
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anticancer and in silico assessment of 8-caffeinyl-triazolylmethoxy hybrid conjugates†
Mohammad Navid Soltani Rad *a,
Somayeh Behrouz*a,
Saleh Aghajania,
Marzieh Behrouza,
Elham Zarenezhadb and
Ali Ghanbariasadc
*a,
Somayeh Behrouz*a,
Saleh Aghajania,
Marzieh Behrouza,
Elham Zarenezhadb and
Ali Ghanbariasadc
aMedicinal Chemistry Research Laboratory, Department of Chemistry, Shiraz University of Technology, Shiraz 71555-313, Iran. E-mail: soltani@sutech.ac.ir; behrouz@sutech.ac.ir; Fax: +98 71 3735 4520; Tel: +98 71 3735 4500
bNon-communicable Diseases Research Center, Fasa University of Medical Sciences, Fasa, Iran
cDepartment of Medical Biotechnology, School of Medicine, Fasa University of Medical Sciences, Fasa, Iran
First published on 19th January 2023
Abstract
In this research the synthesis, characterization, anticancer and the cytotoxicity assessments of novel 8-caffeinyl-triazolylmethoxy hybrid conjugates have been described. These compounds are the first caffeine-1,2,3-triazolyl hybrid molecules that structurally are composed of three compartments comprising caffeinyl, 1,2,3-triazolyl and N-alkyl/aryl residues. The in vitro evaluations of synthesized compounds on cancer cell lines, including two breast cancer cell lines MDA-MB-468 (ATCC HTB-22), MCF-7 (ATCC HTB-22), melanoma cell line A-375 (ATCC CRL-1619) and normal cell line HEK-293 (ATCC CRL-11268) have determined that 22c (IC50 < 12.5 μM) demonstrated potent activity against A375 and its toxicity is even stronger than methotrexate (MTX) as a standard drug. Additionally, 22c involves more selectivity than MTX regarding its non-toxicity for the HEK-293 cell line. Among the tested compounds against two breast cancer cell lines, 22f (IC50 = 136 ± 0.2 and 126 ± 0.6 μM for MCF-7 and MDA-MB-468, respectively) and 22i (IC50 = 165 ± 1.8 and 175 ± 1.4 μM for MCF-7 and MDA-MB-468, respectively) were the most potent compounds but their activities were less than MTX, moreover 22f showed more selectivity regarding its lower toxicity against HEK-293. Overall, 22f displayed general toxicity and selectivity on all tested cancer cell lines. The in silico physicochemical properties, pharmacokinetic profile, and drug likeness predictions were also carried out for all the studied compounds. Most new compounds exhibited zero violation of Lipinski's rule (RO5). A molecular docking study was also conducted to predict the binding mode and the interaction of 22c as the most active anti-melanoma entry with B-RAF V600E kinase enzyme. The docking results determined that 22c exhibited a strong binding affinity to the active site of the enzyme. These findings demonstrated 22c and 22f as potential future anticancer drug candidates.
1 Introduction
Perhaps, after infectious and cardiac diseases, cancer has been one of the most deadly diseases that has occurred throughout the history of human life. Cancer is a public health problem all over the world. This disease affects all people, young and old, rich and poor, men, women and children. Cancer is the uncontrollable growth and expansion of cells that can infect almost all tissues of the body. More than eleven million people are diagnosed with cancer every year. It is estimated that there will be 16 million new cases of cancer annually before 2020. The treatment of cancer causes a high cost to governments and peoples, for example, the total cost of cancer was €199 billion in Europe (EU-27 plus Iceland, Norway, Switzerland, and the United Kingdom) in 2018.1 The high expenditure of cancer treatment is due to various reasons; however, the most important reason for this high cost is the price of anti-cancer drugs. Despite the good advances in the discovery and use of anti-cancer drugs in recent years; nevertheless, these drugs have several shortcomings, such as low drug efficacy, high toxicity, lack of selectivity, high prices and so on. Therefore, there is still a constant need to design and generate new, cheap and effective anti-cancer drugs with specialized performance.2It is not overstating that xanthine alkaloids are the most prominent naturally occurring alkaloids recognized throughout human life.3 Natural xanthines are purine-based scaffolds which possess widespread medicinal properties that are extensively exploited in the remedy of diverse diseases.4 Amongst xanthine alkaloids, methyl xanthines comprising caffeine, theophylline, theobromine and paraxanthine are amazing since these xanthine scaffolds are well-recognized by diverse receptors or enzymes in animal cells that caused their magnificent clinical and therapeutic activity (Fig. 1).5 They are abundantly found in miscellaneous seeds, fruits, and leaves of a variety of plants, like tea, cacao beans, guarana berries and yerba matte leaves.6,7 However, caffeine is more pronounced than the other methylxanthines owing to its widespread popularity and extreme social consumption as tea, coffee and chocolate. Up to now, the parental caffeine and its derivatives are known as super nootropic molecules with remarkable clinical activity like anti-Alzheimer, anti-diabetes, anti-Parkinson, anti-cancer, anti-inflammatory, antitussives and stimulating agents for the central nervous system (CNS).8
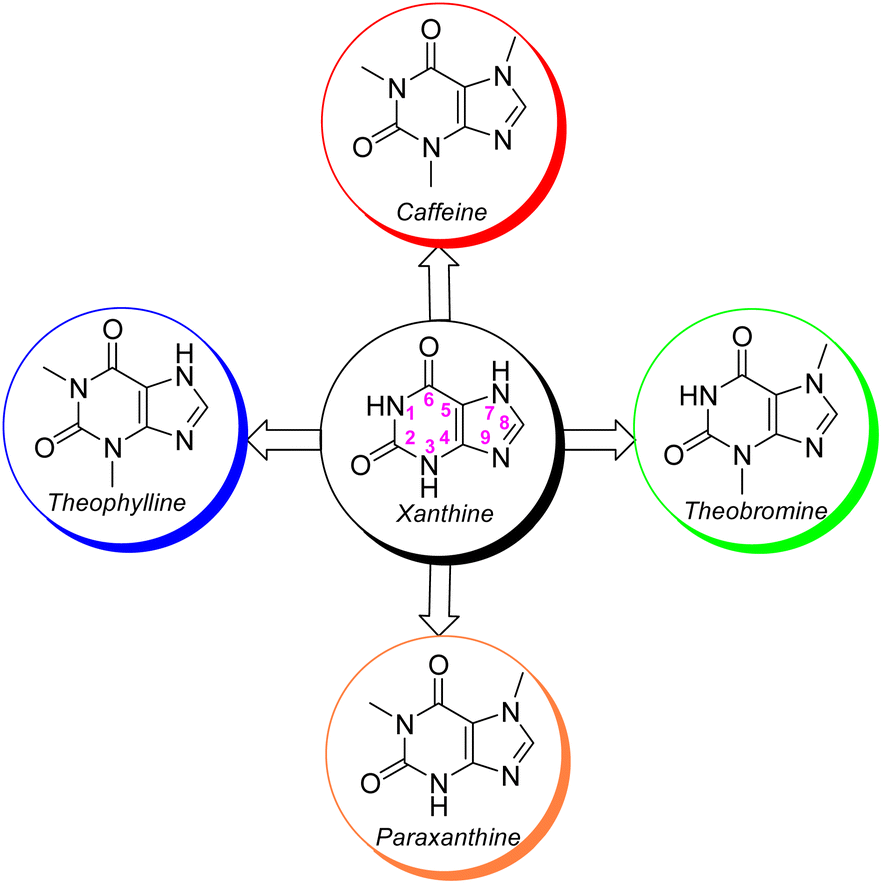 | ||
| Fig. 1 The structures of xanthine and its natural methylated derivatives. | ||
The xanthine as a starting material encompasses multiple active nucleophilic sites (N1, N3, N7 and N9 sites) for the enormous diversification reaction through the nucleophilic substitution. Nevertheless, methyl xanthines like theophylline, theobromine and paraxanthine can merely contribute to the nucleophilic substitution by N7, N1 and N3 atoms, respectively. Conversely, the nucleophilic sites in caffeine are masked by methylated nitrogens and hence the caffeine is unable to undertake the substitution reaction through its masked nitrogens. Indeed, the caffeine can be chemically altered merely via its C8 site with rare electrophiles or active species.9–12 It is worth mentioning that the alteration of methylxanthines at all available sites has emerged the impressive and brilliant pharmacological profiles reviewed in many literature.13,14 In particular, C8-modification of caffeine derivatives extensively improves the biological activities designated in Fig. 2. The most C8-modified caffeine derivatives can be acquired by two main strategies. First, the halogenation (i.e.: bromination and chlorination) followed by SNAr-type reaction via diverse nucleophiles. Second, the coupling of 1,3-dimethyl-5,6-diaminouracil with various carboxylic acids. In general, the reported caffeine's C8-modified adducts can be categorized on the basis of atom's type linked to C8. In this regard, they can be sorted into C8–C, C8–O, C8–S, C8–N caffeine derivatives. The structures of a few examples of C8–C, C8–O, C8–S, C8–N derivatives beside their corresponding biological profiles are summarized in Fig. 3. The C8–C derivatives were mainly prepared through the reaction of 1,3-dimethyl-5,6-diaminouracil with carboxylic acids bearing diverse functionalities.15–20 General structures 1–3 and 5 (Fig. 3) were prepared by this method, however for 3 with n = 0 (i.e. 8-aryl caffeine), the aryl halide and 8-caffeinyl cobalt(I) complex was used.11 Moreover, C8–C derivatives 4 can be obtained by direct reaction of caffeine, alcohol and t-butyl hydroperoxide (TBHP) under microwave irradiation.10 Regarding compounds 6–10 (C8–S),12,21–28 11–19 (C8–N),11,29–34 20 and 21 (C8–O)35–37 (Fig. 3), they were often prepared by the SNAr-type reaction of 8-bromocaffeine (8-BC) or 8-chlorocaffeine (8-CC) with the corresponding nucleophilies in a single or multistep processes.
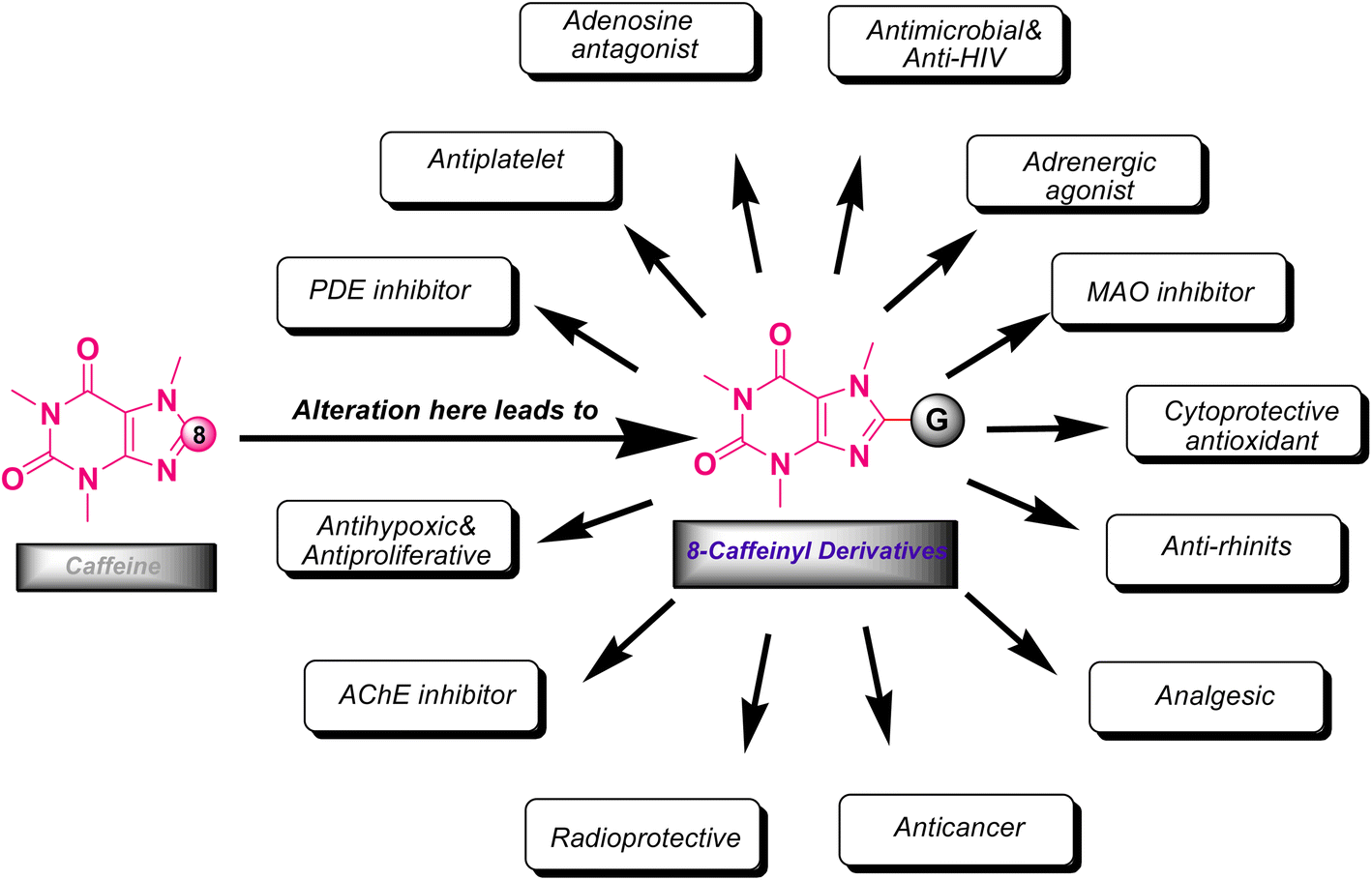 | ||
| Fig. 2 The resulting biological properties by the C8 modification of caffeine. | ||
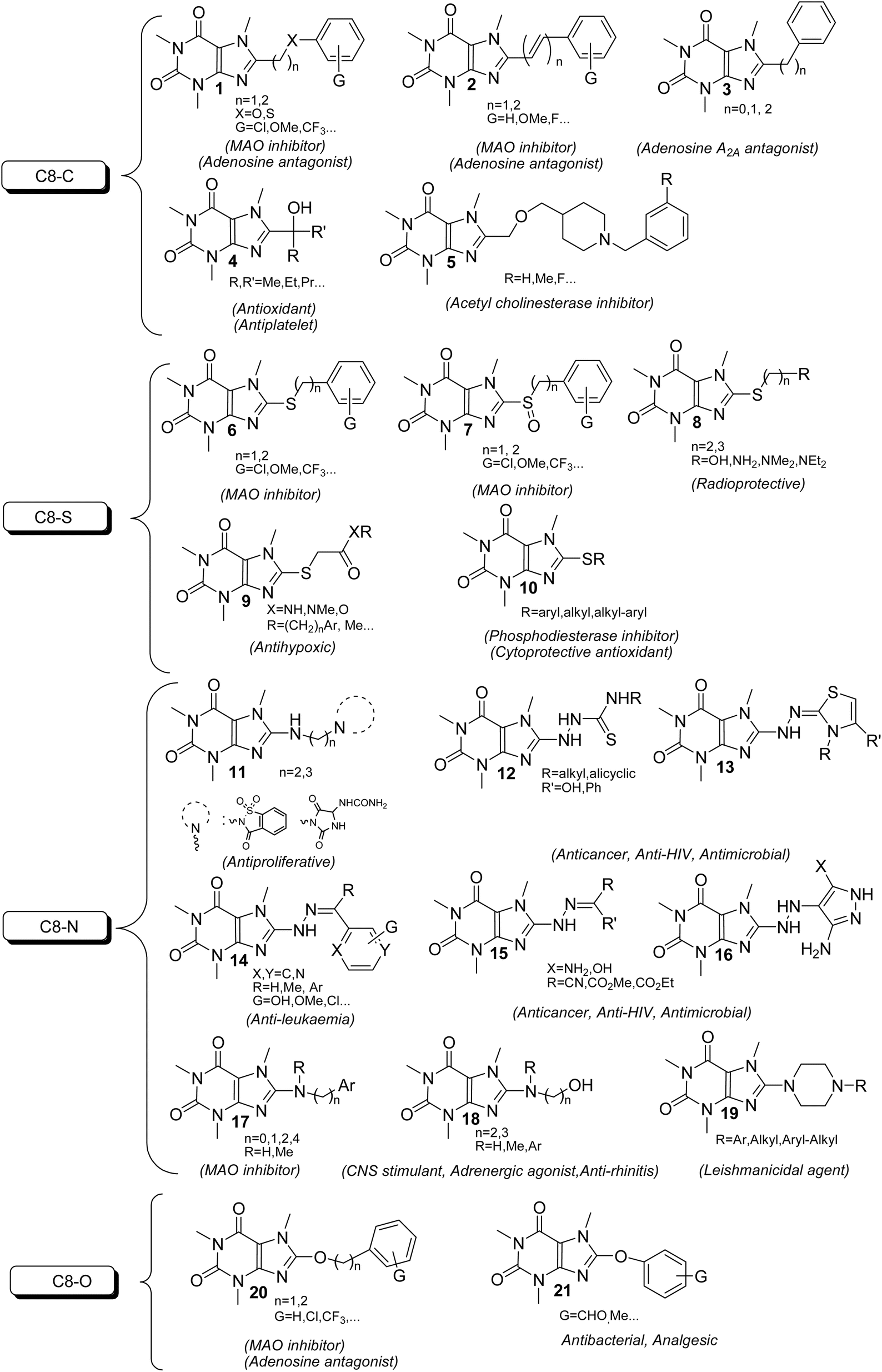 | ||
| Fig. 3 Classification of the C8-modified caffeine derivatives and their corresponding biological activity. | ||
Undoubtedly, the 1H-1,2,3-triazolyl cores are unique and significant scaffolds in medicinal chemistry. Owing to the incorporation of the 1H-1,2,3-triazolyl core into the structure of a bioactive molecule, it extensively increases its pharmacophoric strength by establishing a variety of chemical interactions.38,39 Additionally, the 1,2,3-triazolyl core is known as a main non-hydrolysable bioisostere of the amide bond regarding to its significant electronic and topological similarities.40 In this connection, the ‘Click’ Huisgen azide–alkyne cycloaddition is a reliable and efficient tool for incorporating the 1H-1,2,3-triazolyl cores into the drugs scaffold and also the conjugation of two organic molecules to acquire a hybrid molecule.41
As mentioned earlier and shown in Fig. 3, the C8-modification of caffeine with diverse organic moieties extensively improves its biological activity, however to the best our knowledge no molecule comprising 1H-1,2,3-triazolyl core was considered for conjugating with caffeinyl moiety. In this regard, hereby we would like to report the synthesis, characterization, anticancer and in silico assessment of novel 8-caffeinyl-triazolylmethoxy hybrid conjugates 22a–22j (Fig. 4).
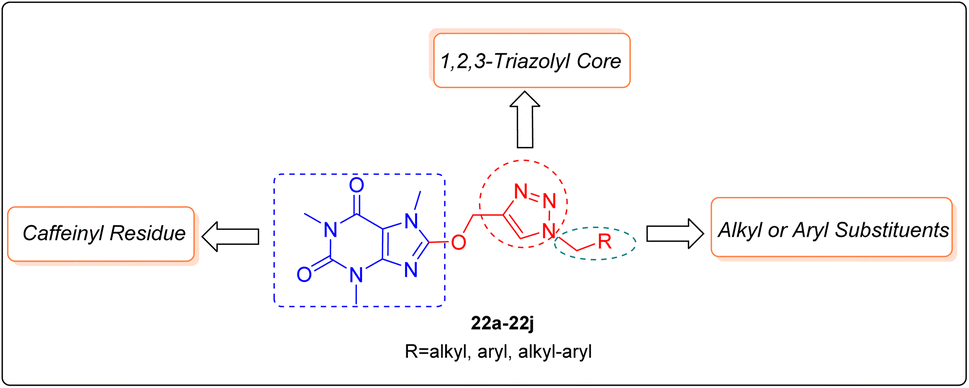 | ||
| Fig. 4 General structure of novel 8-caffeinyl-triazolylmethoxy hybrid conjugates. | ||
2 Results and discussion
2.1. Chemistry
The synthesis of 8-caffeinyl-triazolylmethoxy hybrid conjugates 22 was carried out due to Scheme 1A–D. As indicated in Scheme 1A–D, this synthesis was started by the bromination of caffeine to increase the positive charge density on C(8) for the subsequent SNAr-type reaction (Scheme 1A). The bromination of caffeine was carried out due to our previously reported procedure using NBS as a brominating reagent in a mixture of DCM and water at room temperature.27 Also the bromination of caffeine can be conducted using Br2/H2O2 in glacial HOAC25 or HBr/H2O2;24 however, none of these two current procedures were efficient for synthesis of the 8-BC. Applying the NBS method almost yielded the pure 8-BC, quantitatively. In another reaction the require alkyl azides were readily synthesized using alkyl chlorides or bromides and excess amounts of sodium azide in a mixture of acetone–water at reflux to yield the corresponding alkyl azides almost, quantitatively (Scheme 1B).42 Subsequently, to afford the 8-proparyloxy caffeine as a require alkyne, we attempted to tether the propargyl moiety to caffeinyl residue through the reaction of 8-BC and propargyl alcohol using KOH as a base in DMSO at 100 °C; however, the obtained yield was not better than 25%. The use of other organic solvents, different bases and catalysts had no considerable effect on improving the obtained yield. Thus, 23 was prepared through the ‘Click’ Huisgen alkyl-azide cycloaddition using the propargyl alcohol and desired alkyl azides in the presence of cuprous catalyst and in a mixture of THF–H2O at reflux condition for 6–12 h (Scheme 1C). Ultimately, 23 was coupled with 8-BC through SNAr-type reaction using KOH in DMSO at 100 °C to afford the title products in good yields (Scheme 1D).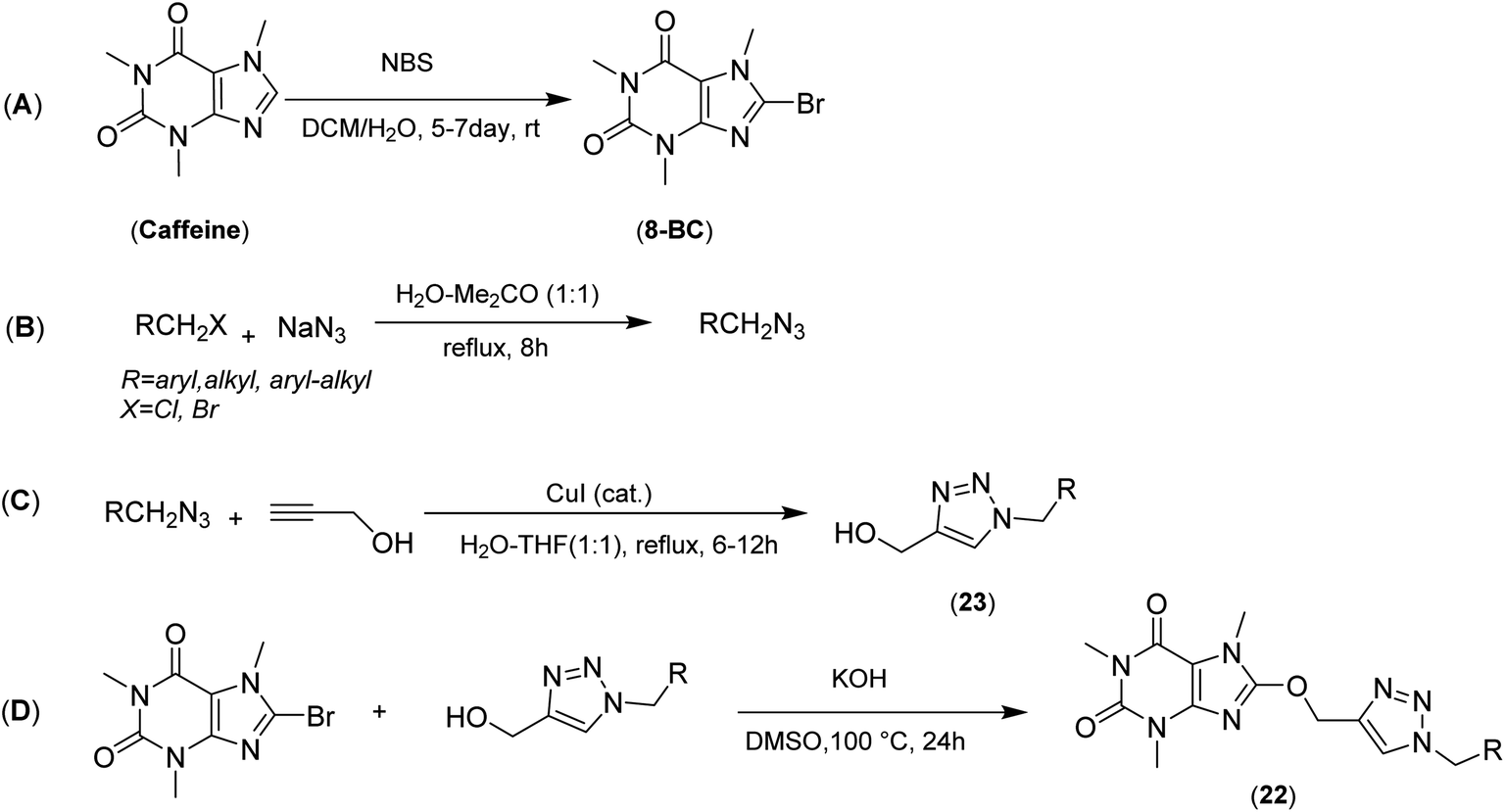 | ||
| Scheme 1 General synthetic pathway (A–D) for preparing 8-caffeinyl-triazolylmethoxy hybrid conjugates (22). | ||
The structures of all target molecules as well as corresponding yields and melting points are demonstrated in Fig. 5. As shown in Fig. 5, compounds are common in caffeinyl and triazolylmethoxy parts but they are different in alkyl residues. Compounds bearing alkyl–aryl (22a–22e) and diverse aliphatic residues having differences in carbon chain length (22f–22j) were synthesized.
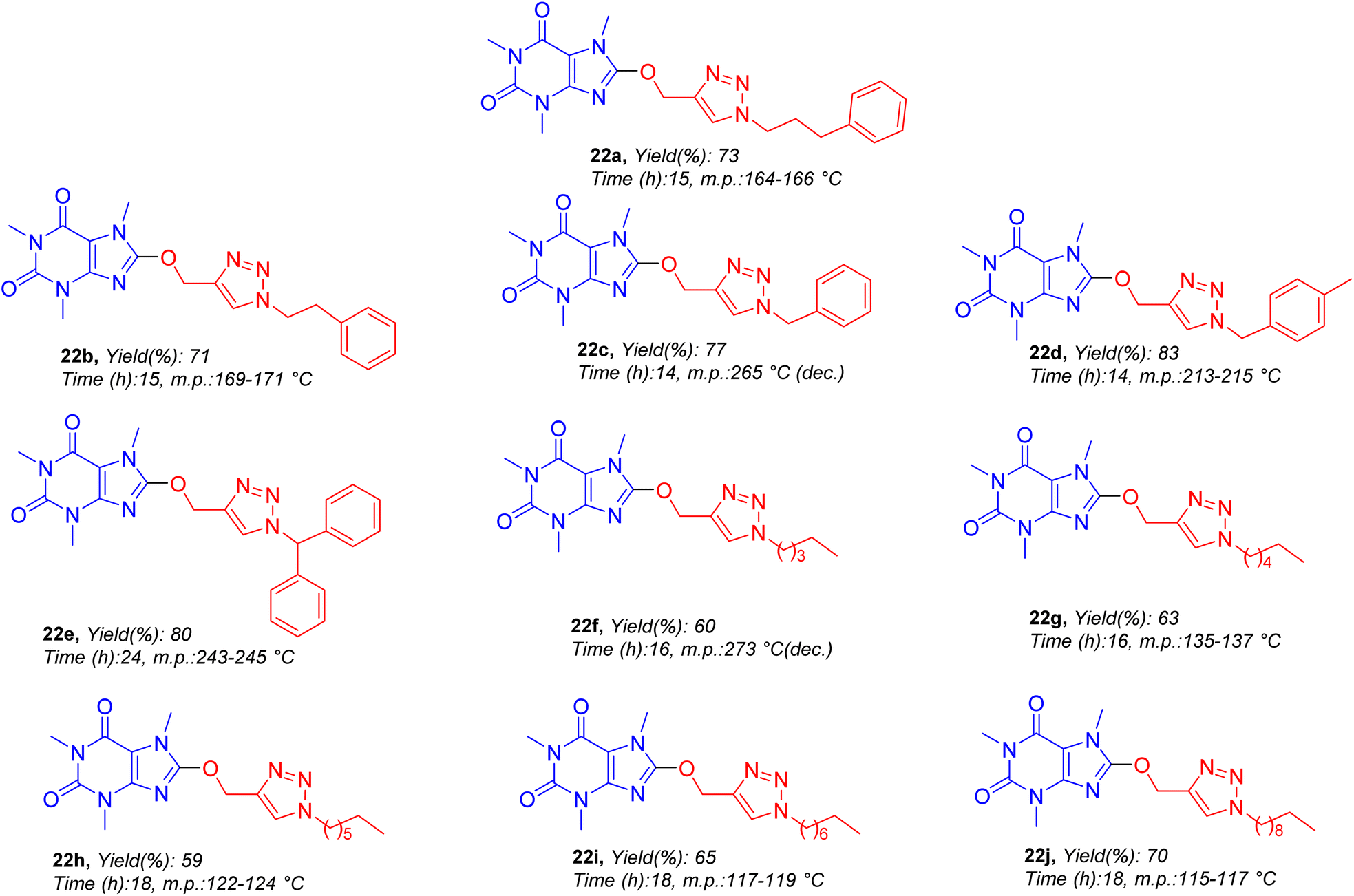 | ||
| Fig. 5 The structures, isolated yields and melting points of compounds 22a–22j. | ||
2.2. In vitro anticancer and cytotoxic activity
The World Health Organization (WHO) reported Melanoma as the most aggressive form of skin cancer that is increasing worldwide. The A375 cell line is a known human melanoma cell line derived from a metastatic melanoma.43 Breast carcinoma is the most frequent malignancy among women and the second leading cause of cancer death in them. The MDA-MB-468 cell line was isolated from women patient with metastatic adenocarcinoma of the breast44 The MCF-7 cell line was established from a pleural effusion from a 69 years-old woman at the Michigan Cancer Foundation in 1973 which is the most common model used for breast cancer. Despite its origin from the metastases of an advanced tumor, the cell line is noninvasive and represents a model of early-stage disease.45 The human embryonic kidney 293 cells (HEK 293) are a normal cell line originally derived from human embryonic kidney cells grown in tissue culture taken from a female fetus.46 The IC50 values pertaining to toxicity tests for all compounds were carried out in three replicates and the final values were reported as mean ± SD. The final values for all samples were compared with SPSS software using one-way ANOVA with a confidence interval of 95% (Table 1). As depicted in Table 1, against normal cell line (HEK-293) all compounds showed IC50 values higher than 300 μM except those of 22e, 22i, 22j and MTX. In the case of A375, 22c (IC50 < 12.5 μM) demonstrated potent activity against A375 and its toxicity is even stronger than MTX as a standard drug. Regarding the ineffectiveness of 22c (IC50 < 300 μM) on HEK-293 and also the remarkable toxicity of MTX (IC50 = 38 ± 1.6 μM) against HEK-293, thus 22c proved to have more selectivity compared to MTX. Additionally, except those of 22b, 22g, 22h and 22j, other compounds were toxic to A375 but their toxicities were considerably less than 22c and MTX. In the case of MCF7 and MDA-MB-468, 22a, 22f and 22i displayed toxicity against both cell lines while 22e was merely effective on MCF7. Among tested compounds against breast cancer cell lines 22f and 22i were the most potent compounds, however 22f showed more selectivity regarding its less toxicity against HEK-293. In general, all compounds were less toxic than MTX against both tested breast cancer cell lines. In overall, 22f displayed general toxicity and selectivity on all tested cancer cell lines.| Compound | Cell line [IC50(μM)] | |||
|---|---|---|---|---|
| A375 | MCF7 | MDA-MB-468 | HEK-293 | |
| a Methotrexate. | ||||
| 22a | 179 ± 1.2 | 268 ± 0.2 | 256 ± 1.4 | >300 |
| 22b | >300 | >300 | >300 | >300 |
| 22c | <12.5 | >300 | >300 | >300 |
| 22d | 194 ± 1.6 | >300 | >300 | >300 |
| 22e | 286 ± 0.2 | 284 ± 0.2 | >300 | 284 ± 1.4 |
| 22f | 160 ± 1.5 | 136 ± 0.2 | 126 ± 0.6 | >300 |
| 22g | >300 | >300 | >300 | >300 |
| 22h | >300 | >300 | >300 | >300 |
| 22i | 146 ± 1.5 | 165 ± 1.8 | 175 ± 1.4 | 194 ± 1.2 |
| 22j | >300 | >300 | >300 | 194 ± 1.2 |
| MTXa | 32 ± 2.2 | 30 ± 2.9 | 37 ± 1.9 | 38 ± 1.6 |
The viability percentages for examined cell lines versus different concentrations of 22a–22j as well as MTX are shown in Fig. 6A–D. A p-value less than 0.05 (typically ≤0.05) is statistically significant that shown with stars in Fig. 6A–D.
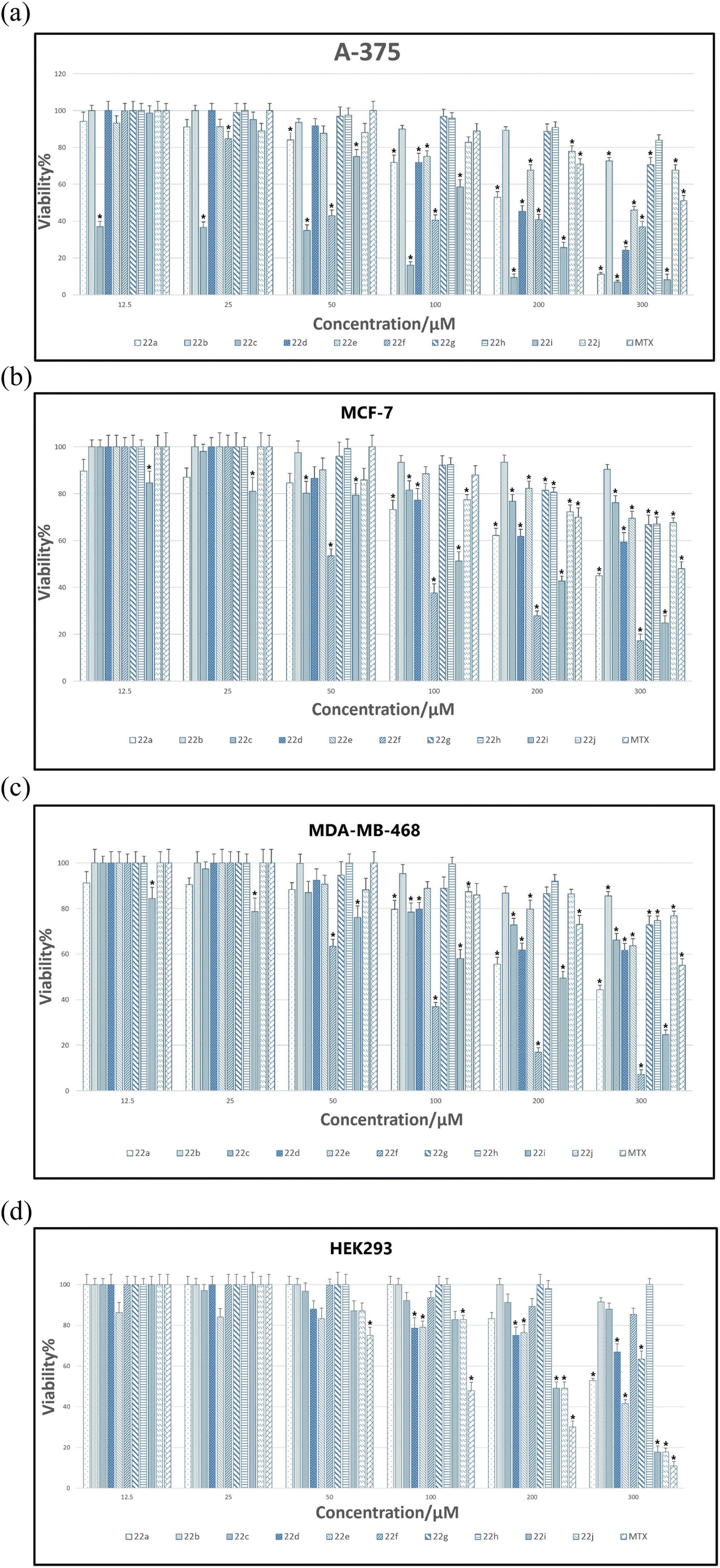 | ||
| Fig. 6 (A) The viability assessment for 22a–22j and MTX against A375. (B) The viability assessment for 22a–22j and MTX against MCF7. (C) The viability assessment for 22a–22j and MTX against MDA-MB-468. (D) The viability assessment for 22a–22j and MTX against HEK-293. | ||
As it is indicated in Fig. 6A, for A375, the lowest viability values and hence the highest toxicity are related to 22c compared to all synthesized compounds. It is also worthy to mention that the viability percentage of 22c at lowest concentration (i.e.: 12.5 μM) is even lower than that of MTX which endorses the higher toxicity of 22c against A375 cancer cell line.
The viability of all compounds and MTX was also assessed for both MCF7 and MDA-MB-468 cell lines (Fig. 6B and C). As determined, among tested compounds, 22f demonstrated lower viability at higher concentrations compared to all entries but its viability was higher than MTX at all tested concentrations. Fig. 6D indicates the viability percentage for HEK-293. As it is well indicated the lowest viability was pertained to MTX at all tested concentrations; however, for two most effective compounds 22c and 22f, the viability was higher than MTX, particularly at lower concentrations which indicate the selectivity of 22c and 22f against HEK-293.
Recently Shahzadi and coworkers have synthesized a series of theophylline-7-acetic acid (acefylline)-derived 1,2,4-triazole hybrids having N-phenyl acetamide moieties and tested for their inhibitory (in vitro) potential against two cancer cell lines, A549 (lung) and MCF-7 (breast), using MTT assay.47 They determined that their most potent compound against MCF-7 breast cancer cell line had the viability value of 31.76 ± 3.16 at effective concentration (100 μg μL−1), whereas current 22f as the most potent entry against MCF-7, demonstrated the viability value of 17.0 ± 3.0 at the most effective concentration of 300 μM (i.e.: 0.12 μg μL−1). This result has clearly shown that 22f inhibited the MCF-7 cells at a much lower concentration than the most potent compound reported by Shahzadi et al.
Methotrexate (MTX, amethopterin) as well as aminopterin (4-aminopteroic acid) are known as 4-amino derivative of folic acid which are different only in a methyl moiety. In fact, these two compounds, which have a very close structure to folic acid, inhibit the dihydrofolate reductase (DHFR) enzyme in a competitive interaction with folic acid.48 This enzyme plays a key role in the reduction of dihydrofolate to tetrahydrofolate (coenzyme F), and its inhibition prevents the biosynthesis of DNA and RNA and ultimately the protein.49 Like folic acid, MTX and aminopterin are composing of three parts comprising pteridine, p-aminobenzoic acid (PABA) and glutamic acid residues. The main pharmacophoric residue in folic acid, MTX and aminopterin is pteridine core which is closely related to xanthine alkaloids and biologically is synthesized from guanine. In same circumstance to MTX and aminopterin, 8-caffeinyl-triazolylmethoxy hybrid conjugates are compose of three parts comprising (i) caffeinyl core as a methyl xanthine that structurally resembles to pteridine, (ii) 1,2,3-triazolyl core is a bioisostere of amide that can mimic the PABA-amide residue in MTX and aminopterin, and ultimately (iii) diverse alkyl or aryl-alkyl residues in 8-caffeinyl-triazolylmethoxy hybrid conjugates that can be considered as a successor for glutamic acid in MTX and aminopterin. Thus, owing to structural similarities of title compounds with MTX and aminopterin, it is proposed that 8-caffeinyl-triazolylmethoxy hybrid conjugates potentially inhibits DHFR enzyme and confines the biosynthesis of DNA (Fig. 7).
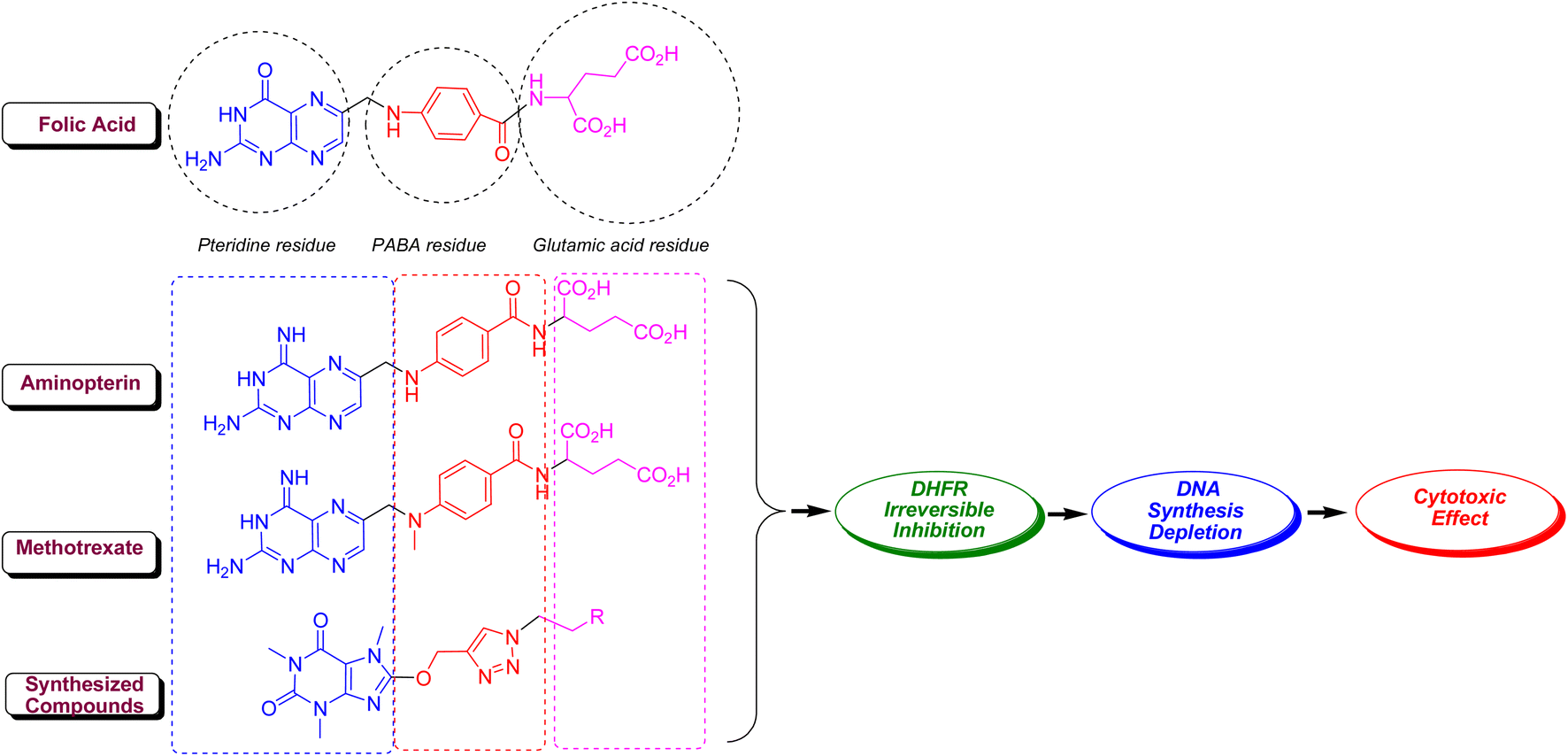 | ||
| Fig. 7 The possible anticancer mechanism of 8-caffeinyl-triazolylmethoxy hybrid conjugates. | ||
2.3. In silico assessments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) value ≤ 5. The in silico physiochemical and pharmacokinetic profile for 22a–22j and MTX was assessed using SwissADME online software,52 OSIRIS Property Explorer,53 and preADMET online server.54 The results of these studies are indicated in Table 2 and 3.
P) value ≤ 5. The in silico physiochemical and pharmacokinetic profile for 22a–22j and MTX was assessed using SwissADME online software,52 OSIRIS Property Explorer,53 and preADMET online server.54 The results of these studies are indicated in Table 2 and 3.
| Compd. | Mw. a | nRB b | nHBD c | nHBA d | cLogP e |
TPSA f | Drug likeness | Drug score |
|---|---|---|---|---|---|---|---|---|
| a Molecular weight.b Number of rotatable bonds.c Number of hydrogen bond donors.d Number of hydrogen bond acceptors.e Logarithm of octanol–water partition coefficient (logP).f Topological polar surface area (Å2).g Methotrexate. |
||||||||
| 22a | 409.44 | 7 | 0 | 6 | 1.45 | 101.76 | −1.87 | 0.46 |
| 22b | 395.42 | 6 | 0 | 6 | 1.00 | 101.76 | −0.74 | 0.56 |
| 22c | 381.39 | 5 | 0 | 6 | 0.66 | 101.76 | −1.12 | 0.54 |
| 22d | 395.42 | 5 | 0 | 6 | 1.00 | 101.76 | −2.32 | 0.46 |
| 22e | 457.48 | 6 | 0 | 6 | 2.45 | 101.76 | −1.39 | 0.29 |
| 22f | 361.40 | 7 | 0 | 6 | 0.92 | 101.76 | −6.46 | 0.44 |
| 22g | 375.43 | 8 | 0 | 6 | 1.37 | 101.76 | −10.88 | 0.43 |
| 22h | 389.45 | 9 | 0 | 6 | 1.83 | 101.76 | −16.02 | 0.42 |
| 22i | 403.48 | 10 | 0 | 6 | 2.28 | 101.76 | −18.89 | 0.40 |
| 22j | 431.53 | 12 | 0 | 6 | 3.19 | 101.76 | −18.89 | 0.35 |
| MTX g | 454.44 | 10 | 5 | 9 | 1.23 | 210.54 | −7.09 | 0.22 |
| Compd. | LogS |
HIA a | LogKp b |
P-gp c | BBB permeability d | CYP2D6 | CYP3A4 | Carcino. e | hERG inhibition f |
|---|---|---|---|---|---|---|---|---|---|
| a Human intestinal absorption (%).b skin permeability (logKp, cm h−1).c P-glycoprotein.d Blood–brain barrier permeability (C. brain/C. blood).e Carcinogenicity (mouse).f Human ether-a-go-go related gene channel.g Methotrexate. S = substrate. WS = weak-substrate. NS = non-substrate. I = inhibitor. NI = non-inhibitor. NC = non-carcinogenic. |
|||||||||
| 22a | −3.30 | 99.28 | −4.13 | NS/I | 0.16 | NS/NI | S/NI | NC | Medium |
| 22b | −3.07 | 99.05 | −4.23 | S/I | 0.12 | NS/NI | S/NI | NC | Medium |
| 22c | −2.88 | 98.77 | −4.30 | S/I | 0.11 | NS/NI | S/NI | NC | Medium |
| 22d | −3.18 | 99.05 | −4.31 | NS/I | 0.13 | NS/NI | S/NI | NC | Medium |
| 22e | −4.40 | 99.37 | −3.51 | S/I | 0.19 | NS/NI | S/NI | NC | Medium |
| 22f | −2.58 | 95.94 | −4.43 | S/I | 0.12 | NS/NI | S/NI | NC | Medium |
| 22g | −2.93 | 96.61 | −4.37 | NS/I | 0.13 | NS/NI | S/NI | NC | Medium |
| 22h | −3.28 | 97.20 | −4.30 | NS/I | 0.15 | NS/NI | S/NI | NC | Medium |
| 22i | −3.63 | 97.71 | −4.22 | NS/I | 0.19 | NS/NI | S/NI | NC | Medium |
| 22j | −4.33 | 98.53 | −4.03 | NS/I | 0.14 | NS/NI | WS/NI | NC | Medium |
| MTX g | −1.19 | 36.61 | −4.63 | S/NI | 0.04 | NS/NI | WS/I | NC | High |
As shown in Table 2, the molecular weights of 22a–22j are between 361.40 and 457.48 Dalton (<500 Dalton). The numbers of rotatable bonds (nRB) for all synthesized compounds except 22j are between 5–10. The number of hydrogen bond donors (nHBD) and hydrogen bond acceptors (nHBA) for 22a–22j are also obeyed from RO5. The lipophilicity parameter of a molecule as one of the most significant features of RO5 is characterized by clogP. The calculated clogP for compounds 22a–22j are between 0.66–3.19. Therefore, all title compounds conform to RO5. The drug transport character can be predicted by total polar surface area (TPSA) which is the summation of surfaces of polar atoms in a molecule. The TPSA contents of most approved drugs are less than 140 Å2. The calculated TPSA contents for all synthesized compounds are less than 140 Å2 which express likely appropriate oral absorption or membrane permeability. A parameter computed by OSIRIS Property Explorer software is drug likenesses which are negative for all compounds as well as MTX. Using the above calculated parameters, the software is able to give the drug score which predicts the chance of a molecule to be selected as a drug candidate. The analysis of computed data indicated that the minimum and maximum drug score values of synthesized compounds belong to 22e and 22b, respectively.
The pharmacokinetic profile of 22a–22j including absorption, distribution, metabolism and toxicity properties are also depicted in Table 3. The aqueous solubility (logS), human intestinal absorption (HIA), skin permeability (logKp), and P-glycoprotein (P-gp) substrate and/or inhibitor are important parameters for prediction of the absorption profile of a drug candidate. The aqueous solubility (logS) of numerous approved and traded drugs is >–4. Due to obtained results, all synthesized compounds except of 22e and 22j may exhibit the desirable water solubility. While the human intestinal absorption of MTX is low; however, all compounds may involve more than 95% human intestinal absorption which would be very ideal. Skin permeability (also known as transdermal delivery) is an important factor in drug delivery which describes the significance of skin absorption. All compounds display skin permeability similar to MTX. P-glycoprotein (P-gp) is a drug transporter which excretes its substrates from the cells. While induction of P-gp function by its substrate reduces the bioavailability of that chemical; however, its inhibition may cause the enhancement in drug bioavailability. According to the obtained data, all compounds are P-gp inhibitors. Additionally, all compounds except those of 22b, 22c, 22e, 22f and MTX are predicted to be the non-substrate of P-gp.
The blood–brain barrier (BBB) permeability indicates the distribution of a compound between the blood (as the most hydrophilic) and the brain (as the most lipophilic) tissues. All compounds possess low BBB permeability which suggests that none of them can easily cross this barrier and thus does not lead to neurotoxicity. Cytochrome P450s, especially CYP2D6 and CYP3A4, play significant roles for drug metabolism. The results have indicated that 22a–22j are non-inhibitor of either CYP2D6 or CYP3A4. MTX acts as non-inhibitor of CYP2D6 and inhibitor of CYP3A4. In addition, all compounds are substrates of CYP3A4 which indicates that they can be metabolized in the liver. Also, the results illustrated that 22a–22j as well as MTX cannot lead to carcinogenicity in mice. Human ether-a-go-go related gene channel (hERG) inhibitors may lead to lengthening the QT and cardiac side effects. As depicted in Table 2, a medium risk of hERG inhibition potency is predicted for 22a–22j while MTX showed high risk of hERG inhibitory activity. Taken all together, 22c can be considered as a promising future drug candidate.
It is reported that vemurafenib is stabilized in the binding site through the hydrogen bonds, π–π* and π–cation interactions as well as the hydrophobic and van der Waals interactions with the residues of amino acids present in the active site of B-RAF V600E kinase enzyme.60 The analysis of the docking protocol revealed that there are four conventional hydrogen bonds between vemurafenib and enzyme through Gln530, Cys532, Phe595, and Gly596. The π–π* stacking interactions were observed between Trp531 and Phe583 with a heterocyclic ring of vemurafenib. The side chain of Lys483 is involved in π–cation interaction with the ligand. Additionally, the hydrophobic and van der Waals interactions of vemurafenib with the enzyme were created by the side chains of Ile463, Ala481, Lys483, Leu505, Leu514, Trp531, Cys532, Phe583, and Phe595.60 As like vemurafenib, 22c is delightfully located in the same binding site and mimics the key interactions with B-RAF V600E kinase enzyme. An overlay view of conformations of co-crystallized vemurafenib, redocked vemurafenib, and 22c at the binding site of B-RAF V600E is shown in Fig. 8.
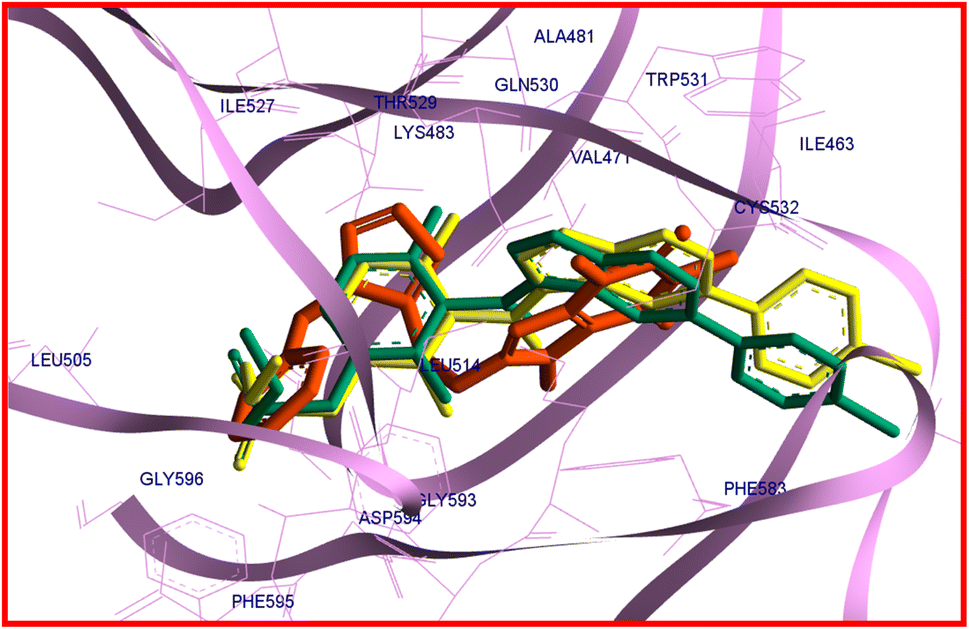 | ||
| Fig. 8 The overlay view of conformations of co-crystallized vemurafenib (green), redocked vemurafenib (yellow), and 22c (orange) at the binding site of B-RAF V600E (PDB code: 3OG7). | ||
Compound 22c provides four hydrogen bond interactions with the active site of B-RAF V600E kinase enzyme including one carbon–hydrogen bond between Gln530 and N(3)-methyl of caffeine, one conventional hydrogen bond between Cys532 and the oxygen atom of 22c which is located on the C(2) atom of caffeine, one π–donor hydrogen bond between Thr529 and triazole ring, and one π–donor hydrogen bond between Asp594 and phenyl ring of benzyl group. The three-dimensional (3D) docked conformation of 22c including hydrogen bonds and its two-dimensional (2D) docked conformation in the active site of enzyme are displayed in Fig. 9 and 10, respectively. The side chains of Trp531, Phe583, and Phe595 were involved in the π–π* interactions with the caffeinyl moiety and phenyl ring of 22c. The Ile463, Leu505, Leu414, Val471, Ala481, Lys483, Trp531, Cys532, and Phe583 create a hydrophobic pocket to stabilize 22c in the active site of enzyme. The van der Waals interactions were also observed between 22c and the amino acids of active site including Phe468, Val482, Ile513, Val528, Ser535, Asn581, Lys591, Ile592, Gly593, and Gly596. The calculated ΔG values for vemurafenib and 22c were −160.41 and −151.02 kcal mol−1, respectively. The total hydrogen bond energies for vemurafenib and 22c were −9.50 and −3.24 (kcal mol−1), respectively. These results indicated that interactions such as π–π*, the hydrophobic and the van der Waals interactions are more important than hydrogen bonding interactions for stabilization of 22c in the active site of B-RAF V600E kinase enzyme.
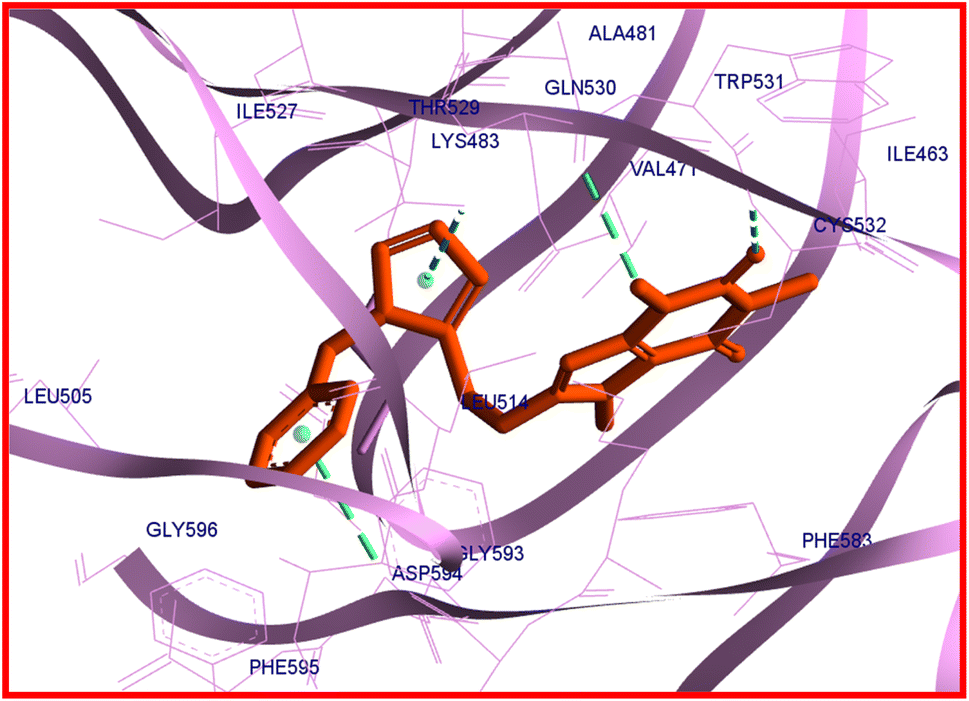 | ||
| Fig. 9 3D docked conformation and hydrogen bonds (green) of 22c with B-RAF V600E kinase enzyme. | ||
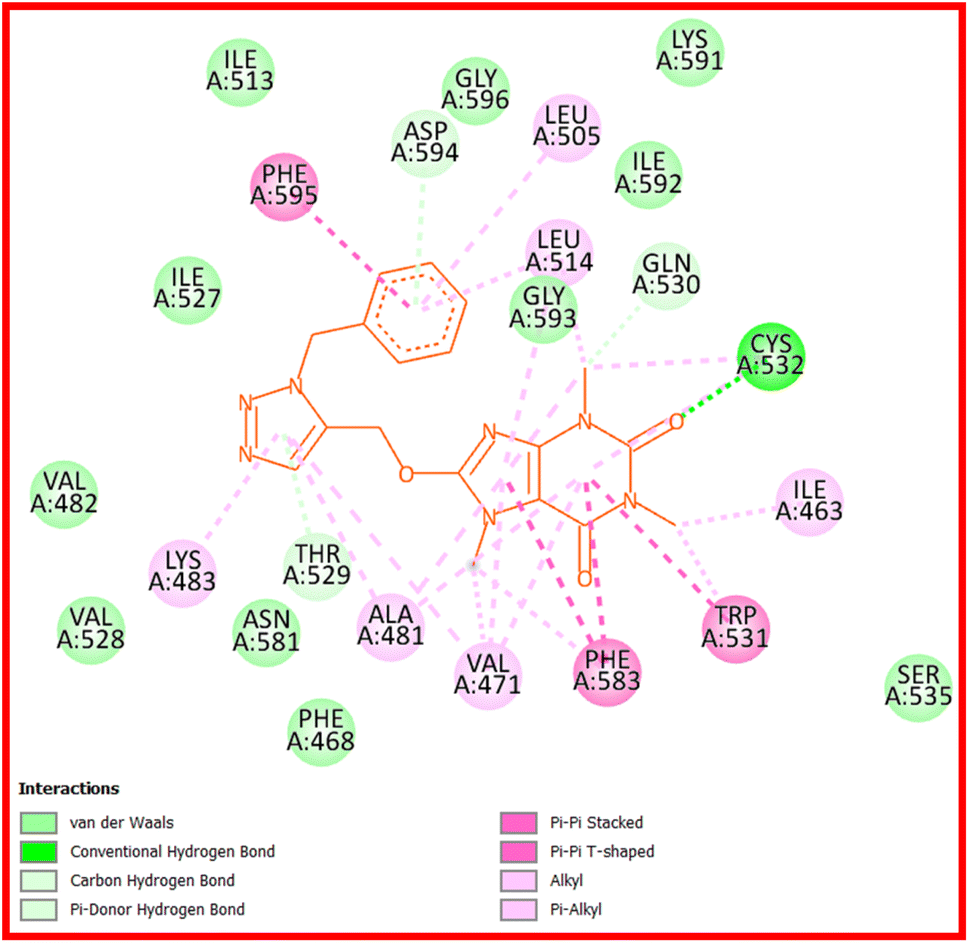 | ||
| Fig. 10 2D docked conformation of 22c at the active site of B-RAF V600E kinase enzyme. | ||
The molecular electrostatic potential (MEP) is a useful parameter to define the susceptible regions of a molecule to electrophilic or nucleophilic interaction with the surface of enzymes and receptors.61 In this regard, MEP of 22c was obtained using the DFT method at the B3LYP/6-31+G** level of theory (Fig. 11). As shown in Fig. 10, the atoms with the positive electrostatic potentials are depicted in blue and the regions carrying the negative electrostatic potentials are displayed in red. The negative electrostatic potentials are located around the triazole ring and some heteroatoms of caffeine moiety. Thus, these regions have high electron density and could interact with the electrophilic groups of B-RAF V600E kinase enzyme as confirmed by docking analysis.
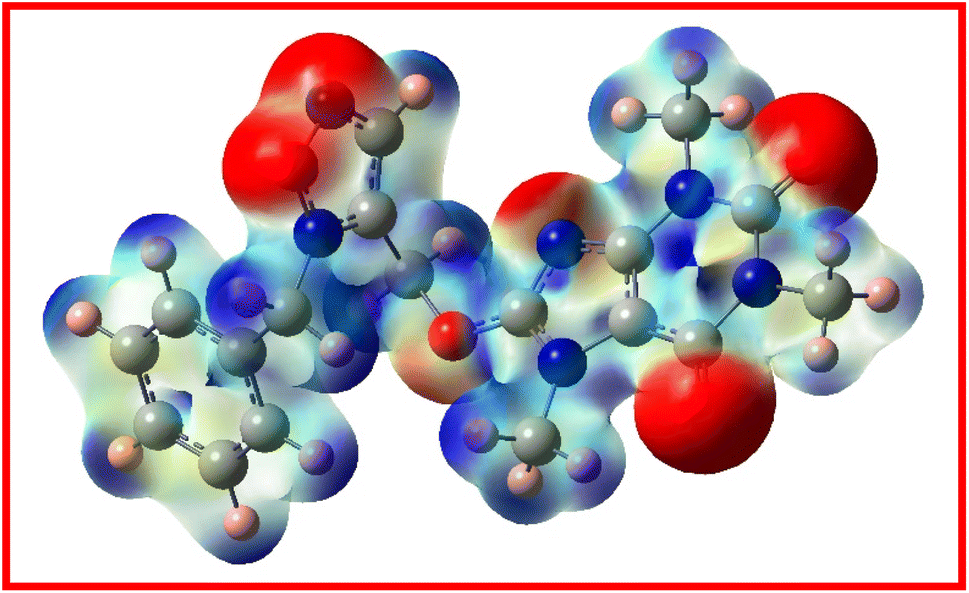 | ||
| Fig. 11 MEP of 22c. | ||
3 Conclusion
In conclusion, it was explained the design and synthesis of the new 8-caffeinyl-triazolylmethoxy hybrid conjugates. These compounds were assessed for the in vitro anticancer and the cytotoxicity against two breast cancer cell lines MDA-MB-468 (ATCC HTB-22), MCF-7 (ATCC HTB-22), melanoma cell line A-375 (ATCC CRL-1619) and normal cell line HEK-293 (ATCC CRL-11268). The results have determined that 22c displayed the highest and lowest toxicity against A375 and HEK-293 compared to methotrexate (MTX) as a standard drug, respectively. Compounds 22f and 22i have demonstrated the most potent activities against MCF-7 and MDA-MB-468 but their activities were weaker than MTX. The in silico studies were performed using the SwissADME online software, the OSIRIS Property Explorer and the preADMET online server to determine the physicochemical properties and pharmacokinetic profile. The obtained data indicated that most designated compounds were matched with established rules such as Lipinski's rule of five (RO5) and etc. The molecular docking study was also conducted by the Molegro Virtual Docker (MVD 6.0) software to predict the binding mode and the interaction of 22c as the most active anti-melanoma compound with B-RAF V600E kinase enzyme. The docking results determined that 22c exhibited a strong binding affinity to the active site of the enzyme.4 Experimental
4.1. Materials and cell lines
Pasteur Institute of Iran supplied the used cancer cell lines, including two breast cancer cell lines MDA-MB-468 (ATCC HTB-22), MCF-7 (ATCC HTB-22), melanoma cell line A-375 (ATCC CRL-1619) and normal cell line HEK-293 (ATCC CRL-11268). Phosphate-buffered saline (PBS) tablets, tetrazolium salt, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazo-lium bromide (MTT) were purchased from Sigma-Aldrich (USA). Penicillin streptomycin, trypsin, dimethyl sulfoxide (DMSO), and Dulbecco's Modified Eagle's Media (DMEM) cell culture medium were obtained from Shellmax (China). Fetal bovine serum (FBS) was supplied from Gibco (USA).4.2. Determination of the anticancer activity
The anticancer activity of 10 new compounds and Methotrexate (MTX) were investigated using MTT assay.62 The various cell lines were cultured in DMEM complete medium (containing FBS 10% and penicillin-streptomycin 1%) and incubated at 37 °C air (95%) and CO2 (5%). Cells (MDA-MB-468, MCF-7, A-375 and HEK-293) were separated by trypsin; after that; they were seeded into 96-well cell culture plates (separately) at a cell density of 5000 cells per well and incubated for 24 h. The culture media was discarded, cells were exposed to serial dilutions of MTX or the tested compounds in the 12.5 to 300 μM range (100 μL per well). After incubation (24 h, 37 °C), the plates' content was discarded, and wells were replaced with 100 μL PBS to wash. Then, 100 μL MTT solution (0.5 mg. mL−1) was added to each well and incubated for another 4 h. In continuation, 100 μL per well DMSO was added to dissolve formed formazan crystals. In each plate, six wells were considered as the control group, which was filled with 200 μL per well of DMEM. Finally, the absorbance (A) was measured at 570 nm on a plate reader (ELISA Plate Reader). The cell viability at each concentration was calculated by eqn (1). IC50 values (μM) were determined from the dose response curves as the concentration of the compound caused a 50% decrease in MTT reduction compared to the control (DMSO) using Prism 7 (GraphPad).
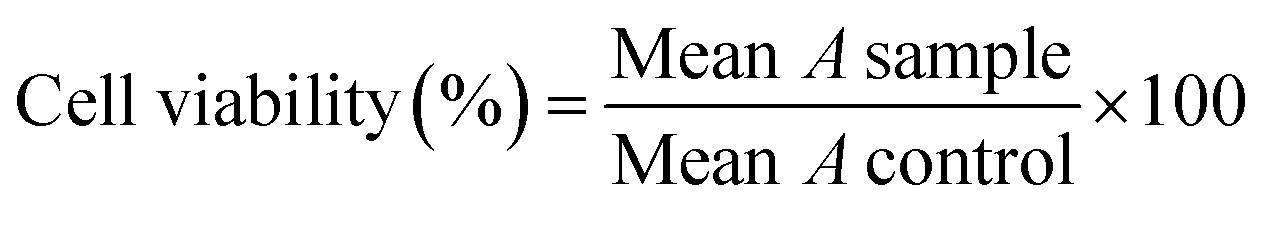 | (1) |
4.3. Chemistry general
All chemicals were purchased from Merck and/or other chemical vendors and straightforwardly applied without further purifications. The 8-bromocaffeine (8-BC) was synthesized via our previously explained procedure in the literature.27,34 The Reactions were monitored by TLC using SILG/UV 254 silica-gel plates. The purifications by a short column chromatography were done on the silica gel 60 (0.063–0.200 mm, 70–230 mesh; ASTM). The melting points are measured on the Electrothermal IA 9000 in open capillary tubes and they are uncorrected. The PerkinElmer 240 B micro-analyzer, Shimadzu GC/MS-QP 1000-EX apparatus (m/z; rel.%), and the Shimadzu FT-IR-8300 spectrophotometer were used to obtain the elemental analyses, GC/MS, and IR spectra, respectively. 1H and 13C-NMR spectrum was recorded on the Brüker Avance-DPX-300 spectrometer operating at 300/75 MHz, respectively. Chemical shifts are given in δ relative to tetramethylsilane (TMS) as an internal standard, coupling constants J are given in Hz. The abbreviations used for 1H-NMR signals are: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad and etc.4.4. General procedure for synthesis of alkyl azides
Alkyl azides were made according to the established method reported in the literature424.5. General procedure for synthesis of (1-alkyl-1H-1,2,3triazol-4-yl)-methanol (23)
In a double-necked round bottom flask (100 mL), it was added a mixture of propargyl alcohol (0.56 g, 10 mmol), desire alkyl azide (12 mmol) and CuI (0.05 g, 0.26 mmol) in THF/H2O (1:1 V/V, 30 mL). The reaction vessel was stirred at reflux until TLC monitoring indicates no further progress of the reaction (6–12 h). Then, the catalyst was separated (sintered glass) and the filtrate was evaporated in vacuo. Afterward, the residue was diluted in CHCl3 (200 mL) and washed with H2O (3 × 100 mL). Finally, the obtained crude product was pure enough that it was used for the subsequent reaction without requiring further purification.
4.6. General procedure for synthesis of 8-caffeinyl-triazolylmethoxy hybrid conjugates (22a–22j)
In a double-necked round bottom flask (100 mL), it was added a mixture of 8-BC (2.73 g, 10 mmol), desire 23 (12 mmol), and KOH (0.56 g, 10 mmol) in DMSO (15 mL). The reaction vessel was stirred at 100 °C until TLC monitoring indicates no further progress of the reaction (24 h). Then, the reaction was diluted in distilled water and the organic phase was extracted by CHCl3 (3 × 100 mL). Finally, the obtained crude product was purified by a simple recrystallization or a short column chromatography using the solvents described below.:10) afforded the pure product as white solid (2.98 g, 73%); m. p. 164–166 °C; IR (KBr): 1150, 1476, 1591, 1690, 1720, 2974, 3100 cm−1.1H NMR (300 MHz, CDCl3) δppm = 2.27–2.32 (m, 2H, PhCH2CH2), 2.65–2.67 (m, 2H, PhCH2), 3.10 (s, 3H, NCH3), 3.22 (s, 3H, NCH3), 3.45 (s, 3H, NCH3), 4.41 (br. s, 2H, NCH2), 5.84 (s, 2H, OCH2), 7.16–7.30 (m, 5H, aryl), 7.71 (s, 1H, C(5)-H of triazole). 13C NMR (75 MHz, CDCl3) δppm = 18.84, 28.18, 28.97, 33.28, 34.79, 53.49, 73.45, 105.79, 121.51, 127.03, 127.81, 128.85, 137.65, 142.50, 149.78, 151.30, 152.57, 155.85. MS (EI): m/z (%) = 409 (20.8) [M+]. Anal. Calc. for C20H23N7O3: C, 58.67; H, 5.66; N, 23.95; found: C, 58.81; H, 5.84; N, 24.06.:10) afforded the pure product as creamy solid (2.80 g, 71%); m. p. 169–171 °C; IR (KBr): 1163, 1482, 1594, 1686, 1715, 2956, 3078 cm−1.1H NMR (300 MHz, CDCl3) δppm = 2.56 (t, J = 7.2 Hz, 2H, PhCH2), 3.35 (s, 3H, NCH3), 3.52 (s, 3H, NCH3), 3.62 (s, 3H, NCH3), 4.44 (t, J = 6.9 Hz, 2H, NCH2), 6.23 (s, 2H, OCH2), 7.17–7.32 (m, 5H, aryl), 8.39 (s, 1H, C(5)-H of triazole). 13C NMR (75 MHz, CDCl3) δppm = 19.19, 28.46, 31.91, 35.21, 58.05, 73.53, 104.79, 121.66, 126.99, 128.60, 129.54, 140.42, 143.20, 150.13, 151.24, 152.63, 156.57. MS (EI): m/z (%) = 395 (22.5) [M+]. Anal. Calc. for C19H21N7O3: C, 57.71; H, 5.35; N, 24.80; found: C, 57.87; H, 5.50; N, 24.98.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
The authors wish to thank Shiraz University of Technology research council for partial support of this work.References
- T. Hofmarcher, P. Lindgren, N. Wilking and B. Jönsson, The cost of cancer in Europe 2018, Eur.J. Cancer, 2020, 129, 41–49, DOI:10.1016/j.ejca.2020.01.011.
- V. Prasad, K. De Jesús and S. Mailankody, The high price of anticancer drugs: origins, implications, barriers, solutions, Nat. Rev. Clin. Oncol., 2017, 14, 381–390, DOI:10.1038/nrclinonc.2017.31.
- A. D. Kinghorn, H. Falk, S. Gibbons and J. Kobayashi, Progress in the chemistry of organic natural products, Springer International Publishing, 2017 Search PubMed.
- C. O. Wilson, O. Gisvold and J. H. Block, Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry, ed. J. H. Block and J. M. Beale Jr., Lippincott Williams & Wilkins, Philadelphia, 11th edn, 2004 Search PubMed.
- B. G. Katzung, Basic & Clinical Pharmacology, Lange Medical Publications, McGraw-Hill Companies, 14 edn, 2018 Search PubMed.
- A. Nehlig, Coffee, Tea, Chocolate, and the Brain, CRC Press, 2004 Search PubMed.
- B. B. Fredholm, Handbook of Experimental Pharmacology (Methylxanthines), Springer-Verlag Berlin Heidelberg, 2011, vol. 200 Search PubMed.
- V. Alagarsamy, Pharmaceutical chemistry of natural products, Elsevier India, 2013 Search PubMed.
- J. Klosa, Caffeno-3-alkanolamines, US Pat. No 3094531, 1963.
- B. R. K. Shyamlal, M. Mathur, D. K. Yadav and S. Chaudhary, Microwave-assisted modified synthesis of C8-analogues of naturally occurring methylxanthines: Synthesis, biological evaluation and their practical applications, Fitoterapia, 2020, 143, 104533, DOI:10.1016/j.fitote.2020.104533.
- T. Zheng, H. Sun, F. Lu, K. Harms and X. Li, Cobalt induced C-H bond activation and C8-arylation of caffeine with aryl bromides, Inorg. Chem. Commun., 2013, 30, 139–142, DOI:10.1016/j.inoche.2013.01.025.
- W. Jiang, J. Zhuge, J. Li, G. Histand and D. Lin, Direct Sulfenylation of the Purine C8–H Bond with Thiophenols, J. Org. Chem., 2020, 85, 2415–2425, DOI:10.1021/acs.joc.9b03115.
- N. Singh, A. K. Shreshtha, M. S. Thakur and S. Patra, Xanthine scaffold: scope and potential in drug development, Heliyon, 2018, 4, e00829, DOI:10.1016/j.heliyon.2018.
- B. Jasiewicz and A. Sierakowska, Caffeine and its analogs, antioxidants and applications, Antioxidants and aging, 2020, pp. 155–164, DOI:10.1016/B978-0-12-818698-5.00015-8.
- T. Okaecwe, A. J. Swanepoel, A. Petzer, J. J. Bergh and J. P. Petzer, Inhibition of monoamine oxidase by 8-phenoxymethylcaffeine derivatives, Bioorg. Med. Chem., 2012, 20, 4336–4347, DOI:10.1016/j.bmc.2012.05.048.
- M. M. Van der Walt, G. Terre'Blanche, A. Petzer, A. C. U. Lourens and J. P. Petzer, The adenosine A2A antagonistic properties of selected C8-substituted xanthines, Bioorg. Chem., 2013, 49, 49–58, DOI:10.1016/j.bioorg.2013.06.006.
- M. M. Van der Walt and G. Terre'Blanche, Selected C8 two-chain linkers enhance theadenosine A1/A2A receptor affinity and selectivity of caffeine, Eur. J. Med. Chem., 2017, 125, 652–656, DOI:10.1016/j.ejmech.2016.09.072.
- J. P. Petzer, S. Steyn, K. P. Castagnoli, J.-F. Chen, M. A. Schwarzschild, C. J. Van der Schyfa and N. Castagnolia, Inhibition of Monoamine Oxidase B by Selective Adenosine A2A Receptor Antagonists, Bioorg. Med. Chem., 2003, 11, 1299–1310, DOI:10.1016/S0968-0896(02)00648-X.
- N. Vlok, S. F. Malan, N. Castagnoli Jr., J. J. Bergh and J. P. Petzer, Inhibition of monoamine oxidase B by analogues of the adenosine A2A receptor antagonist (E)-8-(3-chlorostyryl)caffeine (CSC), Bioorg. Med. Chem., 2006, 14, 3512–3521, DOI:10.1016/j.bmc.2006.01.011.
- M. I. Rodriguez-Franco, M. I. Fernandez-Bachiller, C. Perez, A. Castro and A. Martinez, Design and synthesis of N-benzylpiperidine–purine derivatives as new dual inhibitors of acetyl- and butyrylcholinesterase, Bioorg. Med. Chem., 2005, 13, 6795–6802, DOI:10.1016/j.bmc.2005.07.019.
- S. Mostert, W. Mentz, A. Petzer, J. J. Bergh and J. P. Petzer, Inhibition of monoamine oxidase by 8-[(phenylethyl)sulfanyl]caffeine analogues, Bioorg. Med. Chem., 2012, 20, 7040–7050, DOI:10.1016/j.bmc.2012.10.005.
- G. N. Krutovskikh, M. B. Kolesova, A. M. Rusanov, L. P. Vartanyan and M. G. Shagoyan, radioprotective properties of mercaptocaffeine derivatives, Khim.-Farm. Zh., 1975, 9, 21–23 CAS.
- B. Jasiewicz, A. Sierakowska, N. Wandyszewska, B. Warżajtis, U. Rychlewska, R. Wawrzyniak and L. Mrówczyńskam, Antioxidant properties of thio-caffeine derivatives: Identification of the newly synthesized 8-[(pyrrolidin-1-ylcarbonothioyl)sulfanyl] caffeine as antioxidant and highly potent cytoprotective agent, Bioorg. Med. Chem. Lett., 2016, 26, 3994–3998, DOI:10.1016/j.bmcl.2016.06.091.
- J. Mitkov, N. Danchev, I. Nikolova and A. Zlatkov, Synthesis and brain antihypoxic activity of some aliphatic and arylaliphatic amides of caffeine-8-thioglycolic acid, Acta Pharm., 2007, 57, 361–370, DOI:10.2478/v10007-007-0029-1.
- J. Mitkov, M. Georgieva and A. Zlatkov, Development of an optimized synthetic approach for synthesis of caffeine-8-thioglycolic acid and its ester derivatives, Pharmacy, 2012, 1–4, 17–23 Search PubMed.
- J. Mitkov, L. Nikolova, I. Nikolova, N. Danchev and A. Zlatkov, Synthesis and brain antihypoxic activity of some aminoalcoholic derivatives of caffeine-8-thioglycolic acid, C. R. Acad. Bulg. Sci., 2010, 63, 1075–1082 CAS.
- M. N. Soltani Rad and S. Maghsoudi, Two-step three-component process for one-pot synthesis of 8-alkylmercaptocaffeine derivatives, RSC Adv., 2016, 6, 70335–70342, 10.1039/c6ra17814f.
- S. Sargazi, S. Shahraki, O. Shahraki, F. Zargari, R. Sheervalilou, S. Maghsoudi, M. N. Soltani Rad and R. Saravani, 8-Alkylmercaptocaffeine derivatives: antioxidant, molecular docking, and in-vitro cytotoxicity studies, Bioorg. Chem., 2021, 111, 104900, DOI:10.1016/j.bioorg.2021.104900.
- F. A. Ashour, S. M. Rida, S. A. M. El-Hawash, M. M. ElSemary and M. H. Badr, Synthesis, anticancer, anti-HIV-1, and antimicrobial activity of some tricyclic triazino and triazolo[4,3-e]purine derivatives, Med. Chem. Res., 2012, 21, 1107–1119, DOI:10.1007/s00044-011-9612-6.
- S. M. Rida, F. A. Ashour, S. A. M. El-Hawash, M. M. El-Semary and M. H. Badr, Synthesis of some novel substituted purine derivatives as potential anticancer, anti-hiv-1 and antimicrobial agents, Arch. Pharm. Chem. Life Sci., 2007, 340, 185–194, DOI:10.1002/ardp.200600118.
- H. P. Booysen, C. Moraal, G. Terre'Blanche, A. Petzer, J. J. Bergh and J. P. Petzer, Thio- and aminocaffeine analogues as inhibitors of human monoamine oxidase, Bioorg. Med. Chem., 2011, 19, 7507–7518, DOI:10.1016/j.bmc.2011.10.036.
- R. Kaplánek, M. Jakubek, J. Rak, Z. Kejík, M. Havlík, B. Dolenský, I. Frydrych, M. Hajdúch, M. Kolář, K. Bogdanová, J. Králová, P. Džubák and V. Král, Caffeine-hydrazones as anticancer agents with pronounced selectivity toward T-lymphoblastic leukaemia cells, Bioorg. Chem., 2015, 60, 19–29, DOI:10.1016/j.bioorg.2015.03.003.
- M. Georgieva, J. Mitkov, G. Momekov, B. Zlatkov, P. Peikov and A. Zlatkov, Synthesis, structural and spectral analysis of some 8- substituted derivatives of 1,3,7-trimethylxanthine with antiprolipherative activity, World J. Pharm. Pharma. Sci., 2014, 3, 60–83 CAS.
- M. N. Soltani Rad, S. Behrouz, K. Zokaei, M. Behrouz and A. G. E. Zarenezhad, Synthesis of Some Novel 8-(4-Alkylpiperazinyl) Caffeine Derivatives as Potent Anti-Leishmania Agents, Bioorg. Chem., 2022, 128, 106062, DOI:10.1016/j.bioorg.2022.106062.
- B. Strydom, J. J. Bergh and J. P. Petzer, 8-Aryl- and alkyloxycaffeine analogues as inhibitors of monoamine oxidase, Eur. J. Med. Chem., 2011, 46, 3474–3485, DOI:10.1016/j.ejmech.2011.05.014.
- M. M. van der Walt and G. Terre'Blanche, Selected C8 two-chain linkers enhance the adenosine A1/A2A receptor affinity and selectivity of caffeine, Eur. J. Med. Chem., 2017, 125, 652–656, DOI:10.1016/j.ejmech.2016.09.072.
- A. A. Kadi, K. E. H. El-Tahir, Y. Jahng and A. F. M. Motiur Rahman, Synthesis, biological evaluation and Structure Activity Relationships (SARs) study of 8-(substituted)aryloxycaffeine, Arab. J. Chem., 2019, 12, 2356–2364, DOI:10.1016/j.arabjc.2015.02.021.
- J. E. Moses and A. D. Moorhouse, The growing applications of click chemistry, Chem. Soc. Rev., 2007, 36, 1249–1262, 10.1039/B613014N.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, The 1,2,3-triazole ring as a bioisostere in medicinal chemistry, Drug Discovery Today, 2017, 22, 1572–1581, DOI:10.1016/j.drudis.2017.05.014.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Click Chemistry: 1,2,3-Triazoles as Pharmacophores, Chem.–Asian J., 2011, 6, 2696–2718, DOI:10.1002/asia.201100432.
- B. Meunier, Hybrid molecules with a dual mode of action: Dream or reality?, Acc. Chem.Res., 2008, 41, 69–77, DOI:10.1021/ar7000843.
- M. N. Soltani Rad, S. Behrouz, J. Mohammadtaghi-Nezhad, E. Zarenezhad and M. Agholi, Silica-tethered cuprous acetophenone thiosemicarbazone (STCATSC) as a novel hybrid nano-catalyst for highly efficient synthesis of new 1,2,3-triazolyl-based metronidazole hybrid analogues having potent antigiardial activity, Appl. Organomet. Chem., 2019, 33, e4799, DOI:10.1002/aoc.4799.
- E. Caputo, L. Maiorana, V. Vasta, F. M. Pezzino, S. Sunkara, K. Wynne, G. Elia, F. M. Marincola, J. A. McCubrey, M. Libra, S. Travali and M. Kane, Characterization of human melanoma cell lines and melanocytes by proteome analysis, Cell cycle, 2011, 10, 2924–2936, DOI:10.4161/cc.10.17.17068.
- W. L. Wang, W. Porter, R. Burghardt and S. H. Safe, Mechanism of inhibition of MDA-MB-468 breast cancer cell growth by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin, Carcinogenesis, 1997, 18, 925–933, DOI:10.93/carcin/18.5.925.
- Ş. Comşa, A. M. Cimpean and M. Raica, The story of MCF-7 breast cancer cell line: 40 years of experience in research, Anticancer Res., 2015, 35, 3147–3154 Search PubMed.
- H. D. Soule, J. Vazguez, A. Long, S. Albert and M. Brennan, A human cell line from a pleural effusion derived from a breast carcinoma, J. Natl. Cancer Inst., 1973, 51, 1409–1416, DOI:10.1093/jnci/51.5.1409.
- I. Shahzadi, A. F. Zahoor, A. Rasul, A. Mansha, S. Ahmad and Z. Raza, Synthesis, Hemolytic Studies, and In Silico Modeling of Novel Acefylline−1,2,4-Triazole Hybrids as Potential Anti-cancer Agents against MCF-7 and A549, ACS Omega, 2021, 6, 11943–11953, DOI:10.1021/acsomega.1c00424.
- L. Genestier, R. Paillot, L. Quemeneur, K. Izeradjene and J. P Revillard, Mechanisms of action of methotrexate, Immunopharmacology, 2000, 47, 247–257, DOI:10.1016/s0162-3109(00)00189-2.
- B. Chabner and D. L. Longo, Cancer Chemotherapy and Biotherapy Principles and Practice, Oxford University press, Wolters Kluwer Health/Lippincott Williams & Wilkins, 5th edn, 2011 Search PubMed.
- R. J. Bienstock, Library design, search methods, and applications of fragment-based drug design, American Chemical Society, Washington, 2011 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 2001, 46, 3–25, DOI:10.1016/S0169-409X(96)00423-1.
- SwissADME online software, http://www.swissadme.ch/index.php, Accessed 7 September 2022 Search PubMed.
- OSIRIS Property Explorer, https://www.organic-chemistry.org/prog/peo, Accessed 7 September 2022 Search PubMed.
- PreADMET online server, https://preadmet.webservice.bmdrc.org/adme, Accessed 9 September 2022 Search PubMed.
- R. A. Smith, J. Dumas, L. Adnane and S. M. Wilhelm, Recent Advances in the Research and Development of RAF Kinase Inhibitors, Curr. Top. Med. Chem., 2006, 6, 1071–1089, DOI:10.2174/156802606777812077.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D'Andrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma, Nature, 2010, 467, 596–599, DOI:10.1038/nature09454.
- I. Puzanov, R. K. Amaravadi, G. A. McArthur, K. T. Flaherty, P. B. Chapman, J. A. Sosman, A. Ribas, M. Shackleton, P. Hwu, B. Chmielowski, K. B. Nolop, P. S. Lin and K. B. Kim, Long-term outcome in BRAF(V600E) melanoma patients treated with vemurafenib: Patterns of disease progression and clinical management of limited progression, Eur. J. Cancer, 2015, 51, 1435–1443, DOI:10.1016/j.ejca.2015.04.010.
- N. Aarhus, Molegro Virtual Docker, version 6.0.0, CLC Bio, 8200, Denmark, 2012 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman et al., Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. B. Umar, A. Uzairu, G. A. Shallangwa and S. Uba, Design of potential anti-melanoma agents against SK-MEL-5 cell line using QSAR modeling and molecular docking methods, SN Appl. Sci., 2020, 2, 815, DOI:10.1007/s42452-020-2620-8.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, New York, 5nd edn, 2013 Search PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-90304.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07683g |
| This journal is © The Royal Society of Chemistry 2023 |