
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyether-tethered imidazole-2-thiones, imidazole-2-selenones and imidazolium salts as collectors for the flotation of lithium aluminate and spodumene†
Sophie Ackera,
Jan C. Namysloa,
Martin Rudolphb,
Franziska Strubeb,
Ursula E. A. Fittschenc,
Hao Qiu d,
Daniel Goldmannd and
Andreas Schmidt*a
d,
Daniel Goldmannd and
Andreas Schmidt*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de
bFreiberg, Helmholtz Institute Freiberg for Resource Technology, Chemnitzer Str. 40, D-09599 Freiberg, Germany
cClausthal University of Technology, Institute of Inorganic and Analytical Chemistry, Arnold-Sommerfeld-Str. 4, D-38678 Clausthal-Zellerfeld, Germany
dClausthal University of Technology, Institute of Mineral and Waste Processing, Recycling and Circular Economy Systems, Walther-Nernst-Str. 9, D-38678 Clausthal-Zellerfeld, Germany
First published on 27th February 2023
Abstract
Imidazolium salts were prepared which possess 2-ethoxyethyl pivalate or 2-(2-ethoxyethoxy)ethyl pivalate groups as amphiphilic side chains with oxygen donors as well as n-butyl substituents as hydrophobic groups. The N-heterocyclic carbenes of the salts, characterized by 7Li and 13C NMR spectroscopy as well as by Rh and Ir complex formation, were used as starting materials for the preparation of the corresponding imidazole-2-thiones and imidazole-2-selenones. Flotation experiments in Hallimond tubes under variation of the air flow, pH, concentration and flotation time were performed. The title compounds proved to be suitable collectors for the flotation of lithium aluminate and spodumene for lithium recovery. Recovery rates up to 88.9% were obtained when the imidazole-2-thione was used as collector.
1. Introduction
As a consequence of a steadily increasing demand for critical raw materials (CRMs)1 including elements such as lithium,2 the development of new recycling technologies is constantly being sought and developed. Lithium is considered to be a strategic metal in the world,3 as a considerably increasing demand for this element can be expected in the next years. This is mainly due to its application in lithium-ion batteries for e-mobility, as the share of lithium consumption for batteries was already 71% in 2020. Recycling's contribution to the current lithium demand, however, is close to 0%.1 This is in sharp contrast to the projection of the global lithium demand, which is expected to surpass two million metric tons of lithium carbonate equivalent in 2030. This is more than doubling the forecast for 2025.4 Apart from batteries, lithium is also currently used in ceramics,5 glass,6 lubricants,7 refrigeration8 and the nuclear9 and optoelectronic industries.10 Unfortunately, numerous critical raw materials and other industrially important elements such as lithium are often dissipated in the waste streams. Lithiumaluminate (LiAlO2) formation in slags can be initiated by the addition of Al to the pyrometallurgical process. Minerals which are purposely engineered in a slag to facilitate their recovery have recently been termed engineered artificial minerals (EnAM).11 This strategy has been overlooked as effective method for the recovery of numerous elements in the past. For the recycling of batteries, intense efforts are currently directed toward lithiumaluminate formation during the slag solidification after the thermal decomposition of the batteries. Apart from the removal of heavy metal ions and the simple and rapid dewatering of suspensions by removal of the concentrated sludge fraction along with the froth,12 flotation is an effective method to separate minerals from gangue materials and is the most important and versatile mineral processing technique.13 Flotation is based on the difference in surface hydrophobicity of dispersed particles, obtained by crushing and grinding of raw minerals to liberate valuable components from interlocking particles. After suspending in water and conditioning with a selection of appropriate reagents, air is conducted into the dispersion to separate hydrophobic particles which collide with and attach to the rising air bubbles, whereas hydrophilic particles remain suspended in the pulp. Collectors, frothers, depressants, activators, and regulators take influence on interfacial properties of particles and bubbles affecting their interactions and, besides hydrodynamic parameters, take influence on the success of the flotation.14 Optimization is often directed towards the maximization of the adsorption of the particles on the surfaces to selectively enhance particle's hydrophobicity and thus improving particle-bubble attractive interactions.15 Another key lithium mineral is spodumene (LiAlSi2O6) which is considered to be one of the most critical minerals due to it is high lithium contents and high rate extraction.16 Concerning the flotation of lithium bearing minerals such as lithium aluminate and spodumene, sodium oleate is often employed as surfactant-type collector,17 and other collector formulations and methods have also been developed.18In continuation of a current project dealing with the detection and recycling of metals,19 collector design, and our interest in the chemistry of nitrogen heterocycles including their N-heterocyclic carbenes (NHCs),20 we prepared polyether-tethered imidazolium salts and studied them in metal complex formations with lithium, silver, rhodium and iridium as well as in flotation of lithium minerals. We intended to combine n-butyl-groups as hydrophobic moieties with polyether chains of two different lengths as amphiphilic and relatively electron-rich side chains of an imidazole core (Scheme 1).
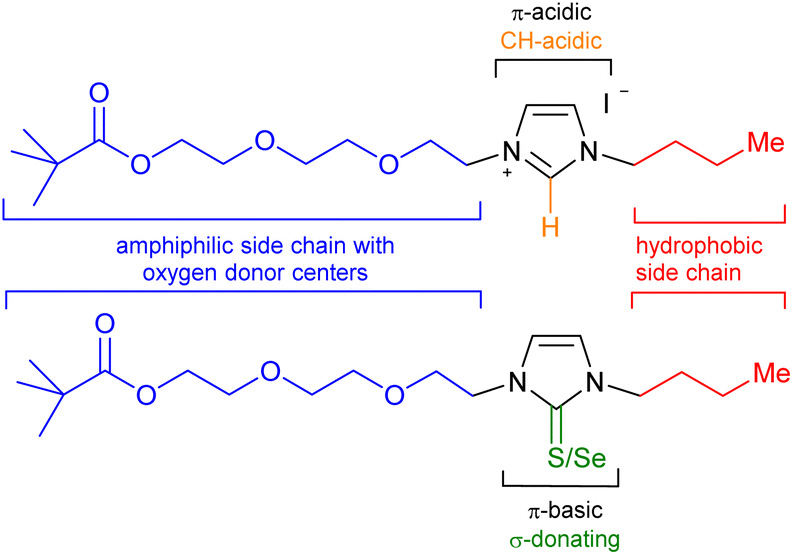 | ||
| Scheme 1 Target imidazolium salts and thiones/selenones. | ||
The central heteroaromatic imidazolium ring of the salts is strongly electron-deficient and π-acidic, and is known to initiate anion–π and π–π-interactions e.g. to copper species,21 σ-hole and π-hole interactions to silicon compounds,22 anion-π and lone-pair-π interactions to halogens,23 and π–π-interactions to adsorb organic substrates on surfaces24 such as anthracite coal.25 The electronic compatibility of the surface of galena (PbS) and π-conjugated systems to gain insight into the flotation's mechanism has been calculated.26 Furthermore, the 2-position of imidazolium salts is CH-acidic so that a structural variation can easily achieved via their N-heterocyclic carbenes. We therefore planned reactions with sulfur and selenium to get imidazole-2-thiones and imidazole-2-selenones as potential new collectors for the flotation of lithium minerals. Imidazole-based ionic liquids such as dodecyl-trimethylimidazolium chloride have i.a. been used for the separation of quartz from phosphorite,27 for the flotation of quartz28 and lithium aluminate.29 Nanoparticles of imidazole functionalized polystyrene are known to bind nickel ions.30 Imidazole-2-thiones combine structure elements of imidazole and thioureas and have been applied in the flotation of copper sulfides and pyrite.31 Thioureas have been used for the flotation of silver,32 gold,33 chalcopyrite,34 copper,35 and many others. In contrast to imidazolium salts, the thiones and selenones are π-bases which are known to undergo cation–π-interactions, as examined in the flotation of molybdenite.36 We applied the imidazolium salts as well as their thiones and selenones in microflotation experiments of lithium aluminate, spodumene and gehlenite in Hallimond tubes under variation of the conditions. Gehlenite is a representative slag gangue material. To the best of our knowledge, the application of selenoureas in flotation have never been described before.
2. Results and discussion
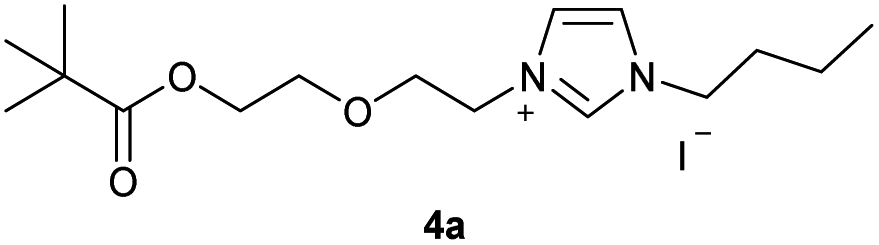
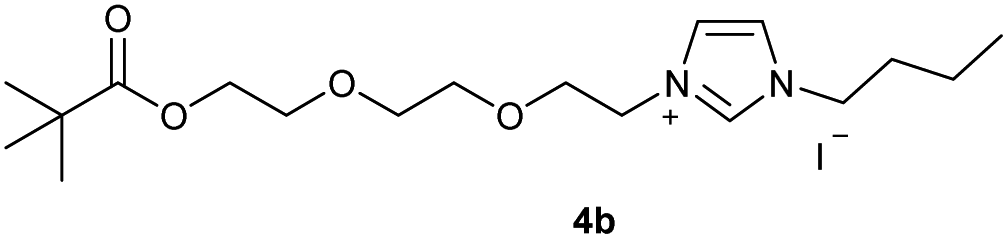
In the first step, the desired imidazolium salts were synthesized via a synthetic route which is shown in Scheme 2. The syntheses were accomplished in several steps. First, EINHORN esterification of the alcohol group of 2-(2-chloroethoxy)ethan-1-ol 1a and of its derivative 1b with pivaloyl chloride in the presence of pyridine protected the side chain toward deprotonation during the carbene generation with bases. Finkelstein reaction induced the halogen exchange of chloride toward iodide (3a,b) which proved to be the better leaving group for the subsequent nucleophilic substitution to n-butylimidazole, which resulted in the formation of the imidazolium salts 4a,b as orange-colored oils in high yields (75% and 80%).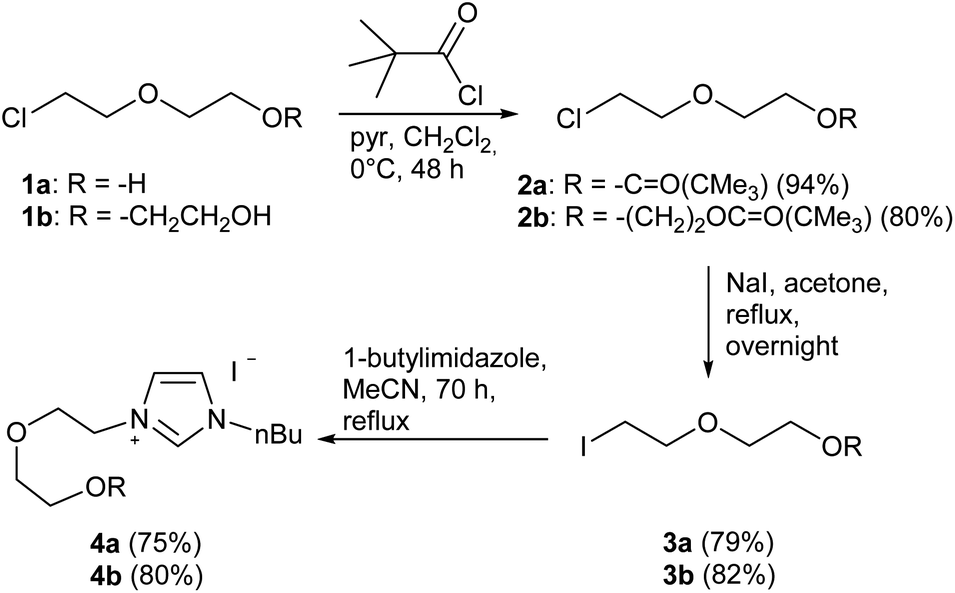 | ||
| Scheme 2 Synthesis of imidazolium salts. | ||
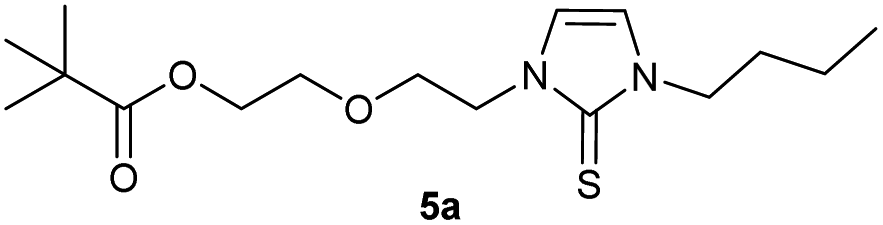
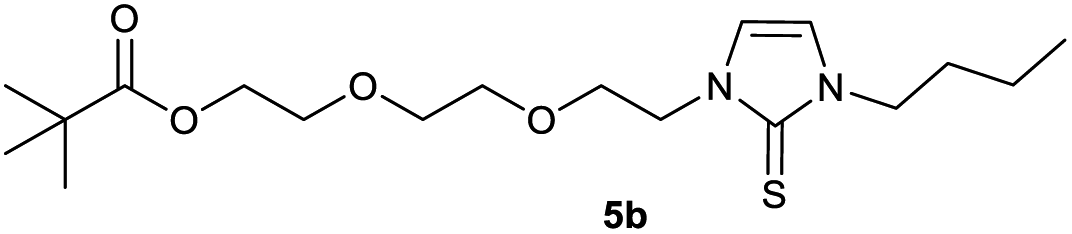
We then proceeded with reactions under water-free conditions in an inert atmosphere. Via DFT calculations on the B3LYP/6-311++G** level we determined the CREF (carbene relative energy of formation)37 value of 4a in advance. The calculated value (0.416) indicates, as expected, a relative ease of carbene formation from its precursor 4a. Indeed, we generated the N-heterocyclic carbenes 4aA and 4bA by addition of bases to a solution of the imidazolium salts 4a,b. The N-heterocyclic carbenes 4aA and 4bA are suitable precursors for the synthesis of thiones 5a,b and selenones 6a,b which we wanted to test as collectors for the flotation of lithium minerals. Thus treatment of the salts 4a,b with elemental sulfur and elemental selenium in the presence of cesium carbonate yielded the target thiones 5a,b and the selenones 6a,b in acceptable yields (77% and 41%), respectively (Scheme 3). In order to characterize the N-heterocyclic carbene we performed the deprotonation of 4b in THF-d8 with lithium bis(trimethylsilyl)amide [LHMDS] as the base and measured the 13C NMR as well as 7Li NMR spectra. The 2-position of the imidazolium salt 4b shifted from 137.9 ppm to 201.7 ppm in the 13C NMR spectra during the carbene formation. The 7Li spectrum of 4bA shows a signal at +0.64 ppm (Fig. 1). The selenones 6a,b display 77Se NMR resonance frequencies at −12.4 ppm, respectively. Reaction of the in situ generated N-heterocyclic carbenes 4aA and 4bA with silver oxide (Ag2O) gave non-isolable silver adducts which were subjected to a silver → iridium and silver → rhodium exchange reaction with bis-(1,5-cyclo-octadiene)dirhodium(I)-dichloride and its iridium analog to give the stable and fully characterizable complexes 7a,b and 8a,b.
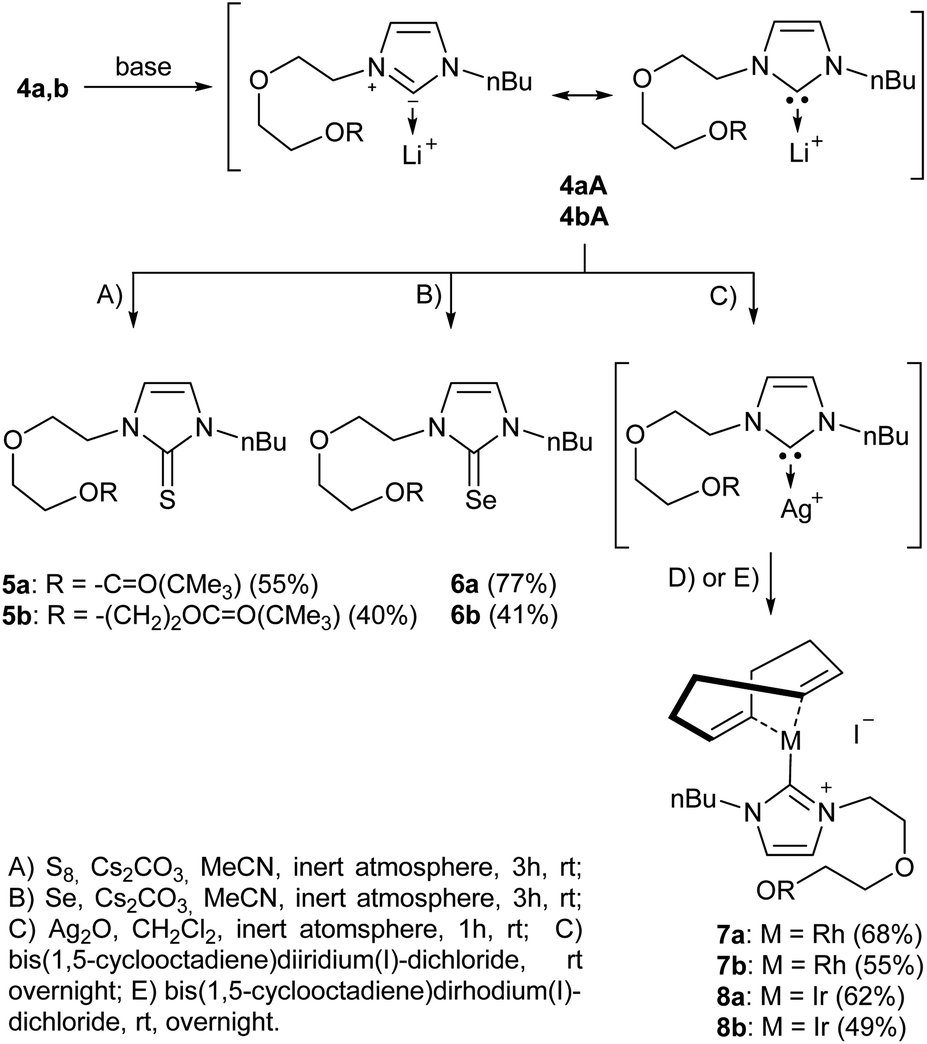 | ||
| Scheme 3 Synthesis of thiones and selenones as potential collectors for lithium mineral flotation. Trapping of the reactive N-heterocyclic carbenes with rhodium and iridium via silver complexes. | ||
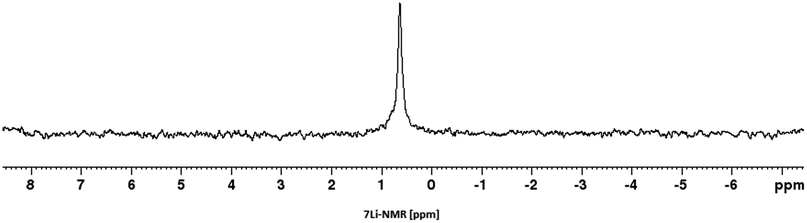 | ||
| Fig. 1 7Li NMR spectrum of the lithium adduct of imidazol-2-ylidene 4bA. | ||
Further DFT calculations (B3LYP/6-311++G**) have been carried out to gain knowledge about the molecular orbitals which contribute to, or even govern the formation of π-complexes and σ-bonds to organic or inorganic substrates including metals. As expected, the lowest unoccupied molecular orbital (LUMO: −5.10 eV) of the salt 4a is located in the imidazolium moiety, whereas the highest occupied molecular orbital (HOMO: −9.34 eV) consists of atomic orbital coefficients of the σ-framework of the pivaloyl group including the non-bonding electron pairs of its carbonyl oxygen (Fig. 2). HOMO−1 (−10.37 eV), HOMO−2 (−10.59 eV), HOMO−3 (−10.80 eV), HOMO−4 (−10.80 eV) as well as HOMO−5 (−11.44 eV) are all located in the polyether chain (see ESI†).
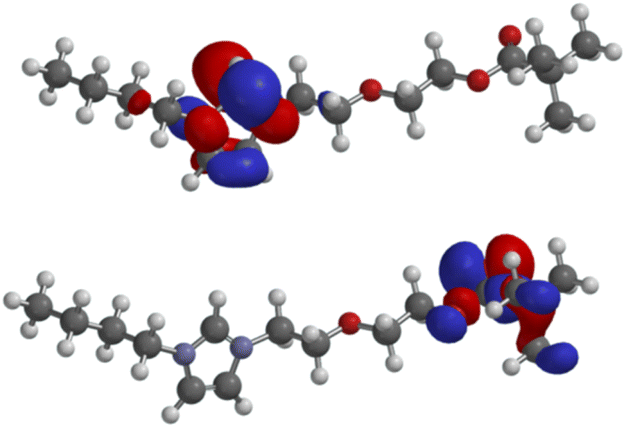 | ||
| Fig. 2 LUMO (above) and HOMO (below) of 4a. | ||
The LUMO of the N-heterocyclic carbene 4aA (−0.35 eV) has very small atomic orbital coefficients, whereas the HOMO (−5.98 eV) displays the characteristic geometry with a large coefficient of the σ-lone pair plus coefficients in the σ-framework of the imidazole ring (Fig. 3). The σ-lone pair is capable of metal complexations as proved by the formation of Ag, Rh and Ir complexes as mentioned before. All other molecular orbitals are shown in the ESI.†
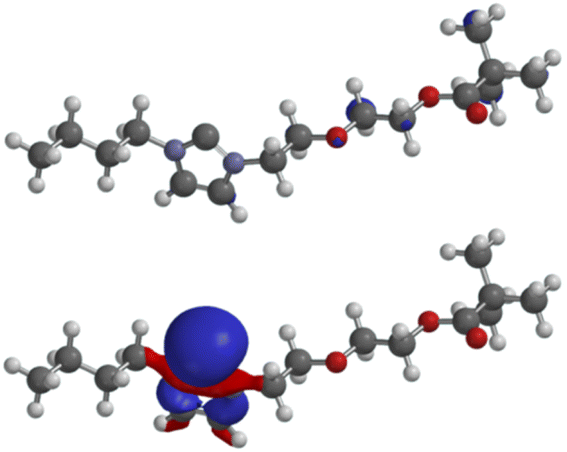 | ||
| Fig. 3 LUMO (above) and HOMO (below) of the non-complexed N-heterocyclic carbene 4aA. | ||
The calculated frontier orbitals of the thione 5a and the selenone 6a are very similar. Thus, the LUMOs of the thione (−0.46 eV) and of the selenone (−0.50 eV) have small atomic orbital coefficients in the imidazole ring (Fig. 4). The HOMO (−5.18 eV) of the thione and the HOMO (−5.37 eV) of the selenone are located in the imidazole moiety; they are π-orbitals. The molecular orbitals of the thione are shown in Fig. 4 and those of the selenone can be found in the ESI.†
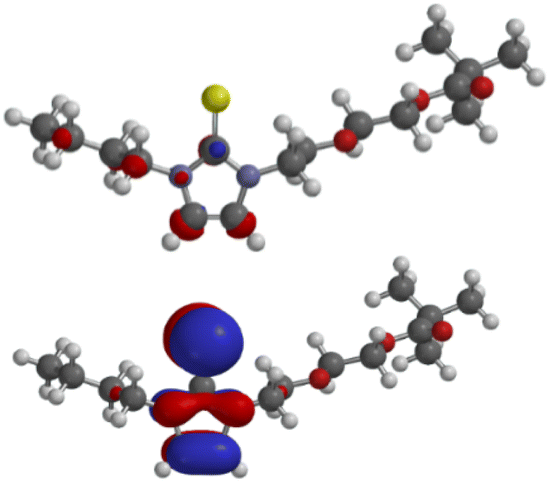 | ||
| Fig. 4 LUMO (above) and HOMO (below) of thione 5a. | ||
Flotation experiments
We applied the imidazolium salts 4a,b, the thiones 5a,b and the selenones 6a,b in microflotation experiments of fine particle fractions, the particle distributions of which are provided in Fig. 5. The chosen three different minerals are, lithium aluminate (LiAlO2), spodumene (LiAlSi2O6) and gehlenite (Ca2Al[AlSiO7]). The latter is a potential gangue material especially of the former mentioned slag formation processes.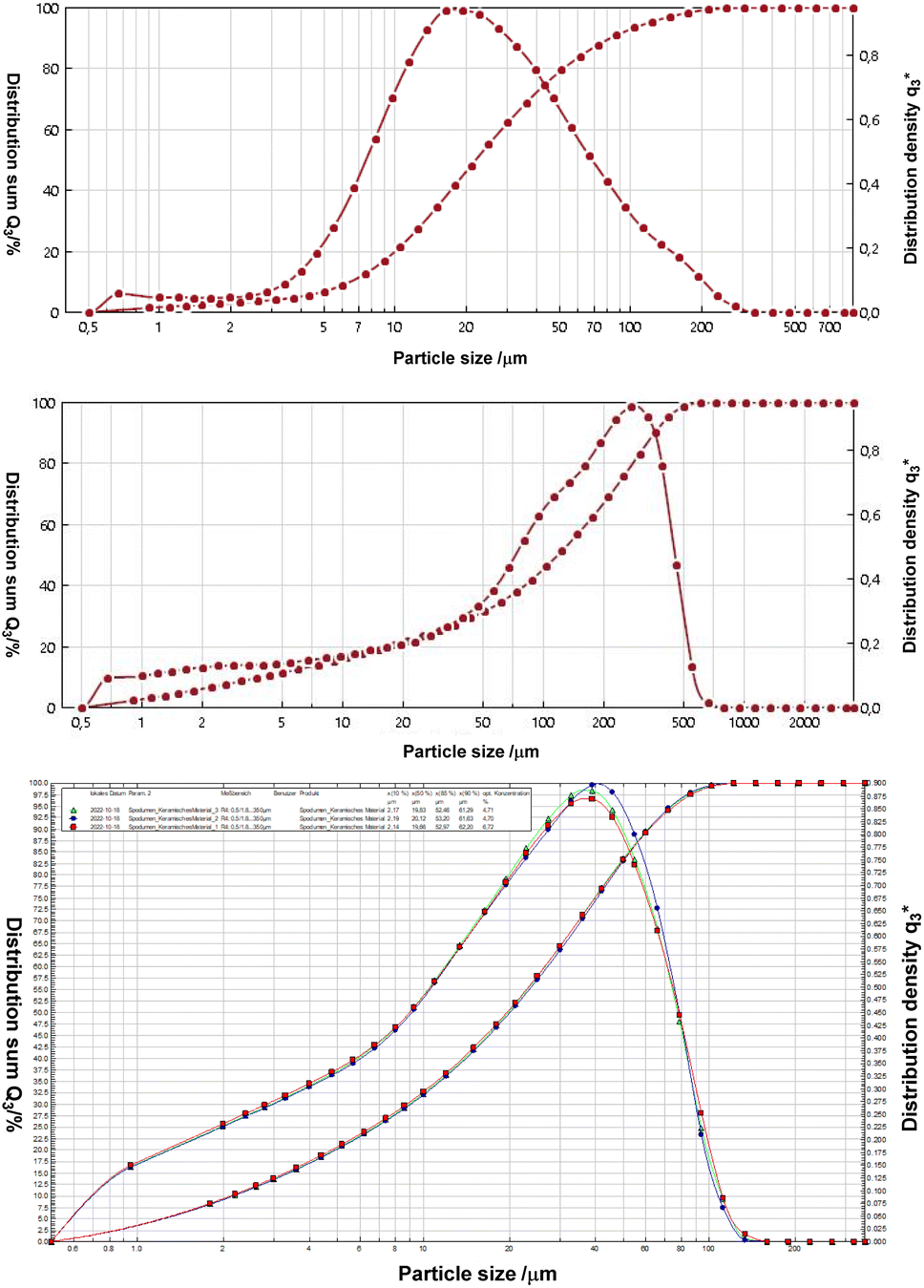 | ||
| Fig. 5 Particle size distributions by means of laser diffraction of lithium aluminate (top), gehlenite (middle) and spodumene (bottom). | ||
In addition, we performed a phase analysis of spodumene by quantitative XRD, showing a composition by weight of 5.1% quartz, 93.5% spodumene and 1.4% topaz. The mineral gehlenite was measured via ICP-OES. Theoretically it consists of 19.7% aluminium, 29.2% calcium and 10.2% silicon. The chemical analysis also revealed a small content (<5%) of iron and magnesium (Fig. 6).
 | ||
| Fig. 6 Chemical analysis of gehlenite. | ||
Different concentrations of the six imidazole derivatives and the frother (pine oil) were used for the flotation. The stock solutions of the individual collectors have a concentration of 1% by mass in distilled water. To achieve better solubility of the collectors, 0.05 ml of tetrahydrofurane (THF) were added. Exclusion of any effects of THF was accomplished by floating 1 wt% solution of THF in distilled water as a blind sample for both spodumene and lithium aluminate. The conversion of the amount of collectors into the molar concentration are shown in Table 1.
| Collector | Vol. added [μl] | Conc. [μmol l−1] |
|---|---|---|
 |
60 | 5.66 |
| 120 | 11.32 | |
| 300 | 28.29 | |
| 600 | 56.59 | |
 |
60 | 5.13 |
| 120 | 10.25 | |
| 300 | 25.63 | |
| 600 | 51.27 | |
 |
60 | 7.31 |
| 120 | 14.63 | |
| 300 | 36.57 | |
| 600 | 73.13 | |
 |
60 | 6.38 |
| 120 | 12.76 | |
| 300 | 31.90 | |
| 600 | 63.81 | |
 |
60 | 6.45 |
| 120 | 12.90 | |
 |
60 | 5.71 |
| 120 | 11.42 |
Flotation under variation of the air flow
First, we performed flotation experiments of lithium aluminate and gehlenite under an air flow of 170 cm3 min−1, respectively. For the experiments, samples of 2 g of each mineral were floated and the recovery yields were calculated from the ratios of the flotation recovery and the residue. As can be seen from the results presented in Table 2, each of the four imidazoles which we examined proved to be a suitable collector for the flotation of lithium aluminate. Best results of the flotation of LiAlO2 (88.9% recovery) were obtained, when 18.28 μmol l−1 of imidazole-2-thione 5a were employed after a flotation time of 10 min and 1.2 × 10−3 g l−1 of frother (pine oil) at the natural pH values of the mineral in water, i.e. between 10 and 11. Lower pH values resulted in lower recovery rates. The yields of the flotation of the gangue material gehlenite are very low (>15%) under the conditions applied and with the material used.| Collector | Mineral | Conditions | Recovery ± confidence interval [%] | |
|---|---|---|---|---|
| a Flotation conditions: A: conditioning time CT = 3 min, flotation time FT = 5 min, 4a: 28.29, 4b: 25.63, 5a: 36.57 and 6a: 31.90 μmol l−1 of collector, 6.0 × 10−3 g l−1 (750 g per ton) frother. B: FT = 10 min, collector 56.59, 51.27, 73.13 and 63.81 μmol l−1, 1.2 × 10−2 g l−1 (1500 g per ton) frother (pine oil). Each measurement consists of three repetitions. 95% confidence interval. | ||||
| 4a | LiAlO2 | pH 2 | A | 61.5 ± 4.31 |
| LiAlO2 | pH 10–11 | A | 68.0 ± 4.29 | |
| LiAlO2 | B | 85.1 ± 1.06 | ||
| Gehlenite | A | 11.1 ± 1.70 | ||
| 4b | LiAlO2 | pH 2 | A | 63.0 ± 1.29 |
| LiAlO2 | pH 10–11 | A | 67.5 ± 7.71 | |
| LiAlO2 | B | 84.4 ± 12.67 | ||
| Gehlenite | A | 10.8 ± 2.40 | ||
| 5a | LiAlO2 | pH 2 | A | 65.2 ± 8.09 |
| LiAlO2 | pH 10–11 | A | 67.7 ± 2.58 | |
| LiAlO2 | B | 88.9 ± 2.47 | ||
| Gehlenite | A | 12.2 ± 7.37 | ||
| 6a | LiAlO2 | pH 2 | A | 61.4 ± 3.56 |
| LiAlO2 | pH 10–11 | A | 64.6 ± 5.60 | |
| LiAlO2 | B | 82.7 ± 3.29 | ||
| Gehlenite | A | 14.0 ± 3.97 | ||
Table 3 summarizes the results of flotation experiments under an air flow of 32 cm3 min−1 under variation of the pH value, the concentrations of the imidazoles as collector as well as of the frother α-pinene, and the flotation time. The blind value for 1 wt% solution of THF in distilled water gave a recovery of 16.7%, indicating no effect of THF on the flotation. The recovery yields under these conditions are slightly lower than those obtained applying a higher air flow. As can be seen, the collectors gave good results at strongly acidic as well as at strongly basic pH values. Again, imidazole-thione 5a gave the highest yields of lithium aluminate recovery. Imidazolium salt 4b, which has a longer side chain, gave slightly better recovery rates than imidazolium salt 4a. Apart from the minerals lithium aluminate and gehlenite, we also examined the flotation of spodumene. A suspension of spodumene in distilled water has a pH value of 8. The results with an air flow of 32 cm3 min−1 are shown in Table 4.
| Collector | Mineral | Conditions | Recovery ± confidence interval [%] | |
|---|---|---|---|---|
| a Flotation conditions: A: conditioning time CT = 3 min, flotation time FT = 3 min, 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.45 (5b) 6.38 (6a), 5.71 (6b) μmol l−1 of collector, 1.2 × 10−3 g l−1 (150 g per ton) frother (pine oil). B: FT = 10 min, 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.45 (5b) 6.38 (6a), 5.71 (6b) μmol l−1 of collector, 1.2 × 10−3 g l−1 (150 g per ton) frother. C: FT = 10 min, collector 11.32 (4a), 10.25 (4b), 14.63 (5a), 12.90 (5b), 12.76 (6a), 11.42 (6b) μmol l−1, 1.2 × 10−3 g l−1 (150 g per ton) frother. Each measurement consists of three repetitions. 95% confidence interval. | ||||
| 4a | LiAlO2 | pH 2 | A | 45.3 ± 7.75 |
| C | 73.5 ± 1.64 | |||
| pH 10–11 | A | 33.6 ± 6.23 | ||
| B | 60.7 ± 14.35 | |||
| C | 70.1 ± 8.10 | |||
| 4b | LiAlO2 | pH 2 | A | 46.0 ± 3.05 |
| C | 76.3 ± 6.32 | |||
| pH 10–11 | A | 33.1 ± 6.28 | ||
| B | 64.7 ± 3.77 | |||
| C | 76.4 ± 4.13 | |||
| 5a | LiAlO2 | pH 2 | A | 47.2 ± 3.75 |
| C | 79.3 ± 4.10 | |||
| pH 10–11 | A | 43.4 ± 8.01 | ||
| B | 75.1 ± 10.78 | |||
| C | 78.9 ± 5.90 | |||
| 5b | LiAlO2 | pH 2 | A | 45.1 ± 5.41 |
| pH 10–11 | A | 35.5 ± 0.84 | ||
| B | 59.9 ± 13.50 | |||
| C | 76.7 ± 4.66 | |||
| 6a | LiAlO2 | pH 2 | A | 43.8 ± 2.62 |
| C | 77.9 ± 4.85 | |||
| pH 10–11 | A | 36.5 ± 21.12 | ||
| B | 63.4 ± 13.01 | |||
| C | 74.2 ± 2.00 | |||
| 6b | LiAlO2 | pH 2 | A | 47.3 ± 7.67 |
| pH 10–11 | A | 31.9 ± 2.64 | ||
| B | 62.6 ± 1.72 | |||
| C | 75.5 ± 8.48 | |||
| Collector | Mineral | Conditions | Recovery confidence interval [%] | |
|---|---|---|---|---|
| a Flotation conditions: A: conditioning time CT = 3 min, flotation time FT = 3 min, 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.45 (5b) 6.38 (6a), 5.71 (6b) μmol l−1 of collector, resp., 1.2 × 10−3 g l−1 (150 g per ton) frother. B: conditioning time CT = 3 min, flotation time FT = 10 min, 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.45 (5b) 6.38 (6a), 5.71 (6b) μmol l−1 of collector, resp., 1.2 × 10−3 g l−1 (150 g per ton) frother (pine oil). Each measurement consists of three repetitions. 95% confidence interval. | ||||
| 4a | Spodumene | pH 2 | A | 45.6 ± 5.82 |
| pH 8 | A | 41.3 ± 11.12 | ||
| B | 60.3 ± 25.04 | |||
| 4b | Spodumene | pH 2 | A | 49.4 ± 4.68 |
| pH 8 | A | 43.7 ± 2.71 | ||
| B | 60.7 ± 7.66 | |||
| 5a | Spodumene | pH 2 | A | 47.3 ± 5.70 |
| pH 8 | A | 46.3 ± 5.81 | ||
| B | 71.8 ± 2.71 | |||
| 5b | Spodumene | pH 2 | A | 48.7 ± 11.01 |
| pH 8 | A | 44.0 ± 4.03 | ||
| B | 71.8 ± 4.15 | |||
| 6a | Spodumene | pH 2 | A | 47.7 ± 1.46 |
| pH 8 | A | 41.9 ± 19.19 | ||
| B | 65.7 ± 28.52 | |||
| 6b | Spodumene | pH 2 | A | 42.2 ± 11.97 |
| pH 8 | A | 39.7 ± 19.69 | ||
| B | 54.2 ± 14.97 | |||
As a test experiment, we performed flotations without the addition of any collector and frother. The flotation of pure spodumene gives a recovery yield of 22.2% after a flotation time of 3 min, and adding 1 wt% solution THF in distilled water gave a recovery of 23.4%, so that the influence of THF can be neglected. All six imidazoles achieve yields between 40 and 50% in combination with the frother with a flotation time of 3 minutes at a pH of 2 and also at a pH of 8 with molar concentrations of 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.45 (5b) 6.38 (6a), 5.71 (6b) μmol l−1. Best results were achieved employing the imidazole-thione 5a and 5b with 71.8% with a flotation time of ten minutes at the same concentration. The values for the long-chained salt 4b are better than those of the shorter-chained salt 4a.
Next, we determined the zeta potentials of lithium aluminate in neat form and of samples of this mineral conditioned with the four imidazoles described here.
Zeta potential
For the neat mineral lithium aluminate, a zeta potential measurement was carried out over a pH range of 2 to 12. It turned out that the mineral does not provide stable values at certain pH values. The fitted trend line shows a large deviation between the individually measured values. Because of this, the zeta potential for the following measurements was determined at a pH value around 8. Fig. 7 shows the diagram for pure lithium aluminate.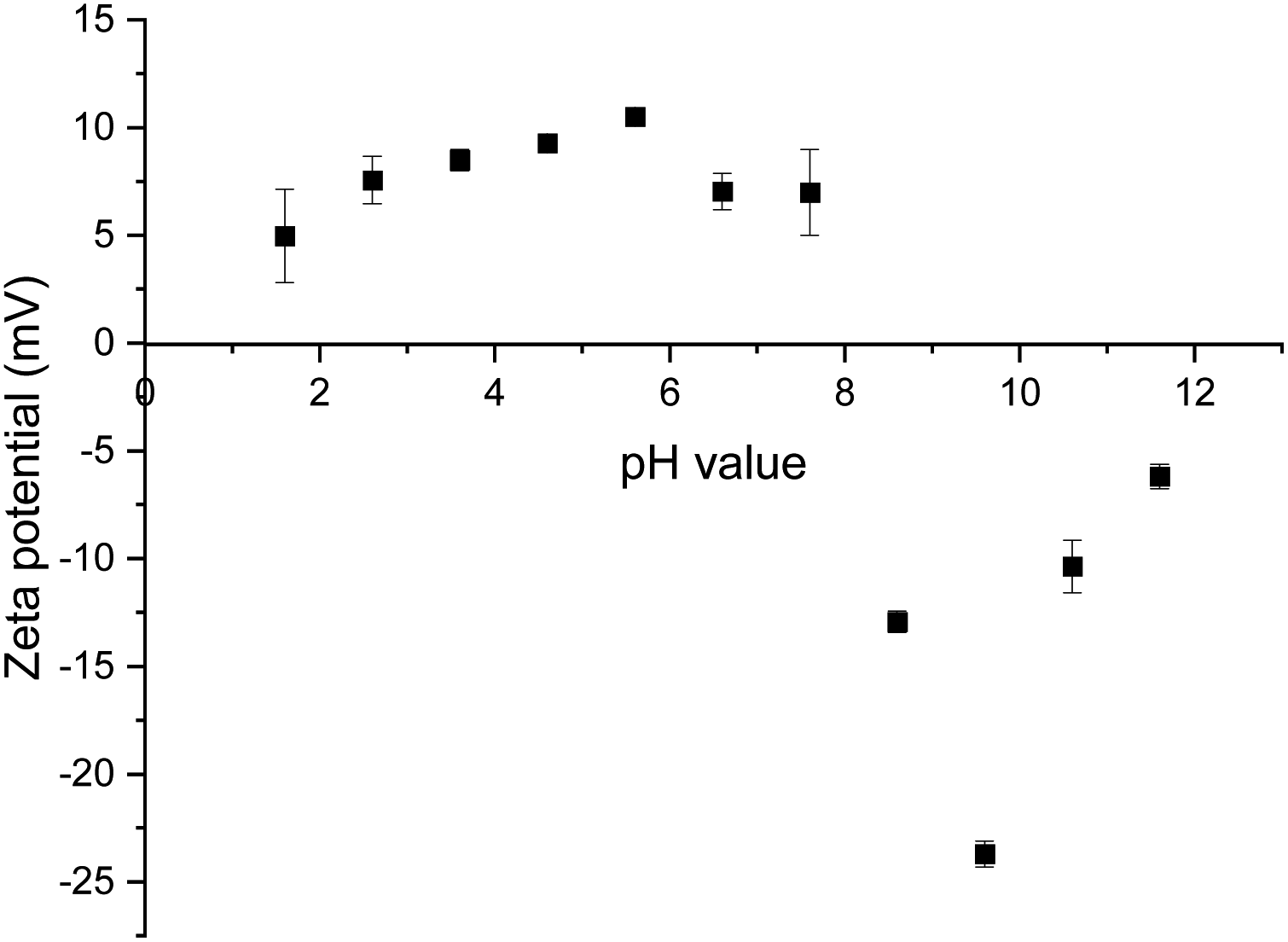 | ||
| Fig. 7 Zeta potential of pure lithium aluminate in distilled water. | ||
The results of the different zeta potential measurements for lithium aluminate conditioned with the collectors are listed in Table 5. The zeta potential of the pure lithium aluminate in 10−3 M KCl solution at a pH value of 8.44 was determined to be −0.75 mV.
| Collector | Amount of collector | pH value | Zeta potential [mV] |
|---|---|---|---|
| 4a | 60 μl | 8.2 | −3.80 |
| 4a | 600 μl | 8.2 | −6.70 |
| 4b | 60 μl | 8.2 | −9.70 |
| 4b | 600 μl | 8.4 | −4.40 |
| 5a | 60 μl | 8.5 | −0.38 |
| 5a | 600 μl | 8.4 | −0.09 |
| 6a | 60 μl | 8.3 | −0.38 |
| 6a | 600 μl | 8.4 | −0.33 |
A comparison of the values shows that an increase in the concentration of the respective collector leads to an increase in the measured zeta potential with the exception of collector 4a. Here the opposite occurs, as the zeta potential becomes even more negative with increased concentration. The measured values for collector 5a and 6a are close to 0 mV in the potential, which indicates that the particles are destabilizing, which is beneficial for flotation.
In conclusion, the prepared imidazolium salts, imidazole-2-thiones and imidazole-2-selenones proved to be suitable collectors for the flotation of lithium aluminate and spodumene for lithium recovery. Best results were achieved employing the imidazole-2-thione, possessing a polyether and an n-butyl side chain. The thione and the selenone were prepared starting from the corresponding imidazolium salts via the N-heterocyclic carbenes, the imidazol-2-ylidenes 4a,bA, which were characterized by means of 7Li and 13C NMR spectroscopy and trapping reactions with rhodium and iridium.
3. Experimental section
General
All used chemicals were purchased from commercial suppliers and used as received unless noted otherwise. The measurements for the 1H and 13C spectra have been performed on Bruker Avance (400 MHz) and Avance III (600 MHz) NMR spectrometer. The multiplicities are given with the following abbreviations: s = singlet, bs = broad singlet, d = doublet, dd = doublet of a doublet, t = triplet, tt = triplet of a triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet. In 13C NMR the following abbreviations are used: * = quaternary carbon atom, + = primary and tertiary carbon atom, − = secondary carbon atom. IR-spectroscopy has been performed by the FT-IR-spectrometer Alpha-T from Bruker with a platinum-ATR-module. The spectra consider a range of 400 to 4000 cm−1. The high-resolution mass spectra were measured with an Impact II from Bruker.The chemical analysis of gehlenite was performed with a Varian model Vista MPX and with an evaluation program from Varian. The sample was digested in two steps. In the first step, aqua regia was used, followed by diluted HF (0.8%) in the second. The value shown is the average of three measurements.
Spodumene was analyzed by quantitative XRD from PANalytical Empyrean by a X-ray diffractometer with a Co X-ray source. A voltage of 35 kV and a current of 35 mA was applied. Prior to the measurement the samples were split into 2.5 ml sample (<400 μm) then were ground in ethanol with zirconium-oxide grinding media in a McCrone mill, ideally producing particles with a narrow size distribution below 1 μm and an intact crystallographic structure. Afterwards the sample was dried overnight and the dried sample homogenized with small steel balls (4 mm diameter) by using the mixer mill MM 400 from Retsch. The diffractogram and quantitative phase determination were performed via Rietveld refinement using Profex 4.1.0 software.
Lithium aluminate (LiAlO2) was bought from Alfa Aesar. Spodumene was provided by the Helmholtz Institute Freiberg for Resource Technology, and gehlenite was provided by the Institute of Mineral and Waste Processing, Recycling and Circular Economy Systems, Clausthal-Zellerfeld.
Flotation experiments
The flotation experiments have been performed in a Hallimond tube (Fig. 8) with a volume of 250 ml. Lithium aluminate and gehlenite were tested as the single minerals (2 g for each measurement). The collector concentrations are in a range of 5.13 μmol l−1 to 73.13 μmol l−1 and for the frother (pine oil) between 1.2 × 10−3 g l−1 and 1.2 × 10−2 g l−1. In addition to the concentrations and the flotation time, the pH-value was varied. The air flows were 32 cm3 min−1 and 170 cm3 min−1. The continuous stirring speed is 500 rpm. The conditioning time was three times one minute for each addition step. The flotation time varied between three and ten minutes as indicated in the tables. | ||
| Fig. 8 Pictorial representation of a Hallimond tube. | ||
The Hallimond tubes used (250 ml) possess a medium-pore fritted glass through which the air can flow into the apparatus. A magnetic stirrer, adjusted to 500 rpm, ensures continuous mixing. The flotation tailings are collected separated in the upper area during flotation. In the first step, the mineral is conditioned in distilled water or buffer (one minute). The buffer consisted of 0.04 M boric acid, 0.04 M phosphoric acid, and 0.04 M acetic acid, and the required pH was achieved by the addition of 0.1 M NaOH. The buffer was used for measurements at a pH of 2. Then the collector is added and, after another minute, the frother. The remaining volume is filled up to 250 ml and floated for 3 or 10 minutes. The natural pH value of lithium aluminate is between 10 -11 and for spodumene at pH 8.
Zeta potential measurements
The measurements were conducted with a Zetasizer Nano from Malvern. The mineral sample (lithium aluminate, 0.5 g) was prepared in 1000 ml of 10−3 M KCl solution. The respective collector was added to the solution with a molar concentration of 5.66 (4a), 5.13 (4b), 7.31 (5a), 6.38 (6a) μmol l−1 for the first measurement and with 56.59 (4a), 51.27 (4b), 73.13 (5a), 63.81 (6a) μmol l−1 for the second measurement. The solution was mixed and sedimented for two hours. The measurements have been performed with the supernatant. Lithium aluminate samples were exclusively measured at the natural pH value.Particle size
The particle sizes for lithium aluminate and gehlenite were measured at the Institute of Mineral and Waste Processing, Recycling and Circular Economy Systems with a HELOS (H2387) & QUIXEL apparatus from Sympatec. Spodumene was measured at the Helmholtz Institute Freiberg for Resource Technology with a HELOS (H2634) & RODOS from Sympatec. For future microflotation testwork it should be considered to have comparable settling velocities of the particles in microflotation, which was not the case in this study. Gehlenite's median size was about six times the median of lithium aluminate. Nonetheless, this does not affect the trend of enhanced flotation ability upon collector adsorption.DFT calculations (molecular geometries, HOMO–LUMO values and orbital pictures, CREF-values) were performed using the Spartan Software (Spartan'20, Wavefunction, Inc., Irvine, CA, available from: http://www.wavefun.com) running on a MS Windows 10 Pro PC system with an AMD Ryzen Threadripper 3970X 32-Core and 128 GB RAM. MMFF optimized structures were used as starting geometries for the geometry optimizations with the B3LYP density functional and the 6-311++G** basis set carried out as vacuum calculations. Subsequent frequency calculations of all final structures evidenced the absence of imaginary frequencies and thus the presence of true minima on the potential energy surface.
Preparation of 2-(2-chloroethoxy)ethyl-2,2-dimethylpropanoate (2a)
Under a nitrogen atmosphere, 1.48 ml (14.00 mmol) of 2-[2-chloroethoxy]ethanol 1a was dissolved in 50 ml of anhyd. diethyl ether and then 10 ml anhyd. pyridine were added. 2.34 ml (19.00 mmol) of pivalic acid chloride was then added at 0 °C. The reaction mixture was stirred at room temperature for 48 hours. The reaction was then quenched with 1 M HCl (ca. 20 ml). The aqueous phase was extracted with three portions of 50 ml of diethyl ether, respectively. The combined organic phases were then washed with 3 × 20 ml of 1 M HCl and 3 × 50 ml water and dried over magnesium sulfate. The solvent was removed on a rotary evaporator and the oil obtained was dried in vacuo. The reaction yielded 2.743 g (94%) of a colorless liquid. 1H NMR (600 MHz, CDCl3): δ = 4.23–4.21 (m, 2H, H-5), 3.72 (t, JH–H = 5.8 Hz, 2H, H-8), 3.72–3.70 (m, 2H, H-6), 3.61 (t, JH–H = 5.8 Hz, 2H, H-9), 1.20 (s, 9H, H-1/1′/1′′) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-3), 71.3 (−, 1C, C-8), 69.3 (−, 1C, C-6), 63.5 (−, 1C, C-5), 42.8 (−, 1C, C-9), 38.8 (*, 1C, C-2), 27.3 (+, 3C, C-1/1′/1′′) ppm. IR (ATR): ν = 2972, 2874, 1809, 1728, 1702, 1481, 1460, 1397, 1366, 1284, 1158, 1130, 1042, 1006, 940, 861, 770, 747, 669, 579, 546 cm−1. HR-ESI-MS: calculated for [C9H17ClO3 + Na]+: 231.0758. Found: 231.0762.Preparation of 2-[2-(2-chloroethoxy)ethoxy]ethyl-2,2-dimethyl-propanoate (2b)
Under a nitrogen atmosphere, 2.04 ml (14.00 mmol) of 2-[2-(2-chloroethoxy)ethoxy]ethanol 1b was dissolved in 50 ml of anhyd. diethyl ether and then 10 ml of anhyd. pyridine were added. 2.34 ml (19.00 mmol) of pivalic acid chloride were added at 0 °C. The reaction mixture was then stirred at room temperature for 48 hours. The reaction was then quenched with 1 M HCl (ca. 20 ml). The aqueous phase was extracted with 3 × 50 ml of diethyl ether, and the combined organic phases were washed with 3 × 20 ml of 1 M HCl and 3 × 50 ml of water, and finally dried over magnesium sulfate. The solvent was removed on a rotary evaporator and the oil obtained was dried in vacuo. The reaction yielded 2.810 g (80%) of a slightly yellow liquid. 1H NMR (600 MHz, CDCl3): δ = 4.22–4.20 (m, 2H, H-5), 3.75 (t, JH–H = 5.9 Hz, 2H, H-11), 3.70–3.69 (m, 2H, H-6), 3.67–3.65 (m, 4H, H-8, H-9), 3.62 (t, JH–H = 5.9 Hz, 2H, H-12), 1.20 (s, 9H, H-1/1′/1′′) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-3), 71.5 (−, 1C, C-11), 70.8 (−, 1C, C-8), 70.7 (−, 1C, C-9), 69.4 (−, 1C, C-6), 63.6 (−, 1C, C-5), 42.8 (−, 1C, C-12), 38.8 (*, 1C, C-2), 27.3 (+, 3C, C-1/1′/1′′) ppm. IR (ATR): ν = 2972, 2873, 1810, 1729, 1701, 1481, 1460, 1398, 1366, 1284, 1200, 1123, 1043, 1006, 940, 863, 769, 745, 668, 589, 547, 524 cm−1. HR-ESI-MS: calculated for [C11H21ClO4 + Na]+: 275.1021. Found: 275.1025.Preparation of 2-(2-iodoethoxy)ethyl-2,2-dimethylpropanoate (3a)
A reaction mixture of 13.506 g (90.11 mmol) of sodium iodide and 1.500 g (7.21 mmol) of 2-(2-chloroethoxy)-ethyl-2,2-dimethyl-propanoate 2a was dissolved in 20 ml of acetone and refluxed overnight. After cooling, 10 ml of water were added. The aqueous phase was then extracted with 3 × 50 ml of diethyl ether. The combined organic phases were washed with 3 × 50 ml of water and dried over magnesium sulfate. The solvent was removed on a rotary evaporator and the oil obtained was dried in vacuo. The reaction yielded 1.714 g of a red liquid which according to the spectra consist of a 0.35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of compound 2b and 3a. This results in a yield of 51% for 3a. However, further purification was not necessary since only the iodo-compound 3a is reactive in the subsequent steps. 1H NMR (600 MHz, CDCl3): δ = 4.22–4.20 (m, 2H, H-5), 3.75–3.73 (m, 2H, H-8), 3.70–3.68 (m, 2H, H-6), 3.23 (t, JH–H = 6.8 Hz, 2H, H-9), 1.20 (s, 9H, H-1/1′/1′′) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-3), 71.9 (−, 1C, C-8), 68.9 (−, 1C, C-6), 63.5 (−, 1C, C-5), 39.2 (*, 1C, C-2), 27.3 (+, 3C, C-1/1′/1′′), 2.7 (−, 1C, C-9) ppm. IR (ATR): ν = 2972, 2873, 1809, 1727, 1702, 1480, 1460, 1397, 1366, 1283, 1156, 1128, 1041, 1003, 940, 864, 770, 746, 669, 618, 579, 523 cm−1. HR-ESI-MS: calculated for [C9H17IO3 + Na]+: 323.0115. Found: 323.0123.
:1 mixture of compound 2b and 3a. This results in a yield of 51% for 3a. However, further purification was not necessary since only the iodo-compound 3a is reactive in the subsequent steps. 1H NMR (600 MHz, CDCl3): δ = 4.22–4.20 (m, 2H, H-5), 3.75–3.73 (m, 2H, H-8), 3.70–3.68 (m, 2H, H-6), 3.23 (t, JH–H = 6.8 Hz, 2H, H-9), 1.20 (s, 9H, H-1/1′/1′′) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-3), 71.9 (−, 1C, C-8), 68.9 (−, 1C, C-6), 63.5 (−, 1C, C-5), 39.2 (*, 1C, C-2), 27.3 (+, 3C, C-1/1′/1′′), 2.7 (−, 1C, C-9) ppm. IR (ATR): ν = 2972, 2873, 1809, 1727, 1702, 1480, 1460, 1397, 1366, 1283, 1156, 1128, 1041, 1003, 940, 864, 770, 746, 669, 618, 579, 523 cm−1. HR-ESI-MS: calculated for [C9H17IO3 + Na]+: 323.0115. Found: 323.0123.
Preparation of 2-[2-(2-iodoethoxy)ethoxy]ethyl-2,2-dimethyl-propanoate (3b)
A reaction mixture of 11.148 g (74.37 mmol) of sodium iodide and 1.500 g (5.95 mmol) of 2-[2-(2-chloroethoxy)ethoxy]ethyl-2,2-di-methylpropanoate 2b was dissolved in 20 ml of acetone and refluxed overnight. After cooling, 10 ml of water were added. The aqueous phase was then extracted with 3 × 50 ml of diethyl ether. The combined organic phases were then washed with 3 × 50 ml of water and dried over magnesium sulfate. The solvent was removed on a rotary evaporator and the oil obtained was dried in vacuo. The reaction yielded 1.688 g (82%) of a red liquid. 1H NMR (600 MHz, CDCl3): δ = 4.21–4.20 (m, 2H, H-5), 3.74 (t, JH–H = 6.8 Hz, 2H, H-11), 3.70–3.68 (m, 2H, H-6), 3.64 (s, 4H, H-8, H-9), 3.24 (t, JH–H = 6.8 Hz, 2H, H-12), 1.19 (s, 9H, H-1/1′/1′′) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-3), 72.1 (−, 1C, C-11), 70.7 (−, 1C, C-8), 70.4 (−, 1C, C-9), 69.4 (−, 1C, C-6), 63.6 (−, 1C, C-5), 38.8 (*, 1C, C-2), 27.3 (+, 3C, C-1/1′/1′′), 2.9 (−, 1C, C-12) ppm. IR (ATR): ν = 2971, 2872, 1809, 1728, 1701, 1481, 1460, 1398, 1365, 1284, 1133, 1042, 1003, 939, 863, 769, 619, 589, 578, 546, 523 cm−1. HR-ESI-MS: calculated for [C11H21IO4 + Na]+: 367.0377. Found: 367.0391.Preparation of 1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethyl)-1H-imidazolium iodide (4a)
0.500 g of crude 3a, which corresponds to 1.11 mmol of 2-(2-iodoethoxy)ethyl-2,2-dimethylpropanoate 3a and 0.13 ml (1.00 mmol) of 1-butylimidazole were dissolved in 10 ml of acetonitrile and refluxed for 70 hours. After the reaction had cooled, the solvent was removed on a rotary evaporator. The oil obtained was washed with 3 × 10 ml of diethyl ether and dried in vacuo. The reaction yielded 0.318 g (75%) of an orange oil. 1H NMR (600 MHz, CDCl3): δ = 9.91–9.88 (m, 1H, H-2), 7.59 (dd, JH–H = 3.6, 1.8 Hz, 1H, H-5), 7.37 (dd, JH–H = 3.6, 1.8 Hz, 1H, H-4), 4.59 (t, JH–H = 4.7 Hz, 2H, H-10), 4.29 (t, JH–H = 7.5 Hz, 2H, H-6), 4.17 (t, JH–H = 4.7 Hz, 2H, H-14), 3.87 (t, JH–H = 4.7 Hz, 2H, H-11), 3.66 (t, JH–H = 4.7 Hz, 2H, H-13), 1.89 (quint, JH–H = 7.5 Hz, 2H, H-7), 1.36 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.15 (s, 9H, H-18/18′/18′′), 0.93 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.4 (*, 1C, C-16), 136.5 (+, 1C, C-2), 123.5 (+, 1C, C-5), 121.6 (+, 1C, C-4), 69.5 (−, 1C, C-13), 68.8 (−, 1C, C-11), 63.0 (−, 1C, C-14), 50.0 (−, 1C, C-6), 50.0 (−, 1C, C-10), 38.7 (*, 1C, C-17), 32.0 (−, 1C, C-7), 27.2 (+, 3C, C-18/18′/18′′), 19.5 (−, 1C, C-8), 13.5 (+, 1C, C-9) ppm. IR (ATR): ν = 3135, 3074, 2962, 2872, 1722, 1623, 1563, 1457, 1395, 1361, 1284, 1161, 1124, 1042, 938, 856, 822, 732, 698, 640, 521, 480 cm−1. HR-ESI-MS: calculated for [C16H29N2O3]+: 297.2173. Found: 297.2178.Preparation of 1-butyl-3-[2-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethoxy)ethyl]-1H-imidazoliumiodide (4b)
0.500 g (1.45 mmol) of 2-[2-(2-iodoethoxy)ethoxy]ethyl-2,2-di-methylpropanoate 3b and 0.17 ml (1.31 mmol) of 1-butylimidazole were dissolved in 10 ml of acetonitrile and refluxed for 70 hours. After the reaction had cooled, the solvent was removed on a rotary evaporator. The oil obtained was washed with 3 × 10 ml of diethyl ether and dried in vacuo. The reaction yielded 0.542 g (80%) of an orange-brown oil. 1H NMR (600 MHz, CDCl3): δ = 9.91–9.88 (m, 1H, H-2), 7.66 (dd, JH–H = 3.6, 1.8 Hz, 1H, H-5), 7.37 (dd, JH–H = 3.6, 1.8 Hz, 1H, H-4), 4.58 (t, JH–H = 4.7 Hz, 2H, H-10), 4.29 (t, JH–H = 7.5 Hz, 2H, H-6), 4.17 (t, JH–H = 5.0 Hz, 2H, H-17), 3.88 (t, JH–H = 4.7 Hz, 2H, H-11), 3.65–3.60 (m, 4H, H-16), 3.60–3.57 (m, 2H, H-13), 3.60–3.58 (m, 2H, H-14), 1.88 (quint, JH–H = 7.5 Hz, 2H, H-7), 1.36 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.15 (s, 9H, H-21/21′/21′′), 0.93 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.5 (*, 1C, C-19), 136.4 (+, 1C, C-2), 123.6 (+, 1C, C-5), 121.7 (+, 1C, C-4), 70.4 (−, 1C, C-13), 70.3 (−, 1C, C-14), 69.2 (−, 1C, C-16), 68.9 (−, 1C, C-11), 63.2 (−, 1C, C-17), 50.0 (−, 1C, C-6), 49.9 (−, 1C, C-10), 38.7 (*, 1C, C-20), 32.1 (−, 1C, C-7), 27.2 (+, 3C, C-21/21′/21′′), 19.5 (−, 1C, C-8), 13.5 (+, 1C, C-9) ppm. IR (ATR): ν = 3136, 3075, 2961, 2871, 1722, 1625, 1563, 1457, 1396, 1359, 1284, 1162, 1120, 1040, 939, 858, 827, 732, 698, 641, 482 cm−1. HR-ESI-MS: calculated for [C18H33N2O4]+: 341.2435. Found: 341.2444.Preparation of 2-[2-(3-butyl-2-sulfanylidene-2,3-dihydro-1H-imidazol-1-yl)ethoxy]ethyl-2,2-dimethylpropanoate (5a)
0.205 g (0.48 mmol) of 1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]ethoxy}ethyl)-1H-imidazolium iodide was dissolved in 15 ml of anhyd. acetonitrile and 0.313 g (0.96 mmol) of cesium carbonate were added. After that, 0.019 g (0.58 mmol) of sulfur were added at room temperature. The reaction mixture was stirred at room temperature for three hours. The solvent was removed on a rotary evaporator and the residue was taken up in dichloromethane. The reaction mixture was filtered through a glass frit filled with Celite and then concentrated. The product obtained was dried in vacuo. The reaction yielded 0.087 g (55%) of a yellow-green oil. 1H NMR (600 MHz, CDCl3): δ = 6.85 (d, JH–H = 2.2 Hz, 1H, H-5), 6.62 (d, JH–H = 2.2 Hz, 1H, H-4), 4.25 (t, JH–H = 4.9 Hz, 2H, H-10), 4.17 (t, JH–H = 4.8 Hz, 2H, H-14), 4.02 (t, JH–H = 7.5 Hz, 2H, H-6), 3.76 (t, JH–H = 4.9 Hz, 2H, H-11), 3.62 (t, JH–H = 4.8 Hz, 2H, H-13), 1.74 (quint, JH–H = 7.5 Hz, 2H, H-7), 1.38 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.17 (s, 9H, H-18/18′/18′′), 0.94 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.5 (*, 1C, C-16), 161.6 (*, 1C, C-2), 118.5 (+, 1C, C-5), 116.2 (+, 1C, C-4), 69.2 (−, 1C, C-11), 69.2 (−, 1C, C-13), 63.5 (−, 1C, C-14), 47.9 (−, 1C, C-10), 47.8 (−, 1C, C-6), 38.8 (*, 1C, C-17), 31.1 (−, 1C, C-7) 27.3 (+, 3C, C-18/18′/18′′), 19.9 (−, 1C, C-8), 13.8 (+, 1C, C-9) ppm. IR (ATR): ν = 2959, 2934, 2869, 1725, 1450, 1414, 1364, 1283, 1229, 1157, 1122, 1044, 936, 857, 764, 714, 674, 527 cm−1. HR-ESI-MS: calculated for [C16H28N2O3S + Na]+: 351.1713. Found: 351.1716.Preparation of 2-[2-(3-butyl-2-selanylidene-2,3-dihydro-1H-imidazol-1-yl)ethoxy]ethyl-2,2-dimethylpropanoate (5b)
0.210 g (0.50 mmol) of 1-butyl-3-(2-{2-[(2,2-dimethylpropa-noyl)oxy]ethoxy}ethyl)-1H-imidazolium iodide was dissolved in 15 ml of dry acetonitrile and 0.326 g (1.00 mmol) of cesium carbonate were added. After that, 0.047 g (0.59 mmol) of selenium were added at room temperature. The reaction mixture was stirred at reflux for three hours. The solvent was removed on a rotary evaporator and the residue was taken up in dichloromethane. The reaction mixture was filtered through a glass frit filled with Celite and then concentrated. The product obtained was dried in vacuo. The reaction yielded 0.144 g (77%) of a yellow oil. 1H NMR (600 MHz, CDCl3): δ = 7.02 (d, JH–H = 2.2 Hz, 1H, H-5), 6.78 (d, JH–H = 2.2 Hz, 1H, H-4), 4.34 (t, JH–H = 4.9 Hz, 2H, H-10), 4.16 (t, JH–H = 4.8 Hz, 2H, H-14), 4.10 (t, JH–H = 7.5 Hz, 2H, H-6), 3.78 (t, JH–H = 4.9 Hz, 2H, H-11), 3.61 (t, JH–H = 4.8 Hz, 2H, H-13), 1.76 (quint, JH–H = 7.5 Hz, 2H, H-7), 1.37 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.16 (s, 9H, H-18/18′/18′′), 0.94 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.5 (*, 1C, C-16), 154.9 (*, 1C, C-2), 120.7 (+, 1C, C-5), 118.2 (+, 1C, C-4), 69.2 (−, 1C, C-11), 69.2 (−, 1C, C-13), 63.4 (−, 1C, C-14), 49.8 (−, 1C, C-10), 49.7 (−, 1C, C-6), 38.8 (*, 1C, C-17), 31.1 (−, 1C, C-7) 27.3 (+, 3C, C-18/18′/18′′), 19.9 (−, 1C, C-8), 13.8 (+, 1C, C-9) ppm. 77Se NMR (114 MHz, CDCl3): δ = −12.4 (s) ppm. IR (ATR): ν = 2960, 2869, 1724, 1565, 1455, 1407, 1363, 1283, 1228, 1161, 1123, 1044, 936, 858, 762, 716, 669, 522, 481 cm−1. HR-ESI-MS: calculated for [C16H28N2O3Se + Na]+: 399.1157. Found: 399.1159.Preparation of 2-{2-[2-(3-butyl-2-sulfanylidene-2,3-dihydro-1H-imidazol-1-yl)ethoxy]ethoxy}ethyl 2,2-dimethylpropanoate (6a)
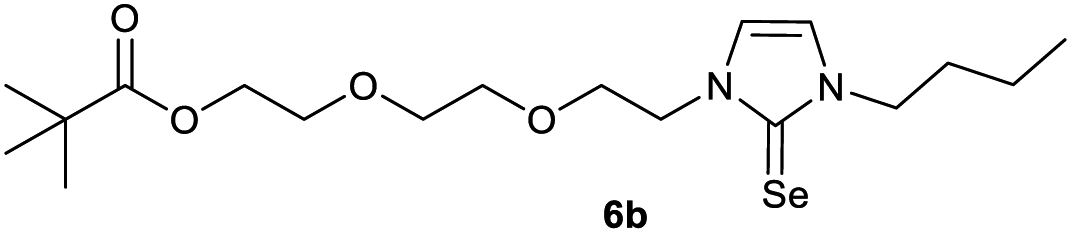
0.100 g (0.21 mmol) of 1-butyl-3-[2-(2-{2-[(2,2-dimethylpropa-noyl)oxy]ethoxy}ethoxy)ethyl]-1H-imidazolium iodide was dis-solved in 15 ml of dry acetonitrile and 0.137 g (0.42 mmol) of cesium carbonate were added. After that, 0.008 g (0.26 mmol) of sulfur were added at room temperature. The reaction mixture was stirred at room temperature for three hours. The solvent was removed on a rotary evaporator and the residue was taken up in dichloromethane. The reaction mixture was filtered through a glass frit filled with Celite and then concentrated. The product obtained was dried in vacuo. The reaction yielded 0.031 g (40%) of a yellow oil. 1H NMR (600 MHz, CDCl3): δ = 6.89 (d, JH–H = 2.1 Hz, 1H, H-5), 6.63 (d, JH–H = 2.1 Hz, 1H, H-4), 4.25 (t, JH–H = 5.0 Hz, 2H, H-10), 4.19 (t, JH–H = 4.9 Hz, 2H, H-17), 4.02 (t, JH–H = 7.3 Hz, 2H, H-6), 3.77 (t, JH–H = 5.0 Hz, 2H, H-11), 3.65 (t, JH–H = 4.9 Hz, 2H, H-16), 3.61–3.57 (m, 4H, H-13, H-14), 1.74 (quint, JH–H = 7.3 Hz, 2H, H-7), 1.37 (sext, JH–H = 7.3 Hz, 2H, H-8), 1.19 (s, 9H, H-21/21′/21′′), 0.95 (t, JH–H = 7.3 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.6 (*, 1C, C-19), 161.6 (*, 1C, C-2), 118.5 (+, 1C, C-5), 116.3 (+, 1C, C-4), 70.5 (−, 2C, C-14, C-13), 69.3 (−, 1C, C-16), 69.1 (−, 1C, C-11), 63.5 (−, 1C, C-17), 47.8 (−, 1C, C-6), 47.7 (−, 1C, C-10), 38.8 (*, 1C, C-20), 31.1 (−, 1C, C-7), 27.3 (−, 3C, C-21/21′/21′′), 19.9 (−, 1C, C-8), 13.8 (+, 1C, C-9) ppm. IR (ATR): ν = 2959, 2934, 2869, 1725, 1450, 1414, 1362, 1284, 1230, 1117, 1044, 938, 861, 764, 715, 675, 528 cm−1. HR-ESI-MS: calculated for [C18H32N2O4S + Na]+: 395.1975. Found: 395.1969.Preparation of 2-{2-[2-(3-butyl-2-selanylidene-2,3-dihydro-1H-imidazol-1-yl)ethoxy]ethoxy}ethyl 2,2-dimethylpropanoate (6b)
0.100 g (0.21 mmol) of 1-butyl-3-[2-(2-{2-[(2,2-dimethylpropa-noyl)oxy]ethoxy}ethoxy)ethyl]-1H-imidazolium iodide was dis-solved in 15 ml of anhyd. acetonitrile and 0.137 g (0.42 mmol) of cesium carbonate were added. After that, 0.021 g (0.26 mmol) of selenium were added at room temperature. The reaction mixture was stirred at reflux for three hours. The solvent was then removed on a rotary evaporator and the residue was dissolved in dichloromethane. The reaction mixture was filtered through a glass frit filled with Celite and then concentrated. The product obtained was dried in vacuo. The reaction yielded 0.036 g (41%) of a yellow oil. 1H NMR (600 MHz, CDCl3): δ = 7.08 (d, JH–H = 1.9 Hz, 1H, H-5), 6.81 (d, JH–H = 1.9 Hz, 1H, H-4), 4.35 (t, JH–H = 5.0 Hz, 2H, H-10), 4.19 (t, JH–H = 4.9 Hz, 2H, H-17), 4.10 (t, JH–H = 7.5 Hz, 2H, H-6), 3.79 (t, JH–H = 5.0 Hz, 2H, H-11), 3.65 (t, JH–H = 4.9 Hz, 2H, H-16), 3.59–3.57 (m, 4H, H-13, H-14), 1.76 (quint, JH–H = 7.5 Hz, 2H, H-7), 1.37 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.18 (s, 9H, H-21/21′/21′′), 0.94 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 178.5 (*, 1C, C-19), 154.5 (*, 1C, C-2), 120.7 (+, 1C, C-5), 118.4 (+, 1C, C-4), 70.5 (−, 1C, C-14), 70.5 (−, 1C, C-13), 69.3 (−, 1C, C-16), 69.2 (−, 1C, C-11), 63.5 (−, 1C, C-17), 49.7 (−, 1C, C-6), 49.6 (−, 1C, C-10), 38.8 (*, 1C, C-20), 31.3 (−, 1C, C-7), 27.3 (−, 3C, C-21/21′/21′′), 19.9 (−, 1C, C-8), 13.8 (+, 1C, C-9) ppm. 77Se NMR (114 MHz, CDCl3): δ = −12.4 (s) ppm. IR (ATR): ν = 2960, 2934, 2870, 1724, 1456, 1408, 1362, 1284, 1229, 1162, 1120, 1044, 912, 726, 670, 645 cm−1. HR-ESI-MS: calculated for [C18H32N2O4Se + Na]+: 443.1420. Found: 443.1417.Preparation of [1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethyl)-1H-imidazolium-2-yl](cycloocta-1,5-diene-1,5-diyl-κ2C1,C5)rhodium(I)-iodide (7a)
Under a nitrogen atmosphere, 0.175 g (0.41 mmol) of 1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]ethoxy}ethyl)-1H-imidazolium iodide was dissolved in 8 ml of dry dichloromethane and 0.049 g (0.21 mmol) of silver oxide were added. The reaction mixture was stirred at room temperature for one hour until the solution decolorized. 0.104 g (0.21 mmol) of bis-(1,5-cyclo-octadiene)dirhodium(I)-dichloride was then added. The reaction was stirred at room temperature overnight. It was then filtered through Celite and purified by column chromatography using a solvent mixture of petroleum ether (PE) and ethyl acetate (EE) = 1:1. The obtained product was dried in vacuo. The reaction yielded 0.177 g (68%) of an orange oil. 1H NMR (600 MHz, CDCl3): δ = 7.04 (d, JH–H = 1.8 Hz, 1H, H-4), 6.76 (d, JH–H = 1.8 Hz, 1H, H-5), 5.03–4.99 (m, 1H, CH-COD), 4.95–4.89 (m, 2H, H-6), 4.54–4.43 (m, 3H, CH-COD, H-10), 4.20–4.17 (m, 2H, H-14), 3.92–3.88 (m, 2H, H-11), 3.65–3.63 (m, 2H, H-13), 3.29–3.21 (m, 2H, CH-COD), 2.40–2.29 (m, 4H, CH2-COD), 1.97–1.73 (m, 6H, H-7, CH2-COD), 1.46 (sext, JH–H = 7.6 Hz, 2H, H-8), 1.17 (s, 9H, H-18/18′/18′′), 1.02 (t, JH–H = 7.6 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 181.8 (*, d, JRh–C = 51.2 Hz, 1C, C-2), 178.5 (*, 1C, C-16), 122.1 (+, 1C, C-4), 119.8 (+, 1C, C-5), 98.5 (+, d, JRh–C = 6.7 Hz, 1C, CH-COD), 98.3 (+, d, JRh–C = 6.7 Hz, 1C, CH-COD), 71.0 (−, 1C, C-11), 69.2 (−, 1C, C-13), 68.4 (+, d, JRh–C = 14.5 Hz, 1C, CH-COD), 67.7 (+, d, JRh–C = 14.5 Hz, 1C, CH-COD), 63.4 (−, 1C, C-16), 50.8 (−, 1C, C-6 or C-10), 50.6 (−, 1C, C-10 or C-6), 38.8 (*, 1C, C-17), 33.4 (−, 1C, CH2-COD), 33.1 (−, 1C, CH2-COD), 32.7 (−, 1C, CH2-COD or C-7), 29.3 (−, 1C, CH2-COD or C-7), 28.6 (−, 1C, CH2-COD or C-7), 27.3 (+, 3C, C-18/18′/18′′), 20.2 (−, 1C, C-8), 13.9 (+, 1C, C-9) ppm. IR (ATR): ν = 2958, 2934, 2872, 2831, 1725, 1561, 1459, 1415, 1398, 1366, 1283, 1227, 1160, 1124, 1080, 1044, 917, 861, 728, 699, 644, 520, 485, 446 cm−1. HR-ESI-MS: calculated for [C24H40RhN2O3]+: 507.2088. Found: 507.2087.
Preparation of {1-butyl-3-[2-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethoxy)ethyl]-1H-imidazolium-2-yl}(cycloocta-1,5-diene-1,5-diyl-κ2C1,C5)-rhodium(I)iodide (7b)
Under a nitrogen atmosphere, 0.200 g (0.43 mmol) of 1-butyl-3-[2-(2-{2-[(2,2-dimethylpropanoyl)oxy]ethoxy}ethoxy)ethyl]-1H-imidazolium iodide was dissolved in 8 ml of dry dichloromethane and 0.050 g (0.21 mmol) of silver oxide were added. The reaction mixture was stirred at room temperature for one hour until the solution decolorized. 0.104 g (0.21 mmol) of bis-(1,5-cyclo-octadiene)dirhodium(I)-dichloride was then added. The reaction was stirred at room temperature overnight. The reaction mixture was then filtered through Celite and finally purified by column chromatography in a solvent mixture of petroleum ether (PE) and ethyl acetate (EE) = 1:1. The obtained product was dried in vacuo. The reaction yielded 0.160 g (55%) of a red oil. 1H-NMR (600 MHz, CDCl3): δ = 7.06 (d, JH–H = 2.0 Hz, 1H, H-4), 6.78 (d, JH–H = 2.0 Hz, 1H, H-5), 5.02–4.99 (m, 1H, CH-COD), 4.96–4.89 (m, 3H, CH-COD, H-6), 4.51–4.44 (m, 2H, H-10), 4.20–4.19 (m, 2H, H-17), 3.94–3.89 (m, 2H, H-11), 3.67–3.65 (m, 2H, H-16), 3.64–3.58 (m, 4H, H-13, H-14), 3.28–3.22 (m, 2H, CH-COD), 2.41–2.29 (m, 4H, CH2-COD), 1.99–1.77 (m, 6H, H-7, CH2-COD), 1.47 (sext, JH–H = 7.5 Hz, 2H, H-8), 1.19 (s, 9H, H-21/21′/21′′), 1.02 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C-NMR (150 MHz, CDCl3): δ = 181.8 (*, d, JRh–C = 50.7 Hz, 1C, C-2), 178.5 (*, 1C, C-19), 122.2 (+, 1C, C-4), 119.8 (+, 1C, C-5), 98.5 (+, d, JRh–C = 6.6 Hz, 1C, CH-COD), 98.3 (+, d, JRh–C = 6.6 Hz, 1C, CH-COD), 71.0 (−, 1C, C-11), 70.5 (−, 1C, C-13), 70.5 (−, 1C, C-14), 69.2 (−, 1C, C-16), 68.4 (+, d, JRh–C = 14.6 Hz, 1C, CH-COD), 67.8 (+, d, JRh–C = 14.6 Hz, 1C, CH-COD), 63.6 (−, 1C, C-17), 50.7 (−, 1C, C-6), 50.6 (−, 1C, C-10), 38.8 (*, 1C, C-20), 33.3 (−, 1C, CH2-COD), 33.1 (−, 1C, CH2-COD), 32.7 (−, 1C, CH2-COD or C-7), 29.2 (−, 1C, CH2-COD or C-7), 28.7 (−, 1C, CH2-COD or C-7), 27.3 (+, 3C, C-21/21′/21′′), 20.2 (−, 1C, C-8), 13.9 (+, 1C, C-9) ppm. IR (ATR): ν = 2957, 2929, 2870, 1725, 1560, 1459, 1398, 1366, 1283, 1228, 1161, 1114, 1041, 938, 861, 732, 689, 517 cm−1. HR-ESI-MS: calculated for [C26H44RhN2O4]+: 551.2351. Found: 551.2355.
Preparation of [1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethyl)-1H-imidazolium-2-yl](cycloocta-4,8-diene-1,4-diyl-κ2C1,C5)iridium(I)iodide (8a)
Under a nitrogen atmosphere, 0.178 g (0.42 mmol) of 1-butyl-3-(2-{2-[(2,2-dimethylpropanoyl)oxy]ethoxy}ethyl)-1H-imidazolium iodide was dissolved in 8 ml of dry dichloromethane and 0.049 g (0.21 mmol) of silver oxide were added. The reaction mixture was stirred at room temperature for one hour until the solution decolorized. 0.141 g (0.21 mmol) of bis-(1,5-cyclo-octadiene)diiridium(I)-dichloride was then added. The reaction was stirred at room temperature overnight. It was then filtered through Celite and finally purified by column chromatography in a solvent mixture of petroleum ether (PE) and ethyl acetate (EE) = 1:1. The obtained product was dried in vacuo. The reaction yielded 0.187 g (62%) of a red oil. 1H NMR (600 MHz, CDCl3): δ = 7.06 (d, JH–H = 2.0 Hz, 1H, H-5), 6.78 (d, JH–H = 2.0 Hz, 1H, H-4), 4.77–4.73 (m, 1H, H-10), 4.62–4.58 (m, 1H, CH-COD), 4.53–4.49 (m, 1H, CH-COD), 4.42–4.39 (m, 1H, H-10), 4.38–4.33 (m, 2H, H-6), 4.20–4.18 (m, 2H, H-14), 3.85 (t, JH–H = 4.5 Hz, 2H, H-11), 3.64 (t, JH–H = 5.0 Hz, 2H, H-13), 2.91–2.85 (m, 2H, CH-COD), 2.25–2.09 (m, 4H, CH2-COD), 1.79–1.72 (m, 2H, H-7), 1.70–1.54 (m, 4H, CH2-COD), 1.49–1.39 (m, 2H, H-8), 1.19 (s, 9H, H-18/18′/18′′), 1.00 (t, JH–H = 7.4 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 179.9 (*, 1C, C-2), 178.5 (*, 1C, C-16), 121.9 (+, 1C, C-5), 119.5 (+, 1C, C-4), 84.6 (+, 1C, CH-COD), 84.1 (+, 1C, CH-COD), 71.0 (−, 1C, C-11), 69.2 (−, 1C, C-13), 63.4 (−, 1C, C-14), 52.0 (+, 1C, CH-COD), 51.4 (+, 1C, CH-COD), 50.5 (−, 1C, C-6 or C-10), 50.3 (−, 1C, C-6 or C-10), 38.8 (*, 1C, C-17), 34.0 (−, 1C, CH2-COD), 33.4 (−, 1C, CH2-COD), 33.1 (−, 1C, C-7), 30.0 (−, 1C, CH2-COD), 29.4 (−, 1C, CH2-COD), 27.3 (+, 3C, C-18/18′/18′′), 20.1 (−, 1C, C-8), 13.9 (+, 1C, C-9) ppm. IR (ATR): ν = 2957, 2931, 2872, 2831, 1725, 1556, 1457, 1414, 1366, 1327, 1283, 1227, 1159, 1125, 1078, 1044, 969, 917, 862, 818, 770, 730, 706, 645, 591, 521, 474 cm−1. HR-ESI-MS: calculated for [C24H40IrN2O3]+: 597.2663. Found: 597.2664.
Preparation of {1-butyl-3-[2-(2-{2-[(2,2-dimethylpropanoyl)oxy]-ethoxy}ethoxy)-ethyl]-1H-imidazolium-2-yl}(cycloocta-1,5-diene-1,5-diyl-κ2C1,C5)-iridium(I)iodide (8b)
Under a nitrogen atmosphere, 0.166 g (0.35 mmol) of 1-butyl-3-[2-(2-{2-[(2,2-dimethylpropanoyl)oxy]ethoxy}ethoxy)ethyl]-1H-imidazolium iodide was dissolved in 8 ml of dry dichloromethane and 0.041 g (0.18 mmol) of silver oxide were added. The reaction mixture was stirred at room temperature for one hour until the solution decolorized. 0.121 g (0.18 mmol) of bis-(1,5-cyclo-octadiene)diiridium(I)-dichloride was then added. The reaction was stirred at room temperature overnight. It was then filtered through Celite and finally purified by column chromatography in a solvent mixture of petroleum ether (PE) and ethyl acetate (EE) = 1:1. The obtained product was dried in vacuo. The reaction yielded 0.131 g (49%) of an orange oil. 1H NMR (600 MHz, CDCl3): δ = 7.07 (d, JH–H = 1.8 Hz, 1H, H-4), 6.78 (d, JH–H = 1.8 Hz, 1H, H-5), 4.77–4.72 (m, 1H, H-6), 4.61–4.57 (m, 1H, CH-COD), 4.52–4.48 (m, 1H, CH-COD), 4.41–4.32 (m, 3H, H-10, H-6), 4.21–4.18 (t, JH–H = 4.8 Hz, 2H, H-17), 3.89–3.83 (m, 2H, H-16), 3.63–3.59 (m, 4H, H-13, H-14), 2.91–2.85 (m, 2H, CH-COD), 2.22–2.12 (m, 4H, CH2-COD), 1.94–1.88 (m, 1H, H-7), 1.76–1.65 (m, 3H, CH2-COD, H-7), 1.63–1.40 (m, 2H, H-8), 1.19 (s, 9H, H-21/21′/21′′), 0.99 (t, JH–H = 7.5 Hz, 3H, H-9) ppm. 13C NMR (150 MHz, CDCl3): δ = 179.8 (*, 1C, C-2), 178.6 (*, 1C, C-16), 121.9 (+, 1C, C-4), 119.5 (+, 1C, C-5), 84.4 (+, 1C, CH-COD), 84.0 (+, 1C, CH-COD), 70.9 (−, 1C, C-11), 70.5 (−, 2C, C-13, C-14), 69.3 (−, 1C, C-16), 63.6 (−, 1C, C-17), 51.9 (+, 1C, CH-COD), 51.4 (+, 1C, CH-COD), 50.3 (−, 1C, C-6 or C-10), 50.3 (−, 1C, C-6 or C-10), 38.8 (*, 1C, C-20), 33.9 (−, 1C, CH2-COD), 33.4 (−, 1C, CH2-COD), 33.1 (−, 1C, CH2-COD or C-7), 29.9 (−, 1C, CH2-COD or C-7), 29.4 (−, 1C, CH2-COD or C-7), 27.3 (+, 3C, C-21/21′/21′′), 20.1 (−, 1C, C-8), 13.9 (+, 1C, C-9) ppm. IR (ATR): ν = 2957, 2931, 2872, 2832, 1725, 1567, 1457, 1414, 1366, 1327, 1284, 1228, 1161, 1119, 1042, 969, 916, 863, 838, 819, 770, 729, 707, 591, 510, 474 cm−1. HR-ESI-MS: calculated for [C26H44IrN2O4]+: 641.2925. Found: 641.2923.
Conflicts of interest
No conflicts of interest to declare.Acknowledgements
Birgit Wawrzinek and Florian Ahrend-Vaiana are gratefully acknowledged for the NMR-spectroscopy. Maike Weigert is gratefully acknowledged for ESI-MS spectra. Maike Gamenik and Sarah Tuchtfeld are gratefully acknowledged for measuring the zeta potentials for the minerals. Petra Lassen is gratefully acknowledged for measuring the chemical composition of lithium and gehlenite. We thank the Deutsche Forschungsgemeinschaft (DFG) for funding of this work within the Priority Programme "Engineered Artificial Minerals (EnAM) – a geo-metallurgical tool to recycle critical elements from waste streams (SPP 2315), project number 470324113.Notes and references
- (a) L. Domaracka, S. Matuskova, M. Tausova, A. Senova and B. Kowal, Sustainability, 2022, 14, 6554 CrossRef CAS; (b) I. Cerny, M. Vanek, E. W. Maruszewska and F. Benes, Resour. Policy, 2021, 74, 102417 CrossRef; (c) P. Ferro and F. Bonollo, Mater. Des., 2019, 177, 107848 CrossRef CAS; (d) M. Hofmann, H. Hofmann, C. Hageluken and A. Hool, Sustainable Mater. Technol., 2018, 17, e00074 CrossRef CAS; (e) M. L. Grilli, T. Bellezze, E. Gamsjäger, A. Rinaldi, P. Novak, S. Balos, R. R. Piticescu and M. L. Ruello, Materials, 2017, 10, 285 CrossRef PubMed; (f) A. H. Tkaczyk, A. Bartl, A. Amato, V. Lapkovskis and M. Petranikova, J. Phys. D: Appl. Phys., 2018, 51, 203001 CrossRef.
- (a) L. C. Zheng, G. Chen, L. T. Liu and Y. Q. Hu, Front. Energy Res., 2022, 10, 992617 CrossRef; (b) G. Martin, L. Rentsch, M. Hock and M. Bertau, Energy Storage Mater., 2017, 6, 171–179 CrossRef; (c) D. Calisaya-Azpilcueta, S. Herrera-Leon, F. A. Lucay and L. A. Cisternas, Minerals, 2020, 10, 604 CrossRef CAS; (d) F. Meng, J. McNeice, S. S. Zadeh and A. Ghahreman, Miner. Process. Extr. Metall. Rev., 2021, 42, 123–141 CrossRef CAS.
- X. Y. Guo, J. X. Zhang and Q. H. Tian, Renewable Sustainable Energy Rev., 2021, 137, 110461 CrossRef CAS.
- (a) M. Garside, https://www.statista.com/statistics/452025/projected-total-demand-for-lithium-globally/; (b) C. Scheller, S. Blömeke, M. Nippraschk, K. Schmidt, M. Mennenga, T. S. Spengler, C. Herrmann and D. Goldmann, Procedia CIRP, 2021, 98, 464–469 CrossRef.
- (a) Y. Di, G. Chen, L. L. Zhang, Z. Chen, R. Zhang, M. I. Asghar, S. J. Geng and P. D. Lund, J. Power Sources, 2021, 503, 120070 Search PubMed; (b) C. Venkateswaran, H. Sreemoolanadhan and R. Vaish, Int. Mater. Rev., 2022, 67, 620–657 CrossRef CAS; (c) S. Bahel, R. Singh, G. Kaur and S. B. Narang, Ferroelectrics, 2016, 502, 49–56 CrossRef CAS.
- (a) V. N. Sigaev, A. S. Lipat'ev, S. S. Fedotov, S. V. Lotarev, A. S. Naumov and D. M. Shevyakina, Glass Ceram., 2022, 79, 45–47 CrossRef CAS; (b) S. J. Huang, W. Z. Wang, H. Jiang, H. F. Zhao and Y. P. Ma, Materials, 2022, 15, 4555 CrossRef CAS PubMed.
- (a) F. M. Yeh, V. Volli, L. W. Bin, P. H. Tung and C. M. Shu, Appl. Therm. Eng., 2019, 150, 1328–1336 CrossRef CAS; (b) Z. H. Song, Y. M. Liang, M. J. Fan, F. Zhou and W. M. Liu, Tribol. Int., 2014, 70, 136–141 CrossRef CAS.
- (a) W. Rivera, G. Moreno-Quintanar, C. O. Rivera, R. Best and F. Martinez, Sol. Energy, 2011, 85, 38–45 CrossRef CAS; (b) K. Sedighi, M. Farhadi and M. Liaghi, Proc. Inst. Mech. Eng., Part C, 2007, 221, 1345–1351 CrossRef CAS.
- (a) E. Stefanelli, M. Puccini, A. Pesetti, R. Lo Frano and D. Aquaro, Nucl. Mater. Energy, 2022, 30, 101131 CrossRef CAS; (b) L. J. Mao, P. R. Zhang, H. Q. Ju, X. L. Zhou, Z. X. Xue, C. M. Wang, J. H. Sun, Y. Z. Jia, F. Shao, X. W. Zou, B. Li and Y. Jing, J. Mol. Liq., 2022, 367, 120357 CrossRef CAS.
- Y. Cao, S. L. Tan, E. J. H. Cheung, S. Y. Siew, C. J. Li, Y. Liu, C. S. Tan, M. Lal, G. Y. Chen, K. Dogheche, P. Yang, S. Pennycock, A. T. S. Wee, S. Chua, E. Dogheche, T. Venkatesan and A. Danner, Adv. Mater., 2021, 33, 2101128 CrossRef CAS PubMed.
- (a) H. Li, H. Qiu, T. Schirmer, D. Goldmann and M. Fischlschweiger, ACS ES&T Engg, 2022, 2, 1883, DOI:10.1021/acsestengg.2c00105; (b) A. Wittkowski, T. Schirmer, H. Qiu, D. Goldmann and U. E. A. Fittschen, Metals, 2021, 11, 188 CrossRef CAS; (c) T. Schirmer, H. Qiu, D. Goldmann, C. Stallmeister and B. Friedrich, Minerals, 2022, 12, 310 CrossRef CAS; (d) T. Schirmer, H. Qiu, H. Li, D. Goldmann and M. Fischlschweiger, Metals, 2020, 10, 1633 CrossRef CAS; (e) T. Elwert, D. Goldmann, T. Schirmer and K. Strauß, World Metall.--Erzmet., 2012, 65, 163–171 CAS.
- (a) M. Y. Prajitno, S. Tangparitkul, H. Zhang, D. Harbottle and T. N. Hunter, J. Hazard. Mater., 2021, 402, 123567 CrossRef CAS PubMed; (b) R. Atkin, V. Craig, E. J. Wanless and S. Biggs, Adv. Colloid Interface Sci., 2003, 103, 219–304 CrossRef CAS PubMed; (c) Y. Huang, M. Takaoka, N. Takeda and K. Oshita, J. Hazard. Mater., 2003, 100, 259–270 CrossRef CAS PubMed; (d) P. Mavros and K. A. Matis, Innovations in Flotation Technology, Springer, Netherlands, Dordrecht, 2013 Search PubMed; (e) H. Wu, W. Wang, Y. Huang, G. Han, S. Yang, S. Su, H. Sana, W. Peng, Y. Cao and J. Liu, J. Hazard. Mater., 2019, 371, 592–602 CrossRef CAS PubMed; (f) M. Zhang, X. Lu, Q. Zhou, L. Xie and C. Shen, J. Hazard. Mater., 2019, 362, 196–205 CrossRef CAS PubMed.
- (a) B. A. Wills and T. Napier-Munn, Mineral processing technology, Elsevier, Amsterdam, 2006, p. 267 Search PubMed; (b) R. M. G. Lima, P. R. G. Brandao and A. E. C. Peres, Miner. Eng., 2005, 18, 267–273 CrossRef CAS; (c) X. Wenig, G. Mei and T. Zhao, Sep. Purif. Technol., 2013, 103, 187–194 CrossRef; (d) A. V. Nguyen and H. J. Schulze, Colloidal Science of Flotation, New York, Marcel Dekker Inc, 2004 Search PubMed; (e) M. Ejtemaei, M. Gharabaghi and M. Irannajad, Adv. Colloid Interface Sci., 2014, 206, 68–78 CrossRef CAS PubMed; (f) S. Aghazadeh, S. K. Mousavinezhad and M. Gharabaghi, Adv. Colloid Interface Sci., 2015, 225, 203–217 CrossRef CAS PubMed; (g) Z. J. Wu, X. M. Wang, H. N. Liu, H. F. Zhang and J. D. Miller, Adv. Colloid Interface Sci., 2016, 235, 190–200 CrossRef CAS PubMed.
- Y. Xing, M. Xu, X. Gui, Y. Cao, B. Babel, M. Rudolph, S. Weber, M. Kappl and H.-J. Butt, Adv. Colloid Interface Sci., 2018, 256, 373–392 CrossRef CAS PubMed.
- (a) T. N. Hunter, R. J. Pugh, G. V. Franks and G. J. Jameson, Adv. Colloid Interface Sci., 2008, 137, 57–81 CrossRef CAS PubMed; (b) H. Polat and D. Erdogan, J. Hazard. Mater., 2007, 148, 267–273 CrossRef CAS PubMed; (c) R. M. Rahman, S. Ata and G. J. Jameson, Int. J. Miner. Process., 2012, 106, 70–77 CrossRef; (d) D. Zamboulis, S. Pataroudi, A. Zouboulis and K. Matis, Desalination, 2004, 162, 159–168 CrossRef CAS.
- (a) C. Dessemond, F. Lajoie-Leroux, G. Soucy, N. Laroche and J. F. Magnan, Minerals, 2019, 9, 334 CrossRef CAS; (b) D. I. Gorves, L. Zhang, I. M. Groves and A. K. Sener, Acta Pet. Sin., 2022, 38, 1–8 Search PubMed.
- (a) G. Zhu, Y. Wang, X. Wang, F. Yu and J. D Miller, Colloids Surf., A, 2018, 558, 313–321 CrossRef CAS; (b) L. Filippov, S. Farrokhpay, L. Lyo and I. Filippova, Minerals, 2019, 9, 372 CrossRef CAS.
- (a) J. Tian, L. Xu, H. Wu, S. Fang, W. Deng, T. Peng, W. Sun and Y. Hu, J. Cleaner Prod., 2018, 174, 625–633 CrossRef CAS; (b) H. Wu, J. Tian, L. Xu, S. Fang, Z. Zhang and R. Chi, Miner. Eng., 2018, 127, 42–47 CrossRef CAS; (c) G. Zhu, Y. Wang, X. Wang, J. D. Miller, D. Lu, X. Zheng, Y. Zhao and H. Zheng, Trans. Nonferrous Met. Soc. China, 2019, 29, 1527–1537 CrossRef CAS.
- (a) H. Taş, J. Adams, J. C. Namyslo and A. Schmidt, RSC Adv., 2021, 11, 36450–36458 RSC; (b) A. Schmidt, C. F. Otto, J. C. Namyslo, A. Fischer and K. Filip, Bull. Mater. Res. Eng., 2021, 10, 601–620 Search PubMed.
- (a) K. Hillrichs, J. C. Namyslo, F. Lederle, E. G. Hübner and A. Schmidt, Synthesis, 2022, 3351–3366 CAS; (b) S. Mummel, F. Lederle, E. Hübner, J. C. Namyslo, M. Nieger and A. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 18882–18887 CrossRef CAS PubMed; (c) T. Freese, J. C. Namyslo, M. Nieger and A. Schmidt, RSC Adv., 2019, 9, 4781–4788 RSC; (d) M. Liu, M. Nieger and A. Schmidt, Chem. Commun., 2015, 51, 477–479 RSC; (e) Z. Guan, S. Wiechmann, M. Drafz, E. Hübner and A. Schmidt, Org. Biomol. Chem., 2013, 11, 3558–3567 RSC; (f) A. Schmidt, M. K. Kindermann, P. Vainiotalo and M. Nieger, J. Org. Chem., 1999, 64, 9499–9506 CrossRef CAS.
- F. Liu, P. F. Hao, T. L. Yu, Q. Guan and Y. L. Fu, J. Mol. Struct., 2016, 1119, 431–436 CrossRef CAS.
- J. J. Wang, L. X. Mo, X. Y. Li, Z. K. Geng and Y. L. Zeng, J. Mol. Model., 2016, 22, 299 CrossRef PubMed.
- H. I. Althagbi, R. R. Bernstein, W. C. Crombie, J. R. Lane, D. K. McQuiston, M. A. Oosterwijk, G. C. Saunders and W. Y. Zou, J. Fluorine Chem., 2018, 206, 61–71 CrossRef CAS.
- B. Xiong, S. S. Mao, F. Ding, T. Shen, J. J. Wang, X. Jin and M. L. Gao, Colloids Surf., A, 2021, 631, 127701 CrossRef CAS.
- (a) Z. Y. Miao, Y. W. Xing, X. H. Gui, Y. J. Cao and T. X. Wang, Int. J. Coal Prep. Util., 2018, 38, 393–401 CrossRef CAS; (b) Y. J. Li, W. C. Xia, Y. L. Peng and G. Y. Xie, J. Cleaner Prod., 2020, 273, 123172 CrossRef CAS.
- G. Yang, G. W. Yang, H. Xu and W. H. Hou, Acta Chim. Sin., 2004, 62, 153–159 CAS.
- Y. Y. Zhao, W. Xu, G. J. Mei, M. M. Yu, S. Y. Yang and Z. He, Physicochem. Probl. Miner. Process., 2022, 58, 159–168 CAS.
- Q. Cheng, G. J. Mei, W. Xu and Q. Z. Yuan, Miner. Eng., 2022, 180, 107491 CrossRef CAS.
- H. Qiu, C. Degenhardt, N. Feuge, D. Goldmann and R. Wilhelm, RSC Adv., 2022, 12, 29562–29568 RSC.
- S. T. Yang, R. Pelton, C. Abarca, Z. F. Dai, M. Montgomery, M. Q. Xu and J. A. Bos, Int. J. Miner. Process., 2013, 123, 137–144 CrossRef CAS.
- P. K. Ackermann, G. H. Harris, R. R. Klimpel and F. F. Aplan, Miner. Metall. Process., 1999, 16, 27–35 Search PubMed.
- X. C. He, Sep. Sci. Technol., 1998, 33, 141–148 CrossRef CAS.
- M. Kanuchova, A. Oravcova, M. Sisol, M. Koscova and L. Kozakova, Acta Montan. Slovaca, 2021, 26, 98–105 CAS.
- S. K. Li, G. H. Gu, G. Z. Qiu and Z. X. Chen, Trans. Nonferrous Met. Soc. China, 2018, 28, 1241–1247 CrossRef CAS.
- G. Y. Liu, H. Zhong, L. Y. Xia, S. A. Wang and Z. H. Xu, Miner. Eng., 2011, 24, 817–824 CrossRef CAS.
- H. Wan, W. Yang, T. S. He, J. B. Yang, L. Guo and Y. J. Peng, Minerals, 2017, 7, 104 CrossRef.
- (a) C. A. Ramsden and W. P. Oziminski, J. Org. Chem., 2016, 81, 10295–10301 CrossRef CAS PubMed; (b) C. A. Ramsden and W. P. Oziminski, J. Org. Chem., 2017, 82, 12485–12491 CrossRef CAS PubMed; (c) W. P. Oziminski and C. A. Ramsden, RSC Adv., 2018, 8, 14833–14837 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, results of calculations, HOMO/LUMO profiles. See DOI: https://doi.org/10.1039/d2ra07627f |
| This journal is © The Royal Society of Chemistry 2023 |