
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA binuclear aluminium complex as a single competent catalyst for efficient synthesis of urea, biuret, isourea, isothiourea, phosphorylguanidine, and quinazolinones†
Kulsum Bano a,
Jyoti Sharmaa,
Archana Jain*b,
Hayato Tsurugi*c and
Tarun K. Panda*a
a,
Jyoti Sharmaa,
Archana Jain*b,
Hayato Tsurugi*c and
Tarun K. Panda*a
aDepartment of Chemistry, Indian Institute of Technology, Kandi-502 285, Sangareddy, Hyderabad, Telangana, India. Web: https://sites.google.com/site/tkpandagroup/homeE-mail: tpanda@chy.iith.ac.in
bDepartment of Physics and Chemistry, Mahatma Gandhi Institute of Technology, Gandipet-500 075, Hyderabad, Telangana, India. E-mail: archanajain_chem@mgit.ac.in
cDepartment of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: tsurugi.hayato.es@osaka-u.ac.jp
First published on 19th January 2023
Abstract
The synthesis and characterisation of two mononuclear aluminium alkyl complexes with the general composition [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2O}] (E = Se (2a); S (2b)), and two binuclear aluminium complexes, [Al(Me)2{Ph2P-(E)N(CH2)2N(CH2CH2)2O}(AlMe3)] (E = Se (3a) and S (3b)), are described. The binuclear aluminium alkyl complex 3a proved to be a proficient catalyst for the addition of simple nucleophiles to heterocumulenes, leading to the synthesis of a variety of products such as urea, biuret, isourea, isothiourea, phosphorylguanidine, and quinazolinone derivatives, in contrast to its mononuclear analogues. Complex 3a is the first example of a single competent catalyst, which is also low-cost and eco-friendly and derived from a main-group metal, under solvent-free conditions either at room temperature or mild temperatures. Complex 3a possessed a wide functional group tolerance including heteroatoms, yielding the corresponding insertion products in good quantities and with high selectivity.
Introduction
There is currently an upsurge of interest in developing efficient hydroelementation reactions to construct C-heteroatom bonds using suitable catalysts. This can be seen as a renaissance in modern organic chemistry because hydroelementation reactions are highly atom-economical; they utilise affordable and easily accessible substrates. Hydroelementation reactions of alkenes and alkynes by a broad range of transition-metal, lanthanide, actinide, and alkali-metal catalysts are well explored to date.1,2 Hydroelementation of heterocumulenes is now gaining considerable attention, owing to its several advantages such as greater synthetic possibilities such as multiple heterocumulene insertions into one E–H moiety, unsaturated bond selectivity, and double hydroelementations of a single heterocumulene substrate.3,4 As a result of the insertion of E–H bonds (E = N, P, O, S) into various heterocumulenes (isocyanates, isothiocyanates, and carbodiimides), we can gain access to a variety of products such as heteroatom-rich urea, thiourea, guanidines, and quinazoline/quinazolinone derivatives. The obtained products have been significantly utilized for multidentate ligands in organometallic chemistry, organic synthon, and pharmaceutical analogues.5–12In contrast to the significantly developed catalytic hydroamination of alkynes, alkenes, and carbodiimides to form C–N bonds,13–15 catalytic hydroamination of isocyanates, leading to urea derivatives, have been explored only by some group 2, titanium, zinc, actinide complexes, etc.16–22 The urea derivatives are useful across a range of biological systems, pharmaceuticals, agrochemicals, synthetic chemistry, supramolecular chemistry, and materials chemistry.23–31 Moreover, the synthesis of derivatives containing multiple urea moieties within one molecule, such as biuret and triuret derivatives from mono-urea, is quite challenging since the nucleophilic nature of urea compared to secondary amines is considerably less. So far, only a few examples have been reported for the synthesis of such compounds by the catalytic transformation of secondary amines with isocyanates. Recently, Tylor et al.32 reported the use of low-coordinate iron pre-catalyst, (2,6-Mes2C6H3)2Fe (where Mes = 2,4,6-Me3C6H2), in the presence of HNPh2 and Ph-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO (1
CO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) afforded a mixture of urea and biuret products in a molar ratio of 88:12. Biuret selectivity was increased (to 90–100%) by employing HNPh2 and Ph-NCO in a ratio of 1:5; however, it was only successful for aryl isocyanates, as alkyl isocyanates consistently yielded corresponding urea derivatives. Despite the importance of biuret and triuret derivatives in molecular sensors, foldamers, self-assembly, etc., there are hardly any direct catalytic routes. Therefore, the development of efficient, yet simple catalytic systems with good functional group tolerance for the catalytic transformation of the urea moieties is a worthwhile endeavour.
:1 ratio) afforded a mixture of urea and biuret products in a molar ratio of 88:12. Biuret selectivity was increased (to 90–100%) by employing HNPh2 and Ph-NCO in a ratio of 1:5; however, it was only successful for aryl isocyanates, as alkyl isocyanates consistently yielded corresponding urea derivatives. Despite the importance of biuret and triuret derivatives in molecular sensors, foldamers, self-assembly, etc., there are hardly any direct catalytic routes. Therefore, the development of efficient, yet simple catalytic systems with good functional group tolerance for the catalytic transformation of the urea moieties is a worthwhile endeavour.
In addition, successive insertion of heterocumulenes into the N–H bonds of amino acid esters followed by cyclisation to form heterocyclic products (quinazolinones) has garnered considerable attention in the area of synthetic organic and pharmaceutical chemistry.33–35 Fused N-heterocyclic compounds (quinazolinones) have been shown to have wide applications in medicine, particularly in anti-cancer, anti-convulsant, hypotensive, and anti-allergy treatments. They are also good precursors for derivatizing naturally occurring alkaloid derivatives only isolated from natural resources.36–40 Synthesis of quinazolinones generally needs long and tedious reaction steps with the use of toxic reagents. Therefore, novel and efficient catalytic systems involving earth-abundant, environment-friendly, non-toxic metals and substrates are highly demanded for the construction of N-heterocyclic quinazolinones: to this purpose, zinc,41 titanium,42 and rare-earth metal-based complexes43 were recently developed to catalyse tandem hydroamination of amino acid esters with carbodiimides.
Although tremendous progress has been made over the last decade on these reactions, novel green and sustainable catalytic chemical approaches for achieving the dual goals, environment protection and cost effectiveness, can be further explored.44 In this context, study on the use of a single catalyst for multi-element hydroelementation reactions of heterocumulenes has attracted attention. Recently, Eisen et al. illustrated multi-element hydroelementation reactions of heterocumulenes using protic E–H bonds containing nucleophiles in the presence of a single actinide pre-catalyst [{(Me3Si)2N}2An{κ2-(C,N)-CH2Si(CH3)2N-(SiMe3)2}] (An = Th, U).20,22,45 However, this reaction needed more time and a relatively high temperature. Our group18 also demonstrated that a binuclear titanium(IV) complex showed remarkable catalytic activity for the addition of E–H bonds (E = NR2, OR, SR, P(O)R2) towards a wide variety of heterocumulenes under mild conditions. However, till date, there are no known examples of single catalytic systems for hydrolelementation of heterocumulenes with various E–H bonds using main group metals. Herein, we report on a main group metal-based single competent catalytic system that can perform versatile hydroelementation reactions through the addition of E–H (E = N, O, S, P, C) moieties to heterocumulenes, producing highly useful urea and biuret derivatives, isourea derivatives, phosphorylguanidine, and quinazolinone derivatives with high functional group tolerance, in which aluminium complexes are found to be suitable in terms of the high catalytic activity, availability, and low toxicity. Our group has established a series of phosphine amines [Ph2PNHR] (A; R = 2,6-Me2C6H3, CHPh2, CPh3) and their chalcogen derivatives [Ph2P(O)NHR] (NPO), [Ph2P(S)NHR] (NPS), and [Ph2P(Se)NHR] (NPSe) containing both hard and soft donor atoms, to understand their coordination behavior with alkali metals, the heavier alkaline-earth metals, and main-group metals.46–48 Further, we aimed to synthesize the protic amidophosphine chalcogenide ligands, [Ph2P(E)-NH(CH2)2N(CH2CH2)2O] [E = Se, 1a-H; S = 1b-H] containing ethylene-linked morpholine fragment which is more flexible due to the presence of ethylene links. Due to their flexible nature as well as the presence of hard and soft chalcogen donor atoms, these ligands have the potential to exhibit a hemilabile nature. Thus, the study of the coordination behavior of such hemilabile ligands with reactive aluminum metal could be very interesting.
We described the synthesis and structural features of mono- and binuclear aluminium complexes with the molecular formula, [Al(Me)2{Ph2P(Se)N(CH2)2N(CH2CH2)2O}] (2a), [Al(Me)2{Ph2P(S)N(CH2)2N(CH2CH2)2O}] (2b), and [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2-O}(AlMe3)] (E = Se (3a); S (3b)) respectively. The catalytic activity of 3a was shown as the facile synthesis of urea, biuret, isourea, isothiourea, phosphorylguanidine, and quinazolinone derivatives (Fig. 1).
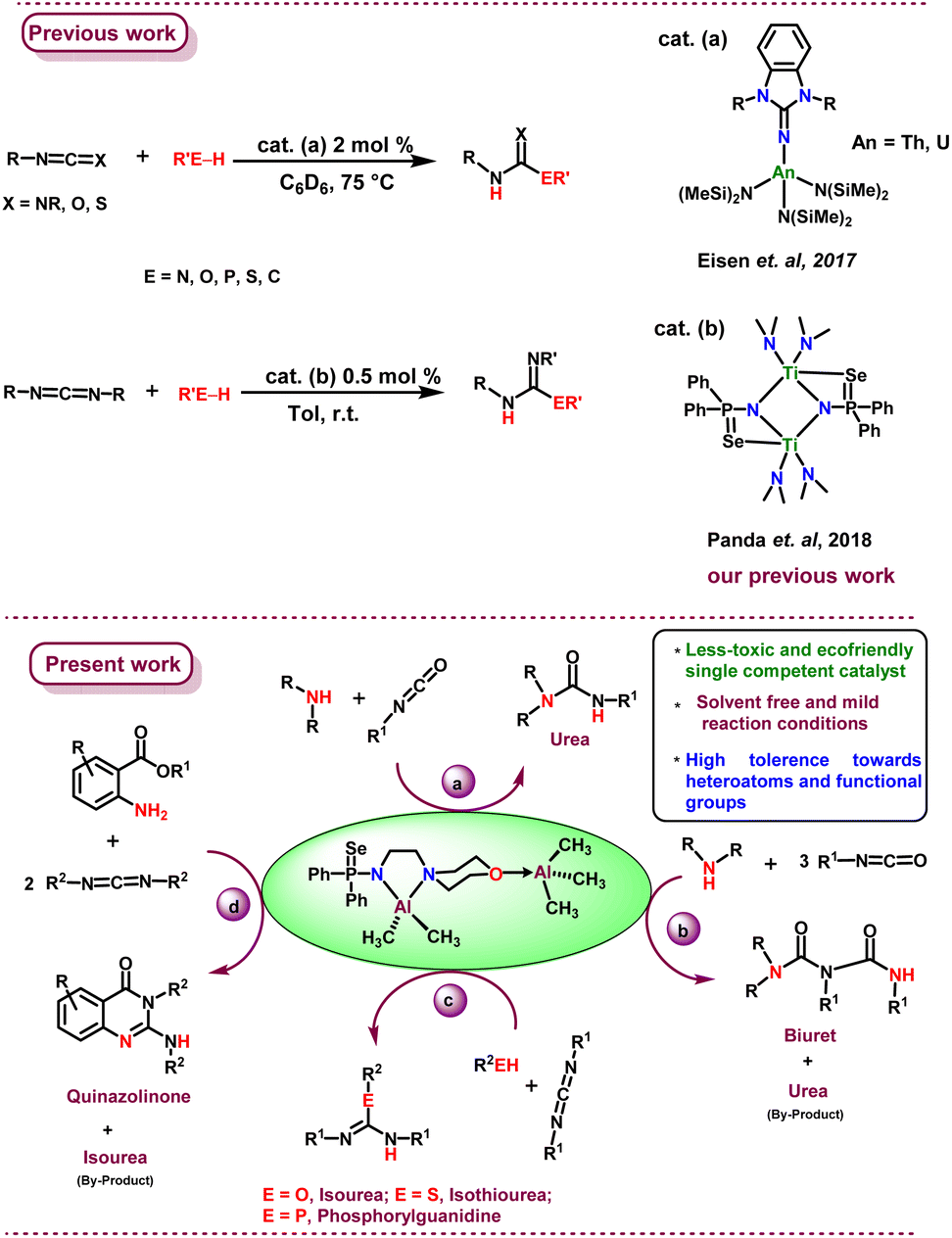 | ||
| Fig. 1 Previous reports on hydroelementation and present work. | ||
Results and discussion
Synthesis of mononuclear and binuclear aluminium complexes (2a, 2b, 3a, and 3b)
Protic amidophosphine chalcogenide ligands [Ph2P(E)-NH(CH2)2N(CH2CH2)2O] [E = Se, 1a-H; S = 1b-H] were synthesised according to published procedure49 by the reaction between 4-(2-aminoethyl)morpholine and chloro-diphenylphosphine (1:1) in the presence of triethylamine in toluene, followed by the addition of elemental sulphur or selenium at 90 °C for 24 hours (Scheme 1). The reaction of ligand 1a-H or 1b-H with trimethylaluminium (AlMe3) in a 1:1 molar ratio in toluene at 60 °C resulted in the formation of the corresponding mononuclear Al complexes [κ2-{Ph2P(E)N(CH2)2N-(CH2CH2)2O}Al(Me)2] [E = Se (2a), S (2b)] as colourless crystalline solids in good yields (Scheme 2). On the other hand, AlMe3 reacted with protic ligand 1a-H/1b-H in a 2:1 molar ratio in toluene at 90 °C for 12 hours, resulting in the formation of a binuclear aluminium complex of molecular formula [Al(Me)2{Ph2P(E)N2C6H12O}(AlMe3)] (E = Se (3a); S (3b)) with a good yield (∼91–95%). Each binuclear aluminium complex 3a/3b was also obtained from the respective mononuclear complex 2a/2b by its reaction with one equivalent of AlMe3 in toluene at 90 °C (Scheme 2). All the Al-alkyl complexes 2a, 2b, 3a, and 3b were fully characterised using multinuclear NMR spectroscopy and combustion analysis.
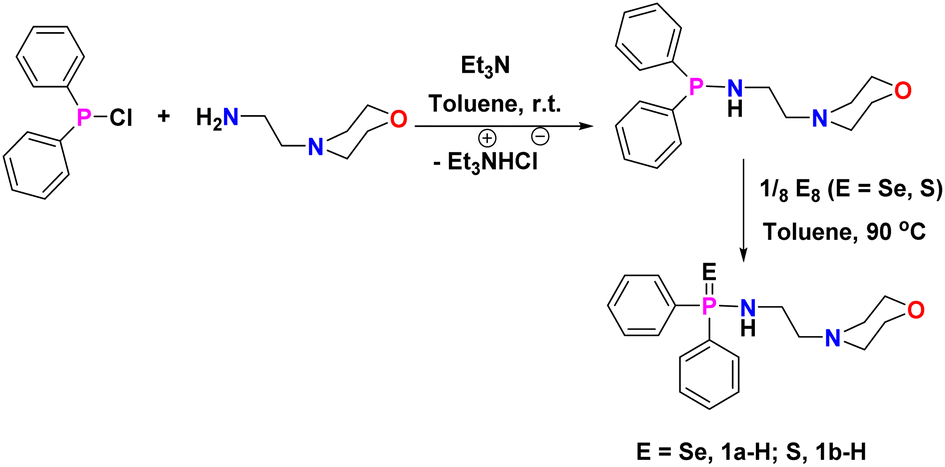 | ||
| Scheme 1 Synthesis of flexible aminophosphine chalcogenide ligands 1a-H and 1b-H. | ||
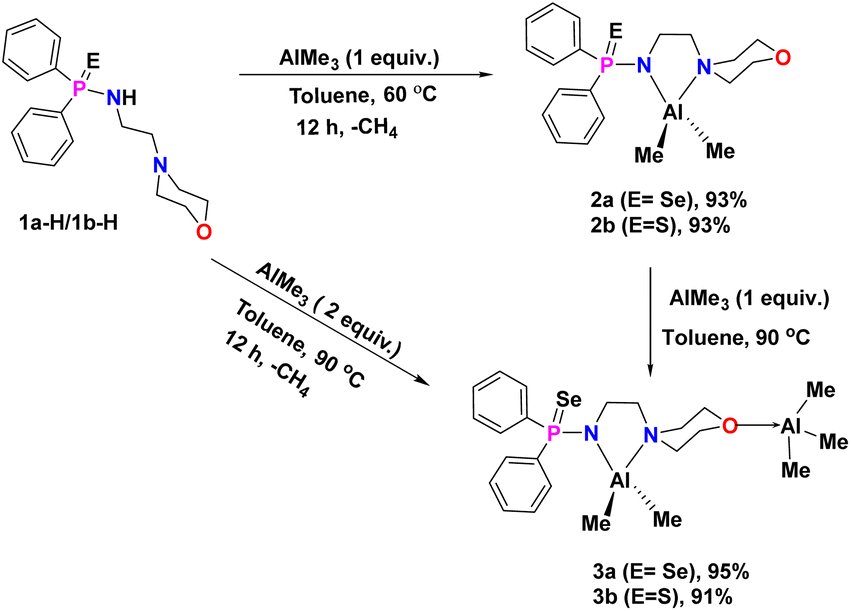 | ||
| Scheme 2 Synthesis of mono- and binuclear aluminium complexes 2a, 2b, 3a, and 3b supported by flexible aminophosphine chalcogenide. | ||
In the 1H NMR spectra of ligands 1a-H and 1b-H, the resonance signal for the –NH group is shown as a broad peak at δH 3.23 ppm and 3.30 ppm, respectively (Fig. FS1 and FS4 in ESI†). The signals are absent in the 1H NMR spectra of Al complexes 2a and 2b, confirming the formation of monoanionic moieties 1a and 1b respectively. Further, the resonance signal of methyl protons attached to the aluminium metal centre for 2a and 2b appeared at δH −0.78 and −0.73 ppm, respectively (Fig. FS7 and FS10, in ESI†). In the 1H NMR spectrum of each binuclear complex, two sharp singlets were obtained in 2:3 ratios at δH −0.89 (for 3a)/−0.98 (for 3b) ppm and −0.45 (for 3a)/−0.35 (for 3b) ppm for two types of methyl protons attached to the two Al centres, which can be assigned to AlMe2 and AlMe3, respectively. The formation of aluminium complexes is also confirmed by the presence of the complex AA′BB′ multiplet spin system of ethylene protons in the range of δH 3.5–1.5 ppm. This occurs due to the restricted rotation of ethylene protons of the anionic ligand upon coordination with aluminium ion. While the –CH2–CH2– unit of the free ligand is expected to rotate freely and hence displayed a simple multiplet signals (A2B2). In the 31P{1H} NMR spectra, the complexes 2a and 3a exhibit sharp singlets at δP 57.1 ppm and 56.0 ppm, respectively, along with two satellite peaks due to coupling with the adjacent selenium atom, whereas complexes 2b and 3b display only a sharp resonance signal at 55.4 and 58.3 ppm, respectively. The molecular structure of ligand 1b-H is already reported in our previous work.49 The molecular structures of 1a-H, 2a, and 3a in the solid states were established using single crystal X-ray diffraction analysis and are presented in Fig. 2, 3, and 4, respectively. Pertinent crystallographic and refinement parameters are given in Table TS1 in ESI.† Colourless crystals of complexes 2a and 3a were obtained from a saturated solution of toluene at −35 °C after one week. Complex 2a crystallises in the monoclinic space group P21/n with four molecules in the unit cell, while complex 3a crystallises in the monoclinic space group P21/c with six molecules in the unit cell.
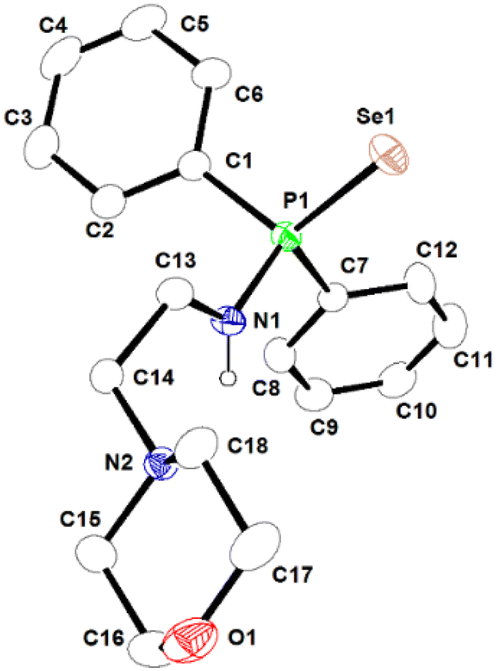 | ||
| Fig. 2 Molecular structure of ligand 1a-H in its solid-state. Selected bond lengths (Å) and angles (°) are given. P1–N1 1.661(3), P1–Se1 2.1026(13), P1–C1 1.812(5), P1–C7 1.815(4), N1–C13 1.465(5), N1–P1–Se1 116.75(13), N1–P1–C1 103.2(2), N1–P1–C7 103.53(18), C1–P1–C7 106.47(19). CCDC no. 2151760. | ||
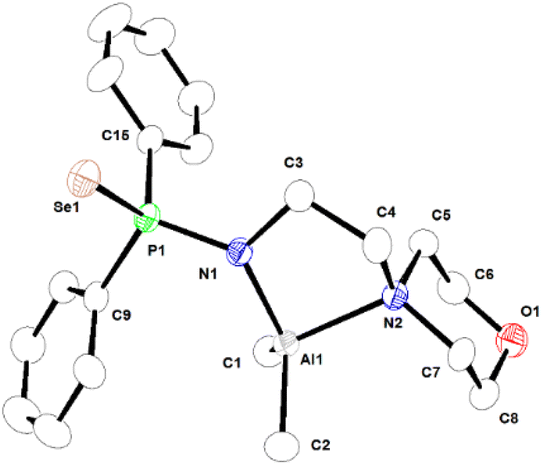 | ||
| Fig. 3 Molecular structure of complex 2a in its solid state. Selected bond lengths (Å) and angles (°) are given. Al1–C1 1.964(3), Al1–C2 1.962(4), Al1–N1 1.883(2), Al1–N2 2.043(2), P1–N1 1.636(2), P1–Se1 2.1230(8), N1–C3 1.479(3), C3–C4 1.507(4), N2–C4 1.496(4), P1–C9 1.812(3), P1–C15 1.817(3), N1–Al1–N2 87.23(10), N1–Al1–C1 114.24(13), N1–Al1–C2 117.69(15), N2–Al1–C1 113.08(14), N2–Al1–C2 104.94(14), N1–P1–Se1 116.15(9). CCDC no. 2151762. | ||
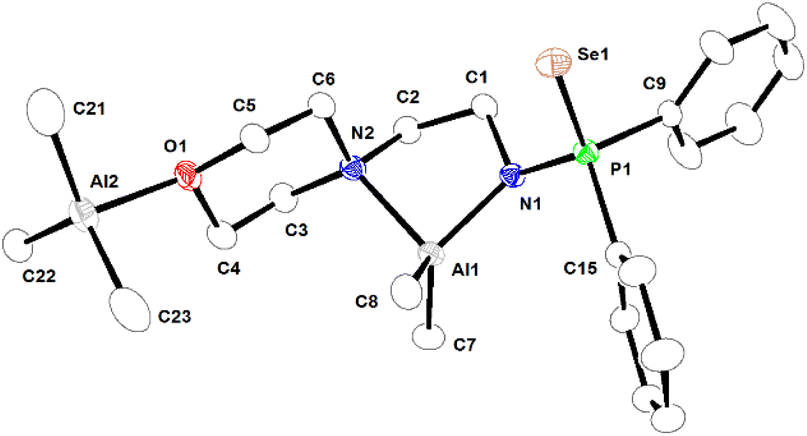 | ||
| Fig. 4 Molecular structure of binuclear complex 3a in the solid state. Selected bond lengths (Å) and angles (°) are given. Al1–N1 1.878(2), Al1–C8 1.949(3), Al1–C7 1.956(3), Al1–N2 2.063(3), Al2–C22 1.956(4), Al2–C21 1.959(5), Al2–C23 1.969(4), Al2–O1 2.001(2), Se1–P1 2.1195 (8), P1–N1 1.644(2), P1–C15 1.807(3), P1–C9 1.818(3); N1–Al1–C8 116.72(14), N1–Al1–C7 116.17(14), C8–Al1–C7 114.86(17), N1–Al1–N2 86.68(10), C8–Al1–N2 113.40(14), C7–Al1–N2 104.80(14), C22–Al2–C21 117.78(19), C22–Al2–C23 117.38(18), C21–Al2–C23 113.4 (2), C22–Al2–O1 100.30(14), C21–Al2–O1 103.06(15), C23–Al2–O1 100.89(16). CCDC no. 2209321. | ||
In complex 2a, ligand 1a is asymmetrically coordinated to the aluminium ion in κ2-fashion through anionic amidophosphine nitrogen and the nitrogen atom of the morpholine moiety, as is evident from the presence of two sets of Al–N distances [Al1–N1 is 1.883(2) Å and Al1–N2 is 2.043(2) Å]. Both Al–N bond distances correspond to the anionic and neutral coordination of the nitrogen atoms to the aluminium metal centre. The selenium atom remains uncoordinated. The Al–C and Al–N bond distances are within the range that we previously reported for the aluminium metal complex [κ2-{2-F-C6H4NP(Se)Ph2}2Al(Me)] and other reported Al complexes.50 The central Al ion is coordinated four-fold via the chelation of one amido nitrogen, one morpholine ring nitrogen from ligand 1a, and two methyl groups, and adopts a distorted tetrahedral geometry around it with the formation of a five-membered metallacycle, Al1–N1–C3–C4–N2. The metallacycle is not coplanar due to the presence of two sp3 carbon atoms C3 and C4 in the ring.51,52
In complex 3a, the ligation of 1a (aminophosphine selenide) is similar to that of complex 2a. Additionally, there is a bond between the oxygen atom of the morpholine moiety of ligand 1a-H and another AlMe3 group, making complex 3a binuclear. The coordination of the oxygen atom present in a multidentate ligand to the AlMe3 group is known in the literature.51,53,54 The Al–N bond distances [Al1–N1 being 1.878(2) Å and Al1–N2 2.063(3) Å] are similar to those in complex 2a. Al–C bond distances [1.946(3)–1.969(4) Å] are also within the ranges previously reported.55 The second Al ion is bonded with the oxygen atom of the morpholine moiety and three methyl groups, adopting a distorted tetrahedral geometry around the metal ion.
Catalytic study
Formation of urea derivatives
Given the importance of urea derivatives, we wanted to examine the catalytic activity of all the mononuclear and binuclear aluminium metal complexes (2a, 2b, 3a, and 3b) in the hydroamination of isocyanates with secondary amines.Initially, all Al-metal complexes were screened as catalysts to meditate the hydroamination reaction of p-tolylisocyanate with diisopropylamine. Catalytic experiments were performed using 5 mol% of the metal complex along with an equimolar amount of p-tolylisocyanate and diisopropylamine under neat conditions at room temperature for one hour (Table 1, entries 1–3). To our delight, all complexes 2a, 2b, 3a, and 3b proved to be active catalysts in this addition reaction, affording the corresponding urea products in excellent yields of up to 99% (Table 1, entries 1–4). We also performed control reactions between p-tolylisocyanate and diisopropylamine at ambient temperature, and when heated to 60 °C without using any catalyst, no detectable product was obtained (Table 1, entries 11 and 12). Among all the catalysts, the binuclear aluminium complexes demonstrated the better reactivity compared to mononuclear complexes in forming the desired urea product. We selected 3a for optimisation of catalytic reaction conditions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Cat. (mol%) | Time (h) | T (°) | Yieldb (%) |
| a Molar ratio of p-methyl-phenylisocyanate:diisopropylamine = 1:1.b Isolated yield. |
||||||
| 1 | 2a | Neat | 5 | 1 | r.t. | 92 |
| 2 | 2b | Neat | 5 | 1 | r.t. | 90 |
| 3 | 3a | Neat | 5 | 1 | r.t. | 99 |
| 4 | 3b | Neat | 5 | 1 | r.t. | 96 |
| 5 | 3a | Neat | 2.5 | 1 | r.t. | 99 |
| 6 | 3a | Neat | 1 | 1 | r.t. | 99 |
| 7 | 3a | Neat | 0.5 | 1 | r.t. | 89 |
| 8 | 3a | Toluene | 1 | 1 | r.t. | 99 |
| 9 | 3a | Hexane | 1 | 1 | r.t. | 75 |
| 10 | 3a | THF | 1 | 1 | r.t. | 40 |
| 11 | — | Neat | — | 12 | r.t. | — |
| 12 | — | Neat | — | 12 | 60 | — |
At first, we examined the influence of catalyst loading on the yield of urea. The yield of urea obtained from the hydroamination of p-tolylisocyanate and diisopropylamine using 1 mol% of complex 3a was almost as much as that obtained from using 5 mol% of catalyst (Table 1, entry 6). When the catalyst loading was decreased to 0.5 mol%, the yield of urea reduced (Table 1, entry 6). Thus, 1 mol% of catalyst was selected as the optimised condition. We also studied the impact on product yields when various solvents, such as toluene, hexane, and THF were used in the hydroamination reaction between p-tolylisocyanate and diisopropylamine using 1 mol% of complex 3a at room temperature for one hour (Table 1, entries 8–10). The lowest yield (40%) of urea obtained was when THF was used as the solvent. This can be attributed to the coordinating nature of this ethereal solvent during the reaction conversion (Table 1, entry 10).
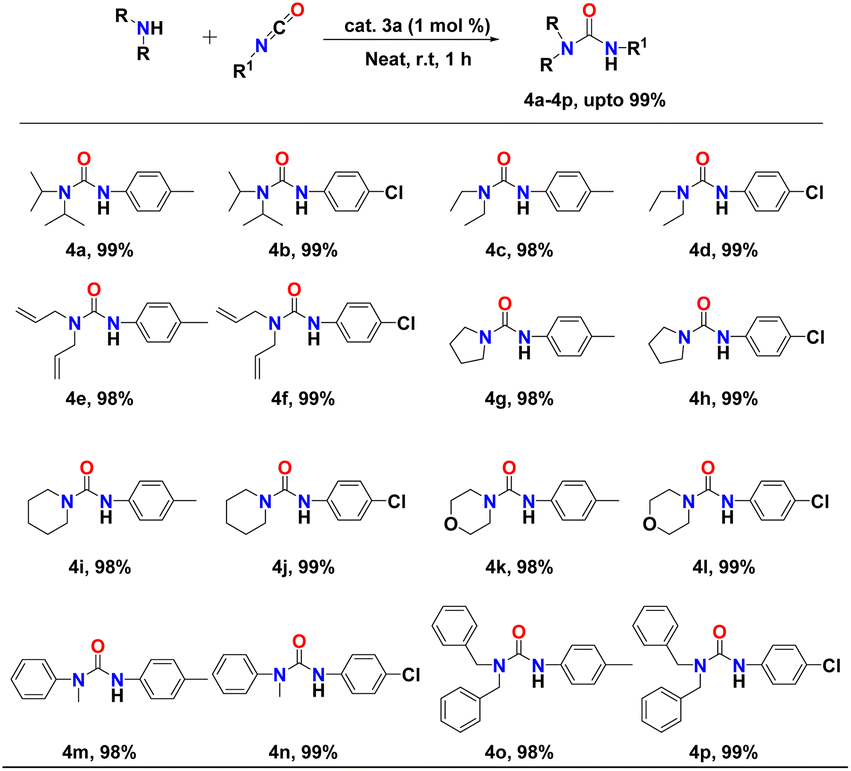
After successful optimisation, we extended our study of complex 3a as a competent catalyst in the addition of N–H bonds of a number of nucleophilic secondary amines to the p-tolylisocyanate/p-chlorophenyl-isocyanate. Catalytic experiments were conducted using 1 mol% of the binuclear aluminium complex (3a) and equimolar amounts of either p-tolylisocyanate or p-chlorophenyl-isocyanate and secondary amines at room temperature for one hour under neat conditions. The reactions displayed a broad substrate scope. In each case, the urea derivatives (4a–p) were isolated and analysed through 1H and 13C NMR spectroscopy (FS19–FS50, see the ESI†). The yields were calculated after isolating pure products. The results of the catalytic hydroamination to p-tolylisocyanate or p-chlorophenyl-isocyanate are presented in Table 2. We observed that the reaction of either p-tolylisocyanate or p-chlorophenyl-isocyanate with dialkyl amines, diallyl amines, dibenzyl amines, and arylalkyl amines was smooth at room temperature and selectively produced the corresponding urea products (with up to 99% yield) (Table 2, entries 4a–f and 4m–p). Even heterocyclic amines such as pyrrolidine, piperidine, and morpholine afforded excellent yields of the corresponding urea derivatives under similar reaction conditions (Table 2, entries 4g–l). The molecular structure of the urea product 4o in the solid state is shown in Fig. 5.
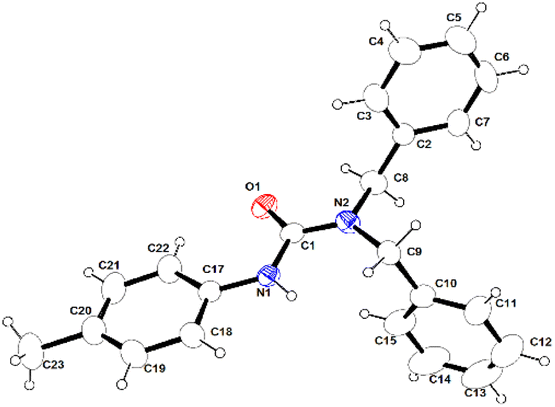 | ||
| Fig. 5 Molecular structure of urea product 4o compound in the solid state. Selected bond lengths (Å) and angles (°) are given. N1–C1 1.368(3), N1–C17 1.418(4), N1–H1 0.8600, O1–C1 1.228(3), C2–C7 1.363(5), C2–C3 1.379(5), C1–N1-C17 127.1(2), C1–N1–H1 116.5, C17–N1–H1 116.5, C7–C2–C3 117.2(3), C7–C2–C8 121.3(3), C3–C2–C8 121.5(3). CCDC No. 2209322.† | ||
Formation of biuret derivatives
The synthesis of biuret and triuret derivatives from mono-urea derivatives is more challenging as it typically requires elevated temperatures due to its reduced reactivity compared to secondary amines,56 owing to the electron-withdrawing effect of the carbonyl group. Reports in literature on the catalytic synthesis of biuret and triuret derivatives are limited to the use of catalysts such as organotin, SOCl2, and ClSO2OH at high temperatures.57,58 After the successful synthesis of urea derivatives, we applied the binuclear aluminium catalyst 3a to the process of directly transforming secondary amines to biuret derivatives under neat reaction conditions. Results of the catalytic transformation of secondary amines to biuret derivatives are set out in Table 3.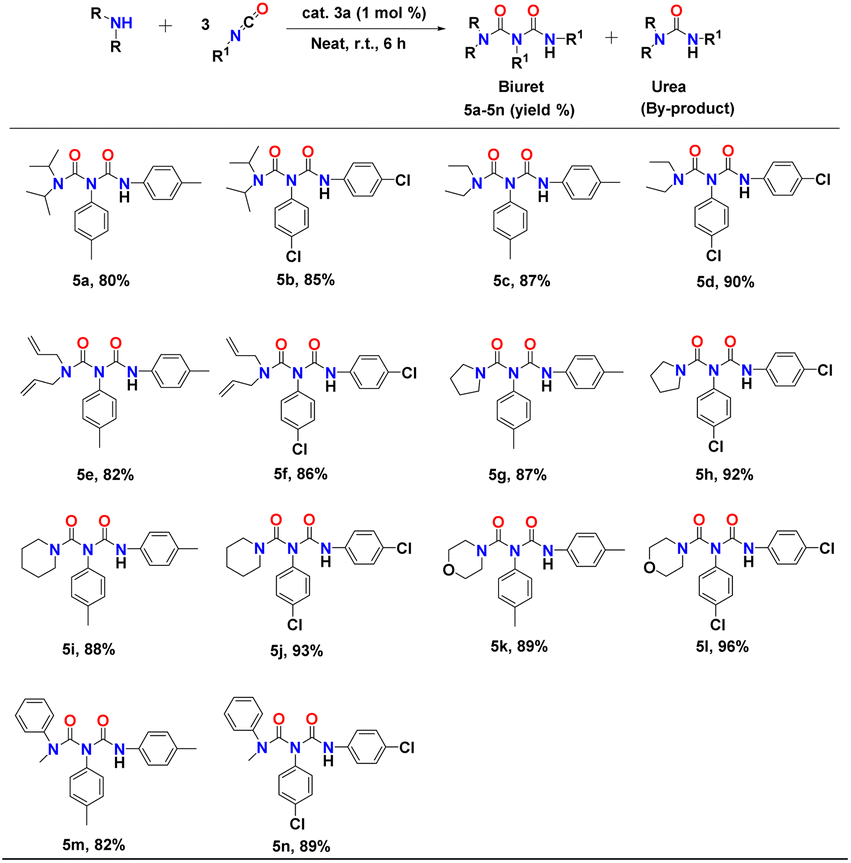
The initial reaction between diisopropylamine and p-tolylisocyanate in a 1:2 molar ratio in the presence of 1 mol% of complex 3a at room temperature after six hours resulted in the formation of the biuret compound (53% yield) along with the corresponding urea derivative as a co-product. However, an increased amount of p-tolylisocyanate, 3 equivalents with respect to the diisopropylamine, afforded 80% of the biuret compound after six hours at room temperature (Table 3, entry 5a). A further increase of isocyanate yielded a mixture of urea, biuret, and triuret derivatives. Thus, we selected the isocyanate and the respective secondary amine in a 3:1 molar ratio to explore the efficiency of catalyst 3a under solvent-free conditions at room temperature. Al catalyst 3a is compatible with both electron-releasing and electron-withdrawing functional groups, as exemplified by methyl and chloro moieties (Table 3, entries 5a–n). In each case, the biuret derivatives were isolated and analysed through 1H and 13C NMR spectroscopy (Fig. FS51–FS77†). Yields were calculated after isolating the pure products.
The reaction between diisopropylamine and p-chloro-phenylisocyanate in a 1:3 molar ratio resulted in good conversion, yielding 85% of biuret under optimum conditions (Table 3, entry 5b). Use of diethylamine, diallylamine, and N-methylaniline with either p-tolylisocyanate or p-chlorophenyl-isocyanate also resulted in smooth conversions. The reactions (conducted at room temperature under neat conditions for six hours) yielded the desired biuret compounds as major products (82–90%) along with corresponding urea derivatives as minor products (Table 3, entries 5c–f). This protocol was also used for heterocyclic secondary amines such as pyrrolidine, piperidine, and morpholine, which showed excellent conversion to the corresponding biuret derivatives under neat reaction conditions (Table 3, entries 5g–l). These results indicate the high proficiency and excellent functional group tolerance of catalyst 3a.
A most plausible mechanism of biurets formation from the secondary amines and isocyanates using Al catalyst 3a is proposed based on previous studies.16,59 It was proposed that in the initial step, more nucleophilic secondary amine reacts on both the Al-centers of electrophilic pre-catalyst to form active catalytic species aluminum amine which further participates in several catalytic steps to give the desired product. The detailed mechanism and scheme are provided in the ESI (Scheme S1†).
Formation of isourea, isothiourea, and phosphorylguanidine
Catalytic intermolecular addition of alcohols to carbodiimides has been developed as an efficient atom-economic tool to construct isourea derivatives, which were found to have many interesting applications, such as being alkylating agents and intermediates.60 Recently, actinide catalysts {(BimR1/R2N)An[N(SiMe3)2]} (An = Th, U)61 and hafnium metal complexes62 were employed to afford higher yields of isourea derivatives from the reaction of alcohols with various carbodiimides in relatively short reaction times. However, preparing the actinide-amide catalysts was rather complicated. Therefore, developing simple and efficient catalysts which demonstrate high tolerance towards diversified carbodiimides and alcohols is highly desirable. Moreover, there are only a few reports in literature of catalysed hydrothiolation (i.e., addition of S–H bond) to carbodiimides at high temperatures which afford products in moderate-to-high yields. Recently, Yao et al.63 reported the addition of alcohol to carbodiimides to obtain isourea derivatives in the presence of commercially available rare-earth metal amide La[N(SiMe3)2]3. The same catalyst system also demonstrated good catalytic activity in the hydrothiolation reaction between phenyl isocyanate and thiols to yield thiocarbamates showing good functional group tolerance.64Encouraged by high activity and chemoselectivity of binuclear aluminium complex [Al(Me)2{Ph2P(Se)N(CH2)2N(CH2CH2)2O}-(AlMe3)] (3a) as a pre-catalyst for the production of urea and biuret derivatives via hydroamination of secondary amines, we explored the activity of aluminium pre-catalysts 2a, 2b, 3a, and 3b in hydroalkoxylation, hydrothiolation, and hydrophosphorylation with carbodiimides too. The intermolecular addition of alcohols, thiols, and diphenylphosphine oxide to carbodiimides results in the formation of isourea, isothiourea, and phosphorylguanidine, respectively. The results of the catalytic addition reactions of a series of REH (E = O, S, P) moieties to N,N′-diisopropylcarbodiimide (DIC) and N,N′-dicyclohexyl-carbodiimide (DCC) are depicted below.
In this context, the reaction of 2-methylphenol with DIC was first investigated using different aluminium pre-catalysts (2a, 2b, 3a, and 3b). As a control experiment, a blank reaction, i.e., a reaction between 2-methylphenol and DIC with no catalyst, was carried out at room temperature for one hour under solvent-free conditions, and no product was seen to be formed (Table 4, entries 1 and 2). In contrast, with the addition of a catalytic amount (5 mol%) of the aluminium complexes 2a, 2b, 3a, and 3b, intermolecular insertion of O–H bond to carbodiimides took place smoothly, affording the corresponding isourea derivatives in high yields (Table 4, entries 3–6). In this case also, the binuclear aluminium(III) catalysts were found to be more efficient. The catalyst 3a was selected for the optimization of reaction conditions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Cat. (mol%) | Time (h) | Temp. (°) | Yieldb (%) |
| a Reactions were performed using a molar ratio of diisopropylcarbodiimides:2-methylphenol = 1:1.b Isolated yield. |
||||||
| 1 | None | Neat | — | 12 | r.t. | 0 |
| 2 | None | Neat | — | 12 | 60 | 0 |
| 3 | 2a | Neat | 5 | 1 | r.t. | 80 |
| 4 | 2b | Neat | 5 | 1 | r.t. | 78 |
| 5 | 3a | Neat | 5 | 1 | r.t. | 92 |
| 6 | 3b | Neat | 5 | 1 | r.t. | 90 |
| 7 | 3a | Neat | 2.5 | 1 | r.t. | 92 |
| 8 | 3a | Neat | 1 | 1 | r.t. | 92 |
| 9 | 3a | Neat | 0.5 | 1 | r.t. | 88 |
| 10 | 3a | Toluene | 1 | 1 | r.t. | 92 |
| 11 | 3a | Hexane | 1 | 1 | r.t. | 75 |
| 12 | 3a | THF | 1 | 1 | r.t. | 40 |
Thereafter, we set out to conduct a routine optimisation of factors that could influence isourea/isothiourea conversion, including catalyst loading and polar/non-polar solvent effect. At first, we investigated the effect of catalyst loading on the product yield. We observed no discernible change in the yield of the isourea product upon reducing the catalyst loading from 5 mol% to 1 mol% (of 3a) at room temperature within one hour (Table 4, entries 5, 7 and 8). When the catalyst loading was reduced further to 0.5 mol%, the conversion decelerated by up to 88% (Table 4, entry 9). Thus, 1 mol% of the catalyst loading was taken as an ideal condition for the model reaction during the optimisation. Further, we screened the effect of the various solvents such as toluene, hexane, and THF (Table 4, entries 10–12). However, the addition of polar and non-polar solvents appeared to be disadvantageous to isourea product formation. Conversion in the THF solvent was sluggish due to the coordinating nature of this solvent during the reaction conversion (Table 4, entry 12).
The scope of the intermolecular addition reaction of various aryl alcohol substrate to dicyclohexylcarbodiimide (DCC) and diisopropylcarbodiimide (DIC) was studied by varying the steric bulkiness and nucleophilicity of the alcohols. All the catalytic experiments were performed using 1 mol% of catalyst 3a at room temperature under neat conditions for one hour. The respective isourea derivatives (Table 5, entries 6a–s) were isolated in each case and characterized by 1H and 13C NMR spectroscopy (FS78-FS111, see the ESI†). The yields were calculated after isolating pure products. The results of the intermolecular coupling reactions are shown in Table 5.
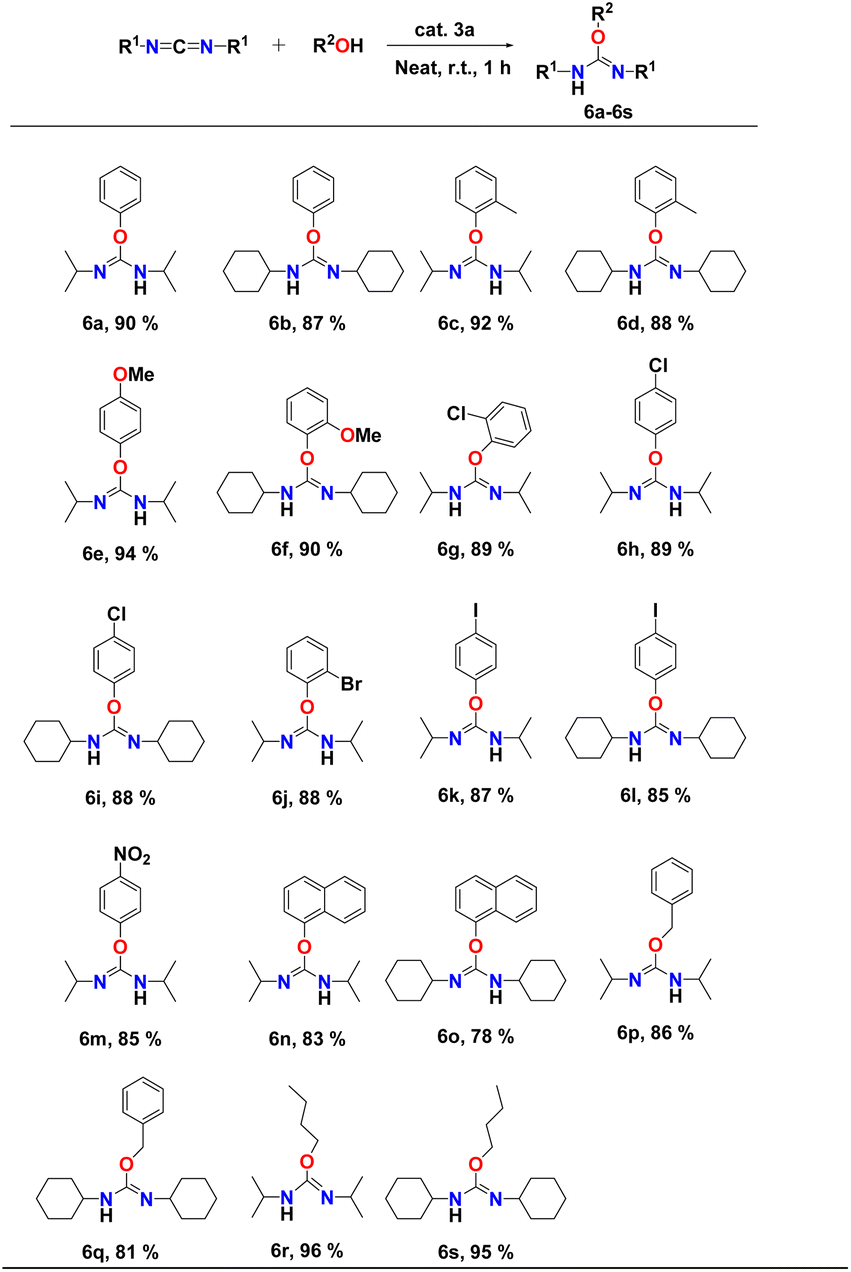
It was observed that aryl alcohols with electron-donating groups, such as simple phenol, 2-methylphenol, 2-methoxyphenol, and 4-methoxyphenol afforded excellent conversion, with yields of 88–94% (Table 5, entries 6a–f). Aryl alcohols containing electron-withdrawing groups such as chloro, bromo, iodo, nitro, and trifluoromethyl, also converted to corresponding isourea derivatives in good yields of 85–90% (Table 5, entries 6g–m) within one hour at room temperature. Even the addition of α-naphthol to DIC resulted in the formation of the respective isourea product with a yield of 83% (Table 5, entry 6n). Reduced reactivity of α-naphthol was observed in case of DCC (Table 5, entry 6o) due to the steric hindrance of both moieties. In the case of α-naphthol, our research group had observed no reactivity when the binuclear Ti-catalyst18 was used, clearly indicating the high reactivity of binuclear aluminium complex 3a. Benzyl alcohol also reacted smoothly with both carbodiimides and yielded the corresponding isourea at 86% (Table 5, entries 6p and q). Aliphatic alcohols such as butanol significantly increased the rate of hydroalkoxylation reaction compared to aryl alcohols (Table 5, entries 6r and s). Dicyclohexylcarbodiimide (DCC) exhibited better reactivity than diisopropylcarbodiimide (DIC) due to the presence of the bulky cyclohexyl group which shows greater electron donation than the isopropyl moieties. Compared to previous hydroalkoxylation reactions,61,63 higher conversions, even at room temperature and in a short amount of time, were observed, suggesting a higher reactivity of aluminium complex 3a.
Encouraged by the high reactivity of complex 3a as a competent catalyst in the case of hydroalkoxylation, we proceeded with its utilisation in the addition reaction of aromatic thiols to carbodiimides. The results of the intermolecular addition reactions of thiophenols to carbodiimides are shown in Table 6. At first, the simple thiophenols were treated with either diisopropylcarbodiimide or dicyclohexylcarbodiimide moieties in the presence of binuclear aluminium pre-catalyst 3a at room temperature under neat conditions and the corresponding isothiourea derivatives were obtained in good yield (∼95%) in each case (Table 6, entries 7a and b). Later, we studied the scope of the reaction with regard to thiol substrates under the same reaction conditions. Aromatic thiols bearing electron-donating groups such as methyl and methoxy groups converted efficiently into the respective isothiourea derivatives in excellent yields of 98% (Table 6, entries 7c and d). Even aromatic thiols bearing electron-withdrawing groups (chloro and trifluoromethyl) reacted smoothly to give respective isothiourea products in yields of up to 92% (Table 6, entries 7f–i). Benzylthiophenol also reacted moderately to yield the corresponding isothiourea product (95%) (Table 6, entries 7j and k). The respective isothiourea derivatives 7a–k were isolated in each case and characterised by 1H and 13C NMR spectroscopy (Fig. FS112–FS132, see the ESI†). Yields were calculated after the pure products were isolated.
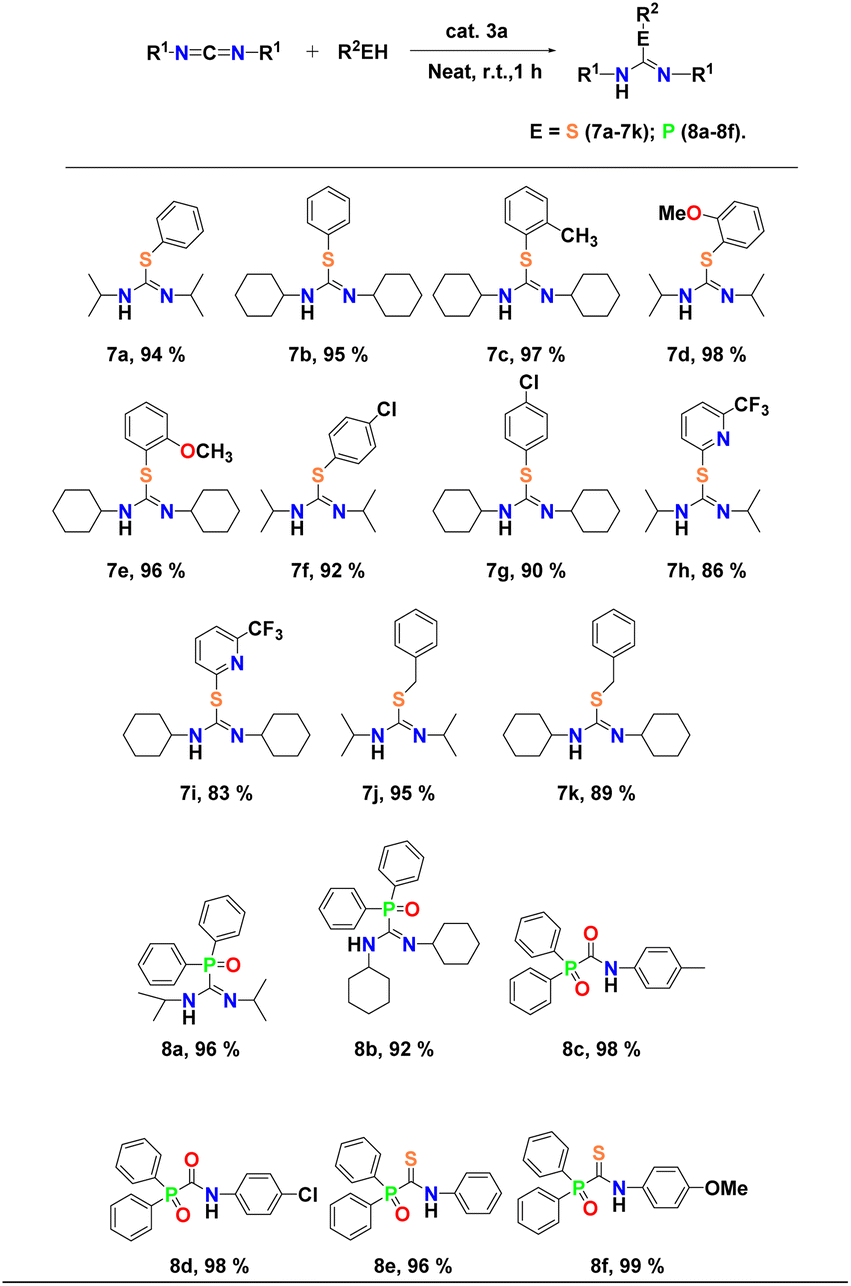
Complex 3a is also found to be a potent catalyst in the hydrophosphorylation of heterocumulenes with diphenylphosphine oxide [Ph2P(O)H] under conditions similar to hydroalkoxylation discussed above. The reaction of DIC and DCC with diphenylphosphine oxide smoothly afforded the corresponding phosphorylguanidine in each case (Table 6, entries 8a and b). In addition, the presence of two resonance peaks in the 31P NMR spectra in the case of DIC indicated the formation of E and Z-isomers of the respective phosphorylguanidine products (Fig. FS134†). Substituting DIC with a bulkier carbodiimide (such as DCC) allowed us to successfully obtain only one isomer of phosphorylguanidine (Fig. FS137†). These results are consistent with those previously reported by our group18 and Westerhausen et al.65 for hydrophosphorylation reaction of carbodiimides catalysed by alkali metals. Analogous reactions of diphenylphosphine oxide with isocyanates and isothiocyanates resulted the facile formation of corresponding 1-(diphenylphosphoryl)-N-(p-tolyl)formamide, N-(4-chlorophenyl)-1-(diphenylphosphoryl)-formamide (Table 6, entries 8c and d) and 1-(diphenyl phosphoryl)-N-(p-tolyl)-methanethioamide, N-(4-chloro-phenyl)-1-(diphenylphos-phoryl)methanethioamide (Table 6, entries 8e and f), indicating the versatility of the catalyst. We were able to crystallise 8f. The molecular structure of 8f in the solid state is already known66 and has been provided in the ESI (see Fig. FS182†).
Formation of quinazolinones
Insertion of heterocumulenes into N–H bonds of amino acid esters, followed by cyclisation, results in the formation of six-membered nitrogen-containing heterocyclic compound quinazolinones. These have been widely used in anti-cancer, anti-malaria, anti-inflammatory, and anti-tuberculosis medication due to their diverse biological properties.67 This class of compounds and their derivatives also have significant uses in the synthesis of natural products.37In continuation of our research, we extended the applicability of aluminium complex 3a as a catalyst in the hydroamination/cyclisation of amino acid esters with carbodiimides to afford corresponding quinazolinones under mild conditions with a broad substrate scope. Initially, the catalytic activity of complex 3a was tested with a catalyst loading of 5 mol% starting with ethyl 2-amino-benzoate and N,N′ diisopropylcarbodiimide (DIC) in a 1:2 molar ratio at 60 °C under solvent-free conditions. To our delight, 95% of the corresponding cyclic aminoquinazolinone product was isolated after 12 hours (Table 7, entry 9a). The use of 2 equivalents of carbodiimides is required for complete conversion of ethyl 2-amino-benzoate, as alcohol is discharged as a side product during this reaction; this reacts further with the second molecule of carbodiimides to form isourea as by-product. This phenomenon was also observed in earlier reports.43
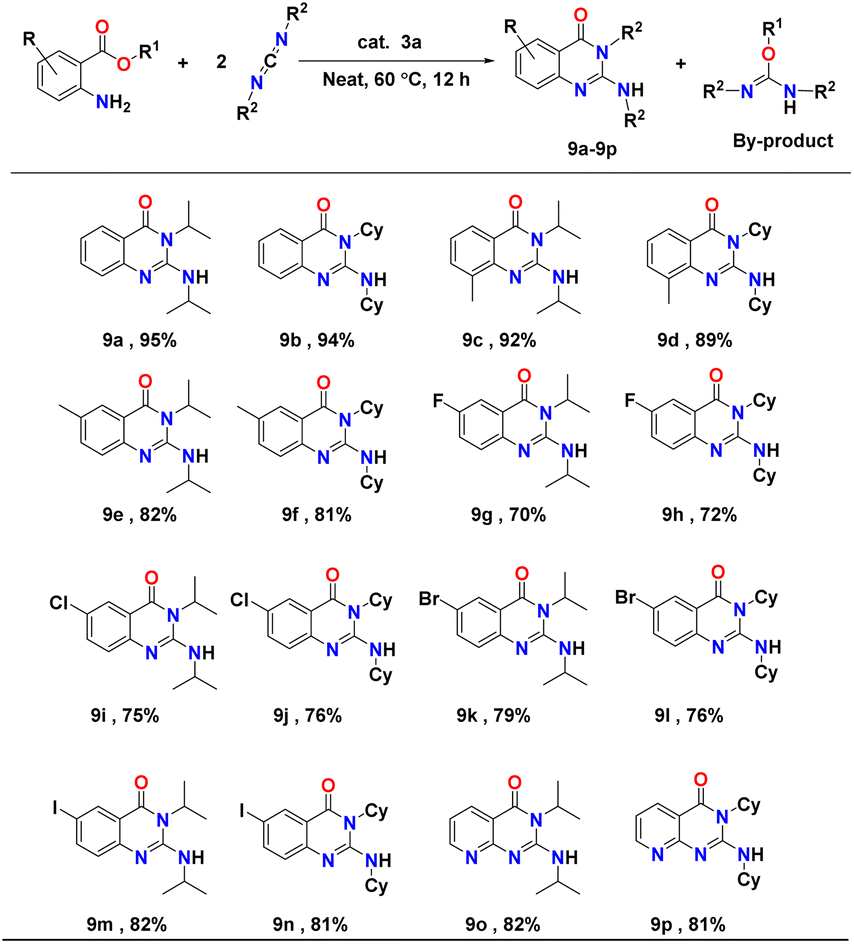
In order to analyse the substrate scope further, the hydroamination/cyclisation reactions of a series of substituted amino acid esters with carbodiimides catalysed by complex 3a were investigated under optimum conditions (i.e., substituted amino acid ester and carbodiimide in a 1:2 molar ratio with 5 mol% of the complex 3a under solvent-free conditions at 60 °C). In each case, the corresponding quinazolinone derivatives (9a–p) were isolated and analysed through 1H and 13C NMR spectroscopy (see Fig. FS150–FS181 in the ESI†). Yields were calculated after isolating the pure products. The reactions evidenced a broad substrate scope. Results of the catalytic hydroamination/cyclisation reactions of aminoesters with carbodiimides are presented in Table 7.
In most cases, very good substrate conversion, producing the corresponding quinazolinone derivatives with excellent chemoselectivity, was achieved. It was observed that the reaction of either diisopropylcarbodiimide (DIC) or N,N′-dicyclohexylcarbodiimide (DCC) with substituted amino acid esters, bearing electron-donating groups (methyl and methoxy) on the aryl ring proceeded smoothly at 60 °C under neat conditions and afforded the respective aminoquinazolinones derivatives in good yields of ∼80–92% (Table 7, entries 9c and f). In contrast, amino acid esters with electron-withdrawing groups (such as fluoro, chloro, bromo, and iodo), showed slightly lower reactivity and conversion (∼70–80%) under similar conditions, due to the deactivation of the aryl moiety (Table 7, entries 9g–n). The heterocyclic amino acid esters also showed excellent conversion to the corresponding aminoquinazolinones derivatives under mild reaction conditions (Table 7, entries 9o and p).
Based on the previous reports,41–43 a most plausible mechanism for the catalytic guanylation/cyclization of amino acid esters using Al catalyst 3a has been proposed. In the first step, aminolysis of the Al-alkyl complex with aminobenzoate takes place to generate active catalytic species (I) with the elimination of two volatile methane molecules, which further participates in several catalytic steps to give the desired product. The detailed mechanism and scheme are provided in the ESI (Scheme S2†).
Experimental section
General
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−4 torr) line or in an argon-filled M. Braun glove box. Hydrocarbon solvents (toluene and n-pentane) were distilled under nitrogen from LiAlH4 and stored in the glove box. 1H NMR (400 MHz), 13C{1H} (100 MHz) NMR 31P{1H} (161.9 MHz) spectra were recorded on a BRUKER AVANCE III-400 spectrometer. BRUKER ALPHA FT-IR was used for FT-IR measurement. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad. The NMR solvent CDCl3, C6D6 and DMSO-d6 were purchased from Sigma Aldrich and used without further purification.Preparation of [Ph2P(E)NH(CH2)2N(CH2CH2)2O] [E = Se (1a-H), S (1b-H)]
In a degassed 25 ml Schlenk tube, triethylamine (0.932 g, 9.2 mmol, 1.2 equiv.) was slowly added to the solution of toluene (10 ml) and 4-(2-aminoethyl)morpholine (1.0 g, 7.68 mmol, 1 equiv.) kept at ice-cold temperature. After stirring for 30 min., chloro-diphenylphosphine (2.03 g, 9.2 mmol, 1.2 equiv.) was added dropwise for 15 minutes to the above solution while it was being vigorously stirred. The resulting reaction mixture was stirred for another 12 hours at room temperature and the formation of a white precipitate of triethylammonium chloride was observed during the reaction. The solution was filtered to remove the triethylammonium chloride salt. The elemental Se (0.73 g, 9.2 mmol, 1.2 equiv.) or S (0.3 g, 9.2 mmol, 1.2 equiv.) was then added to the filtrate under an inert atmosphere. The resulting solution was stirred for another 12 hours at 90 °C. Finally, the reaction mixture was filtered to remove the excess S/Se and the filtrate was evaporated under reduced pressure to give a pale-yellow sticky compound, which resulted in a white powder after being washed with hexane in each case. Colourless crystals of 1a-H were obtained in diethyl ether at room temperature.Preparation of [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2O}] [E = Se (2a), S (2b)]
Inside the glove box in a degassed 25 ml Schlenk flask, ligand 1a (150 mg, 0.381 mmol) or 1b (150 mg, 0.432 mmol) and 5 ml of toluene were added. Thereafter, the reaction solution was taken out from the glovebox. To this solution, trimethylaluminum solution (2.0 M in toluene) (0.19 ml, 0.381 mmol for 1a or 0.22 ml, 0.432 mmol for 1b) was added, and the resulting reaction mixture was stirred continuously for 12 hours at 60 °C. Then the solvent was evaporated under a high vacuum to produce a colourless residue, which resulted in a white solid after being washed with hexane in each case. The white solid was dissolved in 3 ml of toluene and kept aside for re-crystallization at −35 °C. Colourless crystals of 2a were obtained after seven days.Preparation of [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2O}(AlMe3)] (E = Se (3a) and S (3b))
Catalytic experiments
Procedure for synthesis of urea derivatives
Inside the glovebox, in a degassed 25 ml Schlenk tube, a solution of secondary amine (0.384 mmol, 1 equiv.) was added into the mixture of the chosen isocyanate (0.384 mmol, 1 equiv.) and aluminium metal complex (3a) (0.00384 mmol, 1 mol%). A white solid started to form during the reaction at room temperature, and an additional hour was allowed for the reaction to be completed. A white solid compound was obtained in each case which was characterized directly by 1H and 13C{1H} NMR spectroscopy without any further purification. The yields of amines were calculated from isolated pure products.Procedure for synthesis of biuret compounds
Inside the glovebox in a degassed 25 ml Schlenk tube, secondary amine (0.384 mmol, 1 equiv.) was added to the mixture of the chosen isocyanate (1.15 mmol, 3 equiv.) and aluminium complex (3a) (0.00384 mmol, 1 mol%). A white solid started to form during the reaction when it was stirred at room temperature for six hours. A white solid compound was obtained in each case, which was purified by washing with n-hexane several times and the desired product was further purified by the column technique (EtOAc:pet ether:1:10). The yield of biuret was calculated from isolated pure products. The products were characterised using 1H and 13C{1H} NMR spectroscopy.
Procedure for synthesis of isourea and isothiourea derivatives
Inside the glovebox, in a degassed 25 ml Schlenk tube, the chosen aryl alcohol/thiol (0.384 mmol, 1 equiv.) was added into the mixture of carbodiimide (0.384 mmol, 1 equiv.) and aluminium complex (3a) (0.00384 mmol, 1 mol%). A white solid started to form during the reaction, as it was stirred at room temperature for one hour. The corresponding isourea and isothiourea derivatives obtained were characterised directly by 1H and 13C{1H} NMR spectroscopy without any further purification. The yields of isourea and thiourea were calculated from the pure products isolated.Procedure for the synthesis of phosphorylguanidine
Inside the glovebox, in a degassed 25 ml Schlenk tube, diphenylphosphine oxide (0.384 mmol, 1 equiv.) was added to the mixture of carbodiimide (0.384 mmol, 1 equiv.) and aluminium complex (3a) (0.00384 mmol, 1 mol%). A white solid started to form during the reaction, as it was stirred at room temperature for one hour. The reaction mixture was quenched with ethanol and then washed with dichloromethane, followed by Celite filtration to obtain a pure white or yellow solid of phosphoguanidine. The yield of phosphoguanidine was calculated from the isolated product. The isolated products were identified by 1H, 13C{1H}, and 31P{1H} NMR spectroscopy.Procedure for the synthesis of quinazolinones
Inside the glovebox in a degassed 25 ml Schlenk tube, a solution of amino acid ester (0.192 mmol, 1 equiv.) was added dropwise to the mixture of carbodiimide (0.384 mmol, 2 equiv.) and aluminium complex (3a) (0.0095 mmol, 5 mol%). The yellow-coloured reaction mixture was heated at 60 °C for 12 hours to complete the reaction. The progress of reactions was monitored by TLC. Subsequently, the reaction mixture was quenched by adding 20 ml of ethyl acetate solvent. The basic workup was done by adding 5 ml of a saturated sodium bicarbonate solution to the reaction mixture. The organic layer was separated from the product and purified by column chromatography using 5% ethyl acetate as an eluent solvent. The yields of quinazolinones were calculated from pure isolated products. The isolated products were characterised using 1H and 13C{1H} NMR spectroscopy.Conclusion
To summarise, we synthesised and characterised a series of mononuclear and binuclear aluminium complexes with the general composition [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2O}] E = Se (2a); S (2b) and [Al(Me)2{Ph2P(E)N(CH2)2N(CH2CH2)2O}-(AlMe3)] (E = Se (3a) and S (3b)). We demonstrated the superior catalytic efficiency of the binuclear aluminium metal complex 3a in the facile synthesis of a variety of products such as urea, biuret, isourea, isothiourea, phosphorylguanidine, and quinazolinone derivatives. We also demonstrated that the efficiency of the binuclear complex 3a exceeds that of its mononuclear analogues and previously reported catalysts. We noted that the reaction of isocyanates with a variety of secondary amines in a 1:1 molar ratio, catalysed by complex 3a at room temperature, proceeded smoothly and resulted in the selective production of the corresponding urea in up to 99% yield. Biuret derivatives were synthesised in very good yields (of up to 80%) via the direct transformation of secondary amines with isocyanates (1:3 molar ratio) using the same catalyst 3a under solvent-free and mild conditions. This catalyst also exhibited excellent reactivity and selectivity in the addition of E–H bonds (E = OR, SR, P(O)R2) to a wide variety of heterocumulenes under mild conditions. The application of complex 3a as a catalyst was successfully extended to the hydroamination/cyclisation of amino acid esters with carbodiimides to afford the corresponding quinazolinones in yields of up to 95%. In each case, complex 3a displayed a high tolerance toward heteroatoms and functional groups under mild conditions with a broad substrate scope.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan International Cooperation Agency (JICA) under project India-Japan FRIENDSHIP 2 Research Grant AC2022-2 and the Science and Engineering Research Board (SERB)-Teaching Associateship for Research Excellence (TARE), Department of Science and Technology (DST), Government of India, under project no. (TAR/2020/000003). K. B. thanks CSIR, India (09/1001(0028)/2017-EMR-I) and J. S. thanks UGC. India for their PhD fellowships.Notes and references
- In Addition Reactions with Unsaturated Hydrocarbons, John Wiley & Sons, Ltd, 2022, pp. 103–145 Search PubMed.
- In Addition Reactions with Unsaturated Hydrocarbons, John Wiley & Sons, Ltd, 2022, pp. 47–102 Search PubMed.
- C. D. Huke and D. L. Kays, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2021, vol. 75, pp. 1–54 Search PubMed.
- C. J. Weiss, S. D. Wobser and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 2062–2063 CrossRef CAS PubMed.
- E. Ballinger, J. Mosior, T. Hartman, K. Burns-Huang, B. Gold, R. Morris, L. Goullieux, I. Blanc, J. Vaubourgeix, S. Lagrange, L. Fraisse, S. Sans, C. Couturier, E. Bacqué, K. Rhee, S. M. Scarry, J. Aubé, G. Yang, O. Ouerfelli, D. Schnappinger, T. R. Ioerger, C. A. Engelhart, J. A. McConnell, K. McAulay, A. Fay, C. Roubert, J. Sacchettini and C. Nathan, Science, 2019, 363, eaau8959 CrossRef CAS PubMed.
- I.-H. Kim, H.-J. Tsai, K. Nishi, T. Kasagami, C. Morisseau and B. D. Hammock, J. Med. Chem., 2007, 50, 5217–5226 CrossRef CAS PubMed.
- J. M. McCall, R. E. TenBrink and J. J. Ursprung, J. Org. Chem., 1975, 40, 3304–3306 CrossRef CAS PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Polyhedron, 2012, 37, 9–13 CrossRef CAS.
- M. B. Geeson, A. R. Jupp, J. E. McGrady and J. M. Goicoechea, Chem. Commun., 2014, 50, 12281–12284 RSC.
- E. V. Sharova, O. I. Artyushin, Yu. V. Nelyubina, K. A. Lyssenko, M. P. Passechnik and I. L. Odinets, Russ. Chem. Bull., 2008, 57, 1890–1896 CrossRef CAS.
- C. Boehme and G. Wipff, Inorg. Chem., 2002, 41, 727–737 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, in Organic Reactions, ed. John Wiley & Sons, Inc., John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015, pp. 1–554 Search PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- J. Huo, G. He, W. Chen, X. Hu, Q. Deng and D. Chen, BMC Chem., 2019, 13, 89 CrossRef PubMed.
- K. Bano, S. Anga, A. Jain, H. P. Nayek and T. K. Panda, New J. Chem., 2020, 44, 9419–9428 RSC.
- J. Bhattacharjee, S. Das, R. K. Kottalanka and T. K. Panda, Dalton Trans., 2016, 45, 17824–17832 RSC.
- J. Bhattacharjee, A. Harinath, I. Banerjee, H. P. Nayek and T. K. Panda, Inorg. Chem., 2018, 57, 12610–12623 CrossRef CAS PubMed.
- A. Harinath, K. Bano, S. Ahmed and T. K. Panda, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 23–32 CrossRef CAS.
- R. J. Batrice and M. S. Eisen, Chem. Sci., 2016, 7, 939–944 RSC.
- R. J. Batrice, C. E. Kefalidis, L. Maron and M. S. Eisen, J. Am. Chem. Soc., 2016, 138, 2114–2117 CrossRef CAS PubMed.
- I. S. R. Karmel, M. Tamm and M. S. Eisen, Angew. Chem., 2015, 127, 12599–12602 CrossRef.
- W.-X. Zhang, L. Xu and Z. Xi, Chem. Commun., 2015, 51, 254–265 RSC.
- P. Ertl, E. Altmann and J. M. McKenna, J. Med. Chem., 2020, 63, 8408–8418 CrossRef CAS PubMed.
- M. Eto, Biosci., Biotechnol., Biochem., 1997, 61, 1–11 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS.
- X. Liu and J.-R. Hamon, Coord. Chem. Rev., 2019, 389, 94–118 CrossRef CAS.
- J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC.
- P. Selig, Synthesis, 2013, 45, 703–718 CrossRef CAS.
- R. H. Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910–914 CrossRef CAS PubMed.
- A. J. South, A. M. Geer, L. J. Taylor, H. R. Sharpe, W. Lewis, A. J. Blake and D. L. Kays, Organometallics, 2019, 38, 4115–4120 CrossRef CAS.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- K. C. Majumdar, P. Debnath, N. De and B. Roy, Curr. Org. Chem., 2011, 15, 1760–1801 CrossRef CAS.
- P. W. Davies and M. Garzón, Asian J. Org. Chem., 2015, 4, 694–708 CrossRef CAS.
- J. Bartroli, E. Turmo, M. Algueró, E. Boncompte, M. L. Vericat, L. Conte, J. Ramis, M. Merlos, J. García-Rafanell and J. Forn, J. Med. Chem., 1998, 41, 1869–1882 CrossRef CAS PubMed.
- S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS.
- A. Maity, S. Mondal, R. Paira, A. Hazra, S. Naskar, K. B. Sahu, P. Saha, S. Banerjee and N. B. Mondal, Tetrahedron Lett., 2011, 52, 3033–3037 CrossRef CAS.
- N. J. Liverton, D. J. Armstrong, D. A. Claremon, D. C. Remy, J. J. Baldvin, R. J. Lynch, G. Zhang and R. J. Gould, Bioorg. Med. Chem. Lett., 1998, 8, 483–486 CrossRef CAS PubMed.
- N. Malecki, P. Carato, B. Rigo, J.-F. Goossens, R. Houssin, C. Bailly and J.-P. Hénichart, Bioorg. Med. Chem., 2004, 12, 641–647 CrossRef CAS PubMed.
- Y. Chi, L. Xu, S. Du, H. Yan, W.-X. Zhang and Z. Xi, Chem.–Eur. J., 2015, 21, 10369–10378 CrossRef CAS PubMed.
- S. Das, J. Bhattacharjee and T. K. Panda, Dalton Trans., 2019, 48, 7227–7235 RSC.
- C. Lu, C. Gong, B. Zhao, L. Hu and Y. Yao, J. Org. Chem., 2018, 83, 1154–1159 CrossRef CAS PubMed.
- J. Song and B. Han, Natl. Sci. Rev., 2015, 2, 255–256 CrossRef.
- H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2017, 36, 3896–3903 CrossRef CAS.
- K. Naktode, R. K Kottalanka and T. K. Panda, New J. Chem., 2012, 36, 2280–2285 RSC.
- R. K Kottalanka, K. Naktode and T. K. Panda, J. Mol. Struct., 2013, 1036, 189–195 CrossRef.
- R. K Kottalanka, K. Naktode, S. Anga, H. P. Nayek and T. K. Panda, Dalton Trans., 2013, 42, 4947–4956 RSC.
- S. Sagar, K. Bano, A. Sarkar, K. Pal and T. K. Panda, Eur. J. Inorg. Chem., 2022, e202200494 CAS.
- A. Harinath, J. Bhattacharjee and T. K. Panda, Adv. Synth. Catal., 2019, 361, 850–857 CrossRef CAS.
- T.-H. Lin, Y.-R. Cai, W. Chang, C.-H. Hu, T.-Y. Lee, A. Datta, H.-C. Hsiao, C.-H. Lin and J.-H. Huang, J. Organomet. Chem., 2016, 825–826, 15–24 CrossRef CAS.
- K. Bano, D. A. Kisan and T. K. Panda, Eur. J. Inorg. Chem., 2022, 2022, e202200023 CrossRef CAS.
- G.-X. Chen, A. Datta, H.-C. Hsiao, C.-H. Lin and J.-H. Huang, Polyhedron, 2015, 101, 299–305 CrossRef CAS.
- Y.-J. Li, H.-T. Lai, C.-H. Hu, J.-H. Chen, C.-H. Lin and J.-H. Huang, J. Organomet. Chem., 2019, 902, 120957 CrossRef CAS.
- S. Das, H. Karmakar, J. Bhattacharjee and T. K. Panda, Dalton Trans., 2019, 48, 11978–11984 RSC.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- E. Dyer and R. B. Pinkerton, J. Appl. Polym. Sci., 1965, 9, 1713–1729 CrossRef CAS.
- D. R. Park, H. Kim, J. C. Jung, M. S. Shin, S. J. Han and I. K. Song, Korean J. Chem. Eng., 2009, 26, 990–993 CrossRef CAS.
- I. Banerjee, S. Sagar, C. Lorber and T. K. Panda, Z. fur Anorg. Allg. Chem., 2022, 648, e202200188 CAS.
- C. J. Weiss and T. J. Marks, Dalton Trans., 2010, 39, 6576–6588 RSC.
- H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Inorg. Chem., 2017, 56, 3153–3157 CrossRef CAS PubMed.
- M. Khononov, H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2020, 39, 3021–3033 CrossRef CAS.
- L. Hu, C. Lu, B. Zhao and Y. Yao, Org. Chem. Front., 2018, 5, 905–908 RSC.
- L. Hu, C. Lu, B. Zhao and Y. Yao, Organometallics, 2019, 38, 2167–2173 CrossRef.
- S. Krieck, H. Görls, L. Yu, M. Reiher and M. Westerhausen, J. Am. Chem. Soc., 2009, 131, 2977–2985 CrossRef CAS PubMed.
- C. D. Huke, L. J. Taylor, S. P. Argent and D. L. Kays, ACS Sustainable Chem. Eng., 2021, 32, 10704–10709 CrossRef.
- W. Lu, I. A. Baig, H.-J. Sun, C.-J. Cui, R. Guo, I.-P. Jung, D. Wang, M. Dong, M.-Y. Yoon and J.-G. Wang, Eur. J. Med. Chem., 2015, 94, 298–305 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of Al metal complexes (2a, 2b and 3a), NMR data and spectra of all the catalytic insertion products. Full crystallographic details for 1a-H, 2a, 3a and 4o. CCDC reference numbers 2151760 (1a-H), 2151762 (2a), 2209321 (3a), and 2209322 (4o). Crystallographic data and 1H, 13C{1H}, 31P{1H}, NMR spectra pertaining to aluminium metal complexes 2a, 2b, 3a, and 3b, urea products (4a–p), biuret products (5a–n), isourea products (6a–s), isothiourea products (7a–k), phosphorylated products (8a–f). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra07714k |
| This journal is © The Royal Society of Chemistry 2023 |