DOI:
10.1039/D2RA08006K
(Paper)
RSC Adv., 2023,
13, 10873-10883
Identifying novel selective PPO inhibitors through structure-based virtual screening and bio-evaluation†
Received
15th December 2022
, Accepted 6th March 2023
First published on 5th April 2023
Abstract
Protoporphyrinogen oxidase (PPO) is a key enzyme in chlorophyll and heme biosynthesis, and the development of its inhibitors is of great importance both in the pharmaceutical and pesticide industries. However, the currently developed PPO inhibitors have insignificant bio-selectivity and have a serious impact on non-target organisms. In this study, a docking-based virtual screening approach combined with bio-activity testing was used to obtain novel selective inhibitors of PPO. The results of the bio-activity test showed that thirteen compounds showed 10-fold selectivity over human PPO. And the best selective compound, ZINC70338, has a Ki value of 2.21 μM for Nicotiana tabacum PPO and >113-fold selectivity for human PPO. The selectivity mechanism of ZINC70338 in different species of PPO was then analyzed by molecular dynamics simulations to provide a design basis and theoretical guidance for the design of novel selective inhibitors.
1. Introduction
Protoporphyrinogen oxidase (PPO) is the last isozyme in tetrapyrrole biosynthesis and is mainly used in the synthesis of ferrous heme and chlorophyll.1,2 It catalyzes the production of Protoporphyrin IX (PpIX) in the presence of trimolecular oxygen and is widely found in animals, plants, bacteria and fungi.2,3 PpIX is a strong photosensitizer that accelerates the production of singlet oxygen and induces lipid peroxidation and apoptosis. Inhibition of PPO activity leads to excessive accumulation of PpIX and leakage from the mitochondria into the cytoplasm. PpIX in the cytoplasm is rapidly oxidized to PpIX by nonenzymatic autoxidation or by other oxidases (ERase or PMase). The photosensitive PpIX undergoes photodynamics in the presence of light and oxygen, generating singlet oxygen, causing lipid, DNA and protein peroxidation, disrupting cell membrane structure, and ultimately leading to fatal cell damage.4–6 Due to its specific mechanism of action, PPO is a very important target both in the pharmaceutical and pesticide industries.7–10 Studies have shown that PPO inhibitors induce endogenous PpIX enrichment in tumor cells and have been applied in photodynamic therapy for cancer treatment.11 For human, lack of PPO or reduced activity of the enzyme leads to impaired heme synthesis and porphyria.3,12,13 For plants, the development of inhibitors of PPO, an important target for pesticide research in recent years, has led to a new era of pesticide research. The types of herbicides that have been marketed and widely used include diphenyl ethers, pyrimidinones, phenylpyrazoles, oxadiazoles, cyclic imines, and triazolones.3,14
Since protoporphyrinogen oxidase is widely present in plants and animals, if the developed PPO inhibitors have little or no selectivity for human PPO (hPPO),15 they will eventually become toxic to humans, such as inducing porphyria after prolonged use.16 Therefore, it is necessary to design novel and efficient selective PPO inhibitors.
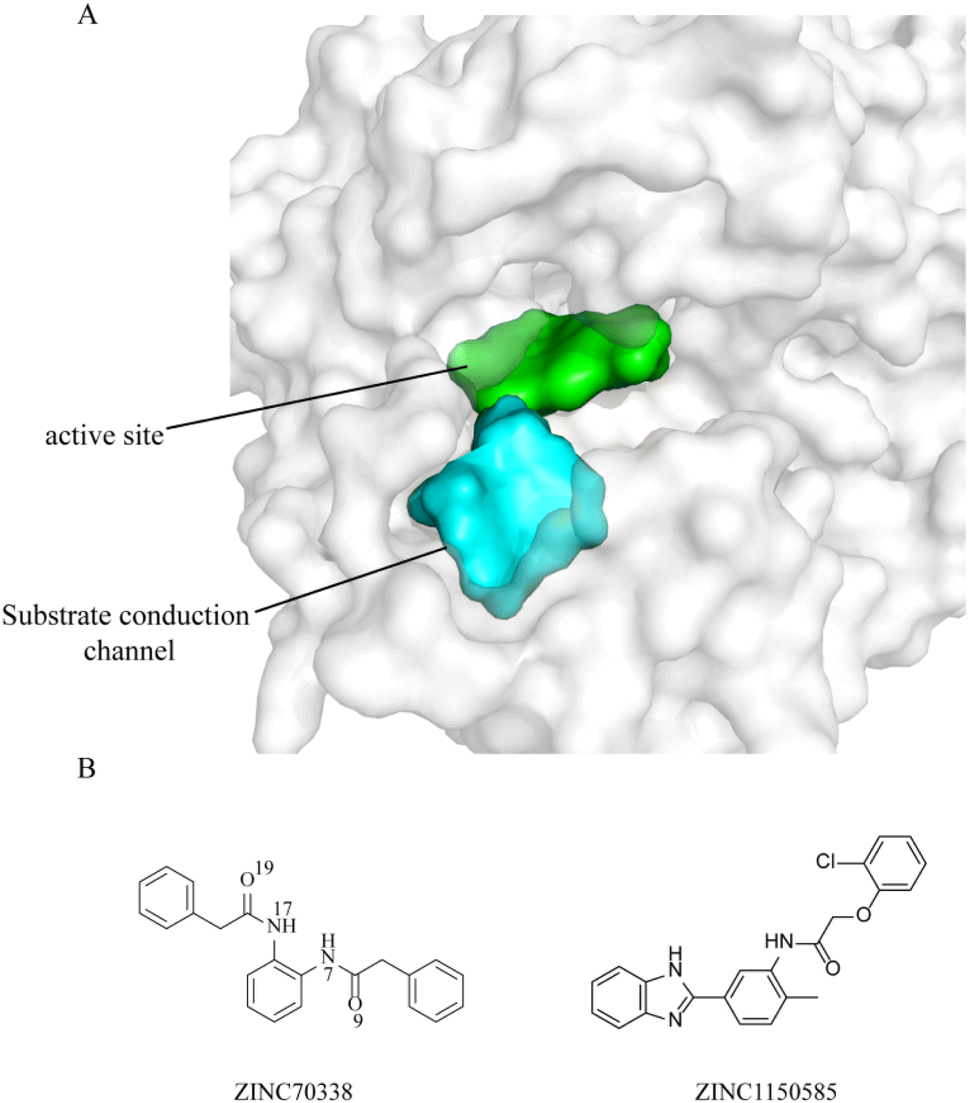
Hao et al.16 explored the selectivity mechanism of commercial PPO inhibitors (Fig. S1†) and developed a novel computational fragment generation and coupling strategy and finally discovered the highest selective PPO inhibitor compound 10 (Ki = 22 nM, 2749-fold selectivity over hPPO) after optimization. Although some meaningful progress has been made in the above studies, the development of selective PPO inhibitors is still lacking. Therefore, in this study, taking into account the structural characteristics of PPO, i.e., there are two connected cavities: the PPO active site and the substrate channel (Fig. 1A), and the compound, when bind in the active site, may also be bound in the substrate channel, and the same inhibition effect could be achieved,17 we designed a screening strategy for the substrate binding site and the conduction channel, taking into account the species selectivity, aiming to discover an effective and selective PPO inhibitor. After multiple screening, combined with bio-activity assays, we identified an effective selective ntPPO inhibitor (Ki = 2.1 μM, >113-fold selectivity over hPPO, Fig. 1B), and finally explored the molecular mechanism of PPO selective inhibitors by molecular dynamics simulations and binding free energy calculations.
 |
| | Fig. 1 Molecular structures. (A) The two channels of PPO; (B) the structure of the top 2 hits. | |
2. Materials and methods
2.1 Preparation of protein and ligands
The crystal structures of tobacco protoporphyrinogen oxidase (PDB:1SEZ)2 and human protoporphyrinogen oxidase (PDB:3NKS)18 were obtained from the RCSB Protein Data Bank (https://www.rcsb.org) with resolutions of 2.9 Å and 1.9 Å, respectively. The co-crystallized proteins were pre-processed using the SYBYL-X-2.0 (Tripos Inc., St. Louis, MO, USA), including deleted all solvent molecules, crystal water and ligands, reserved coenzyme factor FAD, added hydrogen, assigned charge, repaired the missing side chain residues and energy minimization. The number of structure optimization steps was set to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with an energy iteration of 0.005 kcal (mol−1 Å−1). The force field parameter of the protein was generated by using AMBER ff99 force field. The Gasteiger–Huckel charge was used to calculate the partial atomic charge. The optimized protein files were saved in the pdbqt format via AutoDock Tools (v1.5.6)19 and saved in mol2 format via SYBYL-X-2.0 which required for the docking by AutoDock Vina20 and Surflex dock, which was a suit of SYBYL-X 2.0 software,21 respectively.
000 with an energy iteration of 0.005 kcal (mol−1 Å−1). The force field parameter of the protein was generated by using AMBER ff99 force field. The Gasteiger–Huckel charge was used to calculate the partial atomic charge. The optimized protein files were saved in the pdbqt format via AutoDock Tools (v1.5.6)19 and saved in mol2 format via SYBYL-X-2.0 which required for the docking by AutoDock Vina20 and Surflex dock, which was a suit of SYBYL-X 2.0 software,21 respectively.
A database containing 200000 small molecules was obtained from the SPECS and the Maybrige database by applying Lipinski's rule and agrochemicals-like rules22 for the subsequent virtual screening. All ligands were pre-processed using OpenBabel23 and saved in mol2 and pdbqt formats, respectively.
2.2 Molecular docking and virtual screening
When docking with AutoDock Vina, the docking parameters were generated using AutoDock Tools. For the active cavity of hPPO (PDB:3NKS), the center-of-mass of co-crystallized molecule (−26.42, 3.54, 48.37) was selected as the grid center, and the grid box was defined as 20 × 20 × 20 Å. For the substrate conduction channel, the terminal C atom of residue ALA162 was selected as the grid center, and the coordinate was set to 25.93, −2.39, and 57.34, defining the grid size as 20 × 20 × 20 Å. For the active cavity of ntPPO (PDB:1SEZ), like hPPO, the center-of-mass of co-crystallized molecule (−39.91, −6.20, 28.68) was selected as the grid center, and the grid box was defined as 20 × 20 × 20 Å. For the substrate conduction channel, the terminal C atom of residue LEU168 were selected as the grid center, and the coordinates was 44.08, 3.76, 42.46 defining the docking grid size of 20 × 20 × 20 Å. The exhaustiveness was set to eight, and nine modes were generated for per molecule in each docking, while other parameters was set to default values. When docking with Surflex, the docking parameters same as that with Vina were set for virtual screening. Then the docking scores of all compounds were ranked, and the top 40% compounds were selected for clustering based on RMSD. The conformations would be removed when RMSD > 3.1 Å for active cavities and when RMSD > 4.1 Å for substrate conduction channels. The docking parameters details are in Table S1.† PyMOL (https://pymol.org/) and Proteinplus24 was used for product visualizations.
2.3 In vitro evaluation of PPO inhibition kinetics
The expression and purification of recombinant ntPPO and hPPO enzymes were according to the reported methods.2,18,25,26 The product of the enzymatic reaction has a maximum excitation wavelength at 410 nm and a maximum emission wavelength at 630 nm. Thus the PPO activity was assayed by fluorescence as described previously.27,28 In kinetic inhibition assays, dimethyl sulfoxide (DMSO) was used to dissolve the inhibitor. The final concentration ranged from 0.005 μM to 250 μM. The enzymatic reaction rate was tested in 100 mM potassium phosphate (pH = 7.5), 5 mM DTT, 1 mM EDTA, Tween 80 (0.03%, v/v), 200 mM imidazole, 5 μM FAD, and approximately 0–40 μg of protein.
2.4 Molecular dynamics simulations
To investigate the mechanism of inhibitor selectivity, molecular dynamics simulations were performed using AMBER16 (ref. 29) in the following four steps: energy minimized to relax the system and eliminate unreasonable collisions; heated; equilibrated; and performed conventional molecular dynamics simulations. The force field parameters were generated by ff14sb30 for protein and gaff2 (ref. 31) force field for small molecule, and the complex was located in a octahedral solvent box filled with the TIP3P32 water model in which the distance between complex and the box edge was 15 Å, chloride ions were added to neutralize the system and balance the charges according to the charged nature of the solution environment. Next, the energy of above complex systems are minimized by the steepest descent method. Once the initial minimization was completed, the entire system was heated from 10 to 300 K and equilibrated for 1 ns at 300 K and 1 bar in classical (NVT) and isothermal isobaric (NPT) combinations. Finally, a 100 ns timescale MD simulation was performed and repeated three times. During the simulation, the SHAKE33 method is used to constrain the scaling of the chemical bonds attached to the hydrogen atoms, the integration step is set to 2 fs, the PME34 method is used to calculate the long-range electrostatic interactions, and the periodic boundary condition (PBC) is used to eliminate the edge effects of the solvent box. The simulated trajectory files were saved every 2 ps for subsequent processing.
2.5 Data analysis and binding free energy calculations
The Root Mean Square Deviation (RMSD), the Root Mean Square Fluctuations (RMSF), the solvent-available surface area (SASA) analysis, and hydrogen binding analysis were calculated using the CPPTRAJ35 module of the AmberTools 16 software package.
Molecular mechanics generalized Born surface area (MMGBSA)36 was used to calculate the binding free energy (ΔGbind) of the protein–ligand complexes, and residue free energy decomposition was performed to find the key residues that generate the key residues for selectivity. In the MMGBSA method, the binding free energy of the ligand (L) bound to the receptor protein (R) to form the complex (RL) can be expressed by the following equation.37
| | |
ΔGbind = GRL − GR − GL
| (1) |
| | |
ΔGbind = ΔH − TΔS = ΔGgas + ΔGsol − TΔS
| (2) |
| | |
ΔGbind = ΔEele + ΔEvdw + ΔGGB + ΔGsurf − TΔS
| (3) |
TΔS represents the negligible contribution of the entropic change in the ligand–receptor binding process to the binding free energy.
3. Results and discussion
3.1 Structure-based virtual screening
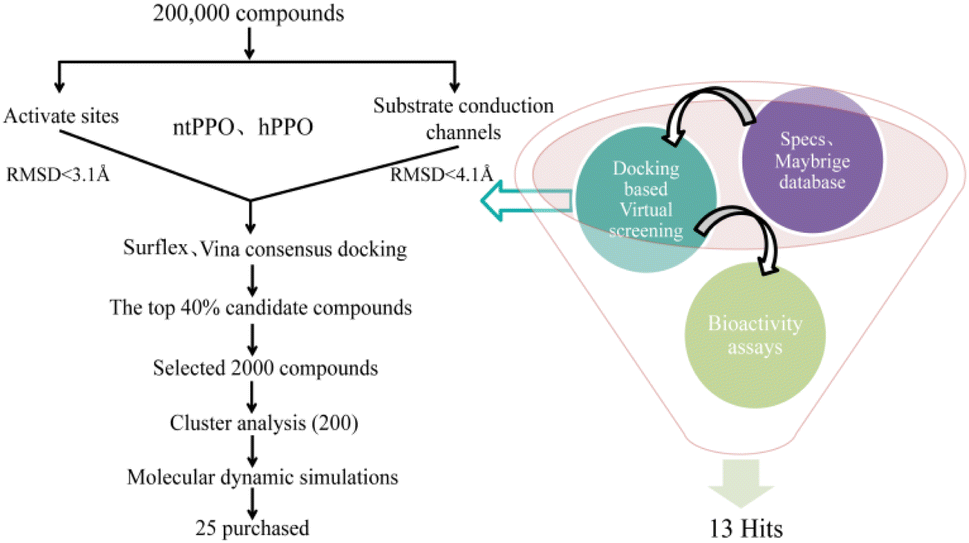
The screening process is shown in Fig. 2. Based on the docking score, we selected the top 40% compounds by considering the docking results of active sites and substrate transmission channels after the consensus docking using Vina and Surflex. Then, according to the RMSD clustering of the binding mode, for the results of the active cavity the compound would be eliminated once the RMSD > 3.1 Å; because of the large cavity of the substrate conduction channel, the compound would be eliminated once the RMSD > 4.1 Å, a total of 2000 compounds were obtained. The 2000 compounds were clustered into 200 classes by ECFP_4 means. These 200 compounds were then subjected to conformational analysis and 1.5 ns molecular dynamics simulations. 25 compounds were selected based on the molecular dynamics simulation trajectories with smaller RMSD fluctuations in ntPPO and larger fluctuations in hPPO and purchased to preliminarily evaluate the biological activity.
 |
| | Fig. 2 Workflow of virtual screening and bio-activity assays for PPO inhibitors. | |
3.2 Bio-activity assays
The Ki of the compounds were determined using fluorometric assay. The bio-activity was determined to evaluate the inhibitory effect and selectivity of the screened inhibitors, in which a total of 13 compounds exhibited more than 10-fold higher bio-selectivity for ntPPO than hPPO. The biological activities and the bio-selectivity of the all screened compounds were shown in Table 1. The Ki of ZINC70338 against ntPPO was 2.21 μM with a >113-fold selectivity over hPPO and it was 2.99 μM for ZINC1150585 with >83-fold selectivity over hPPO. The physical properties of the two compounds with the best bio-activity selectivity multiples are shown in Table 2. Fig. 1B shown the structures of the two compounds and Table 3 summarized the ADMET parameters of ZINC70338 and ZINC1150585 which was calculated by ADMET Lab 2.0.38 The physicochemical properties of the other 11 compounds were shown in Table S2.†
Table 1 The Ki value and the selectivity of all screened compounds
| Compound |
ntPPO Ki (μM) |
hPPO Ki (μM) |
SELE |
| ZINC70338 |
2.21 |
>250 |
>113 |
| ZINC1150585 |
2.29 |
>250 |
>83 |
| ZINC649024 |
4.1 |
>250 |
>60 |
| ZINC2078708 |
4.3 |
172.11 |
>40 |
| ZINC836537 |
6.58 |
>250 |
>40 |
| ZINC16023063 |
5.23 |
194.94 |
>37 |
| ZINC887010 |
3.16 |
112.16 |
>35 |
| ZINC4680768 |
6.01 |
201.62 |
>34 |
| ZINC2079610 |
11.51 |
>250 |
>21 |
| ZINC6147185 |
4.49 |
86.32 |
>19 |
| ZINC10311685 |
6.38 |
>113 |
>17 |
| ZINC686711 |
17.32 |
>250 |
>14 |
| ZINC1069729 |
3.09 |
44.19 |
>14 |
Table 2 The physical characteristics of the top two compounds
| Compound |
MW |
HBD |
HBA |
ROB |
HA |
logP |
TPSA |
logD |
Water solubility |
Formal charges |
The maximum size of rings |
SAscore |
Stereo-centers |
| ZINC70338 |
344.41 |
2 |
2 |
8 |
26 |
3.25 |
58.2 |
3.322 |
3.86 × 10−2 mg ml−1 |
0 |
6 |
1.517 |
0 |
| ZINC1150585 |
391.85 |
2 |
3 |
6 |
28 |
4.665 |
67.1 |
4.018 |
1.00 × 10−3 mg ml−1 |
0 |
9 |
1.98 |
0 |
Table 3 ADMET of the top two compoundsa
| Compound |
HIA |
BBB penetration |
CYP2D6 inhibitor |
T1/2 |
hERG blockers |
H-HT |
| HIA: human intestinal absorption; BBB penetration: blood–brain barrier penetration; CYP2D6: CYP450 inhibitor; T1/2; hERG blockers: drug cardiotoxicity prediction; H-HT: human hepatotoxicity. 0–0.1(−−−), 0.1–0.3(−−), 0.3–0.5(−), 0.5–0.7(+), 0.7–0.9(++), 0.9–1.0(+++). 0–0.3: good; 0.3–0.7: medium; 0.7–1.0: bad. |
| ZINC70338 |
− |
−− |
−− |
0.805 |
−− |
+ |
| ZINC1150585 |
−−− |
−− |
+ |
0.664 |
−− |
++ |
3.3 Binding mode analysis
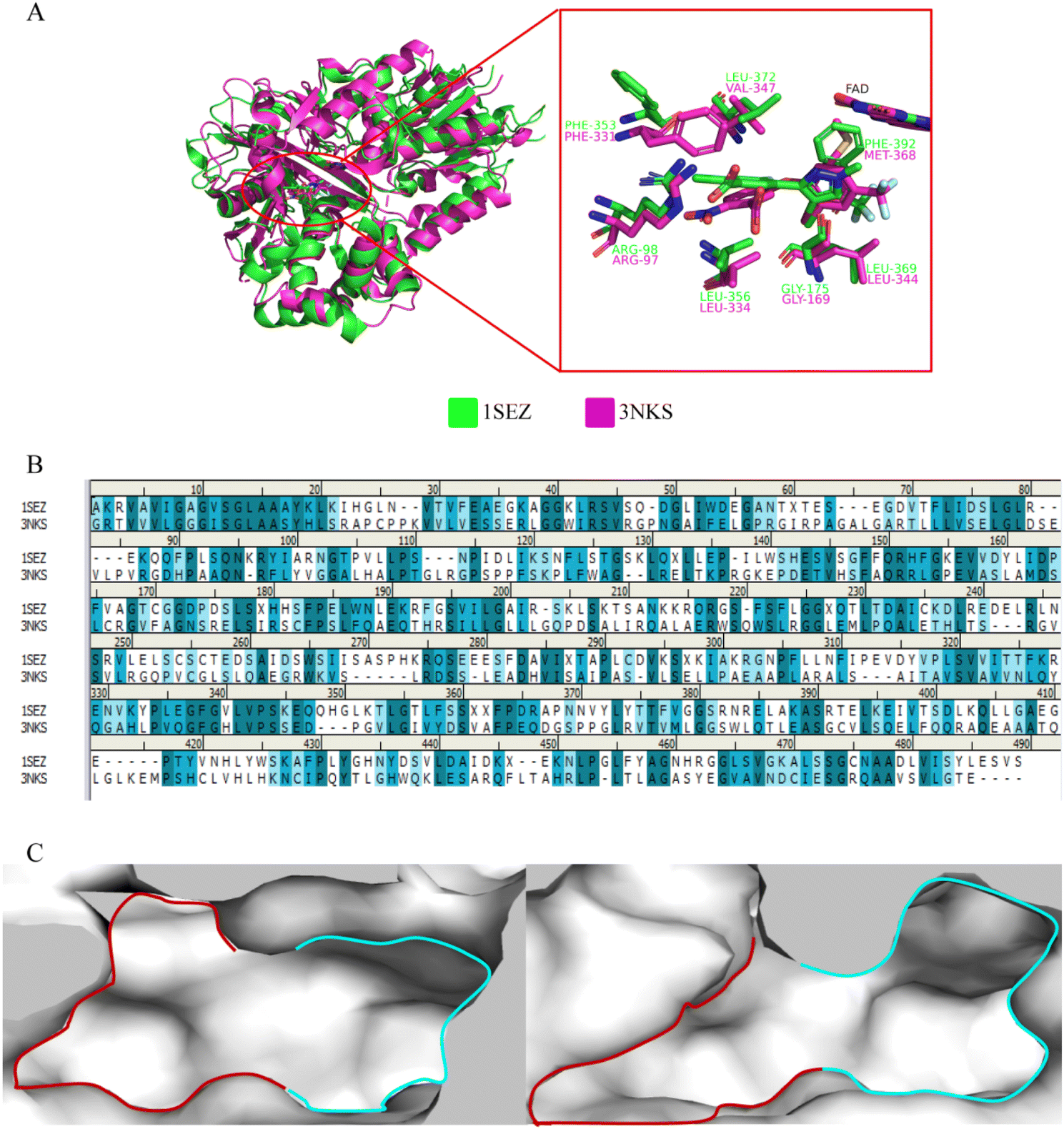
Firstly, a comparative analysis of the protein crystal structure indicated that the sequence similarity between ntPPO and hPPO was only 24.12% (Fig. 3B), but there has a similar sequence of residues in active site. It was found that the active cavity of PPO consists of 2 subsites, site 1 (S1) and site 2 (S2).16 In hPPO, the S1 region consists of non-polar amino acids such as hydrophobic amino acids MET368, GLY169, and LEU344, which are larger in size, while in ntPPO, MET368 is replaced by the larger PHE392, resulting in a smaller S1 subcavity, but the side chain benzene ring of PHE392 forms π–π stacking interactions2,16 with the aromatic ring of the substrate again, enhancing the substrate binding to the protein; the S2 region is exposed to the solvent environment and the basic amino acid ARG98 (ARG97 in hPPO) forms a hydrogen bond with the side chain of the substrate, which is shown to be a highly conserved amino acid here; the side chain structure of PHE331 in the S2 region is downward in hPPO, sandwiching the substrate with LEU334 below, but in ntPPO, the allelic PHE353 structure is upward, resulting in a larger volume of the S2 region of ntPPO compared to hPPO, allowing better binding of the substrate near the S2 region side chain (Fig. 3A and C). This provides theoretical support for the screening of selective inhibitors.
 |
| | Fig. 3 (A) alignment of ntPPO (green) and hPPO (magenta) protein crystal structures; (B) sequence alignment of ntPPO and hPPO; (C) comparison of ntPPO (left) and hPPO (right) active cavities, the red area is the S2 and the cyan area is the S1 region. | |
The structure of ZINC70338 is simple and easy to modify, and it has a 113-fold selectivity for PPO. Based on the above analysis, we used ZINC70338 as the object of study and analyzed its selectivity mechanism. The trend of the docking score that ZINC70338 binding with ntPPO or hPPO is consistent with the Ki values, and the lower docking score means that the compound binds more tightly to the protein, indicating the reasonableness of the selected docking modes (Table 4). The docking conformation of ZINC70338 with ntPPO shown that the compound is located in a hydrophobic pocket surrounded by PHE172, GLY175, LEU334, PHE392, PHE353, LEU356 and LEU372, with the amide bond forming two hydrogen bonds each with ARG98 and GLY175, and the phenylmethyl forming T-shaped interactions with PHE172 and π–π stacking interactions with PHE392, respectively. When binding with hPPO, the amide portion of ZINC70338 forms a hydrogen bond each with AGR97 and GLY169, and the hydrogen bond length is longer than that in ntPPO, and the T-shaped interaction was identified between phenylmethyl and PHE331 due to the down conformation of PHE331 in hPPO (Fig. 4). The similar binding mode could also be observed for ZINC1150585 (Fig. S2†). In summary, the differences in the number and distance of hydrogen bonds and the absence of van der Waals interactions, such as π–π stacking, led to the reduced affinity of the compounds for human protoporphyrinogen oxidase activity.
Table 4 The docking score of ZINC70338 binding with ntPPO and hPPO using Vina and Surflex
| Compound |
Docking score (kcal mol−1) |
| ntPPO |
hPPO |
| Vina |
Surflex |
Vina |
Surflex |
| ZINC70338 |
−9.20 |
8.95 |
−8.00 |
6.59 |
| ZINC1150585 |
−10.0 |
8.71 |
−8.40 |
7.52 |
 |
| | Fig. 4 The binding modes of ZINC70338 with ntPPO (A) and hPPO (C) and 2D interaction diagram of ZINC70338 with ntPPO (B) and hPPO (D). ZINC70338 is shown as cyan sticks, and the key residues is shown as magenta sticks. The yellow dash is hydrogen bond, and the green is the π–π stacking interaction. | |
3.4 Stability analysis of molecular dynamics simulation
Each system was performed three times molecular dynamics simulations with a timescale of 100 ns. The results of the molecular dynamics simulations were expressed by the Root Mean Square Deviation (RMSD), which is an important reference for whether the simulated systems reached equilibrium or not, and could be used to characterize the stability of the system during the molecular dynamics simulations. After 100 ns of simulation, both the apo system and the complex systems reached the equilibrium state (Fig. 5), and the average RMSD value of ntPPO–ZINC70338 was 2.83 Å. The average RMSD value of hPPO–ZINC70338 system was 2.73 Å (Table 5). The RMSDs of the three molecular dynamics simulations of the complexes were shown in Fig. S3.† The Root Mean Square Fluctuation (RMSF) is a measure of the degree of freedom of movement of the atoms, characterizing the flexibility of the structure. As shown in Fig. 5, the RMSF of the complex system was lower than that of the apo system, indicating that the binding of the ligand facilitated the stabilization of the protein conformation; however, the RMSF of ntPPO–ZINC70338 was lower than that of hPPO–ZINC70338 at 100–200 and 350–400 in the substrate binding region, indicating that the binding of the ligand made the conformation of the ntPPO substrate binding region more stable. The solvent accessible surface area (SASA)39,40 of proteins has been considered a key factor in protein folding and stability studies and plays an important role in understanding the structure–function relationships of proteins and the residues. The binding of ligands to proteins is actually the process of replacing water molecules at the protein binding site, when some amino acid residues may change from solvent accessible to solvent inaccessible state, so comparing the changes of SASA before and after binding can compare the ligand binding stability. In this study, the stability of both proteins in solvent was analyzed by calculating the solvent-accessible surface area before and after binding of ZINC70338 to hPPO and ntPPO (Fig. 5). For ntPPO–ZINC70338 system, the SASA of complexes (20505.32 Å2, Table 5) was significantly lower than that of the apo system (21199.38 Å2), while in the hPPO–ZINC70338 system the SASA did not change significantly, being 21187.07 and 21399.79, respectively. Also, the radius of gyration was calculated to assess the compactness and stability of the simulated system. The radius of gyration analysis showed consistent results with SASA. The radius of gyration of the complex in the ntPPO–ZINC70338 system was 23.64 Å lower than that of the apo system which was 23.85 Å (Table 5), but there was no significant differences in height and magnitude between hPPO–apo system and the hPPO–ZINC70338 system. The result indicated that the binding of the inhibitor made the ntPPO structure more compact. By SASA analysis and radius of gyration analysis, ZINC70338 binds more tightly to ntPPO. The average RMSD, RMSF, RoG, and SASA results of the three molecular dynamics simulations are shown in Table S3.†
 |
| | Fig. 5 (A) The RMSD of the complex formed by ntPPO with ZINC70338, the backbond atom of ntPPO and ZINC70338; (B) the RMSF in ntPPO system; (C) the RMSD of the complex formed by hPPO with ZINC70338, the backbond atom of hPPO and ZINC70338; (D) the RMSF in hPPO system; the SASA analysis of ZINC70338 with ntPPO (E) and hPPO (F); the radius of gyration of ZINC70338 with ntPPO (G) and hPPO (H). | |
Table 5 The average RMSD, RMSF, RoG, SASA of PPO–Apo and PPO–ZINC70338 complexes
| Average |
Simulated system |
| ntPPO–apo |
ntPPO–ZINC70338 |
hPPO–apo |
hPPO–ZINC70338 |
| Average RMSD (Å) |
2.83 |
2.92 |
2.73 |
2.64 |
| Average RMSF (Å) |
10.46 |
7.17 |
12.31 |
8.88 |
| Average RoG (Å) |
23.85 |
23.64 |
22.67 |
22.66 |
| Average SASA (Å2) |
21199.38 |
20505.32 |
21187.07 |
21399.79 |
The results of hydrogen bonds generated during the molecular dynamics simulation were generally consistent with the results of docking, further clarifying the reasonableness of the conformation chosen after docking. As shown in Table 6, ZINC70338 formed strong hydrogen bonds interactions with ARG98 with occupancy of 71.45% and 36.36%, and formed hydrogen bonds interactions with occupancy of 67.58% and 39.79% with GLY175 in the molecular dynamic simulations binding with ntPPO. While binding with hPPO, ZINC70338 also formed hydrogen bonds with ARG97 and GLY169, however the number of hydrogen bonds was less than that of ntPPO and the strength of hydrogen bonds formed was also weaker than that of ntPPO. For hPPO–ZINC70338 system, only one hydrogen bond was formed between the inhibitor and ARG97 during docking, but during the molecular dynamic simulation the hydrogen bond interaction was increased with the side chain amino group of ARG97, which indicated that the molecular docking result was only the interaction between the ligand and the protein at one moment and did not reflect the overall state of motion. However, this hydrogen-bonding interaction was not always present during the 100 ns molecular dynamics simulation, and the hydrogen bonding accounted for only 15.78%. Fig. S3C† shown the number of inter molecular hydrogen bonds between protein and ligand during the molecular dynamic simulation, and it could be seen that ntPPO–ZINC70338 exhibited a higher number of inter molecular hydrogen bonds than hPPO–ZINC70338. The above results indicated that ZINC70338 binds more stably to ntPPO.
Table 6 Hydrogen-bonding interactions of ZINC70338 with ntPPO and hPPO
| Complexes |
Hydrogen bonds |
Distance (Å) |
Angle (deg) |
Occupancy |
| ntPPO–ZINC70338 |
ARG98-NE-HE⋯LIG-O9 |
2.84 |
156.39 |
71.45% |
| ARG98-NH2-HH21⋯LIG-O9 |
2.85 |
148.45 |
36.36% |
| LIG-N17-H39⋯GLY175-O |
2.86 |
159.21 |
67.58% |
| LIG-N7-H31⋯GLY175-O |
2.88 |
161.21 |
39.79% |
| hPPO–ZINC70338 |
ARG97-NE-HE⋯LIG-O9 |
2.84 |
156.19 |
46.33% |
| ARG97-NH2-HH21⋯LIG-O9 |
2.86 |
147.85 |
15.78% |
| LIG-N17-H39⋯GLY169-O |
2.84 |
161.9 |
67.58% |
3.5 Binding free energy calculation and residue free energy decomposition
To better understand the interactions between ligand and proteins, identify key residues, and to analyze the reasons for selectivity, the molecular mechanics generalized Born surface area (MMGBSA) method was used to calculate the binding free energy (ΔGbind) and per-residue decomposition analysis based on 1000 snapshots that were extracted from the last 10000 frames after equilibration at a time interval of 10 ps. The binding free energy of the 3 times molecular dynamics simulations are shown in Table 7. The average binding free energy of ntPPO–ZINC70338 and hPPO–ZINC70338 were −48.53 and −37.82 (kcal mol−1), respectively, and was consistent with the bio-activity value, which meant ZINC70338 had better selectivity for ntPPO. van der Waals interaction and electrostatic interaction contributed a favorable force for the binding of ZINC70338 and PPO, and were the main drivers; while polar solvation energy formed an unfavorable interaction for the binding. The main difference of binding free energy was the contribution of electrostatic interaction, i.e. hydrogen-bonding interaction. ΔGEEL is −41.74 kcal mol−1 in ntPPO–ZINC70338 system, while it is −30.80 kcal mol−1 in hPPO–ZINC70338, which was consistent with the previous docking results and hydrogen-bonding analysis, and considered to be the main reason for the selectivity; secondly, van der Waals forces also play an important role for the selectivity. The average value of the three molecular dynamic simulations for each energy of binding free energy is shown in Table S4.†
Table 7 The binding free energy of ZINC70338 to ntPPO and hPPO calculated by MMGBSA method (kcal mol−1)
| Complexes |
Contributions |
ΔGavg |
| ΔEvdw |
ΔEele |
ΔEGB |
ΔEsurf |
ΔEMM |
ΔEsol |
ΔGbind |
| ntPPO–ZINC70338 |
−50.18 ± 0.09 |
−43.89 ± 0.14 |
48.92 ± 0.09 |
−4.42 ± 0.01 |
−94.07 ± 0.15 |
44.50 ± 0.09 |
−49.57 ± 0.12 |
−48.53 |
| −50.12 ± 0.27 |
−39.08 ± 0.51 |
46.09 ± 0.35 |
−4.38 ± 0.01 |
−89.20 ± 0.51 |
41.71 ± 0.35 |
−47.49 ± 0.33 |
| −48.26 ± 0.28 |
−42.26 ± 0.35 |
43.36 ± 0.23 |
−4.37 ± 0.01 |
−90.52 ± 0.32 |
41.99 ± 0.23 |
−48.53 ± 0.32 |
| hPPO–ZINC70338 |
−49.14 ± 0.24 |
−27.27 ± 0.84 |
42.52 ± 0.61 |
−4.35 ± 0.01 |
−76.42 ± 0.79 |
38.17 ± 0.61 |
−38.24 ± 0.33 |
−37.82 |
| −44.50 ± 0.27 |
−30.27 ± 0.76 |
39.34 ± 0.51 |
−3.82 ± 0.02 |
−74.77 ± 0.76 |
35.51 ± 0.50 |
−39.26 ± 0.43 |
| −43.87 ± 0.08 |
−34.86 ± 0.18 |
46.74 ± 0.15 |
−3.96 ± 0.01 |
−78.72 ± 0.19 |
42.78 ± 0.15 |
−35.94 ± 0.10 |
The average conformation after simulated equilibrium was selected to further analyze the difference of key residues in the binding of ZINC70338 with ntPPO and hPPO based on the residue energy decomposition results. The residues with energy contributions over 0.5 kcal mol−1 are listed in Fig. 6. The key residues for the molecular dynamic simulations were all distributed around the binding pocket, and the residue types were relatively similar. The energy contributions of key residues (ARG98, ARG97), (PHE172, LEU166), (LEU372, VAL347), (PHE392, MET368) were identified as the major difference for ZINC70338 binding with ntPPO and hPPO. The contribution of residues forming hydrogen bonds was much larger in ntPPO than in hPPO (Tables S5 and S6†), e.g., the energy contribution of ARG98 and GLY175 (−4.32, −4.10 kcal mol−1) in ntPPO was larger than that of ARG97 and GLY169 (−3.18, −3.46 kcal mol−1) in hPPO. The energy contribution of ARG98 mainly originates from polar interactions with an energy contribution value of −2.58 kcal mol−1, however the polar interaction contribution of AGR97 was −0.8 kcal mol−1 (Tables S5 and S6†). The small size of the S2 region due to the “down” conformation of PHE331 in the S2 subsite of hPPO limited the conformational adjustment of the compound compared to ntPPO, resulting in a smaller number of hydrogen bonds formed in hPPO and a smaller hydrogen bond strength than in ntPPO.
 |
| | Fig. 6 Per-residue decomposition studies and average structural analysis. (A) comparison of the key residues in ntPPO and hPPO; (B and C) a comparison of the average structure generated by extracting 1000 frames of conformation from the two complex traces. | |
In addition, ZINC70338 formed π–π stacking interactions with PHE392 and T-shaped configurations with PHE172 when binding with ntPPO, with contributions of −2.76 and −1.59 kcal mol−1, respectively, of which van der Waals interactions contributed a main favorable force, with contributions of −2.39 and −1.42 kcal mol−1, respectively (Tables S5 and S6†). In contrast, the residues at the intermediate position of hPPO were MET368 and LEU166, respectively, which the hydrophobic interactions was the main contributions with hPPO, and the residue energy contribution values were lower than those of ntPPO in comparison. The residue PHE331 formed a T-shaped configurations with the benzene ring of ZINC70338. Since PHE353 was located in the flexible “loop” structural region and the side chain phenylmethyl was larger in size, it was easy to fluctuate and also forms a weaker T-shaped configurations with ZINC70338 during simulation. The ΔEvdw of PHE331 and PHE353 were −1.72 and −1.17 kcal mol−1, respectively. However, since this was not the dominant conformation of PHE353, the interaction energy was weaker than that of PHE331 (Fig. 6).
In summary, the reduction of hydrogen bonding (mainly hydrogen bonding interactions formed with ARG97) and the non-polar interaction energy contribution of PHE172, PHE392 and LEU372 when binding with hPPO leads to reduced biological activity.
4. Conclusion
In this study, 13 PPO inhibitors with bio-selectivity >10 fold were identified by structure-based virtual screening method. Then based on the analysis of the structural differences between the ntPPO and hPPO binding sites, ZINC70338 with a bio-selectivity over 113-fold was used as the object of the study to analyze the selectivity mechanism using molecular docking molecular dynamics simulation and other methods to provide some ideas for the subsequent design of novel protoporphyrinogen oxidase inhibitors. The results of molecular dynamic simulations and binding free energy calculations indicated that the difference in hydrogen bonding due to the difference in active site cavity volume was the main reason for bio-selectivity; in addition, the π–π stacking interaction formed by PHE392, PHE172 and PHE353 was also the key to selectivity. The molecular docking results and molecular dynamics simulations were consistent with the bio-activity test results. The molecules identified in the study can be used further test and have the potential to be a lead compound.
Data availability
The original contributions presented in the study are included in the article/ESI; † further inquiries can be directed to the corresponding authors.
Author contributions
Sheng-Gang Yang designed the study; Shan-Shan Zhao and Sheng-Gang Yang conducted the computational work and the experimental data analysis; Yu-jie Wang carried out the in vitro test; Shan-Shan Zhao wrote the manuscript; Lei Tang, Bing Guo and Ling Wang reviewed the manuscript; Sheng-Gang Yang and Ji-Quan Zhang revised the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 81860627, 82060625), and the Guizhou Provincial Natural Science Foundation ([2020]1Z073).
References
- G. Layer, J. Reichelt and D. Jahn, et al., Structure and function of enzymes in heme biosynthesis, Protein Sci., 2010, 19(6), 1137–1161 CrossRef CAS PubMed.
- M. Koch, C. Breithaupt and R. Kiefersauer, et al., Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis, EMBO J., 2004, 23(8), 1720–1728 CrossRef CAS PubMed.
- G. F. Hao, Y. Zuo and S. G. Yang, et al., Protoporphyrinogen oxidase inhibitor: an ideal target for herbicide discovery, Chimia, 2011, 65(12), 961–969 CrossRef CAS PubMed.
- H. Gunshin, B. Mackenzie and U. V. Berger, et al., Cloning and characterization of a mammalian proton-coupled metal-ion transporter, Nature, 1997, 388(6641), 482–488 CrossRef CAS PubMed.
- A. Porri, M. Betz and K. Seebruck, et al., Inhibition profile of trifludimoxazin towards PPO2 target site mutations, Pest Manage. Sci., 2023, 79(2), 507–519 CrossRef CAS PubMed.
- K. Iwashita, Y. Hosokawa and R. Ihara, et al., Flumioxazin, a PPO inhibitor: A weight-of-evidence consideration of its mode of action as a developmental toxicant in the rat and its relevance to humans, Toxicology, 2022, 472, 153160 CrossRef CAS PubMed.
- T. D. Sherman, J. M. Becerril and H. Matsumoto, et al., Physiological basis for differential sensitivities of plant species to protoporphyrinogen oxidase-inhibiting herbicides, Plant Physiol., 1991, 97(1), 280–287 CrossRef CAS PubMed.
- J. C. Deybach, H. de Verneuil and Y. Nordmann, The inherited enzymatic defect in porphyria variegata, Hum. Genet., 1981, 58(4), 425–428 CrossRef CAS PubMed.
- P. N. Meissner, A. V. Corrigall and R. J. Hift, Fifty years of porphyria at the University of Cape Town, S. Afr. Med. J., 2012, 102(6), 422–426 CrossRef CAS PubMed.
- G. F. Hao, Y. Tan and S. G. Yang, et al., Computational and experimental insights into the mechanism of substrate recognition and feedback inhibition of protoporphyrinogen oxidase, PLoS One, 2013, 8(7), e69198 CrossRef CAS PubMed.
- V. H. Fingar, T. J. Wieman and K. S. McMahon, et al., Photodynamic therapy using a protoporphyrinogen oxidase inhibitor, Cancer Res., 1997, 57(20), 4551–4556 CAS.
- J. Frank and A. M. Christiano, Variegate porphyria: past, present and future, Skin Pharmacol. Appl. Skin Physiol., 1998, 11(6), 310–320 CrossRef CAS PubMed.
- B. Wang, Z. Zhang and H. Zhu, et al., The hydrogen bonding network involved Arg59 in human protoporphyrinogen IX oxidase is essential for enzyme activity, Biochem. Biophys. Res. Commun., 2021, 557, 20–25 CrossRef CAS PubMed.
- D. W. Wang, H. Zhang and S. Y. Yu, et al., Discovery of a Potent Thieno[2,3-d]pyrimidine-2,4-dione-Based Protoporphyrinogen IX Oxidase Inhibitor through an In Silico Structure-Guided Optimization Approach, J. Agric. Food Chem., 2021, 69(47), 14115–14125 CrossRef CAS PubMed.
- G. F. Hao, Y. Tan and W. F. Xu, et al., Understanding resistance mechanism of protoporphyrinogen oxidase-inhibiting herbicides: insights from computational mutation scanning and site-directed mutagenesis, J. Agric. Food Chem., 2014, 62(29), 7209–7215 CrossRef CAS PubMed.
- G. F. Hao, Y. Zuo and S. G. Yang, et al., Computational Discovery of Potent and Bioselective Protoporphyrinogen IX Oxidase Inhibitor via Fragment Deconstruction Analysis, J. Agric. Food Chem., 2017, 65(28), 5581–5588 CrossRef CAS PubMed.
- K. Wakabayashi and P. Böger, Target sites for herbicides: entering the 21st century, Pest Manage. Sci., 2002, 58(11), 1149–1154 CrossRef CAS PubMed.
- X. Qin, Y. Tan and L. Wang, et al., Structural insight into human variegate porphyria disease, FASEB J., 2011, 25(2), 653–664 CrossRef CAS PubMed.
- G. M. Morris, R. Huey and W. Lindstrom, et al., AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- R. Spitzer and A. N. Jain, Surflex-Dock: Docking benchmarks and real-world application, J. Comput.-Aided Mol. Des., 2012, 26(6), 687–699 CrossRef CAS PubMed.
- C. M. Tice, Selecting the right compounds for screening: does Lipinski's Rule of 5 for pharmaceuticals apply to agrochemicals?, Pest Manage. Sci., 2001, 57(1), 3–16 CrossRef CAS.
- N. M. O'Boyle, M. Banck and C. A. James, et al., Open Babel: An open chemical toolbox, J. Cheminf., 2011, 3, 33 Search PubMed.
- K. Schöning-Stierand, K. Diedrich and R. Fährrolfes, et al., ProteinsPlus: interactive analysis of protein-ligand binding interfaces, Nucleic Acids Res., 2020, 48(W1), W48–W53 CrossRef PubMed.
- I. U. Heinemann, N. Diekmann and A. Masoumi, et al., Functional definition of the tobacco protoporphyrinogen IX oxidase substrate-binding site, Biochem. J., 2007, 402(3), 575–580 CrossRef CAS PubMed.
- A. V. Corrigall, K. B. Siziba and M. H. Maneli, et al., Purification of and kinetic studies on a cloned protoporphyrinogen oxidase from the aerobic bacterium Bacillus subtilis, Arch. Biochem. Biophys., 1998, 358(2), 251–256 CrossRef CAS PubMed.
- L. L. Jiang, Y. Tan and X. L. Zhu, et al., Design, synthesis, and 3D-QSAR analysis of novel 1,3,4-oxadiazol-2(3H)-ones as protoporphyrinogen oxidase inhibitors, J. Agric. Food Chem., 2010, 58(5), 2643–2651 CrossRef CAS PubMed.
- Y. Zuo, S. G. Yang and Y. P. Luo, et al., Design and synthesis of 1-(benzothiazol-5-yl)-1H-1,2,4-triazol-5-ones as protoporphyrinogen oxidase inhibitors, Bioorg. Med. Chem., 2013, 21(11), 3245–3255 CrossRef PubMed.
- D. A. Case, T. E. Cheatham III and T. Darden, et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26(16), 1668–1688 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez and K. Kasavajhala, et al., ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf and J. W. Caldwell, et al., Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- D. Nayar, M. Agarwal and C. Chakravarty, Comparison of Tetrahedral Order, Liquid State Anomalies, and Hydration Behavior of mTIP3P and TIP4P Water Models, J. Chem. Theory Comput., 2011, 7(10), 3354–3367 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- C. Sagui, L. G. Pedersen and T. A. Darden, Towards an accurate representation of electrostatics in classical force fields: efficient implementation of multipolar interactions in biomolecular simulations, J. Chem. Phys., 2004, 120(1), 73–87 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham III, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- B. R. Miller III, T. D. McGee Jr and J. M. Swails, et al., MMPBSA.py: An Efficient Program for End-State Free Energy Calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef PubMed.
- E. Wang, H. Sun and J. Wang, et al., End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design, Chem. Rev., 2019, 119(16), 9478–9508 CrossRef CAS PubMed.
- G. Xiong, Z. Wu and J. Yi, et al., ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49(W1), W5–W14 CrossRef CAS PubMed.
- S. A. Ali, M. I. Hassan and A. Islam, et al., A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states, Curr. Protein Pept. Sci., 2014, 15(5), 456–476 CrossRef CAS PubMed.
- R. Kumari, R. Rathi and S. R. Pathak, et al., Computational investigation of potent inhibitors against YsxC: structure-based pharmacophore modeling, molecular docking, molecular dynamics,
and binding free energy, J. Biomol. Struct. Dyn., 2021, 1–12 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Ji-Quan Zhang
d,
Ji-Quan Zhang