
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular porphyrin as an improved photocatalyst for chloroform decomposition
J. M. S. Lopes *ad,
A. A. Batistab,
P. T. Araujo*c and
N. M. Barbosa Neto*d
*ad,
A. A. Batistab,
P. T. Araujo*c and
N. M. Barbosa Neto*d
aDepartment of Physics, Federal University of Roraima, Boa Vista, RR, Brazil. E-mail: lopesjefferson01@yahoo.com.br
bDepartment of Chemistry, Federal University of São Carlos, São Carlos, SP, Brazil
cDepartment of Physics and Astronomy, University of Alabama, Tuscaloosa, Alabama, USA. E-mail: paulo.t.araujo@ua.edu
dInstitute of Natural Sciences, Graduate Program in Physics, Federal University of Pará, Belém, PA, Brazil. E-mail: barbosaneto@ufpa.br
First published on 13th February 2023
Abstract
In this work, the outlying decoration of the free-base meso-(4-tetra) pyridyl porphyrin (H2TPyP) with the RuCl(dppb)(5,5′-Me-bipy) ruthenium complex (here named Supra-H2TPyP) is observed as an improved molecular photocatalyst for dye-mediated chloroform (CHCl3) decomposition via one-photon absorption operating in the visible spectral range (532 nm and 645 nm). Supra-H2TPyP offers a better option for CHCl3 photodecomposition when compared to the same process mediated by pristine H2TPyP, which requires either excited-state- or UV absorption. The chloroform photodecomposition rates for Supra-H2TPyP as well as its excitation mechanisms are explored as a function of distinct laser irradiation conditions.
1. Introduction
Chlorine-derived disinfectants like sodium hypochlorite, chloramine, and chlorine dioxide are important structures for the treatment of drinking water and swimming pools.1–5 Despite their good effectiveness in killing pathogenic microorganisms, unfortunately, when these substances interact with natural organic matter (e.g., urine, sweat, and saliva) non-negligible amounts of undesired byproducts can be generated. Among them, chloroform (CHCl3) brings significant concerns to health due to its cytotoxic effects.1,6–8 In recent years, two major efforts have been conducted to deal with this problem: (1) the development of novel molecular sensors to detect chloroform in water,9–11 and (2) the creation of methodologies to decompose this molecule in solution. Concerning the effort (2), although the biotechnological12,13 and the pulsed-corona-discharges-assisted14 approaches have been lately tested, the dye-catalyzed photochemical-based approaches stand as low-cost, non-invasive, selective, and controllable alternatives.15–20The excited-state dynamics exhibited by porphyrins21–25 set them as versatile molecular photocatalysts15,26–29 that have been explored in fields like dye-sensitized solar cells,30–33 photomedicine,34–36 and artificial photosynthesis.37–39 In the last decades, these macrocycle molecules have been explored as photocatalysts for dye-mediated decomposition of chloroform (CHCl3) under one-photon absorption (OPA) and/or excited-state absorption (ESA).15,16 Differently from visible light OPA, ultra-violet OPA (λexc = 266 nm) accesses the porphyrin's photooxidative excited states enabling the decomposition of CHCl3 through an oxi-reduction reaction, which yields hydrochloric acid (HCl) and other products.15 More recently, ESA16 became an alternative approach to employ visible radiation for CHCl3 photodecomposition.
The tunning of the excitation energy threshold for photooxidation could allow the photo-reaction to occur at lower- (higher-) energy OPA (ESA) but is still poorly explored. The synthesis of novel porphyrinic structures could facilitate such energy threshold controllability. Examples of synthetic approaches are: (1) the inner and outer macrocycle decoration;21,40,41 (2) oligomer formation involving one or more types of porphyrins;38,42–44 and (3) supramolecular porphyrin structures.26,45–51 The attachment of metallic complexes (e.g., ruthenium-, rhenium- and iridium-derived) at the outer positions of porphyrins generates supramolecular structures that offer novel excited states for achieving the photo-reaction threshold while keeping original states from both the pristine porphyrin and the complexes.46,48,49,52–56
In this work, a supramolecular structure originated from the attachment of the RuCl(dppb)(5,5′-Me-bipy) ruthenium complex at each (4-pyridyl) site of the meso-tetra(4-pyridyl) free-base porphyrin (H2TPyP) is used as a molecular photocatalyst for CHCl3 decomposition, allowing the reaction to occur via visible-light OPA. The influence of the excitation wavelength and mechanisms on the photo-reaction rates is also explored.
2. Materials and methods
2.1 Samples and spectroscopic measurements
The synthesis of H2TPyP and its supramolecular H2TPyP[RuCl(dppb)(5,5′-Me-bipy)]4 structure (Supra-H2TPyP, see Fig. 1) follows the procedures described in the literature.47,57 The spectroscopic measurements were conducted with samples dissolved in CHCl3 stabilized with amylene (NEON Inc.) used as received. Solution concentrations were kept below 10.0 μM to prevent spontaneous aggregation and inner filter effects. Absorption spectra were acquired with a JASCO V-670 spectrophotometer whereas steady-state photoluminescence (PL) spectra were measured using a setup composed of a Xenon lamp (ACTON); a monochromator (ACTON, model 300i) and a portable spectrophotometer (Ocean Optics), with the PL signal being detected in a 90° geometry relative to the excitation beam direction. Time-resolved PL experiments were conducted using a Time-Correlated Single Photon Counting (TCSPC) system (Horiba, Delta-Flex model with 27 ps of temporal resolution), equipped with a pulsed excitation source (λexc = 352 nm with 8.0 MHZ of repetition rate). The PL decays were collected at the maximum of each spectrum. Emission quantum yields58,59 were calculated adopting H2TPyP dissolved in CHCl3 as standard22 and applying eqn (1):
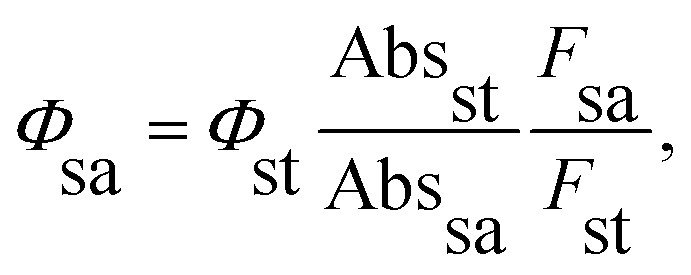 | (1) |
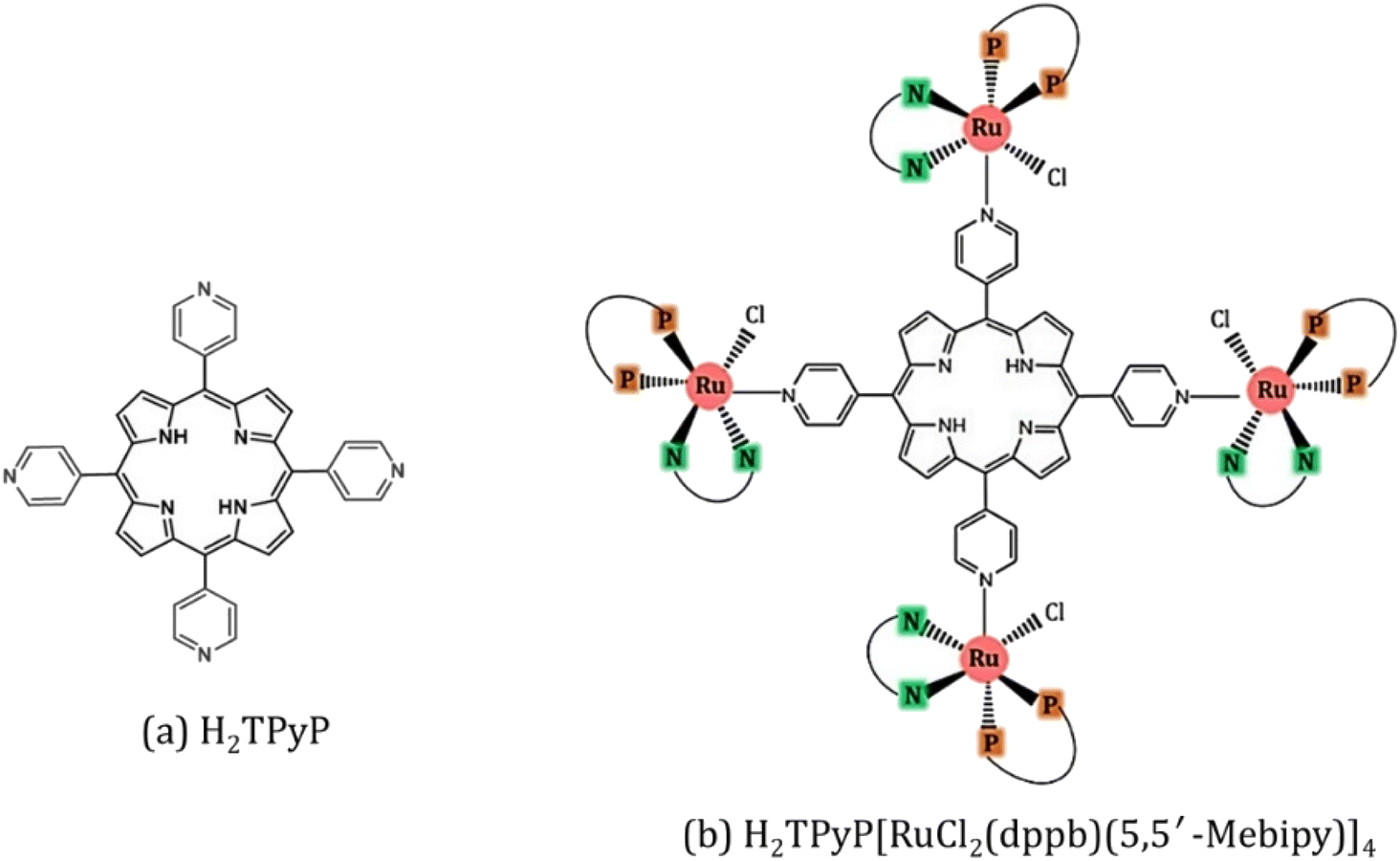 | ||
| Fig. 1 (a) H2TPyP and (b) Supra-H2TPyP, with RuCl2(dppb)(5,5′-Mebipy) ruthenium complexes attached at each (4-pyridyl) site of H2TPyP. Here, 5,5′-Mebipy stands for 5,5′-dimethyl-2,2′-bipyridine (N–N), and dppb stands for 1,4-bis(diphenylphosphine) butane (P–P). | ||
2.2. Photochemistry assays
For OPA in porphyrins, two sources (purchased from Laserline Inc), green-OPA (532 nm; ∼4.0 W cm−2, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 J cm−2 total incident fluence, 90.0 minutes of irradiation) and red-OPA (635 nm; ∼5.0 W cm−2, 30000 J cm−2 total incident fluence, 90.0 minutes of irradiation) were utilized. Here, ESA is promoted by a frequency-doubled Q-switched Nd-YAG laser (Quantel, model Q-smart 100, 6.0 ns FWHM, 532 nm excitation, 20.0 Hz of repetition rate, total incident fluence regime of ∼2500 J cm−2). The quantitative analysis of all irradiation conditions was performed by considering the effectively absorbed fluence instead of incident fluence. The relation between incident (FI), transmitted (FT) and absorbed (FA) fluences is written as FI = FT + FA, or
200 J cm−2 total incident fluence, 90.0 minutes of irradiation) and red-OPA (635 nm; ∼5.0 W cm−2, 30000 J cm−2 total incident fluence, 90.0 minutes of irradiation) were utilized. Here, ESA is promoted by a frequency-doubled Q-switched Nd-YAG laser (Quantel, model Q-smart 100, 6.0 ns FWHM, 532 nm excitation, 20.0 Hz of repetition rate, total incident fluence regime of ∼2500 J cm−2). The quantitative analysis of all irradiation conditions was performed by considering the effectively absorbed fluence instead of incident fluence. The relation between incident (FI), transmitted (FT) and absorbed (FA) fluences is written as FI = FT + FA, or 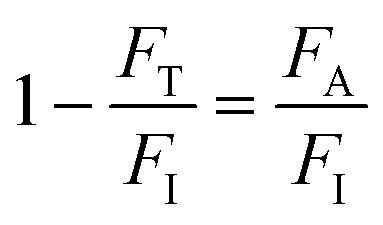 .58 From the Lambert–Beer law,58
.58 From the Lambert–Beer law,58 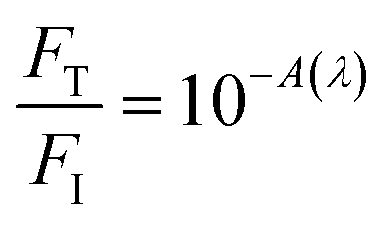 (with A(λ) corresponding to absorbance at the incident wavelength, for the non-irradiated solution) resulting in eqn (2):
(with A(λ) corresponding to absorbance at the incident wavelength, for the non-irradiated solution) resulting in eqn (2):| FA = FI(1 − 10−A(λ)) | (2) |
During all the spectroscopic measurements and irradiation processes, the samples were placed in sealed quartz cuvettes of 1.0 cm path length with four polished windows.
Control samples of H2TPyP and Supra-H2TPyP dissolved in CHCl3 were kept in a dark environment at room temperature (∼22.5 °C) to evaluate their stability. Absorption spectra confirm that no spontaneous modifications occur in both samples for 240.0 minutes of storage, endorsing their stability. This implies their durability is at least three times greater than the time demanded to perform the photo-induced reactions discussed herein.
3. Results and discussions
3.1 Photophysics of Supra-H2TPyP
As shown in Fig. 2, the absorption spectrum of Supra-H2TPyP displays red-shifted B- and Q-bands relative to H2TPyP, in agreement with literature.16,60 Additionally, signatures belonging to the metallic complex are also observed (see intraligand bands located at around 311 nm).47,61–65 Due to their superposition with H2TPyP bands, spectral signatures associated with the complex's MLCT bands centered around 470 nm are not resolved but lead to an overall increase of the spectrum baseline49,52,56 (Fig. 2b).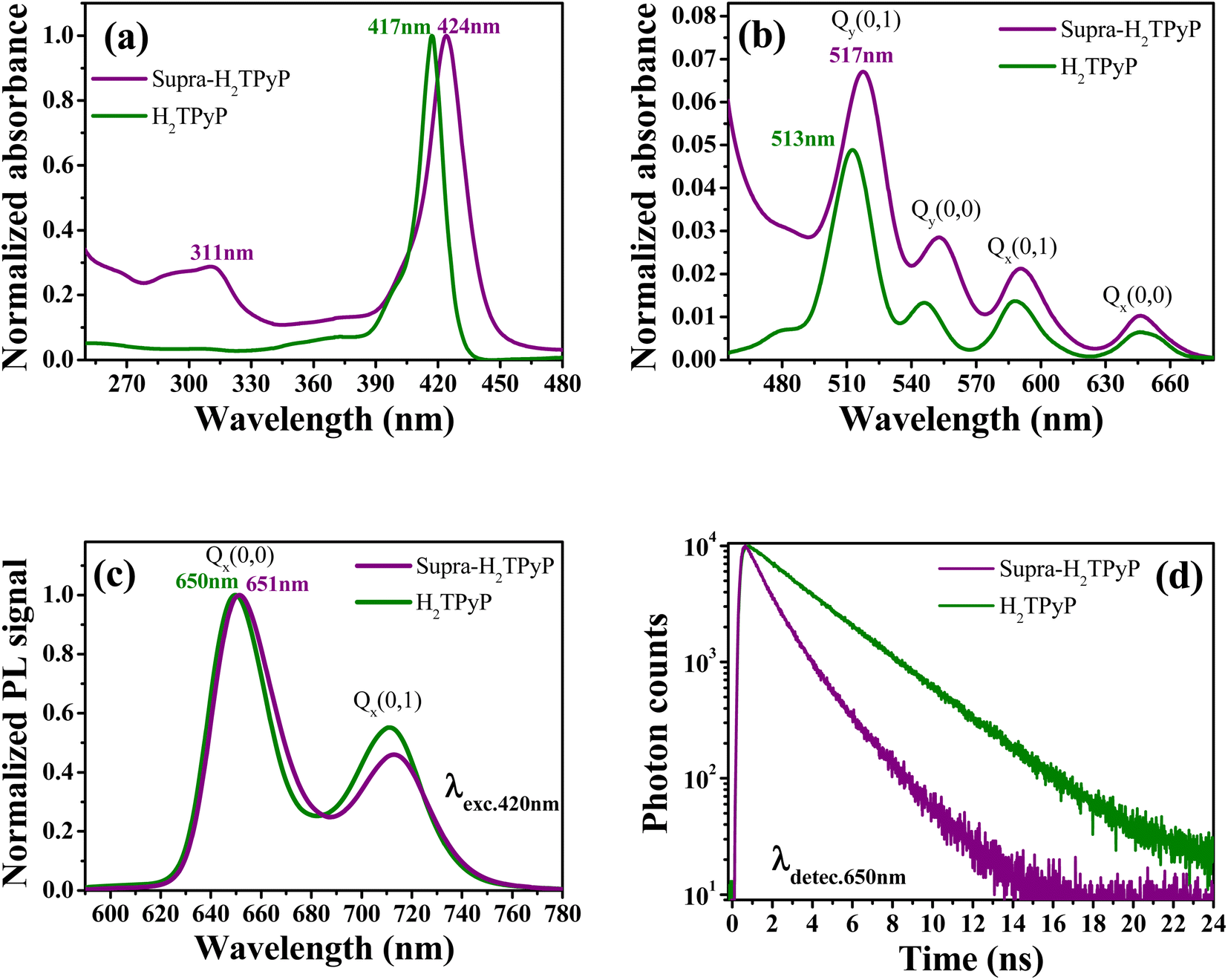 | ||
| Fig. 2 Normalized absorption spectrum at (a) B- and (b) Q-bands. (c) Normalized PL spectrum (λexc. = 420 nm) and (d) fluorescence decays (λdetec. = 650 nm) for H2TPyP (green curves) and Supra-H2TPyP (purple curves). | ||
The Supra-H2TPyP PL spectrum (Fig. 2c) shows a small redshift (∼1 nm) regarding H2TPyP. This effect is accompanied by obtaining a 72% lower emission quantum yield for Supra-H2TPyP concerning H2TPyP22 (see Table 1), in agreement with previous reports.49–52 The Supra-H2TPyP also presents a variation in the relative intensity between electronic (Q(0,0)) and vibronic (Q(0,1)) emission bands with relation to H2TPyP, which indicates that either (1) the involved H2TPyP vibronic progressions are changing52,53 or (2) a new band arises and overlaps with H2TPyP original bands.52,53 From TCSPC experiments it is observed that the mono-exponential decay (7.26 ns) exhibited by H2TPyP in CHCl360 evolves to a bi-exponential decay (τ1 = 2.01 ns (54%) and τ2 = 4.54 ns (46%)) in Supra-H2TPyP (Fig. 2d and Table 1). Based on the literature,49,52,53 and considering that isolated ruthenium complexes do not exhibit emission bands around the probed wavelength (650 nm), τ1 is associated with Supra-H2TPyP, while the lifetime τ2 is associated with the perturbed H2TPyP deactivation pathway.49,52,53 This trend supports the hypothesis of a novel band in the steady-state PL spectrum53 (Fig. 2c).
| Bmax (nm) | Qmax (nm) | PLmax (nm) | τ1 (ns) | τ2 (ns) | Φ (× 10−2) | |
|---|---|---|---|---|---|---|
| H2TPyP | 417 | 513 | 650 | — | 7.26 (100%) | 1.61 |
| Supra-H2TPyP | 424 | 517 | 651 | 2.01 (54%) | 4.54 (46%) | 0.45 |
3.2 Green-OPA irradiation of Supra-H2TPyP
In chloroform, H2TPyP is photostable under green-OPA irradiation.15,16 This is not true for Supra-H2TPyP under the same irradiation condition. Fig. 3 shows that the spectroscopic signatures of Supra-H2TPyP are significantly modified as a function of the incident fluence. Because the modifications occur in different stages, the results are separately discussed in three fluence ranges: (i) 0–2400 J cm2; (ii) 2400–6000 J cm2; and (iii) 6000–19200 J cm−2.
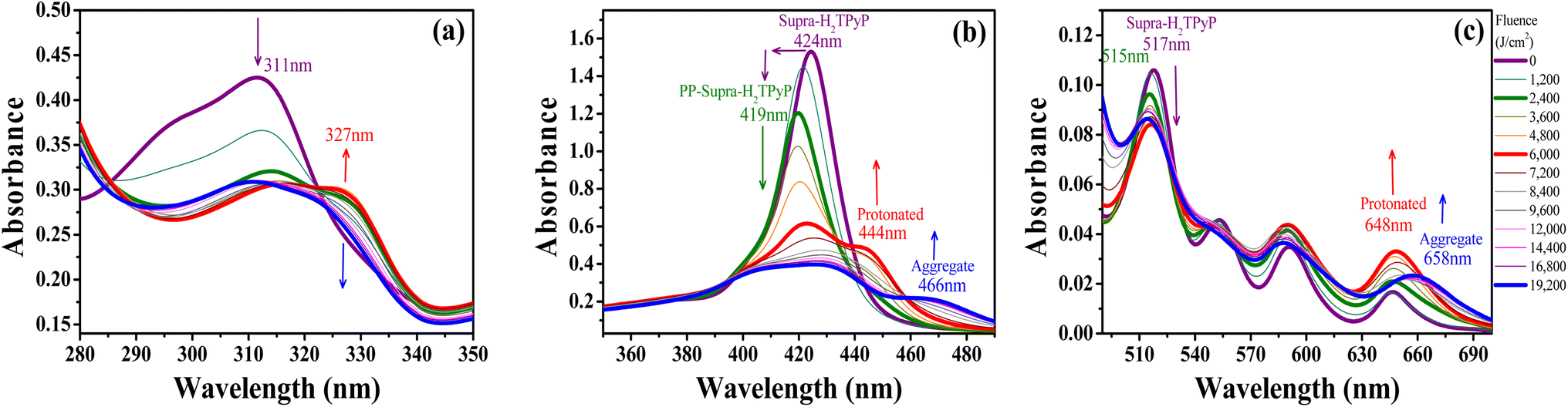 | ||
| Fig. 3 Evolution of the absorption spectrum of Supra-H2TPyP under green-OPA excitation (532 nm, 0–19200 J cm−2) at (a) the UV bands (from 280 nm to 350 nm), (b) the B-band, and (c) the Q-band. | ||
In range (i), both B- and Q-bands undergo a blue shift. The main Supra-H2TPyP B-band, originally centered at 424 nm (0 J cm−2) (purple solid line), downshifts to 419 nm at 2400 J cm−2 (olive solid line) while the Qy(0,1) downshifts from 517 nm to 515 nm. This behavior suggests that Supra-H2TPyP is undergoing a similar photodissociation as reported for the H2TPyP[RuCl2(CO)(PPh3)2]4 tetraruthenated porphyrin.53 The occurrence of photodissociation in the fluence range (i) would imply: (1) the decomposition of ruthenium complexes; and (2) the absence of processes associated with the fluence ranges (ii) and (iii).15,16 However, our data show that (1) and (2) do not happen, see Fig. 3. It is seen that in the fluence range (i), the RuCl(dppb)(5,5′-Me-bipy) complexes in Supra-H2TPyP, with intraligand bands spanning from ∼280–360 nm, undergo a sort of transformation, forming a novel supramolecular species, baptized PP-Supra-H2TPyP. The determination of the type of transformation that is photomodifying RuCl(dppb)(5,5′-Me-bipy) is out of the scope of this work but we understand that photooxidation,66,67 photoisomerization,68–70 and photoinduced ligand loss71,72 are possible processes driving the phenomenon.
In range (ii), the peak at 419 nm no longer blueshifts and starts developing into a novel spectroscopic signature centered at 444 nm, see Fig. 3b. The rise of this new signature occurs simultaneously with the enhancement of the Qx(0,0)-band (around 648 nm). These are signatures associated with photo-protonation of PP-Supra-H2TPyP.15,16,73–75 Moreover, the PP-Supra-H2TPyP 419 nm peak redshifts in ≈2 nm, which is a signature for the protonation of the photomodified RuCl(dppb)(5,5′-Me-bipy).16,76 In fact, a closer inspection of RuCl(dppb)(5,5′-Me-bipy) intraligand bands shows that the peak at ∼327 nm is enhanced (Fig. 3a). It is important to recall that protonation requires acid environments. This is reported for complexes similar to RuCl(dppb)(5,5′-Me-bipy), in which an acidic solution was created through the addition of aliquots of HCl.77,78 Since we do not add any acid to our solutions, the observation of protonation of both the porphyrin and the photomodified RuCl(dppb)(5,5′-Me-bipy) confirms the release of HCl in the solution as a consequence of CHCl3 photodecomposition via green-OPA.
In the range (iii), it is observed that protonated PP-Supra-H2TPyP signatures, B-(≈444 nm) and Qx(0,0)-(≈648 nm) bands, disappear, which is followed by the rise of new bands at ≈466 nm and ≈658 nm, respectively. The new features indicate that the protonated PP-Supra-H2TPyP is forming aggregates (possibly J-aggregates), similar to those previously reported for H2TPyP under ESA excitation.16 It is also observed that the intraligand band of the photomodified RuCl(dppb)(5,5′-Me-bipy) at 327 nm undergoes a decrease in intensity with no indications of further transformations.
The protonation of Supra-H2TPyP also induces new signatures in the steady-state PL spectrum, which are in agreement with the results discussed above for UV-Vis absorption. Fig. 4a shows that the Supra-H2TPyP PL spectrum (purple solid line) evolves to the protonated PL spectrum (red solid line).16 After protonation, the PL signal continuously decreases with increasing fluence, becoming almost null at 19200 J cm−2. This decrease in the PL magnitude is associated with a reduction in the emission quantum yield of the protonated species, which is characteristic of J-aggregation in range (iii).16,74,79–83
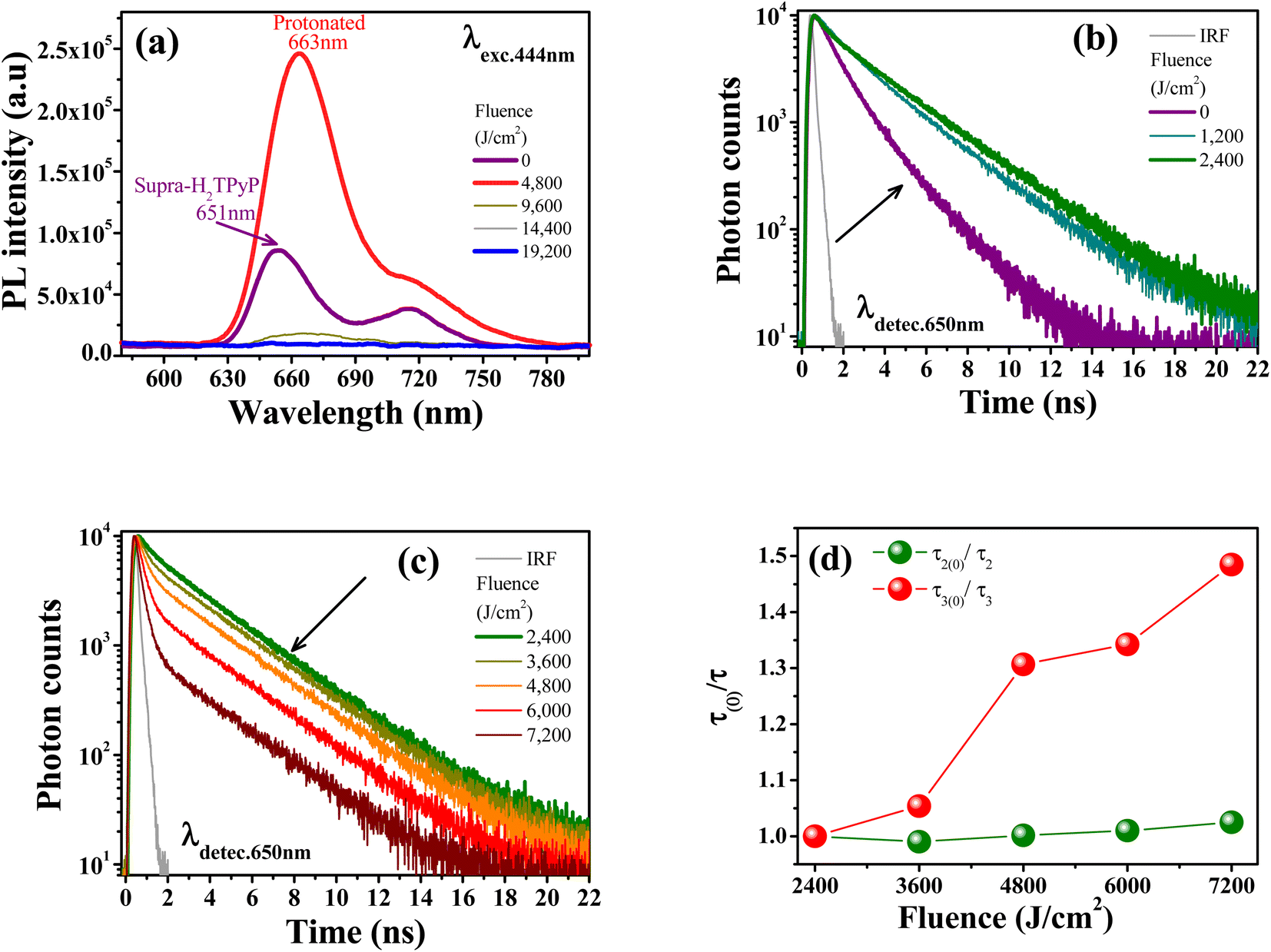 | ||
| Fig. 4 (a) PL spectra evolution with increasing fluence (λexc. = 444 nm). Supra-H2TPyP PL decay (λdetec. = 650 nm) under green-OPA (λexc. = 532 nm) for: (b) fluence range (i), and (c) fluence range (ii). (d) Fluence dependence of the τ(0)/τ ratio for PP-Supra-H2TPyP (green spheres) and its protonated counterpart (red spheres). | ||
Fig. 4b shows that the Supra-H2TPyP PL decay at 0 J cm−2 (purple solid line), which initially displays two characteristic lifetimes (4.54 ns (46%) and 2.01 ns (54%)), continuously evolves into the PP-Supra-H2TPyP decay profile at 2400 J cm−2, with a dominant characteristic lifetime of ≈7.01 ns (91%). A minor contributing lifetime of ≈ 3.17 ns (4%) is also measured and understood to belong to the remaining Supra-H2TPyP. In addition, a new PL decay with a characteristic lifetime of 0.98 ns (5%) associated with the protonated PP-Supra-H2TPyP is observed, see Table 2. The results in Fig. 4c and Table 2 support that in the range (i) both Supra-H2TPyP and PP-Supra-H2TPyP coexist. When the range (ii) starts (2400 J cm−2), PP-Supra-H2TPyP becomes predominant, coexisting with both Supra-H2TPyP and the protonated PP-Supra-H2TPyP. From 3600 J cm−2 on, the percentage contribution of the lifetime associated with Supra-H2TPyP becomes negligible.
| Fluence (J cm−2) | τ1 (ns) | τ2 (ns) | τ3 (ns) | χ2 |
|---|---|---|---|---|
| 0 | 2.01 (54%) | 4.54 (46%) | — | 1.06 |
| 1200 | 2.16 (17%) | 6.39 (83%) | — | 1.03 |
| 2400 | 3.17 (4%) | 7.01 (91%) | 0.98 (5%) | 1.07 |
| 3600 | 7.08 (89%) | 0.93 (11%) | 1.04 | |
| 4800 | 7.00 (82%) | 0.75 (18%) | 1.06 | |
| 6000 | 6.94 (65%) | 0.73 (35%) | 1.02 | |
| 7200 | 6.84 (42%) | 0.66 (58%) | 1.12 |
At 7200 J cm−2, the PP-Supra-H2TPyP (protonated PP-Supra-H2TPyP) lifetime decreases from 7.01 ns (0.98 ns) to 6.84 ns (0.66 ns). Fig. 4d shows that, at 7200 J cm−2, the quenching of these lifetimes, τ2(0)/τ2 (PP-Supra-H2TPyP) and τ3(0)/τ3 (protonated PP-Supra-H2TPyP), are 1.02 and 1.47, which originates from exciplex formation.22 The quenching of the protonated PP-Supra-H2TPyP is 144% more effective in comparison with that for PP-Supra-H2TPyP. This result supports the presence of electrostatically-enhanced exciplex formed between the positively charged protonated PP-Supra-H2TPyP and Cl− ions dispersed in the solution.16 No discussion is addressed for range (iii) because the decrease in PL intensity invisibilized the PL decay measurements.
As discussed in the literature,15,16 free base porphyrins (H2TPyP and H2TPP) under UV-OPA (λexc = 266 nm) and ESA (λexc = 532 nm) excitations are able to photodecompose CHCl3 and form HCl with excitation energy thresholds located above the B-band energy (≈2.97 eV).15,16 This implies that these free-base porphyrins never underwent photoprotonation under OPA operating at 475 nm (2.61 eV) or 532 nm (2.33 eV).15,16 In this work, we were able to photoprotonate PP-Supra-H2TPyP dissolved in CHCl3 under green-OPA (≈2.33 eV), meaning HCl is being formed as a consequence of CHCl3 decomposition. This process should not happen if the ground and the excited states of PP-Supra-H2TPyP were similar in energy to the ground and the excited states of H2TPyP. Since the excitation gap is fixed by the CW laser energy employed in green-OPA (≈2.33 eV), our results indicate two possible scenarios: (1) the involved ground state in PP-Supra-H2TPyP must be higher in energy in comparison to the correspondent state in H2TPyP and/or (2) the photooxidative excited state in PP-Supra-H2TPyP must be lower in energy concerning H2TPyP.
3.3 Other irradiation conditions
Fig. 5a and b show the same photo-induced modifications (PP-Supra-H2TPyP formation, photo-protonation, and photo-aggregation) in the spectroscopic signatures of Supra-H2TPyP for green-ESA and red-OPA irradiations, respectively. The successful decomposition of CHCl3 under red-OPA irradiation (photon energy delivered ≈1.96 eV) sets a new lower limit for the excitation energy required to trigger the process.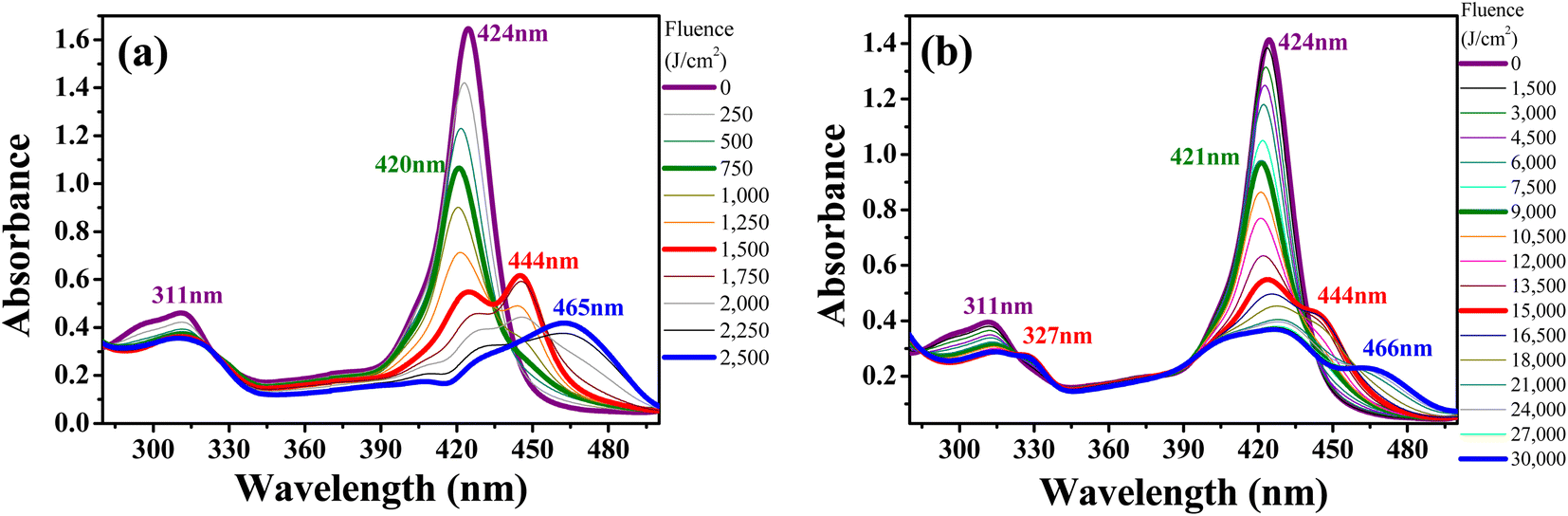 | ||
| Fig. 5 Evolution of the absorption spectrum of Supra-H2TPyP dissolved in CHCl3 and irradiated under (a) green-ESA (532 nm, 6 ns, 0–2500 J cm−2) and (b) red-OPA (633 nm, 0–19200 J cm−2). | ||
To understand the photodecomposition rates under the different laser irradiation conditions, the Supra-H2TPyP B-band integrated area (from 360 to 490 nm) for each laser absorbed fluence (FA) was normalized by the B-band integrated area (also from 360 to 490 nm) of the reference non-irradiated Supra-H2TPyP, and plotted as a function of FA (see Fig. 6). The exponential profiles obtained under each laser irradiation condition were fitted using AF = e(−kFA), where AF and k represent, respectively, the aforementioned normalized areas for each FA and the net photomodification rate (given in cm2 J−1). The fitting results show that the rates obey the following hierarchy: green-ESA (k = 4.32 × 10−3 cm2 J−1) > green-OPA (k = 1.43 × 10−3 cm2 J−1) > red-OPA (k = 0.50 × 10−3 cm2 J−1), which shows that green-ESA leads to more efficient photoreaction processes in comparison with the OPA-based irradiations.
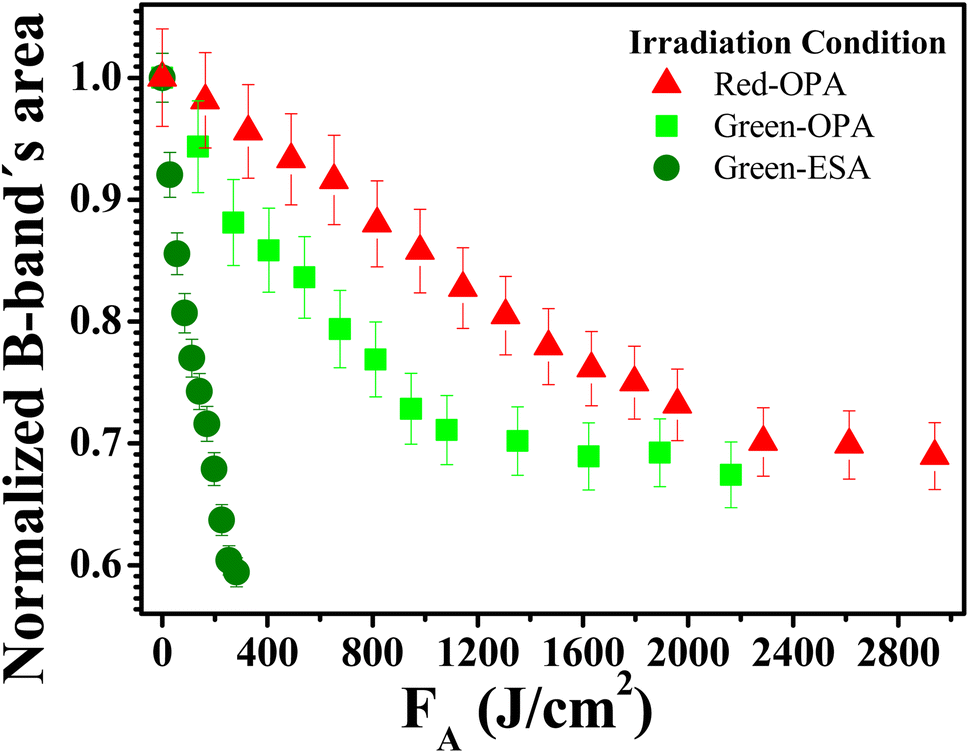 | ||
| Fig. 6 Absorbed fluence-dependent evolutions of normalized B band's area comparing green-ESA (filled olive spheres), green-OPA (filled light-green circles), and red-OPA (filled red triangles) excitations applied to Supra-H2TPyP. | ||
4. Conclusions
Supra-H2TPyP places itself as an excellent candidate for a broad-range OPA-visible-light active molecular photocatalyst in dye-mediated chloroform decomposition. This supramolecular structure displays good photoreaction rates under visible OPA irradiation, which is a simpler and more affordable excitation mechanism than visible ESA and UV-OPA. Our results show that Supra-H2TPyP when combined with different excitation wavelengths can be used to controllably photodecompose CHCl3 and, consequently, controllably photoprotonate Supra-H2TPyP itself, which is also interesting for sensing chloroform in solution and other applications in fields like materials science.Author contributions
J. M. S. Lopes: data curation, writing-original draft, writing – review & editing, investigation, visualization, formal analysis, methodology. A. A. Batista: writing-review & editing, data curation, formal Analysis, project administration, resources, validation. P. T. Araujo: data curation, writing-original draft, writing – review & editing, visualization, investigation, fund acquisition, supervision, project administration, validation, formal analysis, methodology. N. M. Barbosa Neto: data curation, writing – original draft, writing – review & editing, visualization, investigation, validation, formal analysis, methodology, supervision, resources, funding acquisition, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this manuscript.Acknowledgements
The Brazilian authors are indebted to the Brazilian National Council for Scientific and Technological Development (CNPq – processes numbers: 306147/2020-3 and 425124/2018-5), São Paulo Research Foundation (FAPESP – process number: 2014/50869-6) and the Education Ministry (CAPES – Process number: 23038.000776/201754) via the projects of the National Institute for Science and Technology on Organic Electronics and with Amazonian Foundation for the Support of Studies and Research (FAPESPA – process number: 88881.159129/2017-01). N. M. B. N. is especially indebted to the Fulbright Foundation for its Visiting Professor Award Grant. P. T. A. acknowledges the National Science Foundation for supporting this work under the CAREER grant # CHE-1848418. The authors are also grateful to Prof. Sanclayton Moreira, of the Graduate Program in Physics of the Federal University of Para, for granting access to his experimental facilities.References
- C. Hang, B. Zhang, T. Gong and Q. Xian, Occurrence and health risk assessment of halogenated disinfection byproducts in indoor swimming pool water, Sci. Total Environ., 2016, 543, 425–431 CrossRef CAS PubMed.
- S. Chowdhury, K. Al-hooshani and T. Karanfil, Disinfection byproducts in swimming pool: occurrences, implications and future needs, Water Res., 2014, 53, 68–109 CrossRef CAS PubMed.
- H. Kim, J. Shim and S. Lee, Formation of disinfection by-products in chlorinated swimming pool water, Chemosphere, 2002, 46, 123–130 CrossRef CAS PubMed.
- J. Lee, M. J. Jun, M. H. Lee, M. H. Lee, S. W. Eom and K. D. Zoh, Production of various disinfection byproducts in indoor swimming pool waters treated with different disinfection methods, Int. J. Hyg. Environ. Health, 2010, 213, 465–474 CrossRef CAS PubMed.
- M. Panyakapo, S. Soontornchai and P. Paopuree, Cancer risk assessment from exposure to trihalomethanes in tap water and swimming pool water, J. Environ. Sci., 2008, 20, 372–378 CrossRef CAS PubMed.
- L. O. Vajrabhaya, S. K. Suwannawong, R. Kamolroongwarakul and L. Pewklieng, Cytotoxicity evaluation of gutta-percha solvents: chloroform and GP-solvent (limonene), Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod., 2004, 98, 756–759 CrossRef PubMed.
- M. V Templin, K. C. Jamison, C. S. Sprankle, D. C. Wolf, B. A. Wong and B. E. Butterworth, Chloroform-induced cytotoxicity and regenerative cell proliferation in the kidneys and liver of BDF1 mice, Cancer Lett., 1996, 108, 225–231 CrossRef PubMed.
- J. F. Larson, D. C. Wolf and B. E. Butterworth, Induced cytotoxicity and cell proliferation in the hepatocarcinogenicity of chloroform in female b6c3f1 mice: comparison of administration by gavage in corn oil vs. ad Libitum in drinking water, Fundam. Appl. Toxicol., 1994, 22, 90–102 CrossRef CAS PubMed.
- M. Shariati-Rad and F. Fattahi, A simple equipment and colorimetric method for determination of chloroform in water, Anal. Chim. Acta, 2020, 1100, 208–214 CrossRef CAS PubMed.
- V. Choudhary, K. Vellingiri and L. Philip, Potential nanomaterials-based detection and treatment methods for aqueous chloroform, Environ. Nanotechnol., Monit. Manage., 2021, 16, 100487 CAS.
- K. Sheng, H. Lu, A. Sun, Y. Wang, Y. Liu, F. Chen, W. Bian, Y. Li, R. Kuang and D. Sun, A naked-eye colorimetric sensor for chloroform, Chin. Chem. Lett., 2019, 30, 895–898 CrossRef CAS.
- D. Fernández-Verdejo, P. Cortés, A. Guisasola, P. Blánquez and E. Marco-Urrea, Bioelectrochemically-assisted degradation of chloroform by a co-culture of Dehalobacter and Dehalobacterium, Environ. Sci. Ecotechnology, 2022, 12, 5 Search PubMed.
- H. Wang, R. Yu, J. Webb, P. Dollar and D. L. Freedman, Anaerobic Biodegradation of Chloroform and Dichloromethane with a Dehalobacter Enrichment Culture, Appl. Environ. Microbiol., 2022, 88(4), e01970 Search PubMed.
- J. Jose, S. Ramanujam and L. Philip, Applicability of pulsed corona discharge treatment for the degradation of chloroform, Chem. Eng. J., 2019, 360, 1341–1354 CrossRef CAS.
- Z. Muñoz, A. S. Cohen, L. M. Nguyen, T. A. McIntosh and P. E. Hoggard, Photocatalysis by tetraphenylporphyrin of the decomposition of chloroform, Photochem. Photobiol. Sci., 2008, 7, 337–343 CrossRef PubMed.
- J. M. S. Lopes, A. E. H. Machado, A. A. Batista, P. T. Araujo and N. M. Barbosa Neto, Protonation, exciplex, and evidence of aggregate formation in meso-tetra(4-pyridyl) porphyrin triggered by excited-state absorption, J. Photochem. Photobiol., A, 2022, 426, 113759 CrossRef CAS.
- R. Gilbert, M. Karabulut and P. E. Hoggard, Photocatalysis of chloroform degradation by μ-dichlorotetrachlorodipalladate(II), Inorg. Chim. Acta, 2010, 363, 1462–1468 CrossRef CAS.
- L. A. Peña and P. E. Hoggard, Hexachlororhodate(III) and the photocatalytic decomposition of chloroform, J. Mol. Catal. A: Chem., 2010, 327, 20–24 CrossRef.
- L. A. Peña, A. J. Seidl, L. R. Cohen and P. E. Hoggard, Ferrocene/ferrocenium ion as a catalyst for the photodecomposition of chloroform, Transition Met. Chem., 2009, 34, 135–141 CrossRef.
- A. J. Seidl, L. R. Cohen, L. A. Peña and P. E. Hoggard, Chlorochromate ion as a catalyst for the photodegradation of chloroform by visible light, Photochem. Photobiol. Sci., 2008, 7, 1373–1377 CrossRef CAS.
- D. Dolphin, The Porphyrins, Volume III. Physical Chemistry, Part A, The Porphyrins, 1978, vol. 3, p. 640 Search PubMed.
- J. M. S. Lopes, J. R. T. Reis, A. E. H. Machado, T. H. O. Leite, A. A. Batista, T. V. Acunha, B. A. Iglesias, P. T. Araujo and N. M. Barbosa Neto, Influence of the meso-substituents on the spectral features of free-base porphyrin, Spectrochim. Acta, Part A, 2020, 238, 118389 CrossRef CAS PubMed.
- A. K. Mandal, M. Taniguchi, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, Photophysical properties and electronic structure of porphyrins bearing zero to four meso-phenyl substituents: new insights into seemingly well understood tetrapyrroles, J. Phys. Chem. A, 2016, 120, 9719–9731 CrossRef CAS PubMed.
- J. S. Baskin, H. Z. Yu and A. H. Zewail, Ultrafast dynamics of porphyrins in the condensed phase: I. Free base tetraphenylporphyrin, J. Phys. Chem. A, 2002, 106, 9837–9844 CrossRef CAS.
- H. Z. Yu, J. S. Baskin and A. H. Zewail, Ultrafast dynamics of porphyrins in the condensed phase: II. Zinc tetraphenylporphyrin, J. Phys. Chem. A, 2002, 106, 9845–9854 CrossRef CAS.
- J. Jing, J. Yang, Z. Zhang and Y. Zhu, Supramolecular Zinc Porphyrin Photocatalyst with Strong Reduction Ability and Robust Built-In Electric Field for Highly Efficient Hydrogen Production, Adv. Energy Mater., 2021, 11, 1–7 Search PubMed.
- T. Shiragami, J. Matsumoto, H. Inoue and M. Yasuda, Antimony porphyrin complexes as visible-light driven photocatalyst, J. Photochem. Photobiol., C, 2005, 6, 227–248 CrossRef CAS.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, A Full-Spectrum Metal-Free Porphyrin Supramolecular Photocatalyst for Dual Functions of Highly Efficient Hydrogen and Oxygen Evolution, Adv. Mater., 2019, 31, 1–6 Search PubMed.
- Y. Chen, A. Li, Z. H. Huang, L. N. Wang and F. Kang, Porphyrin-based nanostructures for photocatalytic applications, Nanomaterials, 2016, 6, 1–17 Search PubMed.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Porphyrins in bio-inspired transformations: light-harvesting to solar cell, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS.
- T. Higashino and H. Imahori, Porphyrins as excellent dyes for dye-sensitized solar cells: recent developments and insights, Dalton Trans., 2015, 44, 448–463 RSC.
- L. L. Li and E. W. G. Diau, Porphyrin-sensitized solar cells, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, The role of porphyrin chemistry in tumor imaging and photodynamic therapy, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Porphyrin and nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- J. Kou, D. Dou and L. Yang, Porphyrin photosensitizers in photodynamic therapy and its applications, Oncotarget, 2017, 8, 81591–81603 CrossRef PubMed.
- H. Imahori, Porphyrin – fullerene linked systems as artificial photosynthetic mimics, Org. Biomol. Chem., 2004,(2), 1425–1433 RSC.
- Y. Nakamura, N. Aratani and A. Osuka, Cyclic porphyrin arrays as artificial photosynthetic antenna: synthesis and excitation energy transfer, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- N. Zarrabi and P. K. Poddutoori, Aluminum(III) porphyrin: a unique building block for artificial photosynthetic systems, Coord. Chem. Rev., 2021, 429, 213561 CrossRef CAS.
- N. M. Boyle, J. Rochford and M. T. Pryce, Thienyl-Appended porphyrins: synthesis, photophysical and electrochemical properties, and their applications, Coord. Chem. Rev., 2010, 254, 77–102 CrossRef CAS.
- J. M. S. Lopes, R. N. Sampaio, A. S. Ito, A. A. Batista, A. E. H. Machado, P. T. Araujo and N. M. B. Neto, Evolution of electronic and vibronic transitions in metal(II) meso-tetra(4-pyridyl)porphyrins, Spectrochim. Acta, Part A, 2019, 215, 327–333 CrossRef CAS PubMed.
- F. Hajjaj, Z. S. Yoon, M. C. Yoon, J. Park, A. Satake, D. Kim and Y. Kobuke, Assemblies of supramolecular porphyrin dimers in pentagonal and hexagonal arrays exhibiting light-harvesting antenna function, J. Am. Chem. Soc., 2006, 128, 4612–4623 CrossRef CAS PubMed.
- T. Tanaka and A. Osuka, Conjugated porphyrin arrays: synthesis, properties and applications for functional materials, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- E. Alessio, E. Ciani, E. Iengo, V. Y. Kukushkin and L. G. Marzilli, Stepwise assembly of unsymmetrical supramolecular arrays containing porphyrins and coordination compounds, Inorg. Chem., 2000, 39, 1434–1443 CrossRef CAS PubMed.
- H. E. Toma and K. Araki, Supramolecular assemblies of ruthenium complexes and porphyrins, Coord. Chem. Rev., 2000, 196, 307–329 CrossRef CAS.
- H. E. Toma and K. Araki, Exploring the supramolecular coordination chemistry-based approach for nanotechnology, Prog. Inorg. Chem., 2009, 56, 379–485 CrossRef CAS.
- T. H. O. Leite, G. Grawe, J. Honorato, B. N. Cunha, O. R. Nascimento, P. S. De Vargas, C. Donatoni, K. T. Oliveira, J. M. S. Lopes, N. M. Barbosa Neto, W. C. Moreira, L. R. Dinelli and A. A. Batista, Remarkable Electronic Effect on the meso-Tetra(thienyl)porphyrins, Inorg. Chem., 2019, 58, 1030–1039 CrossRef CAS PubMed.
- N. M. B. Neto, S. L. Oliveira, I. Guedes, L. R. Dinelli and L. Misoguti, Reverse Saturable Absorption in 5,10,15,20-Tetra(4-pyridyl)-21H,23H-porphyrin with Ruthenium Outlying Complexes, J. Braz. Chem. Soc., 2006, 17(7), 1377–1382 CrossRef.
- R. N. Sampaio, M. M. Silva, A. A. Batista and N. M. B. Neto, Investigation of the photophysical and electrochemical properties of a free base tetrapyridyl porphyrin with meso carbon linked ruthenium(II) groups, J. Photochem. Photobiol., A, 2016, 315, 98–106 CrossRef CAS.
- A. Prodi, M. Teresa, C. J. Kle and E. Alessio, Energy transfer pathways in pyridylporphyrin metal adducts and side-to-face arrays, Coord. Chem. Rev., 2002, 229(1–2), 51–58 CrossRef CAS.
- A. Prodi, C. J. Kleverlaan, M. T. Indelli, F. Scandola, D. Chimica, E. Alessio, E. Iengo, S. Chimiche and R. V January, Photophysics of Pyridylporphyrin Ru(II) Adducts: Heavy-Atom Effects and Intramolecular Decay Pathways, Inorg. Chem., 2001, 40(14), 3498–3504 CrossRef CAS PubMed.
- R. N. Sampaio, W. R. Gomes, D. M. S. Araujo, A. E. H. Machado, R. A. Silva, A. Marletta, I. E. Borissevitch, A. S. Ito, L. R. Dinelli, A. A. Batista, S. C. Zílio, P. J. Gonçalves and N. M. Barbosa Neto, Investigation of Ground- and Excited-State Photophysical Properties Outlying Complexes, J. Phys. Chem. A, 2012, 116, 18–26 CrossRef CAS PubMed.
- J. M. S. Lopes, S. N. Costa, A. A. Batista, L. R. Dinelli, P. T. Araujo and N. M. B. Neto, Photophysics and visible light photodissociation of supramolecular meso-tetra(4-pyridyl) porphyrin/RuCl2(CO)(PPh3)2 structures, Spectrochim. Acta, Part A, 2020, 237, 118351 CrossRef CAS PubMed.
- J. M. S. Lopes, R. N. Sampaio, L. R. Dinelli, A. A. Batista, P. T. Araujo and N. M. B. Neto, On the excitation dependence of fluorescence spectra of meso-tetrapyridyl zinc(II) porphyrin and its relation with hydrogen bonding and outlying decoration, Spectrochim. Acta, Part A, 2020, 224, 117371 CrossRef CAS PubMed.
- J. M. S. Lopes, S. N. Costa, E. Silveira-Alves Jr, A. A. Batista, P. J. Gonçalves, L. R. Dinelli, P. T. Araujo and N. M. Barbosa Neto, Singlet Oxygen Generation and Spectroscopic Properties of Supramolecular Zinc Meso–tetra (4-pyridyl) Porphyrin Bearing Outlying Ruthenium Groups, Braz. J. Phys., 2022, 38–40 Search PubMed.
- M. I. F. Barbosa, G. G. Parra, R. S. Correa, R. N. Sampaio, L. N. Magno, R. C. Silva, A. C. Doriguetto, J. Ellena, N. M. B. Neto, A. A. Batista and P. J. Gonçalves, Reactive nitrogen/oxygen species production by nitro/nitrosyl supramolecular ruthenium porphyrin complexes, J. Photochem. Photobiol., A, 2017, 338, 152–160 CrossRef CAS.
- N. M. Barbosa Neto, L. De Boni, J. J. Rodrigues Jr, L. Misoguti, C. R. Mendonça, L. R. Dinelli, A. A. Batista and S. C. Zilio, Dynamic saturable optical nonlinearities in free base porphyrins, J. Porphyr. Phthalocyanines, 2003, 7, 452–456 CrossRef CAS.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag & Co, Weinheim, 2nd edn, 2012 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- J. M. S. Lopes, K. Sharma, R. N. Sampaio, A. A. Batista, A. S. Ito, A. E. H. Machado, P. T. Araújo and N. M. Barbosa Neto, Novel insights on the vibronic transitions in free base meso-tetrapyridyl porphyrin, Spectrochim. Acta, Part A, 2019, 209, 274–279 CrossRef CAS PubMed.
- R. M. O'Donnell, P. G. Johansson, M. Abrahamsson and G. J. Meyer, Excited-state relaxation of ruthenium polypyridyl compounds relevant to dye-sensitized solar cells, Inorg. Chem., 2013, 52, 8282 CrossRef.
- D. S. Seneviratne, M. J. Uddin, V. Swayambunathan, H. B. Schlegel and J. F. Endicott, Characteristics and properties of metal-to-ligand charge-transfer excited states in 2,3-bis(2-pyridyl)pyrazine and 2,2′-bipyridine ruthenium complexes. Perturbation-theory-based correlations of optical absorption and emission parameters with electrochemis, Inorg. Chem., 2002, 41, 1502–1517 CrossRef CAS PubMed.
- D. W. Thompson, A. Ito and T. J. Meyer, [Ru(bpy)3]2+* and other remarkable metal-to-ligand charge transfer (MLCT) excited states, Pure Appl. Chem., 2013, 85, 1257–1305 CrossRef CAS.
- X. Y. Wang, A. Del Guerzo and R. H. Schmehl, Photophysical behavior of transition metal complexes having interacting ligand localized and metal-to-ligand charge transfer states, J. Photochem. Photobiol., C, 2004, 5, 55–77 CrossRef CAS.
- X. Han, L. Z. Wu, G. Si, J. Pan, Q. Z. Yang, L. P. Zhang and C. H. Tung, Switching between ligand-to-ligand charge-transfer, intraligand charge-transfer, and metal-to-ligand charge-transfer excited states in platinum(II) terpyridyl acetylide complexes induced by pH change and metal ions, Chem.–Eur. J., 2007, 13, 1231–1239 CrossRef CAS PubMed.
- A. Ülveczky and A. Horváth, Photooxidation of Ru (bpy) (CN)42− and Ru (DMbpy) (CN)42−, Inorg. Chim. Acta, 1995, 236, 173–176 CrossRef.
- Z. Z. Li, Y. L. Niu, H. Y. Zhou, H. Y. Chao and B. H. Ye, Visible-light-induced photooxidation of ruthenium(II) complex with 2,2′-biimidazole-like ligand by singlet oxygen, Inorg. Chem., 2013, 52, 10087–10095 CrossRef CAS PubMed.
- R. Girotti, A. Romerosa, S. Mañas, S. R. Manuel and R. N. Perutz, Visible-light photoisomerization and photoaquation of trans-[Ru(1,3,5-triaza-7-phosphaadamantane)4Cl2] in organic solvent and water, Inorg. Chem., 2009, 48, 3692–3698 CrossRef CAS PubMed.
- S. J. Wezenberg, K. Y. Chen and B. L. Feringa, Visible-Light-Driven Photoisomerization and Increased Rotation Speed of a Molecular Motor Acting as a Ligand in a Ruthenium(II) Complex, Angew. Chem., Int. Ed., 2015, 54, 11457–11461 CrossRef CAS.
- M. O. Santiago, C. L. Donicci Filho, I. de S. Moreira, R. M. Carlos, S. L. Queiroz and A. A. Batista, Photochemical isomerization of trans- to cis-[RuCl2(dppb) (4,4′-X2-2,2′-bipy)] (X = –H, –NO2, –Me, –COOH, –SMe, –O = SMe, –Cl, –OMe) complexes, Polyhedron, 2003, 22, 3205–3211 CrossRef CAS.
- Z. J. Fuller, W. D. Bare, K. A. Kneas, W. Y. Xu, J. N. Demas and B. A. DeGraff, Photostability of luminescent ruthenium(II) complexes in polymers and in solution, Anal. Chem., 2003, 75, 2670–2677 CrossRef CAS PubMed.
- D. W. Thompson, C. N. Fleming, B. D. Myron and T. J. Meyer, Rigid medium stabilization of metal-to-ligand charge transfer excited states, J. Phys. Chem. B, 2007, 111, 6930–6941 CrossRef CAS PubMed.
- J. R. Weinkauf, S. W. Cooper, A. Schweiger and C. C. Wamser, Substituent and solvent effects on the hyperporphyrin spectra of diprotonated tetraphenylporphyrins, J. Phys. Chem. A, 2003, 107, 3486–3496 CrossRef CAS.
- M. Zannotti, R. Giovannetti, B. Minofar, D. Řeha, L. Plačková, C. A. D'Amato, E. Rommozzi, H. V. Dudko, N. Kari and M. Minicucci, Aggregation and metal-complexation behaviour of THPP porphyrin in ethanol/water solutions as function of pH, Spectrochim. Acta, Part A, 2018, 193, 235–248 CrossRef CAS PubMed.
- I. Gupta and M. Ravikanth, Spectroscopic properties of meso-thienylporphyrins with different porphyrin cores, J. Photochem. Photobiol., A, 2006, 177, 156–163 CrossRef CAS.
- Y. Fang, J. Zhu, Y. Cui, L. Zeng, M. L. Naitana, Y. Chang, N. Desbois, C. P. Gros and K. M. Kadish, Protonation and Electrochemical Properties of Pyridyl- and Sulfonatophenyl-Substituted Porphyrins in Nonaqueous Media, ChemElectroChem, 2017, 4, 1872–1884 CrossRef CAS.
- P. A. Anderson, R. F. Anderson, M. Furue, P. C. Junk, F. R. Keene, B. T. Patterson and B. D. Yeomans, Protonation studies of reduced ruthenium(II) complexes with polypyridyl ligands, Inorg. Chem., 2000, 39, 2721–2728 CrossRef CAS PubMed.
- S. Zhang and R. E. Shepherd, Protonation of coordinated 2-methylpyrazine and 4,4′-bipyridine as a probe of n-donor potential of ruthenium(II) polyaminopoly-carboxylate complexes, Transition Met. Chem., 1992, 17, 199–203 CrossRef CAS.
- J. Mosinger, M. Janošková, K. Lang and P. Kubát, Light-induced aggregation of cationic porphyrins, J. Photochem. Photobiol., A, 2006, 181, 283–289 CrossRef CAS.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J- and H-aggregates of porphyrin-surfactant complexes: time-resolved fluorescence and other spectroscopic Studies, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- G. De Luca, A. Romeo and L. M. Scolaro, Role of counteranions in acid-induced aggregation of isomeric tetrapyridylporphyrins in organic solvents, J. Phys. Chem. B, 2005, 109, 7149–7158 CrossRef CAS PubMed.
- Y. Arai and H. Segawa, Cl- complexation induced H- and J-Aggregates of meso-tetrakis-(4-sulfonatothienyl)porphyrin diacid in aqueous solution, J. Phys. Chem. B, 2011, 115, 773–7780 CrossRef PubMed.
- G. De Luca, A. Romeo and L. M. Scolaro, Aggregation properties of hyperporphyrins with hydroxyphenyl substituents, J. Phys. Chem. B, 2006, 110, 14135–14141 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |