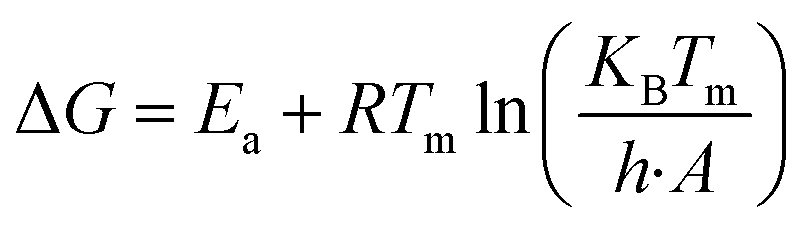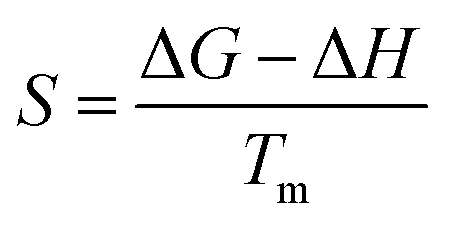DOI:
10.1039/D2RA08223C
(Paper)
RSC Adv., 2023,
13, 6966-6982
Degradation of tetrabromobisphenol A (TBBA) with calcium hydroxide: a thermo-kinetic analysis†
Received
25th December 2022
, Accepted 16th February 2023
First published on 28th February 2023
Abstract
Thermal treatment of bromine-contaminated polymers (i.e., as in e-waste) with metal oxides is currently deployed as a mainstream strategy in recycling and resources recovery from these objects. The underlying aim is to capture the bromine content and to produce pure bromine-free hydrocarbons. Bromine originates from the added brominated flame retardants (BFRs) to the polymeric fractions in printed circuits boards, where tetrabromobisphenol A (TBBA) is the most utilized BFR. Among notable deployed metal oxides is calcium hydroxide, i.e., Ca(OH)2 that often displays high debromination capacity. Comprehending thermo-kinetic parameters that account for the BFRs:Ca(OH)2 interaction is instrumental to optimize the operation at an industrial scale. Herein, we report comprehensive kinetics and thermodynamics studies into the pyrolytic and oxidative decomposition of a TBBA:Ca(OH)2 mixture at four different heating rates, 5, 10, 15, and 20 °C min−1, carried out using a thermogravimetric analyser. Fourier Transform Infrared Spectroscopy (FTIR) and a carbon, hydrogen, nitrogen, and sulphur (CHNS) elemental analyser established the vibrations of the molecules and carbon content of the sample. From the thermogravimetric analyser (TGA) data, the kinetic and thermodynamic parameters were evaluated using iso-conversional methods (KAS, FWO, and Starink), which were further validated by the Coats–Redfern method. The computed activation energies for the pyrolytic decomposition of pure TBBA and its mixture with Ca(OH)2 reside in the narrow ranges of 111.7–112.1 kJ mol−1 and 62.8–63.4 kJ mol−1, respectively (considering the various models). Obtained negative ΔS values suggest the formation of stable products. The synergic effects of the blend exhibited positive values in the low-temperature ranges (200–300 °C) due to the emission of HBr from TBBA and the solid–liquid bromination process occurring between TBBA and Ca(OH)2. From a practical point of view, data provided herein are useful in efforts that aim to fine-tune operational conditions encountered in real recycling scenarios, i.e., in co-pyrolysis of e-waste with Ca(OH)2 in rotary kilns.
1 Introduction
Electronic and electrical waste (e-waste) is accumulating at an alarming rate worldwide. The annual generation of e-waste has exceeded 50 million tons recently.1 Such a figure is projected to significantly increase over the next year owing to the relatively short-life time of electrical devices. While most parts in e-waste could be mechanically recycled, recovery of resources from the non-metallic fraction printed circuit boards (PCBs) in particular requires chemical treatment.2 The valuable (yet minute) metallic loads in PCBs constitute Ag, Pd, Au, Ni, and Pt.3 These constituents are typically recovered via hydrometallurgical-based operations.4 The non-metallic load in waste PCBs (WPCBs) consists of polymeric materials most notably Acrylonitrile Butadiene Styrene (ABS), fibers, and brominated flame retardants (BFRs).5 The latter category is added to delay or reduce the onset of fire when electrical devices are subjected to high thermal stress during their operation. As bromine-containing hydrocarbons, oxidative and pyrolytic decomposition of BFRs produce a wide array of brominated species, including bromo-alkanes/bromo-alkenes, bromobenzenes, bromophenols, and Br-polyaromatic hydrocarbons (Br-PAHs).6 Primitive recycling of e-waste in open fires is still widely practiced in different parts of the world.7 Prevailing conditions in these scenarios (random temperature profiles, insufficient oxygen, presence of particulate matters) provide a perfect recipe for the incomplete combustion; and thus, facilitating the synthesis of notorious polybrominated dibenzo-p-dioxins and furans (PBDD/Fs). Brominated phenols and benzenes serve as direct precursors for the formation of PBDD/Fs via homogenous gas phase pathways and surface-assisted routes.8 Nonetheless, it is generally found that nearly 50% of the initial bromine content in BFRs is emitted as corrosive HBr gases.
Bromine recovery and production of bromine-free hydrocarbons streams is currently implemented through co-pyrolysis of BFRs with transition metal oxides. The metal Mn+–O2- structural linkages in metal oxides serve as Lewis acid-basic sites in the dissociative adsorption and uptake of brominated species.9 Previously deployed debromination agents include iron oxides (Fe2O3/Fe3O4), ZnO, La2O3, CaO, and Al2O3).10,11 Ni- or Fe-decorated zeolites were also utilized.12 The debromination capacity of heterogenous mixtures of metal oxides as in electric arc furnace dust (EAFD)13 or bauxite14 residues have been comprehensively investigated. The debromination capacity is quantified based on the capture of inorganic bromine (HBr) as well as organic bromine (brominated hydrocarbons). The co-pyrolysis process is not a catalytic reaction as the metal oxides is consumed and converted into corresponding metal bromides and oxybromides, evidenced by XRD and XPS measurements. As the most widely utilized BFRs, debromination of Tetrabromobisphenol A (TBBA) is commonly investigated.8
The reported debromination efficiency primarily depend on the deployed formulation of metal oxides additives and the considered BFRs-containing materials. We have recently surveyed literature pertinent to the debromination ability of various BRs-metal oxides mixtures.15 For example, catalytic upgrading of vapor intermediates from pyrolysis of Br–ABS mixture (i.e., bromine in the form of TBBA) over Fe/zeolite catalysts reduced the bromine content in oil by ∼80%. The use of the Fe/zeolite catalysts promoted the formation of single and two ring aromatic molecules.12 In related study,16 we have illustrated that pyrolytic decomposition of 2,4,6-tribromophenol (TBP) over Fe2O3 nanoparticles reduced emission of HBr by 55%. However, Fe2O3 assumes a rather modest cracking capacity of the parent TBP molecule. In a recent study, Liu et al. (2022),17 investigated pyrolysis of iron-loaded WPCBs. It was found that addition of iron promoted the generation of bromine-free gaseous products and decreases the activation energies. However, addition of iron did not completely eliminate brominated compounds as bromophenols and bromobenzenes, in addition to HBr, appear at all investigated temperatures. Analogously, the work by Terakado et al. (2013) demonstrated that ZnO and La2O3 noticeably suppressed the formation of HBr and brominated hydrocarbons. Poor debromination performance by ZnO and CuO was ascribed to their reduction into metallic forms rather than undergoing halogenation reactions.10
Density functional theory (DFT) calculations and thermogravimetric analysis (TGA) experiments provided important mechanistic insight into steps that govern the interaction between the metal oxides additives and the BFRs18,19 Based on DFT computations, conversion of metal oxides into their bromides and oxybromides ensue via three steps mechanisms, dissociative adsorption of HBr (or bromo-hydrocarbons), intramolecular hydrogen transfer forming surface hydroxyl groups, and desorption of water. As in the case of Fe2O3, endothermic mass loss step at elevated temperature was ascribed to evaporation of FeBr2.20 Interaction of metal oxides with hydrocarbons skeletons in BFRs result in their sequential reduction eventually to the metallic form.20 The seminal work by Al-Harahsheh and co-workers provided thermo-kinetic parameters that underpin interaction of BFRs (mainly TBBA) with a wide array of pure and mixture of transition metal oxides.13,21
Among notable metal oxides additives in the debromination process of BFRs or materials laden with BFRs is calcium hydroxides Ca(OH)2. While also the most affordable, Ca(OH)2 entails higher debromination capacity than transition metal oxides (Fe, Zn, Ln, Pd).22–24 Kumagai et al. illustrated that Ca(OH)2 captures ∼94% and 98% of emitted HBr and bromophenols from pyrolysis of TBBA-containing PCBs. Fixation of these bromine entities ensue at temperatures higher than 300 °C. Coupling the TGA with evolved gas analysis (py-GCMS/FTIR) linked the chemical events with the observed mass loss curves of Ca(OH)2:PCBs. Detected compounds mainly consist of neat phenols, dimethylphenol, and aliphatic fragments. It is viewed that Ca(OH)2 catalyzes fission of the C–CH3 linkages in TBBA. However, the aforementioned studies by Kumagai et al. and Gao et al. did not attempt to derive the governing thermo-kinetic parameters germane to the catalytic co-pyrolysis process by Ca(OH)2. It is likely that devising an industrial-scale catalytic upgrading process of plastic contaminated with halogens (or bromine in particular) to reply on the use of Ca(OH)2. For this reason, it is of a prime importance to derive accurate kinetic and thermodynamics parameters that underpin the co-pyrolysis process. Acquiring these values will be instrumental to fine-tube operational parameters. Along the same line of enquiry, representative chemical mechanisms remain unclear. To this end, the aim of this contribution is three-fold; to extract representative kinetic and thermodynamics functions that account for the decomposition of a mixture of TBBA; Ca(OH)2, to map out prevailing reaction pathways in this interaction; and to predict the thermal degradation curve based on artificial networks approach.
2 Characterization methods
2.1 Materials
Tetrabromobisphenol (TBBA), the major BFRs in electronic wastes, was obtained from Sigma Aldrich with a purity of 99%. The ultimate analysis for the elemental composition of the TBBA was determined using the CHNS instrument of vario MACRO cube from elementar. TBBA constitutes 27.36% C, 2.27% H, 0.04% Ni, and 0.01% S. Commonly available calcium hydroxide (Ca(OH)2) was bought from Thermo Scientific (purity-99%), which was used to mix with the TBBA. The nitrogen gas with ultra-high purity of 99.999%, and synthetic zero air gas with composition (21.9% oxygen, CO2, and CO mixture (<1 ppm), hydrocarbons (<0.2 ppm), and remaining nitrogen) were obtained from air-products Gulf Gas LLC AbuDhabi. A spectroscopic grade of potassium bromide (KBr) was purchased from Sigma Aldrich and was added to the samples for obtaining the IR spectrum to avoid baseline interferences.
2.2 Thermogravimetric and differential scanning calorimetric analysis (TGA and DSC)
The two thermo-chemical methods, pyrolysis, and combustion were conducted using the TGA Q200 V20.10 manufactured by TA instruments. The most prevalent technique for examining the thermal decay of any sample constituting carbonaceous material is TGA.25 It determines the change in weight as a function of temperature or time (isothermal and non-isothermal). The nitrogen and synthetic zero air with a flow rate maintained at 50 ml min−1 were operated to perform the pyrolysis and combustion reaction. Nitrogen gas is also known as inert gas, which is considered non-reacting. The samples were loaded into the platinum pan for running the TGA. Before the experiments, the pan was calibrated, and further, the sample was loaded with approximately 8–10 mg of the mixture. The corresponding weight loss of the sample was recorded as the heating process initiated from 25 °C (ambient) to 800 °C. All the reactions were performed at four different heating rates 5, 10, 15, and 20 °C min−1, respectively. Various heating rates were utilized to calculate the thermo-kinetic and thermodynamic parameters by using model fitting and model-free approaches. The differential scanning calorimeter (DSC), model SDT 650 from TA instruments, was utilized to determine the melting point and heat of the reaction involved in both processes. The sample of approximately 6–8 mg was weighed in an aluminum pan and kept in the balance of the instrument, which is connected with the purge gases (N2 and O2) maintained at a flow rate of 100 ml min−1.
2.3 Fourier transform infrared spectroscopic analysis(FTIR)
FTIR equipped with attenuated total reflectance (ATR) from JASCO, was used to investigate the functional groups of the sample. In the present study, FTIR was utilized to study the fluctuations occurring in the chemical bonds by analyzing the sample prior to and after the thermal decomposition. The FTIR from Bruker, model Invenio-S was used to analyze vibrations of the evolved gas emitted from the sample during the different thermal treatments. This FTIR is equipped with a heated gas sampling cell of an optical wavelength 10 cm maintained at a temperature of 250 °C to prevent the condensation of gas inside the cell. The analysis was run within the wave number range of 500–4000 cm−1, maintaining a resolution of 4 cm−1.
3 Model-free and model fitting kinetics
3.1 Kinetic analysis
The base of kinetic analysis starts from Arrhenius equation represented as eqn (1) and basic rate equation, eqn (2), where α signifies the degree of conversion which can be determined by using eqn (3). In this formula wi, wo and wf denote weight loss at the initial, at the moment, and at final stages, respectively. f(α) is reaction model. By combining the Arrhenius equation and basic rate equation, a new rate equation can be represented as eqn (4)| |
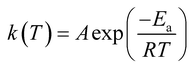 | (1) |
| |
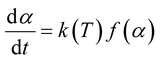 | (2) |
| |
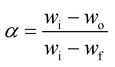 | (3) |
| |
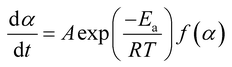 | (4) |
in which A is pre-exponential factor, R is gas constant (0.008314 kJ mol−1 K), Ea represents the activation energy. Non-isothermal behaviour in solid-state reaction rate can be expressed as eqn (5)| |
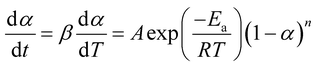 | (5) |
where T represents absolute temperature, β denotes heating rates, t is time, and n represent the order of the reaction. To find the analytical solution of the rate equations, various strategies are adopted. After the integration of eqn (5), a new equation is obtained which leads to a final equation of different models. The final equation after integration translates into eqn (6)| |
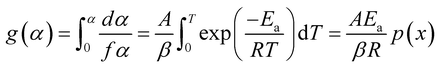 | (6) |
where p(x) is an approximation equation with value of  . To find the analytical solution of the rate equations, distinct different models are utilized.
. To find the analytical solution of the rate equations, distinct different models are utilized.
3.2 Model-free kinetics
The model-free method is one of the important methods which does not need information about the order of reaction and provides the kinetic parameters merely based on different conversion points and calculation can be done by using different heating rates.26,27 Kissinger-Akahira-Sunose (KAS) method, Flynn–Wall–Ozawa (FWO) method, and Starink method are the iso-conversional model-free method, commonly deployed to determine kinetic profiles that dictate pyrolysis and combustion processes.28–30 The equations of KAS, FWO, and Starink method are given from eqn (7)–(9) to determine the activation energies and pre-exponential factors at different conversion points.| |
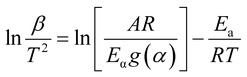 | (7) |
| |
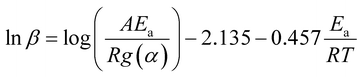 | (8) |
| |
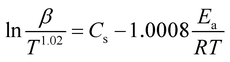 | (9) |
The plot of 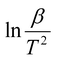 , ln
, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β, and
β, and  vs. 1/T gives a linear line with the slope
vs. 1/T gives a linear line with the slope  ,
, 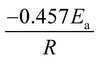 , and
, and  for KAS, FWO, and Starink methods respectively, from which the calculations of activation energies are executed. Intercepts of the straight lines signify the preexponential factors.
for KAS, FWO, and Starink methods respectively, from which the calculations of activation energies are executed. Intercepts of the straight lines signify the preexponential factors.
3.3 Model fitting kinetics
Model-free method or Coats and Redfern model is one of the important methods that provide information pertinent to reaction and reaction mechanism.31 It provides the required kinetic parameters based on different temperature ranges obtained through TGA data. This is carried out by using different considered equations to account for the various plausible mechanisms. The basic equation of the Coats and Redfern model assumes the formula indicated in eqn (10)| |
 | (10) |
The plot of  and 1/T give a straight line in Coats and Redfern method, and the slope and intercept of this line determine the kinetic parameters based on the different reaction mechanisms. The g(α) function varies according to different reaction mechanisms. The g(α) for different mechanism models is listed in Table 1.31,32
and 1/T give a straight line in Coats and Redfern method, and the slope and intercept of this line determine the kinetic parameters based on the different reaction mechanisms. The g(α) function varies according to different reaction mechanisms. The g(α) for different mechanism models is listed in Table 1.31,32
Table 1 Reaction mechanism models used in Coats and Redfern method
| Model name |
Symbol |
f(α) |
g(α) |
| Chemical reaction zero order |
CR0 |
1 |
α |
| Chemical reaction 1st order |
CR1 |
(1 − α) |
−ln(1 − α) |
| Chemical reaction 1.5th order |
CR1.5 |
(1 − α)1.5 |
(2 [(1 − α)−1.5 − 1] |
| Chemical reaction 2nd order |
CR2 |
(1 − α)2 |
[1/(1 − α)] − 1 |
| Chemical reaction 3rd order |
CR3 |
(1 − α)3 |
½[(1 − α)−2− 1]) |
| One-dimensional diffusion |
D1 |
½α |
α2 |
| Two-dimensional diffusion |
D2 |
[−ln (1 − α)]−1 |
(1 − α) − ln(1 − α) + α |
| Three-dimensional diffusion |
D3 |
0.5 × [(1 + α)2/3[(1 + α)1/3 − 1]−1] |
[[−ln(1 − α)]⅓]2 |
| Diffusion control (Crank) |
D4 |
1.5 × [(1 − α)−1/3–1]−1 |
(1 − 2/3α) − (1 − α)⅔ |
| Diffusion control (Jander) |
D5 |
1.5 × [(1 − α)⅔ [1 − (1 − α)⅓]−1] |
[1 − (1 − α)⅓]2 |
| Contracting cylinder |
CS |
2(1 − α)½ |
1 − (1 − α)½ |
| Contracting sphere |
SS |
3(1 − α)⅔ |
1 − (1 − α)⅓ |
| Power law |
P4 |
4α¾ |
α¼ |
| Power law |
P3 |
3α⅔ |
α⅓ |
| Avrami–Erofeev |
AE2 |
2(1 − α)[−ln(1 − α)]½ |
[−ln(1 − α)]½ |
| Avrami–Erofeev |
AE3 |
3(1 − α)[−ln(1 − α)]⅔ |
[−ln(1 − α)]⅓ |
3.4 Thermodynamic Analysis
Thermodynamic parameters (ΔH, ΔG, and ΔS) were calculated with the help of kinetic parameters and TGA data by using the following eqn (11)–(13)| |
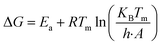 | (12) |
| |
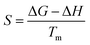 | (13) |
Here, KB is “Boltzmann constant” and h is “Planck constant”, with the value of 1.381 × 10−23 m2 kg s−1−2 K−1 and 6.626 × 10−34 m2 kg s−1. Tm is the peak temperature obtained from DTA data at which maximum conversion occurs during thermal degradation.
3.5 Synergistic effects
The synergistic effect during pyrolysis and combustion of TBBA and TBBA:Ca(OH)2 samples was determined at different temperature ranges by using the following eqn (14)| | |
Wcal = xW1 + yW2 + …zWn
| (14) |
Here, x, y, and z are the blended ratio of individual samples in the mixture, W1, W2 and, Wn are the mass losses of each sample component mixed to make the blend. The variance between experimented and calculated mass loss indicates positive or negative synergy and can be calculated by the following eqn (15)
If the experimental value is lower than the calculated value, the synergy is positive and if the difference in the experimental value is more than that of the calculated value then the synergy entails a negative behaviour.31
3.6 DFT calculations
The methodology of the DFT calculations is fully described in previous publications.9 In a nutshell, the DMol3 code33 is used in calculations based on the Perdew and Wang (PAW) functional along with a double numerical plus polarization (DNP) basis set.34,35 Final energies converge to less than 1 × 10−5 Ha. The surface of Ca(OH)2 was simulated using a cluster model comprising of four calcium atoms.
4 Results and discussion
4.1 Pyrolysis and combustion interpretation
The TGA was used to investigate the degradation behavior of TBBA and TBBA:Ca(OH)2 during the pyrolysis and combustion process. The TBBA:Ca(OH)2 mass ratio was set to 1:2 (mass ratio) based on the experimental studies. To determine the best TBBA:Ca(OH)2 mixture blend, three different ratios of TBBA:Ca(OH)2 were prepared and tested by TGA at 10 °C for the pyrolysis reaction, as shown in the Fig. 1. It was observed that the major degradation step of the 1:1 and 2:1 blend ratio started around 205 °C, whereas in the 1:2 ratio it initiated earlier around 190 °C. Compared to the residue formation, 2:1 has a weight loss of 33% with the highest char, and 1:1 with the lowest char owing a weight loss of 37%. The blend of 1:1 showed a mass loss of 35% which is in between the other two mixtures. Higher char formation resembles the increased debromination ability by forming CaBr2, which indicates a 2:1 with excellent debromination capacity. But considering the char formation as well as the early onset degradation, 1:2 has been opted as the best blending ratio, and hence further experiments were conducted on that specific ratio. Ca(OH)2 was used in a higher proportion than TBBA because previous research has shown that the low heating rate and high ratio of the mixture containing metal oxides and hydroxides resulted in good interaction between the molecules, resulting in the formation of essential products while simultaneously capturing HBr.36
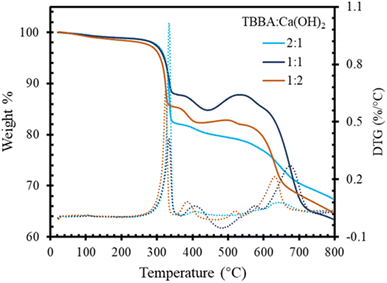 |
| | Fig. 1 TGA curve for different compositions of TBBA:Ca(OH)2 mixture. | |
For the pyrolysis and combustion processes, degradation of pure TBBA features a one-stage decomposition behaviour, which took place in the range of 250–341 °C for the four heating rates as shown in Fig. 2a and c. As the heating rate increased, a slight positive shift was observed in the order of increasing heating rate for both thermal treatments, but there was no significant difference in the TGA curves obtained. These shifts are occurring as the heating rate increases, the rate of degradation of the products ensues in a shorter time, which even can lead to incomplete decomposition of the products. A similar trend was reported in previous studies.36–38 This region of degradation is due to the disintegration of the TBBA compound initiated by the boiling point of TBBA at 250 °C, which results in the emission of HBr around 300 °C39 and other unstable organic compounds due to the breakage of the C–Br bond, which has a low dissociation energy of approximately 55.5–67.5 kcal mol−1.40 The gas phase evolution due to the evaporation of TBBA originates with the scission of C–CH3 bonds.41 After the decomposition of TBBA, the solid residue commonly known as char was formed from the pyrolysis experiments with a 20 wt% compared to the initial sample weight, which can be utilized for reducing the metal oxide into metals.42 Whereas, for the combustion, there was no formation of char due to complete oxidation. Based on the DTG curves, the peak degradation temperature (TP) for all heating rates was found to be between 306 and 311 °C. However, in both processes, no significant differences in mass loss were found across the temperature or heat rate ranges investigated. Hence, there was no influence of oxygen in the decomposition of TBBA compared to pyrolysis and the thermal effects remained unchanged. The initial temperature (Ti) and the final temperature (Tf) of the TGA cycle are represented in the Table 2.
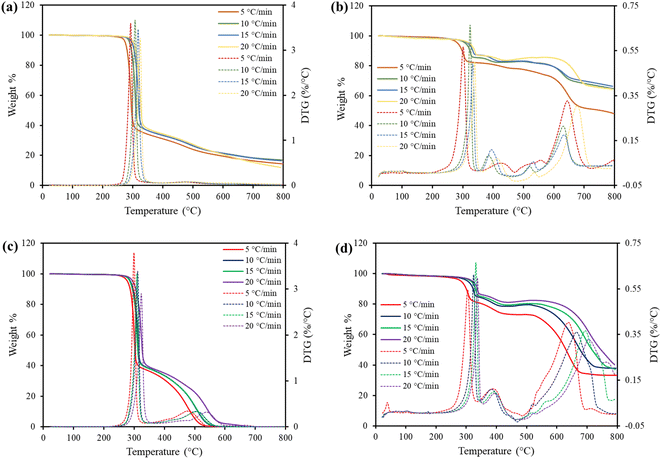 |
| | Fig. 2 TGA and DTG curves for (a) TBBA Pyrolysis, (b) TBBA:Ca(OH)2 Pyrolysis, (c) TBBA combustion and, (d) TBBA:Ca(OH)2 combustion. | |
Table 2 Ti, Tf, and Tp values for the thermal treatments of TBBA and TBBA:Ca(OH)2
| Process |
Sample |
Heating rate |
Ti (°C) |
Tf (°C) |
Tp (°C) |
| Peak 1 |
Peak 2 |
Peak 3 |
| Pyrolysis |
TBBA |
5 |
232 |
567 |
291 |
— |
— |
| 10 |
237 |
576 |
306 |
— |
— |
| 15 |
240 |
601 |
318 |
— |
— |
| 20 |
243 |
611 |
323 |
— |
— |
| TBBA:Ca(OH)2 |
5 |
173 |
716 |
302 |
427 |
650 |
| 10 |
206 |
710 |
323 |
385 |
627 |
| 15 |
219 |
711 |
338 |
391 |
646 |
| 20 |
231 |
761 |
340 |
410 |
682 |
| Combustion |
TBBA |
5 |
214 |
538 |
299 |
— |
— |
| 10 |
233 |
580 |
309 |
— |
— |
| 15 |
240 |
590 |
319 |
— |
— |
| 20 |
254 |
609 |
324 |
— |
— |
| TBBA:Ca(OH)2 |
5 |
192 |
692 |
306 |
386 |
645 |
| 10 |
206 |
767 |
321 |
384 |
663 |
| 15 |
224 |
771 |
331 |
391 |
692 |
| 20 |
238 |
791 |
337 |
401 |
705 |
In the case of a mixture of TBBA and Ca(OH)2, a small weight loss of approximately 2 wt% was observed in the temperature range of 180–250 °C, whereas no mass loss was observed in the case of pure TBBA. TBBA has a melting point of 178 °C, indicating that there was an interaction between the liquid molecules of TBBA and the Ca(OH)2 particles in the mixture. From the earlier studies on other metal oxides, with the help of TG-MS, it has been proven that in this temperature region mass loss is occurring due to the solid–liquid bromination reaction.20 Hence, it is confirmed that the Ca(OH)2 endured to react with the liquid TBBA resulting in the formation of CaBr2 (Calcium bromide) and consequently releasing the water molecules. Al-Harahsheh et al. (2017) also spotted, on leaching the pyrolysis residue of the mixture of metal oxides and TBBA, the metal bromides formed when the pyrolysis reaction was carried out at a temperature of below 250 °C. On the other hand, the TGA identifies three major peaks, P1, P2, and P3, respectively, for both pyrolysis and combustion in all the heating rates explored. The identified peaks were almost the same in both processes, other than a slight shift in temperature ranges in different regions as shown in Table 2. The range of the three decomposition peaks is approximately 250–340 °C, 357–435 °C, and 553–670 °C respectively, for the heating rate of 10 °C min−1 in both the thermal degradation methods as shown in Fig. 2 The corresponding TP values for the three regions obtained from the DTG curves are 323, 385, and 627 °C for pyrolysis and 321, 384, and 663 °C for combustion, as shown in Fig. 2b and d.
For both thermal processes, the first region of degradation that lasted to 327 °C with a mass loss of 20 wt% is due to the decomposition of long-chained TBBA units. This stage commences with the C–Br bond breakage subsequent to the formation of Br radicals. Contrary to pure TBBA, the resultant HBr formation has been completely eliminated in the mixture. This is due to the action of Ca–O linkages that fixes emitted HBr in a process that forms CaBr2, which is confirmed from the upcoming FTIR results. The TP value in this range was raised by 10 °C compared to the TP value of pure TBBA for the different heat rates explored, which could be due to the increase in the interaction between the substrate radicals. This observation coincides with the previous studies.39,44
The second region in the thermal treatments between 357–435 °C corresponds to the dehydration of the precursor Ca(OH)2 because the H2O release from the Ca(OH)2 occurs in this temperature range.45 The weight loss achieved in this region was approximately 3 wt%. As the physically adsorbed water is released from the reactant, it results in the formation of calcium oxide (CaO). Moreover, CO2 formation is expected to take place in this temperature window.39 As such, a carbonation reaction occurs, which ultimately leads to the formation of CaCO3. It is clear that the carbonation reaction occurs at a lower temperature.46 The final third region of decomposition between 553–670 °C can be attributed to the decarbonization of CaCO3 formed in the previous step at a higher temperature to form CaO. Also, it can be seen from the TGA and DTG curves in Fig. 2b and d that CaCO3 breaks down occur over a wider range of temperatures than Ca(OH)2 does (the second region).47
The second and third degradation regions (357–670 °C) from the TGA curve thus show the decomposition of Ca(OH)2, which is consistent with previous findings.48 The weight loss was around 15–25 wt%, as the lower the heating rate, the more weight loss was detected. As a result, char formation was found to be less in the case of a 5 °C min−1 heating rate, while the residue remaining was nearly the same in the other three heating rates. A low heating rate is commonly preferred due to less char formation in both combustion and pyrolysis because a lower yield of char means the formation of gas products is enhanced. However, in this study, Ca(OH)2 is used to capture the HBr, so the formation of more char can be attributed to HBr fixation and a reduction in TBBA evaporation, which is the main source of toxic HBr formation.43 This means that pyrolysis reaction with Ca(OH)2 at 10 °C min−1 is the best and most efficient way of thermally treating the TBBA, taking into account the formation of char, the amount of heat used, and the formation of required products.
The DSC experiments were run for the pure and blend under pyrolysis and oxidation conditions with a heating rate of 10 °C min−1 as shown in ESI† (SM) Fig. S1. For all environmental conditions, the first curve indicated the melting process of TBBA at around 113 °C, but the heat of reaction (ΔHr) values for the pure and blend were slightly different as 36–39 J g−1 and 16–18 J g−1 with endothermic reactions. For pure TBBA, a second endothermic peak with ΔHr 124.45 J g−1 was observed, which is due to the degradation and vaporization of the TBBA compound, and the results were in agreement with previous studies.49 In the case of blended TBBA and Ca(OH)2 sample after the melting point of TBBA, a small exothermic peak in pyrolysis in the range of 319–340 °C with a peak temperature of 334.61 °C and ΔHr of 237.43 J g−1, whereas in oxidation this peak formed with ΔHr of 84.214 J g−1 and temperature range of 331–347 °C with maxima temperature of 337.72 °C. This exothermic peak is due to the formation of CaBr2 due to the reaction amongst emitted HBr and Ca(OH)2, which was in agreement with the results of the previous studies in the formation of metal bromides such as ZnBr2, SbBr3, FeBr2.50–52 There are also third and fourth endothermic peaks in the sample mixture, with the third due to the conversion of Ca(OH)2 into CaO and the fourth due to physically adhered water molecules and disintegration of CaCO3 into CaO.45 The DSC curves obtained are consistent with the weight loss determined from the TGA profiles for all samples. The two phenomena of metal bromide formation and Ca(OH)2 decomposition appears almost simultaneously in the same temperature range and therefore they interfere with each other as shown in the Fig. S1† of SM.
4.2 IR spectrum analysis
The functional group analysis was done using FT-IR as shown in Fig. 3. In Fig. 3a, the short broad peak at 3444 cm−1 is due to the alcoholic OH stretching and those at 2964 and 2870 cm−1 are from the strong symmetrical and asymmetrical stretching of CH3 groups present in pure TBBA.53 Whereas the short strong peak at 1755 cm−1 corresponds to the weak C–H bending in an aromatic compound and the one at 1583 cm−1 is due to the cyclic alkene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching in a benzene ring. The fingerprint region for pure TBBA starts from a strong absorption peak at 1454 cm−1 owing to the presence of aromatic C–C bonds, a strong phenolic O–H bending at 1378 cm−1, strong aromatic C–O stretching at 1256 cm−1, strong C–C–O asymmetric stretch of alcohols at 1020 cm−1, strong CC bending at 980 cm−1, strong C–H aromatics bending at 873 and 735 cm−1 along with strong C–Br and aromatic C–Br stretching vibrations at 673 and 564 cm−1 respectively.15,53–56 The biochar analysis of pyrolyzed TBBA in Fig. 3b shows that there is a substantial reduction in the absorption peaks notably in the fingerprint region. The biochar was left with a minimal amount of C–H bending at 741, 803, and 874 cm−1. A similar reduction in intensity is observed for the aromatic C–C bonds at 1453 cm−1. The peaks at 3436 and 1620 cm−1 are attributed to hydroxyl groups and alkene CC respectively which are the characteristic peaks of any typical biochar.57,58 Fig. 3c portrays the distinctive peaks of the 1:2 mixture of TBBA and Ca(OH)2 wherein the sharp peak at 3642 cm−1 is assigned in particular to the hydroxyl group of Ca(OH)2.59 Along with the alcoholic OH stretching at 3433 cm−1, the short strong stretching of CH3 groups at 2968 and 2868 cm−1 is from TBBA. However, the strong small peaks at 2512 and 1795 cm−1 have been assigned to the symmetric vibrations of Ca–O bonds.60,61 The mixture resulted in producing a saturated band around 1425 cm−1 which made peak assignment uneasy. The vibration at 1255 cm−1 is due to strong aromatic C–O stretching, strong alcoholic C–C–O asymmetric stretch at 1023 cm−1, and the strong CC bending at 980 cm−1 is from TBBA. Nevertheless, the strong peaks at 874 and 712 cm−1 belong to the in-plane bending of Ca–O bonds followed by the C–Br stretching vibrations at 577 cm−1.62 The co-pyrolysis char in Fig. 3d uncovers the presence of hydroxyl peaks at 3426 cm−1, alkyl peaks at 2977 and 2868 cm−1, alkene CC peaks at 1634 cm−1, and methylene bending at 1439 cm−1 pertaining to the organics present in the biochar. The strong peaks at 2513, 1796, 874, and 712 cm−1 are attributed to the Ca–O bonds from the unreacted Ca(OH)2. It also portrays the complete removal of C–Br vibrations in the right shoulder below 600 cm−1 which justifies an expected 100% debromination capacity of Ca(OH)2.
C stretching in a benzene ring. The fingerprint region for pure TBBA starts from a strong absorption peak at 1454 cm−1 owing to the presence of aromatic C–C bonds, a strong phenolic O–H bending at 1378 cm−1, strong aromatic C–O stretching at 1256 cm−1, strong C–C–O asymmetric stretch of alcohols at 1020 cm−1, strong CC bending at 980 cm−1, strong C–H aromatics bending at 873 and 735 cm−1 along with strong C–Br and aromatic C–Br stretching vibrations at 673 and 564 cm−1 respectively.15,53–56 The biochar analysis of pyrolyzed TBBA in Fig. 3b shows that there is a substantial reduction in the absorption peaks notably in the fingerprint region. The biochar was left with a minimal amount of C–H bending at 741, 803, and 874 cm−1. A similar reduction in intensity is observed for the aromatic C–C bonds at 1453 cm−1. The peaks at 3436 and 1620 cm−1 are attributed to hydroxyl groups and alkene CC respectively which are the characteristic peaks of any typical biochar.57,58 Fig. 3c portrays the distinctive peaks of the 1:2 mixture of TBBA and Ca(OH)2 wherein the sharp peak at 3642 cm−1 is assigned in particular to the hydroxyl group of Ca(OH)2.59 Along with the alcoholic OH stretching at 3433 cm−1, the short strong stretching of CH3 groups at 2968 and 2868 cm−1 is from TBBA. However, the strong small peaks at 2512 and 1795 cm−1 have been assigned to the symmetric vibrations of Ca–O bonds.60,61 The mixture resulted in producing a saturated band around 1425 cm−1 which made peak assignment uneasy. The vibration at 1255 cm−1 is due to strong aromatic C–O stretching, strong alcoholic C–C–O asymmetric stretch at 1023 cm−1, and the strong CC bending at 980 cm−1 is from TBBA. Nevertheless, the strong peaks at 874 and 712 cm−1 belong to the in-plane bending of Ca–O bonds followed by the C–Br stretching vibrations at 577 cm−1.62 The co-pyrolysis char in Fig. 3d uncovers the presence of hydroxyl peaks at 3426 cm−1, alkyl peaks at 2977 and 2868 cm−1, alkene CC peaks at 1634 cm−1, and methylene bending at 1439 cm−1 pertaining to the organics present in the biochar. The strong peaks at 2513, 1796, 874, and 712 cm−1 are attributed to the Ca–O bonds from the unreacted Ca(OH)2. It also portrays the complete removal of C–Br vibrations in the right shoulder below 600 cm−1 which justifies an expected 100% debromination capacity of Ca(OH)2.
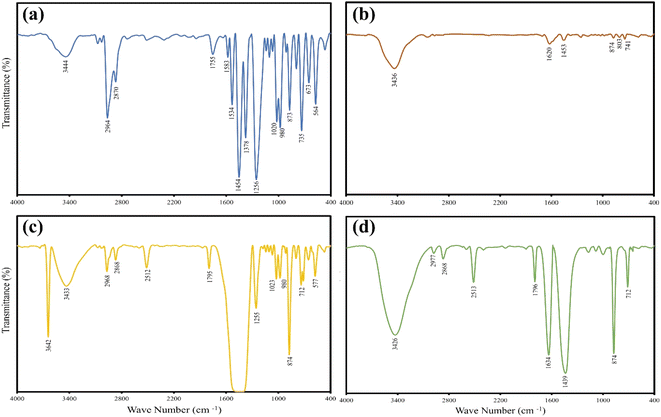 |
| | Fig. 3 FT-IR analysis for (a) pure TBBA, (b) TBBA biochar, (c) 1:2 mixture of TBBA:Ca(OH)2, and (d) co-pyrolysis biochar of TBBA:Ca(OH)2. | |
The FTIR analysis of the evolved gas from the degradation of TBBA was investigated at four different temperature ranges 200, 300, 400, and 500 °C as shown in the Fig. 4. The opted temperature ranges are based on the TGA curve since the major decomposition of TBBA occurs in these selected regions. The peaks at 2930–3100 cm−1 resemble the C–H bonds from aromatic structures, whereas the peak at 1473 cm−1 indicated the stretching vibration of aliphatic C–H bonds from the degradation of TBBA due to the formation of methyl and methylene groups.56 The formation of HBr is indicated by the characteristic band in the range of 2414–2680 cm−1.24,63 The HBr formation initiated from 300 °C with the highest intensity which further decreased as the temperature increased, which agrees with the TGA results as the decomposition of TBBA starts around 250 °C which results in the emission of carcinogenic HBr. The Lambert–Beer law declares that the intensity of the peaks is directly related to the concentration, which signifies the HBr formation was more in the range of 300 °C. The peaks at 2280–2380 cm−1, and 650–800 cm−1 represent the formation of CO2, and further, the C–O vibration in the evolved CO2 is observed in the range of 2052–2072 cm−1.64 The C–O–C stretching vibrations are shown at the peaks of 1236–1305 cm−1.65 During the decomposition of TBBA, aromatic C–O bonds from the phenolic and alcohol compounds are shown by the absorption peaks at 2165–2220 cm−1 and 2700 cm−1.66,67
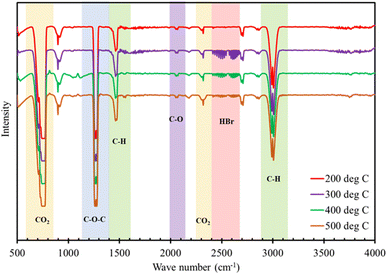 |
| | Fig. 4 FTIR analysis for evolved gas during thermal treatment of TBBA at different temperatures. | |
4.3 Kinetic analysis
A comprehensive knowledge of kinetic and thermodynamic parameters is required to scale up and process optimization of complex processes like pyrolysis and combustion, and to design it with proper safety. The kinetic analysis of TBBA with and without Ca(OH)2 was carried out using iso-conversional model-free methods (KAS, FWO, and Starink) to find kinetic parameters like activation energy (Ea), pre-exponential factor (A), and linear regression (R2), as suggested by the International Confederation for Thermal Analysis and Calorimetry (ICTAC) kinetic committee.68 KAS, FWO, and Starink plots were drawn from eqn (7)–(9) by plotting ln(β/T2), ln(β), and ln(β/T1.92) vs. (1/T), respectively. The three models were predicted utilizing the data obtained for both samples at four different heating rates from the TGA analysis. Ea and A were obtained from linear lines at conversion by the slope (−Ea/R) and intercept (c), the values are shown in Table S1† for pyrolysis and Table S2† for combustion in SM.
In pyrolysis, the average activation energies for TBBA are 111.65, 121.99, 112.07 kJ mol−1, and for TBBA:Ca(OH)2 are 61.86, 74.01, 62.34 kJ mol−1 for KAS, FWO, and starink methods, respectively. In combustion, the corresponding values of Ea are 137.94, 148.61, 138.37 kJ mol−1 for individual TBBA and are 62.79, 76.51, 63.36 kJ mol−1 for TBBA:Ca(OH)2 blends in all three iso-conversional models. It can be seen that the values of Ea for the blend are significantly lower than that of the pure TBBA sample in both the pyrolysis and combustion processes. On the other hand, the pyrolysis process entails lower activation energies as compared to the combustion process. Thus, pyrolysis of TBBA:Ca(OH)2 could be more energy efficient than that of combustion based on the activation energies reported. The activation energies from all three models (KAS, FWO, and Starink) for TBBP showed an approximately similar trend to the previous studies.50,69 In the pyrolysis of TBBA, the activation energies remained almost constant till 0.5 conversion, then decreased to 0.65 conversion, and then increased to 0.75 conversion, and again decreased till the end. The increasing trend of Ea in the end during the conversion range of 0.65 to 0.75 is the result of heavy component initiation reactions during the char development, which used up about 140 kJ mol−1.70 In the combustion of TBBA, the value of activation energies decreased till 0.5 conversion, then slightly increased at 0.55 conversion, and again decreased till the end. During the combustion process, it was observed a complete decomposition of the sample. Hence, no char formation was witnessed and there was no change in the trend of activation energy. For both processes, the increasing trends in Ea show the presence of an endothermic reaction, while the decreasing trend indicates an exothermic process.68 Instead of utilizing the energy to break the bonds of the reactant, exothermic reactions take place because of the formation of bonds in the product, and vice versa for an endothermic reaction. In both pyrolysis and combustion, the activation energies of TBBA:Ca(OH)2 blends dropped to 0.2 conversion and then went up until the end. The different activation energy profiles for individual and blended samples are caused by differences in their composition, the breaking of chemical bonds, and competitive reactions that happen during thermal degradation. Overall, the decrease in activation energy in the mixture revealed a great way to capture the HBr given off by the TBBA when Ca(OH)2 is added. This HBr is given off when the carbon-halogen bonds in the parent compound and its fragments extinguish. Variations of Ea with conversion for all the studied model-free iso-conversional methods are shown in Fig. 5 for both samples.
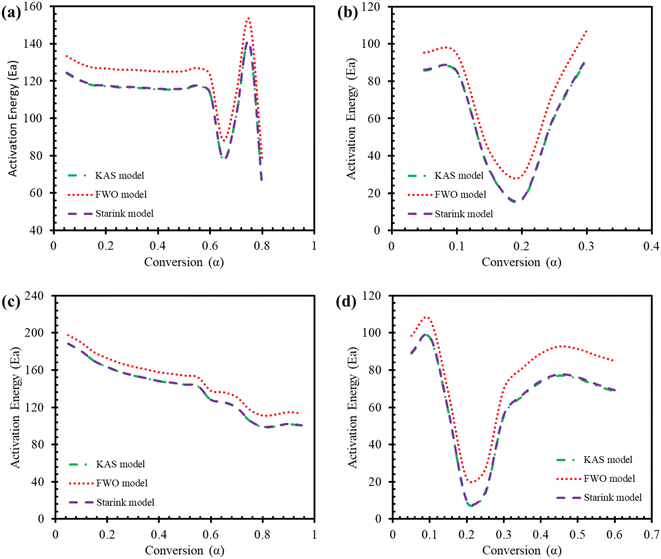 |
| | Fig. 5 Activation energies during pyrolysis of (a) TBBA, (b) TBBA:Ca(OH)2 and during combustion of (c) TBBA, (d) TBBA:Ca(OH)2. | |
Pre-exponential factor A for TBBA was in the range of 106 to 1013 min−1 in pyrolysis and was in the range of 109 to 1018 min−1 in the combustion process. On the other hand, the values of A for TBBA:Ca(OH)2 were in the range of 101 to 109 min−1 in pyrolysis and the range of 101 to 1010 min−1 in the combustion process. This indicates the pyrolysis and combustion of TBBA with and without blend are complex reaction processes. The value of A implies the collision frequency between reactant molecules to proceed with the reaction. The value of A less than 109 confirms the presence of only surface reaction taking place, the range from 1010 to 1012 indicates that degradation of reactant takes place, and the value of A more than 1014 indicates high energy demand due to high collision of molecules during the thermal process.68 The A values were reduced significantly with the help of Ca(OH)2, demonstrating that Ca(OH)2 aids in reducing the demand for energy during the thermal treatments of TBBA. In the case of the mixture, it was observed that the Ea and A values were high during the conversion range of 0.05–0.2. This is due to the formation of CaBr2 which implies a high collision of molecules at the initial range of degradation as discussed in the previous section due to the solid–liquid bromination. Similarly, high particle impact occurs during the early stages of the conversion in pure TBBA, as evidenced by the A values. The value of A for combustion is much higher than that in pyrolysis in both individual and blended samples, which reveals that the pyrolysis process is much simpler and faster than combustion in terms of energy requirement and reactivity. The R2 also shows the best agreement in the determined range in the literature (0.90–0.99) for both samples in both processes, but pyrolysis exposes more appropriate agreement than the values obtained for Ea and A during the combustion process. The isoconversional curves for the Starink, KAS, and FWO models were plotted as a function of conversion for the pyrolysis and combustion processes of TBBP and TBBP: Ca(OH)2, using the four different heating rates to determine linearity, as shown in Fig S2 and S3† (SM).
The validation of activation energies obtained from the iso-conversional model-free method was also carried out by using the “Coats and Redfern” method based on different reaction mechanism models to find out the best-fitted reaction mechanism happening during the degradation of TBBA and its blend in oxygen and nitrogen environments. At least 16 models were used to check the values in two temperature regions based on kinetic data at all heating rates 5, 10, 15, and 20 °C as shown in Table 3. The best mechanism was opted for comparing the rate of heating and selected temperature zone based on the attained R2 values. The TBBA during the pyrolysis showed good correlation with all orders of chemical reaction (CR0, CR1.5, CR2, and CR3), diffusion models (D1, D3, D4, and DF), cylinder and spherical contraction, power law P3, and avrami-erofeev AE3 models at the four heating rates and both temperature ranges selected. At the heating rate of 5 and 10 °C min−1, the diffusion models (D1, D4, and D5) proved to be the best-fitting ones when TBBA and Ca(OH)2 were blended. Moreover, it was noticed that the mixtures showed a good relationship of the linear regression at the heating rates of 5 and 10 °C min−1 in the temperature zone of (250–350 °C), whereas 15 and 20 °C min−1 were in good agreement at the higher temperature zone (550–650 °C). In the case of combustion, pure TBBA exhibited an acceptable regression coefficient for chemical reaction orders (CR1, CR1.5, CR2, and CR3), and diffusion model D5 at different heating rates and both temperature ranges, whereas for the mixture during oxidation there was no model fitted for both temperature zone for the same model. In the mixture, the first temperature region (200–400 °C) illustrated the best fit for the chemical reaction order models (CR1, CR1.5, CR2, and CR3), diffusion mode (D1, D4, and D5), and contracting sphere models at the heating rates of 5 and 10 °C min−1. The fitted regression models for the second temperature zone (450–650 °C) follow the power laws (P3 and P4) for the four heating rates. Overall, the linear regression coefficient in Coats–Redfern method also fits best in most pyrolysis reactions with a heating rate of 10 °C min−1 and at a lower temperature region. This trend reconciles very well with TGA's data interpretation that the HBr formation and its capture by Ca(OH)2 ensue at the same temperature zone.
Table 3 Kinetic parameters of TBBA and TBBA:Ca(OH)2 via Coats and Redfern method in pyrolysis and combustion process
| Model name |
Sample |
Heating rate (°C min−1) |
Pyrolysis |
Combustion |
| Temperature (250–350 °C) |
Temperature (550–650 °C) |
Temperature (200–400 °C) |
Temperature (450–650 °C) |
| Ea (kJ mol−1) |
R2 |
Ea (kJ mol−1) |
R2 |
Ea (kJ mol−1) |
R2 |
Ea (kJ mol−1) |
R2 |
| CR0 |
TBBA |
5 |
113.90 |
0.79 |
11.71 |
0.99 |
79.11 |
0.87 |
8.42 |
0.82 |
| 10 |
147.94 |
0.91 |
11.83 |
0.99 |
80.56 |
0.90 |
4.95 |
0.57 |
| 15 |
144.85 |
0.94 |
11.61 |
0.99 |
80.09 |
0.88 |
4.95 |
0.57 |
| 20 |
148.61 |
0.93 |
9.96 |
0.99 |
77.43 |
0.90 |
2.35 |
0.39 |
| TBBA + Ca(OH)2 |
5 |
63.83 |
0.90 |
15.09 |
0.75 |
35.63 |
0.90 |
7.69 |
0.41 |
| 10 |
58.33 |
0.90 |
17.31 |
0.8 |
33.81 |
0.88 |
3.68 |
0.14 |
| 15 |
47.23 |
0.85 |
12.26 |
0.75 |
32.68 |
0.84 |
4.24 |
0.34 |
| 20 |
45.38 |
0.83 |
9.39 |
0.54 |
29.67 |
0.82 |
7.43 |
0.63 |
| CR1 |
TBBA |
5 |
137.81 |
0.83 |
1.42 |
0.73 |
88.65 |
0.88 |
32.46 |
0.82 |
| 10 |
171.73 |
0.91 |
1.55 |
0.66 |
89.42 |
0.89 |
40.40 |
0.94 |
| 15 |
166.19 |
0.93 |
2.10 |
0.87 |
89.07 |
0.90 |
43.32 |
0.95 |
| 20 |
165.76 |
0.93 |
3.57 |
0.91 |
85.66 |
0.90 |
28.02 |
0.93 |
| TBBA + Ca(OH)2 |
5 |
73.19 |
0.91 |
60.90 |
0.69 |
38.12 |
0.90 |
20.04 |
0.57 |
| 10 |
65.67 |
0.89 |
41.93 |
0.83 |
36.57 |
0.88 |
9.83 |
0.35 |
| 15 |
52.70 |
0.84 |
34.28 |
0.82 |
35.10 |
0.84 |
1.73 |
0.05 |
| 20 |
44.93 |
0.79 |
8.38 |
0.31 |
31.88 |
0.81 |
5.97 |
0.41 |
| CR1.5 |
TBBA |
5 |
185.83 |
0.87 |
54.69 |
0.98 |
106.41 |
0.89 |
270.47 |
0.95 |
| 10 |
218.65 |
0.92 |
57.02 |
0.98 |
105.69 |
0.90 |
273.49 |
0.97 |
| 15 |
207.76 |
0.92 |
57.88 |
0.98 |
105.61 |
0.90 |
302.84 |
0.96 |
| 20 |
197.84 |
0.91 |
50.93 |
0.98 |
100.66 |
0.90 |
139.60 |
0.94 |
| TBBA + Ca(OH)2 |
5 |
89.26 |
0.92 |
205.26 |
0.56 |
43.54 |
0.91 |
47.26 |
0.61 |
| 10 |
78.01 |
0.87 |
99.11 |
0.82 |
41.01 |
0.87 |
21.65 |
0.51 |
| 15 |
61.76 |
0.83 |
86.07 |
0.82 |
38.98 |
0.84 |
2.81 |
0.06 |
| 20 |
51.99 |
0.77 |
20.37 |
0.51 |
35.40 |
0.81 |
3.37 |
0.11 |
| CR2 |
TBBA |
5 |
168.28 |
0.86 |
33.77 |
0.98 |
100.04 |
0.89 |
270.47 |
0.95 |
| 10 |
201.58 |
0.92 |
35.25 |
0.97 |
99.88 |
0.89 |
183.96 |
0.97 |
| 15 |
192.69 |
0.93 |
35.97 |
0.98 |
99.70 |
0.90 |
203.54 |
0.96 |
| 20 |
186.33 |
0.92 |
32.33 |
0.98 |
95.32 |
0.90 |
96.01 |
0.94 |
| TBBA + Ca(OH)2 |
5 |
83.66 |
0.92 |
149.41 |
0.58 |
41.67 |
0.91 |
37.09 |
0.61 |
| 10 |
73.72 |
0.88 |
77.53 |
0.83 |
39.49 |
0.88 |
17.37 |
0.47 |
| 15 |
58.63 |
0.83 |
66.48 |
0.82 |
37.65 |
0.84 |
1.19 |
0.01 |
| 20 |
49.56 |
0.78 |
16.03 |
0.46 |
34.20 |
0.82 |
4.29 |
0.11 |
| CR3 |
TBBA |
5 |
204.73 |
0.87 |
76.77 |
0.98 |
113.18 |
0.90 |
363.38 |
0.96 |
| 10 |
236.98 |
0.92 |
79.94 |
0.98 |
111.87 |
0.90 |
365.91 |
0.97 |
| 15 |
223.91 |
0.92 |
80.99 |
0.98 |
111.89 |
0.90 |
405.05 |
0.96 |
| 20 |
210.11 |
0.91 |
70.91 |
0.98 |
106.33 |
0.88 |
185.99 |
0.94 |
| TBBA + Ca(OH)2 |
5 |
95.16 |
0.92 |
265.39 |
0.58 |
45.47 |
0.91 |
58.33 |
0.62 |
| 10 |
82.47 |
0.87 |
122.76 |
0.82 |
42.59 |
0.87 |
26.25 |
0.53 |
| 15 |
65.02 |
0.82 |
107.56 |
0.82 |
40.35 |
0.83 |
4.53 |
0.11 |
| 20 |
54.49 |
0.77 |
25.03 |
0.55 |
36.63 |
0.81 |
2.40 |
0.05 |
| D1 |
TBBA |
5 |
12.64 |
0.78 |
12.04 |
0.99 |
167.61 |
0.89 |
3.24 |
0.16 |
| 10 |
305.39 |
0.91 |
9.16 |
0.99 |
170.52 |
0.90 |
3.67 |
0.16 |
| 15 |
299.22 |
0.94 |
8.71 |
0.99 |
169.57 |
0.90 |
3.67 |
0.17 |
| 20 |
306.73 |
0.94 |
5.43 |
0.99 |
164.25 |
0.91 |
8.87 |
0.72 |
| TBBA + Ca(OH)2 |
5 |
137.11 |
0.91 |
44.68 |
0.86 |
79.01 |
0.92 |
28.96 |
0.69 |
| 10 |
126.16 |
0.91 |
49.11 |
0.88 |
77.03 |
0.91 |
20.95 |
0.56 |
| 15 |
103.97 |
0.87 |
39.01 |
0.88 |
74.75 |
0.87 |
5.09 |
0.14 |
| 20 |
90.76 |
0.83 |
18.79 |
0.53 |
68.73 |
0.85 |
1.27 |
0.01 |
| D2 |
TBBA |
5 |
23.61 |
0.78 |
2.71 |
0.99 |
4.80 |
0.39 |
22.58 |
0.82 |
| 10 |
24.15 |
0.76 |
2.53 |
0.89 |
3.99 |
0.31 |
27.20 |
0.94 |
| 15 |
21.11 |
0.66 |
2.21 |
0.92 |
4.15 |
0.32 |
29.75 |
0.94 |
| 20 |
16.05 |
0.54 |
1.84 |
0.82 |
3.16 |
0.20 |
15.08 |
0.88 |
| TBBA + Ca(OH)2 |
5 |
5.67 |
0.62 |
32.15 |
0.54 |
3.67 |
0.82 |
1.67 |
0.03 |
| 10 |
2.67 |
0.17 |
14.29 |
0.62 |
33.81 |
0.88 |
3.68 |
0.14 |
| 15 |
0.24 |
0.00 |
10.95 |
0.57 |
5.09 |
0.88 |
9.86 |
0.95 |
| 20 |
2.09 |
0.16 |
5.88 |
0.59 |
5.45 |
0.90 |
11.36 |
0.97 |
| D3 |
TBBA |
5 |
88.60 |
0.82 |
3.89 |
0.98 |
55.90 |
0.87 |
17.08 |
0.74 |
| 10 |
111.19 |
0.91 |
3.80 |
0.96 |
56.42 |
0.88 |
22.37 |
0.92 |
| 15 |
107.51 |
0.93 |
3.44 |
0.98 |
4.15 |
0.32 |
29.75 |
0.94 |
| 20 |
107.22 |
0.92 |
2.45 |
0.93 |
53.92 |
0.88 |
14.12 |
0.90 |
| TBBA + Ca(OH)2 |
5 |
45.57 |
0.89 |
35.72 |
0.64 |
22.25 |
0.87 |
8.80 |
0.37 |
| 10 |
40.56 |
0.88 |
23.08 |
0.77 |
21.21 |
0.85 |
2.00 |
0.05 |
| 15 |
31.92 |
0.82 |
17.99 |
0.74 |
20.24 |
0.80 |
5.69 |
0.56 |
| 20 |
26.75 |
0.76 |
0.73 |
0.01 |
18.09 |
0.77 |
8.51 |
0.77 |
| D4 |
TBBA |
5 |
26.64 |
0.87 |
4.70 |
0.99 |
175.53 |
0.89 |
11.60 |
0.41 |
| 10 |
324.81 |
0.92 |
2.28 |
0.97 |
177.91 |
0.91 |
22.98 |
0.76 |
| 15 |
316.66 |
0.94 |
1.58 |
0.98 |
177.06 |
0.90 |
23.36 |
0.77 |
| 20 |
320.95 |
0.94 |
2.61 |
0.96 |
171.15 |
0.91 |
26.37 |
0.91 |
| TBBA + Ca(OH)2 |
5 |
145.14 |
0.91 |
74.51 |
0.83 |
81.87 |
0.92 |
38.58 |
0.71 |
| 10 |
132.48 |
0.91 |
67.90 |
0.88 |
79.41 |
0.90 |
26.00 |
0.58 |
| 15 |
108.68 |
0.87 |
55.67 |
0.87 |
76.86 |
0.87 |
7.19 |
0.22 |
| 20 |
94.49 |
0.83 |
23.91 |
0.57 |
70.65 |
0.85 |
0.04 |
0.01 |
| D5 |
TBBA |
5 |
267.75 |
0.83 |
4.58 |
0.97 |
179.93 |
0.89 |
31.16 |
0.71 |
| 10 |
335.79 |
0.92 |
4.45 |
0.95 |
181.98 |
0.91 |
45.08 |
0.90 |
| 15 |
326.54 |
0.94 |
5.27 |
0.98 |
181.19 |
0.90 |
46.64 |
0.91 |
| 20 |
328.85 |
0.93 |
9.39 |
0.98 |
174.93 |
0.91 |
41.51 |
0.94 |
| TBBA + Ca(OH)2 |
5 |
149.39 |
0.91 |
96.79 |
0.79 |
83.37 |
0.92 |
44.40 |
0.70 |
| 10 |
135.79 |
0.90 |
79.55 |
0.87 |
80.65 |
0.90 |
28.84 |
0.59 |
| 15 |
111.17 |
0.87 |
66.168 |
0.87 |
77.95 |
0.87 |
8.35 |
0.24 |
| 20 |
96.44 |
0.83 |
26.81 |
0.58 |
71.64 |
0.85 |
0.62 |
0.00 |
| CS |
TBBA |
5 |
125.04 |
0.81 |
7.32 |
0.99 |
83.64 |
0.88 |
1.77 |
0.05 |
| 10 |
159.09 |
0.91 |
7.42 |
0.99 |
84.79 |
0.89 |
7.84 |
0.55 |
| 15 |
154.89 |
0.94 |
7.03 |
0.99 |
84.37 |
0.88 |
8.19 |
0.59 |
| 20 |
156.76 |
0.93 |
4.97 |
0.99 |
81.37 |
0.90 |
8.55 |
0.79 |
| TBBA + Ca(OH)2 |
5 |
68.36 |
0.90 |
33.10 |
0.75 |
36.43 |
0.90 |
13.28 |
0.51 |
| 10 |
61.91 |
0.90 |
28.27 |
0.82 |
35.17 |
0.88 |
6.58 |
0.26 |
| 15 |
49.91 |
0.85 |
22.02 |
0.8 |
33.87 |
0.84 |
3.04 |
0.17 |
| 20 |
42.74 |
0.80 |
5.08 |
0.19 |
30.76 |
0.82 |
6.72 |
0.52 |
| SS |
TBBA |
5 |
129.11 |
0.82 |
4.96 |
0.99 |
85.26 |
0.88 |
8.78 |
0.43 |
| 10 |
163.14 |
0.91 |
5.02 |
0.99 |
86.29 |
0.89 |
15.75 |
0.79 |
| 15 |
158.51 |
0.94 |
4.57 |
0.99 |
85.90 |
0.89 |
16.52 |
0.82 |
| 20 |
159.67 |
0.93 |
2.55 |
0.96 |
82.77 |
0.90 |
13.96 |
0.88 |
| TBBA + Ca(OH)2 |
5 |
69.94 |
0.90 |
41.15 |
0.74 |
36.99 |
0.90 |
15.41 |
0.54 |
| 10 |
63.14 |
0.89 |
32.52 |
0.82 |
35.63 |
0.88 |
7.62 |
0.29 |
| 15 |
50.83 |
0.85 |
25.83 |
0.81 |
34.28 |
0.84 |
2.61 |
0.12 |
| 20 |
43.46 |
0.80 |
6.14 |
0.23 |
31.13 |
0.82 |
6.48 |
0.48 |
| P4 |
TBBA |
5 |
1.94 |
0.64 |
13.81 |
0.99 |
12.73 |
0.74 |
12.30 |
0.98 |
| 10 |
29.85 |
0.86 |
13.83 |
0.99 |
13.09 |
0.77 |
11.42 |
0.98 |
| 15 |
29.08 |
0.91 |
13.77 |
0.99 |
12.98 |
0.76 |
11.42 |
0.98 |
| 20 |
30.02 |
0.91 |
13.36 |
0.99 |
12.31 |
0.78 |
10.77 |
0.99 |
| TBBA + Ca(OH)2 |
5 |
8.82 |
0.72 |
7.09 |
0.92 |
1.65 |
0.23 |
8.26 |
0.94 |
| 10 |
7.45 |
0.71 |
6.54 |
0.91 |
1.41 |
0.18 |
9.26 |
0.95 |
| 15 |
4.68 |
0.50 |
7.81 |
0.95 |
1.12 |
0.11 |
11.25 |
0.98 |
| 20 |
3.02 |
0.28 |
10.34 |
0.96 |
3.87 |
0.01 |
12.04 |
0.99 |
| P3 |
TBBA |
5 |
31.59 |
0.73 |
13.57 |
0.99 |
20.08 |
0.80 |
11.87 |
0.98 |
| 10 |
42.92 |
0.88 |
13.61 |
0.99 |
20.56 |
0.83 |
10.71 |
0.97 |
| 15 |
41.89 |
0.92 |
13.53 |
0.99 |
20.40 |
0.81 |
10.71 |
0.97 |
| 20 |
43.14 |
0.92 |
12.99 |
0.99 |
19.52 |
0.83 |
9.84 |
0.98 |
| TBBA + Ca(OH)2 |
5 |
14.90 |
0.80 |
4.64 |
0.74 |
5.32 |
0.64 |
6.50 |
0.84 |
| 10 |
13.08 |
0.81 |
3.90 |
0.67 |
4.99 |
0.61 |
7.83 |
0.90 |
| 15 |
9.38 |
0.69 |
5.59 |
0.86 |
4.62 |
0.51 |
10.47 |
0.97 |
| 20 |
7.19 |
0.55 |
8.96 |
0.91 |
3.62 |
0.40 |
11.53 |
0.98 |
| AE2 |
TBBA |
5 |
64.15 |
0.81 |
3.54 |
0.99 |
39.62 |
0.86 |
9.43 |
0.61 |
| 10 |
81.11 |
0.91 |
6.47 |
0.99 |
40.01 |
0.87 |
13.40 |
0.87 |
| 15 |
78.34 |
0.93 |
6.19 |
0.99 |
39.84 |
0.86 |
14.86 |
0.91 |
| 20 |
78.13 |
0.92 |
5.46 |
0.99 |
38.14 |
0.87 |
7.22 |
0.79 |
| TBBA + Ca(OH)2 |
5 |
31.84 |
0.87 |
23.20 |
0.58 |
14.36 |
0.84 |
3.22 |
0.12 |
| 10 |
28.08 |
0.86 |
13.71 |
0.68 |
13.59 |
0.81 |
1.87 |
0.08 |
| 15 |
21.59 |
0.79 |
9.89 |
0.61 |
12.85 |
0.75 |
7.65 |
0.81 |
| 20 |
17.71 |
0.71 |
3.06 |
0.19 |
11.24 |
0.70 |
9.77 |
0.90 |
| AE3 |
TBBA |
5 |
39.55 |
0.78 |
9.20 |
0.99 |
23.25 |
0.82 |
1.74 |
0.10 |
| 10 |
50.84 |
0.89 |
9.15 |
0.99 |
23.51 |
0.84 |
4.39 |
0.62 |
| 15 |
49.02 |
0.92 |
8.97 |
0.99 |
23.39 |
0.83 |
5.36 |
0.72 |
| 20 |
48.85 |
0.91 |
8.48 |
0.99 |
222.65 |
0.84 |
0.27 |
0.01 |
| TBBA + Ca(OH)2 |
5 |
18.03 |
0.84 |
10.61 |
0.39 |
6.43 |
0.70 |
2.38 |
0.15 |
| 10 |
15.53 |
0.81 |
4.29 |
0.32 |
5.91 |
0.65 |
5.78 |
0.66 |
| 15 |
11.21 |
0.67 |
1.74 |
0.1 |
5.42 |
0.55 |
9.63 |
0.94 |
| 20 |
8.62 |
0.57 |
6.88 |
0.74 |
4.35 |
0.45 |
11.05 |
0.96 |
4.4 Thermodynamic analysis
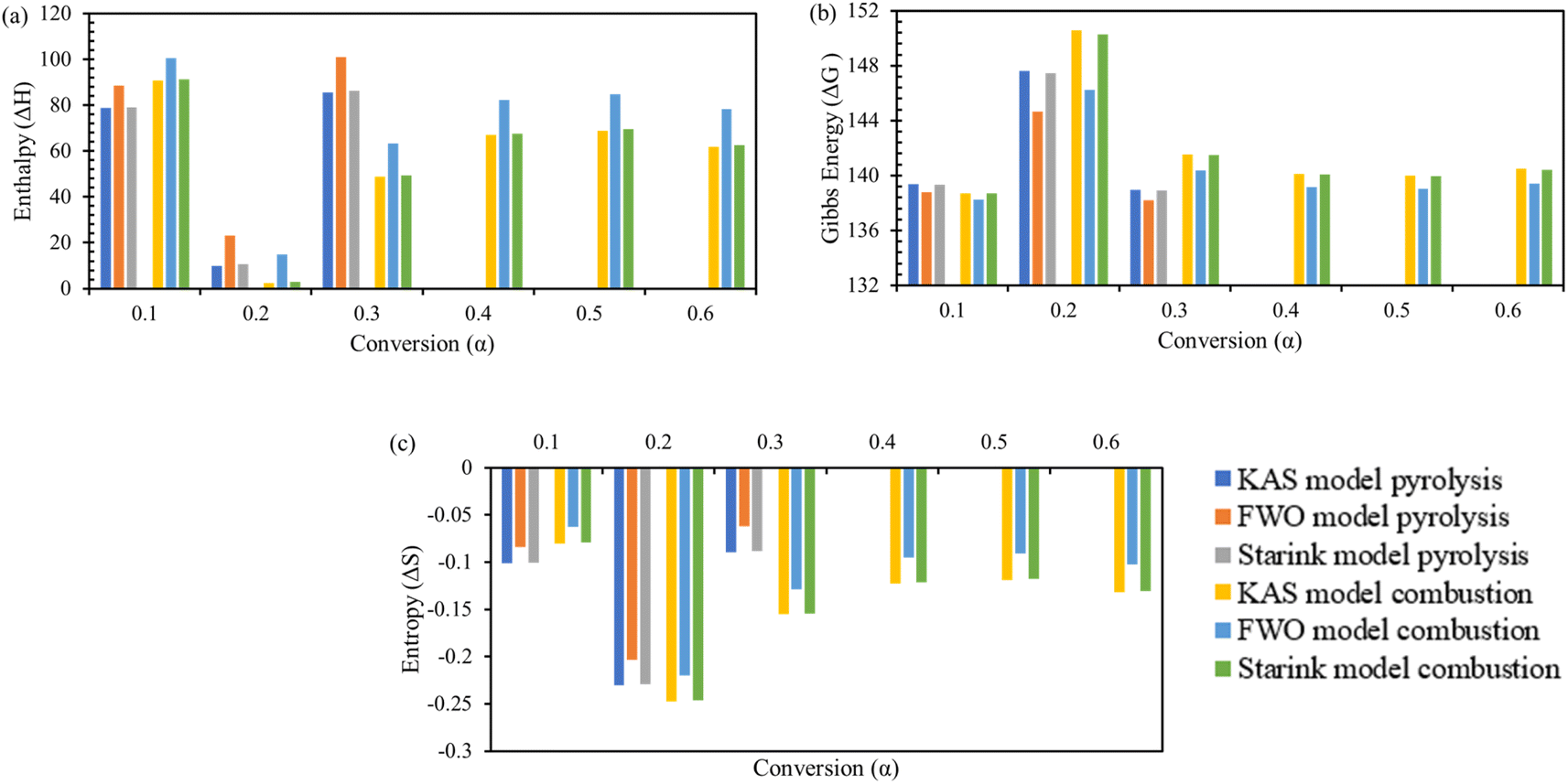
Change in enthalpy ΔH, change in Gibbs free energy ΔG, and change in entropy ΔS are key parameters to understand the thermodynamic profile of TBBA with or without its mixture with Ca(OH)2 under pyrolysis and combustion conditions by using iso-conversional model-free methods equations. Tables S3 and S4† (SM) shows the variation of ΔH, ΔG, and ΔS with each conversion point for pyrolysis and combustion, respectively, for all three KAS, FWO, and Starink methods. Change in enthalpy ΔH is a state function that indicates the endothermic or exothermic behavior of a reaction at fixed pressure.71 The highest value of ΔH for TBBA was 135.47, 148.41, 135.98 kJ mol−1 at 0.75 conversion in the pyrolysis process while it was 182.32, 191.63, and 182.69 kJ mol−1 in the combustion process for KAS, FWO, and Starink method, respectively. The highest value of ΔH for the TBBA:Ca(OH)2 blend in the nitrogen environment was calculated to be in the range 85.63–101.0.0 kJ mol−1 at a conversion of 0.30. Under oxidative conditions, corresponding ΔH values reside at 90.86, 100.67, and 91.25 kJ mol−1 for the KAS, FWO, and Starink methods, respectively. In pyrolysis of TBBA, the ΔH remained almost constant till 0.5 conversion, then decreased to 0.65 conversion, and then increased to 0.75 conversion and again decreased till the end. Values of ΔH in the combustion of TBBA fell until 0.5 conversion, then slightly increased at 0.55 conversion, then again declined until the end. In both pyrolysis and combustion of TBBA:Ca(OH)2 blends, the value of ΔH decreased till 0.2 conversion and then increased till the end. The decrease in the ΔH indicated that the reaction was changing to exothermic due to the formation of CaBr2 as discussed earlier in the DSC section. The positive values of ΔH resemble both thermal treatments were endothermic reactions.
The feasibility and favourability of the reaction were predicted by calculating the difference between the calculated ΔH and Ea. Less of a difference between Ea and ΔH indicates that the product will develop more preferentially.72 The values obtained for the pure and mixture were 4.76 and 6.09 kJ mol−1 for pyrolysis, whereas the higher difference of 5.75 and 6.53 kJ mol−1 was estimated for the combustion process, which implies more thermodynamic driving strength during pyrolysis treatment and hence was the preferred treatment method.
Changes in Gibbs free energy ΔG give information about the overall increase in energy during the breakage of reactant substitutes to convert them into the activated complex products.73 This brings a broad vision of distortedness, changes in the heat stream, and its high values signify the least favourability of reactions.74 In pyrolysis, the change in ΔG for TBBA is 134.50, 134.01, 134.44 kJ mol−1 and for TBBA:Ca(OH)2 is 141.73, 140.52, 141.68 kJ mol−1 for KAS, FWO, and Starink methods; respectively. In combustion, the values of ΔG are 133.60, 133.21, 133.58 kJ mol−1 for individual TBBA and are 141.77, 140.68, 141.69 kJ mol−1 for TBBA:Ca(OH)2 blends in all three iso-conversional models. Because of the occurrence of several chemical reactions such as dehydration of Ca(OH)2, C–Br scission, and recombination of HBr with the calcium to form CaBr2, and decarbonization of CaCO3, the values of blended TBBA with Ca(OH)2 are higher than the individual TBBA. In the pyrolysis of TBBA, the ΔG remained almost constant till 0.5 conversion, then increased to 0.65 conversion, and then decreased to 0.75 conversion, and again increased till the end. In the combustion of TBBA, ΔG raised till 0.5 conversion, then slightly reduced on 0.55 conversion, and again increased till the end. The value of ΔG increased until 0.2 conversion in both pyrolysis and combustion of TBBA:Ca(OH)2 blends and then decreased until the end. All values of ΔG were found to be positive, which signifies that the reactant expects more energy to initiate the reaction. According to the ΔG analysis, the reactions happening in TBBA and mix are non-spontaneous or endergonic reactions since products have greater energy than reactants.53 The change in entropy ΔS for TBBA and TBBA:Ca(OH)2 in Tables S1 and S2† showed negative values in both pyrolysis and combustion processes. This showed that the disorderness of the end products was much lower than that of the initial reactants.75 The ΔS negative values in all conversion represent that the products formed during the decomposition of TBBA have a more stable structure. As a result, ΔS values confirm that the formation of an activated complex is more organized in TBBA:Ca(OH)2 than in TBBA alone. The increase at some conversion points during the combustion of pure TBBA in the early stages is an indication of reaction spontaneity increasing with temperature.76 Fig. 6 and 7 show the ΔH, ΔG, and ΔS variations concerning each conversion for TBBA and TBBA:Ca(OH)2 during the pyrolysis and oxidation.
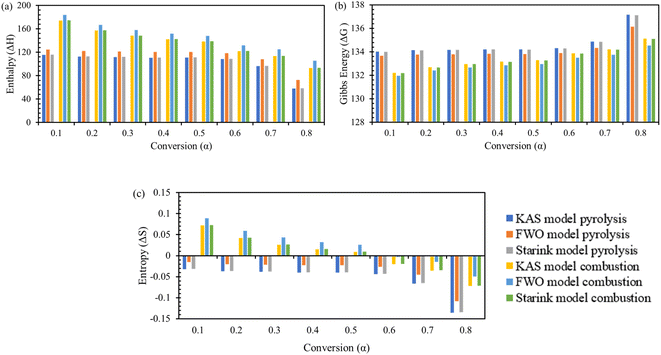 |
| | Fig. 6 Thermodynamic parameters of TBBA via KAS, FWO and Starink model; (a) enthalpy change, (b) Gibbs energy change, and (c) entropy change. | |
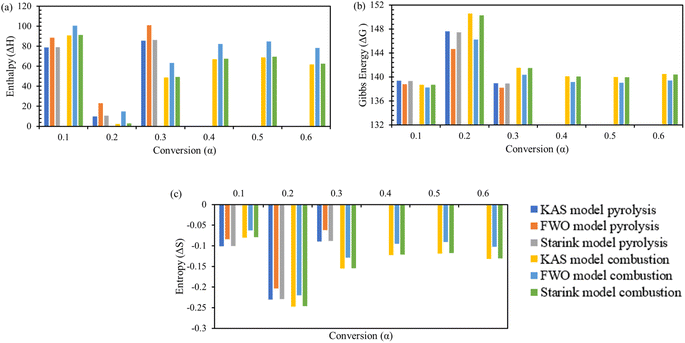 |
| | Fig. 7 Thermodynamic parameters of TBBA:Ca(OH)2 via KAS, FWO and Starink model; (a) enthalpy change, (b) Gibbs energy change, and (c) entropy change. | |
4.5 Synergistic effects
The difference in weight loss obtained by TGA experiments and weight loss calculated by mathematical method reveals the synergetic effect of blended sample TBBA:Ca(OH)2 during both pyrolysis and combustion environments. Table 4 shows the synergetic effect on different temperature ranges ranging from 25 °C to 800 °C and demonstrates that TBBA and Ca(OH)2 both have a strong interaction at a specific temperature range. It can be clearly observed that in both pyrolysis and combustion processes, 25–300 °C, showed a positive synergy, which is the main conversion zone according to TGA data at all heating rates of 5, 10, 15, and 20°C min−1. A higher temperature range (>300 °C) usually resulted in negative synergy for both processes at all heating rates. Positive synergy indicates that TBBA and Ca(OH)2 act as a single component during the early stages of conversion, but when the temperature rises, they act as separate components and do not react with each other. This indicates that TBBA and Ca(OH)2 can be converted at a lower temperature range, which can save energy and make the process more efficient. The TGA results and kinetic analyses show that at lower temperature ranges, loss of Br atoms take place in the form of HBr. This emitted HBr is captured by Ca(OH)2 as a solid–liquid bromination process, resulting in the formation of CaBr2. The negative trend of synergy with increasing temperature is primarily due to hydrogen bond breakage from Ca(OH)2 during the dehydration reaction, which then leads to the carbonisation reaction. The latter can be attributed to Ca(OH)2 reacting solely and separately.
Table 4 Syneristic effect of TBBA and TBBA:Ca(OH)2 during pyrolysis and combustion (25–800 °C)
| Blends |
Heating rate, (°C min−1) |
Temperature range (°C) |
Interactions |
Synergy effect |
| Pyrolysis |
| TBBA + Ca(OH)2 |
5 |
25–220 °C |
Overlapped |
Neutral |
| 221°C–300 °C |
Exp. lower than cal. |
Positive |
| 301–800 °C |
Exp. higher than cal. |
Negative |
| 10 |
25–290 °C |
Exp. lower than cal. |
Positive |
| 301–800 °C |
Exp. higher than cal. |
Negative |
| 15 |
25–300 °C |
Exp. lower than cal. |
Positive |
| 301–800 °C |
Exp. higher than cal. |
Negative |
| 20 |
25–300 °C |
Exp. lower than cal. |
Positive |
| 301–800 °C |
Exp. higher than cal. |
Negative |
|
| Combustion |
| TBBA + Ca(OH)2 |
5 |
25–280 °C |
Exp. lower than cal. |
Positive |
| 281–670 °C |
Exp. higher than cal. |
Negative |
| 671–800 °C |
Exp. lower than cal. |
Positive |
| 10 |
25–290 °C |
Exp. lower than cal. |
Positive |
| 291–800 °C |
Exp. higher than cal. |
Negative |
| 15 |
25–440 °C |
Exp. lower than cal. |
Positive |
| 441–750 °C |
Exp. higher than cal. |
Negative |
| 750–800 °C |
Exp. lower than cal. |
Positive |
| 20 |
25–300 °C |
Exp. lower than cal. |
Positive |
| 301–800 °C |
Exp. higher than cal. |
Negative |
4.6 A mechanistic insight
TBBA and its fragments react with Ca(OH)2 in the debromination process. The Mechanisms of HBr fixation by metal oxides is very-well understood as described in the Introduction section. For instance, we have reported recently a surface kinetic model for the selective halogenation of the zinc content in franklinite. Thus, HBr uptake over Ca(OH)2 proceeds via analogous pathways that are characterized by sequential surface-assisted dissociation of HBr followed by desorption of water molecules. Reaction of brominated aromatic molecules with metal oxides exclusively proceed via direct abstraction of the bromine atom by the M+ centers. The formed phenyl-like radicals are subsequently adsorbed by surface O2− sites. In addition to HBr and brominated aromatics, brominated olefins cuts originating from degradation of BFRs are expected to readily interact with the present metal oxides. As an illustrative example, Fig. 8 portrays potential energy surface for the production of 1-propyene from debromination of 1-bromo-1-propene. Direct elimination of HBr molecule over the Ca–O linkage demands only 25.2 kJ mol−1. Dissociative adsorption (fission of the C–Br bond) requires higher activation energy at 40.2 kJ mol−1. Formation of the 1-propyene is slightly endothermic by 15.3 kJ mol−1.
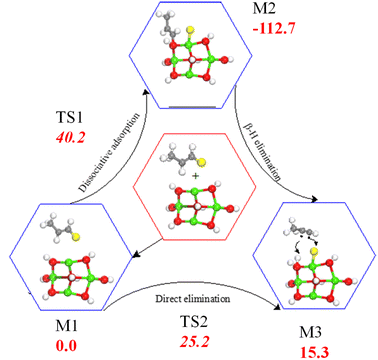 |
| | Fig. 8 Reaction of 1-bromo-1-propene with the Ca(OH)2 cluster. Values are in kcal mol−1 in reference to the physiosorbed 1-bromo-1-propene (M13). | |
5 Conclusion
The thermal degradation of TBBA was investigated using TGA at four different heating rates 5, 10, 15, and 20 °C min−1 under pyrolysis and combustion environments. TGA data for pure TBBA revealed a single-stage decomposition, whereas decomposition of TBBA:Ca(OH)2 involves multiple stages due to Ca(OH)2 dehydration and carbonization. The formation of HBr from pure TBBA was observed during the treatments. The DTG curves were plotted for all heating rates and processes to obtain the peak degradation occurring in each step, which was further utilized in the estimation of kinetic and thermodynamic parameters. In contrast to pyrolysis, combustion had no effect on TBBA and TBBA:Ca(OH)2 degradation. The reduction of brominated compounds was also authenticated by FTIR analysis of the post-pyrolysis products by the disappearance of the C–Br bond vibrations in the right shoulder below 600 cm−1. FTIR analysis identifies the prevailing functional groups in investigated solid solution. The three isoconversional methods (KAS, FWO, and Starink) were employed to compute the kinetic and thermodynamic parameters at different conversion rates. The R2 values computed for all the models in pyrolysis showed a good correlation (0.9–0.99). For pure TBBA, the pyrolysis showed lower Ea compared to the oxidation process. Similarly, all models indicated that the presence of Ca(OH)2 in TBBA aided in reducing values of activation energies. Compared to combustion, the pyrolysis of the mixture revealed the lowest Ea values out of all the treatments studied. The 16 models of the Coats and Redfern method were used further to validate the Ea values obtained by the iso conversional method. Obtained thermo-kinetic data recommend carrying out the co-pyrolysis process at a heating rate of 10 °C min. ΔG values were found to be positive for all the samples under both environments, which signifies that during the reaction, the reactant requires more energy to initiate the reaction. Computing parameters that underline synergetic effect provide insightful information in regard to the plausible behavior of the two components in the blended mixture to act independently or otherwise. It was discovered that the mixture behaved as a single material and participated equally during the reaction, which occurred at a low-temperature range of 25–300 °C.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study has been supported by the 2019 Abu Dhabi Award for Research Excellence (AARE) – (by ASPIRE, part of the Advanced Technology Research Council (ATRC, Abu Dhabi, United Arab Emirates), grant number: 21N225-AARE2019 – ADEK-103). DFT calculations were carried out at the HCP cluster of the United Arab Emirates University (UAEU).
References
- L. Zhao, Y. Lu, H. Zhu, Z. Cheng, Y. Wang, H. Chen, Y. Yao, J. Zhang, X. Li, Z. Sun, C. Zhang and H. Sun, Environ. Int., 2022, 165, 107299 CrossRef CAS PubMed.
- Q. Yuan, Y. Gu, M. Yang, Y. Wu, G. Hu and G. Zhou, J. Cleaner Prod., 2022, 380, 134855 CrossRef.
- X. Tian, J. Xie, L. Hu, H. Xiao and Y. Liu, Resour., Conserv. Recycl., 2022, 187, 106640 CrossRef.
- E. Sahle-Demessie, B. Mezgebe, J. Dietrich, Y. Shan, S. Harmon and C. C. Lee, J. Environ. Chem. Eng., 2021, 9, 104943 CrossRef CAS PubMed.
- S. Ilyas, R. Ranjan Srivastava, V. K. Singh, R. Chi and H. Kim, Waste Manage., 2022, 154, 175–186 CrossRef CAS PubMed.
- M. Altarawneh, J. Hazard. Mater., 2022, 422, 126879 CrossRef CAS PubMed.
- Y. Kojima, T. Fujimori, A. Goto, K. Shiota, T. Kunisue and M. Takaoka, J. Hazard. Mater., 2021, 403, 123878 CrossRef CAS PubMed.
- M. Altarawneh, A. Saeed, M. Al-Harahsheh and B. Z. Dlugogorski, Prog. Energy Combust. Sci., 2019, 70, 212–259 CrossRef.
- M. Altarawneh, O. H. Ahmed, Z. T. Jiang and B. Z. Dlugogorski, J. Phys. Chem. A, 2016, 120, 6039–6047 CrossRef CAS PubMed.
- O. Terakado, R. Ohhashi and M. Hirasawa, J. Anal. Appl. Pyrolysis, 2013, 103, 216–221 CrossRef CAS.
- T. Rieger, J. C. Oey, V. Palchyk, A. Hofmann, M. Franke and A. Hornung, Process, 2021, 9, 530 CrossRef CAS.
- C. Ma, J. Yu, T. Chen, Q. Yan, Z. Song, B. Wang and L. Sun, Fuel, 2018, 230, 390–396 CrossRef CAS.
- M. Al-Harahsheh, J. Al-Nu’Airat, A. Al-Otoom, I. Al-Hammouri, H. Al-Jabali, M. Al-Zoubi and S. Abu Al'Asal, J. Environ. Chem. Eng., 2019, 7, 102856 CrossRef CAS.
- H. Wu, Y. Shen, N. Harada, Q. An and K. Yoshikawa, Energy Environ. Res., 2014, 4, p105 Search PubMed.
- N. A. Mousa, L. Ali, M. S. Kuttiyathil, H. A. Mousa and M. Altarawneh, Chem. Eng. J. Adv., 2022, 11, 100334 CrossRef CAS.
- L. Ali, M. S. Kuttiyathil and M. Altarawneh, Waste Manage., 2022, 154, 283–292 CrossRef CAS PubMed.
- J. Liu, H. Wang, W. Zhang, T. Wang, M. Mei, S. Chen and J. Li, J. Hazard. Mater., 2022, 431, 128612 CrossRef CAS PubMed.
- M. Altarawneh, O. H. Ahmed, M. Al-Harahsheh, Z. T. Jiang and B. Z. Dlugogorski, Appl. Surf. Sci., 2021, 562, 150105 CrossRef CAS.
- O. H. Ahmed, M. Altarawneh, M. Al-Harahsheh, Z.-T. Jiang and B. Z. Dlugogorski, Phys. Chem. Chem. Phys., 2018, 20, 1221 RSC.
- S. Oleszek, M. Grabda, E. Shibata and T. Nakamura, Thermochim. Acta, 2013, 566, 218–225 CrossRef CAS.
- M. Al-Harahsheh, M. Altarawneh, M. Aljarrah, F. Rummanah and K. Abdel-Latif, J. Anal. Appl. Pyrolysis, 2018, 134, 503–509 CrossRef CAS.
- R. Gao, B. Liu, L. Zhan, J. Guo, J. Zhang and Z. Xu, J. Hazard. Mater., 2020, 392, 122447 CrossRef CAS PubMed.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2015, 19, 282–293 CrossRef.
- L. Ali, M. S. Kuttiyathil, O. H. Ahmed and M. Altarawneh, Sep. Purif. Technol., 2023, 307, 122836 CrossRef CAS.
- I. Ali, H. Bahaitham and R. Naibulharam, Bioresour. Technol., 2017, 235, 1–11 CrossRef CAS PubMed.
- I. Ali, R. Tariq, S. R. Naqvi, A. H. Khoja, M. T. Mehran, M. Naqvi and N. Gao, J. Energy Inst., 2021, 95, 30–40 CrossRef CAS.
- L. Ali, M. S. Kuttiyathil, M. Al-Harahsheh and M. Altarawneh, Arabian J. Chem., 2023, 104540 CrossRef CAS.
- H. E. Kissinger, J. Res. Natl. Bur. Stand., 1956, 57, 217–221 CrossRef CAS.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- M. J. Starink, Thermochim. Acta, 2003, 404, 163–176 CrossRef CAS.
- R. Tariq, Y. M. Zaifullizan, A. A. Salema, A. Abdulatif and L. S. Ken, Renewable Energy, 2022, 198, 399–414 CrossRef CAS.
- X. Xu, R. Pan and R. Chen, Appl. Biochem. Biotechnol., 2021, 193, 1427–1446 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1998, 92, 508 CrossRef.
- M. Al-Harahsheh, M. Aljarrah, A. Al-Otoom, M. Altarawneh and S. Kingman, Thermochim. Acta, 2018, 660, 61–69 CrossRef CAS.
- M. Grabda, S. Oleszek-Kudlak, M. Rzyman, E. Shibata and T. Nakamura, Environ. Sci. Technol., 2009, 43, 1205–1210 CrossRef CAS PubMed.
- A. Factor, J. Polym. Sci., Part A-1: Polym. Chem., 1973, 11, 1691–1701 CAS.
- S. Kumagai, G. Grause, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2017, 19, 282–293 CrossRef CAS.
- J. Chem, M. Szwarc, B. N. Ghosh, A. H. Sehon, E. G. Williams and C. N. Hinshelwood, Proc R Soc Lond A Math Phys Sci., 1951, 209, 110–131 Search PubMed.
- M. Altarawneh and B. Z. Dlugogorski, J. Phys. Chem. A, 2014, 118, 9338–9346 CrossRef CAS PubMed.
- M. Al-Harahsheh, A. Al-Otoom, M. Al-Jarrah, M. Altarawneh and S. Kingman, Metall. Mater. Trans. B, 2018, 49, 45–60 CrossRef CAS.
- M. Al-Harahsheh, M. Aljarrah, F. Rummanah, K. Abdel-Latif and S. Kingman, J. Anal. Appl. Pyrolysis, 2017, 125, 50–60 CrossRef CAS.
- F. Barontini, K. Marsanich, L. Petarca and V. Cozzani, Ind. Eng. Chem. Res., 2004, 43, 1952–1961 CrossRef CAS.
- Z. Mirghiasi, F. Bakhtiari, E. Darezereshki and E. Esmaeilzadeh, J. Ind. Eng. Chem., 2014, 20, 113–117 CrossRef CAS.
- B. Acharya, A. Dutta and P. Basu, CFB Technol., 2011, 2011, 1–8 Search PubMed.
- T. Kim and J. Olek, Transp. Res. Rec., 2012, 10–18 CrossRef CAS.
- B. El-Jazairi and J. M. Illston, Cem. Concr. Res., 1977, 7, 247–257 CrossRef CAS.
- F. Barontini, K. Marsanich, L. Petarca and V. Cozzani, Ind. Eng. Chem. Res., 2004, 43, 1952–1961 CrossRef CAS.
- M. Al-Harahsheh, M. Aljarrah, A. Al-Otoom, M. Altarawneh and S. Kingman, Thermochim. Acta, 2018, 660, 61–69 CrossRef CAS.
- M. Rzyman, M. Grabda, S. Oleszek-Kudlak, E. Shibata and T. Nakamura, J. Anal. Appl. Pyrolysis, 2010, 88, 14–21 CrossRef CAS.
- M. Grabda, S. Oleszek-Kudlak, M. Rzyman, E. Shibata and T. Nakamura, Environ. Sci. Technol., 2009, 43, 1205–1210 CrossRef CAS PubMed.
- L. Ali, M. S. Kuttiyathil and M. Altarawneh, J. Environ. Chem. Eng., 2022, 10, 106978 CrossRef CAS.
- M. Klaumünzer, J. Hübner, D. Spitzer and C. Kryschi, ACS Omega, 2017, 2, 52–61 CrossRef PubMed.
- B. Smith, Infrared Spectral Interpretation, CRC Press, 2018 Search PubMed.
- L. Ali, H. A.Mousa, M. Al-Harahsheh, S. Al-Zuhair, B. Abu-Jdayil, M. Al-Marzouqi and M. Altarawneh, Waste Manage., 2022, 137, 283–293 CrossRef CAS PubMed.
- G. Li, W. Zhu, L. Zhu and X. Chai, Korean J. Chem. Eng., 2016, 33, 2215–2221 CrossRef CAS.
- Z. Zeng, S. Da Zhang, T. Q. Li, F. L. Zhao, Z. L. He, H. P. Zhao, X. E. Yang, H. L. Wang, J. Zhao and M. T. Rafiq, J. Zhejiang Univ., Sci., B, 2013, 14, 1152 CrossRef CAS PubMed.
- D. Pokorná, P. Cuřínová and J. Pola, Polycyclic Aromat. Compd., 2021, 41, 841–850 CrossRef.
- G. Bin Cai, S. F. Chen, L. Liu, J. Jiang, H. Bin Yao, A. W. Xu and S. H. Yu, CrystEngComm, 2010, 12, 234–241 RSC.
- M. Galvan-Ruiz, L. Baños and M. E. Rodriguez-Garcia, Sens Instrum Food Qual Saf., 2007, 1, 169–175 CrossRef.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- M. Tsuge, S. Berski, M. Räsänen, Z. Latajka and L. Khriachtchev, J. Chem. Phys., 2013, 138(10), 104314 CrossRef PubMed.
- Q. Song, H. Zhao, Q. Ma, L. Yang, L. Ma, Y. Wu and P. Zhang, Fuel, 2022, 313, 122667 CrossRef CAS.
- A. Meng, H. Zhou, L. Qin, Y. Zhang and Q. Li, J. Anal. Appl. Pyrolysis, 2013, 104, 28–37 CrossRef CAS.
- N. E. Cooke, O. M. Fuller and R. P. Gaikwad, Fuel, 1986, 65, 1254–1260 CrossRef CAS.
- X. Mu, Y. Wang, J. Huang, L. Lan, H. Wang, W. Xu and X. Li, Chemosphere, 2023, 138045 CrossRef CAS PubMed.
- R. K. Singh, D. Pandey, T. Patil and A. N. Sawarkar, Bioresour. Technol., 2020, 310, 123464 CrossRef CAS PubMed.
- Y. Li, D. Han, Y. Arai, X. Fu, X. Li and W. Huang, Chem. Eng. J., 2019, 373, 95–103 CrossRef CAS.
- A. Marongiu, G. Bozzano, M. Dente, E. Ranzi and T. Faravelli, J. Anal. Appl. Pyrolysis, 2007, 80, 325–345 CrossRef CAS.
- S. R. Naqvi, R. Tariq, Z. Hameed, I. Ali, M. Naqvi, W. H. Chen, S. Ceylan, H. Rashid, J. Ahmad, S. A. Taqvi and M. Shahbaz, Renewable Energy, 2019, 131, 854–860 CrossRef CAS.
- E. Müsellim, M. H. Tahir, M. S. Ahmad and S. Ceylan, Appl. Therm. Eng., 2018, 137, 54–61 CrossRef.
- A. A. Shagali, S. Hu, H. Li, H. Chi, H. Qing, J. Xu, L. Jiang, Y. Wang, S. Su and J. Xiang, Fuel, 2023, 331, 125724 CrossRef CAS.
- S. R. Naqvi, Z. Hameed, R. Tariq, S. A. Taqvi, I. Ali, M. B. K. Niazi, T. Noor, A. Hussain, N. Iqbal and M. Shahbaz, Waste Manage., 2019, 85, 131–140 CrossRef CAS PubMed.
- X. Yuan, T. He, H. Cao and Q. Yuan, Renewable Energy, 2017, 107, 489–496 CrossRef CAS.
- V. Dhyani, J. Kumar and T. Bhaskar, Bioresour. Technol., 2017, 245, 1122–1129 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kaushik Sivaramakrishnan
a,
Kaushik Sivaramakrishnan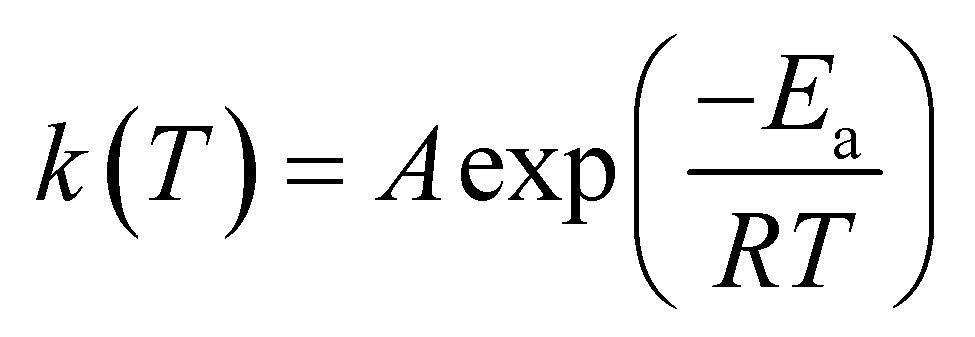





 . To find the analytical solution of the rate equations, distinct different models are utilized.
. To find the analytical solution of the rate equations, distinct different models are utilized.


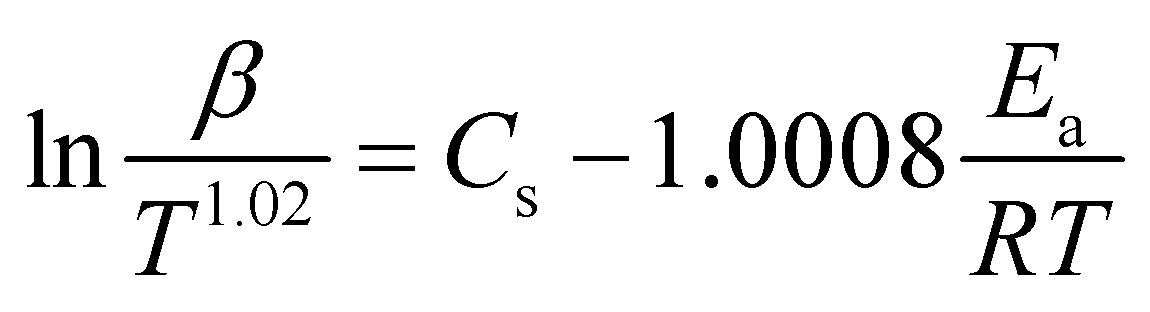
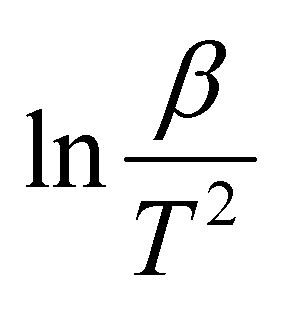 , ln
, ln vs. 1/T gives a linear line with the slope
vs. 1/T gives a linear line with the slope 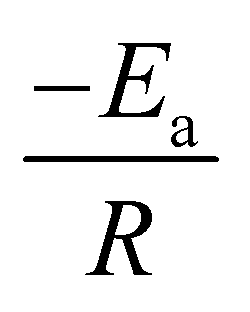 ,
, 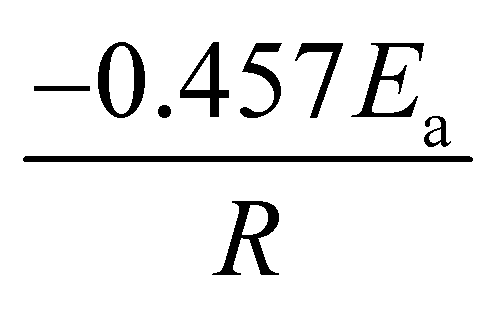 , and
, and  for KAS, FWO, and Starink methods respectively, from which the calculations of activation energies are executed. Intercepts of the straight lines signify the preexponential factors.
for KAS, FWO, and Starink methods respectively, from which the calculations of activation energies are executed. Intercepts of the straight lines signify the preexponential factors.
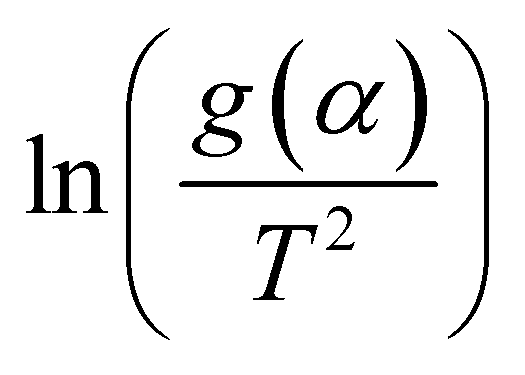 and 1/T give a straight line in Coats and Redfern method, and the slope and intercept of this line determine the kinetic parameters based on the different reaction mechanisms. The g(α) function varies according to different reaction mechanisms. The g(α) for different mechanism models is listed in Table 1.31,32
and 1/T give a straight line in Coats and Redfern method, and the slope and intercept of this line determine the kinetic parameters based on the different reaction mechanisms. The g(α) function varies according to different reaction mechanisms. The g(α) for different mechanism models is listed in Table 1.31,32