DOI:
10.1039/D2RA08118K
(Paper)
RSC Adv., 2023,
13, 4910-4916
A panoramic view on synthetic applications of α-oxothioamides: a highly regioselective synthesis of 2-acyl-4-(het)arylthiazoles and thioethers†
Received
20th December 2022
, Accepted 25th January 2023
First published on 8th February 2023
Abstract
Highly regioselective synthesis of 2-acyl-4-(het)arylthiazoles and thioethers by the reaction between α-oxothioamides and α-bromoketones in the absence of base in DMF and in the presence of triethylamine in acetonitrile, respectively, has been reported. This thiazole synthesis is an important extended work of the Hantzsch thiazole synthesis, which overcomes the drawbacks of earlier reported methods. The probable mechanisms for the formation of thiazoles and thioethers are also presented.
Introduction
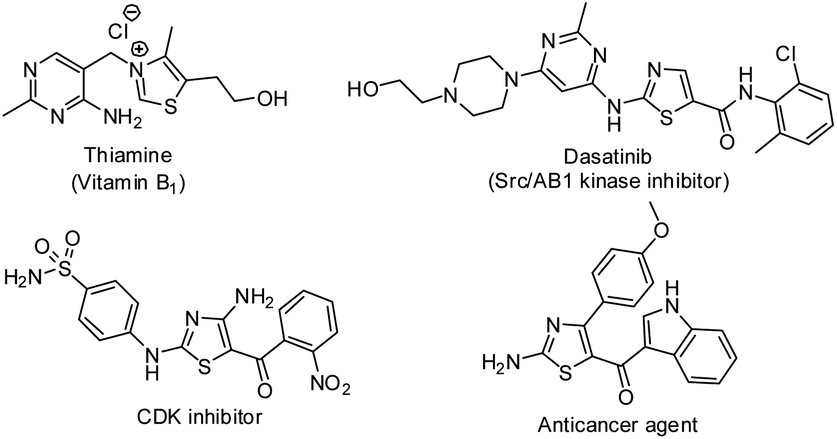
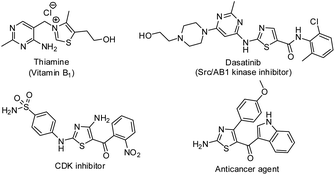
Thiazoles occupy a prominent position in medicinal chemistry due to their various biological activities such as anti-bacterial,1 antihypertensive,2 anti-inflammatory,3 anti-oxidant,4 antitumor,5 anti-HIV,6 antihyperlipidemic,7 antiprotozoal,8 antitubercular9 and antimalarial.10 Besides, they are present in natural products,11 agrochemicals,12 drugs13 and materials.14 Some biologically important thiazole derivatives are listed in Fig. 1. On the other hand, it is noteworthy to mention that thioethers are present in pharmaceuticals and natural products.15 Because of their vast existence and applications, development of new methods for the synthesis of thiazoles and sulfides have gained paramount importance.
 |
| | Fig. 1 Biologically important thiazole derivatives. | |
Our literature survey on the synthesis of thiazoles reflected that the Hantzsch synthesis16 is the conventional method, which entails the reaction between thioamides and α-halocarbonyl compounds. Since an enormous number of methods for the synthesis of thiazole have been reported, we converged our presentation on modified Hantzsch thiazole reactions. Thus, thiourea,17 thioamides18–20 and substituted thioureas21 gave thiazoles when reacted with α-halocarbonyl compounds. Alternatively, reaction of ethyl 3-bromo-2-oxopropanoate with thioamides furnished thiazoles.22,23 Other methods involve reaction of thioamides with alkynyl(aryl)iodonium salts24 and 1H-1-(1′-alkynyl)-5-methyl-1,2,3-benziodoxathiole 3,3-dioxides.25 Substrate modified approaches are the reaction of β-keto esters with tribromoisocyanuric acid followed by cyclization with thiourea26 and α-halogenation of β-keto esters with N-bromosuccinimide followed by reaction with thiourea.27 In addition, α-halocarbonyl compounds in the Hantzsch synthesis are replaced by α-tosyloxy ketones.28 Recently, reaction between α-oxo-1H-indole-3-thioacetamides and 2-bromo-1-(1H-indol-3-yl)-ethanones produced (4-(1H-indol-3-yl)thiazol-2-yl)(1H-indol-3-yl)methanones.29 Unfortunately, this report suffers from the generality of the protocol and long reaction time. To the best of our knowledge, a detailed or a general examination of α-oxothioamides with α-halocarbonyl compounds is not reported. As a consequence, we present the modified Hantzsch thiazole synthesis in this article, which fills the aforementioned void.
On the other hand, thioethers are present in pharmaceuticals and natural products.30,31 They can be synthesized classically by the thiol alkylation or arylation.32,33 The most recent methods include reaction of redox-active esters with thiosulfonates,34 thiolation of alkyl oxalates,35 reaction between 4-alkyl-1,4-dihydropyridines and thiosulfonates,36 and others.37 These methods suffer from limitations such as use of malodorous thiols, formation of stoichiometric amounts of thiol byproducts and disulfides as impurities in the presence of air. We present herein α-oxothioamides as thionating agent from which thioethers are formed upon reaction with α-bromoketones.
In continuation of our efforts in organic synthesis38–44 we have recently reported the synthesis of quinoxalines,45,46 thiazoles47 and benzoxazoles45 from α-ketodithioesters. In this direction, we have recently reported the synthesis of α-ketothioamides and explored their applications for the synthesis of 3,5-bis(acyl)-1,2,4-thiadiazoles.48 In extension of this work, we report herein an important extended Hantzsch thiazole synthesis by the reaction of α-ketothioamides with α-bromoketones in the absence of a base. Besides, we have also demonstrated α-ketothioamides as an excellent thionating reagent for the synthesis of thioethers from α-bromoketones in the presence of a base.
Results and discussion
We prepared required α-ketothioamides according to our earlier reported protocol.48 Thus, α-ketodithioesters45,47 1 on treatment with stoichiometric amounts of ammonium chloride and anhydrous sodium acetate in acetonitrile furnished α-ketothioamides 2 (Scheme 1).
 |
| | Scheme 1 Synthesis of α-ketothioamides. | |
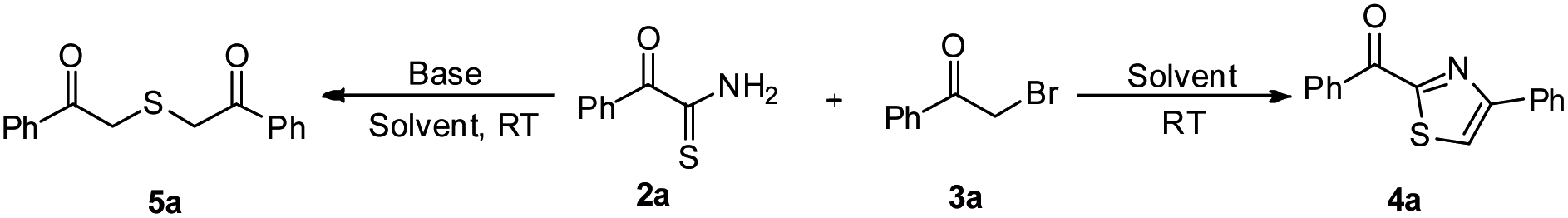
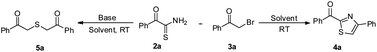
With the key intermediates in hand, we started optimization of reaction conditions for the synthesis of phenyl(4-phenylthiazol-2-yl)methanone 4a by reacting 2-oxo-2-phenylethanethioamide 2a with phenacyl bromide 3a in acetone in the presence of triethylamine (Table 1, Scheme 2). A product was obtained after an hour which was found to be 2,2′-thiobis(1-phenylethanone) 5a based on spectroscopic data. Later, we tried the same reaction in the absence of triethylamine, which afforded anticipated product phenyl(4-phenylthiazol-2-yl)methanone 4a in 90% yield (Table 1, entry 2). To improve the yield, we performed the same reaction in DMF, which furnished 4a in 93% yield (Table 1, entry 3). Further, reactions in acetonitrile and dichloromethane resulted in decreased yield of 4a (Table 1, entries 4 and 5). To screen the best condition for the formation of 5a, we did the reaction 2a with 3a in the presence of triethylamine in acetonitrile, which produced 5a in 95% yield (Table 1, entry 6). Change of solvent to DMF and methanol reduced the yield of 5a (Table 1, entries 7 and 8). Change of base to potassium carbonate in acetonitrile gave 5a in 90% yield (Table 1, entry 9). A reaction in pyridine as a solvent as well as base gave 5a in trace amounts (Table 1, entry 10). Further, DMAP in acetonitrile furnished 5a in 40% yield (Table 1, entry 11). Finally, a reaction with diisopropylethylamine (DIPEA) furnished 5a in 80% yield (Table 1, entry 12). Thus, DMF and acetonitrile were found to be the best solvents for the formation of 4a and 5a in the absence and presence of triethylamine respectively.
Table 1 Optimization of reaction conditions for the synthesis of phenyl(4-phenylthiazol-2-yl)methanone 4a and 2,2′-thiobis(1-phenylethanone) 5a
| Entry |
Solvent |
Base |
Time (h) |
Yielda (%) of 4a |
Yieldb (%) of 5a |
| Reaction conditions: 2a (1 mmol), 3a (1 mmol), solvent (5 mL). Reaction conditions: 2a (0.5 mmol), 3a (1 mmol), base (1 mmol), solvent (5 mL). |
| 1 |
Acetone |
Et3N |
1 |
|
93 |
| 2 |
Acetone |
— |
2 |
90 |
— |
| 3 |
DMF |
— |
1 |
93 |
— |
| 4 |
MeCN |
— |
3 |
80 |
— |
| 5 |
CH2Cl2 |
— |
2 |
60 |
— |
| 6 |
MeCN |
Et3N |
0.5 |
— |
95 |
| 7 |
DMF |
Et3N |
1 |
— |
90 |
| 8 |
MeOH |
Et3N |
1 |
— |
80 |
| 9 |
MeCN |
K2CO3 |
1 |
— |
90 |
| 10 |
Pyridine |
Pyridine |
2 |
— |
Trace |
| 11 |
MeCN |
DMAP |
2 |
|
40 |
| 12 |
MeCN |
DIPEA |
1.5 |
|
80 |
 |
| | Scheme 2 Synthesis of phenyl(4-phenylthiazol-2-yl)methanone 4a and 2,2′-thiobis(1-phenylethanone) 5a. | |
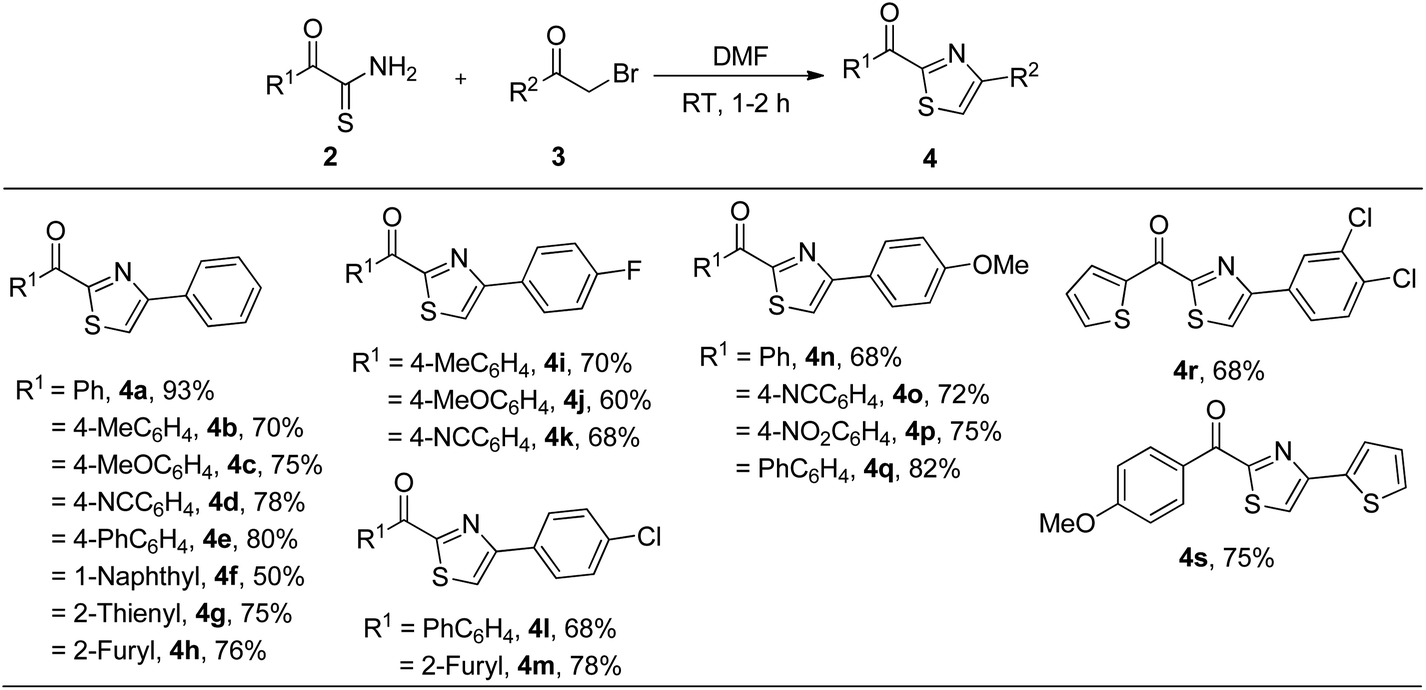
With the optimized reaction conditions in hand, we next explored the generality of the method for the synthesis of 2-acyl-4-(het)arylthiazoles 4 from α-ketothioamides 2 and α-bromoketones 3 (Table 2). Thus, various α-oxodithioesters 2 containing electron donating groups (methyl and methoxy), electron withdrawing groups (cyano and phenyl), 1-naphthyl and heteroaryl groups (2-thienyl and 2-furyl) reacted smoothly with phenacyl bromide (3a) to furnish 2-acyl-4-phenylthiazoles 4a–h in 50–93% yield. Similarly, electronically rich and deficient α-ketothioamides 2 underwent cyclization reaction with p-fluorophenacyl bromide (3b) to give respective thiazoles 4i–k in 60–70% yield. In the same fashion, p-chlorophenacyl bromide (3c) furnished 2-acyl-4-(4-chlorophenyl)arylthiazoles 4l and 4m in 68% and 78% yield respectively by undergoing reaction with α-oxodithioesters substituted with biphenyl and 2-furyl moieties. Likewise, p-methoxyphenacyl bromide (3d) underwent cyclocondensation with electronically diverse α-oxodithioesters to produce 2-acyl-4-(4-methoxyphenyl)thiazoles 4n–q in 68–82% yield. In parallel, 2-oxo-2-(thiophen-2-yl)ethanethioamide reacted with 3,4-dichlorophenacyl bromide (3e) to form (4-(3,4-dichlorophenyl)thiazol-2-yl)(thiophen-2-yl)methanone 4r in 68% yield. Finally, a reaction between 2-oxo-2-(4-methoxyphenyl)ethanethioamide and 2-bromo-1-(thiophen-2-yl)ethanone (3f) yielded 4-methoxyphenyl(4-(thiophen-2-yl)thiazol-2-yl)methanone 4s in 75%. Thus, α-haloketones containing electron donating and withdrawing groups did not affect the yield of products.
Table 2 Substrate scope for the synthesis of 2-acyl-4-(het)arylthiazoles
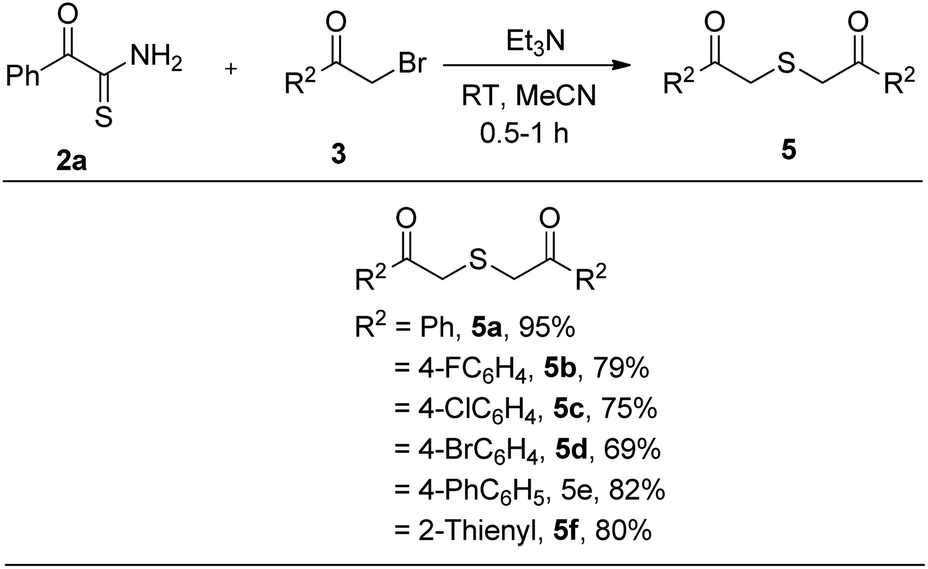
On the other hand, we reacted 2-oxo-2-phenylethanethioamide 2a with various substituted phenacyl bromides which afforded thioethers 5a–e in 69–95% yield (Table 3). Besides, 2-bromo-1-(thiophen-2-yl)ethanone reacted with 2a to furnish 2,2′-thiobis(1-(thiophen-2-yl)ethanone) 5f in 80% yield.
Table 3 Substrate scope for the synthesis of 2,2′-thiobis(1-(het)arylethanones)
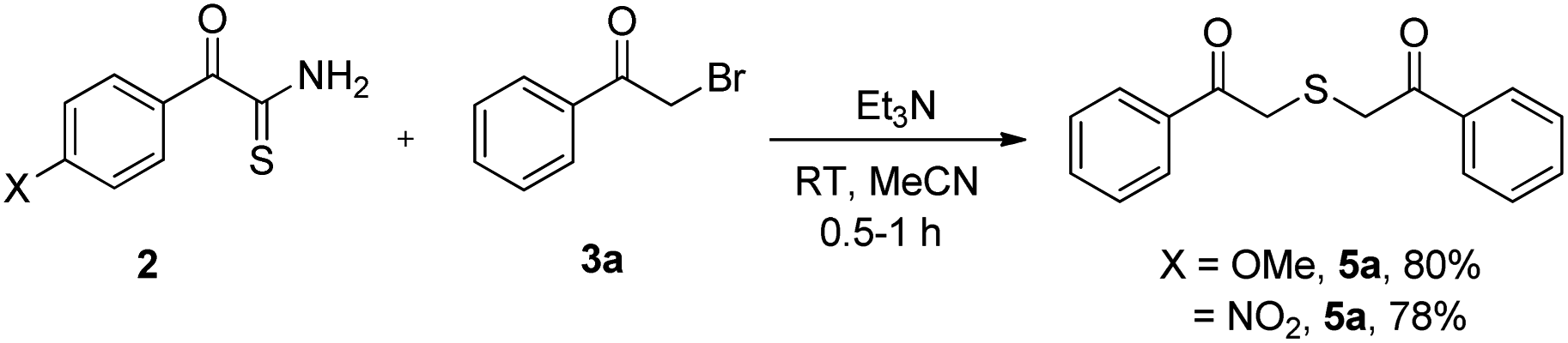
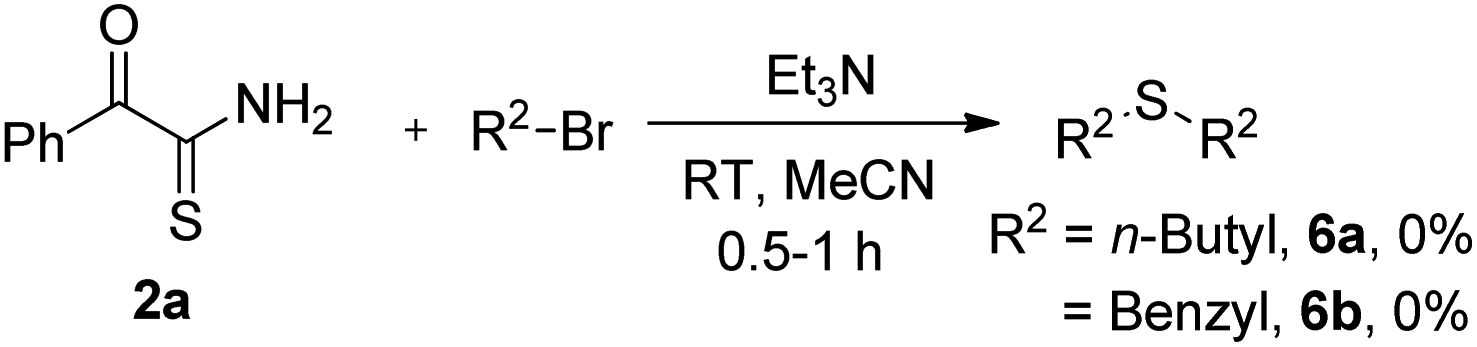
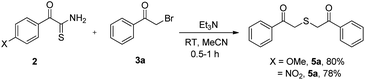
Later, we demonstrated that other electronically diverse α-oxothioamides 2 bearing methoxy and nitro groups behaved as excellent thionating agents for the synthesis of 2,2′-thiobis(1-phenylethanone) 5a in 80% and 78% yield respectively (Scheme 3). To determine the generality of this protocol, we tested few reactions with n-butyl bromide and benzyl bromide, which did not produce anticipated thioethers 6 (Scheme 4).
 |
| | Scheme 3 Other thionating α-oxothioamides. | |
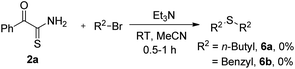 |
| | Scheme 4 Reaction of an α-oxothioamide with n-butyl and benzyl bromide. | |
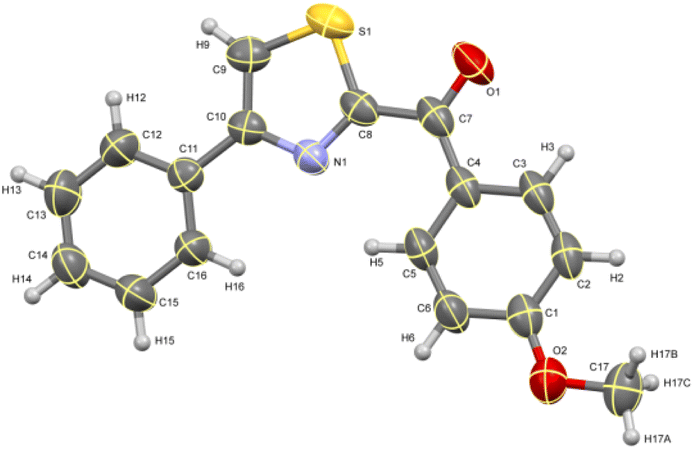
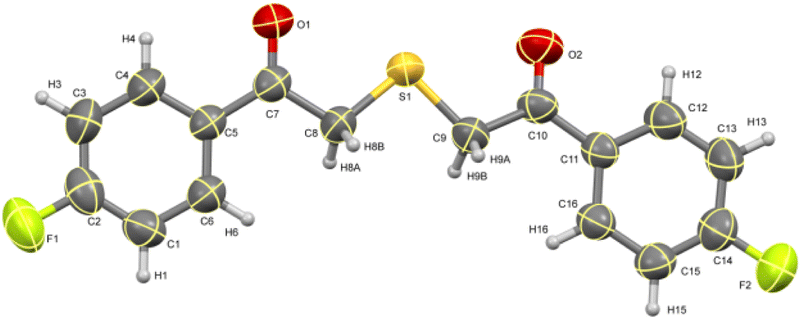
The ORTEP diagram for one of the thiazoles (4-methoxyphenyl)(4-phenylthiazol-2-yl)methanone 4c is given in Fig. 2.49 Similarly, the diagram for one of the thioethers 2,2′-thiobis(1-(4-fluorophenyl)ethanone) 5b is given in Fig. 3.50
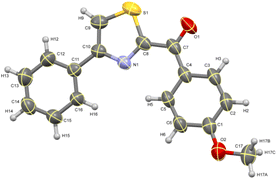 |
| | Fig. 2 ORTEP diagram of thiazole 4c. | |
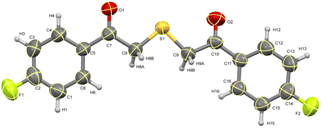 |
| | Fig. 3 ORTEP diagram of thioether 5b. | |
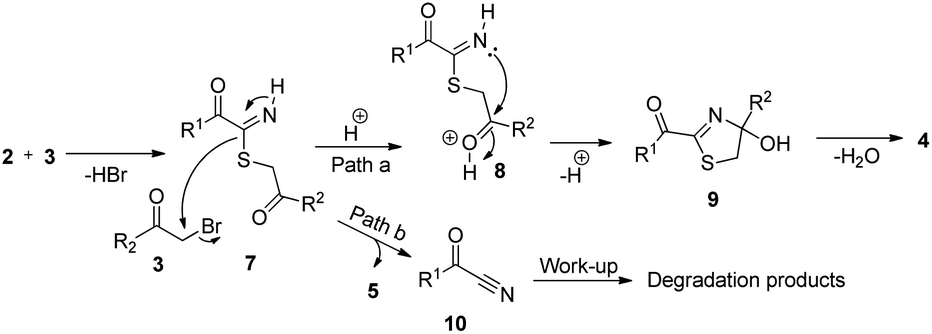
The probable mechanisms for the formation of 2-acyl-4-(het)arylthiazoles 4 and thioethers 5 are given in Scheme 5. For thiazoles, it involves nucleophilic attack of α-oxothiamide 2 to α-bromoketone 3 to give intermediate 7, which is protonated to give cation 8. This undergoes intramolecular cyclization to furnish dihydrothiazole intermediate 9. Finally, dehydration of 9 affords products 4 (path a). On the other hand, second nucleophilic substitution reaction by 7 onto 3 furnishes thioethers 5 and α-ketonitrile 10 (path b). Notably, we observed the degradation of byproduct 10 during work-up and isolation.
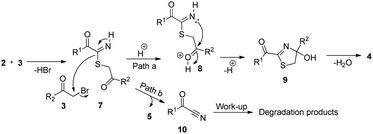 |
| | Scheme 5 Probable mechanism for the formation of 2-acyl-4-(het)arylthiazoles 4. | |
Experimental
Materials and methods
All reagents and solvents were purchased from commercial suppliers in India and were used without further purification. Reactions were monitored by TLC (commercially available precoated plates, MERCK 60F254, 0.25 mm thickness) and UV light was used for visualization. NMR spectra were recorded with a Agilant NMR spectrometer. Chemical shift (δ) values are given in ppm. CDCl3 was used as solvent with reference to TMS. Coupling constant (J) values are given in Hz. Mass spectra were recorded by using Water-SynaptG2 mass spectrometer. The single-crystal X-ray diffraction data for the compounds were obtained with a Bruker Apex II CCD diffractometer using a Cu and Mo source at room temperature with the monochrome beam method. The structures were established via full-matrix least-squares methods using the SHELKS program.
Synthetic procedures
General procedure for the synthesis of 2-acyl-4-(het)arylthiazoles 4a–s. Equimolar ratio of α-oxothioamides 2 (1 mmol) and α-bromoketones 3 (1 mmol) were dissolved in DMF (5 mL) and allowed to stir at room temperature. The reactions were monitored by thin layer Chromatography (TLC). After the completion of the reaction, brine (25 mL) was added to the reaction mixture. It was then extracted with ethyl acetate twice (25 mL). The combined ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure. Later, the crude products were purified by column chromatography using a mixture of ethyl acetate in hexane (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane: 80:20).
:hexane: 80:20).
Phenyl(4-phenylthiazol-2-yl)methanone (4a). Yellow solid; 93% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 145–147 °C; IR (KBr): νmax (Nujol) cm−1 3104, 1599, 1249, 1031, 782; 1H NMR (CDCl3, 400 MHz) δ: 8.59 (d, J = 7.6 Hz, 2H, Ar–H), 7.94 (d, J = 7.6 Hz, 4H, Ar–H), 7.82 (s, 1H, Ar–H), 7.64 (d, J = 7.6 Hz, 1H, Ar–H), 7.52–7.38 (m, 2H, Ar–H), 7.36 (t, J = 7.6 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 191.2, 183.7, 167.3, 157.5, 135.0, 133.7, 131.4, 131.1, 128.9, 128.6, 126.5, 119.7; HRMS (ESI): m/z [M + H]+ calculated for C16H11NOS: 266.0640; found: 266.0647.
(4-Phenylthiazol-2-yl)(p-tolyl)methanone (4b). Yellow solid; 70% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 136–138 °C; IR (KBr): νmax(Nujol) cm−1 3096, 1594, 1274, 1176, 846; 1H NMR (CDCl3, 400 MHz) δ: 8.52 (d, J = 8.0 Hz, 2H, Ar–H), 7.96 (d, J = 8.0 Hz, 2H, Ar–H), 7.82 (s, 1H, Ar–H), 7.46 (d, J = 4.0 Hz, 2H, Ar–H), 7.34 (t, J = 8.0 Hz, 3H, Ar–H), 2.44 (s, 3H, ArMe); 13C NMR (CDCl3, 100 MHz) δ: 195.3, 187.4, 171.8, 161.5, 148.8, 137.9, 135.7, 135.4, 133.3, 132.9, 130.6, 123.9, 25.9; HRMS (ESI) [M + H]+ calculated C17H13NOS 280.0796 found 280.0798.
(4-Methoxyphenyl)(4-phenylthiazol-2-yl)methanone (4c). White solid; 75% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 100–102 °C; IR (KBr): νmax (Nujol) cm−1 3093, 1597, 1172, 844; 1H NMR (CDCl3, 400 MHz) δ: 8.68 (d, J = 9.2 Hz, 2H, Ar–H), 7.95 (d, J = 7.2 Hz, 2H, Ar–H), 7.79 (s, 1H, Ar–H), 7.43 (m, 2H, Ar–H), 7.39 (t, J = 7.2 Hz, 1H, Ar–H), 7.01 (d, J = 8.8 Hz, 2H, Ar–H), 3.88 (s, 3H. MeO); 13C NMR (CDCl3, 100 MHz) δ: 181.8, 168.1, 164.1, 157.2, 133.8, 133.7, 128.7, 127.7, 126.4, 126.2, 119.5, 113.7, 55.5; HRMS (ESI) [M + H]+ calculated C17H13NO2S 296.0745 found 296.0748.
4-(4-Phenylthiazole-2-carbonyl)benzonitrile (4d). Yellow solid; 78% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 130–132 °C; IR (KBr): νmax (Nujol) cm−1 3109, 1297, 755; 1H NMR (CDCl3, 400 MHz) δ: 8.66 (d, J = 12.0 Hz, 2H, Ar–H), 7.93 (d, J = 8.0 Hz, 2H, Ar–H), 7.90 (s, 1H, Ar–H), 7.82 (d, J = 8.0 Hz, 2H, Ar–H), 7.47 (d, J = 4.0 Hz, 2H, Ar–H), 7.41 (t, J = 6.0 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 186.4, 170.3, 162.1, 142.4, 137.4, 136.2, 135.9, 135.4, 133.1, 130.6, 125.0, 122.1, 120.7; HRMS (ESI) [M + H]+ calculated C17H10N2OS 291.0592 found 291.0596.
[1,1′-Biphenyl]-4-yl(4-phenylthiazol-2-yl)methanone (4e). Yellow solid; 80% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 100–102 °C; IR (KBr):νmax(Nujol) cm−1 3092, 1474, 1249, 952, 748; 1H NMR (CDCl3, 400 MHz) δ: 8.72 (d, J = 8.4 Hz, 2H, Ar–H), 8.01 (t, J = 6.8 Hz, 2H, Ar–H), 7.89 (s, 1H, Ar–H), 7.80 (d, J = 7.8 Hz, 2H, Ar–H), 7.71 (t, J = 7.2 Hz, 2H, Ar–H), 7.50–7.48 (m, 4H, Ar–H), 7.42 (d, J = 7.2 Hz, 2H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ:183.6, 167.9, 157.9, 146.6, 140.2, 134.1,132.3, 132.1, 129.2, 128.9, 127.4, 127.2, 126.9, 126.7, 120.3, 120.1; HRMS (ESI) [M + H]+ calculated C22H15NOS 342.0953 found 342.0958.
Naphthalen-1-yl(4-phenylthiazol-2-yl)methanone (4f). Brown solid; 50% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 126–128 °C; IR (KBr): νmax (Nujol) cm−1 3094, 1560, 1267, 884; 1H NMR (CDCl3, 400 MHz) δ: 8.54 (d, J = 12.0 Hz, 1H, Ar–H), 8.40 (d, J = 8.0 Hz, 1H, Ar–H), 8.06 (d, J = 8.0 Hz, 1H, Ar–H), 7.94 (t, J = 8.0 Hz, 2H, Ar–H), 7.89 (d, J = 8.0 Hz, 1H, Ar–H), 7.86 (s, 1H, Ar–H), 7.60–7.52 (m, 4H, Ar–H), 7.38 (d, J = 4.0 Hz, 1H, Ar–H), 7.34 (d, J = 8.0 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ:191.2, 186.8, 168.1, 157.6, 133.8, 133.6, 133.1, 132.4, 132.0, 129.1, 128.6, 127.6, 126.6, 126.3, 125.6, 124.0, 120.4, 120.1. HRMS (ESI) [M + H]+ calculated C20H13NOS 316.0796 found 316.0789.
(4-Phenylthiazol-2-yl)(thiophen-2-yl)methanone (4g). Yellow solid; 75% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 130–132 °C; IR (KBr): νmax (Nujol) cm−1 3110, 1474, 1262, 860, 640; 1H NMR (CDCl3, 400 MHz) δ: 8.66 (t, J = 2.8 Hz, 1H, Ar–H), 8.0 (d, J = 1.2 Hz, 2H, Ar–H), 7.83 (s, 1H, Ar–H), 7.80 (d, J = 4.0 Hz, 1H, Ar–H), 7.47 (t, J = 6.8 Hz, 2H, Ar–H), 7.39 (d, J = 8.0 Hz, 1H, Ar–H), 7.23 (t, J = 5.6 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 191.2, 175.6, 166.5, 157.4, 139.3, 136.8, 136.6, 133.6, 128.8, 128.3, 126.5, 119.8; HRMS (ESI) [M + H]+ calculated C14H9NOS2 272.0204 found 272.0209.
Furan-2-yl(4-phenylthiazol-2-yl)methanone (4h). Yellow solid; 76% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 126–128 °C; IR (KBr): νmax (Nujol) cm−1 2923, 1476, 1189, 860; 1H NMR (CDCl3, 400 MHz) δ: 8.45 (s, 1H, Ar–H), 8.05 (d, J = 8.0 Hz, 2H, Ar–H), 7.93 (s, 1H, Ar–H), 7.89 (s, 1H, Ar–H), 7.56 (t, J = 8.0 Hz, 2H, Ar–H), 7.49 (d, J = 7.2 Hz, 1H, Ar–H), 6.77 (d, J = 1.6 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 175.0, 170.1, 161.6, 153.8, 152.7, 137.7, 132.9, 130.5, 130.4, 128.6, 123.6, 116.8; HRMS (ESI) [M + H]+ calculated C14H9NO2S 256.0432 found 256.0438.
(4-(4-Fluorophenyl)thiazol-2-yl)(p-tolyl)methanone (4i). Yellow solid; 70% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 140–142 °C; IR (KBr): νmax (Nujol) cm−1 3102, 1527, 1262, 1121, 835; 1H NMR (CDCl3, 400 MHz) δ: 8.40 (d, J = 12.0 Hz, 2H, Ar–H), 7.82 (dd, J = 8.0 Hz, 4.0 Hz, 2H, Ar–H), 7.66 (s, 1H, Ar–H), 7.25 (d, J = 8.0 Hz, 2H, Ar–H), 7.05 (dd, J = 4.0 Hz, 8.0 Hz, 2H, Ar–H), 2.36 (s, 3H, Ar-Me); 13C NMR (CDCl3, 100 MHz) δ: 189.7, 183.2, 167.8, 156.3, 144.7, 132.3, 131.4, 129.2, 128.2, 128.1, 119.1, 115.8, 21.9; HRMS (ESI) [M + H]+ calculated C17H12FNOS 298.0702 found 298.0706.
(4-(4-Fluorophenyl)thiazol-2-yl)(4-methoxyphenyl)methanone (4j). Yellow solid; 60% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 128–130 °C; IR (KBr):νmax (Nujol) cm−1 3097, 1438, 1173, 880; 1H NMR (CDCl3, 400 MHz) δ: 8.65 (d, J = 9.2 Hz, 2H, Ar–H), 7.93–7.89 (m, 2H, Ar–H), 7.73 (s, 1H, Ar–H), 7.13 (d, J = 12.0 Hz, 2H, Ar–H), 7.01 (d, J = 12.0 Hz, 2H, Ar–H), 3.89 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 181.8, 168.2, 164.1,156.2, 133.8, 130.1, 128.2, 128.1, 127.7, 118.9, 115.8, 113.8, 55.6; HRMS (ESI) [M + H]+ calculated C17H12FNO2S 314.0651 found 314.0678.
4-(4-(4-Fluorophenyl)thiazole-2-carbonyl)benzonitrile (4k). Yellow solid; 68% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 134–136 °C; IR (KBr): νmax (Nujol) cm−1 3093, 1449, 1278, 1031, 832; 1H NMR (CDCl3, 400 MHz) δ: 8.65 (d, J = 8.4 Hz, 2H, Ar–H), 7.91 (d, J = 8.0 Hz, 2H, Ar–H), 7.88 (d, J = 8.8 Hz, 2H, Ar–H), 7.82 (s, 1H, Ar–H), 7.15 (d, J = 8.4 Hz, 2H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 182.3, 166.3, 164.3, 161.9, 156.9, 138.2, 131.9, 131.4, 128.3, 128.0, 120.3, 117.9, 116.6; HRMS (ESI) [M + H]+ calculated C17H9FN2OS 310.0531 found 310.0538.
[1,1′-Biphenyl]-4-yl(4-(4-chlorophenyl)thiazol-2-yl)methanone (4l). Yellow solid; 68% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 90–92 °C; IR (KBr): νmax (Nujol) cm−1 3091, 1477, 1258, 958, 746; 1H NMR (CDCl3, 400 MHz) δ: 8.59 (d, J = 12.0 Hz, 2H, Ar–H), 7.82 (d, J = 8.0 Hz, 1H, Ar–H), 7.79 (d, J = 8.8 Hz 2H, Ar–H), 7.75 (s, 1H, Ar–H), 7.69 (d, J = 8.4 Hz, 1H, Ar–H), 7.59 (d, J = 6.8 Hz, 2H, Ar–H), 7.39 (d, J = 8.0 Hz, 2H, Ar–H), 7.34 (d, J = 3.2 Hz, 2H, Ar–H), 7.33 (t, J = 2.4 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 196.8, 182.9, 167.7, 156.2, 146.2, 132.1, 131.9, 131.6, 130.8, 129.7, 129.2, 128.5, 127.7, 127.4, 120.2, 119.8; HRMS (ESI)[M + H]+ calculated C22H14ClNOS 376.0563 found 376.0569.
(4-(4-Chlorophenyl)thiazol-2-yl)(furan-2-yl)methanone (4m). Yellow solid; 78% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 134–136 °C; IR (KBr): νmax (Nujol) cm−1 3126, 1463, 1280, 1012, 756; 1H NMR (CDCl3, 400 MHz) δ: 8.23 (d, J = 3.2 Hz, 1H, Ar–H), 7.80 (d, J = 7.2 Hz, 2H, Ar–H), 7.74 (d, J = 6.0 Hz, 2H, Ar–H), 7.35 (d, J = 7.6 Hz, 2H, Ar–H), 6.60 (s, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 170.7, 166.2, 156.2, 149.2, 148.8, 134.6, 132.1, 129.0, 127.5, 124.5, 119.8, 112.9; HRMS (ESI) [M + H]+ calculated C14H8ClNO2S 290.0043 found 290.0049.
(4-(4-Methoxyphenyl)thiazol-2-yl)(phenyl)methanone (4n). Yellow solid; 68% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 136–138 °C; IR (KBr): νmax (Nujol) cm−1 3104, 1599, 1249, 1031, 786; 1H NMR (CDCl3, 400 MHz) δ: 8.58 (d, J = 7.2 Hz, 2H, Ar–H), 7.85 (d, J = 9.2 Hz, 2H, Ar–H), 7.67 (s, 1H, Ar–H), 7.54 (t, J = 7.6 Hz, 1H, Ar–H), 7.51 (d, J = 7.6 Hz, 2H, Ar–H), 6.94 (d, J = 8.8 Hz, 2H, Ar–H), 3.81 (s, 3H, MeO); 13C NMR (CDCl3,100 MHz) δ: 183.6, 167.1, 160.0, 157.4, 135.1, 131.3, 128.2, 127.8, 127.7, 126.6, 118.4, 114.2, 55.4; HRMS (ESI) [M + H]+ calculated C17H13NO2S 296.0745 found 296.0749.
4-(4-(4-Methoxyphenyl)thiazole-2-carbonyl)benzonitrile (4o). Yellow solid; 72% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 146–148 °C; IR (KBr): νmax (Nujol) cm−1 3103, 1600, 1226, 1073, 835; 1H NMR (CDCl3, 400 MHz) δ: 8.65 (d, J = 8.0 Hz, 2H, Ar–H), 7.82 (dd, J = 8.0 Hz, 8 Hz, 4H, Ar–H), 7.76 (s, 1H, Ar–H), 6.97 (d, J = 8.0 Hz, 2H, Ar–H), 3.83 (s, 3H, MeO); 13C NMR (CDCl3, 100 MHz) δ: 182.3, 165.9, 160.2, 157.9, 138.4, 132.2, 131.6, 127.9, 126.2, 119.3, 118.0, 116.5, 114.3, 55.2; HRMS (ESI) [M + H]+ calculated C18H12N2O2S 321.0698 found 321.0685.
(4-(4-Methoxyphenyl)thiazol-2-yl)(4-nitrophenyl)methanone (4p). Yellow solid; 75% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 138–140 °C; IR (KBr): νmax (Nujol) cm−1 3103, 1600, 1226, 1073, 832; 1H NMR (CDCl3, 400 MHz) δ: 8.73 (d, J = 8.0 Hz, 2H, Ar–H), 8.37 (d, J = 9.2 Hz, 2H, Ar–H), 7.86 (d, J = 8.0 Hz, 2H, Ar–H), 7.79 (s, 1H, Ar–H), 6.98 (d, J = 12.0 Hz, 2H, Ar–H), 3.85 (s, 3H, MeO); 13C NMR (CDCl3, 100 MHz) δ: 182.2, 165.8, 160.2, 158.0, 140.0, 132.2, 127.9, 127.3, 126.1, 123.3, 119.4, 114.3, 55.4; HRMS (ESI) [M + H]+ calculated C17H12N2O4S 341.0596 found 341.0597.
[1,1′-Biphenyl]-4-yl(4-(4-methoxyphenyl)thiazol-2-yl)methanone (4q). Yellow solid; 82% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 102–104 °C; IR (KBr): νmax (Nujol) cm−1 3094, 1476, 1260, 943, 782; 1H NMR (CDCl3, 400 MHz) δ: 8.69 (d, J = 8.0 Hz, 1H, Ar–H), 7.54 (d, J = 12.0 Hz, 2H, Ar–H), 7.91 (d, J = 8.0 Hz, 1H, Ar–H), 7.81–7.71 (m, 3H, Ar–H), 7.64 (d, J = 8.0 Hz, 2H, Ar–H), 7.47 (d, J = 8.0 Hz, 2H, Ar–H), 7.41 (t, J = 8.0 Hz, 1H, Ar–H), 7.28 (s, 1H, Ar–H), 6.99 (d, J = 8.0 Hz, 1H, Ar–H), 3.85 (s, 3H, MeO); 13C NMR (CDCl3, 100 MHz) δ: 183.5, 160.0, 157.4, 155.3, 146.1, 139.9, 131.8, 131.5, 128.6, 127.5, 127.2, 126.8, 126.5, 123.7, 116.8, 114.3, 55.4; HRMS (ESI) [M + H]+ calculated C23H17NO2S 373.1092 found 373.1011.
(4-(3,4-Dichlorophenyl)thiazol-2-yl)(thiophen-2-yl)methanone (4r). Yellow solid; 68% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 142–144 °C; IR (KBr): νmax (Nujol) cm−1 3097, 1507, 1356, 1035, 774; 1H NMR (CDCl3, 400 MHz) δ: 8.60 (t, J = 4.0 Hz, 1H, Ar–H), 8.0 (s, 1H, Ar–H), 7.83 (s, 1H, Ar–H), 7.81 (d, J = 8.8 Hz, 1H, Ar–H), 7.78 (d, J = 2.0 Hz, 1H, Ar–H), 7.76 (d, J = 2.0 Hz, 1H, Ar–H), 7.24 (d, J = 2.4 Hz, 1H, Ar–H); 13C NMR (CDCl3, 100 MHz) δ: 175.3, 167.1, 154.8, 139.0, 137.1, 136.8, 133.5, 130.1, 128.3, 128.1, 125.6, 125.4, 120.9, 120.6; HRMS (ESI) [M + H]+ calculated C14H7Cl2NOS2 339.9424 found 339.9465.
(4-Methoxyphenyl)(4-(thiophen-2-yl)thiazol-2-yl)methanone (4s). Yellow solid; 75% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 126–128 °C; IR (KBr): νmax (Nujol) cm−1 2928, 1432, 1235, 1171, 843; 1H NMR (CDCl3, 400 MHz) δ: 8.59 (d, J = 7.2 Hz, 2H, Ar–H), 7.56 (s, 1H, Ar–H), 7.42 (t, J = 2.4 Hz, 1H, Ar–H), 7.23 (d, J = 1.2 Hz, 1H, Ar–H), 7.0 (d, J = 1.2 Hz, 1H, Ar–H), 6.93 (d, J = 8.8 Hz, 2H, Ar–H), 3.81 (s, 3H, MeO); 13C NMR (CDCl3, 100 MHz) δ: 181.5, 168.0, 164.2, 151.7, 137.6, 134.0, 133.7, 127.6, 118.3, 117.8, 113.8, 113.6, 55.3; HRMS (ESI) [M + H]+ calculated C15H11NO2S2 302.3913 found 302.3918.
General procedure for the synthesis of thioethers 5a–f
A mixture of α-oxothioamides 2 (0.5 mmol), α-bromoketones 3 (1 mmol) and triethylamine (1 mmol) were stirred in acetonitrile (5 mL) at room temperature. The reactions were monitored by TLC. After the completion of the reaction, water (25 mL) was added to the reaction mixture. It was then extracted with ethyl acetate twice (25 mL). The combined ethyl acetate layer was washed with brine (25 mL) and dried over anhydrous sodium sulphate and concentrated under reduced pressure. Later, the crude products were purified by column chromatography using a mixture of ethyl acetate in hexane (EtOAc:hexane::80:20).
2,2′-Thiobis(1-phenylethanone) (5a). Yellow solid; 95% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 140–142 °C; IR (KBr): νmax (Nujol) cm−1 2982, 1176, 678; 1H NMR (CDCl3, 400 MHz) δ: 7.87 (d, J = 8.0 Hz, 4H, Ar–H), 7.49 (t, J = 6.8 Hz, 2H, Ar–H), 7.36 (t, J = 6.8 Hz, 4H, Ar–H), 3.89 (s, 4H, (CH2)2); 13C NMR (CDCl3, 100 MHz) δ: 193.7, 134.9, 133.2, 128.3, 128.2, 37.2; HRMS (ESI) [M + H]+ calculated C16H14O2S 271.0793 found 271.0798.
2,2′-Thiobis(1-(4-fluorophenyl)ethanone) (5b). Yellow solid; 79% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 134–136 °C; IR (KBr): νmax (Nujol) cm−1 2954, 1216, 1279, 670; 1H NMR (CDCl3, 400 MHz) δ: 7.92 (dd, J = 9.2 and 8.8 Hz, 4H, Ar–H), 7.08 (d, J = 8.4 Hz, 4H, Ar–H), 3.87 (s, 4H, (CH2)2; 13C NMR(CDCl3, 100 MHz) δ: 192.5, 167.2, 164.6, 131.6 (J = 30.0 Hz), 116.0, 37.4; HRMS (ESI) [M + H]+ calculated C16H12F2O2S 307.0604 found 307.0612.
2,2′-Thiobis(1-(4-chlorophenyl)ethanone) (5c). Yellow solid; 75% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 130–132 °C; IR (KBr): νmax (Nujol) cm−1 2930, 1399, 1009, 987, 683; 1H NMR (CDCl3, 400 MHz) δ: 7.81 (d, J = 8.0 Hz, 4H, Ar–H), 7.35 (d, J = 8.0 Hz, 4H, Ar–H), 3.85 (s, 4H, (CH2)2); 13C NMR (CDCl3, 100 MHz) δ: 193.4, 140.8, 134.2, 130.8, 129.7, 38.0; HRMS (ESI) [M + H]+ calculated C16H12Cl2O2S 339.0013 found 339.0018.
2,2′-Thiobis(1-(4-bromophenyl)ethanone) (5d). Yellow solid; 69% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 138–140 °C; IR (KBr): νmax (Nujol) cm−1 2952, 1289, 988, 643; 1H NMR (CDCl3, 400 MHz) δ: 7.75 (d, J = 8.8 Hz, 4H, Ar–H), 7.55 (d, J = 8.4 Hz, 4H, Ar–H), 3.85 (s, 4H, (CH2)2); 13C NMR (CDCl3, 100 MHz) δ: 193.7, 134.6, 132.7, 130.7, 129.5, 38.0; HRMS (ESI) [M + H]+ calculated C16H12Br2O2S 428.8983 found 428.8985.
2,2′-Thiobis(1-([1,1′-biphenyl]-4-yl)ethanone) (5e). Yellow solid; 82% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 158–160 °C; IR (KBr): νmax (Nujol) cm−1 2923, 1184, 688; 1H NMR (CDCl3, 400 MHz) δ: 7.99 (d, J = 8.0 Hz, 4H, Ar–H), 7.63 (d, J = 8.8 Hz, 4H, Ar–H), 7.57–7.55 (m, 5H, Ar–H), 7.42–7.39 (m, 5H, Ar–H), 3.97 (s, 4H, (CH2)2); 13C NMR (CDCl3, 100 MHz) δ: 194.3, 146.8, 140.2, 134.6, 129.8, 129.5, 128.9, 127.9, 127.8, 38.2; HRMS (ESI) [M + H]+ calculated C28H22O2S 423.1419 found 423.1423.
2,2′-Thiobis(1-(thiophen-2-yl)ethanone) (5f). Yellow solid; 80% yield (Rf = 0.525 in hexane/EtOAc 80:20 v/v); mp: 145–146 °C; IR (KBr): νmax (Nujol) cm−1 3012, 1289, 992, 688; 1H NMR (CDCl3, 400 MHz) δ: 7.72 (d, J = 4.0 Hz, 2H, Ar–H), 7.62 (d, J = 4.8 Hz, 2H, Ar–H), 7.09 (t, J = 4.0 Hz, 2H, Ar–H), 3.86 (s, 4H, (CH2)2); 13C NMR (CDCl3, 100 MHz) δ: 187.7, 142.8, 135.0, 129.0, 128.3, 38.0; HRMS (ESI) [M + H]+ calculated C12H10O2S3 282.9921 found 282.9926.
Conclusions
In conclusion, we have developed a highly regioselective synthesis of 2-acyl-4-(het)arylthiazoles by the reaction between α-oxothioamides and α-bromoketones in the absence of any base. It should be noted with main concern that the reaction reported herein is an important extended work of the Hantzsch thiazole synthesis, which overcomes the limitations of earlier reported protocols. On the other hand, the same reaction, in the presence of a base yielded thioethers in a highly regioselective fashion. Further studies on synthetic applications of α-oxodithioesters and α-oxothioamides are underway in our laboratory.
Author contributions
R. N. Suresh: methodology, conceptualization, analysis. T. R. Swaroop: writing – review and editing. D. Gowda: writing – methodology, conceptualization, analysis. K. Mantelingu: writing – review and editing. K. S. Rangappa: writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
RNS thanks IOE Project (Vide No. MVV/IOE/PROJECT FELLOW/684/2019–20) Dated 01.01.2020. DG thanks CSIR for junior and senior research fellowship. KSR thanks Indian Science Congress Association (ISCA) for providing Asutosh Mookerjee Fellowship and CSIR for Emeritus scientist fellowship. KM thanks VGST for financial support.
Notes and references
- K. Tsuji and H. Ishikawa, Bioorg. Med. Chem. Lett., 1994, 4, 1601–1606 CrossRef CAS.
- Y. Tsuruni, H. Ueda, K. Hayashi, S. Takase, M. Nishikawa, S. Kiyoto and M. Okuhara, J. Antibiot., 1995, 48, 1066–1072 CrossRef PubMed.
- S. Miwatashi, Y. Arikawa, E. Kotani, M. Miyamoto, K. I. Naruo, H. Kimura, T. Yanaka, S. Asahi and S. Ohkawa, J. Med. Chem., 2005, 48, 5966–5979 CrossRef CAS PubMed.
- K. S. Rakesh, S. Jagadish, T. R. Swaroop, N. Ashwini, K. B. Harsha and K. S. Rangappa, Asian J. Biochem. Pharm. Res., 2014, 4, 316–327 Search PubMed.
- H. I. Ei-Subbagh and A. M. Al-Obaid, Eur. J. Med. Chem., 1996, 31, 1017–1021 CrossRef.
- F. W. Bell, A. S. Cantrell, M. Hoegberg, S. R. Jaskunas, N. G. Johansson, C. Jordan, M. D. Kinnick, P. Lind, J. M. Morin Jr., R. Noveen, B. Oberg, J. A. Palkowitz, C. A. Parrish, P. Pranc, C. Sahlberg, R. J. Ternansky, R. T. Vasileft, L. Vrang, S. J. West, H. Zhang and X.-Y. Zhou, J. Med. Chem., 1995, 38, 4929–4936 CrossRef CAS PubMed.
- R. Pereira, C. Gaudon, B. Iglesias, P. Germain, H. Gronemeyer and A. R. de Lera, Bioorg. Med. Chem. Lett., 2006, 16, 49–54 CrossRef CAS PubMed.
- C. Nava Zuazo, F. Chávez-Silva, R. Moo-Puc, M. J. Chan-Bacab, B. O. Ortega-Morales, H. Moreno-Diaz, D. Díaz-Coutiño, E. Hernández-Núñez and G. Navarrete-Vázquez, Bioorg. Med. Chem., 2014, 22, 1626–1633 CrossRef CAS PubMed.
- M. R. Shiradkar, K. K. Murahari, H. R. Gangadasu, T. Suresh, C. A. Kalyan, D. Panchal, R. Kaur, P. Barange, J. Ghogare, V. Mokale and M. Raut, Bioorg. Med. Chem., 2007, 15, 3997–4008 CrossRef CAS PubMed.
- A. Hamzé, E. Rubi, P. Arnal, M. Boisbrun, C. Carcel, X. SalomRoig, M. Maynadier, S. Wein, H. Vial and M. Calas, J. Med. Chem., 2005, 48, 3639–3643 CrossRef PubMed.
- I. Diaz-Padilla and A. M. Oza, Future Oncol., 2011, 7, 559–568 CrossRef CAS PubMed.
- P. Maienfisch and A. J. F. Edmunds, in Heterocyclic Chemistry in the 21st Century, ed. E. F. V. Scriven and C. A. B. T.-A. H. C. Ramsden, Academic Press, 2017, vol. 121, pp. 35–88 Search PubMed.
- R. N. Brogden and R. C. Heel, Drugs, 1986, 31, 96–130 CrossRef CAS PubMed.
- T. Tao, B. B. Ma, Y. X. Peng, X. X. Wang, W. Huang and X. Z. You, J. Org. Chem., 2013, 78, 8669–8679 CrossRef CAS PubMed.
- M. R. Prinsep, In Studies in Natural Products Chemistry, ed. A. Rahman, Elsevier Science, B.V., Amsterdam, The Netherlands, 2003; vol. 28, pp. 617−751 Search PubMed.
- A. Hantzsch and J. H. Weber, Ber. Dtsch. Chem. Ges., 1887, 20, 3118–3132 CrossRef.
- V. Facchinetti, M. M. Avellar, A. C. S. Nery, C. R. B. Gomes, T. R. A. Vasconcelos and M. V. N. de Souza, Synthesis, 2016, 48, 437–440 CAS.
- M. C. Bagley, J. W. Dale, R. L. Jenkins and J. Bower, Chem. Commun., 2004, 102–103 RSC.
- Q. Qiao, S. S. So and R. A. Goodnow, Org. Lett., 2001, 3, 3655–3658 CrossRef CAS PubMed.
- A. R. Katritzky, J. Chen and Z. J. Yang, J. Org. Chem., 1995, 60, 5638–5642 CrossRef CAS.
- S. E. Bramley, V. Dupplin, D. G. C. Goberdhan and G. D. Meakins, J. Chem. Soc., Perkin Trans. 1, 1987, 639–643 RSC.
- E. Aguilar and A. I. Meyers, Tetrahedron Lett., 1994, 35, 2473 CrossRef CAS.
- C. W. Holzapfel and G. R. Pettit, J. Org. Chem., 1985, 50, 2323–2327 CrossRef CAS.
- P. Wipf and S. Venkatraman, J. Org. Chem., 1996, 61, 8004–8005 CrossRef CAS PubMed.
- Y. Ishiwata and H. Togo, Synlett, 2008, 2637–2641 CAS.
- V. S. C. de Andrade and M. C. S. de Mattos, Synthesis, 2018, 50, 4867–4874 CrossRef CAS.
- M. Narender, M. S. Reddy, V. P. Kumar, B. Srinivas, R. Sridhar, Y. V. D. Nageswar and K. R. Rao, Synthesis, 2007, 3469–3472 CAS.
- R. M. Moriarty, B. K. Vaid, M. P. Duncan, S. G. Levy, O. Prakash and S. Goyal, Synthesis, 1992, 845–846 CrossRef CAS.
- C. G. L. Veale, K. A. Lobb, R. Zoraghi, J. P. Morrison, N. E. Reiner, R. J. Andersen and M. T. Davies-Coleman, Tetrahedron, 2014, 70, 7845–7853 CrossRef CAS.
- M. R. Prinsep, In Studies in Natural Products Chemistry, ed. A. Rahman, Elsevier Science, B.V., Amsterdam, The Netherlands, 2003, Vol. 28, pp. 617−751 Search PubMed.
- O. A. Rakitin, Sci. Synth., 2007, 31, 975–1000 Search PubMed.
- J. Yin and C. Pidgeon, Tetrahedron Lett., 1997, 38, 5953–5954 CrossRef CAS.
- I. P. Beletskaya and D. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- Y. Dong, P. Ji, Y. Zhang, C. Wang, X. Meng and W. Wang, Org. Lett., 2020, 22, 9562–9567 CrossRef CAS PubMed.
- Y. Gong, Z. Zhu, Q. Qian, W. Zong and H. Gong, Org. Lett., 2021, 23, 1005–1010 CrossRef CAS PubMed.
- J. Li, X.-E. Yang, S.-L. Wang, L.-L. Zhang, X.-Z. Zhou, S.-Y. Wang and S.-J. Ji, Org. Lett., 2020, 22, 4908–4913 CrossRef CAS PubMed.
- L. Li and Y. Ding, Mini-Rev. Org. Chem., 2017, 14, 407–431 CAS.
- K. Ravi Singh, C. Santhosh, T. R. Swaroop and M. P. Sadashiva, Org. Biomol. Chem., 2022, 20, 5771–5778 RSC.
- C. S. P. Kumar, K. B. Harsha, K. Mantelingu and K. S. Rangappa, RSC Adv., 2016, 5, 61664–61670 RSC.
- T. R. Swaroop, K. S. Rangappa and L. Torun, ChemistrySelect, 2021, 6, 177–180 CrossRef CAS.
- Dukanya, T. R. Swaroop, K. S. Rangappa and Basappa, Curr. Org. Chem., 2020, 24, 2792–2799 CrossRef CAS.
- N. Rajeev, T. R. Swaroop, S. M. Anil, K. R. Kiran, Chandra, P. E Georghiou, K. S. Rangappa and M. P. Sadashiva, Beilstein J. Org. Chem., 2020, 16, 159–167 CrossRef CAS PubMed.
- A. B. Ramesha, C. S. P. Kumar, N. C. Sandhya, M. N. Kumara and K. Mantelingu, RSC Adv., 2016, 6, 48375–48378 RSC.
- N. Rajeev, T. R. Swaroop, S. M. Anil, K. R. Kiran, K. S. Rangappa and M. P. Sadashiva, J. Chem. Sci., 2018, 130, 150 CrossRef.
- K. R. Kiran, T. R. Swaroop, K. P. Sukrutha, J. B. Shruthi, S. M. Anil, K. S. Rangappa and M. P. Sadashiva, Synthesis, 2019, 51, 4205–4214 CrossRef CAS.
- K. R. Kiran, T. R. Swaroop, C. Santhosh, K. S. Rangappa and M. P. Sadashiva, ChemistrySelect, 2021, 6, 7262–7265 CrossRef CAS.
- K. R. Kiran, T. R. Swaroop, N. Rajeev, S. M. Anil, K. S. Rangappa and M. P. Sadashiva, Synthesis, 2020, 52, 1444–1450 CrossRef CAS.
- R. N. Suresh, T. R. Swaroop, V. Shalini, K. Mantelingu and K. S. Rangappa, Tetrahedron Lett., 2023, 116, 154302 CrossRef CAS.
- CCDC 2217740 contains the ESI† for compound 4c.
- CCDC 2221383 contains the ESI† for compound 5b.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Darshini Gowdaa,
Kempegowda Mantelingu
b,
Darshini Gowdaa,
Kempegowda Mantelingu