
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical bonding and dynamic structural fluxionality of a boron-based Al2B8 binary cluster: the robustness of a doubly 6π/6σ aromatic [B8]2− molecular wheel†
Rong-Xin Yuea,
Shu-Juan Gaoab,
Peng-Fei Hana and
Hua-Jin Zhai *a
*a
aNanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: hj.zhai@sxu.edu.cn
bDepartment of Chemistry and Chemical Engineering, Lvliang University, Lvliang 033000, China
First published on 11th January 2023
Abstract
Despite the isovalency between Al and B elements, Al-doping in boron clusters can deviate substantially from an isoelectronic substitution process. We report herein on a unique sandwich di-Al-doped boron cluster, Al2B8, using global structural searches and quantum chemical calculations. The cluster features a perfectly planar B8 molecular wheel, with two isolated Al atoms symmetrically floating above and below it. The two Al atoms are offset from the center of the molecular wheel, resulting in a C2v symmetry for the cluster. The Al2B8 cluster is shown to be dynamically fluxional even at far below room temperature (100 K), in which a vertical Al2 rod slides or rotates freely within a circular rail on the B8 plate, although there is no direct Al–Al interaction. The energy barrier for intramolecular rotation is only 0.01 kcal mol−1 at the single-point CCSD(T) level. Chemical bonding analysis shows that the cluster is a charge–transfer complex and can be formulated as [Al]+[B8]2−[Al]+. The [B8]2− molecular wheel in sandwich cluster has magic 6π/6σ double aromaticity, which underlies the dynamic fluxionality, despite strong electrostatic interactions between the [Al]+, [B8]2−, and [Al]+ layers.
1. Introduction
Over the past 30 years, extensive theoretical and experimental studies have been devoted to boron clusters,1–15 which show unusual structural and electronic properties, as well as exotic chemical bonding. The uniqueness in physical chemistry of boron clusters is governed by the intrinsic electron-deficiency of boron.16 Consequently, boron clusters can maintain planar or quasi-planar structures in a wide range of sizes, up to some 40 atoms,17 which is unknown in any alternative cluster systems. New chemical bonding concepts are developed to elucidate boron clusters, such as π/σ aromaticity, antiaromaticity, multifold aromaticity and conflicting aromaticity.12–15 Essential to the above bonding concepts is electron delocalization, which effectively compensates for boron's electron deficiency.Owing also to the electron deficiency, boron-based clusters are magic nanosystems to develop dynamic structural fluxionality. The peculiar spiderweb structure of B19− cluster18 stimulated and triggered computational exploration of a molecular Wankel motor.19 Similar dynamic fluxionality was subsequently extended to an array of circular boron clusters, such as B182− and B13+.20–22 Furthermore, nanotank-type of dynamic fluxionality was reported in elongated boron clusters like B11, B11−, and B15+,23–25 in which a peripheral boron ring glides near freely around an elongated boron core.
Mixing or alloying a metal element with boron leads to boron-based alloy clusters, whose structures can be delicately tailored and electronic properties tuned. In particular, intramolecular charge–transfers allow precise control of the electron counting in alloy clusters, therefore offering opportunities to rationally design new-types of cluster structures and to further explore their bonding and dynamic properties. For example, compass-like clusters B8X2 (X = Mg, Zn, Cd), MB7X2, and MB8X2 (X = Zn, Cd; M = Be, Ru, Os; Be for the Zn-based cases only) were reported with an X2 needle rotating on a baseplate.26,27 The systems have rotation energy barriers of 0.1–0.6 kcal mol−1. Furthermore, Na-doped three-layered Na6B7− and Na8B7+ rotor clusters and a Li-doped propeller B7Li4− cluster were revealed, in which the Na or Li units twist relative to boron wheel with an energy barrier of less than 0.1 eV.28,29 A tubular molecular rotor, B2–Ta@B18−,30 was reported with a B2 unit rotating around molecular axis of a Ta@B18 drum. Its dynamic barrier is 1.13 kcal mol−1. Zhai and co-workers discovered two subnanoscale earth-moon systems, that is, Be6B11− and Be6B102− clusters,31,32 which have dual dynamic modes of rotation and revolution. The outer B11 ring in Be6B11− cluster orbits relative to the Be6 unit and two Be3 rings can also twist against each other.31 The dynamic barriers are 0.21 versus 4.70 kcal mol−1, respectively.
Intuitively, it can be challenging to reach a dynamically fluxional system for boron-based alloy clusters. It can be also challenging to minimize the dynamic barriers for such systems. Ultimately, is it possible to completely diminish the dynamic barrier for a boron-based alloy cluster, that is, to reach an energy barrier of zero? To this end, we have computationally designed a di-Al-doped boron-based Al2B8 cluster via computer global searches and electronic structure calculations. The Al2B8 cluster turns out to be a simple sandwich system with a heptacoordinate B8 molecular wheel. The Al2 component is divided into two isolated Al atoms, which are situated offset from the center of B8 wheel at above and below, collectively serving as a penetrating Al2 rod. The alloy cluster therefore represents an intriguing system. A transition-state (TS) structure is readily located with an energy barrier of 0.01 kcal mol−1 at the single-point CCSD(T) level, which is indeed close to zero and virtually barrierless. Molecular dynamics simulation confirms the dynamic structural fluxionality of the cluster, even at far below room temperature. Chemical bonding analysis suggests that the cluster can be formulated as a charge-transfer [Al]+[B8]2−[Al]+ complex, whose three [Al]+, [B8]2−, and [Al]+ layers are held together via electrostatics. The [B8]2− molecular wheel shows double 6π/6σ aromaticity. This unique bonding picture underlies the dynamic fluxionality of the sandwich cluster.
2. Methods
The global-minimum (GM) and low-lying isomeric structures of Al2B8 cluster were searched using the Coalescence Kick (CK) algorithm,33,34 which was also aided with manual structural constructions. A total of 3500 stationary points were probed on the potential energy surface, including 2000 singlet and 1500 triplet states. The candidate low-lying structures were subsequently reoptimized at the PBE0/6-311+G(d) level.35,36 Frequency calculations were carried out at the same level to ensure that the reported structures are true minima. In order to benchmark the relative energies, the top five low-lying isomers, as well as the TS structure, were further assessed at the single-point CCSD(T)/6-311+G(d) level37–39 on the basis of their PBE0/6-311+G(d) geometries. The B3LYP/6-311+G(d) and single-point CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) calculations were also done for top five isomers to check for consistency of different functionals in terms of structures and energetics. Overall, the electronic structure calculations in this work were accomplished at a total of four levels of theory.Natural bond orbital (NBO 6.0) analyses40 were performed to obtain the Wiberg bond indices (WBIs) and natural atomic charges. Chemical bonding was elucidated through canonical molecular orbital (CMO) analysis and adaptive natural density partitioning (AdNDP).41 Iso-chemical shielding surfaces (ICSSs)42 were calculated to evaluate π/σ aromaticity. The dynamic properties were confirmed by the Born-Oppenheimer molecular dynamics (BOMD) simulations, which were carried out at a set of selected temperatures (100, 300, and 600 K). All the above calculations were performed at the PBE0/6-311+G(d) level. The ICSSs, orbital compositions, and AdNDP analyses were accomplished using the Multiwfn program.43 All electronic structure calculations and the BOMD simulations were done using the Gaussian 09 package.44 The computational results were visualized using the GaussView, CYLview, and VMD programs.45–47
3. Results
3.1. Global-minimum structure
The GM C2v (1A1) structure of Al2B8 cluster is shown in Fig. 1(a) and those of the top 20 low-lying isomers are presented in the ESI (Fig. S1, ESI†). Their relative energies are listed at the PBE0/6-311+G(d) level, including zero-point energy (ZPE) corrections. Further benchmarking of top 5 structures is performed using single-point CCSD(T) calculations. The GM cluster turns out to be 5.28 kcal mol−1 lower than its nearest competitor at single-point CCSD(T)/6-311+G(d)//PBE0/6-311+G(d). The same is true at the complementary CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level, by 4.87 kcal mol−1. Thus, the GM cluster is reasonably well-defined on its potential energy surface at three out of all four levels of theory presented in the paper, although it is marginally competitive at PBE0/6-311+G(d). We consider the single-point CCSD(T) data to be the ultimate energetics of the system, and therefore the C2v (1A1) structure is assigned as the GM cluster. The triplet-state structures are unimportant for the present system (Fig. S1, ESI†).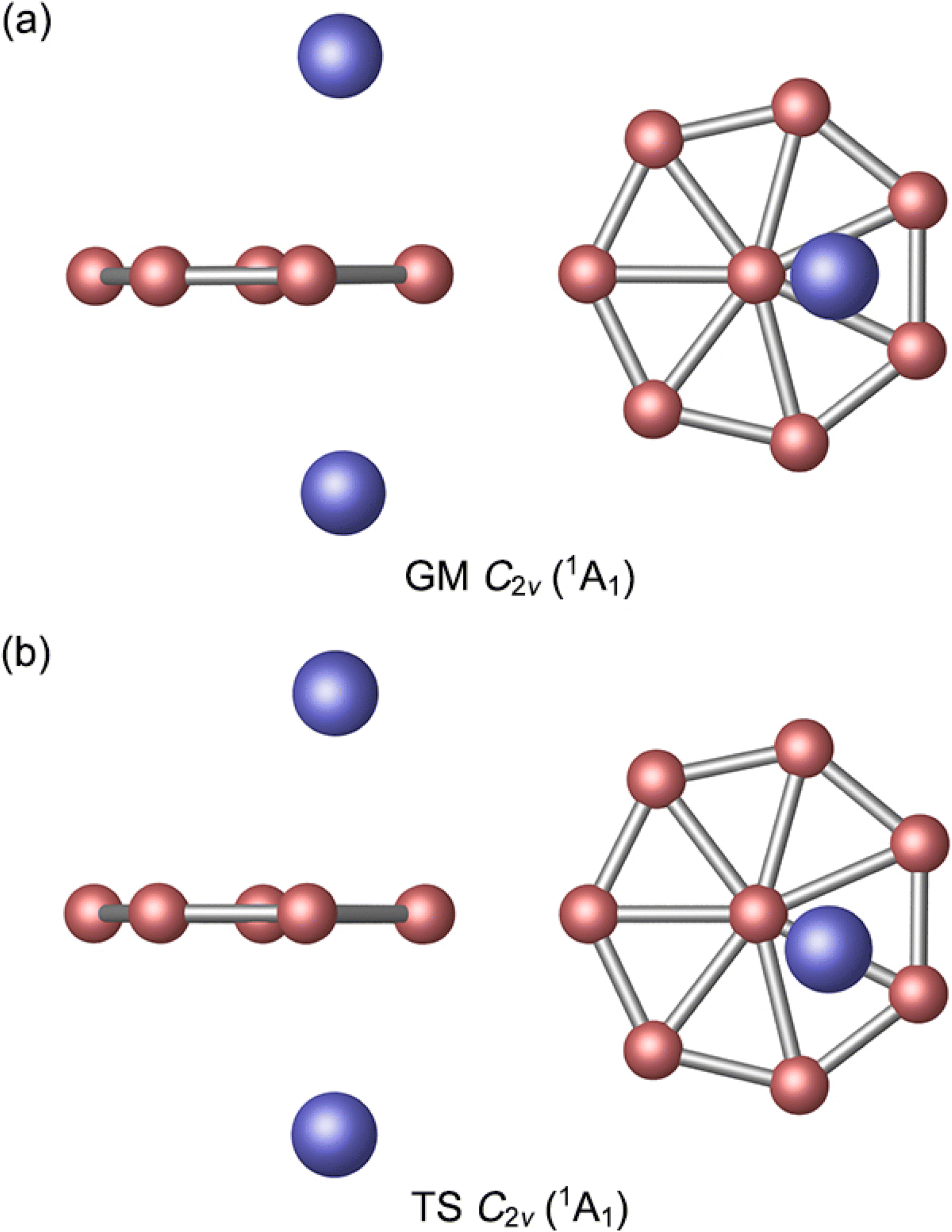 | ||
| Fig. 1 Optimized (a) C2v (1A1) global-minimum (GM) and (b) C2v (1A1) transition-state (TS) structures of Al2B8 cluster at the PBE0/6-311+G(d) level. Both top- and side-views are presented. | ||
The GM Al2B8 cluster assumes a closed-shell C2v (1A1) geometry. Two Al atoms are isolated from each other, floating symmetrically above and below the B8 molecular wheel. Its top- and side-views are illustrated in Fig. 1(a). Basically, it is among the simplest form of three-layered sandwiches. The Al atoms are offset from the center of molecular wheel, by a horizontal distance of 0.82 Å, which is probably due to steric hindrance (between the Al and central B atoms). Relevant D2d or D8h structures are also located in the CK searches (Fig. S1, ESI†), but these are 4.82 and 21.47 kcal mol−1 higher in energy at the PBE0 level, respectively.
By rotating the Al2 atoms slightly and tangentially, by about 25.7°, one reaches another C2v (1A1) structure as shown in Fig. 1(b). This is a true TS structure. It has an imaginary vibrational frequency of 8.5i cm−1 at PBE0, as well as 9.3i cm−1 at B3LYP. For comparison, the corresponding frequency for GM cluster is 7.2 cm−1 at PBE0 and 10.1 cm−1 at B3LYP. Thus, the assignments of GM and TS structures in this work are rather solid. Their optimized cartesian coordinates at PBE0/6-311+G(d) are listed in Table S1 (ESI†). These two structures differ in that the Al2 unit is located in the middle of a B3 triangle in the GM cluster, whereas it overlaps with one radial B–B link in the TS structure.
3.2. Bond distances, Wiberg bond indices, and natural atomic charges
The optimized bond distances of GM C2v (1A1) Al2B8 cluster at PBE0 are presented in Fig. 2(a). The B8 molecular wheel is perfectly planar and its peripheral B–B distances are virtually uniform (1.55 Å), which are to be compared to the recommended upper-bound values of B–B single (1.70 Å) and double (1.56 Å) bonds.48 The peripheral B–B links are clearly beyond single bonds. In contrast, the radial B–B distances are slightly uneven (1.74–1.83 Å), owing to disturbation associated to vertical Al2 unit. The radial B–B links are weaker than single bonds. The calculated WBIs show that peripheral and radial B–B links indeed have distinct bond orders: 1.27–1.34 versus 0.48–0.62 (Fig. 2(a)). The above structural data are remarkably similar to those of a bare D7h B82− cluster,7 except for certain distortions. The distance between an Al atom to its nearest B atom amounts to 2.42 Å, which is significantly longer than the upper limit of B–Al single bond (2.11 Å),48 indicating that the B–Al bonding in GM cluster has a relatively minor covalent component. It is noted that two Al atoms are 4.55 Å apart from each other and there is definitely no Al–Al bonding in the cluster, although they appear collectively as an Al2 unit either structure-wise or dynamically (vide infra).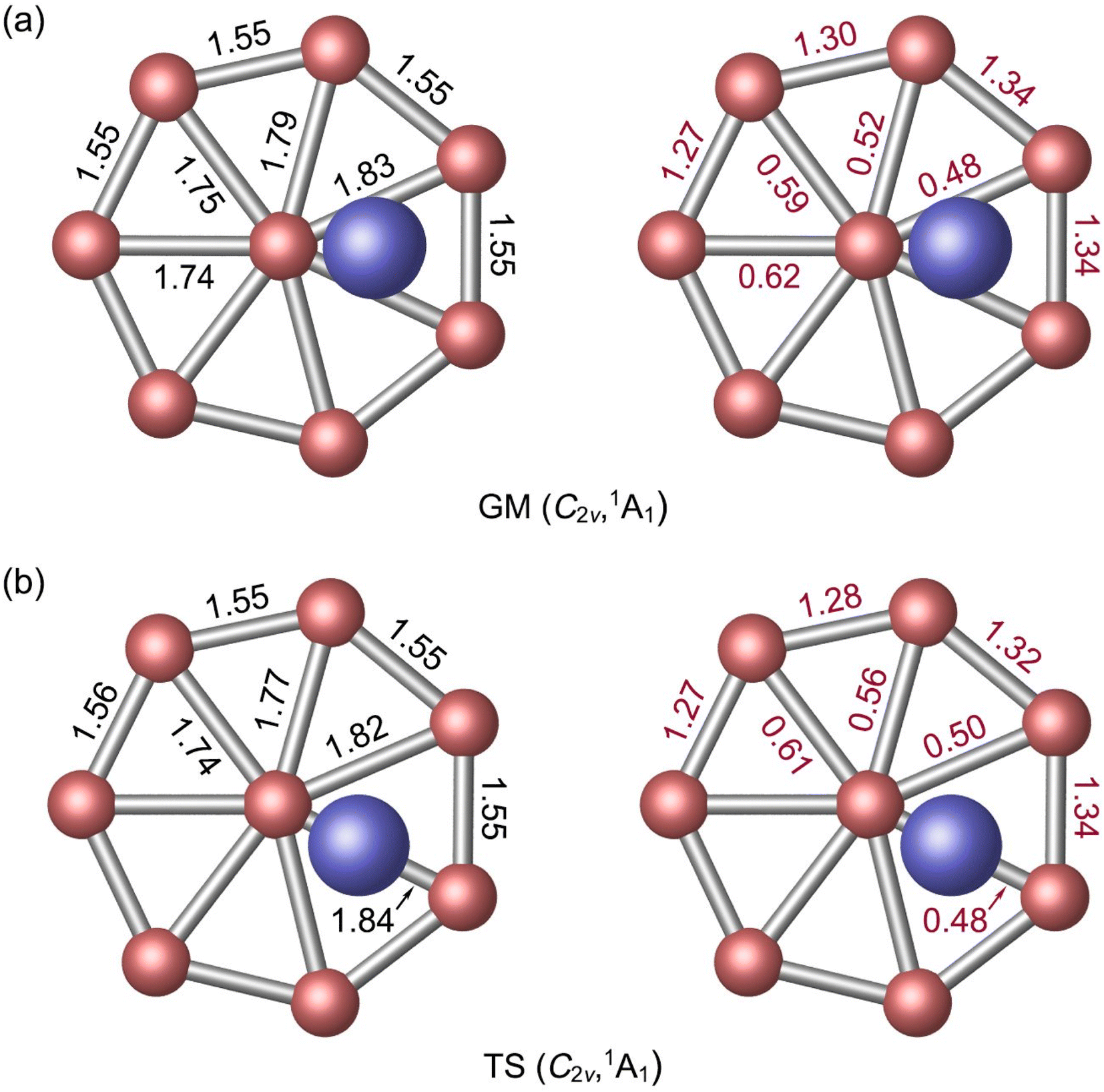 | ||
| Fig. 2 Calculated bond distances (in Å; black color) and Wiberg bond indices (WBIs, in red color) for (a) GM and (b) TS structures of Al2B8 cluster at the PBE0/6-311+G(d) level. The WBIs are obtained from natural bond orbital (NBO) analysis. | ||
As for the natural atomic charges in GM cluster (Fig. 3(a)), two Al atoms both have a positive charge of +0.75 |e|. Three B sites in the vicinity of Al atoms each carries a negative charge from −0.38 to −0.39 |e|. The remaining B site are close to neutral (from −0.01 to −0.13 |e|). This general pattern suggests that intramolecular charge transfer in the cluster is a relatively local process. For example, the B3Al2 or B5Al2 fragment has a collective net charge of +0.35 or +0.09 |e| only, that is, +0.12 or +0.02 |e| per B site. As a consequence of intramolecular charge transfer, the B–B bonding in the vicinity of Al sites are moderately enhanced, either for peripheral or radial B–B links (Fig. 2(a)). The calculated bond distances, Wiberg bond indices, and natural atomic charges for the TS structure are closely similar to those discussed above (Fig. 2(b) and 3(b)). Both the GM and TS clusters may be described as charge–transfer complexes and formally formulated as [Al]+[B8]2−[Al]+.
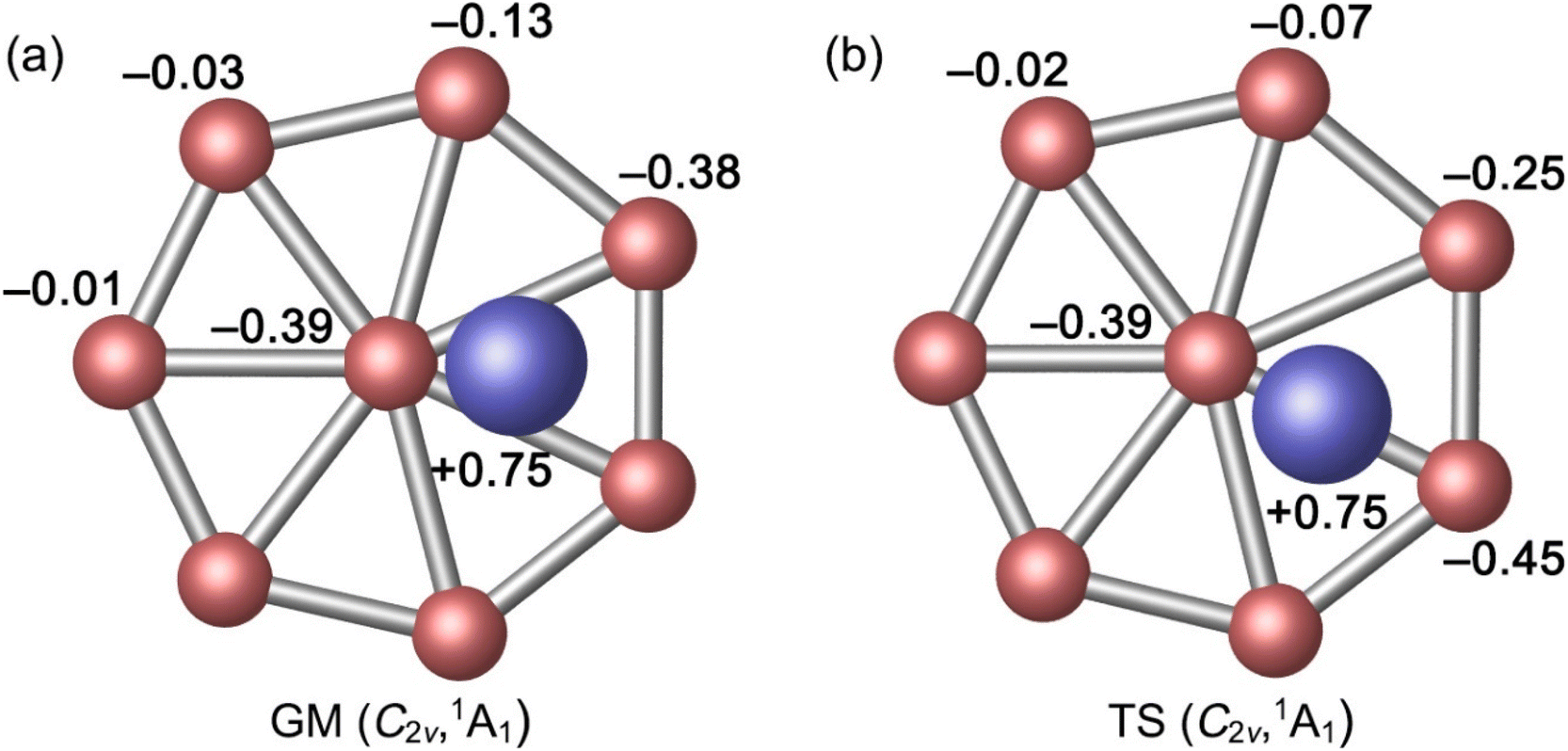 | ||
| Fig. 3 Natural atomic charges (in |e|) for (a) C2v (1A1) GM and (b) C2v (1A1) TS structures of Al2B8 cluster, as obtained from the NBO analysis at PBE0/6-311+G(d). | ||
4. Discussion
4.1. Dynamic structural fluxionality
The similarity between of the GM and TS structures of Al2B8 cluster (Fig. 1) suggests the possibility of dynamic fluxionality. A schematic presentation of structural evolution during such a dynamic process is shown in Fig. 4. The vertical Al2 unit in GM cluster (labeled as GM1) is located at the B1–B2–B8 triangle of the molecular wheel. Let the Al2 unit rotates clockwise with respect to molecular wheel by 25.7°, and the cluster reaches its TS structure (labeled as TS1−2). In TS structure, the Al2 unit overlaps with radial B2–B8 link. Further rotate the Al2 unit clockwise by 25.7°, and the system recovers its GM structure (labeled as GM2). In GM2, the Al2 unit is situated on the B2–B3–B8 triangle. The whole process is simple and straightforward.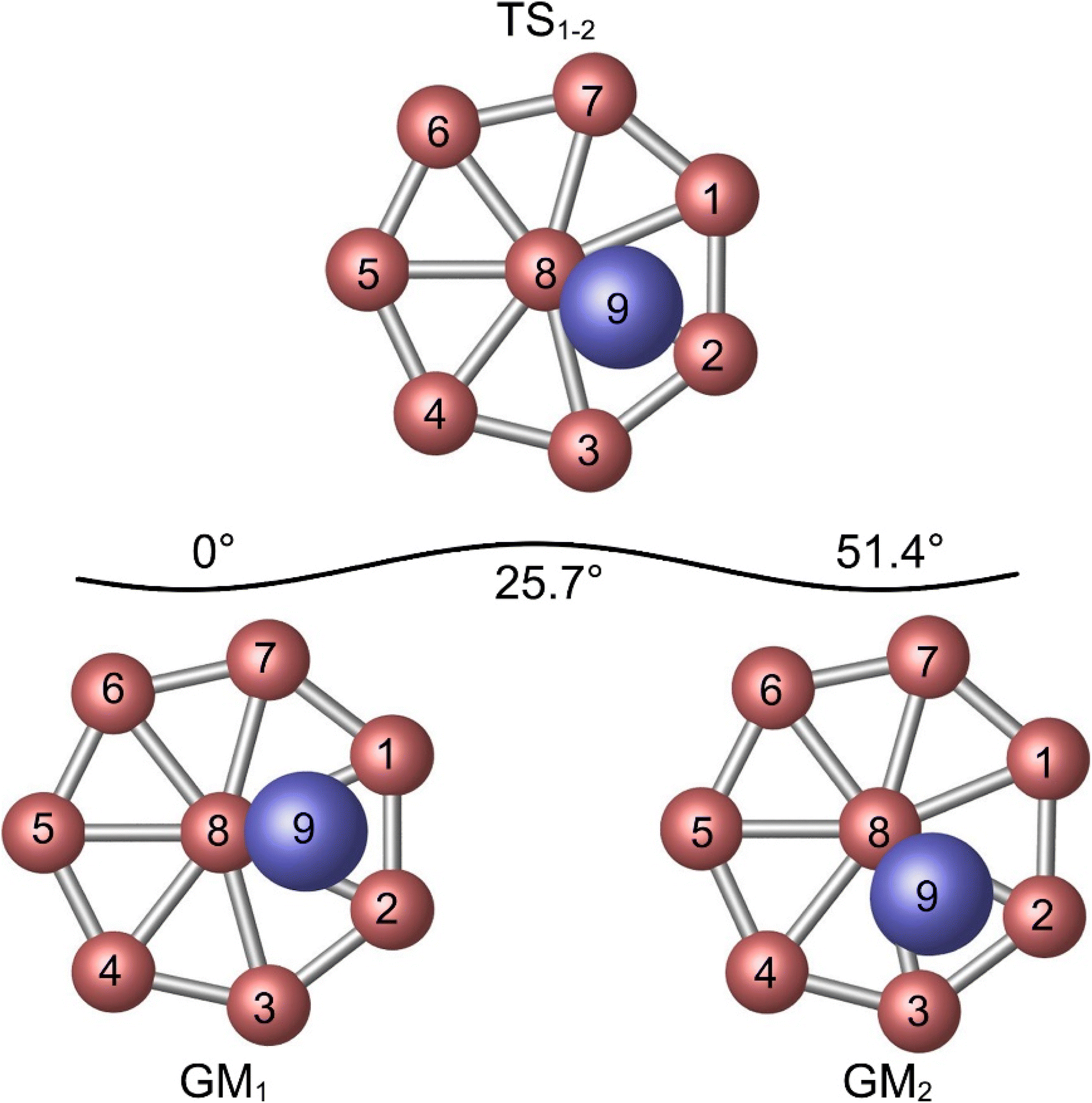 | ||
| Fig. 4 Structural evolution of Al2B8 cluster during the intramolecular dynamic rotation of the Al2 rod with respect to B8 molecular wheel. | ||
Our vibrational frequency analysis reveals a soft mode of 7.2 cm−1 for GM cluster at the PBE0 level, as illustrated in Fig. S2(a) (ESI†). The soft mode is relevant to the collective rotation of peripheral B ring against Al2 unit; and vice versa. A similar soft imaginary mode (8.5i cm−1; Fig. S2(b), ESI†) is revealed for the TS structure at PBE0. The above soft modes are also confirmed at B3LYP, whose calculated values are 10.1 and 9.3i cm−1, respectively. These soft modes facilitate dynamic structural fluxionality of the Al2B8 cluster. As for the dynamic barrier, we can evaluate using the energetics data at the single-point CCSD(T)/6-311+G(d)//PBE0/6-311+G(d) level, including ZPE corrections at PBE0. The energy barrier thus obtained is 0.01 kcal mol−1, which is virtually zero, suggesting that dynamic fluxionality of the cluster is barrierless. This observation is quite unusual in particular for an alloy cluster system.
To vividly demonstrate the dynamic fluxionality of Al2B8 cluster, we have run the BOMD simulations at a selected set of temperatures of 100, 300, and 600 K for about 50 ps. An animation extracted from the BOMD simulation at 300 K is provided in the ESI,† which covers a time span of about 10 ps. It is noted that the cluster is dynamically fluxional even at 100 K, that is, far below room temperature. The latter observation is in line with a virtually zero value for the dynamic barrier.
It is of interest to comment that sandwich GM Al2B8 cluster behaves sort of like a magnetic levitation system. In the former, the three layers are held together by quite strong electrostatics, and yet the Al2 unit seems to be completely floating above and below B molecular wheel. The intramolecular dynamic motion turns out to be absolutely barrierless. We propose to describe this cluster as an “electrostatic levitation system”. We believe the key to this phenomenon is the three-layered sandwich geometry, in which electrostatic repulsion and attraction are ideally balanced.
4.2. Chemical bonding
For an in-depth understanding of the unique structure and dynamic fluxionality of GM Al2B8 cluster, it is essential to perform a chemical bonding analysis. The GM Al2B8 cluster is a closed-shell cluster with 30 valence electrons. Its occupied CMOs are presented in Fig. 5, which are classified into four subsets based on their constituent atomic orbitals (AOs). The two CMOs in subset (a) are composed mainly of 3s AOs from two Al atoms, in their constructive versus destructive combinations. According to the CMO construction principles, these two CMOs can be recombined approximately as two Al 3s2 lone pairs. The remaining 13 CMOs of the cluster turn out to be boron-based, thus validating the bonding picture of a charge–transfer [Al]+[B8]2−[Al]+ complex.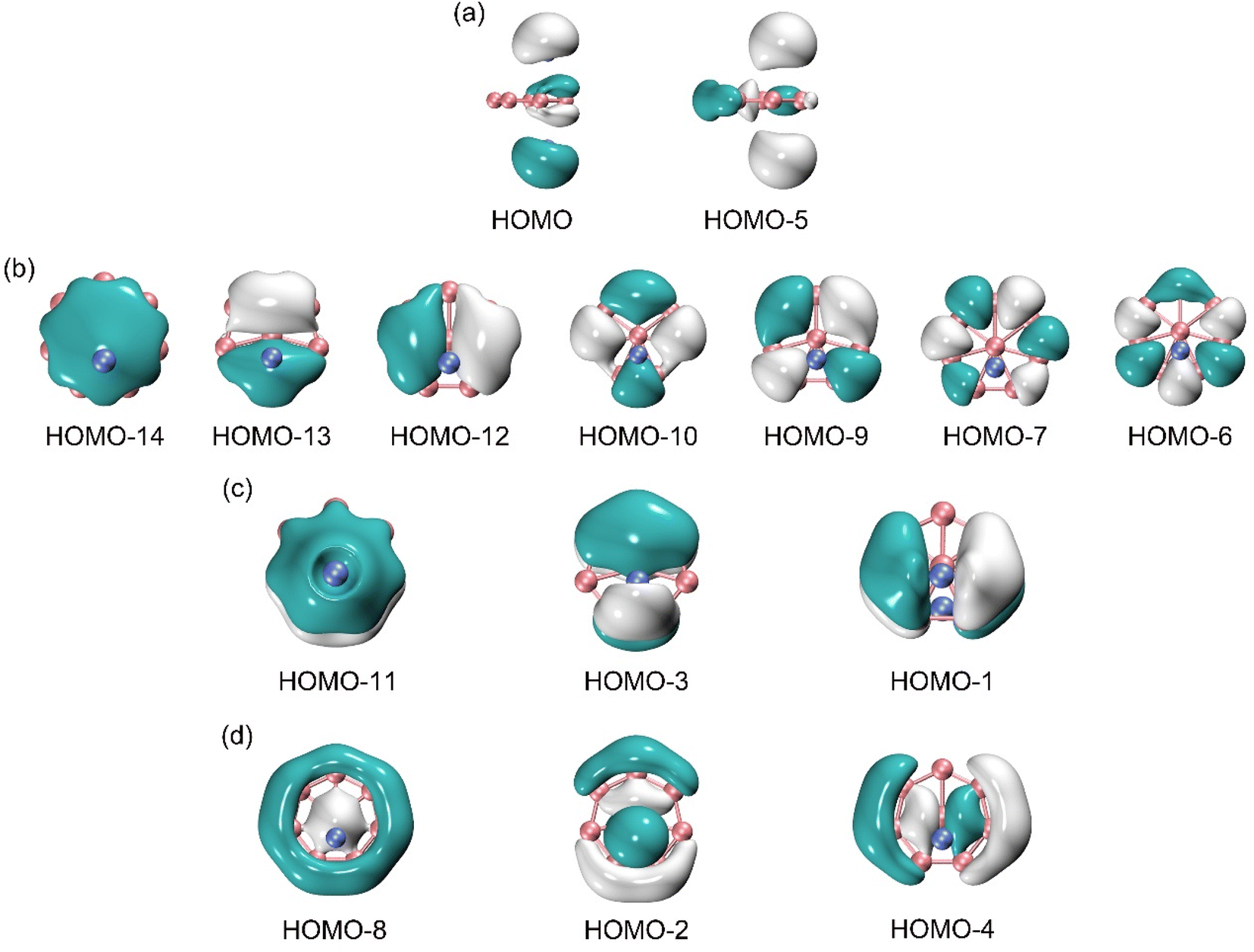 | ||
| Fig. 5 Pictures of canonical molecular orbitals (CMOs) of GM C2v (1A1) Al2B8 cluster. (a) Two CMOs for the Al-based lone pairs. (b) Seven CMOs for Lewis B–B σ single bonds along the periphery of B8 wheel. (c) Three delocalized π CMOs. (d) Three delocalized σ CMOs. | ||
Specifically, the seven CMOs in subset (b) are contributed largely from B 2s/2p AOs on the periphery. They strictly follow the CMO construction principles with 0 to 3 nodal planes from left to right, including three quasi-degenerate pairs. This subset can directly recombine and generate seven two-center two-electron (2c-2e) B–B σ bonds, one for each peripheral B–B edge. The above Lewis-type elements form the structural skeleton of the cluster, collectively consuming 18 electrons out of a total of 30 in the system.
Subset (c) in Fig. 5 shows the π framework, whose three CMOs are contributed primarily from B 2p AOs of the molecular wheel. The overall pattern closely resemble the π sextet in benzene. Furthermore, the pseudo-heptagonal symmetry of the molecular wheel suggests that the π framework is intrinsically delocalized and cannot be reduced to Lewis-type elements. Thus, the π sextet renders the sandwich cluster π aromaticity. The 6π electron counting conforms to the (4n + 2) Hückel rule. Note that the Al2 unit does contribute to these π CMOs, by 7.5%, 15.0%, and 14.8%, respectively, according to the orbital composition analysis (Table S2, ESI†). The Al2 contributions originate from either Al 3s or 3p AOs, in their destructive and constructive combinations, respectively. This is the minor component of Al–B covalency in the system.
Likewise, the three CMOs in subset (d) parallel those in subset (c) in terms of spatial patterns, except that the former CMOs are σ in nature. For a technical note, one of these σ CMOs, that is, HOMO−2, has about 30% contribution from Al 3s AOs (Table S2, ESI†). The latter component may recombine with HOMO/HOMO−2 to help fully recover two Al 3s lone-pairs in the system. Again, this σ framework is truly delocalized and cannot be transformed to Lewis-type σ bonds. The σ sextet renders σ aromaticity to the sandwich cluster, following the (4n + 2) Hückel rule. In short, GM Al2B8 cluster features double π/σ aromaticity, with the magic 6π/6σ electron counting. This unique bonding pattern clearly underlies the stability of Al2B8 cluster, as well as facilitates its intriguing dynamic fluxionality.
The above bonding picture is perfectly borne out from the AdNDP analysis.41 The AdNDP scheme of GM Al2B8 cluster is presented in Fig. 6, which recovers two Al lone-pairs, seven peripheral B–B σ single bonds, as well as double 6π/6σ aromaticity. The occupation numbers (ONs) are generally close to ideal. It is stressed that the appearance of Al 3s lone-pairs, which have ONs of as large as 1.97 |e|, in GM Al2B8 cluster (Fig. 5(a) and 6(a)) is compelling evidence that Al/B substitution in binary Al–B clusters can deviate substantially from an isoelectronic substitution process. Indeed, the Al2B8 cluster does not resemble a bare B10 cluster.49 The Al sites in the former are essentially valence one (rather than three) in terms of chemical bonding.
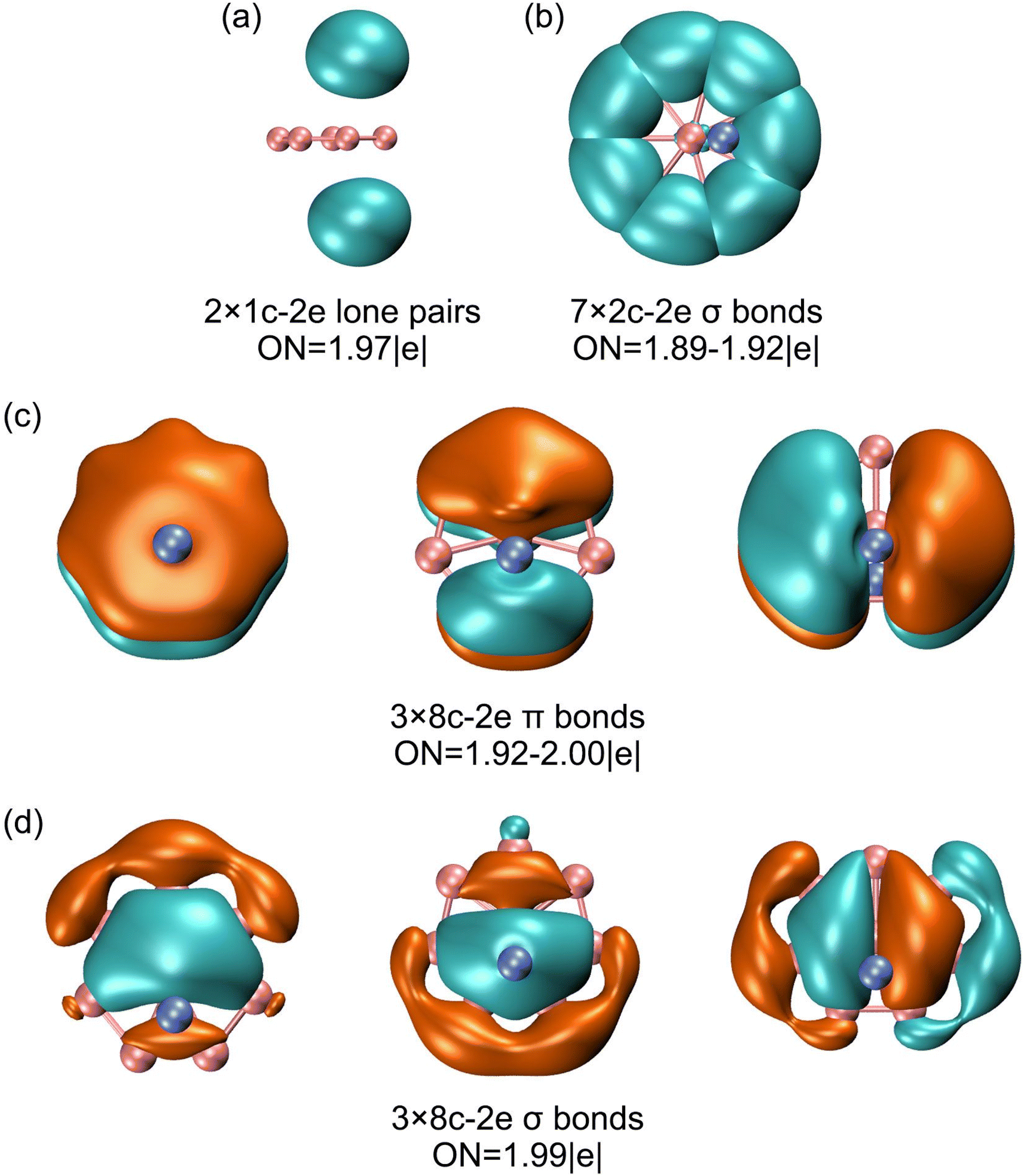 | ||
| Fig. 6 AdNDP bonding scheme for GM C2v (1A1) Al2B8 cluster. Occupation numbers (ONs) are shown. | ||
In the TS structure, the CMOs, AdNDP scheme, orbital compositions virtually do not alter (Fig. S3 and S4, Table S3, ESI†), which explain why the dynamic fluxionality process has no energy barrier. Basically, the covalent component in GM and TS structures are the same (Fig. 5 versus Fig. S3, ESI†), whereas their ionic component differ by a slight shift in spatial charge distributions (Fig. 3). The negative charges in B wheel intimately follow the positively charged Al2 sites. We note that the Al2 unit collectively participates in chemical bonding, as well as in dynamic fluxionality, although there is no direct Al–Al bonding. This is why the Al2 unit orients perpendicularly to the molecular wheel.
To further assess double 6π/6σ aromaticity in GM Al2B8 cluster, we have performed the ICSS calculations. The results are visualized in Fig. 7. Here ICSSzz(0) and ICSSzz(1)50 are probed at the molecular plane and at 1 Å above it, respectively, which roughly differentiate between σ and π aromaticity. The green areas in (a) and (b) within the molecular wheel, in which the shielding effect is primarily concentrated, are in line with σ and π aromaticity of the cluster, respectively.
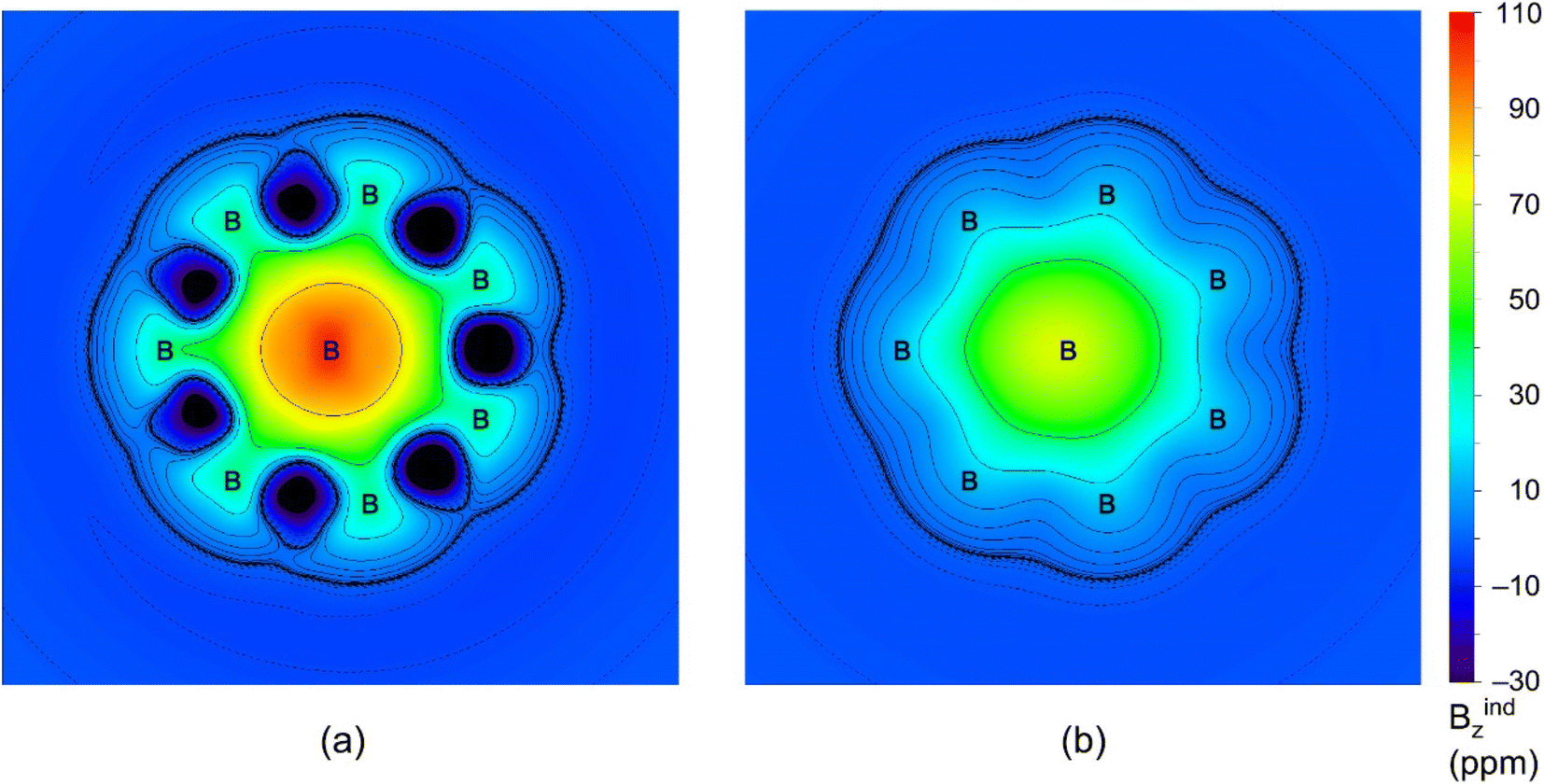 | ||
| Fig. 7 The iso-chemical shielding surfaces (ICSSs) of GM Al2B8 cluster. (a) ICSS(0)zz is calculated at the molecular plane. (b) ICSS(1)zz is calculated at 1 Å above the molecular plane. For ICSSs, a positive value indicates aromaticity, and vice versa. | ||
It should be noted that multifold π/σ aromaticity is prevalent in boron clusters, the latter being also magic molecular systems for dynamic structural fluxionality. The Al2B8 cluster matches the three key factors proposed for a dynamically fluxional species,27 that is, intramolecular charge–transfer, interlayer electrostatic interaction, and completely delocalized 6π/6σ frameworks. Beyond this, its virtually barrier-free dynamics (0.01 kcal mol−1) and unique conformation help further distinguish it from other clusters. Such a binary cluster should be easy to make in a molecular beam machine, following which gas-phase spectroscopic characterizations can be carried out.
5. Conclusions
We have elaborated an ideal boron-based Al2B8 nanorotor cluster with unique sandwich structure. The sandwich cluster is established using computer global searches, electronic structure calculations, and molecular dynamics simulations. It features dynamic structural fluxionality even at far below room temperature (100 K). The dynamic process is virtually barrierless. Chemical bonding analysis suggests that the cluster can be described as a charge-transfer complex and formulated as [Al]+[B8]2−[Al]+, whose three charged layers are bound via quite strong electrostatic forces. The core [B8]2− molecular wheel features magic 6π/6σ double aromaticity. This bonding pattern underlies the stability of the sandwich cluster, as well as facilitates its dynamic structural fluxionality. The work also highlights the idea that the Al/B substitution in binary Al–B clusters can deviate markedly from an isoelectronic process, thus offering opportunities for the rational design of new types of Al–B alloy clusters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21873058 and 21573138).References
- E. Oger, N. R. M. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503–8506 CrossRef CAS PubMed.
- B. Kiran, S. Bulusu, H. J. Zhai, S. Yoo, X. C. Zeng and L. S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 961–964 CrossRef CAS PubMed.
- J. i. Aihara, H. Kanno and T. Ishida, J. Am. Chem. Soc., 2005, 127, 13324–13330 CrossRef CAS PubMed.
- A. P. Sergeeva, D. Y. Zubarev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 7244–7246 CrossRef CAS PubMed.
- W. L. Li, Q. Chen, W. J. Tian, H. Bai, Y. F. Zhao, H. S. Hu, J. Li, H. J. Zhai, S. D. Li and L. S. Wang, J. Am. Chem. Soc., 2014, 136, 12257–12260 CrossRef CAS PubMed.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed.
- T. B. Tai, A. Ceulemans and M. T. Nguyen, Chem.–Eur. J., 2012, 18, 4510–4512 CrossRef CAS PubMed.
- I. Boustani, Int. J. Quantum Chem., 1994, 52, 1081–1111 CrossRef CAS.
- J. E. Fowler and J. M. Ugalde, J. Phys. Chem. A, 2000, 104, 397–403 CrossRef CAS.
- R. Li, X. R. You, K. Wang and H. J. Zhai, Chem.–Asian J., 2018, 13, 1148–1156 CrossRef CAS PubMed.
- S. Jalife, L. Liu, S. Pan, J. L. Cabellos, E. Osorio, C. Lu, T. Heine, K. J. Donald and G. Merino, Nanoscale, 2016, 8, 17639–17644 RSC.
- D. Y. Zubarev and A. I. Boldyrev, J. Comput. Chem., 2007, 28, 251–268 CrossRef CAS PubMed.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS.
- H. J. Zhai, B. Kiran, J. Li and L. S. Wang, Nat. Mater., 2003, 2, 827–833 CrossRef CAS PubMed.
- W. N. Lipscomb, Science, 1977, 196, 1047–1055 CrossRef CAS PubMed.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- J. O. C. Jiménez-Halla, R. Islas, T. Heine and G. Merino, Angew. Chem., Int. Ed., 2010, 49, 5668–5671 CrossRef PubMed.
- G. Martínez-Guajardo, A. P. Sergeeva, A. I. Boldyrev, T. Heine, J. M. Ugalde and G. Merino, Chem. Commun., 2011, 47, 6242–6244 RSC.
- D. Moreno, S. Pan, L. L. Zeonjuk, R. Islas, E. Osorio, G. Martínez-Guajardo, P. K. Chattaraj, T. Heine and G. Merino, Chem. Commun., 2014, 50, 8140–8143 RSC.
- M. R. Fagiani, X. W. Song, P. Petkov, S. Debnath, S. Gewinner, W. Schöllkopf, T. Heine, A. Fielicke and K. R. Asmis, Angew. Chem., Int. Ed., 2017, 129, 515–519 CrossRef.
- Y. J. Wang, X. R. You, Q. Chen, L. Y. Feng, K. Wang, T. Ou, X. Y. Zhao, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 15774–15782 RSC.
- Y. J. Wang, X. Y. Zhao, Q. Chen, H. J. Zhai and S. D. Li, Nanoscale, 2015, 7, 16054–16060 RSC.
- Y. J. Wang, J. C. Guo and H. J. Zhai, Nanoscale, 2017, 9, 9310–9316 RSC.
- Y. J. Wang, L. Y. Feng, J. C. Guo and H. J. Zhai, Chem.–Asian J., 2017, 12, 2899–2903 CrossRef CAS PubMed.
- R. Yu, J. Barroso, M. H. Wang, W. Y. Liang, C. Chen, X. Zarate, M. Orozco-Ic, Z. H. Cui and G. Merino, Phys. Chem. Chem. Phys., 2020, 22, 12312–12320 RSC.
- Y. J. Wang, L. Y. Feng and H. J. Zhai, Chem.–Asian J., 2019, 14, 2945–2949 CrossRef PubMed.
- Y. J. Wang, L. Y. Feng and H. J. Zhai, Phys. Chem. Chem. Phys., 2019, 21, 18338–18345 RSC.
- W. L. Li, T. Jian, X. Chen, H. R. Li, T. T. Chen, X. M. Luo, S. D. Li, J. Li and L. S. Wang, Chem. Commun., 2017, 53, 1587–1590 RSC.
- J. C. Guo, L. Y. Feng, Y. J. Wang, S. Jalife, A. Vásquez-Espinal, J. L. Cabellos, S. Pan, G. Merino and H. J. Zhai, Angew. Chem., Int. Ed., 2017, 56, 10174–10177 CrossRef CAS PubMed.
- L. Y. Feng, J. C. Guo, P. F. Li and H. J. Zhai, Phys. Chem. Chem. Phys., 2018, 20, 22719–22729 RSC.
- P. P. Bera, K. W. Sattelmeyer, M. Saunders, H. F. Schaefer III and P. v. R. Schleyer, J. Phys. Chem. A, 2006, 110, 4287–4290 CrossRef CAS PubMed.
- M. Saunders, J. Comput. Chem., 2004, 25, 621–626 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Y. Jin, B. Chen, X. Y. Kuang, C. Lu, W. G. Sun, X. X Xia and G. L. Gutsev, J. Phys. Chem. C, 2019, 123, 6276–6283 CrossRef CAS.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382–7387 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer III, J. Chem. Phys., 1989, 90, 3700–3703 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- P. L. Cao, W. Zhao, B. X. Li, B. Song and X. Y. Zhou, J. Phys.: Condens. Matter, 2001, 13, 5065–5076 CrossRef CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 468–475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A short movie extracted from the molecular dynamics simulation for Al2B8 cluster at 300 K. Cartesian coordinates for GM and TS structures of Al2B8 cluster at the PBE0/6-311+G(d) level (Table S1); orbital composition analyses for GM and TS structures of Al2B8 cluster (Tables S2 and S3); top low-lying structures of Al2B8 cluster and their relative energies at four levels of theory (Fig. S1); soft vibrational modes for GM and TS structures (Fig. S2); and CMOs and AdNDP bonding scheme for TS structure (Fig. S3 and S4). See DOI: https://doi.org/10.1039/d2ra07268h |
| This journal is © The Royal Society of Chemistry 2023 |