
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiperonal chalcone derivative incorporating the pyrazolo[3,4-b]pyridine moiety; crystal structure, spectroscopic characterization and quantum chemical investigations†
Efraín Polo-Cuadrado a,
Karoll Ferrer*b,
Edison Osorioc,
Iván Britod,
Jonathan Cisternae,
Luis Espinozaf,
Joel B. Alderete*g and
Margarita Gutiérrez*a
a,
Karoll Ferrer*b,
Edison Osorioc,
Iván Britod,
Jonathan Cisternae,
Luis Espinozaf,
Joel B. Alderete*g and
Margarita Gutiérrez*a
aLaboratorio Síntesis Orgánica y Actividad Biológica (LSO-Act-Bio), Instituto de Química de Recursos Naturales, Universidad de Talca, Casilla 747, Talca 3460000, Chile. E-mail: mgutierrez@utalca.cl
bLaboratory of Growth Regulators, Institute of Experimental Botany, The Czech Academy of Sciences, Palacký University, Šlechtitelů 27, 78371 Olomouc, Czech Republic. E-mail: karoll.ferrerpertuz@upol.cz
cFacultad de Ciencias Naturales y Matemáticas, Universidad de Ibagué, Carrera 22 Calle 67, Ibagué 730001, Colombia
dDepartamento de Química, Facultad de Ciencias Básicas, Universidad de Antofagasta, Campus Coloso, Antofagasta 02800, Chile
eDepartamento de Química, Facultad de Ciencias, Universidad Católica del Norte, Sede Casa Central, Av. Angamos 0610, Antofagasta, Chile
fDepartamento de Química, Universidad Técnica Federico Santa María, Av. España No. 1680, Valparaíso 2340000, Chile
gInstituto de Química de Recursos Naturales (IQRN), Universidad de Talca, Avenida Lircay S/N, Casilla 747, Talca, Chile. E-mail: joel.alderete@utalca.cl
First published on 9th February 2023
Abstract
A single crystal of a piperonal chalcone derivative was obtained, fully characterized, and crystallized by a slow evaporation technique. The synthesized compound was characterized by UV-Visible, FT-IR, HRMS, 1H NMR, and 13C NMR spectroscopic studies and X-ray crystallography, revealing that the crystal belongs to a triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, Z = 2. In the present work, we focus on molecular modeling studies such as Hirshfeld surface analysis, energy framework calculations, frontier molecular orbital analysis, natural bond orbital analysis, and NLO properties of a π-conjugate system combining the chalcone and the pyrazole[3,4-b]pyridine scaffolds to describe the in-depth structural analysis thereof. Good agreement was found between the calculated results and experimental data. In addition, Hirshfeld surface analysis of the crystal structure showed that the intermolecular stabilization in the crystal packing comes mainly from H⋯H bond interactions. The chalcone crystal exhibits significant NLO properties suggesting that it could be considered a potential candidate for application in nonlinear optical devices.
space group, Z = 2. In the present work, we focus on molecular modeling studies such as Hirshfeld surface analysis, energy framework calculations, frontier molecular orbital analysis, natural bond orbital analysis, and NLO properties of a π-conjugate system combining the chalcone and the pyrazole[3,4-b]pyridine scaffolds to describe the in-depth structural analysis thereof. Good agreement was found between the calculated results and experimental data. In addition, Hirshfeld surface analysis of the crystal structure showed that the intermolecular stabilization in the crystal packing comes mainly from H⋯H bond interactions. The chalcone crystal exhibits significant NLO properties suggesting that it could be considered a potential candidate for application in nonlinear optical devices.
1 Introduction
Nonlinear optical (NLO) materials possess optical properties that depend on the intensity of incident light.1 NLO materials significantly impact different fields, such as medicine and photonics. Additionally, they are widely used in industrial applications such as second harmonic generation (SHG), optical modulation, optical switching, laser technology, optical data storage, electro-optical switches, optical logic, signal processing, ultra-fast optical communication, and for optical limiting purposes.2–8The NLO properties of a material depend on the degree of charge separation (molecular hyperpolarizability) induced by light and the photoinductive changes of the molecular dipole moment.1,9 Thus, in general, the NLO properties of inorganic substances depend on their constituent ions. In contrast, the NLO properties of organic substances depend on the intermolecular donor–acceptor interaction, delocalized π electron systems, and their ability to crystallize non-symmetrically to the center.8 Current research trends have been focused on organic materials, mainly those with chromophore groups and widely delocalized π electron structures, since their NLO coefficients are higher than those of inorganic materials.
From the perspective of optical device applications, organic materials could be considered the “materials of the future” due to unique properties such as ultra-fast optical response, high resistance to damage, broader transparency down to the blue region, ease, and low cost of production. Furthermore, their architectural flexibility allows modification at the molecular level of these compounds, which is suitable for obtaining large crystals of high optical quality and desired optical properties.8,10 Molecular improvements such as increased planar conformations and chromophore groups in organic structures have attracted attention, as they have been reported to enhance NLO activity. In this regard, increased planarity contributes to electron mobility within the π-conjugated molecular system, while chromophore groups increase the hyperpolarizability of the molecule.11,12
Among the organic compounds, chalcones stand out due to their high tendency to crystallize, resistance to laser damage, excellent second and third-order NLO responses, and extensive applications in biological and pharmaceutical fields.5,13–15 Several experimental studies have been conducted on the NLO properties of various chalcones, and encouraging results have been reported for future applications such as nonlinear frequency conversion, nonlinear refraction, optical limiters, and SHG (15 times greater than urea).5,8,16–20
However, although chalcones are an essential group of molecules in terms of NLO properties, structural building blocks such as pyrazoles and pyridines are relevant groups that confer planarity and extension of the delocalized π-electron conjugation to the systems into which they are incorporated. Specifically, it has been found that the asymmetric electronic distribution can be improved in either or both the ground state and excited state configurations by adding the appropriate functionality at the ends of the conjugated chalcone systems, leading to an increase in NLO. Electron donor and acceptor components result in a considerable change in hyperpolarizability.9,12,21–24
Hence, prompted by the above observations and our interest in pyrazolopyridine derivatives, we have focused on the structural properties of a piperonal chalcone derivative incorporating the pyrazolo[3,4-b]pyridine moiety. Apart from XRD, UV-Visible, FT-IR, HRMS, and NMR experimental techniques, confirmation of the stable crystal structure has been based on the results of quantum chemistry such as geometry optimization, Hirshfeld surface analysis, energy framework calculations, frontier molecular orbitals (FMOs) analysis, natural bond orbital (NBO) analysis, and nonlinear optical (NLO) properties of the title compound.
2 Experimental
2.1 Materials and methods
All chemical reagents and organic solvents were obtained from commercial suppliers and used without further purification. The experiments were performed in a Discover microwave apparatus (CEM Corporation, USA) and a Branson 1510 ultrasonic cleaning bath with a mechanical timer and a heater switch, 47 kHz. Thin-layer chromatography (TLC) was performed to check the purity of compounds on silica gel 60 HF254 plates (Merck, Germany). The melting point ranges (mp) were taken on an Electrothermal IA9100 apparatus (Stone, UK) using the one-end open capillary method and were uncorrected. IR spectra (KBr discs, 500–4000 cm−1) were recorded on a NEXUS 670 FT-IR spectrophotometer (Thermo Nicolet, USA). 1H and 13C NMR spectra were recorded using DMSO-d6 and CDCl3 as solvents and tetramethylsilane (TMS) as an internal reference on an AM-400 spectrometer (Bruker, Germany) at working frequencies 400 and 100 MHz, respectively. High-resolution mass spectrometry (HRMS) analyses were carried out using a Bruker “Compact” quadrupole time-of-flight mass spectrometry (qTOF-MS, Germany) coupled with an Apollo II ion funnel electrospray ionization (ESI) source. The UV-Vis spectrum was recorded using a Spectroquant UV/VIS Pharo 300 Spectrophotometer (Merck, Germany) in the wavelength range of 160–750 nm.2.2 Synthesis of 1-(3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridin-5-il)etanone (4)
Pyrazolo[3,4-b]pyridine 4 was synthesized according to our previously reported procedure (Scheme 1).25 A catalytic amount of InCl3 (15% mol) was added to a solution of equimolar amounts of 5-aminopyrazole 1 (1 mmol), p-formaldehyde 2 (1 mmol), and acetylacetone 3 (1 mmol) in water (3 mL). A focused microwave reactor irradiated the mixture at 300 MW and 210 °C for 17 min. The reaction mixture was poured into water and extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated to afford the title compound 4 (238.7 mg, 90% yield) as a yellow solid. mp: 100–102 °C; IR (KBr, cm−1): 3065, 2960, 2924, 2853, 1679, 1611, 1592, 1503, 1436, 1236, 1119, 1026, 944, 755, 692, 585; 1H NMR (DMSO-d6, 400 MHz) δH: 2.63 (s, 3H, CH3), 2.67 (s, 3H, CH3), 2.77 (s, 3H, CH3), 7.31 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 8.0 Hz, 2H), 8.25 (d, J = 7.7 Hz, 2H), 8.87 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δC: 12.2 (CH3), 25.8 (CH3), 29.4 (CH3), 114.4 (C), 120.1 (2 × CH), 125.7 (CH), 127.2 (C) 129.1 (2 × CH), 133.3 (CH), 138.9 (C), 144.3 (C), 149.5 (C), 158.5 (C), 199.6 (C); HRMS (ESI, m/z): calculated for C16H16N3O [M + H]+ 266.1293 found 266.2751.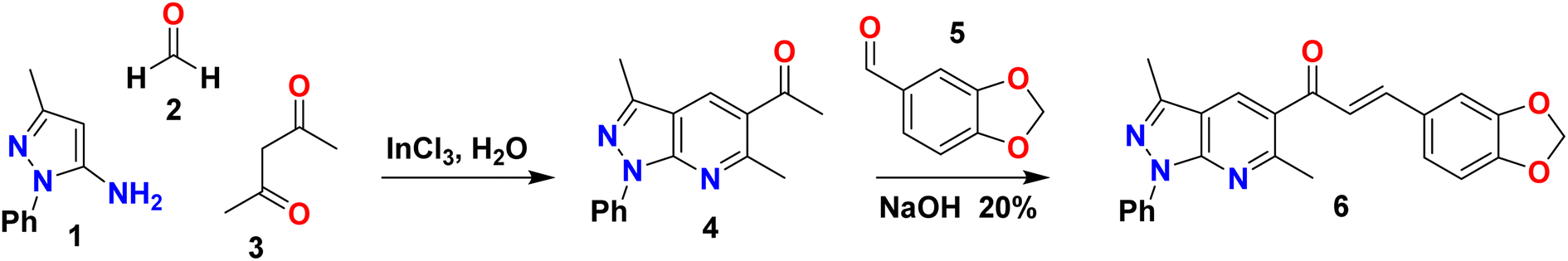 | ||
| Scheme 1 Synthetic route of (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridin-5-yl)prop-2-en-1-one (6). | ||
2.3 Synthesis and crystallization of (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridin-5-yl)prop-2-en-1-one (6)
Piperonal chalcone derivative 6 was prepared according to our previously reported experimental procedure (Scheme 1).25 KOH (1 mmol) was added to a solution of pyrazolo[3,4-b]pyridine 4 (1 mmol) and piperonal 5 (1 mmol) in ethanol (2 mL). The mixture was sonicated for 10 min in an ultrasonic cleaning bath. The end of the reaction was detected by TLC using a mixture of solvent (dichloromethane/ethyl acetate 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. The reaction mixture was allowed to cool down in an ice-cold water bath. A yellow precipitate was collected by vacuum filtration and washed with cold water, followed by recrystallization from ethanol to obtain the desired compound 6 (250.3 mg, 63% yield) as a yellow solid. mp: 186–188 °C; IR (KBr, cm−1): 3082, 2990, 2894, 1655, 1589, 1577, 1444, 1308, 1033, 984, 754; 1H NMR (CDCl3, 400 MHz) δH: 2.63 (s, 3H, CH3), 2.81 (s, 3H, CH3), 6.01 (s, 2H, CH2), 6.82 (d, J = 8.0 Hz, 1H), 7.12–7.04 (m, 3H), 7.28 (t, J = 7.5 Hz, 1H), 7.52–7.45 (m, 3H), 8.16 (s, 1H), 8.32 (d, J = 7.6 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δC: 12.43 (CH3), 24.75 (CH3), 101.64 (CH2), 106.53 (CH), 108.59 (CH), 114.20 (C), 120.60 (2 × CH), 123.97 (CH), 125.34 (CH), 125.57 (CH), 128.72 (C), 128.76 (C), 128.91 (2 × CH), 129.87 (CH), 139.28 (C), 143.36 (C), 145.77 (CH), 148.41 (C), 150.12 (C), 150.16 (C), 158.01 (C), 193.85 (C
:1) as an eluent. The reaction mixture was allowed to cool down in an ice-cold water bath. A yellow precipitate was collected by vacuum filtration and washed with cold water, followed by recrystallization from ethanol to obtain the desired compound 6 (250.3 mg, 63% yield) as a yellow solid. mp: 186–188 °C; IR (KBr, cm−1): 3082, 2990, 2894, 1655, 1589, 1577, 1444, 1308, 1033, 984, 754; 1H NMR (CDCl3, 400 MHz) δH: 2.63 (s, 3H, CH3), 2.81 (s, 3H, CH3), 6.01 (s, 2H, CH2), 6.82 (d, J = 8.0 Hz, 1H), 7.12–7.04 (m, 3H), 7.28 (t, J = 7.5 Hz, 1H), 7.52–7.45 (m, 3H), 8.16 (s, 1H), 8.32 (d, J = 7.6 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δC: 12.43 (CH3), 24.75 (CH3), 101.64 (CH2), 106.53 (CH), 108.59 (CH), 114.20 (C), 120.60 (2 × CH), 123.97 (CH), 125.34 (CH), 125.57 (CH), 128.72 (C), 128.76 (C), 128.91 (2 × CH), 129.87 (CH), 139.28 (C), 143.36 (C), 145.77 (CH), 148.41 (C), 150.12 (C), 150.16 (C), 158.01 (C), 193.85 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); HRMS (ESI, m/z): calculated for C24H20N3O3+ [M + H]+ 398.1499, found 398.1499.
O); HRMS (ESI, m/z): calculated for C24H20N3O3+ [M + H]+ 398.1499, found 398.1499.
2.4 X-ray crystal structure determination
Suitable crystals of the piperonal chalcone derivative 6 were chosen for the X-ray crystallographic analysis. X-ray intensity data were collected at 296 K in a Bruker D8 Venture diffractometer equipped with a bidimensional CMOS Photon 100 detector, using Mo-Kα radiation of wavelength 0.71073 Å. The diffraction frames were integrated using the APEX3 package26 and were corrected for absorption effects using the multi-scan method (SADABS). The crystal structure was solved by intrinsic phasing27 using the OLEX2 program.28 Refinement was carried out by full-matrix least-squares methods based on F2 (SHELXL-2014).27 The figures of the X-ray structure of the title compound were generated with the help of OLEX2 and ORTEP program packages. Crystallographic information files were deposited in the Cambridge Crystallographic Data Centre (CCDC) with the deposition number 2218131.†3 Computational methods
3.1 Theoretical calculations
The X-ray diffraction experimental result of the title compound was directly taken for an initial molecular geometry. The geometry optimization of the monomeric structure was carried out using the M062X exchange–correlation functional and 6-311++G(d,p) basis set29,30 in the gas phase. M062X is a hybrid meta exchange–correlation functional with double the nonlocal exchange (2×), which is parametrized for nonmetal and long noncovalent range interactions.31 The calculations were performed using Gaussian 09 software.32Optimized structural parameters were used to calculate the 1H and 13C NMR chemical shifts in chloroform with the Gauge-Independent Atomic Orbital (GIAO)33 method at M062X, TPSS, and B3LYP levels using 311++G(d,p) basis set. The solvent effect on theoretical NMR parameters was included using the Conductor polarizable Continuum Model (CPCM)34–37 provided by Gaussian 09 program.32
The theoretical electronic spectrum of the monomeric structure was performed through the time-dependent density-functional theory (TDDFT) calculations38,39 at the M062X/6-311++G(d,p) level. The solvent effect of methanol on the calculated electronic properties of the studied compound was investigated using the polarizable continuum model (PCM).40,41
The frontier molecular orbitals (FMOs),42 the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), which account for the ability to donate and accept electrons, were calculated at DFT/M062X/6-311++G(d,p) level. Some global reactivity descriptors, such as HOMO–LUMO gap, chemical hardness (η), chemical potential (μ), electrophilicity index (ω), and chemical softness (S), which are based on the HOMO and LUMO energies were studied to measure the stability of the synthesized molecule.
The natural bond orbital (NBO)43 calculations of the title compound were achieved at M062X/6-311++G(d,p) level of theory using the NBO 3.1 program within the Gaussian 09 software.32 Chemcraft program package (https://www.chemcraftprog.com) was used to draw the molecular graphs.
3.2 Nonlinear optical (NLO) details
The electric dipole moment, linear polarizability, and the first and second hyperpolarizability tensors were evaluated using FF approach to explore the non-lineal optical (NLO) properties of the title compound with a field frequency of 0.0010 au at the DFT/DCAM-B3LYP/6-311G++(d,p) level.DFT functional has shown results close to CCSD and MP2 methodologies,44,45 indicating efficiency and accuracy for calculation of second order hyperpolarizability. In the finite field (FF) method, when a molecule is subjected to a static field (F), the energy (E) of the molecule can be expressed as:
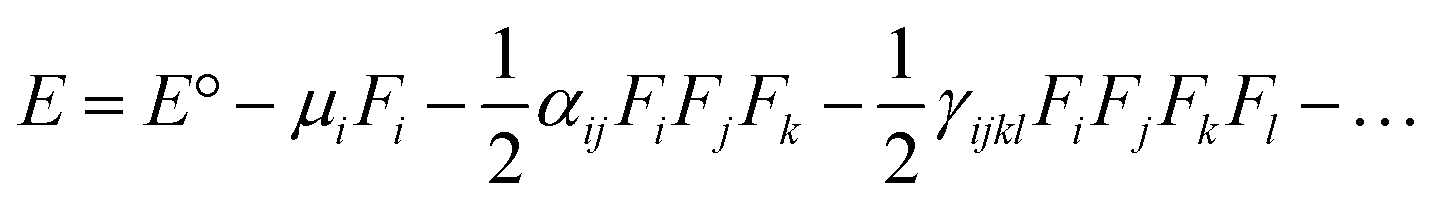
| μ = (μx2 + μy2 + μz2)1/2 |
On the other hand, α corresponds to the linear polarizability tensor, while β and γ are the first and second hyperpolarizability tensors, with i, j, and k as the labels for x, y, and z components, respectively. The tensors equations are given as follows:
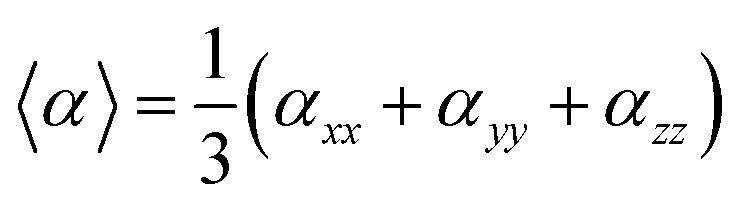
| 〈β〉 = [(βxxx + βxyy + βxzz)2 + (βyyy + βyzz + βyxx)2 + (βzzz + βzxy + βzyy)2]1/2 |
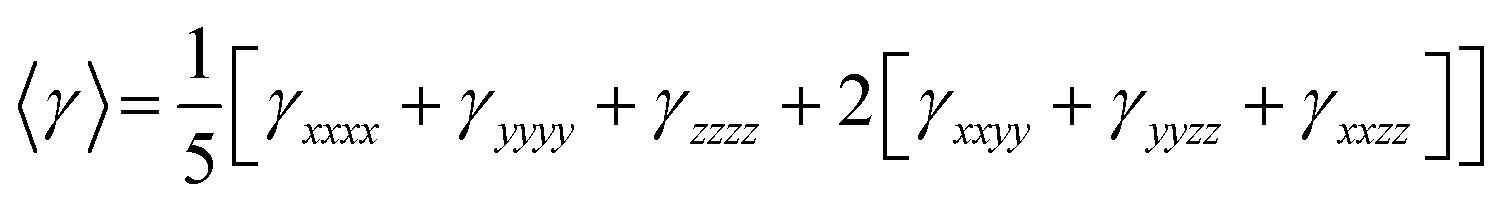
3.3 Hirshfeld surface analysis details
Hirshfeld surface analysis and 2D-fingerprint plots46 were carried out using the CrystalExplorer 21.3 software47 to determine significant intermolecular interactions and contacts in the crystal structure of the studied compound. For computations, the crystallographic information file (*.cif) was initially used for the analysis. Two distances were calculated for each point on the Hirshfeld isosurface, de and di, representing distances from a point on the surface to the nearest nucleus outside and inside the surface. All measures were normalized using dnorm distance (normalized contact distance), which is defined in eqn (1) and is based on de, di and the van der Waals radius (vdW) for de and di atoms within the compound.48,49
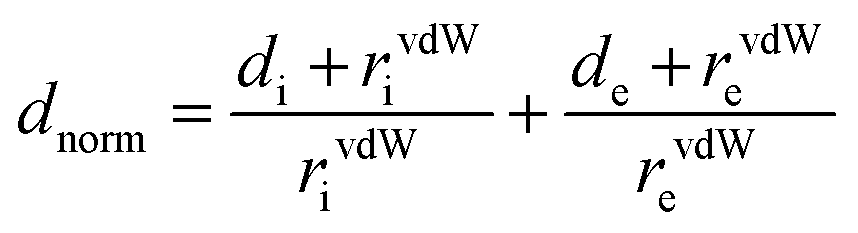 | (1) |
The electrostatic potentials were mapped on the Hirshfeld surface at B3LYP/6-31G(d,p) level of theory over a range of ±0.002 au.50 The bond lengths of hydrogen atoms involved in interactions were normalized to standard values from neutron diffraction measurements (C–H = 1.083 Å, N–H = 1.009 Å, O–H = 0.983 Å)51 for the generation of fingerprint plots. The intermolecular energies of the molecular pairs in the crystal packing were calculated at B3LYP/6-31G(d,p) level of theory in a cluster of radius 3.8 Å around the molecule.52,53
4 Results and discussion
4.1 Chemistry and characterization
Piperonal chalcone derivative 6 was synthesized according to our previously reported experimental procedure outlined in Scheme 1.25 The one-pot condensation of 5-amino-3-methyl-1-phenylpyrazole 1, p-formaldehyde 2 and β-diketone 3 under microwave irradiation in aqueous media and catalyzed by InCl3 generated the pyrazolo[3,4-b]pyridine derivative 4 (90% yield) followed by treatment with piperonal 5 to give the title compound 6 (63% yield). The structure of the piperonal chalcone derivative 6 was characterized by 1H NMR, 13C NMR, FT-IR and HRMS. The 1H- and 13C-NMR assignments have been completed by 1D and 2D heteronuclear correlation HSQC and HMBC techniques.The 1H NMR spectrum of compound 6 was measured in deuterated chloroform and revealed the presence of nineteen protons (see Fig. S1 and Table S1†). The two singlets due to methyl groups linked to the pyrazole and pyridine rings were observed at δH = 2.63 ppm (s, 3H, CH3-6) and δH = 2.81 ppm (s, 3H, CH3-17), respectively. The methylene protons were found at δH = 6.01 ppm (s, 2H, CH2). At the same time, the doublet corresponding to H-24 was shown at δH = 6.82 (d, J = 8.0 Hz, 1H). The presence of signals at δH = 8.32 ppm (d, J = 7.6 Hz, 2H, H-12 and H-16), 7.52–7.45 (m, 2H, H-13 and H-15) and 7.28 (t, J = 7.5 Hz, 1H, H-14) were assigned to aromatic protons of the phenyl group attached to the pyrazolo[3,4-b]pyridine system. A singlet observed at δH = 8.16 ppm (s, 1H) was due to the resonance of the pyridine ring proton corresponding to H-11.
The 13C NMR spectrum of the title compound was measured in deuterated chloroform and revealed the presence of twenty-four carbon atoms consistent with the target compound (see Fig. S2 and Table S1†). 13C NMR showed a signal at δC = 193.85 ppm assigned to the carbonyl carbon of chalcone, and the signals at δC = 12.43 and 24.75 ppm were attributed to methyl groups CH3-6 and CH3-17, respectively.
Additional evidence for the structure of piperonal chalcone derivative (6) was provided from 2D HMBC spectrum, which showed significantly correlated couplings of the α,β-unsaturated ketone system (see Fig. 1). The α-H (H-18) showed correlations with carbonyl group (13C, at 193.85 ppm) and C-22 (13C, at 128.76 ppm). In contrast, the β-H (21) showed correlations with both C-23 (13C, at 123.97 ppm) and C-27 (13C, at 106.53 ppm). On the other hand, the methyl group CH3-6 (1H, at 2.63 ppm) showed correlations with C-3 (13C, at 143.36 ppm) and C-2 (13C, at 114.20 ppm), whereas the H-11 signal (1H, at 8.16 ppm) showed HMBC correlation with both C-1 (13C, at 150.16 ppm) and C-9 (13C, at 158.01 ppm).
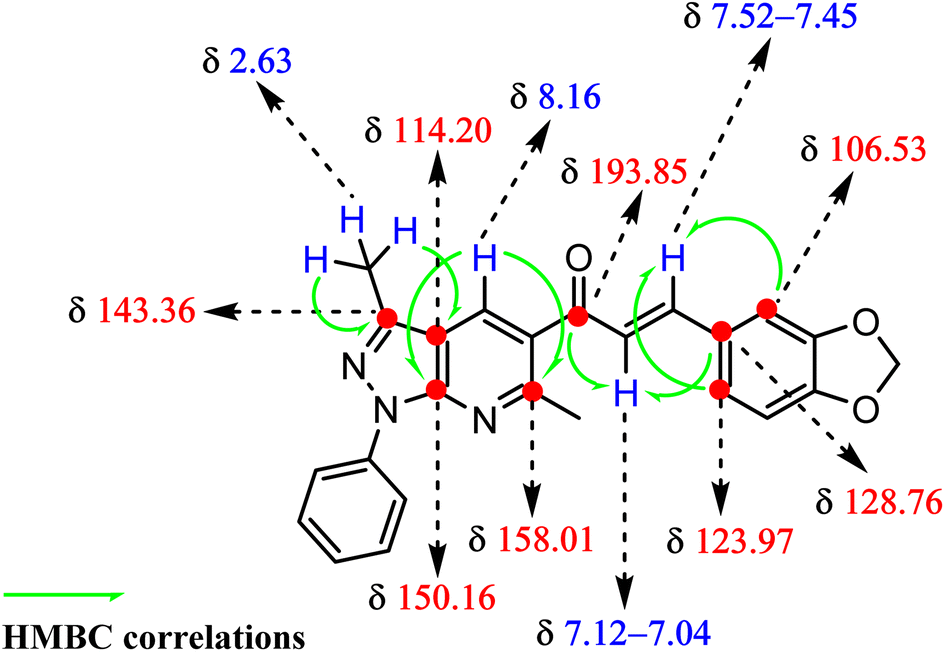 | ||
| Fig. 1 HMBC-assisted NMR-based structural determination of compound 6. | ||
The IR spectrum of the synthesized chalcone is presented in Fig. S3† and showed the presence of absorption bands at 3082, 2990 and 2894 cm−1 due to aromatic and aliphatic C–H vibrations, the CO, CN and CC stretching vibrations were identified as distinguishing absorption bands at 1655, 1589 and 1577 cm−1 (see Table S2†).
The electrospray ionization-mass spectrometry (ESI-MS) of the title compound showed an [M + H]+ peak of 398.1499, corresponding to the molecular formula C24H20N3O3+. Thus, based on the above spectral data, the structure of the synthesized chalcone was confirmed (see Fig. S4†).
All computed and experimental 1H and 13C NMR chemical shift values are listed in Tables S3 and S4.† M062X and B3LYP systematically overestimated the chemical shift values in 13C, with high errors, greater than 20 ppm for some atoms. We have calculated the absolute errors in 13C and found the following trend in the sum of the absolute errors: M062X > B3LYP > TPSS. In the case of the 1H spectra, the next trend was found: M062X > B3LYP > TPSS.
Correlation graphs were prepared and given in Fig. S5 and S6.† A good linear correlation between experimental and theoretical 1H and 13C NMR chemical shift values of the title compound was observed as indicated by the R2 values (see corresponding graphs in ESI†).
4.2 X-ray single crystal studies
The molecular structure of the title compound corresponds to a chalcone containing 2,2-dimethyl-1,3-benzodioxole and pyrazole[3,4-b]pyridine moieties. The piperonal chalcone shows a centrosymmetric setting with normal bond distances and angles.51 The compound shows the E isomer in the solid state, being the most common conformation for this type of compound.54,55 The ORTEP diagram of the title compound drawn at 30% probability thermal displacement ellipsoids with the atom numbering scheme is presented in Fig. 2. The molecule is essentially planar, which encourages us to study the NLO properties54 of the title compound.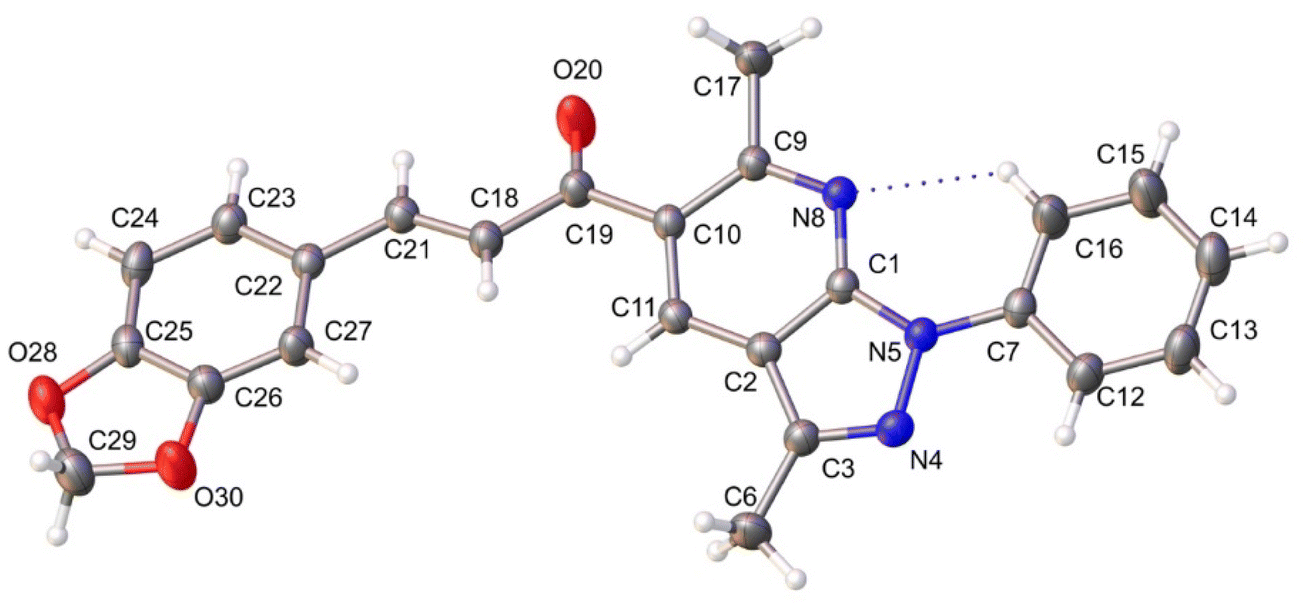 | ||
| Fig. 2 ORTEP view of the title compound drawn at 30% of probability thermal displacement ellipsoids. | ||
For the title compound, non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were included in their calculated positions, assigned fixed isotropic thermal parameters, and constrained to ride on their parent atoms. The crystallographic data and the parameters of the refinement process of the title compound are given in Table 1.
| Empirical formula | C24H19N3O3 | Crystal size (mm) | 0.446 × 0.269 × 0.184 |
| Formula mass, g mol−1 | 397.42 | F(000) | 416.0 |
| Collection T, K | 295.09 | Abs coeff. (mm−1) | 0.090 |
| Crystal system | Triclinic | 2θ range (°) | 5.692 to 54.052 |
| Space group | P |
Range h, k, l | −11/10, −13/13, −13/14 |
| a (Å) | 8.669(6) | No. total refl. | 14220 |
| b (Å) | 10.856(7) | No. unique refl. | 4213 [Rint = 0.0373, Rsigma = 0.0350] |
| c (Å) | 11.092(7) | Comp. θmax (%) | 98.1 |
| α (°) | 95.470(16) | Max/min transmission | 0.984/0.971 |
| β (°) | 97.788(14) | Data/restraints/parameters | 4213/0/274 |
| γ (°) | 106.365(14) | Final R [I > 2σ(I)] | R1 = 0.0521, wR2 = 0.1209 |
| V (Å3) | 982.5(11) | R indices (all data) | R1 = 0.0926, wR2 = 0.1544 |
| Z | 2 | Goodness of fit/F2 | 1.021 |
| ρcalcd (g cm−3) | 1.343 | Largest diff. peak/hole (e Å−3) | 0.19/−0.19 |
In the crystal structure, one C–H⋯N intramolecular hydrogen bond interaction is observed with a graph-set motif56 S11(6). Additionally, the molecules are linked by π–π interactions, observed between benzodioxole rings, and benzodioxole and pyridine rings with mean distance of 3.945 and 3.962 Å, distance plane-centroid of 1.602 and 1.872 Å, and angles between rings α, β and γ of 15.77(10)° and 0.02(11)°; 21.5° and 24.0°; 28.2° and 24.0°, respectively (see Fig. 3).
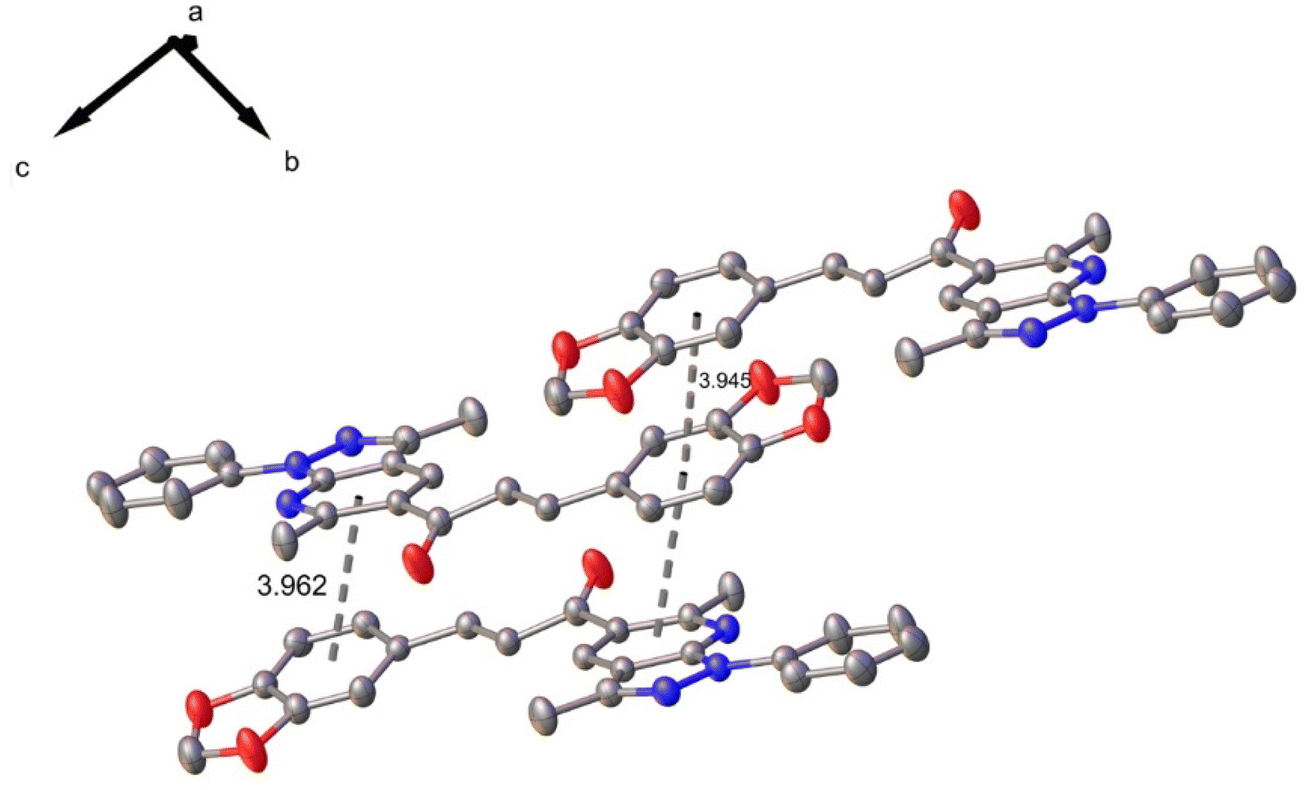 | ||
| Fig. 3 π–π interactions in compound 6. | ||
4.3 Comparison of geometry data
The root mean squared (RMS) value predicted by Chemcraft software, which accounts for the difference between the monomer from X-ray diffraction experiment and the theoretically optimized geometry, was relatively low: 0.550 Å. Then, a good agreement was found between the optimized molecular geometrical parameters and experimental ones. Only a slight difference in the dihedral angle (C11, C10, C19 and C18) was shown between the monomer of X-ray diffraction experiment and the optimized geometry, which have values of 20.62 and 32.34, respectively. However, these results demonstrate the structural validity of the optimized geometry. A complete list of coordinates, bond lengths, bond angles and dihedral angles obtained by X-ray diffraction and DFT/M062X/6-311++G(d,p) of the title compound are presented in the ESI (see Tables S6–S8†).4.4 UV-Vis studies and electronic properties
The comparison of the experimental and simulated theoretical UV-Vis spectra of piperonal chalcone has been studied. The experimental spectrum scanned in methanol solution within the range of 190–410 nm (Fig. 4a) includes two principal bands localized around 280 and 345 nm. These two bands were assigned to n → π* and π → π* electronic transitions. The theoretical electronic spectrum (Fig. 4b) shows two principal π → π* electronic transitions located around 334 and 329 nm near the experimental signal. On the other hand, the theoretical electronic spectrum presents two local maximums with oscillator strengths (f) of 0.2993 and 0.2574, which are localized around 282 and 255 nm, respectively, and agree with the experimental UV-Vis spectra. As can also be seen in Fig. 4, the theoretical spectrum presents only values higher than 219 nm. Those values lower than this wavelength could not be simulated in our TDDFT calculations. Overall, the theoretical UV-Vis spectrum shows that the absorption values are at the exact location of the experimental average (x-axis in Fig. 4). However, the calculated absorbance value does not match the experimental value because this measurement is directly proportional to the sample concentration, which cannot be calculated in our Gaussian experiment. To obtain absorbance values close to the experimental ones, it is necessary to model different geometrical conformations using explicit solution methods involving a long computational process, which is an unintended goal of this work. | ||
| Fig. 4 Experimental (a) and calculated (b) electronic absorption spectra of title compound. | ||
4.5 Frontier molecular orbitals (FMOs) analysis and molecular reactivity
The frontier molecular orbitals (FMOs), HOMO and LUMO, are the main orbitals responsible for chemical stability or reactivity. The HOMO–LUMO plots for the title compound is shown in Fig. 5. The molecular form of HOMO shows a high electron density located over N–C groups. In the case of LUMO, the highest probability regions are in the same area as the HOMO, thus an allowed electronic transition could occur between these two states.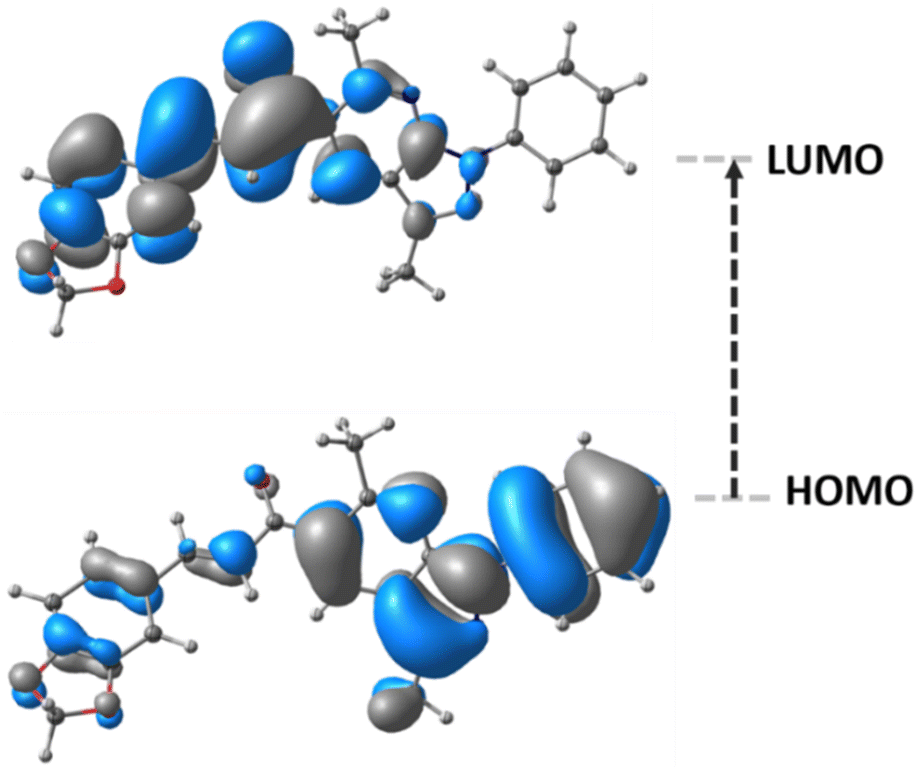 | ||
| Fig. 5 Frontier molecular orbitals of title compound. | ||
HOMO energy is associated with the electron-donating ability of a compound, whereas LUMO energy with electron-accepting. The frontier molecular orbital energy gap provides information about the chemical reactivity and kinetic stability. A molecule with a small frontier orbital gap is associated with a high chemical reactivity and low kinetic stability.
The HOMO and LUMO energies, and frontier orbital energy gap of the title compound were found to be 7.2861, −1.5934, and 5.6926 eV, respectively. The calculated values of the chemical hardness, chemical potential, electrophilicity index, and chemical softness parameters for the compound described above are 2.84, −4.43, 3.46 eV, and 0.17 eV−1, respectively.
The piperonal chalcone derivative is expected to be less polarizable and associated with a low chemical reactivity due to its larger molecular gap and chemical hardness values, indicating a hard molecule with low polarizability with high kinetic stability.
4.6 Natural bond orbital (NBO) analysis
The charge transfer and intramolecular interactions between the occupied and unoccupied molecular orbitals of the title compound were analyzed through the natural bond orbital (NBO) analysis. The analysis of second-order perturbation energy57 (E(2)) showed that the two most important donor–acceptor intermolecular interactions (Fig. 6) presented in the compound are interactions between CC π and CC π antibonding orbitals, with energies values of 1138.4 (Fig. 6a) and 161.2 (Fig. 6b) kcal mol−1, respectively. This mixing of donors and acceptors leads to an overall decrease in stabilization energy.
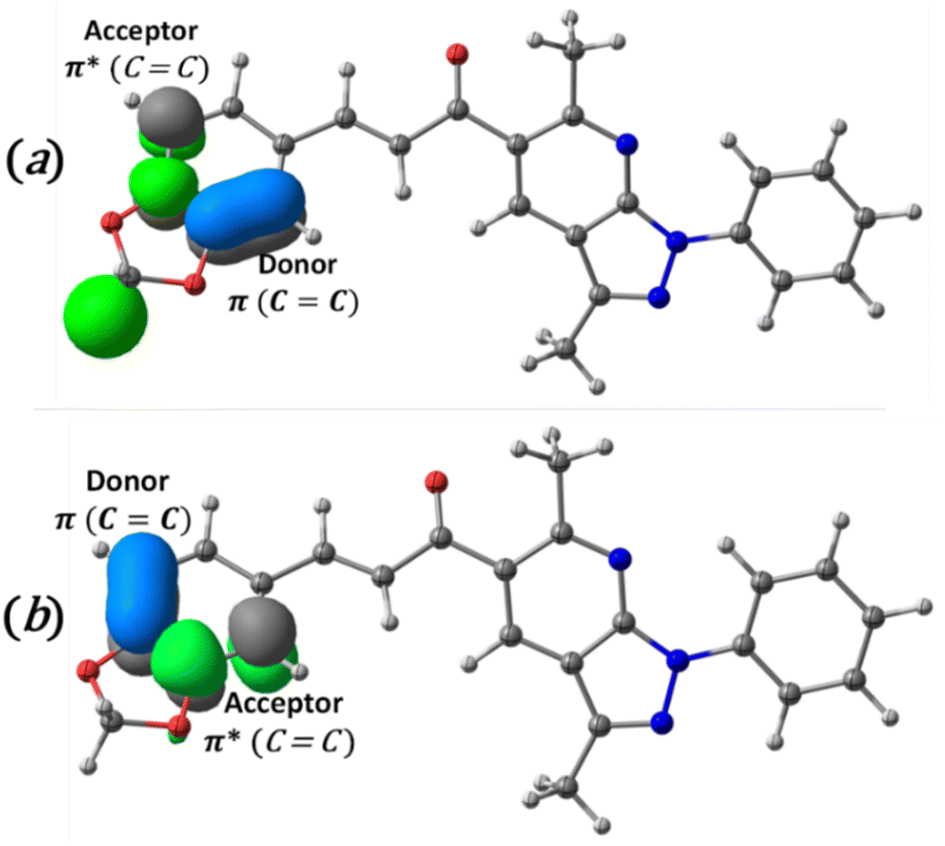 | ||
| Fig. 6 Schematic diagram of intramolecular charge transfer from CC π bond to CC π antibonding and the second-order perturbation energies (E(2)). (a) 1138.4 kcal mol−1 and (b), 161.2 kcal mol−1. | ||
4.7 Nonlinear optical (NLO) properties
The dipole moment, polarizability and hyperpolarizability results are presented in Table 2. Urea has been used as a prototype molecule for a comparative approach to NLO properties.58| μ (D) | α (×10−24 esu) | β (×10−30 esu) | γ (×10−36 esu) | |
|---|---|---|---|---|
| Urea | 3.875 | 4.823 | 0.414 | 2.941 |
| Studied compound (6) | 5.007 | 50.830 | 27.464 | 137.399 |
| (E)-1-(1,3-Benzodioxol-5-yl)-4,4-dimethylpent-1-en-3-one59 | 3.546 | 27.625 | 22.413 | 44.189 |
| (2E)-1-(1,3-Benzodioxol-5-yl)-3-phenylprop-2-en-1-one (SSP1)60 | 3.011 | 31.346 | 8.344 | 67.879 |
The relevance of polarizability and hyperpolarizability of a molecular system depends on electronic communication between two different parts of a molecule. The results showed non-zero μ values for the title compound, which should cause microscopic quadratic and cubic hyperpolarizabilities, with resulting non-zero values derived by the second numerical derivatives of electric dipole moments to the field implemented (see Table 2). The calculated dipole moment (μ) for the title compound is 1.29 times higher than that obtained from urea. The non-zero 〈α〉, 〈β〉 and 〈γ〉 tensors values for the title compound predict the ability to acquire second- and third-order microscopic NLO responses. The of α, β and γ calculated values for the title compound are 10.5, 66.3 and 46.7 times higher than those obtained from urea. Since we lack experimental data on NLO properties, the implemented theoretical analyses provide a predictive tool to show that the studied compound can be usefully applied in this field. To be more confident of these results, we have added the same theoretical analysis for two reported molecules with optical properties and structures similar to our studied compound (Table 2). Surprisingly the α, β and γ calculated values of our compound present better results than reference molecules. Hence, compared with urea and the other two molecules with NLO properties, the title compound may fulfill many of the quadratic optical nonlinearity requirements and could have potential applications in NLO and electro-optic devices.
4.8 Hirshfeld surface analysis and 2D fingerprint plots
Visualization of intermolecular contacts through the crystal structure was achieved by Hirshfeld surface analysis and was performed as a complement to XRD analysis. The dnorm surfaces were visualized with three colors, white indicates the contacts with distances equal to the sum of the van der Waals radii (dnorm = VdW radii), blue areas show the positive electrostatic potential (hydrogen-bond donors, dnorm > VdW radius), and red regions indicate the negative electrostatic potential (hydrogen-bond acceptors, dnorm < VdW radii).50 All the contacts contributing to the total area of the surface are illustrated in Fig. 7.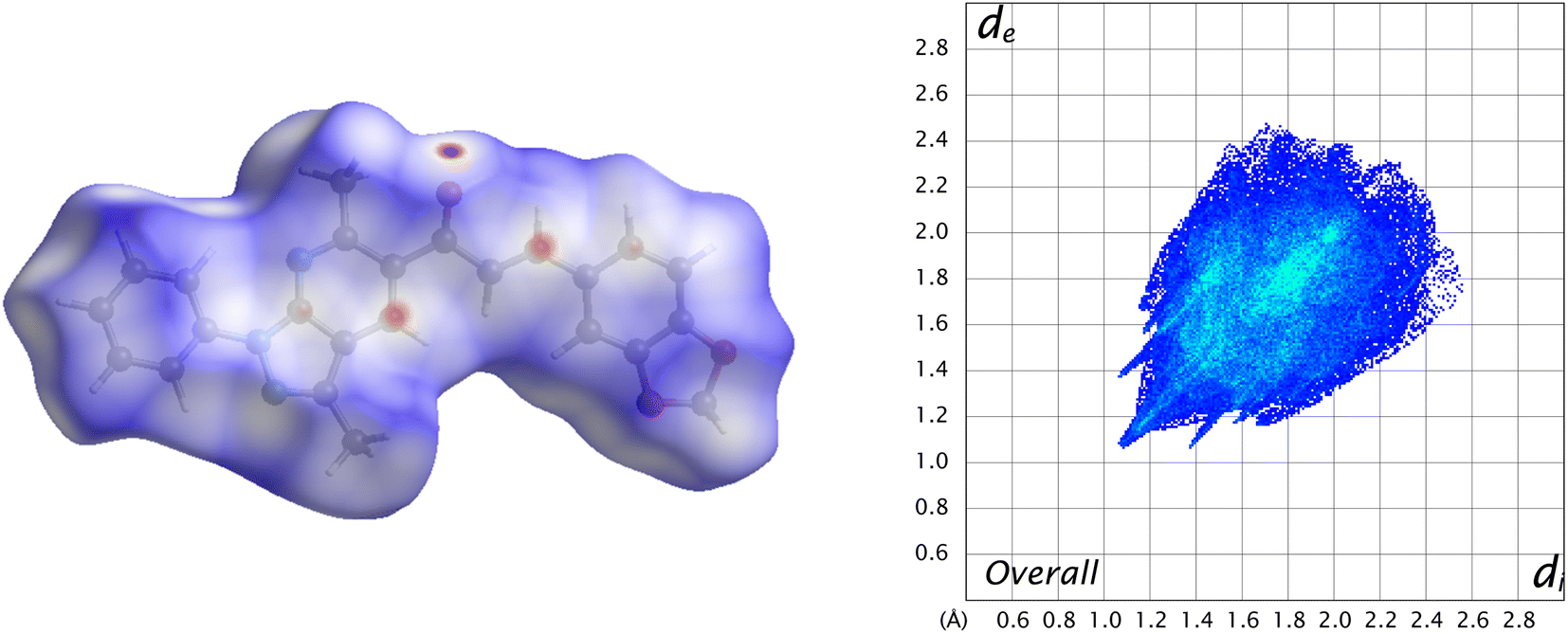 | ||
| Fig. 7 Overall Hirshfeld surface (left) of the title compound and its fingerprint plot (right). | ||
A 48.2% contribution with pair of sharp needles and the tip at di + de ≈ 2.2 Å was noticed from H⋯H contacts, covering a significant region of the entire Hirshfeld surface. These interactions generate an important effect on the molecular packing in the intermolecular stabilization. These short H⋯H contact links adjacent molecules into inversion related generating dimers (Fig. 8). The second important interaction is C–H⋯C, contributing 18.8% to the overall crystal packing. Moreover, C–H⋯O and C–H⋯N bond interactions have contributions of 15% each.
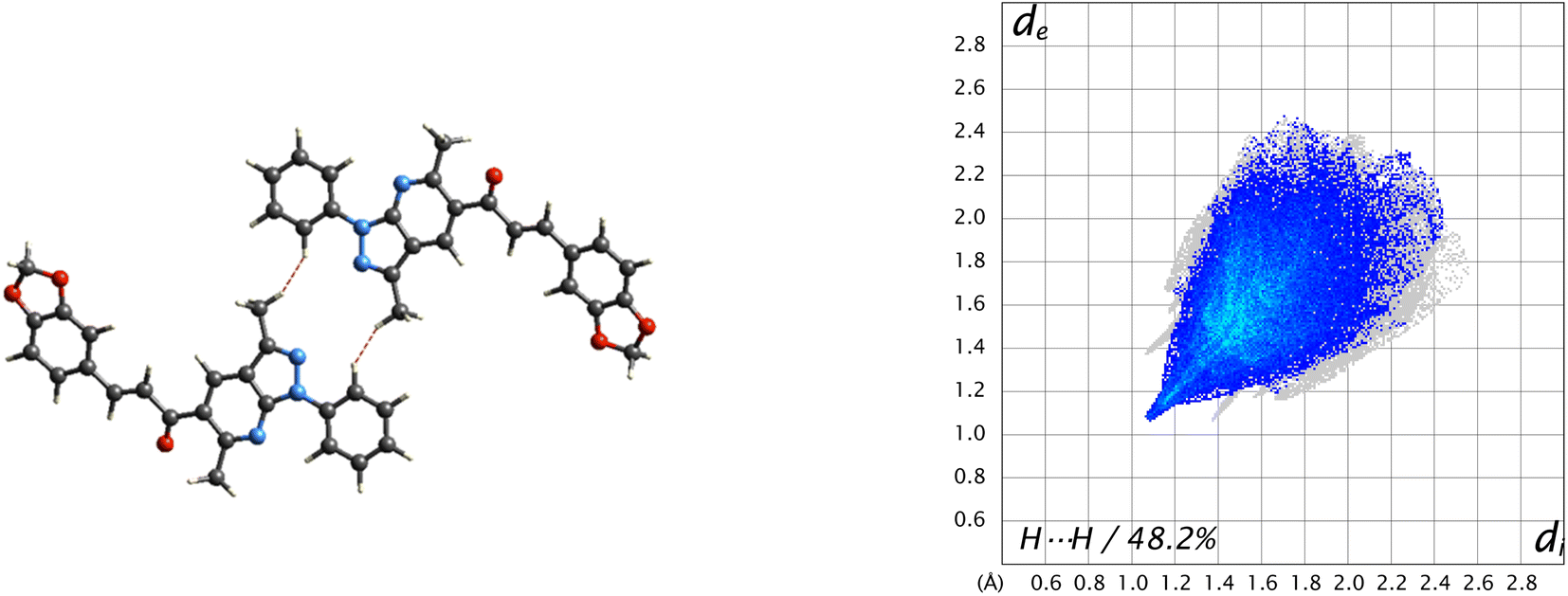 | ||
| Fig. 8 Short H⋯H contact interactions in the crystal structure stabilization of the title compound. | ||
Another type of weak C⋯C contact gives only 7.5% of the surface with the tip at de + di ≈ 3.4 Å, which helps in understanding interactions π⋯π stacking (Fig. 9).
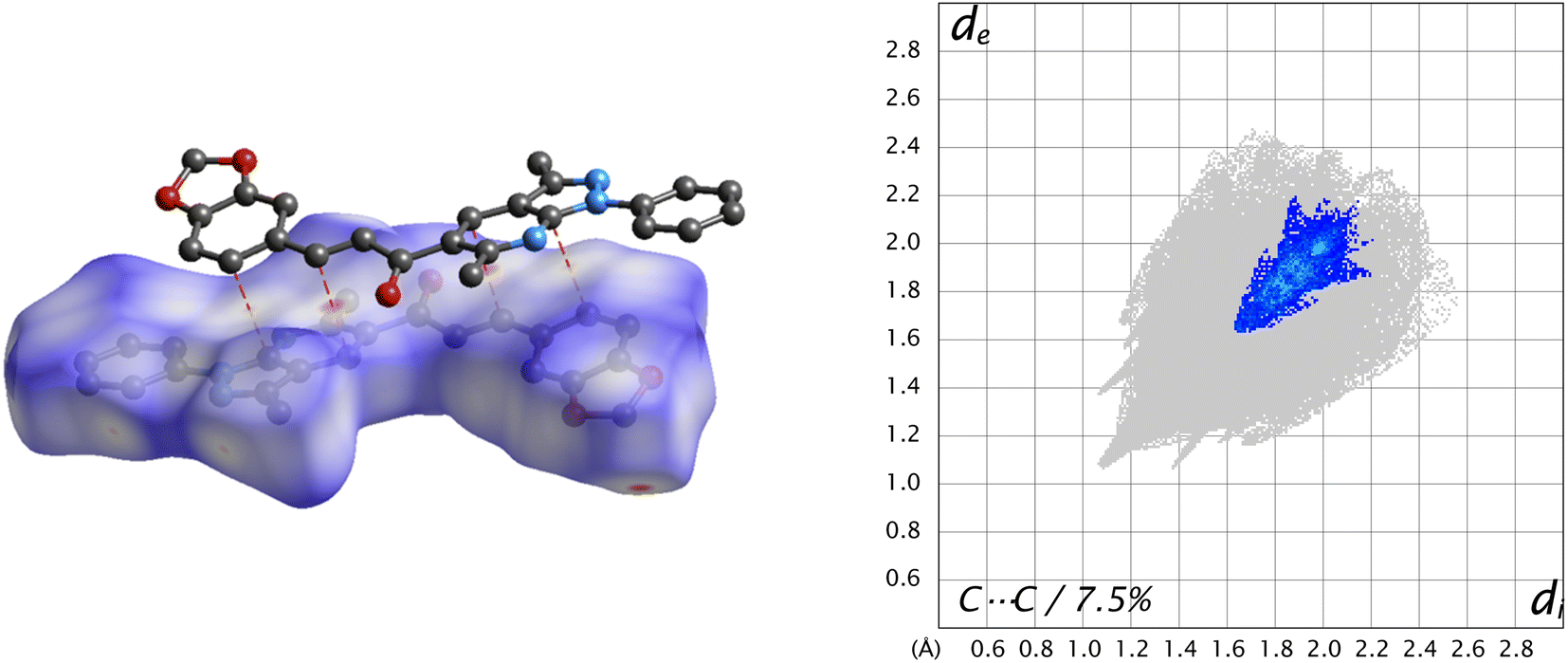 | ||
| Fig. 9 dnorm surface of the title compound (left) and its fingerprint plot (right), showing π⋯π stacking interactions in the title compound. | ||
4.9 Energy frameworks
The energy framework61 was performed to obtain the intermolecular interaction energies. This method allows analysis and visualization of the three-dimensional crystal packing to analyze the three-dimensional crystal topology. The interaction energy is calculated and compared using the formula Etot = Eele + Epol + Edis + Erep (where Eele is the electrostatic component, Epol the polarization energy, Edis the dispersion energy, and Erep the exchange repulsion energy) based on the anisotropy of the topology of pairwise intermolecular interaction energies. The thickness of the cylinder radius indicates the grade of interaction energies.62According to the tube direction, it can be concluded that the formation of the framework is directed by the translational or centrosymmetric elements. However, this rearrangement allows the formation of other weak interactions in the crystal structure. The energy framework is presented in Fig. 10. The dispersion (Edis) energy shows a zig-zag rod shape energy topologies along c-axis, as a component of the framework energy being less dominating than Eele. In this case, the rearrangement is reinforced by the presence of weak interactions in the crystal structure, such as π⋯π and classic hydrogen bond interactions, but not enough for the better stabilization of the framework.
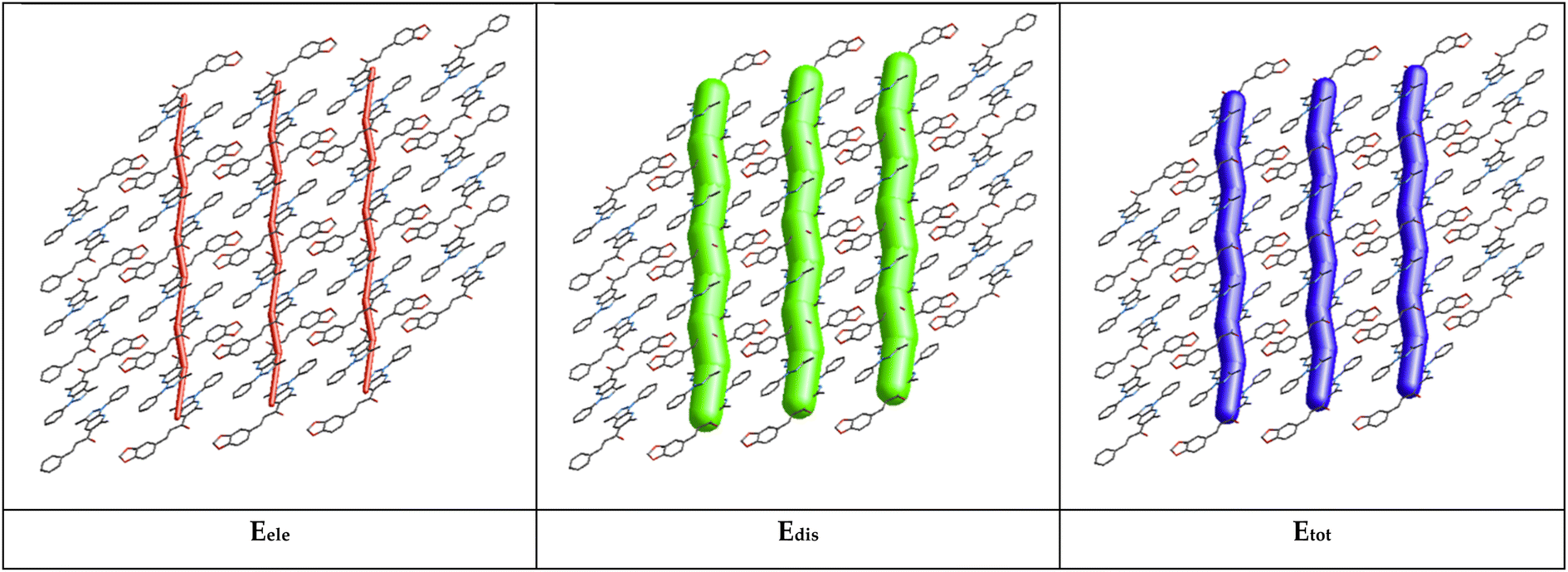 | ||
| Fig. 10 Energy framework diagram for electrostatic (Eele, red), (b) dispersion (Edis, green), and (c) total interaction energy (Etot, blue) of the title molecule. | ||
The total interaction energies are electrostatics (Eele = −40.1 kJ mol−1), polarization (Epol = −7.7 kJ mol−1), dispersion (Edis = −210.2 kJ mol−1), repulsion (Erep = 104.7 kJ mol−1), and total interaction energy (Etot = −153.3 kJ mol−1). The sum of dispersion energies (−210.2 kJ mol−1) is greater than that of electrostatic energies (−40.1 kJ mol−1) for the title compound and this low value is attributed to the absence or few numbers of classical hydrogen bonds (see Table 3). According to the result obtained, the dispersion energy framework has a significant contribution over the electrostatic energy framework.
| No. | Symop | R | Eele | Epol | Edis | Erep | Etot |
|---|---|---|---|---|---|---|---|
 |
−x, −y, −z | 4.43 | −23 | −3.3 | −107 | 56.3 | −77 |
 |
x, y, z | 10.9 | 1.4 | −0.2 | −3.2 | 0 | −2.0 |
 |
−x, −y, −z | 6.71 | −18.5 | −4.2 | −100 | 48.4 | −74.3 |
| Total energies | −40.1 | −7.7 | −210.2 | 104.7 | −153.3 | ||
5 Conclusions
In this work, we have presented the data and results used in confirming the structural properties of a π-conjugate system combining the chalcone and the pyrazole[3,4-b]pyridine scaffolds. The chalcone crystal has been synthesized and fully characterized by UV-Visible, FT-IR, HRMS, 1H NMR, and 13C NMR spectroscopic studies and X-ray crystallography. The X-ray findings showed that the studied molecule has planar molecular geometry and crystallizes in the triclinic system with a P space group, Z = 2, and unit cell parameters a = 8.669(6) Å; b = 10.856(7) Å, c = 11.092(7) Å, β = 97.788(14)° and V = 982.5(11) Å3. In general, good agreement was found between all the theoretical properties investigated (structural, electronic, and spectroscopic) and the experimental ones. Molecular stability from hyper conjugative interactions and charge delocalization was analyzed using NBO analysis. The FMOs analysis and chemical reactivity descriptors revealed that the piperonal chalcone is a less reactive and more stable molecule. Hirshfeld surface analysis of the crystal structure indicated that the largest contribution to the intermolecular stabilization in the crystal packing comes from the H⋯H (48.2%) bond interactions. Furthermore, the calculated interaction energy suggested that the dispersion energy framework significantly contributes to the total energy framework. According to the computed α, β, and γ values, the chalcone crystal exhibits significant NLO properties. Thus, we concluded that the piperonal chalcone is a promising candidate for future applications in nonlinear optical devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A FONDECYT project number 1200531 supported this research. The authors also acknowledge to FONDEQUIP program (EQM 130021, 180024 and 170172). E. P.-C. thanks FONDECYT Post-Doctoral Fellowship No. 3220681. “Powered@NLHPC: This research/thesis was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02)”.References
- S. Basu, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 183–186 CrossRef CAS.
- A. Abudurusuli, J. Li and S. Pan, Dalton Trans., 2021, 50, 3155–3160 RSC.
- S. Tao, T. Miyagoe, A. Maeda, H. Matsuzaki, H. Ohtsu, M. Hasegawa, S. Takaishi, M. Yamashita and H. Okamoto, Adv. Mater., 2007, 19, 2707–2710 CrossRef CAS.
- I. D. Borges, J. A. V. Danielli, V. E. G. Silva, L. O. Sallum, J. E. Queiroz, L. D. Dias, I. Iermak, G. L. B. Aquino, A. J. Camargo, C. Valverde, C. Valverde, F. A. P. Osório, F. A. P. Osório, B. Baseia, B. Baseia, H. B. Napolitano and H. B. Napolitano, RSC Adv., 2020, 10, 22542–22555 RSC.
- S. Raghavendra, K. V. Anil Kumar, T. Chandra Shekhara Shetty and S. M. Dharmaprakash, J. Mol. Struct., 2014, 1074, 653–659 CrossRef CAS.
- I. Polyzos, G. Tsigaridas, M. Fakis, V. Giannetas, P. Persephonis and J. Mikroyannidis, Chem. Phys. Lett., 2003, 369, 264–268 CrossRef CAS.
- R. L. Sutherland, D. G. Mclean and S. Kirkpatrick, Handbook of Nonlinear Optics, 2nd edn, Revised and Expanded, 2003 Search PubMed.
- K. Naseema, V. Rao, K. B. Manjunatha, G. Umesh, K. V. Sujith and B. Kalluraya, J. Opt., 2010, 39, 143–148 CrossRef.
- M. Makowska-Janusik, E. Gondek, I. V. Kityk, J. Wisła, J. Sanetra and A. Danel, Chem. Phys., 2004, 306, 265–271 CrossRef CAS.
- J. Chen, Q. Ren, X. Wang, T. Li, H. Yang, J. Zhang and G. Li, Opt., 2012, 123, 1761–1764 CAS.
- J. P. Abraham, D. Sajan, V. Shettigar, S. M. Dharmaprakash, I. Němec, I. Hubert Joe and V. S. Jayakumar, J. Mol. Struct., 2009, 917, 27–36 CrossRef CAS.
- K. Ozga, E. Gondek, A. Danel and K. Chaczatrian, Opt. Commun., 2004, 231, 437–446 CrossRef CAS.
- L. Shen, Z. Li, X. Wu, W. Zhou, J. Yang and Y. Song, RSC Adv., 2020, 10, 15199–15205 RSC.
- E. D. Dsilva, D. Narayan Rao, R. Philip, R. J. Butcher, Rajnikant and S. M. Dharmaprakash, J. Phys. Chem. Solids, 2011, 72, 824–830 CrossRef CAS.
- S. R. Marder, W. E. Torruellas, M. Blanchard-Desce, V. Ricci, G. I. Stegeman, S. Gilmour, J. L. Brédas, J. Li, G. U. Bublitz and S. G. Boxer, Science, 1997, 276, 1233–1236 CrossRef CAS PubMed.
- V. Shettigar, P. S. Patil, S. Naveen, S. M. Dharmaprakash, M. A. Sridhar and J. Shashidhara Prasad, J. Cryst. Growth, 2006, 295, 44–49 CrossRef CAS.
- B. Gu, W. Ji, X.-Q. Huang, P. S. Patil and S. M. Dharmaprakash, Opt. Express, 2009, 17, 1126 CrossRef CAS PubMed.
- H. J. Ravindra, A. J. Kiran, S. M. Dharmaprakash, N. Satheesh Rai, K. Chandrasekharan, B. Kalluraya and F. Rotermund, J. Cryst. Growth, 2008, 310, 4169–4176 CrossRef CAS.
- P. Poornesh, K. Ravi, G. Umesh, P. K. Hegde, M. G. Manjunatha, K. B. Manjunatha and A. V. Adhikari, Opt. Commun., 2010, 283, 1519–1527 CrossRef CAS.
- B. Gu, W. Ji, P. S. Patil, S. M. Dharmaprakash and H. T. Wang, Appl. Phys. Lett., 2008, 92, 091118 CrossRef.
- S. Y. Priya, R. K. Rao, V. P. Chalapathi and C. Author, J. Clin. Med. Images Short Reports, 2018, 1, 1–9 Search PubMed.
- R. Kumar, T. Karthick, P. Tandon, P. Agarwal, A. P. Menezes and A. Jayarama, J. Mol. Struct., 2018, 1164, 180–190 CrossRef CAS.
- A. P. Menezes and A. Jayarama, J. Mol. Struct., 2014, 1075, 246–253 CrossRef CAS.
- A. J. Kiran, H. W. Lee, H. J. Ravindra, S. M. Dharmaprakash, K. Kim, H. Lim and F. Rotermund, Curr. Appl. Phys., 2010, 10, 1290–1296 CrossRef.
- E. Polo, K. Ferrer-Pertuz, J. Trilleras, J. Quiroga and M. Gutiérrez, RSC Adv., 2017, 7, 50044–50055 RSC.
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery, T. Vreven, K. Kudin, J. Burant, J. Millam, S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. Knox, H. Hratchian, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Ayala, K. Morokuma, G. Voth, P. Salvador, J. Dannenberg, V. Zakrzewski, S. Dapprich, A. Daniels, M. Strain, O. Farkas, D. Malick, A. Rabuck, K. Raghavachari, J. Foresman, J. Ortiz, Q. Cui, A. Baboul, S. Clifford, J. Cioslowski, B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Martin, D. Fox, T. Keith, A. Laham, C. Peng, A. Nanayakkara, M. Challacombe, P. Gill, B. Johnson, W. Chen, M. Wong, C. Gonzalez, D. J. Fox and J. Pople, Gaussian 09, Revision A.08, Gaussian, Inc., 2009 Search PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. L. Pascual-ahuir, E. Silla and I. Tuñon, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- F. W. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, 2013.
- Y.-Y. Hu, S.-L. Sun, S. Muhammad, H.-L. Xu and Z.-M. Su, J. Phys. Chem. C, 2010, 114, 19792–19798 CrossRef CAS.
- H.-B. Zhao, Y.-Q. Qiu, C.-G. Liu, S.-L. Sun, Y. Liu and R.-S. Wang, J. Organomet. Chem., 2010, 695, 2251–2257 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Gromwood, P. R. Spackman, D. J. - Alitaka and M. A. Spackman, Crystal Explorer, University of Western Australia, 2017 Search PubMed.
- S. S. Batsanov, Inorg. Mater. Transl. from Neorg. Mater. Orig. Russ. Text, 2001, 37, 871–885 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. A. Zainuri, M. Abdullah, S. Arshad, M. S. A. Aziz, G. Krishnan, H. Bakhtiar and I. A. Razak, Opt. Mater., 2018, 86, 32–45 CrossRef CAS.
- B. Evranos Aksöz and R. Ertan, FABAD J. Pharm. Sci., 2011, 36, 223–242 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. -L Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- C. Cassidy, J. M. Halbout, W. Donaldson and C. L. Tang, Opt. Commun., 1979, 29, 243–246 CrossRef CAS.
- R. I. Al-Wabli, K. S. Resmi, Y. Sheena Mary, C. Yohannan Panicker, M. I. Attia, A. A. El-Emam and C. Van Alsenoy, J. Mol. Struct., 2016, 1123, 375–383 CrossRef CAS.
- S. Shettigar, P. Poornesh, G. Umesh, B. K. Sarojini, B. Narayana and K. Prakash Kamath, Opt. Laser Technol., 2010, 42, 1162–1166 CrossRef CAS.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- H. A. Khamees, K. Chaluvaiah, N. A. El-khatatneh, A. Swamynayaka, K. H. Chong, J. P. Dasappa and M. Madegowda, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 1620–1626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2218131. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra08101f |
| This journal is © The Royal Society of Chemistry 2023 |