
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic performance of Ni–Co/HZSM-5 catalysts for aromatic compound promotion in simulated bio-oil upgrading†
Liyuan Qin *ab,
Jiani Lia,
Shengming Zhangab,
Zhongyuan Liua,
Shuang Lia and
Lina Luoa
*ab,
Jiani Lia,
Shengming Zhangab,
Zhongyuan Liua,
Shuang Lia and
Lina Luoa
aCollege of Engineering, Northeast Agricultural University, Harbin, 150030, China. E-mail: qinliyuan2006@163.com
bKey Laboratory of Pig-breeding Facilities Engineering, Ministry of Agriculture and Rural Affairs, Harbin 150030, China
First published on 8th March 2023
Abstract
Bio-oil can be used as a substitute for fossil fuels after it is upgraded. Bimetal-modified HZSM-5 catalysts with various Ni-to-Co ratios were prepared to address catalysis problems, including deactivation of the catalysts and low hydrocarbon yields. The catalytic performance of Ni–Co/HZSM-5 in upgrading the simulated bio-oil was investigated with a fixed-bed reactor, and the influence of the loaded duplex metal ratio was also discussed. The new moderately strong/strong acid sites of Ni–Co/HZSM-5 changed according to the Ni/Co loading ratios, which substantially affected the acidity, catalytic activity and selectivity of the Ni–Co/HZSM-5. However, incorporating Co and Ni into the zeolite did not alter the structure of the HZSM-5. The interactions of the loaded bimetallic oxides reached equilibrium in 6Ni–4Co/HZSM-5, in which moderately strong acid sites and strong acid sites were formed after loading with Co3O4 and NiO. With a suitable acid site ratio, 6Ni–4Co/HZSM-5 exhibited excellent performance, with a lower coke deposition of 3.29 wt% and stable catalytic activity, and the conversion remained at 83–73% during 360 min of uninterrupted catalysis. Periodic changes in the acid sites and interfacial protons were the critical factors that improved the properties of the 6Ni–4Co/HZSM-5 and enhanced its selectivity for aromatic compounds.
1. Introduction
Searches for alternative energy sources have been strongly motivated by increasing environmental problems and the consumption of fossil fuels.1–3 Biomass-derived fuel has been widely explored due to its short CO2 cycles, high energy density and abundance of natural feedstocks.4 Accordingly, many economically viable processes have been developed to convert biomass.5 Pyrolysis is a promising and simple thermochemical process for directly converting biomass feedstock into bio-oil, biochar and noncondensable gas at relatively low costs.6 Bio-oil is a potential biofuel and source for producing biobased chemicals.7 However, it is usually difficult to utilize bio-oil directly because of its high oxygen content and low stability. In addition, bio-oil contains some highly viscous organic molecules; these molecules (heavy portion) lead to a low heating value, high viscosity and corrosiveness.8–10 Thus, bio-oils must be upgraded before application.An effective method of upgrading bio-oil is catalytic cracking.11 Many types of catalysts have been prepared and studied for this process; however, these catalysts are usually deactivated due to coke deposition. Despite this problem, HZSM-5 is still a common catalyst for upgrading tar, heavy bio-oil and vapor due to its ideal pore size, high acidity and high selectivity for aromatic hydrocarbons.12 Some studies claim that HZSM-5 is deactivated because it exhibits smaller micropores, cannot easily diffuse internally and easily forms coke deposition on the Brønsted (B) acid site of HZSM-5, leading to channel blockage and acid site poisoning.13 Therefore, adjusting the B and Lewis (L) acidity, promoting the decomposition of macromolecules on the surface and increasing the pore size of HZSM-5 should effectively compensate for the above inadequacy.
The accessibility of acid sites and mesoporosity are important parameters for the catalytic performance of HZSM-5. The source of acidity in the HZSM-5 catalysts depends on the Si/Al ratio, in which the Si–OH group represents weak acid sites and the Si–OH–Al group represents strong acid sites.14,15 Previous studies have demonstrated that HZSM-5 catalysts with lower Si/Al ratios exhibit higher concentrations of B acidity, which are beneficial for treatment of condensed aromatics; thus, the ability of the catalyst to perform deoxygenation and the selectivity for aromatic compounds are enhanced.16,17 However, coke deposition will obviously decrease over longer catalytic lifetimes when the HZSM-5 catalysts have higher Si/Al ratios. Therefore, a suitable Si/Al ratio should be important for the catalytic performance of HZSM-5. In addition, introducing mesopores in HZSM-5 can reduce the formation of coke to a certain degree.18
HZSM-5 can also be modified with metals, and it has been demonstrated that this method can significantly improve the activity and lifetime of a parent catalyst and decrease the formation of coke on the catalytic surface.19 In particular, HZSM-5 catalysts modified with the transition metals Ni and Co typically achieved higher yields of aromatic hydrocarbon products.20–22 Moreover, compared with HZSM-5 catalysts modified with single metals, bimetal-modified catalysts, such as Cu–Ni/HZSM-5, Co–Zn/HZSM-5 and Ni–Mo/HZSM-5 catalysts, displayed better performance due to the synergistic or bifunctional effect of different modified metals.23–25 Of course, the bimetallic Ni–Co/HZSM-5 catalyst has also been investigated, and adding Co to Ni/HZSM-5 efficiently enhanced the dispersion of Ni and stabilized the active Ni sites.26 However, no further details have been provided regarding the effect of the Ni and Co ratio and its metal oxide on catalytic activity and selectivity during the catalytic upgrading process.
Therefore, in this work, the performance of Ni–Co/HZSM-5, which exhibits a hierarchical microporous–mesoporosity structure, a high Si/Al ratio (80) of the HZSM-5 and various loading ratios for Ni and Co, was investigated in a simulated upgrading process for bio-oil.
2. Experimental
2.1 Catalyst preparation
HZSM-5 (SiO2/Al2O3 = 80) was used as a support and was purchased from Nankai University Catalyst Corp., China. An impregnation method was used to prepare Ni–Co/HZSM-5 with different Ni-to-Co loading ratios. The total mass loading content of metals was maintained at 10 wt% based on an assumption of 100% impregnated metal loading, and the Ni-to-Co mass ratios were 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 8:2, 6:4, 5:5, 4:6, 2:8 and 0:10. HZSM-5 was first calcined for 4 h in air at 500 °C and then impregnated with aqueous solutions containing different concentrations of the corresponding metal nitrates Ni(NO3)2·6H2O and Co(NO3)2·6H2O at room temperature for 2 h. Then, the catalyst was dried and calcined for 3 h at 550 °C. For example, 10 g of HZSM-5 was added to a 150 ml aqueous solution containing 4.9550 g of Ni(NO3)2·6H2O to prepare Ni/HZSM-5 with 10 wt% Ni loading, and 2.9730 g of Ni(NO3)2·6H2O and 1.9755 g of Co(NO3)2·6H2O were added to prepare Ni–Co/HZSM-5 with 6 wt% Ni loading and 4 wt% Co loading, by parity of reasoning. The modified HZSM-5 catalysts are denoted as 10Ni/HZSM-5, 8Ni–2Co/HZSM-5, 6Ni–4Co/HZSM-5, 5Ni–5Co/HZSM-5, 4Ni–6Co/HZSM-5, 2Ni–8Co/HZSM-5 and 10Co/HZSM-5, where 10Ni means 10 wt% Ni loading, 8Ni–2Co means 8 wt% Ni, 2 wt% Co loading, etc.
:0, 8:2, 6:4, 5:5, 4:6, 2:8 and 0:10. HZSM-5 was first calcined for 4 h in air at 500 °C and then impregnated with aqueous solutions containing different concentrations of the corresponding metal nitrates Ni(NO3)2·6H2O and Co(NO3)2·6H2O at room temperature for 2 h. Then, the catalyst was dried and calcined for 3 h at 550 °C. For example, 10 g of HZSM-5 was added to a 150 ml aqueous solution containing 4.9550 g of Ni(NO3)2·6H2O to prepare Ni/HZSM-5 with 10 wt% Ni loading, and 2.9730 g of Ni(NO3)2·6H2O and 1.9755 g of Co(NO3)2·6H2O were added to prepare Ni–Co/HZSM-5 with 6 wt% Ni loading and 4 wt% Co loading, by parity of reasoning. The modified HZSM-5 catalysts are denoted as 10Ni/HZSM-5, 8Ni–2Co/HZSM-5, 6Ni–4Co/HZSM-5, 5Ni–5Co/HZSM-5, 4Ni–6Co/HZSM-5, 2Ni–8Co/HZSM-5 and 10Co/HZSM-5, where 10Ni means 10 wt% Ni loading, 8Ni–2Co means 8 wt% Ni, 2 wt% Co loading, etc.
2.2 Catalytic experiment
Toluene, cyclopentanone, acetic acid, furfural and guaiacol were mixed with a volume ratio of 1:1:1:1:1 to represent simulated bio-oil. All these reagents were analytically pure. The catalytic tests were conducted on a fixed bed reactor, as shown in Fig. S1.† For each run, 5 g of catalyst was placed on the insulating spacers of the catalyst reaction bed, and the reaction temperature was 400 °C. Model bio-oil (10 mL) was gasified and introduced into the 400 °C preheated reactor using a peristaltic pump at a flow rate of 20 ml h−1 for 30 min with N2 as the carrier. N2 flushing was maintained for another 20 min after the experiment was complete to ensure that all condensable gas was cooled and collected. The liquid was dissolved in chloroform, and the mass of the liquid after removing chloroform was reported as ML. The yield of liquid after upgrading was defined as follows:
 | (1) |
The conversion of each model component was calculated as follows:
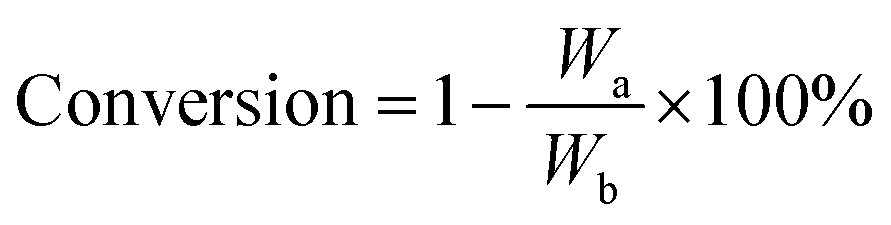 | (2) |
2.3 Characterization
Scanning electron microscopy (SEM, S-3400, Hitachi, Japan) was used to investigate the surface morphology of the fresh, spent and regenerated catalysts, and the elements were detected by energy dispersive X-ray spectroscopy (EDS, NC250). X-ray diffraction (XRD, MiniFlex-600, Rigaku, Japan) was employed to analyze the crystalline structure. The porous structure of the catalysts was investigated by means of an adsorption–desorption instrument (3H-2000PS1, BeiShiDe Instrument, China). The N2 adsorption–desorption instrument had a repeatability error of less than 1.5%. The total pore volume (Vtotal), micropore pore volume (Vmic) and specific surface area (SBET) were determined from the absorbed volume of nitrogen at a relative pressure (P/P0) of 0.99, the t-plot method, and the multipoint BET equation, respectively.X-ray photoelectron spectroscopy (XPS, Thermo, Thermo Electron Corporation, USA) was used to detect and analyze the variations in metal loading of the modified HZSM-5 catalyst. Temperature-programmed desorption of ammonia (NH3-TPD) was conducted by means of a chemisorption analyzer (AutoChem 2920, Micromeritics, USA). Pyridine adsorption Fourier transform infrared spectrometry (Py-IR, VERTEX 70, Bruker, Germany) was used to detect the B acid and L acid centers of the catalysts. For each test, 0.1 g of catalyst was heated to 300 °C for 2 h and then cooled to 50 °C.
Coke deposition was measured by thermogravimetric analysis (TGA, STA449C Jupiter, NETZSCH-Gerätebau GmbH, Germany) and analyzed by Fourier transform infrared spectroscopy (FT-IR, Nicolet 50, Thermo Fisher, USA). A gas chromatography–mass spectrometry (GC–MS) system (6890, Agilent, USA) was used to analyze the liquid products using a DB-5MS column with a film thickness of 0.25 μm, column length of 30 meters, and diameter of 250 μm. The viscosity was measured at 40 °C using a viscometer (NDJ-5S, YUEPING, China). The higher heating value (HHV) and pH were measured with an oxygen bomb calorimeter (YX-ZR9302, LI-THERM, China) and a pH meter (PHS-3CW, BANTE, China), respectively. Additional parameter details were reported in our previous work.27–29
The products that formed on the surface of Ni–Co/HZSM-5 during the conversion of the model substrates were investigated at 400 °C through in situ FTIR experiments (Frontier FT-IR Spectrometer, PerkinElmer, USA). The catalyst was placed in a flow-through IR cell reactor with CaF2 windows. The composition of the reagent was 1000 ppm, and the feed was held at 30 min. FTIR spectra were measured with an MCT detector, which continuously recorded data during the reaction and processed data every 5 min. The spectrum of the calcined catalyst was subtracted from the monitored spectra that were recorded under the in situ conditions of the catalytic reaction.
3. Results and discussion
3.1 Characterization of catalysts
The XRD results are shown in Fig. 1a. All modified catalyst plots exhibited the characteristic peaks of the original HZSM-5, which means that incorporating the metal species did not destroy the structure of HZSM-5. Moreover, the loaded metals were present in their oxide forms in both the mono- and bimetal-modified catalysts. Only peaks corresponding to NiO appeared in the 10Ni and 8Ni–2Co/HZSM-5 catalysts when the amount of cobalt species was too low for detection.30 For 6Ni–4Co/HZSM-5, Co3O4 peaks appeared and were accompanied by NiO peaks, and there were no peaks for CoO species. After further increasing the loading amount of Co and decreasing the Ni content, the NiO peaks disappeared, and only Co3O4 peaks were detected in the 4Ni–6Co/HZSM-5 and 2Ni–8Co/HZSM-5 catalysts; the same results were observed for 5Ni–5Co/HZSM-5. This means that the incorporation of Co might enhance the interaction between Ni and HZSM-5 and improve the Ni dispersion on HZSM-5.24 According to the XRD analysis, the crystallite sizes for the NiO and Co3O4 in the modified catalysts were approximately 6.1 nm and 9.3 nm, respectively. | ||
| Fig. 1 (a) XRD and (b) NH3-TPD results for fresh Ni–Co/HZSM-5 catalysts. | ||
The characteristics of the acidic site in the catalysts were determined, and the results are shown in Fig. 1b and Table 1. For the parent HZSM-5, two broad peaks at approximately 135 °C and 363 °C (Fig. 1b) corresponded to weak acid (<300 °C) and moderately strong acid sites (300–450 °C), and no peaks that corresponded to strong acid sites (>450 °C) were detected at higher temperatures.31,32 After Ni and Co modifications were performed with the HZSM-5, the intensity of the peak at approximately 135 °C decreased slightly, and the peak at 362 °C changed according to the Ni-to-Co loading ratio; this was due to the difference in electronegativity for Co and Ni, which affected the acidity, catalytic activity and selectivity. The acidities of the bimetal-modified HZSM-5 catalysts were related to the differences in the Ni-to-Co loading ratios, affecting the interaction between metal ions and the interaction between metal ions and HZSM-5, and strong acid sites formed more easily in the Ni-modified HZSM-5 catalyst.33 Moreover, previous research reported that metal loading exhibits a preference for new L acid sites due to the formation of the corresponding oxides, i.e., NiO and Co3O4 (XRD results in Fig. 1a), and exchange with the partial protons responsible for the B acid sites in HZSM-5; a reduction in B acidity and an increase in L acidity occurred as a result.34 Thus, the B/L value of the modified HZSM-5 catalysts decreased to 0.08–0.11 compared to 0.18 for HZSM-5 (in Table 1). This indicates that the acidity was mainly due to L acids in Ni–Co/HZSM-5. In addition, HZSM-5 was used in this study with a Si/Al of 80 and led to a significantly lower B acid content (B/L = 0.18); thus, the effect of metal doping on decreasing the acidity was less, and the modified HZSM-5 catalyst showed a greater total acidity than the parent HZSM-5 catalyst.
| Catalysts | Pore properties | Acidity (mmol g−1) | AreaB/areaL | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SBETa (m2 g−1) | Vmicb (cm3 g−1) | Vtotalc (cm3 g−1) | Vmesod (cm3 g−1) | Total | Weak acid sites | Moderately strong acid sites | Strong acid sites | ||
| a SBET is the specific surface area determined by the BET method.b Vmic is the micropore volume determined by the t-plot method.c Vtotal is the total pore volume at P/P0 = 0.99.d Vmeso is mesopore volume calculated by Vtotal − Vm. | |||||||||
| HZSM-5 | 284.41 | 0.13 | 0.38 | 0.25 | 0.62 | 0.51 | 0.11 | — | 0.18 |
| 10Ni/HZSM-5 | 274.24 | 0.12 | 0.35 | 0.23 | 0.78 | 0.50 | 0.28 | — | 0.11 |
| 8Ni–2Co/HZSM-5 | 266.86 | 0.12 | 0.34 | 0.22 | 0.76 | 0.46 | 0.30 | — | 0.11 |
| 6Ni–4Co/HZSM-5 | 263.04 | 0.11 | 0.32 | 0.21 | 0.79 | 0.44 | 0.22 | 0.13 | 0.11 |
| 5Ni–5Co/HZSM-5 | 265.55 | 0.12 | 0.34 | 0.22 | 0.76 | 0.43 | 0.33 | — | 0.11 |
| 4Ni–6Co/HZSM-5 | 262.70 | 0.12 | 0.33 | 0.21 | 0.74 | 0.40 | 0.34 | — | 0.11 |
| 2Ni–8Co/HZSM-5 | 260.03 | 0.12 | 0.32 | 0.20 | 0.74 | 0.38 | 0.36 | — | 0.08 |
| 10Co/HZSM-5 | 265.22 | 0.13 | 0.33 | 0.20 | 0.66 | 0.33 | 0.33 | — | 0.10 |
For the HZSM-5 catalysts that were modified by monometallic 10 wt% Ni and 10 wt% Co, peaks attributed to moderately strong acid sites were observed at 430 °C and 395 °C, respectively. The desorption peaks of the bimetal-modified HSZM-5 catalysts occurred at approximately 135 °C for the weak acid and moderately strong/strong acid sites, at approximately 350–400 °C for the Co dominant modifications, and at 430–460 °C for the Ni dominant modifications, which consisted of both B acid sites and L acid sites.17 The two loaded metals competed for the proton positions in the HZSM-5.24 In addition, the interactions between Co and Ni species in the bimetal-modified HSZM-5 shifted the desorption peaks at 350–400 °C to lower temperatures, and the peaks at 430–460 °C were shifted to higher temperatures compared to those of the monometallic HZSM-5 catalysts. However, only the 6Ni–4Co/HZSM-5 gave rise to a peak at 458 °C (>450 °C), which suggested formation of a minority of strong L acid sites.31 This region of strong acid sites was considered to be the main catalytic center and to act as the dominant acid sites during the acid-catalyzed and deoxygenation reactions.12 In conclusion, the mono- and bimetal-modified HZSM-5 catalysts in this study all displayed a characteristic combination of weak and strong acid sites with lower B/L values, which is expected to lead to a reduction in the chance of coke formation.35
Fig. S2† and Table 1 show the porosity characteristics of the catalysts, and the pore sizes were mainly concentrated at 2.3 nm and 8.5 nm. All catalysts showed a textural hierarchical micropore–mesopore structure. Because the total metal loading amounts for all the bimetal-modified HZSM-5 catalysts in this study were the same, the pore parameters of SBET, Vmic, Vmeso and Vtotal of the Ni–Co/HZSM-5 catalysts were similar, and all decreased compared to that of the parent HZSM-5 because some pores were blocked by the accumulation of metal species (Ni and Co) on the surface and within the channels of the HZSM-5.24,36 However, the pore change results were not the same as those for monometallic HZSM-5 because of the interactions between the different metal ions. This did not gradually increase or decrease with changes in one of the metal loading amounts. Moreover, the micropores were less affected, Vmic decreased by only 0.01, and the loaded metal oxides were mainly on the surface and in the mesopores. Thus, macromolecule decomposition occurred mostly on the surface during the catalytic process. The surface morphologies of the modified catalysts were similar, as shown in Fig. 2. The metal oxide aggregated on the catalyst surface, and nanoparticle voids were observed. The Co and Ni contents of each modified catalyst were measured, and the results are shown in Fig. S3.† The metal contents of the modified catalysts deviated slightly from the calculated values, as Ni and Co were not equally distributed on the HZSM-5 support.
 | ||
| Fig. 2 SEM images of the fresh catalysts: (a) HZSM-5; (b) 10Ni/HZSM-5; (c) 6Ni–4Co/HZSM-5; (d) 10Co/HZSM-5. | ||
3.2 Catalytic performance of the Ni–Co/HZSM-5 catalysts
Fig. 3 shows the catalytic properties of the catalysts in upgrading model compounds. For the Ni and Co modified catalysts, the conversions of the model compounds were much higher, especially for toluene and acetic acid. The corresponding liquid product yields decreased because the Ni and Co modified HZSM-5 catalysts not only promoted the cracking of large-molecule organic components but also accelerated the cracking of small-molecule components, which produced more gases.37 For the 6Ni–4Co/HZSM-5 catalyst, the conversion of each component was much higher than that of the other catalysts; the conversions of toluene, acetic acid and cyclopentenone were all higher than 50%. Notably, the conversion of cyclopentenone remained basically unchanged for the co dominant catalysts from 5Ni–5Co/HZSM-5 to 10Co/HZSM-5. This demonstrated that the moderately strong/strong acid sites from NiO loaded on HZSM-5 had higher selectivity for the cyclopentenone conversion. Moreover, the coke deposition of Ni–Co/HZSM-5 was improved, as a very low value (3.68–4.19%) was obtained (Fig. 3b). | ||
| Fig. 3 (a) Conversion of the model compound and (b) the yields of liquid and coke for the fresh Ni–Co/HZSM-5 catalysts. | ||
A lower Si/Al ratio in the HZSM-5 led to more B acidity, which enhanced the conversion of oxygenates and promoted the production of aromatic compounds during the catalytic upgrading process.16 In addition, the acid yield of the upgraded product was increased when the Si/Al ratio of the HZSM-5 was enhanced.17 Thus, HZSM-5 samples with Si/Al ratios of 25 and 30 were usually chosen, but the opposite and disappointing result was obtained for coke deposition. However, as mentioned above, the 6Ni–4Co/HZSM-5 catalyst showed an ideal catalytic performance and a lower coke yield when the Si/Al ratio was 80, the acidity was appropriate and the structure exhibited hierarchical microporous–mesoporosity, as the HZSM-5 catalyst produces a lower B acidity.
Fig. 4 shows the coke deposits on the surfaces of the spent catalysts, and the result is consistent with the trend seen in Fig. 3b. The amount of coke deposited on the spent Ni–Co/HZSM-5 catalyst was much less than that on the HZSM-5 and Ni/HZSM-5 catalysts. One reason is that the loaded metallic oxides decreased the B acidity of the HZSM-5 catalysts. Previous studies have shown that the coke mass is directly proportional to the concentration of B acid sites.38 Moreover, compared to other transition metals, loading with Co can reduce coke deposition to a larger degree.39
 | ||
| Fig. 4 SEM images of spent catalysts: (a) HZSM-5, (b) 10Ni/HZSM-5, and (c) 6Ni–4Co/HZSM-5. | ||
Fig. 5 shows the components of the mixed model were upgraded over the 6Ni–4Co/HZSM-5 and included esters, alcohols, ketones, phenols, aliphatic hydrocarbons, furans, and aromatic hydrocarbons. Aromatic hydrocarbons, aliphatic hydrocarbons and alcohols are the main components of the upgraded oil, and when the contents of these species increase, the HHV of the product is enhanced.40 According to the product distribution, it is speculated that the reaction during the catalytic process involves cyclization, aromatization and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O addition reactions to form alcohols because the loaded metal oxides act as L acid sites to promote hydrogen transfer and hydrogenation functions.32
O addition reactions to form alcohols because the loaded metal oxides act as L acid sites to promote hydrogen transfer and hydrogenation functions.32
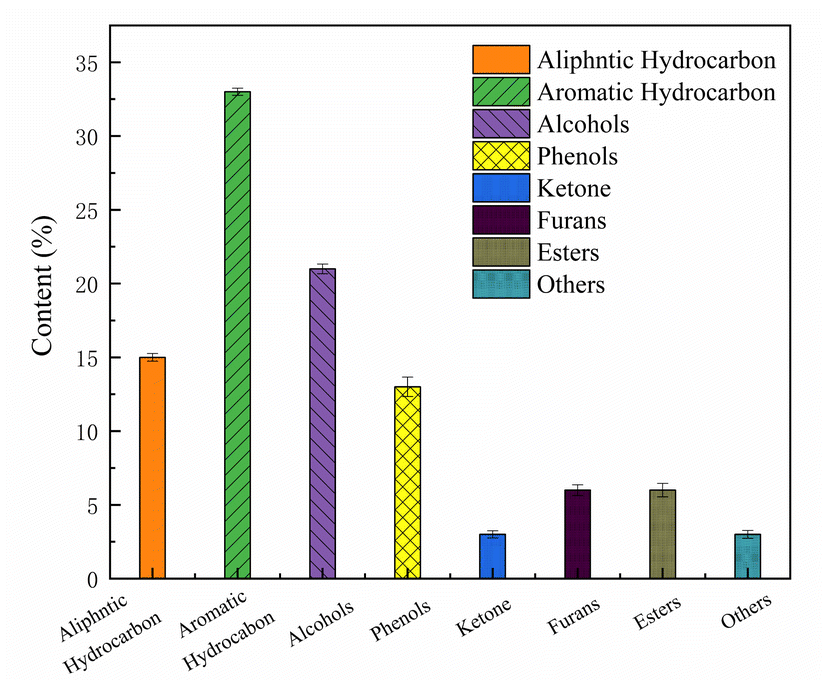 | ||
| Fig. 5 Composition of the liquid product catalyzed by 6Ni–4Co/HZSM-5. | ||
Fig. 6 shows the FT-IR results for the coke on the spent catalysts. For these spent catalysts, the FT-IR peaks of the chemical groups were almost identical. The peak at 1878 cm−1 was attributed to the ramification of benzene. The peaks at 1590 cm−1, 1503 cm−1, 1107 cm−1 and 808 cm−1 are related to aromatic stretching vibrations and correlate with the coke formed by the aromatization of long-chain alkanes. The infrared band at 800–500 cm−1 was attributed to the presence of aromatic compounds. These results illustrate that the coke formed on the catalyst mainly contains aromatic compounds. The 6Ni–4Co/HZSM-5 catalyst retained good selectivity for aromatic compounds.
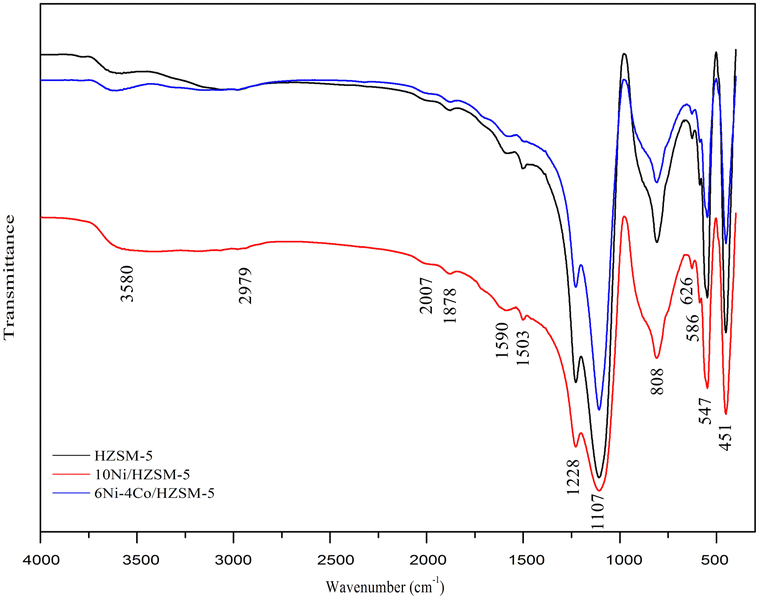 | ||
| Fig. 6 FT-IR patterns of coke on the spent catalysts. | ||
3.3 Lifetimes and regeneration of the bimetallic-modified HZSM-5 catalysts
Catalytic experiments were conducted using fresh and regenerated 6Ni–4Co/HZSM-5 catalysts to test the lifetime and activity of the catalyst in comparison with those of 10Ni/HZSM-5, and the results are shown in Fig. 7 and S4.† Toluene was selected as the reactant because it induces significant conversion (in Fig. 3a), and the catalytic tests were conducted without interruption for 360 min. As shown in Fig. 7, fresh 6Ni–4Co/HZSM-5 displayed little deactivation during the 360 min long-term cycle and maintained a toluene conversion of 83–73%, which remained over 80% for 180 min. Notably, the conversion during the reaction time sometimes showed a trend of decreasing and then increasing. The decline in conversion was mainly attributed to the deposition of coke that occurred during the reaction, which is a common problem. The subsequent increase in conversion occurred because a portion of the deposited coke precursors on the catalyst surface was consumed as the reaction time increased, which exposed some active sites; as a result, the activity was restored, and the lifetime of the catalysts was prolonged.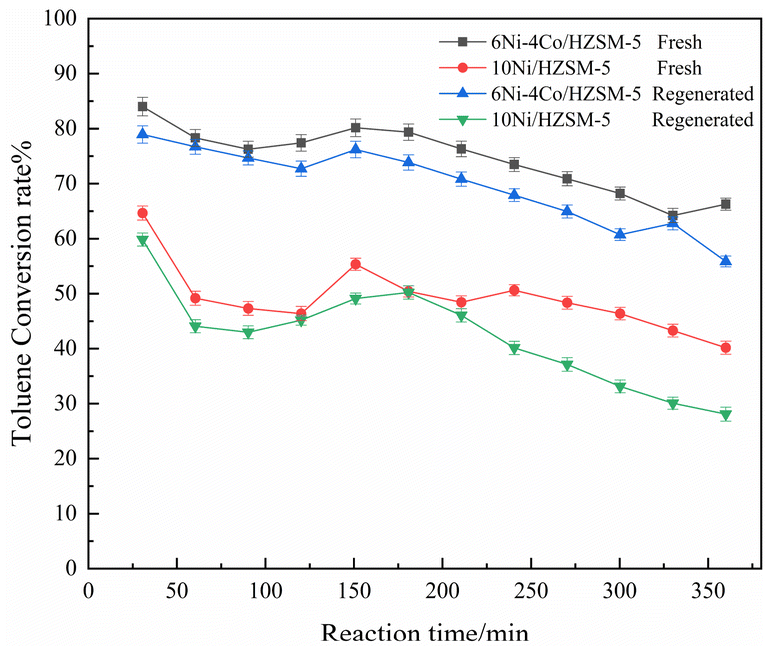 | ||
| Fig. 7 Activity of fresh and regenerated catalysts for toluene conversion. | ||
After 360 min of uninterrupted catalysis, 6Ni–4Co/HZSM-5 was regenerated, and the lifetime test was conducted once more. The conversion of the regenerated catalyst for toluene was 80–60%, which is a slight decrease compared with that of the fresh catalyst. A decrease in activity should have occurred because the metal centers and acid sites in the catalysts were lost during regeneration, which has also been reported in previous studies.35 Overall, the 6Ni–4Co/HZSM-5 catalyst exhibited better catalytic activity and lifetime during continuous catalysis.
The interaction between the loaded Ni and Co in bimetal-modified catalysts can benefit the catalytic activity, stability and regeneration of the catalysts, and these factors need to be further explored. The catalytic mechanism and synergy of different kinds of acid sites in the 6Ni–4Co/HZSM-5 catalyst will be discussed in the following section.
3.4 Analysis of the synergistic effect of loaded bimetallic oxides on catalyst activity
Fig. 8 shows the Ni 2p and Co 2p XPS spectra of 6Ni–4Co/HZSM-5 during the catalytic upgrading process. As seen in Fig. 8a, the fresh catalyst generated peaks at 856.6 eV and 874.4 eV that corresponded to Ni2+, and these peaks were accompanied by two satellite peaks at 862.7 eV and 881.2 eV.41 During the 30 min reaction, the peaks observed for the catalysts were the same as those for the fresh catalyst, and only peaks attributed to NiO were obtained; the details are shown in Table S1.† As seen in Fig. 8b, the Co 2p spectra showed two main peaks at 782.1 eV and 797.4 eV, and spin–orbit splitting was correlated with the Co 2p3/2 and Co 2p1/2 states. Moreover, previous studies have reported that the spin–orbital splitting values of Co3+ and Co2+ compounds are 15.0 eV and 16.0 eV, respectively; the mixed valence Co3O4 spin–orbital values were 15.3–15.4 eV.42,43 According to the data in Table S1,† the spin–orbital values of Co 2p in fresh 6Ni–4Co/HZSM-5 were all within this range. Therefore, the Co species of the fresh catalyst consisted of Co3O4. However, after 10 min of reaction, the Co 2p spectra for spent 6Ni–4Co/HZSM-5 catalysts showed peaks at a binding energy of 776.5 eV, which corresponded to Co0. This Co0 originated from the reduction of Co3O4 in the catalysts (Co3O4 → CoO → Co0), which is beneficial to the hydrogen transfer reaction.33 Moreover, similar Co0 peaks at 776.5 eV occurred for the catalyst after 30 min of reaction. | ||
| Fig. 8 XPS diagram of 6Ni–4Co/HZSM-5 during the catalytic process: (a) Ni 2p and (b) Co 2p. | ||
The acetic acid catalysis with 6Ni–4Co/HZSM-5 was studied by means of in situ FTIR, and the results in 30 min are shown in Fig. 9. The broad peak at 3458 cm−1 was attributed to –OH in moisture, and the peaks at 2346 cm−1 were assigned to CO2 adsorption.44 The bands at 1714 cm−1 correspond to the stretching vibration of CO in carbonyl, those at 1206 cm−1 correspond to the C–O stretching vibration, and those at 1365 cm−1 correspond to the –CH3 deformation vibration, which are assigned to adsorbed acetone.45–47 These results indicated that acetone should form from the condensation of acetic acid to produce H2O and CO2 according to previous studies.46,48 Moreover, these signal peaks decreased and disappeared after 10 min of reaction as acetic acid was consumed, and the bands at 2871 cm−1 and 1788 cm−1 originating from acetic acid decreased as well. The bands at 1089 cm−1, 1123 cm−1, 1321 cm−1 and 1392 cm−1 were associated with the stretching and deformation vibrations of C–O, out-of-plane C–H vibrations and –CH3 vibrations in the dissociative adsorption of isopropanol.49 In addition, a weak, out-of-plane C–H vibration band at 934 cm−1 and a CC stretching vibration at 1650 cm−1 from propylene were observed.45 This supported that propylene was converted from acetone by isopropanol as an intermediate in previous studies.49,50 The intensity of the signal attributed to acetone, isopropanol and propylene decreased with the reaction time, indicating that the compounds were converted to other products. The –CH3 and C–H groups were attributed to the conversion of these compounds to CO.47 The band at 1592 cm−1 corresponds to the CC skeletal vibration of the benzene ring, and the band at 910–665 cm−1 corresponds to the out-of-plane deformation of aromatic groups (C–H).51 Based on these results and previous studies, it is suggested that the alkene undergoes polymerization and cyclization to form an aromatic hydrocarbon.18,52 The peak signal of aromatic compounds increased with the reaction time, as this signal is related to the formation of more aromatic products.
 | ||
| Fig. 9 The in situ FTIR spectra for the conversion of acetic acid on the 6Ni–4Co/HZSM-5 catalyst at 400 °C. | ||
Based on the analysis and discussion of the in situ FTIR and XPS results, a possible pathway of acetic acid conversion on the 6Ni–4Co/HZSM-5 catalyst is provided in Fig. 10. From the XRD and NH3-TPD results (Fig. 1), it can be seen that a proper ratio for the Ni and Co loading generated strong acid sites, moderately strong acid sites and weak acid sites simultaneously after metal oxide loading, which were coexisted in the 6Ni–4Co/HZSM-5. Moreover, the loaded metal oxides act as L acid sites to promote hydrogen transfer and hydrogenation, which removes the B acid site in HZSM-5 (in Table 1). The incorporation of Co obviously enhanced the ability of HZSM-5-loaded Ni to perform hydrogenation.52 In addition, the Si/Al ratio of 80 for the parent HZSM-5 catalysts led to lower B acidity. These factors all resulted in the 6Ni–4Co/HZSM-5 catalyst having a dominant L acid site combined with an appropriate strong acidity.
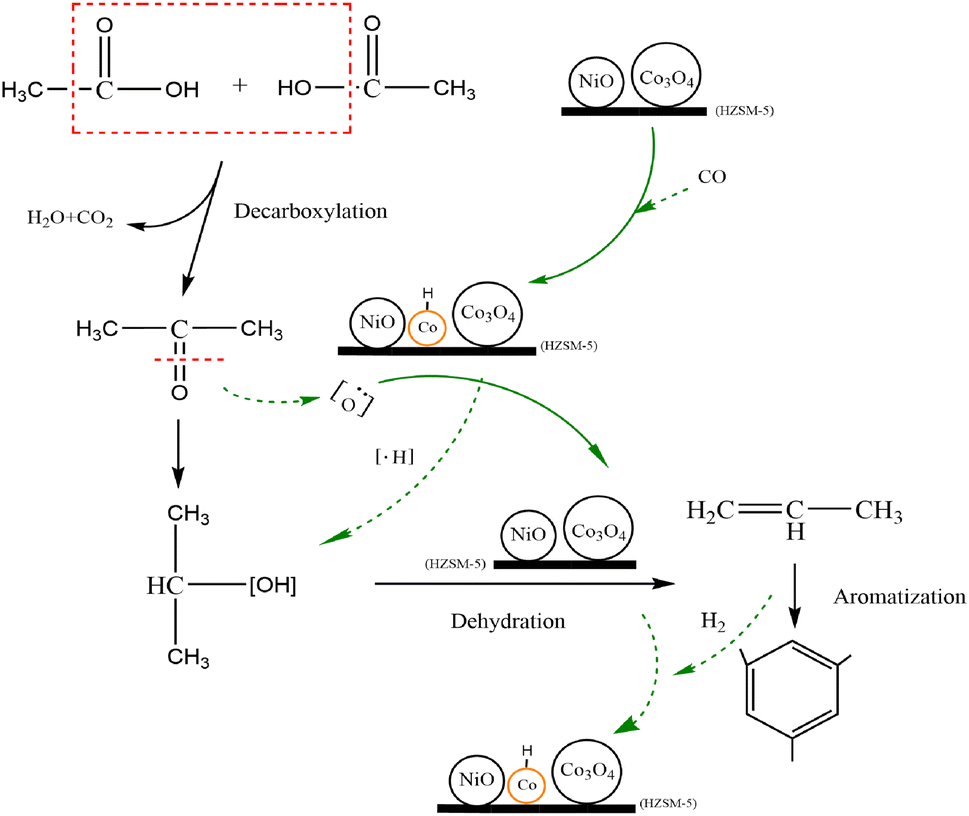 | ||
| Fig. 10 Possible pathway for acetic acid conversion on 6Ni–4Co/HZSM-5. | ||
When the 6Ni–4Co/HZSM-5 catalyst exhibited strong acid sites, it performed oxidation–reduction reactions to convert acetic acid to acetone and produce H2O and CO2.46 Simultaneously, some of the Co3O4 on HZSM-5 was reduced to Co0 metal by the reducing gas generated during the process. As previously reported,26 the Co3O4 reduction to Co0 in the modified HZSM-5 catalysts underwent two steps, that is, Co3O4 → CoO and CoO → Co0. Two successive reduction reactions occurred at 563 K (290 °C) and 653 K (380 °C) in Co/HZSM-5, and the reduction temperatures shifted to 583 K (310 °C) and 713 K (440 °C) in Ni–Co/HZSM-5; Si/Al = 25 in HZSM-5. The higher reduction temperatures for Ni–Co/HZSM-5 than for Co/HZSM-5 indicate greater stability of O for the Ni-containing sample and stronger interaction between the smaller particle components (particle size of NiO < Co3O4) and the HZSM-5 support.26 Thus, when no extra reducing gas was added, only Co0 derived from the Co3O4 was observed in the catalyst during the reaction process (Fig. 8). Moreover, Co0 provided more protons on the acid sites of the modified HZSM-5 and generated the Co/CoOx or Co/NiO interface in 6Ni–4Co/HZSM-5, which may be responsible for the higher selectivity for alcohol synthesis, as stated in previous reports.53–56
In addition, the synergistic effects of the strong L acid sites and B acid sites of the 6Ni–4Co/HZSM-5 catalyst supplied abundant sites for acetone adsorption and hydrogenation; therefore, a reaction to generate isopropanol occurred instead of a condensation reaction.49 Thus, acetone was converted to isopropanol and consequently dehydrated to produce propene. The small chain hydrocarbons showed a higher tendency to undergo aromatization due to the strong L acid sites in the modified HZSM-5 catalyst.57 However, the corresponding Co0 returned to an oxidized state at this stage. Then, the acid site distribution on 6Ni–4Co/HZSM-5 changed accordingly. In this process, the periodic changes in acid sites and the interface of Co/CoOx or Co/NiO were the critical factors that improved the catalytic properties of 6Ni–4Co/HZSM-5, and these improvements reduced coke deposition, prolonged the lifetime of the catalyst and enhanced the selectivity of aromatic compounds.
4. Conclusion
In this work, bimetal-modified Ni–Co/HZSM-5 catalysts were prepared to improve catalytic activity and coke deposition during the process of upgrading model bio-oil. The metal modification and high Si/Al ratio of the HZSM-5 support resulted in suitable acidity for Ni–Co/HZSM-5. Various Ni-to-Co loading ratios were compared to illustrate the competition and synergy between the loaded metals and the protons of the acid sites, which affected the activity and selectivity of the catalysts. NiO modification favored the conversion of toluene, acetic acid and cyclopentanone. The moderately strong acid sites and strong acid sites reached equilibrium in 6Ni–4Co/HZSM-5 after metal loading. Co incorporation facilitated periodic changes in the acid sites and provided more protons in 6Ni–4Co/HZSM-5, which resulted in excellent performance. Specifically, a lower amount of coke deposition (3.29 wt%) and a higher conversion rate (83–73%) were achieved in 360 min of uninterrupted catalysis with simulated bio-oil; in addition, the selectivity of alcohols and aromatics was enhanced. Although many bimetal-modified HZSM-5 catalysts have been prepared and their catalytic performance has been studied, it is expected that 6Ni–4Co/HZSM-5 will be used to upgrade true pyrolysis bio-oil rich in acids to obtain a high yield of aromatic hydrocarbons.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was supported by the National Natural Science Foundation of China (51706040) and the China Postdoctoral Science Foundation (2019M651246).References
- I. Hanif, S. M. Faraz Raza, P. Gago-de-Santos and Q. Abbas, Energy, 2019, 171, 493–501 CrossRef.
- F. Jabari, B. Mohammadi-ivatloo, M. B. Bannae-Sharifian and H. Ghaebi, Energy Convers. Manage., 2018, 169, 371–382 CrossRef.
- X. P. Li, G. Y. Chen, C. X. Liu, W. C. Ma, B. B. Yan and J. G. Zhang, Renewable Sustainable Energy Rev., 2017, 71, 296–308 CrossRef CAS.
- Q. Y. Liu, C. H. Zhang, N. Shi, X. H. Zhang, C. G. Wang and L. L. Ma, RSC Adv., 2018, 8, 13686–13696 RSC.
- B. Changmai, C. Vanlalveni, A. P. Ingle, R. Bhagat and S. L. Rokhum, RSC Adv., 2020, 68(10), 41625–41679 RSC.
- S. Y. Oh and T. C. Seo, RSC Adv., 2019, 9, 28284–28290 RSC.
- S. S. Qiang and W. C. Wang, J. Cleaner Prod., 2020, 277, 124136 CrossRef CAS.
- Q. Dong, H. Li, S. Zhang, X. Li and W. Zhong, RSC Adv., 2018, 8, 40873–40882 RSC.
- S. Oh, H. S. Choi, I. G. Choi and J. W. Choi, RSC Adv., 2017, 7, 15116–15126 RSC.
- H. Y. Zhao, X. Hu, J. X. Hao, N. Li, K. D. Zhi, R. X. He, Y. F. Wang, H. C. Zhou and Q. S. Liu, Appl. Catal., A, 2020, 591, 117378 CrossRef CAS.
- L. Atanda, G. L. L. Fraga, M. H. M. Ahmed, Z. A. Alothman, J. Na and N. Batalha, J. Hard Mater., 2020, 402, 123539 CrossRef PubMed.
- Z. Y. Luo, K. Y. Lu, Y. Yang, S. M. Li and G. X. Li, RSC Adv., 2019, 9, 31960–31968 RSC.
- X. Wang, J. P. Cao, X. Y. Zhao, S. N. Liu, X. Y. Ren and M. Zhao, Bioresour. Technol., 2019, 278, 116–123 CrossRef PubMed.
- Z. Y. Zakaria, J. Linnekoski and N. S. Amin, Chem. Eng. J., 2012, 207, 803–813 CrossRef.
- X. Niu, J. Gao, K. Wang, Q. Miao, M. Dong, G. Wang and J. Wang, Fuel Process. Technol., 2017, 157, 99–107 CrossRef CAS.
- Z. Z. Zhang, B. Du, H. Zhu, C. Chen, Y. Sun and X. Wang, Biomass Convers. Biorefin., 2023, 13, 2017–2028 CrossRef CAS.
- Y. Zhang, P. Chen and H. Lou, J. Energy Chem., 2016, 25, 427–433 CrossRef.
- X. Y. Ren, J. P. Cao, X. Y. Zhao, Z. Yang, T. L. Liu and X. Fan, Fuel, 2018, 218, 33–40 CrossRef CAS.
- Y. Zhang, X. Xu and H. Q. Jiang, J. Energy Chem., 2020, 51, 161–166 CrossRef.
- A. Veses, B. Puértolas, M. S. Callén and T. García, Microporous Mesoporous Mater., 2015, 209, 189–196 CrossRef CAS.
- Y. W. Zheng, F. Wang, X. Q. Yang, Y. B. Huang, C. Liu, Z. F. Zheng and J. Y. Gu, J. Anal. Appl. Pyrolysis, 2017, 126, 169–179 CrossRef CAS.
- S. Wang, Q. Yin, J. Guo, B. Ru and L. Zhu, Fuel, 2013, 108, 597–603 CrossRef CAS.
- R. Kumar, V. Strezov, E. Lovell, T. Kan, H. Weldekidan, J. He, B. Dastjerdi and J. Scott, Bioresour. Technol., 2019, 279, 404–409 CrossRef CAS.
- S. Y. Cheng, L. Wei, J. Julson, K. Muthukumarappan and P. R. Kharel, Energy Convers. Manage., 2017, 147, 19–28 CrossRef CAS.
- J. J. Zhang, B. Fidalgo, S. Wagland, D. K. Shen, X. L. Zhang and S. Gu, Fuel, 2019, 238, 257–266 CrossRef CAS.
- S. R. Wang, Q. Q. Yin, J. F. Guo, B. Ru and L. J. Zhu, Fuel, 2013, 108, 597–603 CrossRef CAS.
- L. Y. Qin, Y. Shao, Z. W. Hou, Y. W. Jia and E. C. Jiang, Energy Fuels, 2019, 33, 8640–8648 CrossRef CAS.
- L. Y. Qin, Y. Wu, Z. W. Hou and E. C. Jiang, Bioresour. Technol., 2020, 313, 123682 CrossRef CAS PubMed.
- L. Y. Qin, Z. W. Hou, S. H. Zhang, W. Zhang and E. C. Jiang, Electroanal. Chem., 2020, 866, 114140 CrossRef CAS.
- M. Ammar, Y. Cao, P. He, L. G. Wang, J. Q. Chen and H. Q. Li, Chin. J. Chem. Eng., 2017, 25, 1760–1770 CrossRef.
- Q. T. Cheng, B. X. Shen, H. Sun, J. G. Zhao and J. C. Liu, RSC Adv., 2019, 9, 20818–20828 RSC.
- X. Y. Ren, J. P. Cao, X. Y. Zhao, W. Z. Shen and X. Y. Wei, Anal. Appl. Pyrolysis, 2018, 130, 190–197 CrossRef CAS.
- B. Gu, J.-P. Cao, F. Wei, X.-Y. Zhao, X.-Y. Ren and C. Zhu, Fuel, 2019, 244, 151–158 CrossRef CAS.
- S. S. M. Konda, S. Caratzoulas, D. G. Vlachos and G. Vlachos Dionisios, ACS Catal., 2016, 6, 123–133 CrossRef CAS.
- X. Xu, Y. Zhang, X. Xia, F. Liang and H. Jiang, Catal. Lett., 2020, 150, 3495–3504 CrossRef CAS.
- Y. M. Chen, B. C. Qiu, Y. Liu and Y. Zhang, Appl. Catal., B, 2020, 269, 118801 CrossRef CAS.
- A. R. Stanton, K. Iisa, M. M. Yung and K. A. Magrini, J. Anal. Appl. Pyrolysis, 2018, 135, 199–208 CrossRef CAS.
- V. Paasikallio, C. Lindfors, J. Lehto, A. Oasmaa and M. Reinikainen, Top. Catal., 2013, 56, 800–812 CrossRef CAS.
- S. Y. Cheng, L. Wei and M. Rabnawaz, Fuel, 2018, 223, 252–260 CrossRef CAS.
- V. Rameshwari, K. V. Santosh, V. Shekhar, J. Wang, J. Liu and B. Jing, J. Cleaner Prod., 2021, 282, 124488 CrossRef.
- T. Chen, J. Zhang, L. L. Wang, Y. Jiao, Q. Y. Zhang, J. L. Wang, Y. Q. Chen and X. Y. Li, J. Anal. Appl. Pyrolysis, 2019, 141, 104642 CrossRef CAS.
- T. V. Larina, L. S. Dovlitova, V. V. Kaichev, V. V. V. Malakhov, T. S. Glazneva, E. A. Paukshtis and B. S. Bal'zhinimaev, RSC Adv., 2015, 97, 79898–79905 RSC.
- B. Zhao, B. H. Zhang, C. X. Lu, Z. Cai and L.-D. Li, J. Alloys Compd., 2020, 833, 155130 CrossRef CAS.
- L. Ohlin, P. Bazin, F. Thibault-Starzyk, J. Hedlund and M. Grahn, J. Phys. Chem. C, 2013, 117, 16972–16982 CrossRef CAS.
- T. A. Palankoev, K. I. Dement’ev, D. V. Kuznetsova, G. N. Bondarenko and A. L. Maximov, ACS Sustainable Chem. Eng., 2020, 8, 10892–10899 CAS.
- A. Gumidyala, T. Sooknoi and S. Crossley, J. Catal., 2016, 340, 76–84 CrossRef CAS.
- W. Jiang, J. Chu, S. Wu and L. A. Lucia, Fuel Process. Technol., 2018, 176, 221–229 CrossRef CAS.
- M. Bjørgen, Karl-Petter Lillerud, Unni Olsbye, Silvia Bordiga, and Adriano Zecchina, J. Phys. Chem., 2004, 108, 7862–7870 CrossRef.
- L. L. Xu, R. R. Zhao and W. P. Zhang, Appl. Catal., B, 2020, 279, 119389 CrossRef CAS.
- C. Matheus, L. Chagas, G. Gonzalez, E. Falabella, S. Aguiar and L. Appel, ACS Catal., 2018, 8, 7667–7678 CrossRef.
- N. shu, Y. Li and R. Liu, J. Energy Inst., 2022, 101, 111–121 CrossRef.
- Y. Xu, J. Liu, G. Ma, J. Wang, J. Lin and H. Wang, Fuel, 2018, 228, 1–9 CrossRef.
- J. Su, W. Mao, X.-C. Xu, Z. Yang, H. Li and J. Xu, AIChE J., 2014, 60, 1797–1809 CrossRef CAS.
- H. A. J. van Dijk, J. H. B. J. Hoebink and J. C. Schouten, Chem. Eng. Sci., 2001, 56, 1211–1219 CrossRef CAS.
- Y.-P. Pei, J.-X. Liu, Y.-H. Zhao, Y.-J. Ding, T. Liu, W.-D. Dong, H.-J. Zhu, H.-Y. Su, L. Yan, J.-L. Li and W.-X. Li, ACS Catal., 2015, 5, 3620–3624 CrossRef CAS.
- T.-Y. Chen, J. Su, Z. Zhang, C. Cao, X. Wang and R. Si, ACS Catal., 2018, 8, 8606–8617 CrossRef CAS.
- M. Li, J. Fu, S. Xing, L. Yang, X. Zhang and P. Lv, Appl. Catal., B, 2020, 260, 118114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07706j |
| This journal is © The Royal Society of Chemistry 2023 |