DOI:
10.1039/D2RA07552K
(Paper)
RSC Adv., 2023,
13, 16311-16320
Molecular dynamic simulation reveals the molecular interactions of epidermal growth factor receptor with musk xylene are involved in the carcinogenicity
Received
28th November 2022
, Accepted 24th May 2023
First published on 31st May 2023
Abstract
Musk xylene (MX), a kind of personal care product, has become a new type of environmental contaminant in recent years. Long-term exposure to MX is associated with a variety of cancers, but the mechanism is still unclear. Meanwhile, our previous research showed that MX exposure could lead to malignant transformation of human liver cells L02 and up-regulation of multi genes which are involved in the MAPK signaling pathway, such as the epidermal growth factor receptor (EGFR). These findings indicated that the MAPK signaling pathway might be involved in the malignant transformation caused by MX, but the mechanism is also unclear. In this study, the underlying interaction mechanisms between EGFR and MX were investigated using molecular dynamics (MD) simulation. Results revealed that MX bound to the ECD of EGFR in four binding sites, which was mainly driven by van der Waals and nonpolar interactions, and the affinity of MX toward ECD was sIII > sI > sII > sIV. Further analysis through MD simulation found that s III, the site with the strongest binding, was coincidentally located at the binding area of EGF, which is the natural ligand of EGFR. Therefore, we speculated that MX may activate the MAPK signaling pathway by binding to EGFR in a similar way to EGF, and finally lead to tumorigenesis. In addition, the MM/PBSA method could also be utilized to calculate the hot residues in each binding site. The prediction of hot residues would provide some theoretical guidance for further study of the carcinogenesis mechanisms of MX both in MD simulation and experimental research.
Introduction
Nitro musk represented by musk xylene (MX) is a substitute for natural musk.1 These synthetic musks enter into the environment through various ways and became a new environmental pollutant in recent years. Since 1981, synthetic musk has been gradually detected in various environmental medi a including the atmosphere, indoor dust, water, soil sludge, fish, shrimp, shellfish and even human adipose tissue, blood and breast milk.2–5 Since synthetic musk is widely present in environmental matrices and human samples, its potential toxicological effects have attracted many researchers' attentions. Previous research showed that the residues of synthetic musk can cause certain harm to organisms, which mainly show as neurotoxicity, liver toxicity and enzyme toxicity.6–9 Studies on mice have shown that long-term exposure with MX would cause liver cancer.10 Our previous research also found that MX exposure can induce malignant transformation of human liver cell line L02 and up-regulate some genes of the MAPK signaling pathway.11
MAPK (mitogen-activated protein kinase) signaling pathway is one of the important signal transduction systems in cells, that plays an important role in complex cellular programs like proliferation, differentiation, development, transformation, apoptosis, angiogenesis and other physiological and pathological processes.12–15 Dysregulation of controlled MAPK signaling has been shown to be important in the development and progression of many diseases including various types of tumor and neurodegenerative diseases. In mammalian cells, four MAPK families have been clearly characterized: ERK1/2 (extracellular signal regulated kinases), JNK (c-Jun N-terminal kinase), p38 kinase and ERK5.16–18 Though each of the MAPK pathway mediates different biological effects, there are also extensive interactions, which play an important role in the occurrence and development of tumor in a synergistic or inhibitory way. In our previous work, an affirmative effect of MX on cell malignant transformation in vitro was observed,11 and highly expression of some MAPK signaling pathway related genes in MX-treated cells were also found according to the microarray results. Therefore, we speculated that MAPK pathway may be involved in the carcinogenesis of MX. However, how does MX activate MAPK pathway? This issue is not clear yet, and more research is needed.
MAPK pathways can be activated by diverse extracellular and intracellular stimuli including cytokines, growth factors, hormones, as well as various cellular stressors. EGFR, epidermal growth factor receptor, binding ligands of the EGF family and activating several signaling cascades to convert extracellular cues into appropriate cellular responses, such as MAPK signaling pathway. Studies have shown that the high expression of EGFR in cells can provide binding sites for downstream signaling molecules.19,20 EGFR can be activated by the potential ligands such as EGF and transforming growth factor α (TGFα), which led to the formation of receptor dimers and activated the intrinsic kinase domain.21 The human EGFR consists of three regions: extracellular domain (ECD), transmembrane (TM) domain and intracellular tyrosine kinase domain,22,23 and the ECD can be further divided into four domains (dI, dII, dIII and dIV).23,24 In this paper, molecular docking technology and molecular dynamics methods were used to construct and simulate the binding process of ECD with MX. Conformational change and binding energy were analysed to predict the possible binding site of ECD with MX (Fig. 1).
 |
| | Fig. 1 The 2D structure of EGFR ECD (a) and MX (b). | |
Materials and methods
BIAcore assay
Surface plasmon resonance binding experiment was performed using Biacore T200 system to analyse the binding affinity between ECD and MX. Human EGFR ECD protein was purchased from Sino Biological (China), and the series s sensor chip CM5 were purchased from Bio. Co. (USA). The ECD protein was immobilized on a CM5 sensor chip and the binding of MX to ECD was accomplished by injecting concentration of MX (250–2000 μM) into the sensor chip under the buffer condition of PBS-buffered saline at 25 °C.
Homology modeling
The primary EGFR protein sequence of homo sapiens (accessio-n ID: P00533) was retrieved from UniProt Database (https://www.uniprot.org/) in FASTA format. In order to construct the whole structure of extracellular domain (ECD) of EGFR, the X-ray crystal structural of ECD (PDBID: 4UV7) was retrieved from RCSB Protein Data Bank (http://www.rcsb.org/).The ECD was homology modeled using Modeller 9v24 according to the methods descripted by Kuntal et al. (2010) and Webb et al. (2017).25,26 Firstly, perform sequence alignment between the target sequence and the template ECD PDB to generate ali and pap files. Then, modify the corresponding file name and required number of models in model-default.py, and run python3 model-default.py to obtain the modeled structure. Finally, the structural domain IV was supplemented through homologous modeling.
Ligand preparation and molecular docking
The 2D structure of MX was retrieved from PubChem Database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format and conver-ted to 3D in MOE v2014 (ref. 27) through energy minimization. In order to keep ligand flexible, all rotable bonds in ligand were set and the residues in domain I and domain III of ECD receptor inside the binding grid box were retained using the AutoDockTools package.28,29 The grid size was set while x, y and z axes were 50, 50 and 50 with 1 Å, respectively. The center of the grid box was x = −30.466, y = −22.109 and z = 16.303, respectively (domain I), and x = 8.641, y = 16.838 and z = 27.808, respectively (domain III). Then Protein and ligand structure in PDBQT format for molecular docking were prepared by AutoDock Vina.30 PyMOL was used to visualize the interactions in the top modes between the ECD and MX.31,32
Molecular dynamics simulation
Partial atomic charges of MX were first assigned based on the AM1-BCC method33,34, using the ANTECHAMBER program of AmberTools.35 Parameters of MX were taken from the general AMBER force field (GAFF).36 ACPYPE script in the AmberTools package was used to convert the AMBER format files of MX to the GROMACS format.37 The MD simulations were performed using GROMACS 2018 program with the AMBER99SB-ILDN force field for all receptors.38 All systems were solvated using TIP3P39 water molecules in a cubic box with at least 1.2 nm distance around the complex. The net charge on systems was neutralized by adding counterions as required. The systems carried out the energy minimization by using the steepest descent algorithm, then heated from 0 K to 300 K during a 200 ps constant volume simulation with 1 fs time step. The pressure was equilibrated to 1 atm during 100 ps NPT simulation with 2 fs time step. Both temperature and pressure equilibration were balanced using the Berendsen algorithm40 during the equilibration step. In all simulations, the heavy atoms of protein were position restrained with a force constant of 1000 kJ mol−1 nm−2, the cut-off distance was set to 1.4 nm, while Colulomb cut-off and neighbour list were set to 1.4 nm. All MD simulations were performed for 200 ns with 2 fs time step. The temperature and pressure were maintained at 300 K and 1 bar by using the V-rescale temperature41 and Parrinello–Rahman pressure42 coupling method in the production step, respectively. During the simulation, periodic boundary conditions were applied, and the particle-mesh Ewald (PME) method was used to handle all electrostatic interactions.43 All bonds were constrained using the LINCS algorithm.44,45
Calculation of binding free energy
Binding free energies between EGFR and MX in four systems were calculated by the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method implemented in GROMACS program using g_mmpbsa tool.46 The trajectory of last 50 ns was used to calculate the binding energies. Thus, a total of 250 snapshots were extracted from four system. For each snapshot, the binding free energy can be summarized as:47
| ΔGbinding = GComplex − (GProtein + GLigand) |
where GComplex, GProtein,and GLigand are the total free energy of the complex, receptor, and ligand molecules, respectively. The free energy (G) was estimated by:
| EMM = Ebonded + Enonbonded = Ebonded + EvdW + Eelec |
where x is the complex, protein, or ligand. EMM is the average molecular mechanics potential energy in vacuum and defined as the sum of the internal interaction (Ebonded), van der Waals (EvdW) and the electrostatic (Eelec). Ebonded is always taken as zero.35
The free energy of solvation (GSolvation) was estimated as the sum of the electrostatic solvation free energy (Gpolar) and the non-electrostatic solvation free energy (Gnopolar):
| GSolvation = Gpolar + Gnopolar |
where
Gpolar is calculated by solving the Poisson–Boltzmann(PB) equation and
Gnopolar is estimated by the solvent-accessible surface area (SASA) as equation following:
where the solvent-accessible surface area (SASA) determined a water probe radius of 1.4 nm. The surface tension constant
γ and the fitting parameter
b were set to 0.02267 and 3.849 (kJ mol
−2 Å
−2) respectively.
48
Result and discussion
BIAcore assay of the interaction between ECD and MX in vitro
In this study, the ECD protein was purified and BIAcore assay was applied to characterize the binding constants in vitro to verify the supposition that MX exposure may lead to tumorigen-esis by binding to EGFR. The binding affinity between MX and ECD was evaluated according to the dissociation constant KD value. The RU values revealed that MX bound to ECD in a dose-dependent manner, and the determined equilibrium dissociate-on constant KD was about 259 μM (KD = 2.59 × 10–6 M) (Fig. 2). This result suggested that binding with EGFR and then activating MAPK pathway may be a possible carcinogenesis mechanism of MX.
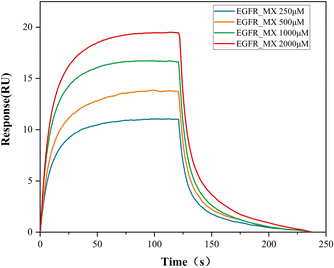 |
| | Fig. 2 The response value of ECD/MX complex quantified by BIAcore assay. | |
Molecular docking
Docking simulation studies were carried out to investigate the binding modes of MX with the domain I and domain III of ECD. The docking scores and the interactions of binding site were shown in Table 1. All binding modes of ECD with MX were illustrated in Fig. 3. The docking results showed that MX bound to ECD domain I and III with four possible different sites: site I (sI), site II (sII), site III (sIII) and site IV (sIV).
Table 1 The docking results between EGFR and MX
| Binding mode |
Affinity (kcal mol−1) |
H-bond interaction |
Hydrophobic interaction |
| sI |
−5.4 |
K454, K464, N452 |
W453, I451, I466, K463, F457 |
| sII |
−5.4 |
E388 |
P387, W386, F352, P365, R353 |
| sIII |
−5.1 |
H409, Q408, H346 |
F380, D344, I316, L325 |
| sIV |
−5.3 |
R74 |
Y50, D51, L52, K56, P76, V72, E73 |
 |
| | Fig. 3 (a)Whole binding mode of four sites between ECD and MX, the binding mode of ECD with MX in (b) sI, (c) sII, (d) sIII and (e) sIV. | |
In order to explain detailed interactions between ECD and MX, the structure of the four complexes were analysed systematically. From the binding mode in sI, MX was embedded in hydrophobic interaction with residues including F457, T464, I466, I451, W453, K463, W453 and N452 and last stabilized in binding pocket by hydrogen bonding. The binding mode in sII showed that five residues (R353, F352, P365, W386 and E388) stabilized the MX through hydrophobic interaction, and MX also kept bond with P387 through hydrogen bonding. MX was stabilized by four hydrophobic residues (I316, L325, D344 and F380) through hydrophobic interactions in sIII binding mode, and three residues (H346, H409 and Q408) also played important role through hydrogen bonding. The binding mode in sIV was found that MX was stabilized by seven hydrophobic residues (Y50, D51, L52, K56, P76, V72, E73) through hydrophobic interactions and R74 residue also played essential role in the binding of MX through hydrogen bonding.
Analysis of molecular dynamic simulation
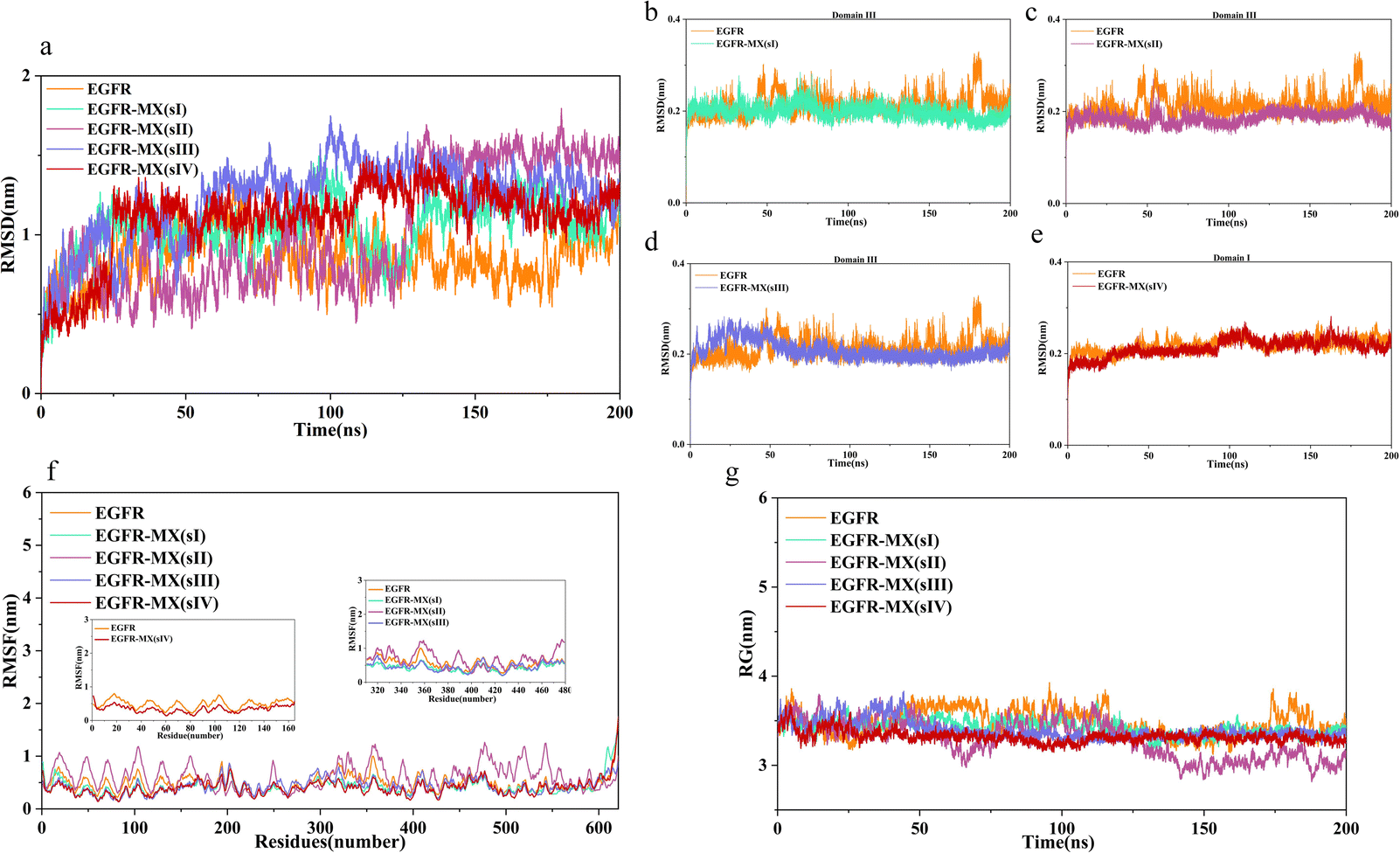
Analysis of root mean square deviation. In MD simulation, RMSD is often used to quickly determine whether a certain trajectory reaches equilibrium for subsequent simulation or analysis. The height of RMSD reflects the overall deviation of the track from the reference molecule. In present study, the RMSD values of ECD were calculated. As results showed in Fig. 4a, the position of ECD shifted at the beginning of the binding process, and then vibrated stably near a certain height. However, owing to the relative deflection of domain I and domain III caused by MX binding, the RMSD values of all complexes except for the EGFR-MX (sIII) are higher than the unbound ECD. At the same time, the RMSD values of domain III or domain I were calculated independently to verify the stability of ligand binding domains. Results showed that the RMSD curves of all the binding domains except when MX binded in sIII, slightly inclined upward but not higher than the unbound ECD during a short period of time at the beginning of the simulation, and that then vibrated stably near a certain height, which indicated that the systems reached equilibrium (Fig. 4b, c and e). As for the EGFR-MX (sIII), the RMSD curve significantly inclined upward during about 25 ns simulation, which indicated that some significant movement may occur in the system conformation in response to the binding of MX. Then the curve also vibrated stably near a certain height, suggesting a stable system (Fig. 4d).
 |
| | Fig. 4 RMSD plots of unbound and bound ECD (a–e), RMSF plots of unbound and bound ECD (f), RG plots of unbound and bound ECD (g). | |
Analysis of root mean square fluctuation. Root mean square fluctuation (RMSF) of the residues in both unbound and bound ECD were calculated to characterize the flexibility of molecular structure. As the results showed in Fig. 4f, residues in EGFR-MX (sI, sIII, sIV) systems exhibited approximately fluctuation trend and amplitude compared to the unbound ECD. However, in EGFR-MX (sII) system, most of the residues highlight the maximum fluctuations, and the amplitude was noticeably higher than that in unbound ECD. These results suggested that there are some high flexibility regions in EGFR-MX (sII) system, which might play an important role in protein function.Unlike the position change of whole molecular structure during the simulation characterized by RMSD, RMSF calculates the fluctuation of each atom, which is an index to measure the degree of freedom of atomic motion. Based on the results of RMSD and RMSF, it indicated that after MX bound to the ECD, structural domain III was more stable, and structural domain II was more flexibility. However, neither of them could explain which domain is better combined, and more analyses are needed.
Analysis of radius of gyration. Radius of gyration (RG) was analysed to investigate the compactness of complexes' structure. According to the results showed in Fig. 4g, the unbound EGFR had certain flexibility, and the RG curve fluctuated around 3.3 nm, but the structural compactness was decreased, and slight fluctuations of RG curve were observed in EGFR-MX (sI, sIII, sIV) systems. Interestingly, the fluctuation of RG curve in EGFR-MX (sII) system seemed to have great flexibility. The RG value was comparable to that of other systems before 110 ns, but it decreased drastically from 125 ns and then stabilized again, suggesting that structure adjustment occurred when MX bounded to ECD in site II. The average RG in five systems were 3.48 ± 0.15 nm, 3.42 ± 0.08 nm, 3.28 ± 0.21 nm, 3.39 ± 0.10 nm, 3.32 ± 0.07 nm, compared well with the experimental value of 3.54 ± 0.011 nm found from SAXS.49
Analysis of ECD conformational change. Snapshots of the five systems were captured at 200 ns, and the conformations of EGFR-MX (sI, sII, sIII, sIV) complexes were overlapped with the ECD of unbound EGFR, respectively. Results showed that the conformational shift trend of ECD in EGFR-MX (sII) system was significantly different from that of EGFR-MX (sI, sIII and sIV) systems (Fig. 5a–d). In EGFR-MX (sII) system, domain II bent and led to the proximity of domain I and domain III, which ultimately led to dimerization. These conformational changes further verified that it had the maximum flexibility when MX binded in sII of domain III.
 |
| | Fig. 5 The 200 ns snapshot of ECD confirmation with four binding sites: sI (a), sII (b), sIII (c) and sIV (d), the definition of two vectors in domain I and III (e), the angle distribution between two vectors (f). | |
In order to further analyse the conformational changes between the domains in each EGFR-MX complexes, the angle distribution was calculated to define the orientations of domain I and III. Amino acid residues Val36, Glu118 in domain I and Ser340, Glu431 in domain III were selected as vector markers to reduce the influence of intramolecular movement. As the results showed in Fig. 5f, varying degrees of angle deviations were observed in all EGFR-MX complexes, especially EGFR-MX (sII) system highlighted the maximum angle change (Fig. 5f). Based on these results, it is considered that the binding of MX might activate the dimerization trend of EGFR ECD, which plays a key role in the process of signal pathway activation.
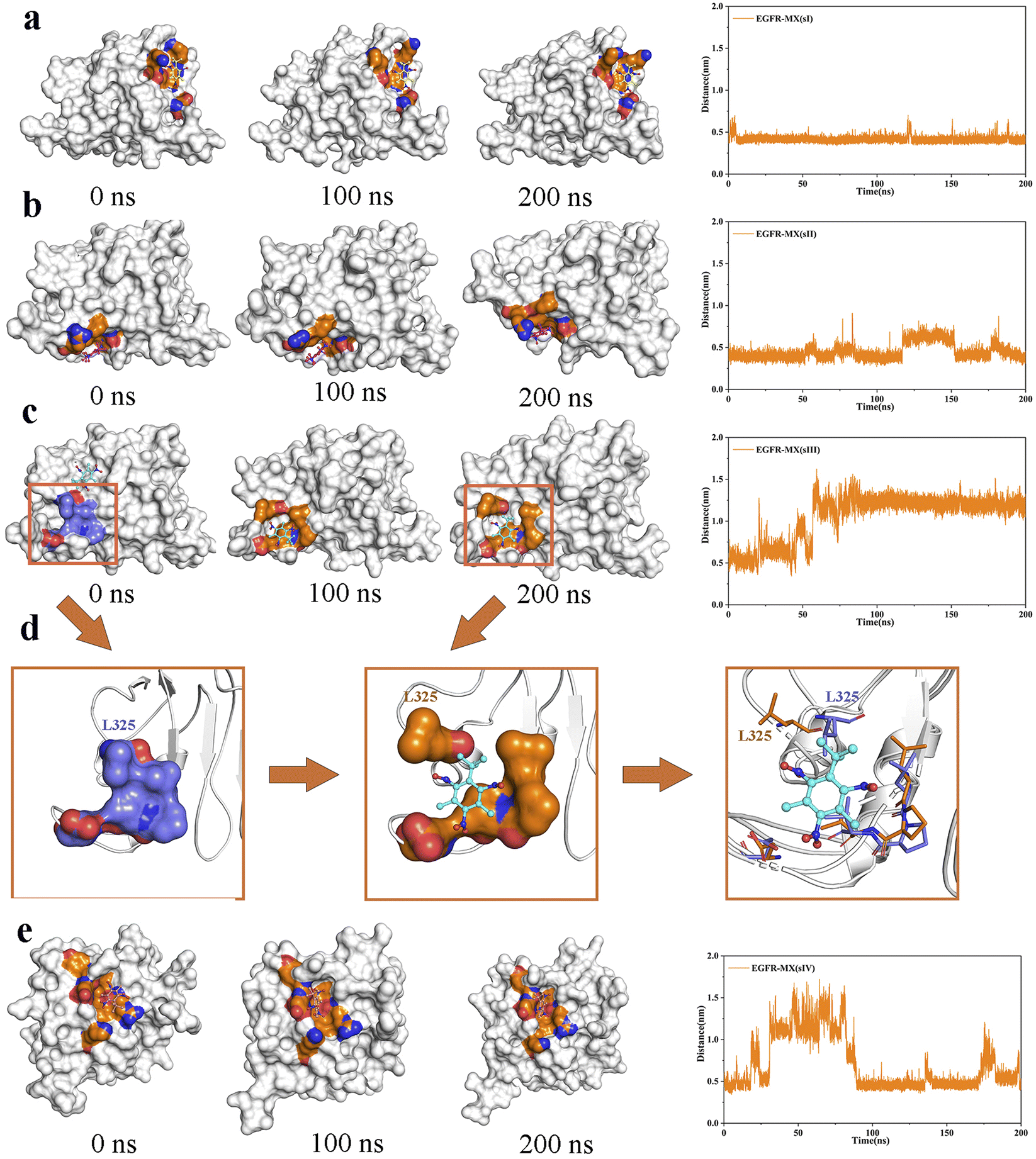
Analysis of MX binding pocket comparison. The snapshot of MX in the binding pocket on 0 ns, 100 ns and 200 ns were extracted to evaluate the binding stability of MX in the binding pocket intuitively. The center of binding amino acid residues in initial states was defined to be the center of initial binding pocket, and its distance between MX at final state was calculated. It can be seen that the movement of MX in EGFR-MX (sI) system and EGFR-MX (sII) was quite smooth, indicating a strong binding ability to the two binding sites (Fig. 6a, b). However, in EGFR-MX (sIII) system, MX was initially located in the β sheet area where has strong rigidity, but it left the current binding pocket after 50 ns and bound to a new site III (sIII) (Fig. 6c). At the same time, it can be seen from Fig. 6d that the amino acid residue L325 blocked the entrance in the initial state of the new binding region. Then the side chain of L325 swung up and opened the entrance as the simulation proceeded, which allowed MX to embed in closely and finally be attracted firmly in the hydrophobic cavities formed by residues L325, l348, P349 and v350 (Fig. 6d). As for EGFR-MX (sIV) system the strong flexible fluctuation of the binding pocket made the binding of MX extremely unstable in this region until 100 ns, and slight flexible fluctuation could also be observed according to the distance between MX and the center of binding amino acid residues during 100 ns to 200 ns (Fig. 6e). Through visual observation of the bonding process in four EGFR-MX systems, it could be found that MX would choose the appropriate binding area, and the cavity in the region would also be adjusted to make the chimerism more stable. In addition, it interestingly found that the binding site sII and new sIII were coincidentally located at the binding area of EGF, which is the natural ligand of EGFR.
 |
| | Fig. 6 The snapshot of binding domain with MX on 0 ns, 100 ns and 200 ns, and the distance of MX from the center of initial binding pocket was calculated in EGFR-MX (sI) system (a), EGFR-MX (sII) system (b), EGFR-MX (sIII) system (c), and EGFR-MX (sIV) system (e). (d) Pocket confirmational change on 0 ns, 100 ns and 200 ns. | |
Hydrogen bond analysis
Hydrogen-bonded interaction provided a measurement of interaction power and played an essential role in stabilizing ECD-MX complexes. In order to authenticate the stability of four bound systems, the hydrogen bonds and pairs within 0.35 nm between ECD-MX were calculated in a solvent environment from 100 to 200 ns. As Fig. 7 (a–d) showed, the average number of hydrogen bonds between MX and ECD in the binding site was 4, 8, 9 and 10, respectively. Fig. 7 (e–h) showed the structure of the active site in the EGFR-MX systems, and schematically identified the most significant H-bond contacts between ECD and MX. MX mainly created two hydrogen bonds with K454 (58.4% occupancy) and T464 (89.3% occupancy) in EGFR-MX (sI) system (Fig. 7e), one hydrogen bond with W386 (33.6% occupancy) in EGFR-MX (sII) system (Fig. 7f), one hydrogen bond with V350 (29.9% occupancy) in EGFR-MX (sI) system (Fig. 7g), and one hydrogen bond with L52 (33.9% occupancy) in the EGFR-MX (sIV) system (Fig. 7h). In addition, there were still some weak hydrogen bond bindings owing to the adjustment of pocket position in the binding region.
 |
| | Fig. 7 The hydrogen numbers and the hydrogen binding mode of four binding system including EGFR-MX (sI) (a and e), EGFR-MX (sII) (b and f), EGFR-MX (sIII) (c and g) and EGFR-MX (sIV) (d and h). | |
Binding characterization of MX
Though MM-PBSA method cannot accurately reproduce the binding free energy in the experiment, it has good correlation with the values in the experiment and provides semiquantitat-ive evaluation.50 The binding free energy was calculated in this study, and the affinity of MX toward ECD ranked as follows: sIII > sI > sII > sIV. Then, various energies were decomposed using MM-PBSA method, and results showed that van der Waals(ΔGvdW), electrostatics interactions (ΔGelec) and nonpolar solvation terms (ΔGnonpolar) played positive role on the binding behaviour, whereas polar solvation terms (ΔGpolar) made an opposite reaction (Table 2). On the other hand, the total binding free energy was further decomposed to each residue to screen the amino acids residues that with important contribution. As the results showed in Fig. 8, there were several residues made great contribution to the binding behaviours between MX and ECD in four binding sites. In the binding pocket, residues Trp453, Lys454, Phe457, Lys463 and I1e466 in sI, residues Pro349, Phe352, Pro365 and Pro387 in sII, residues Leu325, leu 348, Pro349 and Val350 in sIII, residues Ser52, Ser53, Gln73 and Pro76 in sIV, constituted hydrophobic core in each binding site, which then generate strong van der Waals and hydrophobic interactions with the MX. Among them, residues Trp453 sI, Phe352 in sII, and Leu348, Pro349, Val350 in sIII contributed more than 1 kcal mol−1 energy in each binding site, and so be defined as hot residues. The discovery of hot residues would provide guidance for the experimental study on the interaction mechanism between MX and EGFR.
Table 2 Individual energy components of the calculated binding free energies of ECD/MX complex
| Ligand binding site contribution (kcal mol−1) |
sI |
sII |
sIII |
sIV |
| ΔGvdW |
Mean |
−28.33 |
−26.09 |
−25.91 |
−23.18 |
| Std. |
2.70 |
2.11 |
2.10 |
4.52 |
| ΔGelec |
Mean |
−2.80 |
0.12 |
−1.98 |
−1.12 |
| Std. |
1.06 |
1.48 |
0.79 |
1.12 |
| ΔGpolar |
Mean |
14.01 |
9.11 |
10.17 |
13.18 |
| Std. |
2.32 |
2.98 |
1.63 |
4.88 |
| ΔGnonpolar |
Mean |
−2.60 |
−2.42 |
−2.57 |
−2.27 |
| Std. |
0.18 |
0.22 |
0.16 |
0.30 |
| ΔGbinding |
Mean |
−19.71 |
−19.27 |
−20.29 |
−13.39 |
| Std. |
2.53 |
2.56 |
2.13 |
4.63 |
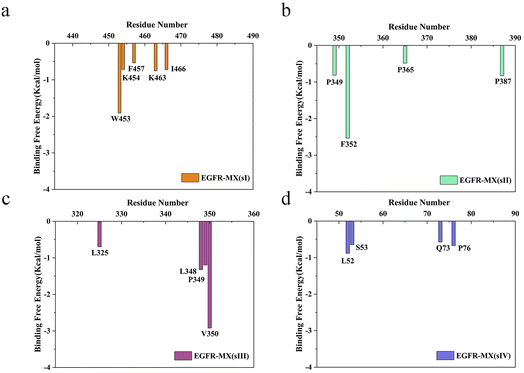 |
| | Fig. 8 The energy contribution of significant residues in EGFR-MX (sI) system (a), EGFR-MX (sII) system (b), EGFR-MX (sIII) system (c) and EGFR-MX (sIV) system (d). | |
Conclusion
The binding mode of EGFR and MX was studied through molecular docking. Results revealed that MX bound to the ECD of EGFR in four binding sites, which was mainly driven by van der Waals and nonpolar interactions, and the affinity of MX toward ECD was sIII > sI > sII > sIV. Further analysis through molecular dynamics simulation found that the binding of MX stabilized the skeleton fluctuation of its binding domain, and different conformational changes of ECD were observed in each binding sites. Moreover, we interestingly found that sIII, the site with the strongest binding, were coincidentally located at the binding area of EGF, which is the natural ligand of EGFR. Therefore, we speculated that MX may activate MAPK signaling pathway by binding to EGFR in a similar way to EGF, and finally lead to tumorigenesis. This is one of the possible carcinogenesis mechanisms of MX. In addition, MM/PBSA method also be utilized to decompose the binding free energy, aiming to calculate the hot residues in each binding site. The prediction of hot residues provided some theoretical guidance for further study of the carcinogenesis mechanisms of MX both in MD simulation and experimental research.
Data availability
All data generated or analysed during this study are included in this article. All the initial structures and input files for molecular dynamics simulations are available at the following GitHub repository: https://github.com/scarlettzyy/generaldata. All the software used in this article is listed in the materials and methods section. Software, named Autodock vina, is an open-source program for Molecular docking. And gromacs is also an open-source program for MD simulation.
Author contributions
Wen Li and Huaxing Fei contributed equally to this work.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Shenzhen Science and Technology Project (JCYJ20180306173301083). Wecomput Tech. Co. Ltd is thanked for the sharing of the PyMOL-software for the molecular graphics production.
References
- K. Bester, Analysis of musk fragrances in environmental samples, J. Chromatogr. A, 2009, 1216, 470–480 CrossRef CAS PubMed.
- M. Llompart, C. Garcia-Jares, C. Salgado, M. Polo and R. Cela, Determination of musk compounds in sewage treatment plant sludge samples by solid phase microextraction, J. Chromatogr. A, 2003, 999, 185–193 CrossRef CAS PubMed.
- Y. Lv, T. Yuan, J. Hu and W. Wang, Simultaneous determination of trace polycyclic and nitro musks in water samples using optimized solid-phase extraction by gas chromatography and mass spectrometry, Anal. Sci., 2009, 25, 1125–1130 CrossRef CAS PubMed.
- J. L. Reiner, C. M. Wong, K. F. Arcaro and K. Kannan, Synthetic musk fragrances in human milk from the United States, Environ. Sci. Technol., 2007, 41, 3815–3820 CrossRef CAS PubMed.
- H. Zhang, S. Bayen and B. C. Kelly, Co-extraction and simultaneous determination of multi-class hydrophobic organic contaminants in marine sediments and biota using gc-ei-ms/ms and lc-esi-ms/ms, Talanta, 2015, 143, 7–18 CrossRef CAS PubMed.
- A. Api, R. Ford and R. San, An evaluation of musk xylene in abattery of genotoxicity tests, Food Chem. Toxicol., 1995, 33, 1039–1045 CrossRef CAS PubMed.
- A. Api, E. Pfitzer and R. San, An evaluation of genotoxicity tests with musk ketone, Food Chem. Toxicol., 1996, 34, 633–638 CrossRef CAS PubMed.
- G. Carlsson and L. Norrgren, Synthetic musk toxicity to early life stages of zebrafish (danio rerio), Arch. Environ. Contam. Toxicol., 2004, 46, 102–105 CrossRef CAS PubMed.
- M. Schlumpf, R. Suter-Eichenberger, M. Conscience-Egli and W. Licht ensteiger, Bioaccumulation and induction of cyp450 liver enzymes by synthetic musk fragrances in developing and adult rats, Toxicol. Lett., 1998, 95, 210 CrossRef.
- Y. Matsushima, A. Maekawa, H. Onodera, M. Shibutani, J. Yoshida, Y. Kurokawa and Y. Hayashi, Toxicity and carcinogenicity studies of musk xylol in b6c3f1 mouse, Bull. Natl. Inst. Hyg. Sci., 1990, 89–94 CAS.
- Y. Zhang, L. Huang, Y. Zhao and T. Hu, Musk xylene induces malignant transformation of human liver cell line l02 via repressing the tgf-β signaling pathway, Chemosphere, 2017, 168, 1506–1514 CrossRef CAS PubMed.
- B. A. Ballif and J. Blenis, Molecular mechanisms mediating mammalian mitogen-activated protein kinase (mapk) kinase (mek)- mapk cell survival signals, Cell Growth Differ., 2001, 12, 397–408 CAS.
- L. Barault, N. Veyrie, V. Jooste, D. Lecorre, C. Chapusot, J. M. Ferraz, A. Lièvre, M. Cortet, A. M. Bouvier and P. Rat, et al. Mutations in the ras-mapk, pi (3) k (phosphatidylinos-itol-3-oh kinase) signaling network correlate with poor survival in a population-based series of colon cancers, Int. J. Cancer, 2008, 122, 2255–2259 CrossRef CAS PubMed.
- M. Dimri, A. Humphries, A. Laknaur, S. Elattar, T. J. Lee, A. Sharma, R. Kolhe and A. Satyanarayana, Nqo1 ablation inhibits activation of the pi3k/akt and mapk/erk pathways and blocks metabolic adaptation in hepatocellular carcinoma, Hepatology, 2020, 71, 549 CrossRef CAS PubMed.
- E. F. Wagner and Á. R. Nebreda, Signal integration by jnk and p38 mapk pathways in cancer development, Nat. Rev. Cancer, 2009, 9, 537–549 CrossRef CAS PubMed.
- B. Dérijard, M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin and R. J. Davis, Jnk1: a protein kinase stimulated by uv light and ha-ras that binds and phosphorylates the c-jun activation domain, Cell, 1994, 76, 1025–1037 CrossRef PubMed.
- R. Seger and E. G. Krebs, The mapk signaling cascade, FASEB J., 1995, 9, 726–735 CrossRef CAS.
- C. Widmann, S. Gibson, M. B. Jarpe and G. L. Johnson, Mitogen-9 activated protein kinase: conservation of a three-kinase module from yeast to human, Physiol. Rev., 1999, 79, 143–180 CrossRef CAS PubMed.
- R. N. Jorissen, F. Walker, N. Pouliot, T. P. Garrett, C. W. Ward and A. W. Burgess, Epidermal growth factor receptor: mechanisms of activation and signalling, The EGF receptor family, 2003, pp. 33–55 Search PubMed.
- K. Komposch and M. Sibilia, Egfr signaling in liver diseases, Int. J. Mol. Sci., 2016, 17, 30 CrossRef PubMed.
- M. A. Olayioye, R. M. Neve, H. A. Lane and N. E. Hynes, The erbb signaling network: receptor heterodimerization in development and cancer, EMBO J., 2000, 19, 3159–3167 CrossRef CAS PubMed.
- T. P. Garrett, N. M. McKern, M. Lou, T. C. Elleman, T. E. Adams, G. O. Lovrecz, H. J. Zhu, F. Walker, M. J. Frenkel and P. A. Hoyne, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α, Cell, 2002, 110, 763–773 CrossRef CAS PubMed.
- H. Ogiso, R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J. H. Kim, K. Saito, A. Sakamoto, M. Inoue and M. Shirouzu, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains, Cell, 2002, 110, 775–787 CrossRef CAS PubMed.
- A. W. Burgess, H. S. Cho, C. Eigenbrot, K. M. Ferguson, T. P. Garrett, D. J. Leahy, M. A. Lemmon, M. X. Sliwkowski, C. W. Ward and S. Yokoyama, An open-and-shut case? recent insights into the activation of egf/erbb receptors, Mol. Cell, 2003, 12, 541–552 CrossRef CAS PubMed.
- B. K. Kuntal, P. Aparoy and P. Reddanna, Easymodeller: A graphical interface to modeller, BMC Res. Notes, 2010, 3, 1–5 CrossRef PubMed.
- B. Webb and A. Sali, Protein structure modeling with modeller, in: Functional genomics, Springer, 2017, pp. 39–54 Search PubMed.
- [MOE], M.O.E., 2014, Molecular operating environment (moe), 09, 2014 Search PubMed.
- D. S. Goodsell, G. M. Morris and A. J. Olson, Automated docking of flexible ligands: applications of autodock, J. Mol. Recognit., 1996, 9, 1–5 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. Huey, W. E. Hart, S. Halliday, R. Belew and A. J. Olson, Autodock. Automated docking of flexible ligands to receptor-User Guide, 2001 Search PubMed.
- O. Trott and A. J. Olson, Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- W. L. DeLano, et al., Pymol, 2002 Search PubMed.
- D. Seeliger and B. L. de Groot, Ligand docking and binding site analysis with pymol and autodock/vina, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generationof high-quality atomic charges. am1-bcc model: Ii. parameterization and validation, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- D. A. Case, V. Babin, J. Berryman, R. Betz, Q. Cai, D. Cerutti, T. Cheatham Iii, T. Darden, R. Duke, H. Gohlke, et al., Amber 14, 2014 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. W. S. Da Silva and W. F. Vranken, Acpype-antechamber python parser interface, BMC Res. Notes, 2012, 5, 1–8 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple amber force fields and development of improved protein backbone parameters, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. Berendsen, J. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- S. Nosé and M. Klein, Constant pressure molecular dynamics for molecular systems, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- M. Deserno and C. Holm, How to mesh up ewald sums. i. a theoretical and numerical comparison of various particle mesh routines, J. Chem. Phys., 1998, 109, 7678–7693 CrossRef CAS.
- B. Hess, P-lincs: A parallel linear constraint solver for molecular simulation, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, Lincs:a linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, g_mmpbsa a gromacs tool for high-throughput mmp-bsa calculations, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- N. Homeyer and H. Gohlke, Free energy calculations by the molecular mechanics poisson- boltzmann surface area method, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- I. Massova and P. A. Kollman, Combined molecular mechanical and continuum solvent approach (mm-pbsa/gbsa) to predict ligandbinding, Perspect. Drug Discovery Des., 2000, 18, 113–135 CrossRef CAS.
- J. P. Dawson, Z. Bu and M. A. Lemmon, Ligand-induced structural transitions in erbb receptor extracellular domains, Structure, 2007, 15, 942–954 CrossRef CAS PubMed.
- J. M. Sanders, M. E. Wampole, M. L. Thakur and E. Wickstrom, Molecular determinants of epidermal growth factor binding: a molecular dynamics study, PLoS One, 2013, 8, e54136 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab