
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect generation of polypyrrole-coated palladium nanoparticles inside a metal-organic framework for a semihydrogenation catalyst†
Yohei Takashima *,
Seiko Tetsusashi,
Shintaro Tanaka,
Takaaki Tsuruoka and
Kensuke Akamatsu*
*,
Seiko Tetsusashi,
Shintaro Tanaka,
Takaaki Tsuruoka and
Kensuke Akamatsu*
Department of Nanobiochemistry, Frontiers of Innovative Research in Science and Technolgoy (FIRST), Konan University, 7-1-20 Minatojimaminamimachi, Chuo-ku, Kobe 650-0047, Japan. E-mail: takashim@konan-u.ac.jp; akamatsu@konan-u.ac.jp
First published on 8th March 2023
Abstract
Herein, the direct synthesis of polypyrrole (PPy)-coated palladium nanoparticles (PdNPs) inside a metal–organic framework (MIL-101) was successfully demonstrated. Owing to the PPy coating of PdNPs, the resulting composites exhibited higher semihydrogenation capability (selectivity: up to 96%) than the analog composite without PPy coating.
Introduction
Metal–organic frameworks (MOFs) are porous materials generated by metal ions and organic linkers via coordination bonding.1–4 By tuning their pore structures, MOFs can accommodate not only small gas molecules but also larger guest entities with various functionalities.5–13 Among them, metal nanoparticles (NPs) have garnered significant attention as functional guests. For instance, Au or Ag NPs can be immobilized inside MOF pores as guests for plasmonic applications, and other metal NPs, such as Pd and Ru, have been used to introduce catalytic activity.14–17 Especially, the MOF composites with PdNP guests (PdNPs@MOFs) have been vigorously studied as catalysts for various reactions including semihydrogenation. For examples, Jiang et al. has reported unique PdNPs@MOFs with high catalytic activities by surface hydrophobization and pore wall engineering etc.18–22The semihydrogenation reaction in which alkynes are selectively converted to the corresponding alkenes has been a crucially important reaction in chemical industry not only for bulk chemicals, such as C2–C4 olefins but also for fine chemicals.23,24 However, it is difficult to use simple PdNPs@MOFs as catalysts for semihydrogenation reactions because the resulting bare PdNPs in MOFs strongly induce overhydrogenation. Therefore, some modifications have been applied for PdNPs@MOFs until now. For instance, Wu et al. and Jiang et al. incorporated alloyed PdNPs in MOF pores to tune their catalytic activities.25,26 Pd@Ag NPs in an MOF, where the Pd core was covered by a Ag shell, could also exhibit high selectivity.27 Luo et al. succeeded in cogenerating PdNPs and iron oxides on MOF crystals, where iron oxides function as support for PdNPs to enhance catalytic activity and selectivity.28
Herein, we synthesized polypyrrole (PPy)-coated PdNPs inside an MOF for use as a semihydrogenation catalyst (Fig. 1). In this system, PPy is expected to coordinate to PdNPs in the MOF pores to control its catalytic activity. Controlling the catalytic properties of metal NPs in MOFs using organic molecules has seldom been reported thus far, but we believe that this method would be promising because organic molecules can be easily designed with a synthetic strategy to finely tune the catalytic properties of metal NPs.29,30
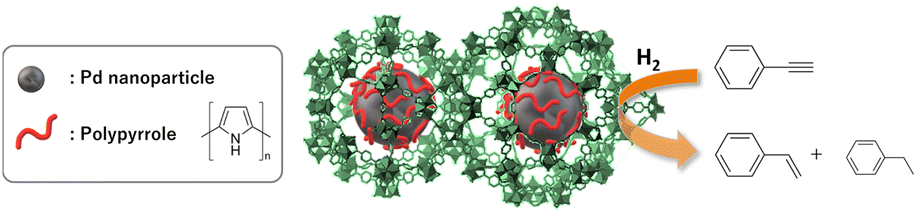 | ||
| Fig. 1 Schematic illustration of this research. | ||
Experimental
General methods
All reagents unless otherwise stated were obtained from commercial sources and were used without further purification. X-ray powder diffraction data were collected on a Rigaku RINT-2200 Right System (Ultima IV) diffractometer with CuKα radiation. Thermogravimetric analyses were recorded on a Rigaku Thermo plus TG-8120 apparatus in the temperature range between 298 and 773 K at a heating rate of 5 K min−1. The TEM observations were performed with a JEOL JEM-1400 transmission electron microscopy (TEM) system operating at 120 kV. ICP analyses were conducted by ICP AES (SPS 7800, Seiko Instruments). 1H NMR spectra were recorded on a JEOL JNM-A500 spectrometer. The 1HNMR chemical shifts are referenced to the residual internal CDCl3. FT-IR spectra were obtained using a Spectrum Two spectrometer (PerkinElmer) using ATR technique. Sorption isotherm measurements were recorded using an automatic volumetric adsorption apparatus (autosorb iQ, Quantachrome). Samples were evacuated under high vacuum (<10−2 Pa) at 393 K for 2 h to remove guest molecules. XPS spectra were collected by X-ray photoelectron spectrometer (JEOL JPS-9010MC) with Mg Kα radiation. MOF powders were placed on carbon substrate, and Pd 3d peaks were observed.Synthesis of MIL-101
Chromium(III) nitrate (4.00 g, 10.0 mmol), benzene-1,4-dicarboxylic acid (1.64 g, 10.0 mmol) and HNO3 (10.0 mmol) were dispersed in water (50 ml). The mixture was transferred to Teflon-lined stainless steel autoclave. The resulting suspension was heated at 220 °C for 8 h under hydrothermal condition. The reaction product was finally obtained after washing three times with DMF under sonication.Pd(OAc)2 accommodation in MIL-101
To 5 ml of Pd(OAc)2 acetone solutions (12 mM, 24 mM and 36 mM), was added 25 mg of MIL-101 and the resulting suspensions were stirred at 26 °C for 15 h. After filtration, washing with acetone under sonication and drying in vacuo, powder samples (Pd(OAc)2@MIL-101(x) x = 12, 24, 36) were obtained.Synthesis of PdNPs/PPy@MIL-101
To 10 mg of Pd(OAc)2@MIL-101(x) (x = 12, 24, 36) was added 1 ml of pyrrole and the resulting suspensions were stirred at 26 °C for 5 min. After filtration, washing with acetone under sonication and drying in vacuo, powder samples (PdNPs/PPy@MIL-101(x) x = 12, 24, 36) were obtained.Hydrogenation reaction of phenylacetylene
To PdNPs/PPy@MIL-101 (Pd: 0.05 mol%), added phenylacetylene (2.7 mmol) and heated at 60 °C under H2 (5 atm). Phenylacetylene conversion and styrene selectivity of each step were determined as 1HNMR yields using the integrals of both reactant and products.Results and discussions
As an MOF, we have selected Cr-based MIL-101 that has two large cage-like pores (diameters: 2.9 and 3.4 nm) to accommodate PdNPs (Fig. 2A and B).31,32 PPy-coated PdNPs were generated inside the MOF pores using Pd(OAc)2 as PdNP precursor and pyrrole (Py) as reductant; Py exhibits oxidative polymerization during the reduction of Pd(OAc)2 to generate PdNPs (Fig. 2C).33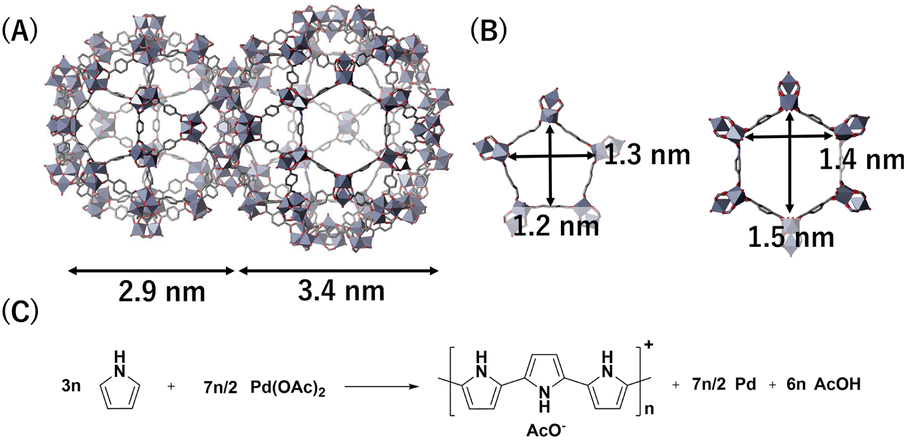 | ||
| Fig. 2 Structures of (A) MIL-101 pores and (B) pore apertures. (C) Reaction scheme to generate PPy-coated PdNPs. | ||
The accommodation of Pd(OAc)2 inside the MOF pores was achieved by mixing the MOF with Pd(OAc)2 acetone solution at 26 °C. The amount of Pd(OAc)2 inside the MOF was determined using inductively coupled plasma – atomic emission spectrometry (ICP-AES) after digesting the resulting composite with strong acids. The ICP-AES results of Pd/Cr (atom/atom) clearly showed that the amount of Pd(OAc)2 could be reasonably controlled by changing the concentration of Pd(OAc)2 acetone solution (Fig. 3A).34 From powder X-ray diffraction analysis (PXRD) and transmission electron microscope (TEM) observations, we have also confirmed that MOF structures were maintained, and PdNPs were not produced by the Pd(OAc)2 accommodation (Fig. 3B and S1†).
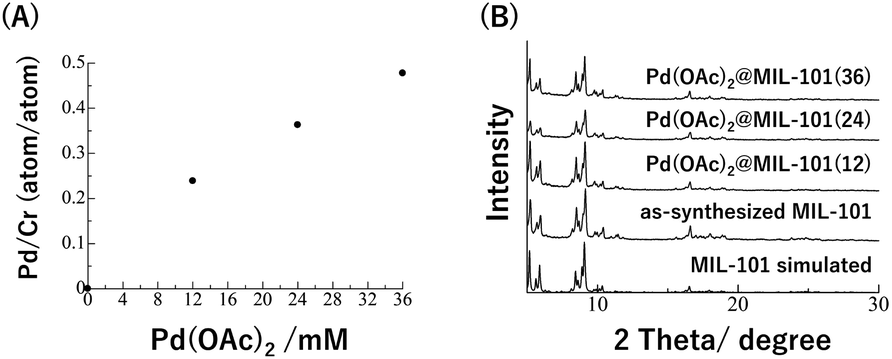 | ||
| Fig. 3 (A) Pd/Cr ratios and (B) XRD patterns of Pd(OAc)2@MIL-101. | ||
The resulting MOF composite (Pd(OAc)2@MIL-101(x): x denotes the concentration (mM) of the Pd(OAc)2 acetone solution used for Pd(OAc)2 accommodation) was reduced by neat Py at 26 °C. From the TEM observations, in all cases, PdNPs were homogeneously generated in the MOFs, though a few PdNPs were observed on the MOFs (Fig. 4A–C). Additionally, their particle sizes were similar, indicating that the nucleation for PdNP generation was sufficiently fast owing to the use of neat Py (Fig. 4D–F). From the results of the PXRD and thermogravimetric analysis (TG), we confirmed that the MOF structures were maintained with high thermal stability (Fig. S2†). The presence of PPy was confirmed via ultraviolet-visible diffuse reflectance spectroscopy; broad and featureless absorption corresponding to PPy was observed only in the case of the samples after reduction with Py (Fig. S3†). The peaks corresponding to PPy could not be observed in the IR measurements, presumably because the amount was extremely small to detect (Fig. S4†).
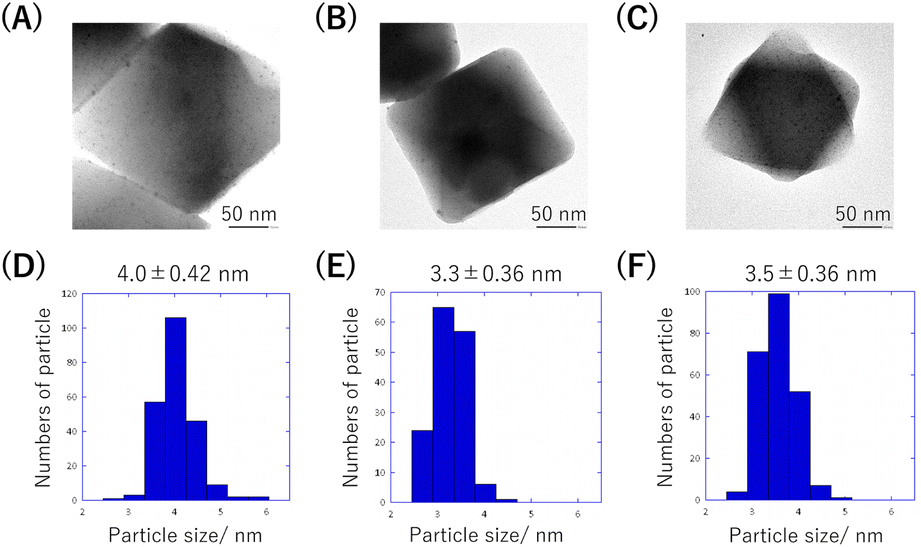 | ||
| Fig. 4 TEM images of (A) PdNPs/PPy@MIL-101(12), (B) PdNPs/PPy@MIL-101(24), and (C) PdNPs/PPy@MIL-101(36). PdNP size distribution histograms of (D) PdNPs/PPy@MIL-101(12), (E) PdNPs/PPy@MIL-101(24), and (F) PdNPs/PPy@MIL-101(36). | ||
The obtained MOF composites (PdNPs/PPy@MIL-101(x): x denotes the concentration (mM) of the Pd(OAc)2 acetone solution used for Pd(OAc)2 accommodation) were used as catalysts for the hydrogenation of phenylacetylene, where styrene was obtained via semihydrogenation. To determine the effects of PPy on the semihydrogenation selectivity, an analog MOF composite without PPy (PdNPs@MIL-101(24)) was also generated via H2 reduction (Fig. S5†).
Fig. 5A and B show the results of the time-course experiments of the hydrogenation reaction (reaction conditions: [Pd] 0.05 mol%, 5 atm H2, 60 °C) in the presence of PdNPs@MIL-101(24) or PdNPs/PPy@MIL-101(24) as the catalyst. In the case of PdNPs@MIL-101(24), the semihydrogenation selectivity was relatively low (81%) from the beginning of the reaction and thereafter significantly decreased to 0% because of the further hydrogenation of styrene to generate ethylbenzene, indicating that PdNPs in the composite are excessively active for semihydrogenation. Conversely, in the case of PdNPs/PPy@MIL-101(24), the semihydrogenation selectivity was maintained at 96% when the conversion of phenylacetylene was 79%, indicating that PPy could suppress the catalytic activity of PdNP to enhance its selectivity. Note that bulk PdNPs–PPy composite without MIL-101 showed very low catalytic activity because of large aggregation to hamper the access of reactants (Fig. S7†).
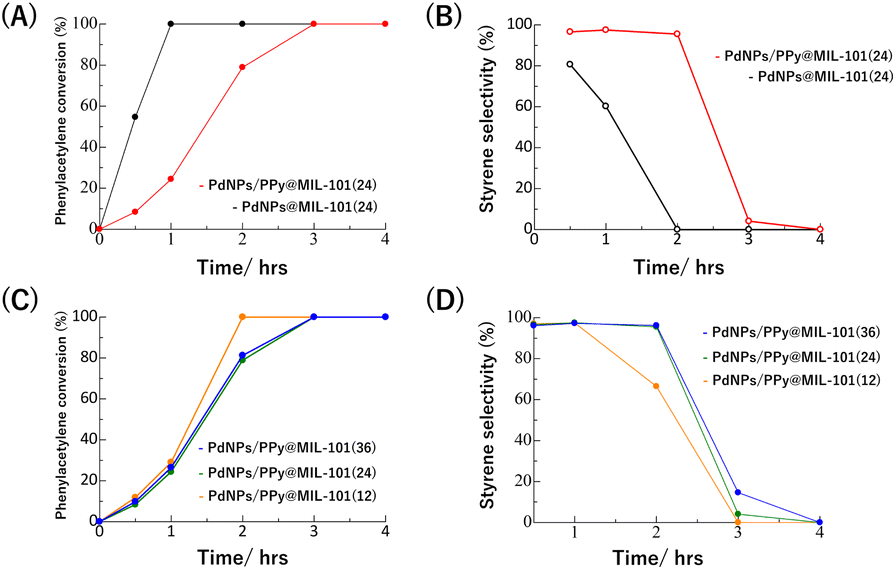 | ||
| Fig. 5 Time course analyses of (A) phenylacetylene conversion and (B) styrene selectivity with PdNPs@MIL-101(24) or PdNPs/PPy@MIL-101(24). Time course analyses of (C) phenylacetylene conversion and (D) styrene selectivity with PdNPs/PPy@MIL-101(x) (x = 12, 24, and 36). | ||
Fig. 5C and D show the results of the similar experiments using PdNPs/PPy@MIL-101(12) and PdNPs/PPy@MIL-101(36) (reaction conditions: [Pd] 0.05 mol%, 5 atm H2, 60 °C), respectively, where different amounts of PdNPs were accommodated in the MOF pores. In the case of PdNPs/PPy@MIL-101(12), the reaction became faster, but the selectivity decreased, indicating that PdNPs/PPy@MIL-101(12) was catalytically more active than PdNPs/PPy@MIL-101(24). Conversely, the catalytic activity and selectivity of PdNPs/PPy@MIL-101(36) were similar to those of PdNPs/PPy@MIL-101(24), which demonstrates that the high PdNP loading in the MOF pores could suppress its catalytic activity and enhance its selectivity. Although the reason for this observation remains unclear, the low mobility or reactivity of phenylacetylene because of the congestion of PdNPs in MOF pores, may be one possible explanation.
Finally, to further understand the improvement of the selectivity of semihydrogenation by PPy, the electronic states of PdNPs in PdNPs@MIL-101(24) and PdNPs/PPy@MIL-101(24) were examined via X-ray photoelectron spectroscopy (XPS) (Fig. 6A). For PdNPs@MIL-101(24), the oxidation state of Pd was determined as zero from the binding energies of the peaks corresponding to Pd 3d3/2 and Pd 3d5/2. In contrast, two Pd catalysts with different oxidation states were observed for PdNPs/PPy@MIL-101(24): one was in the zero oxidation state, and the other was in the oxidation state between 0 and 2 (Fig. 6B). Although the oxidized PdNPs could originate from PdO generated through the oxidation of PdNPs or surface Pd atoms on PdNPs coordinated by the N atom in PPy, we have finally determined the origin to be the latter because PdO was not observed for PdNPs@MIL-101(24), and the previous study on the bulk synthesis of the PdNPs–PPy composite also revealed the same species via XPS.35 The possibility that the surface Pd atom coordinated by PPy has higher semihydrogenation selectivity could be explained by considering a previous study on the theoretical calculation in which a positive Pd surface could more preferentially induce desorption than the hydrogenation of styrene.36
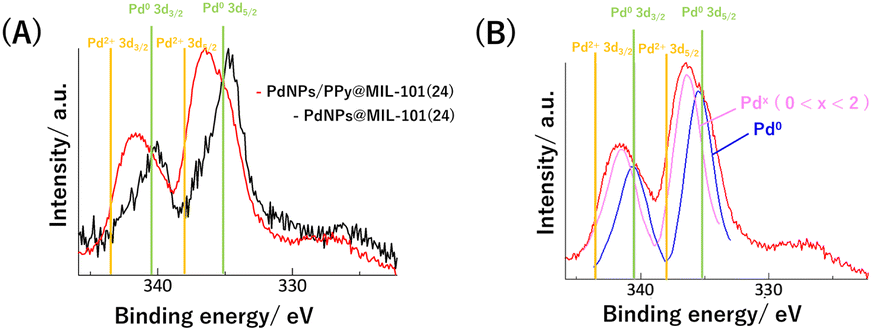 | ||
| Fig. 6 (A) Pd 3d peaks in XPS spectra of PdNPs@MIL-101(24) and PdNPs/PPy@MIL-101(24). (B) Fitted curves to Pd 3d peaks in XPS spectrum of PdNPs/PPy@MIL-101(24). | ||
Conclusions
Herein, we have successfully demonstrated the one-pot synthesis of a PdNP–PPy composite inside MOF pores, where Py acts as both a reductant for PdNP generation and a PPy monomer. Owing to the cogeneration of PdNP and PPy inside the MOF pores, they could be appropriately hybridized for application as a catalyst; the stepwise generation of PdNP and PPy could afford inhomogeneous PdNP–PPy composites inside the MOF pores. The resulting PdNPs/PPy@MIL-101 exhibited higher selectivity for semihydrogenation than PdNPs@MIL-101 because of the positive Pd surface generated by coordination with PPy. Significantly, we also confirmed that the spatial density of PdNP–PPy in the MOF affected the selectivity for the semihydrogenation reaction. We are currently attempting to synthesize new PdNP-based MOF composites with functionalized Py or thiophene to demonstrate the versatility and applicability of this methodology for various catalytic systems.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was carried out with support from JSPS KAKENHI Grant Number 19K05661.Notes and references
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- T. Uemura, N. Yanai and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228–1236 RSC.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- R. W. Larsen, L. Wojtas, J. Perman, R. L. Musselman, M. J. Zaworotko and C. M. Vetromile, J. Am. Chem. Soc., 2011, 133, 10356–10359 CrossRef CAS PubMed.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- A. Grigoropoulos, G. F. S. Whitehead, N. Perret, A. P. Katsoulidis, F. M. Chadwick, R. P. Davies, A. Haynes, L. Brammer, A. S. Weller, J. Xiao and M. J. Rosseinsky, Chem. Sci., 2016, 7, 2037–2050 RSC.
- H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351–3370 RSC.
- C. Rosler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- H. Kobayashi, Y. Mitsuka and H. Kitagawa, Inorg. Chem., 2016, 55, 7301–7310 CrossRef CAS PubMed.
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS PubMed.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed.
- D. Chen, W. Yang, L. Jiao, L. Li, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2020, 32, 2000041 CrossRef CAS PubMed.
- L. Li, Z. Li, W. Yang, Y. Huang, G. Huang, Q. Guan, Y. Dong, J. Lu, S.-H. Yu and H.-L. Jiang, Chem, 2021, 7, 686–698 CAS.
- M.-L. Gao, L. Li, Z.-X. Sun, J.-R. Li and H.-L. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202211216 CAS.
- Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- M.-J. Dong, X. Wang and C.-D. Wu, Adv. Funct. Mater., 2020, 30, 1908519 CrossRef CAS.
- L. Li, W. Yang, Q. Yang, Q. Guan, J. Lu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2020, 10, 7753–7762 CrossRef CAS.
- L. Chen, B. Huang, X. Qiu, X. Wang, R. Luque and Y. Li, Chem. Sci., 2015, 7, 228–233 RSC.
- Y. Tao, H. Q. Wu, J. Q. Li, L. X. Yang, W. H. Yin, M. B. Luo and F. Luo, Dalton Trans., 2018, 47, 14889–14892 RSC.
- L. Peng, S. Yang, S. Jawahery, S. M. Moosavi, A. J. Huckaba, M. Asgari, E. Oveisi, M. K. Nazeeruddin, B. Smit and W. L. Queen, J. Am. Chem. Soc., 2019, 141, 12397–12405 CrossRef CAS PubMed.
- V. V. Karve, D. T. Sun, O. Trukhina, S. Yang, E. Oveisi, J. Luterbacher and W. L. Queen, Green Chem., 2020, 22, 368–378 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- T. Zhao, F. Jeremias, I. Boldog, B. Nguyen, S. K. Henninger and C. Janiak, Dalton Trans., 2015, 44, 16791–16801 RSC.
- S. Bahraeian, K. Abron, F. Pourjafarian and R. A. Majid, Adv. Mater. Res., 2013, 795, 707–710 Search PubMed.
- Y. Takashima, Y. Sato, N. Kubo, T. Tsuruoka and K. Akamatsu, Chem. Lett., 2021, 50, 244–247 CrossRef CAS.
- S. V. Vasilyeva, M. A. Vorotyntsev, I. Bezverkhyy, E. Lesniewska, O. Heintz and R. Chassagnon, J. Phys. Chem. C, 2008, 112, 19878–19885 CrossRef CAS.
- K. Choe, F. Zheng, H. Wang, Y. Yuan, W. Zhao, G. Xue, X. Qiu, M. Ri, X. Shi, Y. Wang, G. Li and Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 3650–3657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra08190c |
| This journal is © The Royal Society of Chemistry 2023 |