
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLWJ-M30, a conjugate of DM1 and B6, for the targeted therapy of colorectal cancer with improved therapeutic effects†
Qiu-Yan Zhang‡
 a,
Qing-Long Yu‡a,
Wei-Jing Luan‡a,
Tong-Fang Lia,
Ya-Ni Xiaoa,
Li Zhanga,
Yi Li*a,
Rong Rong*a and
Chun-Guang Ren*ab
a,
Qing-Long Yu‡a,
Wei-Jing Luan‡a,
Tong-Fang Lia,
Ya-Ni Xiaoa,
Li Zhanga,
Yi Li*a,
Rong Rong*a and
Chun-Guang Ren*ab
aYantai Key Laboratory of Nanomedicine & Advanced Preparations, Yantai Institute of Materia Medica, Shandong, China. E-mail: yli@simmyt.ac.cn; rrong@yimm.ac.cn
bCollege of Life Sciences, Yantai University, Yantai 264005, China. E-mail: cgren@ytu.edu.cn
First published on 5th April 2023
Abstract
Colorectal cancer (CRC) is one of the most prevalent cancers worldwide as well as a significant cause of mortality. The conventional treatment could cause serious side effects and induce drug resistance, recurrence and metastasis of cancers. Hence, specific targeting of cancer cells without affecting the normal tissues is currently an urgent necessity in cancer therapy. The emerging of peptide–drug conjugates (PDC) is regarded as a promising approach to address malignant tumors. LWJ-M30, a conjugate of DM1 and B6 peptide, targeted transferrin receptors (TfRs) on the surface of the CRC cells, showing a powerful anti-cancer effect. LWJ-M30 significantly inhibited the HCT116 cells proliferation and migration in vitro. LWJ-M30 also dramatically decreased the level of polymeric tubulin, while the disruption of microtubules caused the cell cycle to be arrested in the G2/M phase. LWJ-M30 induced the HCT116 cells apoptosis both in vivo and in vitro. The results in vivo demonstrated that LWJ-M30 could inhibit the HCT116 growth without affecting the mouse body weight. Taking these results together, our data indicated that LWJ-M30 could improve the therapeutic effects of DM1 while reducing the systemic toxicity in normal tissues.
1. Introduction
Colorectal cancer (CRC) is a common malignancy as well as a significant cause of mortality.1,2 The yearly incidence of CRC is nearly 1.4 million.3 According to new research, it is estimated that the mortality of CRC will increase by 71.5% until 2035.4 In China, CRC is the second leading cause of cancer and the incidence has increased rapidly over the past two decades.5 The primary risk factors for CRC include older age, family history, unhealthy diet, smoking and alcohol.6 But the aetiology of CRC has not been completely illustrated and the immediate causes remain unknown. The treatment of CRC is closely associated with the stage of the cancer. The staging of CRC depends on the extent of tumour invasion, the size of the tumour, metastasis, size and location of involved lymph nodes. Due to medical advances, the optional treatments for CRC are developing rapidly such as surgery, radiation therapy, chemotherapy, immunotherapy and cell therapy.6 Despite advances in these cancer therapies, there are still no clinically approved effective drugs targeting CRC as the first-line drug treatment and the mortality from CRC has not decreased as much as we expected in recent decades. It is the urgent need for a novel approach in treating CRC.7 Peptide–drug conjugates (PDC) is regarded as a new modality for cancer targeted treatment with high efficacy and low toxicity. PDC consists of three main components: the tumor-homing peptides, cytotoxic drug, and a linker. The peptides do not have pharmacological action, but they can penetrate membranes and bind to the receptors which overexpress on the tumor cell surface. This attribute enables the targeted delivery of the cytotoxic payload.8 The targeting peptide used in the conjugates must be highly specific and lead to receptor-mediated endocytosis of the PDC by the target cells only. The highly toxic payloads, for instance, maytansine, camptothecin derivatives, auristatin, or doxorubicin are recommended to choose in the conjugates.9 Finally, the linker which links the former two is selected to gain sufficient circulation time for the PDC to reach the target cell. The three components work together then contribute to increase the efficacy and selectivity of the PDC.9 Overall, the PDC should be stable enough to reach the target cell before getting cleaved or metabolized and have sufficient concentration to kill the tumor. Recently, many PDCs are already in clinical or preclinical stages, like ANG1005, EP-100, BT1718 and so on. ANG1005 can target low-density lipoprotein receptor-related protein-1 (LRP-1) to induce receptor-mediated paclitaxel entry into brain tissue through transcytosis, which is currently in clinical phase III. EP-100 and BT1718 had entered phase II clinical trial for the treatment of ovarian cancer and breast cancer or the advanced solid tumors with high MT-1 expression, respectively.10–12 Transferrin receptor (TfR) is a hopeful target in cancer therapy due to its overexpress on the surface of most solid tumors. TfR belongs to type II transmembrane glycoprotein with a special extracellular structure. It expresses highly in proliferating cells while at low levels in human normal tissues, which may facilitate the increase of the uptake of the drug and the reduction of toxic effect on normal tissues.13 In this regard, TfR is supposed to be an ideal target for the delivery of cytotoxic payload to tumor selectively.14 Maytansine (DM1), isolated from Maytenus ovatus, is a potent anticancer drug through inhibiting tubulin assemble into microtubules.15 Its antitumor mechanism is similar to vinblastine and paclitaxel, but the DM1 exhibits 10–1000 times higher cytotoxicity than some tubulin inhibitors such as taxanes, vincristine and vinblastine.16–18 Therefore, DM1 is evaluated for its powerful cytotoxicity as a potential anticancer drug against most of malignancies like breast cancer, liver cancer, multiple myeloma, and lung cancer.19 However, the disadvantages of DM1 gradually emerged with the subsequent experiments. Researchers didn't get the promising results, on the contrary, due to narrow therapeutic window, neurotoxicity and poor water solubility which hampered DM1 to be an anticancer therapy.20 Given these findings, we employed tissue-specific drug delivery approaches—PDC to overcome those adverse effects. Several TfR-targeted peptides sequence with different affinity have been reported, so we synthesized a series of TfR-targeted peptide–DM1 conjugates for screening the effects of TfR-targeted drug candidates. It is speculated that the conjugates can specifically target tumor with high expression of TfR and can be uptake more than that of the normal cells. We succeeded found LWJ-M30, a conjugate of DM1 and B6, had a significant therapeutic effect after preliminary screening. In order to further reveal the improved therapeutic effects and mechanism of the LWJ-M30 on cancer with high TfR expression, in this study, we investigated the role of the LWJ-M30 in the targeted therapy for colorectal cancer in vitro and in vivo and explored the therapeutic mechanism.2. Material and methods
2.1 Chemicals and materials
The peptide NH2-GHKAKGPRKC-CONH2 was synthesized by a solid-phase synthesis method.21 Rink amide-MBHA resin (loading: 0.661 mmol g−1) was purchased from GL Biochem (Shanghai) Ltd. Piperidine (99.50%) was purchased from Sinopharm Chemical Reagent Co. Ltd. N,N-Dimethylformamide (DMF, 99.50%) was purchased from Shanghai Titan Scientific Co. Ltd.N′,N-Diisopropylethylamine (DIPEA, 99.00%) was purchased from Shanghai Aladdin Biochemical. Benzotriazole-n ‘N’ n-tetramethylurea hexafluorophosphate ester (HBTU, 99.00%) was purchased from Shanghai Aladdin Biochemical. Dichloromethane (99.50%) was purchased from Shanghai Titan Scientific Co. Ltd. Fmoc-Gly-OH (99.89%), Fmoc-His(Trt)-OH (99.60%), Fmoc-Lys(Boc)-OH (99.26%), Fmoc-Ala-OH (99.81%), Fmoc-Pro-OH (99.37%), Fmoc-Arg(Pbf)-OH (99.74%) and Fmoc-Cys(Trt)-OH (99.48%) were purchased from CSBio Co. Ltd. Trifluoroacetic acid (TFA, 99.00%) was purchased from Shanghai Aladdin Biochemical. Triisopropylsilane (98.00%) was purchased from Sinopharm Chemical Reagent Co. Ltd. 1,2-Ethanedithiol (95.00%) was purchased from ThermoFisher Scientific. Ether (99.50%) was purchased from Sinopharm Chemical Reagent Co. Ltd. Acetonitrile (99.90%) was purchased from Sinopharm Chemical Reagent Co. Ltd. N2′-deacetyl-N2'-(3-mercapto-1-oxopropyl)-maytansine (DM1, 98.0%) was purchased from Bright Gene Bio-Medical Technology Co. Ltd. Dimethyl sulfoxide (DMSO, 99.50%) was purchased from Sinopharm Chemical Reagent Co. Ltd. 2,2′-Dithiodipyridine (diPyr, 98.00%) was purchased from Shanghai Aladdin Biochemical. All the chemical reagents were used without further purification. HCT116 cells were purchased from China infrastructure of cell line resource (Beijing, China) and cultured with high-glucose DMEM medium (Gibco) added 100 U penicillin, 100 mg mL−1 streptomycin and 10% fetal bovine serum (FBS, Gibco). MTT was obtained from Sigma-Aldrich (St. Louis, MO, USA). The primary antibodies against PARP, p53, caspase-3, Bcl2, Bax, CDK1 and cyclin B1 were purchased from Cell Signaling Technology (Danvers, MA, USA); antibodies against Ki67 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The Annexin V-FITC Assay kit, Cell Cycle and Apoptosis Analysis Kit, Hoechst Staining Kit, BCA protein assay kit and TUNEL Apoptosis Assay Kit were purchased from Beyotime Institute of Biotechnology (Shanghai, China).
2.2 Peptide synthesis
The peptide NH2-GHKAKGPRKC-CONH2 (B6) was synthesized on a Fmoc-rinkamide MBHA resin (0.661 mmol, 1135 mg) according to the standard Fmoc procedure with a 4-fold excess of amino acids (3.0 mmol).22 Deprotection of the coupled amino acids was performed by using 20% piperidine in DMF. HBTU (0.3 M in DMF) and DIPEA (0.6 M in DMF) were used to activate the protected amino acids. After completion of the sequence, the resin was washed with dichloromethane and dried under vacuum. The peptide was cleaved from the resin by using a TFA-1, 2-ethanedithiol–water–triisopropylsilane (94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:2) mixture (1 h) and precipitated with cold ether. Crude product was confirmed by a mass spectroscope (Agent Technologies, 6120 Quadrupole LC/MS + 1269 Infinity) and the characteristic m/z value was found: 541.0 [M + 2H]2+.
:2:2:2) mixture (1 h) and precipitated with cold ether. Crude product was confirmed by a mass spectroscope (Agent Technologies, 6120 Quadrupole LC/MS + 1269 Infinity) and the characteristic m/z value was found: 541.0 [M + 2H]2+.
2.3 Pyr-B6 synthesis
Briefly, B6 (600 mg, 0.55 mmol) and diPyr (245 mg, 1.10 mmol) were dissolved in 10 mL DMSO and stirred for 24 h at room temperature. Crude product was purified by using a preparative RP-HPLC (Waters, MA, USA, 2545 Binary Gradient Module + 2707 Auosampler + 2489 UV/Vis detector + Fraction Collector III) and the purity was confirmed by analytical HPLC (Waters, MA, USA,e2695 Separations Module + 2489 UV/Vis detector). The molecular mass of Pyr-GHK was determined by mass spectroscope (Agent Technologies, 6120 Quadrupole LC/MS + 1269 Infinity) and the characteristic m/z value was found: 595.4 [M + 2H]2+ and 397.3 [M + 3H]3+.2.4 DM1-B6 synthesis
DM1-B6 was synthesized with modified method from a protocol reported previously.23 Briefly, DM1 (93 mg, 126 μmol) and Pyr-B6 (100 mg, 84 μmol) were dissolved in 10 mL DMSO and reacted for 24 h at room temperature. The mixture was then purified with preparative RP-HPLC (Waters, MA, USA, 2545 Binary Gradient Module + 2707 Auosampler + 2489 UV/Vis detector + Fraction Collector III) and the purified DM1-B6 was analyzed with HPLC and ESI-MS for purity and molecular weight. The collected fractions with the target molecule were combined, concentrated and lyophilized by a freeze dryer (Labconco, Kansas City, FreeZone 6 Plus).2.5 HCT116 cell culture and MTT
The HCT116 cell line cells were plated in 10 cm2, 96-well or 6-well plates for different experiments. All the cells were cultured at 37 °C in 95% O2 and 5% CO2 in the incubator. The cells were seeded in 96-well format in triplicate incubated for 24 h then treated with different concentration of DM1 and LWJ-M30. The viability was measured by MTT-based assay after 48 h of treatment. And the IC50 was calculated by SPSS software.2.6 Wound-healing assay
The wound-healing assay was performed to estimate the cell migration capacity.24 HCT116 cells were plated in 6-well plate and grown for 24 h to nearly 70% confluence. The cells were scraped with a sterile tip to create a “scratch”, then washed with PBS and treated with DM1 (4 nM) and LWJ-M30 (1, 5, 10 nM). The images of the cells were photographed at different times.2.7 Colony formation assays
Approximately 250–300 HCT116 cells were seeded into the 6-well plates. The medium was replaced with different concentrations of DM1 and LWJ-M30 after 24 h. After incubated for 48 h, the cells were cultured with fresh DMEM media for another 10 days, then fixed by 4% paraformaldehyde, and stained with 0.1% crystal violet staining solution. The cell colonies (more than 50 cells) were counted and photographed.252.8 Transwell assay
The transwell migration assay was performed using a transwell membrane (8 μm pores, corning).26,27 The upper chambers were seeded with HCT116 cells (2 × 104 cells per well), the lower chambers were filled with 600 μL of 10% FBS to induce migration. After 24 h incubation, cells that migrated to the bottom membrane were fixed then stained with 0.1% crystal violet staining solution. The cells were counted and photographed.2.9 Hoechst 33258 staining
The Hoechst 33258 staining was used to assess the cell apoptosis according to the manufacturer's protocol. Briefly, the cells were seeded in confocal dish for 24 h then treated with DM1 and LWJ-M30 for another 24 h. The cells were washed with PBS then fixed overnight. The cells washed twice with PBS and stained with 0.5 mL Hoechst 33258 dye for 5 minutes. The cells were washed with PBS again before observing by a fluorescence microscope (Ti-U, Nikon, Japan).282.10 Flow cytometry analysis
Apoptosis caused by DM1 and LWJ-M30 was evaluated by Annexin V-FITC/PI double staining apoptosis detection kit.29 After 24 h treatment, the cells were harvested, washed with PBS and incubated with Annexin V-FITC/PI for 15 min. Apoptosis was analyzed by flow cytometry.For cell cycle analysis, HCT116 cells were treated for 24 h, harvested, washed by PBS and fixed in 70% ethanol for 4 h. The fixed cells were washed twice and incubated with 500 μL buffer containing 5 μL PI for 30 min at 37 °C. Finally, cell cycle was analyzed by flow cytometry.30
2.11 Immunohistochemistry and TUNEL staining
Tumor samples were fixed with 4% paraformaldehyde then embedded with paraffin. Tumor sections were used for immunohistochemistry and TUNEL staining. As for immunohistochemistry staining the sections were deparaffinized and antigen retrieval then blocked at room temperature for 0.5 h. The tumor sections were incubated with the primary antibody overnight at 4 °C. After washing with PBS, the secondary antibody was used for 1 h. To measure the apoptosis of the cells in vivo, TUNEL assay was used according to the manufacturer's protocol. All the images were obtained by microscopy (Leica DM500, Germany). The results were analyzed as previously described.252.12 Western blot
We performed western blot analyses as previously described. In briefly, we collected HCT116 cells, then lysed with RIPA buffer containing 1% phenylmethanesulfonyl fluoride (100 mM). The BCA protein assay kit was used to detect the protein concentration. An equal quantity of protein (approximately 80 μg) was loaded in a 10% or 12% SDS-PAGE (Genscript SurePAGE, China) then transferred on a PVDF transfer membrane (Millipore, USA). Each membrane was incubated with primary antibodies against PARP or Tublin (1:1000) for the night at 4 °C. After washing away unbound primary antibodies, we incubated the membrane with HRP secondary antibodies for 1 h at room temperature. The expression of each protein was normalized to GAPDH density by densitometry.
2.13 Xenograft tumor studies
Four- to five- week-male nude mice were purchased from Gem Pharmatech Co., Ltd and housed on a 12 h light–dark cycle under pathogen-free conditions. The animal experiment was approved by the Committee on the Ethics of Animal Experiments of the Yantai Institute of Materia Medica, and was performed in strict accordance with the guidelines of the National Research Council's Guide for the Care and Use of Laboratory Animals. HCT-116 cells were harvested, 2 × 106 cells per 200 μL were injected on the flank of the mice. Tumor growth was measured, when the average tumor volume reached to 200 mm3, the mice were randomized into six groups (n = 5). The mice in the treatment groups were treated with DM1 (1 mg kg−1), LWJ-M30 (1 mg DM1 equiv. kg−1) twice a week in the tail. The mice in control group were administrated with 5% glucose solution. Tumor size was measured three times a week using a caliper and the volume was calculated as| V = longest diameter × shortest diameter2/2. |
After 21 days treatment, the mice were euthanized and the tumors were excised and weighed.
2.14 Statistical analysis
All the data are expressed as mean values ± SD. The IC50 values were analyzed by SPSS software (version 13.0). Groups were compared by a one-way analysis of variance (ANOVA) followed by Dunnett's test using GraphPad Prism7. p < 0.05 was considered as statistical significance.3. Results
3.1 Synthesis and characterization of DM1-B6
To create a TfR targeted prodrug of DM1, the TfR binding peptide B6 was designed to conjugate with DM1 through the side chain of cysteine at the C-terminus via a disulfide bond (Fig. 1A). We first synthesized B6 according to the standard Fmoc procedure, which is the crude peptide with correct molecular weight and without further purification (Fig. S2†). The Pyr-B6 was obtained by reacting the crude B6 with diPyr, the purified product had a purity of >98% with correct molecular weight (Fig. S3†). The obtained Pyr-B6 then reacted with DM1 in DMSO to give the DM1-HKD. After prep-HPLC purification, DM1-HKD with correct molecular weight and high purity (>98%) was obtained, which was confirmed by ESI-MS and analytical HPLC (Fig. 1B and C). | ||
| Fig. 1 Molecular structure (A), ESI-MS spectra (B), and HPLC (C) of DM1-B6. | ||
3.2 LWJ-M30 regulated HCT116 cell viability and migration
To determine the activity of the compounds, we analyzed the effects of them on cell growth and migration in HCT116 cells. We found that LWJ-M30 displayed significant inhibition of cell proliferation (Fig. 2A) and clone formation ability (Fig. 2B and E). The IC50 of LWJ-M30 was 0.047 μM which was close to DM1 (0.026 μM). According to the results of wound-healing assay (Fig. 2C and F) and transwell migration assay (Fig. 2D and G), LWJ-M30 also dramatically showed a inhibitory effect on cells migration. These results suggested that LWJ-M30 could inhibit the cells proliferation and migration.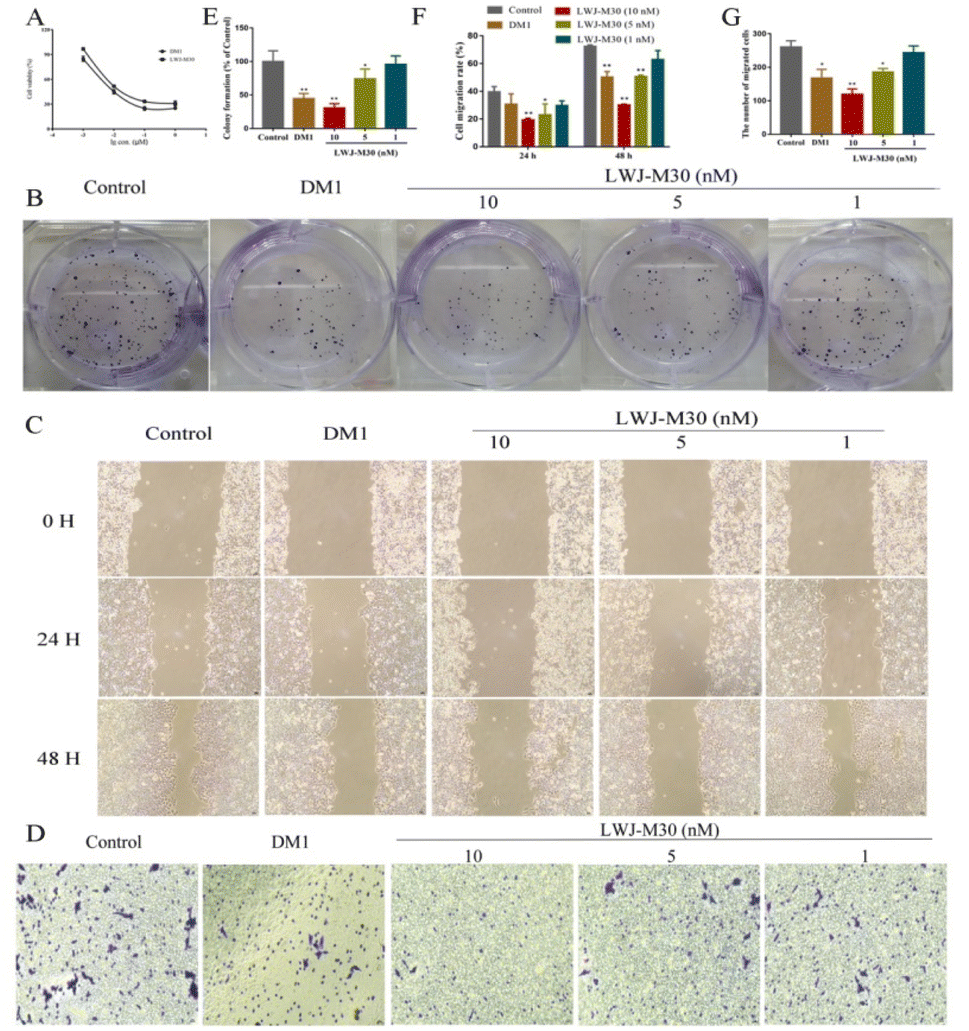 | ||
| Fig. 2 Effect of LWJ-M30 on the proliferation of HCT116 cells. (A) The effect of LWJ-M30 on cell viability of HCT116. Cell viability was evaluated by MTT assay. DM1 and LWJ-M30 significantly inhibited the cell viability in a dose-dependent manner. (B) LWJ-M30 inhibited the cell proliferation by colony forming assay. The colonies (>50 cells) were counted under the microscope. The cell migration ability was measured by the wound-healing assay (C) and transwell migration assay (D). Scale bar = 10 μm. LWJ-M30 also inhibited the HCT116 cells migration. (E)–(G) were the statistical results of colony forming, wound-healing assay and transwell migration assay, respectively. *p < 0.05, **p < 0.01 compared with control. Results were represented as mean values ± SD of triplicate independent experiments. | ||
3.3 LWJ-M30 induced the cells apoptosis
To confirm the pro-apoptotic effect of LWJ-M30 on HCT116 cells, Annexin V/PI flow cytometry, Hoechst staining and western blot were performed. As shown in Fig. 3A and B, LWJ-M30 induced the apoptosis of HCT116 cells in a dose-dependent manner, the early apoptosis increased from 8.92% ± 2.84 to 56.50% ± 4.24 while the late apoptosis increased from 7.48% ± 2.54 to 16.55% ± 1.91 by the flow cytometry analysis. The free DM1 induced the early and late apoptosis rate were 30.53% ± 2.40 and 12.02% ± 2.16, respectively. Furthermore, the Hoechst staining data showed that LWJ-M30 (5 and 10 nM) significant induced the HCT116 cells apoptosis (Fig. 3C). Subsequently, we used western blot to analysis the expression of apoptosis-associated protein. The results revealed that LWJ-M30 dramatically increased the expression of cleaved-PARP (Fig. 3D). These results indicated that LWJ-M30 could significantly inhibit the cell apoptosis. | ||
| Fig. 3 LWJ-M30 induced the apoptosis of HCT116 cells. (A) Flow cytometry of apoptotic HCT116 cells after being treated with LWJ-M30 and DM1 or not treated. The histogram (C) displayed the proportions of the apoptotic HCT116 cells. (B) The Hoechst-stained cells were captured by fluorescence microscope. LWJ-M30 induced typical apoptotic bodies in colorectal cancer cells. Scale bar = 20 μm. (D) Western blot analysis of PARP, in HCT116 cells. *p < 0.05, **p < 0.01 compared with control. Results were represented as mean values ± SD of triplicate independent experiments. | ||
3.4 LWJ-M30 induced cell cycle arrest in HCT116 cells
As we know, DM1 could inhibit tubulin polymerization.31 To investigate whether LWJ-M30 could inhibit the tubulin polymerization in HCT116 cells, we examined the expression of polymeric tubulin. Fig. 4A displayed that LWJ-M30 (10 nM) significantly decreased polymerization of tubulin, which showed better efficacy than the equal equivalent of DM1. The disruption of microtubules results in the cell cycle arrests in the G2/M phase.32 Thus, the cell cycle was measured by flow cytometry. As expected, the results suggested that LWJ-M30 induced G2/M-phase accumulation in dose-dependent manners (Fig. 4B and C). | ||
| Fig. 4 LWJ-M30 induced the disruption of microtubules promoting cell cycle arrest of HCT116 cells. (A) Western blot analyzed the level of polymeric tubulin in HCT116 cells treated with LWJ-M30 and DM1 or not treated. (B) The cell cycle distribution was analyzed by flow cytometry in HCT116 cells. (C) The proportions of G0/G1, S and G2/M phase were showed in the histogram. *p < 0.05, **p < 0.01 compared with control. Results were represented as mean values ± SD of triplicate independent experiments. | ||
3.5 Anti-cancer efficiency of LWJ-M30 against HCT116 in vivo
We further investigated the role of LWJ-M30 in preventing HCT116 progression in vivo. The results displayed that LWJ-M30 dramatically suppressed the tumor growth (Fig. 5A). As shown in Fig. 5B and C, the tumor volume and weight in LWJ-M30 treated mice were significantly decreased compared with the control group without affecting the body weight (Fig. 5D). Then we evaluated whether LWJ-M30 suppressed the tumor growth through anti-proliferation capability and apoptosis induction. As expected, LWJ-M30 significantly down-regulated the expression of Ki67 by immunohistochemical analysis. Moreover, we observed abundant apoptotic cells in LWJ-M30-treated mice using TUNEL staining (Fig. 5E and F). These outcomes indicated that LWJ-M30 could inhibit the tumor proliferation and promote apoptosis in vivo. | ||
| Fig. 5 LWJ-M30 showed anti-cancer efficiency against CRC in vivo. (A) Photographs of tumor tissues collected from HCT116 bearing nude mice in different treatment groups. (B) Tumor volumes of HCT116 cells bearing mice were measured 3 times a week after treating with DM1 and LWJ-M30. Tumor weight (C) and the mice body weight (D) derived from different groups. (E) The immunofluorescence staining of Ki67 and TUNEL staining were photographed. Scale bar = 20 μm. (F) The histogram displayed the proliferation index and apoptosis index. *p < 0.05, **p < 0.01 compared with control. Results were represented as mean values ± SD of triplicate independent experiments. | ||
4. Discussion
CRC is the leading cause of mortality among the various cancers.33 The conventional treatment displayed non-specific cytotoxicity which affected the healthy tissues and caused serious side effects. At the same time, the drug resistance, the recurrence and metastasis of cancer still seriously threaten the health of the patients.34 Therefore, it is urgent to design highly specific therapies to increase the drug concentration in the tumor while decrease the exposure to healthy cells. To achieve this, antibody–drug conjugates (ADC) and peptide–drug conjugates (PDC) have been extensively exploited. They consist of chemotherapeutic or cytotoxic agents and target ligands like peptides or antibodies via the specific linker. The conjugates can deliver higher concentration of drugs to the tumor by targeting the specific tumor-surface receptor.9 The ADC and PDC are viewed as promising therapeutic means for cancer treatment. Until now, at least 8 ADC, such as Adcetris® and Kadcyla® have been approved by the FDA.35 However, with the widely study, due to the size and complexity of the ADC, the disadvantages of ADC have been exposed gradually such as immunogenicity, insufficient drug load, high manufacturing costs, off-target toxicities and so on.36 The majority of these drawbacks can be overwhelmed by using peptides. The peptides contain about 5–50 amino acids which can be easily synthesized. The low molecule weight contributes to their high penetrability into the solid tumor, generating better anti-tumor effects.9,37The peptide NH2-GHKAKGPRKC-CONH2 (B6) we used is resulting from a phage display library which could target TfR and can cross the blood–brain barrier (BBB).38,39 It has been reported that the B6 can be used as targeted vector for cancer gene therapy.32 What's more, PLGA-PEG nanoparticles loaded curcumin that conjugated with B6 peptide showed potential application in Alzheimer's disease.38 DM1 is a potent anti-mitotic agent which displays powerful cytotoxicity by depolymerizing microtubules and inducing mitotic arrest. Its powerful anti-cancer activity makes it a promising candidate for inhibiting the proliferation of the tumor cells. But the human clinical trials suggested that DM1 have general toxicity to the normal tissues.
We designed the LWJ-M30 by conjugating the B6 peptide with DM1, expecting it could ameliorate serious side effects of DM1 and improve anti-cancer activities. LWJ-M30 could significantly inhibit the cells proliferation by MTT and clone formation assay, and the cells migration by wound-healing assay and transwell migration assay. The pro-apoptotic ability of LWJ-M30 was analyzed by flow cytometry, the results revealed that LWJ-M30 induced the apoptosis of HCT116 in dose-dependent manners. This was confirmed by the Hoechst stain. We also determined the expression of apoptosis-related protein by western blot, we found that LWJ-M30 could significantly increase the amounts of cleaved PARP. We also found that LWJ-M30 dramatically decreased the level of polymeric tubulin, and then made the cell cycle arrested in the G2/M phase.40 This may be the reason that LWJ-M30 significantly inhibited the cells migration and enhanced the apoptosis.
Encouraged by previous in vitro results, we further assessed the anti-cancer effect of LWJ-M30 in vivo. As shown in our results, treatment with LWJ-M30 significantly inhibited the tumor growth without affecting the body weight of the mice. The B6 peptide could specifically target TfR receptors on the cell surface, causing the release of DM1 in tumor tissue and reducing the damage to normal tissue. These consequences were supported by immunohistochemistry and TUNEL staining. LWJ-M30 down-regulated the expression of Ki67 while increased the number of apoptotic cells. Whatever in vivo or in vitro, the anti-cancer activity of LWJ-M30 was better than DM1 with the same equivalent.
5. Conclusions
Taken these results together, our data indicated that LWJ-M30 targeted TfR, dramatically decreased the level of polymeric tubulin, while the disruption of microtubules made the cell cycle arrested in the G2/M phase thus caused cells apoptosis. LWJ-M30 could improve the therapeutic effects of DM1 while reducing the systemic toxicity in normal tissues. Meantime, LWJ-M30 will be a promising drug candidate with the advantages of simple synthesis method, low cost, low toxicity, and high efficiency.Author contributions
Chun-Guang Ren, Qiu-Yan Zhang, Tong-Fang Li, Ya-Ni Xiao, and Li Zhang: the conception and design of the study, or acquisition of data, or analysis and interpretation of data, Yi Li and Qing-Long Yu: drafting the article or revising it critically for important intellectual content, Qiu-Yan Zhang and Rong Rong: final approval of the version to be submitted.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
All the work were financial supported by the project ZR2020QH192 supported by Shandong Provincial Natural Science Foundation and the project 2021MSGY035 supported by Yantai Science and Technology Bureau.Notes and references
- A. L. Zygulska and P. Pierzchalski, Novel Diagnostic Biomarkers in Colorectal Cancer, Int. J. Mol. Sci., 2022, 23(2), 852 CrossRef CAS PubMed.
- K. Chen, et al., Pathological Features and Prognostication in Colorectal Cancer, Curr. Oncol., 2021, 28(6), 5356–5383 CrossRef PubMed.
- A. Lewandowska, et al., Risk Factors for the Diagnosis of Colorectal Cancer, Cancer Control, 2022, 29, 1–15 CrossRef PubMed.
- T. Sawicki, et al., A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis, Cancers, 2021, 13(9), 2025 CrossRef CAS PubMed.
- J. Shen, et al., Assessing Individual Risk for High-Risk Early Colorectal Neoplasm for Pre-Selection of Screening in Shanghai, China: A Population-Based Nested Case-Control Study, Cancer Manage. Res., 2021, 13, 3867–3878 CrossRef PubMed.
- H. Y. Hsu, et al., Increased standardised incidence ratio of cardiovascular diseases among colorectal cancer patients, Int. J. Colorectal Dis., 2022, 37(4), 887–894 CrossRef PubMed.
- V. R. Silva, et al., Emerging agents that target signaling pathways to eradicate colorectal cancer stem cells, Cancer Commun., 2021, 41(12), 1275–1313 CrossRef PubMed.
- B. Balogh, et al., ConjuPepDB: a database of peptide-drug conjugates, Nucleic Acids Res., 2021, 49(D1), D1102–D1112 CrossRef CAS PubMed.
- M. Alas, A. Saghaeidehkordi and K. Kaur, Peptide-Drug Conjugates with Different Linkers for Cancer Therapy, J. Med. Chem., 2021, 64(1), 216–232 CrossRef CAS PubMed.
- F. Li and S. C. Tang, Targeting metastatic breast cancer with ANG1005, a novel peptide-paclitaxel conjugate that crosses the blood-brain-barrier (BBB), Genes Dis., 2017, 4(1), 1–3 CrossRef CAS PubMed.
- W. Hansel, F. Enright and C. Leuschner, Destruction of breast cancers and their metastases by lytic peptide conjugates in vitro and in vivo, Mol. Cell. Endocrinol., 2007, 260–262, 183–189 CrossRef CAS PubMed.
- N. Cook, et al., Pharmacokinetic (PK) assessment of BT1718: A phase I/II a study of BT1718, a first in class bicycle toxin conjugate (BTC), in patients (pts) with advanced solid tumours, Ann. Oncol., 2019, 30, 174 CrossRef.
- M. Santi, et al., Rational Design of a Transferrin-Binding Peptide Sequence Tailored to Targeted Nanoparticle Internalization, Bioconjugate Chem., 2017, 28(2), 471–480 CrossRef CAS PubMed.
- S. Li, et al., Design, synthesis, and in vitro antitumor activity of a transferrin receptor-targeted peptide-doxorubicin conjugate, Chem. Biol. Drug Des., 2020, 95(1), 58–65 CrossRef CAS PubMed.
- M. Lopus, Antibody-DM1 conjugates as cancer therapeutics, Cancer Lett., 2011, 307(2), 113–118 CrossRef CAS PubMed.
- J. B. Venghateri, et al., Ansamitocin P3 depolymerizes microtubules and induces apoptosis by binding to tubulin at the vinblastine site, PLoS One, 2013, 8(10), e75182 CrossRef CAS PubMed.
- G. Rinnerthaler, S. P. Gampenrieder and R. Greil, HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer, Int. J. Mol. Sci., 2019, 20(5), 1115 CrossRef CAS PubMed.
- W. C. Widdison, et al., Semisynthetic maytansine analogues for the targeted treatment of cancer, J. Med. Chem., 2006, 49(14), 4392–4408 CrossRef CAS PubMed.
- X. Yu, et al., Zein nanoparticles as nontoxic delivery system for maytansine in the treatment of non-small cell lung cancer, Drug Delivery, 2020, 27(1), 100–109 CrossRef CAS PubMed.
- E. Oroudjev, et al., Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability, Mol. Cancer Ther., 2010, 9(10), 2700–2713 CrossRef CAS PubMed.
- I. Martin, et al., Solid-phase-assisted synthesis of targeting peptide-PEG-oligo(ethane amino)amides for receptor-mediated gene delivery, Org. Biomol. Chem., 2012, 10(16), 3258–3268 RSC.
- S. Abramkin, et al., Solid-phase synthesis of oxaliplatin-TAT peptide bioconjugates, Dalton Trans., 2012, 41(10), 3001–3005 RSC.
- W. Ran, et al., Self-assembling mertansine prodrug improves tolerability and efficacy of chemotherapy against metastatic triple-negative breast cancer, J. Controlled Release, 2020, 318, 234–245 CrossRef CAS PubMed.
- C. Zhang, et al., KMT2A regulates cervical cancer cell growth through targeting VDAC1, Aging, 2020, 12(10), 9604–9620 CrossRef CAS PubMed.
- Y. Li, et al., XHL11, a novel selective EGFR inhibitor, overcomes EGFR(T790M)-mediated resistance in non-small cell lung cancer, Eur. J. Pharmacol., 2021, 907, 174297 CrossRef CAS PubMed.
- J. Wang, et al., Pleckstrin-2 as a Prognostic Factor and Mediator of Gastric Cancer Progression, Gastroenterol Res Pract., 2021, 2021, 5527387 Search PubMed.
- L. Wang, et al., Zoledronic acid inhibits the growth of cancer stem cell derived from cervical cancer cell by attenuating their stemness phenotype and inducing apoptosis and cell cycle arrest through the Erk1/2 and Akt pathways, J. Exp. Clin. Cancer Res., 2019, 38(1), 93 CrossRef PubMed.
- Y. C. Ma, et al., Effect of Furin inhibitor on lung adenocarcinoma cell growth and metastasis, Cancer Cell Int., 2014, 14, 43 CrossRef PubMed.
- A. Das, et al., A novel triazole, NMK-T-057, induces autophagic cell death in breast cancer cells by inhibiting gamma-secretase-mediated activation of Notch signaling, J. Biol. Chem., 2019, 294(17), 6733–6750 CrossRef CAS PubMed.
- X. Wang, et al., The RNA polymerase III repressor MAF1 is regulated by ubiquitin-dependent proteasome degradation and modulates cancer drug resistance and apoptosis, J. Biol. Chem., 2019, 294(50), 19255–19268 CrossRef CAS PubMed.
- J. Yang, et al., Covalent modification of Cys-239 in beta-tubulin by small molecules as a strategy to promote tubulin heterodimer degradation, J. Biol. Chem., 2019, 294(20), 8161–8170 CrossRef CAS PubMed.
- Y. Nie, et al., Dual-targeted polyplexes: one step towards a synthetic virus for cancer gene therapy, J. Controlled Release, 2011, 152(1), 127–134 CrossRef CAS PubMed.
- B. C. Szeglin, et al., A SMAD4-modulated gene profile predicts disease-free survival in stage II and III colorectal cancer, Cancer Rep., 2022, 5(1), e1423 CAS.
- Y. Li, et al., Colorectal cancer stem cell-derived exosomal long intergenic noncoding RNA 01315 (LINC01315) promotes proliferation, migration, and stemness of colorectal cancer cells, Bioengineered, 2022, 13(4), 10827–10842 CrossRef CAS PubMed.
- Y. Wang, et al., Peptide-drug conjugates as effective prodrug strategies for targeted delivery, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS PubMed.
- J. Lindberg, et al., Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy, Molecules, 2021, 26(19), 6042 CrossRef CAS PubMed.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies, Front Chem, 2020, 8, 571 CrossRef CAS PubMed.
- S. Fan, et al., Curcumin-loaded PLGA-PEG nanoparticles conjugated with B6 peptide for potential use in Alzheimer's disease, Drug Delivery, 2018, 25(1), 1091–1102 CrossRef CAS PubMed.
- H. Xia, et al., Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium, J. Virol., 2000, 74(23), 11359–11366 CrossRef CAS PubMed.
- H. Chen, et al., Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy, Molecules, 2017, 22(8), 1281 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07758b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |