
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn donor–acceptor-bridging intramolecular hydrogen bonds in NIR-TADF molecules†
Fabian Weber and
Hirotoshi Mori*
and
Hirotoshi Mori*
Chuo University, Department of Applied Chemistry, Kasuga 1-13-27, 112-8551, Tokyo-to, Bunkyo-ku, Japan. E-mail: fweber13790@gmail.com; qc-forest.19d@g.chuo-u.ac.jp
First published on 27th January 2023
Abstract
Following a report that highlights the importance of intramolecular hydrogen bonds for improved NIR-TADF efficiency in CAT-1 [Leng et al., J. Phys Chem. A, 2021, 125, 2905], an intramolecularly doubly hydrogen-bonded base design is investigated and compared to singly and non-bonded derivatives. It is found that the potential to form more intramolecular hydrogen bonds correlates with decreasing internal reorganization energies in most of the designed structures. In addition, our proposed base design facilitates the desired interlocking of charge transfer donor-, linker- and acceptor-planes through steric gauche-interactions to both linker-plane sides, allowing to trap also smaller heterocyclic linkers.
Introduction
Nearly one decade has passed since the realization of the first application oriented, metal-free thermally-activated delayed fluorescence (TADF) molecules1 and ever since, design concepts have been successfully applied to conquer more and more regions of the electromagnetic spectrum. This way, TADF molecules like CAT-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 (4-(4-(diphenylamino)phenyl)acenaphtho[1,2-b]pyrazine-3,8,9-tricarbonitrile) and CZ-MPS3 (2,7-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-9,9-dimethyl-9H-thioxanthene 10,10-dioxide) with emitter wavelengths as low-energetic as 950 nm and as high-energetic as 389 nm have been reported, reaching from the near-infrared (NIR) all the way into the ultraviolet (UV) region. One recurring challenge in the development of TADF designs is to ensure a high rigidity of the structure across excited states to minimize the activation energy associated with the overall reverse intersystem crossing (rISC) process. Following the semi-classical expression of Marcus' theory, the rISC rate can be expressed as4
2 (4-(4-(diphenylamino)phenyl)acenaphtho[1,2-b]pyrazine-3,8,9-tricarbonitrile) and CZ-MPS3 (2,7-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-9,9-dimethyl-9H-thioxanthene 10,10-dioxide) with emitter wavelengths as low-energetic as 950 nm and as high-energetic as 389 nm have been reported, reaching from the near-infrared (NIR) all the way into the ultraviolet (UV) region. One recurring challenge in the development of TADF designs is to ensure a high rigidity of the structure across excited states to minimize the activation energy associated with the overall reverse intersystem crossing (rISC) process. Following the semi-classical expression of Marcus' theory, the rISC rate can be expressed as4
Besides the spin–orbit coupling term HSOC, there are two major expressions that are direct molecular properties that can be optimized for. The first, ΔEST, represents the vertical energetic singlet–triplet gap at the reactant excited triplet state geometry. The second, λ, is the sum of internal and external reorganization energy after the transition from the triplet to the singlet state. Here, the external term λext captures energetic changes in the surrounding (i.e. solvent relaxation), while the internal portion λint is limited to the reactant's structural relaxation. The latter is defined as the energy difference between the relaxed final state (i.e. the excited singlet at its relaxed structure) and the respective target energy, as seen from the reactant structure (i.e. the excited singlet energy at the relaxed triplet structure). Note for increasing the rISC rate in this expression, the typical focus lies with optimizing the exponential to become as close to 1 as possible by achieving small ΔEST and λ.
Comparing two Jablonski diagrams with high and low internal reorganization energies (see Fig. 1), one can find that there is an indirect connection between ΔEST and λint, such that lower λint may also result in lower ΔEST at the reactant geometry.
 | ||
| Fig. 1 Jablonski diagrams of a molecular system with high (left) and low (right) reorganization energy with ground state S0 (black), excited singlet state S1 (blue) and excited triplet state T1 (red). | ||
Especially in the low energetic regime where electronic excitation energies start to match those of vibrational excitation energies, low internal reorganization energies λint between the electronically excited state structures become decisive for the performance of TADF candidates.5 One possible strategy, how this can be achieved in the CAT-1 structure (see left side of Fig. 2) has recently been reported to lie in the use of intramolecular hydrogen bonds to fixate the relative positions and rotations of charge-transfer (CT) donor and acceptor units.6 In comparing CAT-1 with its non hydrogen-bonded counterpart TPAAP,7 (3-(4-(diphenylamino)phenyl)acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile) Leng et al. found a lowered λint that may be responsible for its more efficient NIR-TADF process compared to TPAAP.
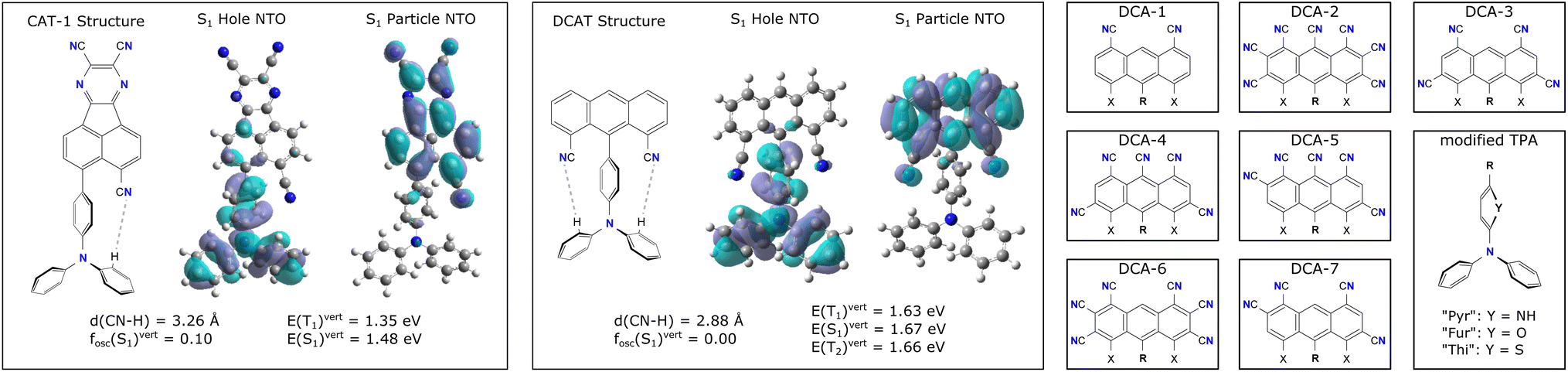 | ||
| Fig. 2 Excited state properties and Lewis structures of CAT-1 (left) and unfunctionalized DCAT (middle). Dashed grey lines highlight potential hydrogen bonds. On the right, other building blocks considered for DCA acceptor and TPA-like donor units are shown. Note that the groups X on the acceptors may be either hydrogen or cyano groups, giving rise to the respective singly or zero hydrogen-bonded versions [1]DCA and [0]DCA. | ||
To build on top of this design principle, in this work we apply theoretical calculations to investigate the properties of potentially doubly hydrogen-bonded 4,5-dicyanoanthracene-10-triphenylamine (DCAT) derivatives. In a first step, we use (time-dependent) density-functional theory (TD-DFT) to optimize the structures at their electronic ground and selected excited states. Using these structures we then evaluate reorganization energies λint, spin–orbit coupling matrix elements (SOCME) between states that are relevant for TADF and correlations with the hydrogen bond properties. Computational details are provided in the ESI.†
Results and discussion
Fig. 2 shows the Lewis representations of CAT-1 and the base DCAT structure with their respective hole and particle natural transition orbitals (NTO)8 that describe the first excited singlet state in TD-DFT quality at the ground state geometry. At these geometries, CAT-1 and DCAT exhibit vertical excitation energies of E(S1) = 1.35 eV and 1.67 eV, respectively. As can be seen from NTO analysis, the S1 state of both molecules describes a charge transfer (CT) in which the triphenylamine (TPA) unit acts as the charge donor9 and the respective other half of the molecule acts as the acceptor. For CAT-1, a vertical singlet–triplet gap ΔEST of 0.13 eV is found, whereas DCAT shows a gap of 0.01 eV. While small ΔEST in principle promote the rate of rISC, it is also known that too low gap energies indicate a strong spatial separation of the frontier molecular orbitals involved in the excitation,10 thus leading to dark singlet states with transition dipole moments of zero. Finally, both molecules feature groups of atoms that may form hydrogen bonds (CN–HC) between the acceptor unit and TPA at distances of 3.26 Å and 2.88 Å for CAT-1 and DCAT respectively. Note, that natural bond orbital (NBO) analysis11 (cf. Fig S1 in the ESI†) confirms that there were indeed electrostatic binding interactions in all investigated structures in the range of 0.4 up to 3.6 kcal mol−1 per site, suggesting that these are weakly to moderately hydrogen bonding groups. For the sake of simplicity, in the following these shall be, however, just referred to as hydrogen bonds.Starting from the base DCAT architecture, three types of modifications were made in order to find candidate structures that achieve both non-zero transition dipole moment singlet states, as well as red-shift the excitation energies further into the NIR regime (i.e. below 1.65 eV, or longer than 750 nm12). Firstly, the 4,5-dicyanoanthracene (DCA) unit was decorated with further cyano-groups, since a higher CT acceptor strength was expected to reduce the excitation energy.5 Such modifications will be expressed by a number code, like DCA-1 and DCA-2 (see right panels of Fig. 2). Secondly, the phenyl ring that connects the nitrogen of the TPA unit with the anthracene was substituted for small heterocycles such as pyrrole, furanyl and thiophenyl. These modified donors will be referred to as TPA-like and the ring in question is referred to as the (π-)linker. Their substitutions will be expressed by name extensions “Pyr”, “Fur” and “Thi” (e.g. DCAT-1-Pyr). Finally, some DCA-like acceptors with only one or zero cyano-groups pointing towards the donor were modeled to investigate the effects of a step wise introduction of hydrogen bonds. Such structures carry a superscript prefix [1] or [0] to denote a smaller number of possible bonds (e.g. [1]DCAT). Note that by this logic, the zero-bonded variant of CAT-1 is the TPAAP molecule.7
With this three-fold functionalization strategy we arrive at a small family of molecules in which several were identified as potential candidates for NIR-TADF in theoretical calculations. Fig. 3 shows an overview of the excited singlet and triplet excitation energies of all investigated derivatives, color coded by the respective linker used. Note that higher opacity of the circles further serves as a rough scale of relatively higher S1 transition dipole moments. While all markers show the respective molecule's S1 state, the triplet state energy was chosen to be the closest lying to the S1 state (that is Tn, i.e. either T1 or T2). This choice was made as in many instances more than one triplet state was in energetic vicinity to the S1 state and supported a useful excited state character based on its spin-free NTOs and spin–orbit coupling matrix elements (SOCME). Also, previous studies have shown that the T1 state does not have to be the TADF-active state, and that higher excitations may also serve as the major TADF-active triplet state.13
 | ||
| Fig. 3 Overview of excited state energies. Note that the triplet state energy was taken from the triplet state with the smallest relative singlet–triplet gap to the S1 state. The diagonal (dashed) line marks the case of E(S1) = E(Tn) (±0.2) and circles are color coded with respect to their linker. Higher opacity indicate molecules with higher transition dipole moments for S1. | ||
Considering all DCAT derived molecules contained in this study, at the ground state geometry we find vertical S1 transition dipole moments up to 0.59 and singlet excitation energies in the range of 0.68 to 2.88 eV. Table 1 shows the detailed properties of a few selected molecules at the ground state and T1 optimized structures that serve as a reference or may be interesting for NIR-TADF application. A full version of the studied molecules' structures and their related data can be found in detail in Table S1 and at the end of the ESI.†
| Structure | ET1 [eV] | ET2 [eV] | ES1 [eV] | ΔEST [eV] | fosc(S1) | λint [kcal mol−1] | SOCME(S1/T1) | SOCME(S1/T2) [cm−1] |
|---|---|---|---|---|---|---|---|---|
| a Numbers in bold font indicate the states presumably relevant for TADF at the T1 structure. | ||||||||
| CAT-1 | 1.35 (0.98) | 2.06 (1.88) | 1.48 (0.99) | 0.13 (0.01) | 0.20 (0.00) | 0.026 | (0.543) | (0.242) |
| DCAT | 1.63 (0.91) | 1.66 (1.56) | 1.67 (1.56) | 0.01 (0.00) | 0.00 (0.01) | 6.179 | (0.124) | (0.180) |
| DCAT-1 | 1.26 (0.89) | 1.49 (1.21) | 1.27 (0.89) | 0.01 (0.00) | 0.00 (0.00) | 0.005 | (0.803) | (0.300) |
| [0]DCAT-2-Thi | 0.86 (0.36) | 1.59 (1.11) | 1.35 (0.37) | 0.24 (0.01) | 0.38 (0.00) | 0.004 | (3.039) | (0.426) |
| DCAT-4-Thi | 0.64 (0.06) | 1.52 (1.01) | 1.19 (0.06) | 0.33 (0.00) | 0.34 (0.00) | 0.002 | (3.087) | (0.165) |
| DCAT-5-Thi | 0.60 (0.05) | 1.48 (0.99) | 1.12 (0.06) | 0.36 (0.01) | 0.33 (0.00) | 0.002 | (3.130) | (0.108) |
Following Kasha's rule,14 the lowest excited singlet state S1 is presumably responsible for the main portion of fluorescence in TADF. Likewise, for the manifold of triplet states, the one that is expectedly the most populated over time is the lowest excited triplet state T1 – which, as mentioned earlier, is however not necessarily also the one that is best suited for rISC. Hence, to get a more in-depth view which molecules of the DCAT family may be suitable for NIR-TADF, all structures were optimized and re-evaluated at the respective lowest lying excited singlet and triplet states. Fig. 4 shows a comparative excerpt of the relevant data for selected excited state structures.
 | ||
| Fig. 4 Excitation properties grouped with dashed lines by acceptor unit (a) using different TPA(-like) donors at position R; and zero, one or two cyano groups at positions X. (b) Excited state energies of S1 (blue) and closest lying Tn (red) and their resulting singlet–triplet-gap (c) at the excited state geometries of T1 (clean bars, diamonds) and S1 (hatched bars, circles). (d) Spin–orbit coupling matrix elements of the S1/T1 (left bar) and S1/T2 (right bar) transitions with respect to excited state optimized structures (clean and hatched). | ||
From the excitation energies (Fig. 4b), two trends within each subgroup of acceptor unit can be identified. Firstly, the structures that are incapable of forming intramolecular hydrogen bonds between the CT acceptor/donor units (TPAAP, [0]DCAT, [0]DCAT-1) always yield the highest excitation energies compared to any of the other derivatives of the same subgroup. Adding one cyano group decreases the excitation energy by roughly 0.5 eV in all cases (CAT-1, [1]DCAT, [1]DCAT-1), whereas the addition of the second cyano group has slightly less impact. This is in line with the expectation that cyano groups increase the CT-acceptor strength, thus lowering the energy required for the charge transfer to occur. Secondly, all non-phenyl linkers further lower the excitation energies since the electron-rich heterocycles allow for overall stronger CT-donor units compared to the unmodified TPA donor unit. Here, pyrrole shows the strongest red-shift over furanyl and thiophenyl, which is in agreement with their relative reactivity in electrophilic substitution and thus reflects their succession of donating ability.15 Overall, our findings suggest a systematic tunability of excitation properties through the choice of linker and substitution pattern in the acceptor.
Focusing on ΔEST (Fig. 4c), it can be noted that at the S1 geometry (hatched circles), all of the shown structures feature relatively low gap energies, whereas the behaviour is somewhat different for the TPA-like modifications of the base DCAT structure at the T1 geometries (hollow diamonds). However, all structures based on DCA-1 indeed show almost the same behaviour at either S1 or T1 structure. Note that structures like [0]DCAT-1, which are unable to form hydrogen bonds between CT donor and acceptor units are still found generally suitable with a gap energy below 0.1 eV at both S1 and T1 structures.
When comparing the SOCMEs of DCAT molecules with CAT-1, it can be noted that most structures show S1/T1 transition (left bar of each pair) of the same order of magnitude (0.1 to 1.0 cm−1; note the logarithmic scale) as the CAT-1 reference. While the pyrrole and furanyl linker versions generally have weaker S1/T2 transitions (right bar of each pair), the thiophenyl variants show SOCMEs in the order of 1.0 to 10.0 cm−1. This latter increase is a cause of the heavy atom effect of the sulfur heteroatom4 and similar behaviour was found for other sulfur containing molecules in Table S1 of the ESI.†
Next, we investigated the internal reorganization energy λint for the reverse intersystem crossing (rISC) of each structure as the energy difference between the S1 state's total energy at the respective T1 and S1 optimized structures. Fig. 5(a) and (b) show a double logarithmic plot of λint versus the overall structural change between the singlet and triplet optimized structures in terms of the RMSD value and change in mean hydrogen bond length, respectively.
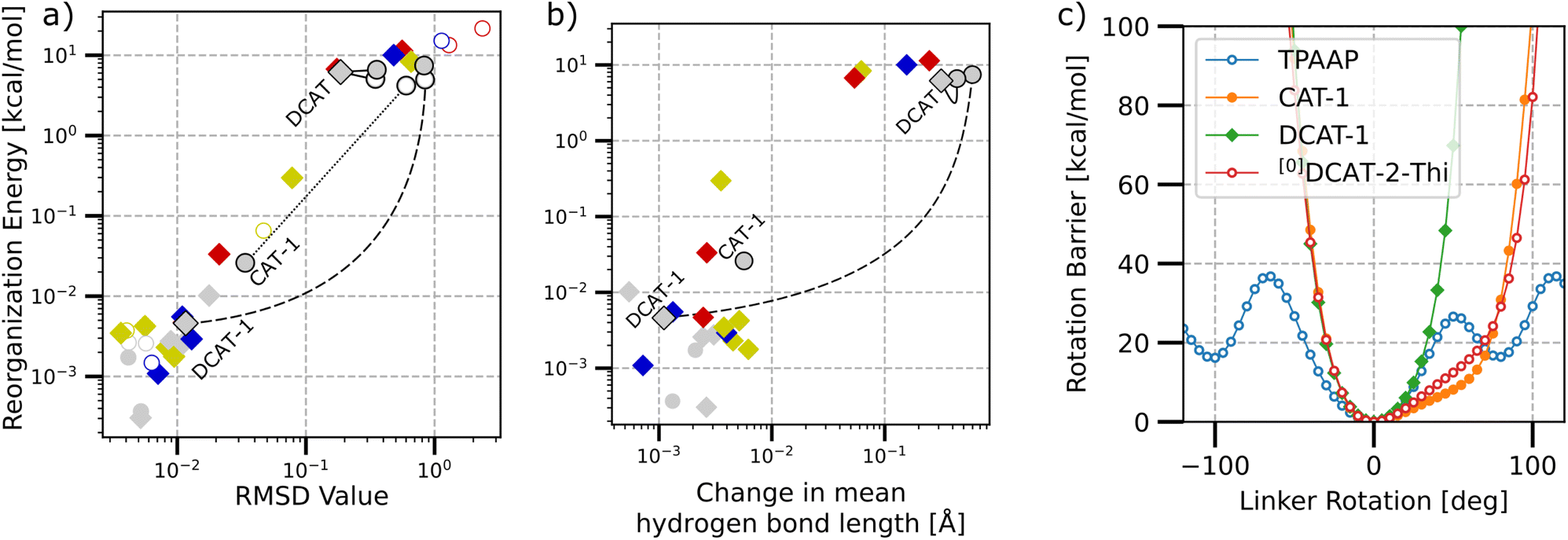 | ||
| Fig. 5 Double logarithmic plots of internal reorganization energy between the S1 and T1 optimized structures and respective structural change in terms of RMSD values (a), change in mean hydrogen bond lengths (b) and primary rotation barrier valley around relaxed structures of selected molecules (c). Diamonds, circles and hollow circles represent structures with two, one and zero hydrogen bonds, respectively. (a and b) Color code applies to different linker types. Annotated and connected data highlights series of two, one and zero bonds. | ||
First of all, it is found that there is a strict correlation between RMSD and reorganization energies, with a large portion of the investigated structures featuring small reorganization energies below 0.1 kcal mol−1 and RMSD values below 0.1. Further analyzing the change in the mean hydrogen bond lengths between the S1 and T1 structures (cf. Fig. 5b) also shows that higher length changes also generally coincide with higher reorganization energies. However, judging by the number of possible hydrogen bonds alone (highlighted by the type of marker) these bonds may not necessarily be the only factor that lead to low reorganization energies in both (a) and (b), as there exist structures with both one as well as zero possible hydrogen bonds that show very low reorganization energies. Considering a series of zero, singly and doubly hydrogen-bonded variants of the same acceptor unit (some examples highlighted by connecting lines in Fig. 5), we typically observe that the step-wise addition of bonds either has only little impact or is strongly stabilizing. The specific behavior is strongly dependent on the acceptor unit though, as for example, the energies in the series for DCAT does not change at all, while DCAT-1 gets stabilized by three orders of magnitude when going from the singly hydrogen-bonded [1]DCAT-1 to the doubly bonded counterpart.
Finally, we investigated the rotation barrier of the π-linker plane between the donor and acceptor units for a sample of molecules (cf. Fig. 5c). Here, the linker unit was rotated around its connection axis (see ESI Fig. S2† for a detailed explanation), while the rest of the molecule was frozen at the relaxed ground state geometry. It is found that the phenyl ring in TPAAP may practically rotate freely above 40 kcal mol−1 (i.e. 1.73 eV, which is similar to the upper bound of NIR to give a perspective). For all other structures, there is a steep gauche-interaction barrier to both directions, as the linker plane eventually would become co-planar and gets repelled by either CN-groups or H-groups of the acceptor and donor units. Surprisingly, CAT-1 and [0]DCAT-2-Thi show almost the same energy profile, despite their rather different designs. While the acceptor in CAT-1 relies on the one CN-group for providing a steric barrier, the acceptor in [0]DCAT-2-Thi architecture features steric barriers through hydrogen atoms to both sides of the linker plane, seemingly leading to a similar trapping situation, despite also the different linker planes themselves. Finally, DCAT-1 features a CN-group on either rotation side, such that the barrier to the right is reached much earlier.
Conclusion
In this study, we investigated the expandability and generality of designing NIR-TADF molecules with one or more intramolecular hydrogen bonds to minimize their rISC-related internal reorganization energy. We conclude that the use of such hydrogen bonds indeed seems to be a useful design strategy for the challenge of designing more rigid NIR-TADF structures, since most bonded structures indeed show an increased rigidity over their non- or singly-bonded counterparts. There is an indication of expandability from one to two bonds, as several of the proposed doubly hydrogen-bonded 4,5-dicyanoanthracene-10-triphenylamine (DCAT) designs reached lower internal reorganization energies than the singly-bonded NIR-TADF molecule CAT-1,2 that was used as reference for its already exceptional performance.Nonetheless, the design strategy does unfortunately not hold as a universal design rule, as we identified also structures that did not experience any stabilization through the introduction of one or more intramolecular hydrogen bonds. Therefore, the amount of stabilization is also strongly dependent on the acceptor unit's general design, revealing that the lowering of λint is correlated but not caused solely by the introduction of hydrogen bonds. Further, studying the rotation barrier of the linker unit's plane between DCA and TPA, we find that the broader DCA acceptor unit and the second CN-group seems to enable even better interlocking of donor and acceptor units. This trapping of the linker also remains effective when substituting for smaller five-membered heterocycles like thiophene, opening the possibility of achieving higher spin–orbit-coupling and transition dipoles through the use of heavy-atom modified TPA donors in exchange for only little rigidity.
Going forward, a more general combined study on both different types and amounts of intramolecular dative bonds (including e.g. hydrogen- and halogen-σ-bonds) and their resulting rotational barriers in twisted donor–π–acceptor TADF molecules is desirable, as it might allow to gain a clearer understanding of the relative importances of steric hindrance and intramolecular bonding. Further, the choice of linker size should be systematically investigated too, as smaller linkers (that are, however, more difficult to trap) in principle allow for shorter effective ranges between the donor and acceptor units which has the potential to improve both the brightness of the S1 state, as well as the spin–orbit-coupling between the T1 and S1 states.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding through the Japanese Society for the Promotion of Science (JSPS – fellowship number P20703) and through Kakenhi (20F20703). We further express our gratitude to the Humboldt Foundation that promoted this project. Calculations were carried out at the Okazaki Research Center for Computational Science (Japan, project number 22-IMS-C015).Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef CAS PubMed.
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu and Z. Lu, Adv. Mater., 2020, 32, 2001248 CrossRef CAS PubMed.
- M. Mońka, I. E. Serdiuk, K. Kozakiewicz, E. Hoffman, J. Szumilas, A. Kubicki, S. Y. Park and P. Bojarski, J. Mater. Chem. C, 2022, 10, 7925–7934 RSC.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- C. Leng, S. You, Y. Si, H.-M. Qin, J. Liu, W.-Q. Huang and K. Li, J. Phys. Chem. A, 2021, 125, 2905–2912 CrossRef CAS PubMed.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242 CrossRef PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T.-T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Beilstein J. Org. Chem., 2018, 14, 282–308 CrossRef CAS PubMed.
- X. Liang, Z.-L. Tu and Y.-X. Zheng, Chem.–Eur. J., 2019, 25, 5623–5642 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- A. Badr Eldin, Wide Spectra of Quality Control, IntechOpen, Rijeka, 2011, ch. 13, pp. 237–248 Search PubMed.
- S. Li, X. Jin, Z. Yu, X. Xiao, H. Geng, Q. Liao, Y. Liao, Y. Wu, W. Hu and H. Fu, J. Mater. Chem. C, 2021, 9, 7400–7406 RSC.
- M. Kasha, Discuss. Faraday Soc., 1950, 14–19 RSC.
- L. I. Belen’kii, I. A. Suslov and N. Chuvylkin, Chem. Heterocycl. Compd., 2003, 39, 36–48 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07450h |
| This journal is © The Royal Society of Chemistry 2023 |