai, T. Kodama and M. Tobisu, J. Org. Chem., 2020, 85, 7588 CrossRef CAS PubMed .
- For selected examples of Phen-containing PMO, see:
(a) X. Guo, X. Wang, H. Zhang, L. Fu, H. Guo, J. Yu, L. D. Carlos and K. Yang, Microporous Mesoporous Mater., 2008, 116, 28 CrossRef CAS ;
(b) X. Guo, H. Guo, L. Fu, R. Deng, W. Chen, J. Feng, S. Dang and H. Zhang, J. Phys. Chem. C, 2009, 113, 2603 CrossRef CAS ;
(c) B. Yan and Y.-J. Gu, Inorg. Chem. Commun., 2013, 34, 75 CrossRef CAS ;
(d) B. Xiao, J. Zhao, X. Liu, P. Wang and Q. Yang, Microporous Mesoporous Mater., 2014, 199, 1 CrossRef CAS .
-
(a) C. Eaborn, Pure Appl. Chem., 1969, 19, 375 CrossRef ;
(b) W. Yao, R. Li, H. Jiang and D. Han, J. Org. Chem., 2018, 83, 2250 CrossRef CAS ;
(c) H. Matsukawa, M. Yoshida, T. Tsunenari, S. Nozawa, A. Sato-Tomita, Y. Maegawa, S. Inagaki, A. Kobayashi and M. Kato, Sci. Rep., 2019, 9, 15151 CrossRef PubMed ;
(d) S. Shirai and S. Inagaki, New J. Chem., 2021, 45, 6120 RSC .
- D. J. Brondani, R. J. P. Corriu, S. El Ayoubi, J. J. E. Moreau and M. Wong Chi Man, J. Organomet. Chem., 1993, 451, C1 CrossRef CAS .
- M. Waki and S. Inagaki, Inorg. Chem. Front., 2022, 9, 3669 RSC .
- For selected reviews, see:
(a) J.-W. Park, Chem. Commun., 2022, 58, 491 RSC ;
(b) L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550 CrossRef CAS PubMed ;
(c) J. Sun and L. Deng, ACS Catal., 2016, 6, 290 CrossRef CAS .
- For selected examples of α-selective Co-catalysed hydrosilylation, see:
(a) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835 CrossRef CAS PubMed ;
(b) Z. Zuo, J. Yang and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 10839 CrossRef CAS PubMed .
- For selected examples of β-trans-selective Co-catalysed hydrosilylation, see:
(a) C. Wu, W. J. Teo and S. Ge, ACS Catal., 2018, 8, 5896 CrossRef CAS ;
(b) G. Wu, U. Chakraborty and A. J. von Wangelin, Chem. Commun., 2018, 54, 12322 RSC ;
(c) Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414 CrossRef CAS .
- For selected examples of β-cis-selective Co-catalysed hydrosilylation, see:
(a) W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328 CrossRef CAS PubMed ;
(b) X. Du, W. Hou, Y. Zhang and Z. Huang, Org. Chem. Front., 2017, 4, 1517 RSC .
- Y. Saitoh, T. Koizumi, K. Osakada and T. Yamamoto, Can. J. Chem., 1997, 75, 1336 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yoshifumi Maegawac,
Katsuhiko Takeuchi
a,
Yoshifumi Maegawac,
Katsuhiko Takeuchi