
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of a redox-active non-innocent ligand in additive-free C–C Glaser–Hay and Suzuki coupling reactions by an o-aminophenol palladium(II) complex†
Fatemeh Pakpour a,
Elham Safaei*a,
S. Mohammad Azamib,
Andrzej Wojtczakc and
Karolina Kaldunskac
a,
Elham Safaei*a,
S. Mohammad Azamib,
Andrzej Wojtczakc and
Karolina Kaldunskac
aDepartment of Chemistry, College of Sciences, Shirazu University, Shiraz, 71454, Iran. E-mail: e.safaei@shirazu.ac.ir
bDepartment of Chemistry, College of Sciences, Yasouj University, Yasouj, 75918-74934, Iran. E-mail: azami@yu.ac.ir
cFaculty of Chemistry, Nicolaus Copernicus University in Torun, 87-100 Torun, Poland
First published on 20th January 2023
Abstract
A novel mononuclear palladium complex with 2-(3,5-di-tert-butyl-2-hydroxyphenyl amino) benzonitrile as a non-innocent ligand (abbreviated as PdIIL2NIS) was synthesized, and characterized by IR, UV-Vis, 1H and 13C NMR spectroscopies and elemental analysis. The crystal structure clearly showed that the metal center was in a square planar environment. The bond lengths obtained from X-ray structure analysis revealed that both ligands are in the o-iminobenzosemiquinone radical form. The neutral complex showed strong absorptions in the NIR region, corresponding to the ILCT (intra-ligand charge transfer). Catalytic tests performed for the coupling reaction of terminal alkynes showed that the palladium PdIIL2NIS complex acts as a highly effective catalyst for the base-free C–C coupling reactions, leading to diyne derivatives with excellent yields. The PdIIL2NIS complex in ethanol, as a green solvent, is demonstrated to be an exceptionally active phosphine-free catalyst for the Suzuki reaction of aryl iodides and bromides. The reaction can be carried out under mild conditions (room temperature) with high yields without using a microwave or phosphine ligands. This catalyst exhibits an interesting application of redox non-innocent ligands, the electron reservoir behavior, which makes it needless to use additional reagents. The theoretical calculation provides more details about the complex structure, molecular orbitals, and electronic state.
Introduction
Scientists have long been working on extending bioinorganic catalytic systems with redox-active “non-innocent” ligands as electron sources.1 Such ligand-originated redox activity for conversions of organic materials is unknown. Part of the attractiveness in these compounds originated from the similar energy to frontier orbitals of the transition metal and ligand, resulting in strong mixing between these orbitals. However, this leads to difficulty assigning oxidation states to independent metal and ligand components.2 Canonical examples of non-innocent ligands are catecholates and o-phenylenediamine derivatives (Scheme 1).3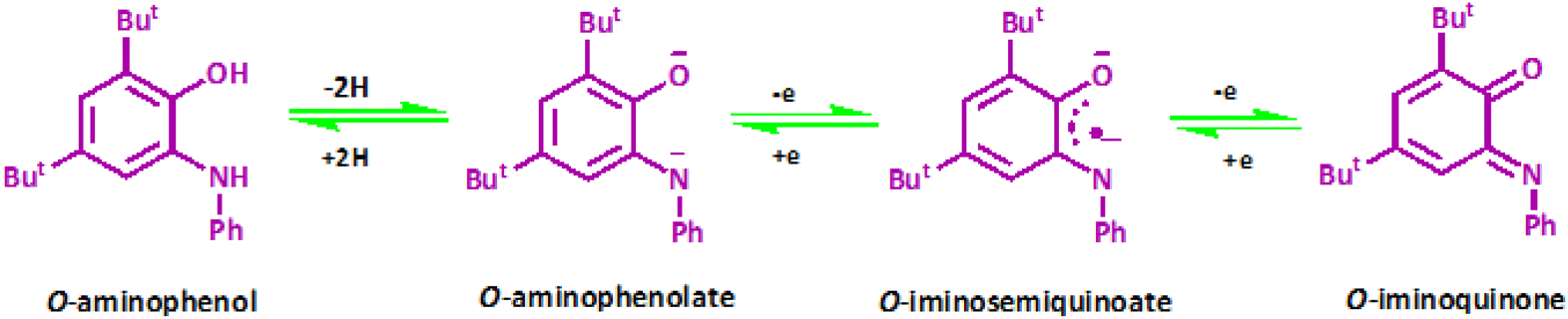 | ||
| Scheme 1 Different oxidation states of o-aminophenol. | ||
Redox-active ligands play a critical role in expanding inorganic, bioinorganic, and catalytic chemistry capabilities. In recent years, some new spectroscopic techniques have permissibly deciphered the electronic states of such metal-radical systems. It was established that metalloradical species play an active role in several metalloenzymes. Scientists have been engrossed in great endeavors to assign and characterize such species.4
Transition metals play a critical role in organic transformations, which consequently cause the development of many catalytic reactions for producing varieties of valuable molecules. Transition metals have a specific capability to make different organic compounds get closer and activate, and via this activation, they can catalyze new bond formation. Palladium is one of the most important metals used for different catalytic proposes. Transition metals, especially palladium, have been essential for various organic transformations, especially forming carbon–carbon bonds.5
For the first time, organoboron compounds were reported as co-catalysts in the cross-coupling with vinyl and aryl halides using palladium-catalysts by Suzuki and colleagues.
In these reactions, organoboron compounds as boronate intermediates promote the organic group transfer from boron to palladium which is known as transmetallation.6 The considerable feasibility of this reaction can be attributed to the nucleophilic character of organoboron compounds. A broad range of functional groups and extreme chemoselectivity have been reported in these reactions. In addition, some advantages of using boron compounds include non-toxicity and feasibility of reaction under mild conditions.5 As a consequence, the Suzuki reaction is one of the most powerful and adaptable method for the synthesis of biaryls.7 These units are known as one of the most applicable chemical compounds in pharmaceuticals, herbicides, and natural products, as well as in material chemistry.8
The typical water- and air-sensitive Suzuki reaction is often catalyzed by palladium in the presence of phosphine ligands as additives.9 Phosphine-free Pd-catalysed reaction conditions are a subject of considerable interest due to economic and environmental reasons.10
Acetylenic compounds containing a series of conjugated acetylenic motifs are notable central structures in different natural products. In particular, conjugated 1,3-diynes have found wide applications11 in bioactive molecules,12 conducting polymer materials,13 optical light emitting materials,14 and supramolecular chemistry.15 Glaser and Hay developed the most straightforward approach for direct coupling of two terminal alkynes to obtain 1,3-diynes.16
The Glaser oxidative homocoupling reaction, mediated by copper, is widely utilized in modern organic synthesis. Also, several reports about the Pd-catalyzed homocoupling of alkynes under homogeneous catalytic conditions using I2, chloroacetone, or ethyl bromoacetate as oxidants have been published.17
In the present work, we prepared and characterized a new palladium complex containing two O,N-coordinated o-iminobenzosemiquinonato(1−) ligands (Scheme 2). The non-innocent ligand used is 2-(3,5-di-tert-butyl-2-butyl-2-hydroxyphenylamino) benzonitrile. Synthesis of the symmetrical 1,3-diynes in a feasible homogeneous version of the Glaser–Hay homocoupling reaction was performed using PdIIL2NIS as an effective catalyst. In addition, the studied complex is used as an impressive catalyst in the Suzuki reaction.
 | ||
| Scheme 2 The structure of PdIIL2NIS complex. | ||
Results and discussion
Preparation and characterization of H2LNAP
The construction of H2LNAP ligand is illustrated in Scheme 3. The H2LNAP ligand was synthesized according to a published procedure.18 | ||
| Scheme 3 Synthesis H2LNAP ligand. | ||
A reaction between 2-aminobenzonitrile and 3,5-di-tert-butylcatechol in the presence of NEt3 generated pure H2LNAP with 69% efficiency. This ligand is air-stable in the solid state. The elemental analysis results of the ligand are consistent with the proposed structure. The ligand architecture was confirmed by spectroscopic and elemental analysis. The IR spectrum of H2LNAP revealed the presence of one strong ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) stretching vibration at 2222 cm−1. The bands at ν = 3421 cm−1 and ν = 3355 cm−1, respectively, correspond to ν(O–H) and ν(N–H) vibrational stretches. The brilliant, sharp bands in the range 2951–2865 cm−1 are specific for tert-butyl group vibrations. Two bands at ν = 760 cm−1 and ν = 1601 cm−1 are attributed to
N) stretching vibration at 2222 cm−1. The bands at ν = 3421 cm−1 and ν = 3355 cm−1, respectively, correspond to ν(O–H) and ν(N–H) vibrational stretches. The brilliant, sharp bands in the range 2951–2865 cm−1 are specific for tert-butyl group vibrations. Two bands at ν = 760 cm−1 and ν = 1601 cm−1 are attributed to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H bending and CC, respectively.
C–H bending and CC, respectively.
Preparation and characterization of PdIIL2NIS
The palladium(II) monomeric complex containing two O,N-coordinated, bidentate ligands derived from H2LNAP was synthesized by applying stoichiometric amounts of the ligand with palladium(II) acetate salt in dichloromethane and methanol solutions. Also, Et3N was used for phenol group deprotonation leading to phenolate at room temperature (eqn (1)). Dark green complex precipitated instantly from the mother solution in excellent yields.
 | (1) |
The complex was fully characterized by elemental analysis and IR, 1H NMR, 13C NMR spectra, and electronic spectroscopic techniques. In addition, single crystal X-ray analysis determined the structure of this complex. Elemental analysis confirms that there is consistency between theoretical and proposed experimental values of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of metal-to-ligand in the complex.
:2 molar ratio of metal-to-ligand in the complex.
IR spectrum
Examination of the complex IR spectrum shows the appearance of all the ligand's vibrations, proving the ligand's presence in the complex. The lack of the strong v(O–H) stretching band in the IR spectrum of the complex proves the ligand deprotonation and coordination of the resulting phenolate to the Pd(II) centre. The phenolic ν(C–O) stretching vibration was observed at ν = 1264 cm−1. The bands with medium/weak intensity in the range 2954–2870 cm−1 correspond to νs(C–H) and νas(C–H) vibrations, while bands with medium intensity due to ν(C–H from Ar) appear at ca. 700 cm−1. In the IR spectrum of a complex, the strong band appears at ν = 2229 cm−1, inferring the presence of the –CN group not interacting with the Pd center.As seen in the IR spectrum of the ligand, the ν(C–C) and ν(CC) stretching bands appear at 1479 cm−1 and 1610 cm−1, respectively. A significant property in the IR spectrum of PdIIL2NIS is the lack of a strong and sharp band of the N–H group in the ligand at ν = 3355 cm−1, which shows this group was involved in metal bonding with Pd(II) ion. The observed band at 3449 cm−1 belongs to O–H of MeOH or H2O, despite the analysis being done in very dry conditions.
Electronic spectroscopy
The UV-visible absorption spectrum was obtained for complex PdIIL2NIS in CH2Cl2 solvent at 298 K (Fig. 1). The electronic spectrum showed absorption bands in 247 and 268 nm attributed to the π–π* intra-ligand charge transfer. The spectra of the complex exhibited absorbance maxima at 429 and 517 nm, which are assigned to the ligand-to-metal charge transfer (LMCT) from phenolate orbitals to the Pd(II) d orbitals. The broad band at low energy and higher wavelengths (789 nm) corresponds to the MLCT charge transfer.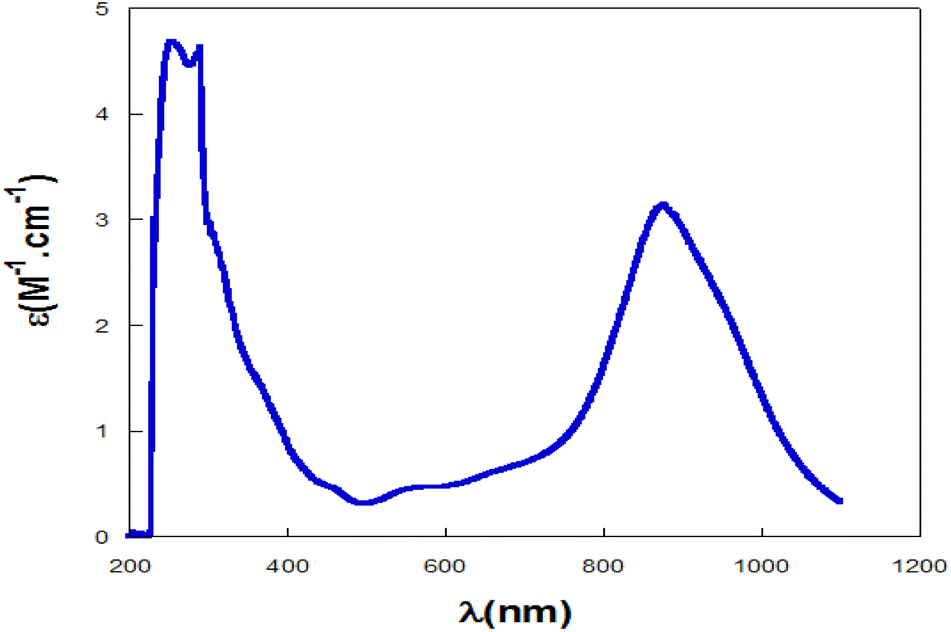 | ||
| Fig. 1 Electronic spectrum of 2 mM CH2Cl2 solutions of PdIIL2NIS. | ||
Crystal structure
Dark green crystals were obtained from the mixture of methanol–dichloromethane (1:1). Details of the diffraction experiment and the structure refinement are summarized in Table 1. Selected bond distances and angles are inscribed in Table 2. The molecular structure of PdIIL2NIS is displayed in Fig. 2. Complex PdIIL2NIS crystallizes in the monoclinic unit cell with space group P21/c, with half of the molecule constituting the asymmetric unit. The complex has a Ci symmetry, with the Pd ion placed at the center of symmetry. Therefore, the symmetry operation [−x + 1, −y + 1, −z + 1] generates the other half of the complex molecule. The X-ray crystal structure reveals that the complex adopts the coordination sphere PdN2O2 of a square-planar geometry, with bi-dentate ligands coordinated via aminophenolate O and N atoms. Two chelate 5-membered rings formed by LNAP are flat. The Pd1–O1 and Pd1–N1 bond lengths are almost equal, the distances being 1.965(5) and 1.963(6) Å, respectively. The N1–Pd1–O1 and N1–Pd1–O1[−x + 1, −y + 1, −z + 1] angles are 81.4(2) and 98.6(2), respectively. Comparison with the similar Ni complex reported previously3 reveals a significant difference between these complexes. In [Ni(LNIS)2], the coordination bonds Ni–O and Ni–N were 0.1 Å longer than reported here for [Pd(LNIS)2], and the Ni–O distance was 0.016 Å shorter than Ni–N. The nitrile group does not interact with any metal center in the structure.
| Formula | C42H48N4O2Pd |
|---|---|
| Formula wt | 747.24 |
| Temperature | 293(2) K |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a (Å) | 16.674(2) |
| b (Å) | 8.0975(7) |
| c (Å) | 16.837(2) |
| α (deg) | 90° |
| β (deg) | 119.020(16)° |
| γ (deg) | 90° |
| Density (calculated) (g cm−3) | 1.248 |
| Absorption coefficient (μ, mm−1) | 0.505 |
| F(000) | 780 |
| Z | 2 |
| Crystal size (mm3) | 0.605 × 0.456 × 0.177 |
| Volume (Å3) | 1987.8(5) |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0813, wR2 = 0.2229 |
| R indices (all data) | R1 = 0.1242, wR2 = 0.2610 |
| Bond length | Value |
|---|---|
| a Symmetry transformations used to generate equivalent atoms: #1 −x + 1, –y + 1, –z + 1. | |
| Pd1–N1 | 1.963(6) |
| Pd1–O1 | 1.965(5) |
| O1–C1 | 1.309(9) |
| C1–C2 | 1.425(10) |
| C2–C3 | 1.371(11) |
| C3–C4 | 1.434(11) |
| C4–C5 | 1.368(11) |
| C5–C6 | 1.409(11) |
| C1–C6 | 1.424(10) |
| N1#1–Pd1–N1 | 180.0(3) |
| O1#1–Pd1–O1 | 180.0 |
| N1#1–Pd1–O1#1 | 81.4(2) |
| N1–Pd1–O1#1 | 98.6(2) |
| N1#1–Pd1–O1 | 98.6(2) |
| N1–Pd1–O1 | 81.4(2) |
| N2–C21–C12 | 178.7(10) |
 | ||
| Fig. 2 Crystal structure of complex PdIIL2NIS. Atoms in the asymmetric unit are labeled. ADPs are plotted at 30% probability level. | ||
In the LCN ligand, the distance of C1–O1 1.309(9) Å is considerably shorter than C6–N1 1.369(10) Å. The corresponding bond distances reported for [Ni(LNIS)2]3 were 1.317(2) and 1.353(2) Å, while in [Cu(LNIS)2] 1.298(3) and 1.340(3) Å. In the complex reported here, the six C–C bonds in the aminobenzoate C1–C6 ring are not equal distances. In particular, the double bonds in the aminophenolate moiety seem to be localized at C2–C3 and C4–C5, while other endocyclic bonds have a significant single bond character (Table 2), which corresponds to the standard pattern of the iminobenzosemiquinone radicals of a short-long-short for three abutting long bonds. Such geometry indicates the ligand form of iminosemiquinone radical monoanion (LISQ)˙−, analogous to that detected for Ni and Cu complexes.3,19 Two nitrogen transferor atoms of the (LISQ)˙− ligands are in the trans positions of the coordination sphere (as are the two oxygen donors). The nitrogen atom N1 is sp2 hybridized with C7–N1–Pd1 124.2(5)°, C6–N1–Pd1 114.4(5)°, and C6–N1–C7 121.2(6)°, and thus the overall geometry of the nitrogen is planar, and ligand is not protonated. The geometrical parameters for this complex are equal within empirical errors to those for the iminobenzosemiquinonate radical ligands stated for alternative relevant complexes in the literature.20
1H NMR
The 1H NMR spectrum of PdIIL2NIS complex in CDCl3 displays a singlet peak in the range 1.0–1.2 ppm, assigned to the tert-butyl group protons. Four hydrogens in the benzonitrile ring were detected with two doubles and two triplet peaks at 7.48, 7.78, and 7.40, 7.68 ppm, respectively. The absence of a singlet near 8.3 ppm, related to the ligand OH group, reflects the coordination of the oxygen atom from this group to the palladium(II) ion.13C NMR
The peaks consistent with the ligand architecture were detected in the 13C NMR spectrum. The 13C NMR spectrum of complex PdIIL2NIS in CDCl3 shows many peaks corresponding to the different carbons present in the complex. The nitrile carbon atom resonance was found at the predicted position, at around 117 ppm for this complex. Singlet signal appearing in the range 29–31 ppm can be attributed to the tert-butyl groups.Computational details
Complexes' electronic structure and geometry are optimized via B3LYP/DGDZVP theoretical level21,22 utilizing Gaussian09 computational chemistry software.23 Polarizable continuum model24 is used to include implicit solvation effects where dichloromethane is considered as solvent. Harmonic vibrational frequency calculations are also performed to locate minimum structures, and time-dependent density functional theory25 is employed to calculate electronic transitions and the corresponding UV-Vis spectrum.Description of the PdIIL2NIS
Fig. 3 displays the DFT-optimized molecular structure of the palladium complex, where bond lengths (in Angstroms) are shown on the corresponding chemical bonds. The complex possesses a center of symmetry, coinciding with the palladium atom The difference between Pd–O and Pd–N bond lengths is negligible (they differ by 0.01 Å), and the two pairs of chelating nitrogen and oxygen atoms form a square-planar environment, while the palladium atom is located at the center of the square.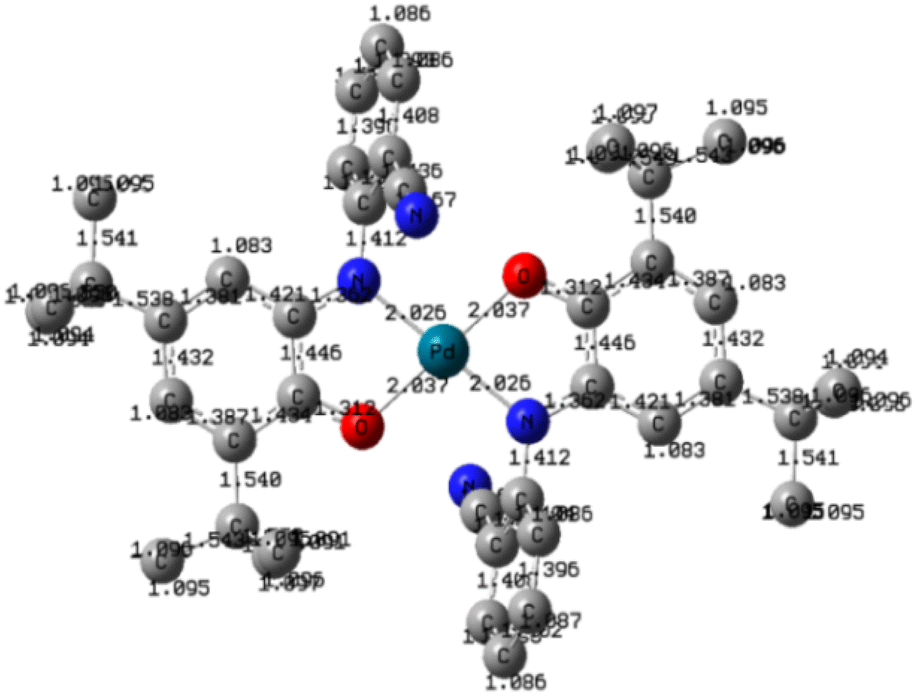 | ||
| Fig. 3 Molecular structure of palladium complex and bond lengths (Å). Hydrogen atoms are not shown for clarity. | ||
Analysis of the UV-Vis transitions for PdIIL2NIS complex
Fig. 4 presents experimental and theoretical UV-Vis spectra of the palladium complex, and shows a strong correlation between experimental and theoretical results, as both exhibit two λmax at ∼300 and ∼900 nm regions with similar qualitative curvatures.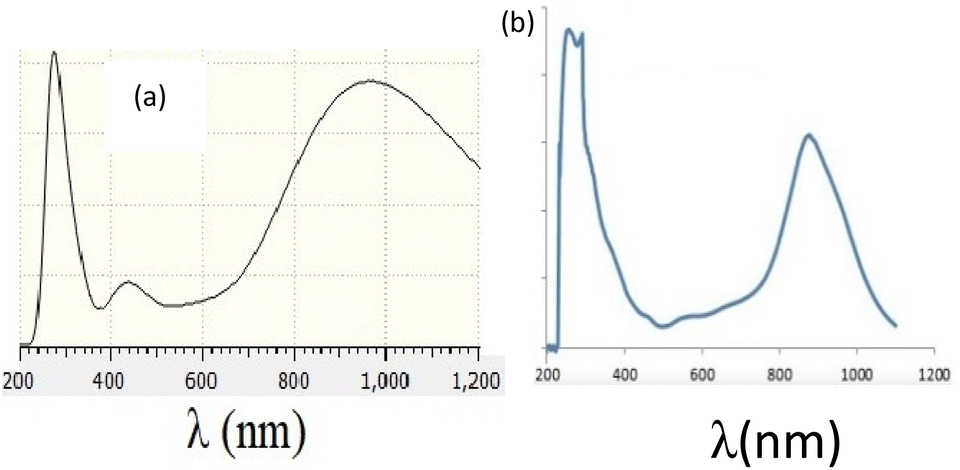 | ||
| Fig. 4 Experimental (a) and theoretical (b) UV-Vis spectra of the palladium complex. | ||
Fig. 5 presents the assignment of electronic excitations to orbital excitations, where the red arrows correspond to the orbital excitation with the major contribution. Since too many possible excitations exist, only those with amplitude (A) greater than 0.05 au are selected. As this figure shows, four excitations exist with A >0.05 au, where HOMO → LUMO transition occurs at λ = 1086 nm, which contributes to the second peak in Fig. 2. The strongest transition corresponds mainly to (HOMO−9) → (LUMO+1) and is responsible for the first peak in Fig. 2 (287 nm).
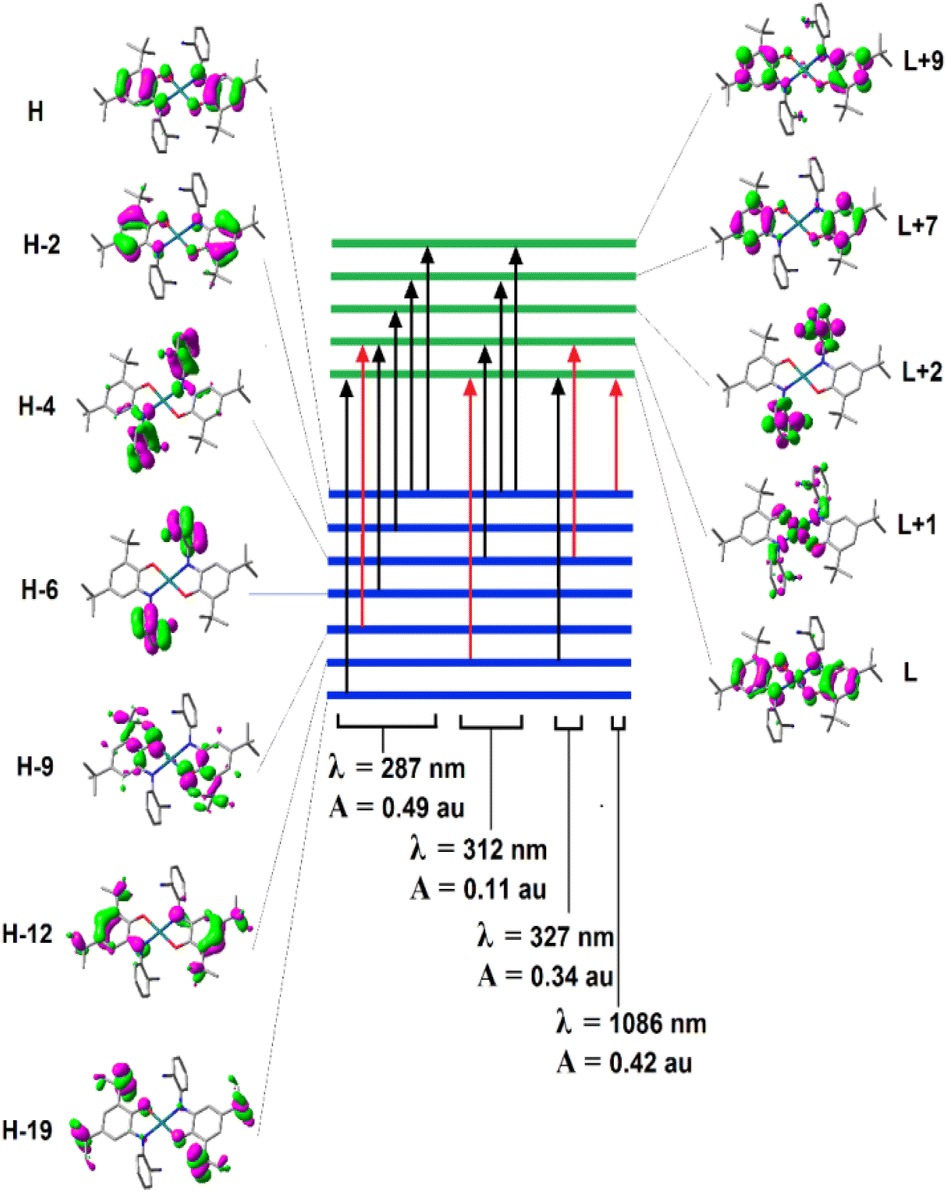 | ||
| Fig. 5 Electronic excitations with amplitude (A) greater than 0.05 au. The red arrows correspond to the orbital excitation with the major contribution to the overall excitation, while H and L correspond to HOMO and LUMO orbitals. | ||
Catalytic activities of PdIIL2NIS complex
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Catalyst | Time (h) | Yield (%) |
| 1 | 2 mmol KOH | Tetrahydrofuran | 2 mol% | 2 | 98 |
| 2 | 2 mmol Cs2CO3 | Tetrahydrofuran | 2 mol% | 2 | 40 |
| 3 | 2 mmol Na2CO3 | Tetrahydrofuran | 2 mol% | 2 | 26 |
| 4 | — | Tetrahydrofuran | 2 mol% | 2 | 20 |
| 5 | 1 mmol KOH | Tetrahydrofuran | 2 mol% | 2 | 27 |
| 6 | 2 mmol KOH | Toluene | 2 mol% | 2 | 23 |
| 7 | 2 mmol KOH | Tetrahydrofuran | 1 mol% | 2 | 16 |
| 8 | 2 mmol KOH | Acetonitrile | 2 mol% | 2 | 22 |

Homocoupling of several terminal alkynes that produced the symmetrical dienes in excellent efficiency without any by-product was then inquired under the optimized conditions. Table 4 shows the abstracted gained results. Despite other reports, we have neither applied dangerous pyridine nor other amines like the costly base of DABCO, etc., as reaction Additive materials, nor harsh conditions. Table 5 compares the activity of PdIIL2NIS and some other catalysts for phenylacetylene homo-coupling reactions. All products were characterized by 1H NMR spectroscopy.
|
|||
|---|---|---|---|
| Substrate | Product | Time (h) | Yield (%) |
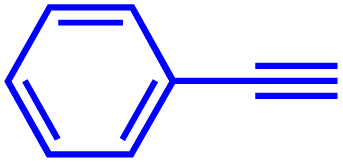 |
 |
2 | 100 |
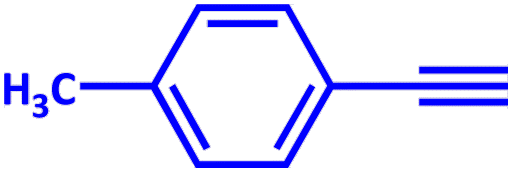 |
 |
2 | 100 |
 |
 |
3 | 100 |
 |
 |
3 | 100 |

|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Solvent/T (°C) | Time (h) | Yield (%) | Additive | Ref. (year) | |
| 1 | 2,2′-Bipyridylpalladium(II)/CuI | Water–TBAB/RT | 48 | 76 | Et3N/I2 | RSC Adv.,26 2014 |
| 2 | Pd(PPh3)4/CuI | Benzene/RT | 18 | 94 | ClCH2COCH3/Et3N | Tetrahedron Lett.,27 1985 |
| 3 | Pd(PPh3)2Cl2/CuI | Diisopropylamine/RT | 2 | 88 | I2 | Tetrahedron Lett.,28 1997 |
| 4 | Pd(PPh3)2Cl2/CuI | Diisopropylamine/RT | 12 | 30 | — | Tetrahedron Lett.,28 1997 |
| 5 | Pd/C, PPh3, CuI | Dimethyl ether–water/80 | 0.5 | 100 | 3-Iodopyrazolo[1,5a]pyrimidine/K2CO3 | Synthesis,29 2005 |
| 6 | NiIIL2NIS | Tetrahydrofuran/RT | 3.5 | 100 | KOH | RSC Adv.,3 2020 |
| 7 | PdIIL2NIS | Tetrahydrofuran/RT | 2 | 100 | KOH | This work |

The suggested mechanism for this reaction has been demonstrated in Scheme 4. In the first step, acetylene must be deprotonated by the base. The coupling process is inefficient in the absence of an extra base. Then two acetylide molecules are coordinated to Pd(II) center concomitant with a change of the o-iminobenzosemiquinone ligands oxidation state to o-iminobenzoquinone form. Therefore, tuning the oxidation state could concentrate on the ligands instead of the central metal. Thus, contrary to other catalysts, we have not used any additive in this reaction. The principal role of palladium in this catalysed cross-coupling is that two molecules are convened on it by creating metal–carbon bonds. Therefore, the carbon atoms attached to palladium are placed closer to each other. In the following step, they couple together, forming a new carbon–carbon single bond. In the last step, the reductive elimination process produces the product with altering the oxidation state of two o-iminobenzoquinone to o-iminobenzosemiquinone ligands again. Hence, adjusting the oxidation state will be focused on the ligands rather than the central metal.
 | ||
| Scheme 4 Suggested mechanism for homo-coupling reaction with PdIIL2NIS | ||
Previously, we reported the catalytic activity of NiIIL2NIS complex3 in the acetylene homo-coupling reaction. Therefore it is possible to compare these data with those obtained for PdIIL2NIS. Table 6 includes the formation energies of NiIIL2NIS and PdIIL2NIS complexes depicted in Scheme 4. The DFT calculations revealed a slight difference between Pd(II) and Ni(II) complexes in the formation energies of o-iminobenzocatechol to o-iminobenzosemiquinone and o-iminobenzosemiquinone to o-iminobenzoquinone. For the above conversions, Pd(II) complex shows formation energies lower by 7.8 and 2.5 kcal mol−1 compared to the analogous energies for Ni(II) complex. It may be concluded that these slight differences are responsible for the performance of the palladium complex better than the nickel one. Experiments revealed that phenylacetylene C–C homo-coupling reaction takes 1.5 hours for palladium(II) catalyst against 3 hours for Ni(II) one. In other words, the reaction rate for the Pd(II) complex is two times higher than the Ni(II) one. Hence, the activity of these two complexes is slightly different in the homo-coupling reaction. The type of metal can affect the redox potential and simplicity of conversion of different oxidation states of non-innocent ligands. Coordination of non-innocent ligands to Pd(II) and Ni(II) results in slight changes in o-iminobenzocatechol to o-iminobenzosemiquinone and o-iminobenzosemiquinone to o-iminobenzoquinone formation energies.
 |
Fig. 6 displays a molecular orbital energy diagram of NiIIL2NIS and PdIIL2NIS complexes in the range of −7 to −1 eV, corresponding to the mechanism in Scheme 4. For clarity, HOMO and LUMO energies of both complexes are boxed in dotted lines. However, HOMO and LUMO of NiIIL2NIS correspond to two- and three-fold degenerate states, respectively, while there is no degeneration in PdIIL2NIS.
 | ||
| Fig. 6 Molecular orbital energy diagram of Ni and Pd complexes in the range of −7 to −1 eV corresponding to mechanism in Scheme 4. Green and purple colors stand for occupied and virtual orbitals, respectively. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Catalyst | Time (h) | Yield (%) |
| 1 | 2 mmol K2CO3 | Ethanol | — | 3 | 17 |
| 2 | — | Ethanol | 4 mol% | 3 | 40 |
| 3 | 2 mmol Et3N | Ethanol | 4 mol% | 3 | 11 |
| 4 | 2 mmol Cs2CO3 | Ethanol | 4 mol% | 3 | 92 |
| 5 | 2 mmol K2CO3 | Ethanol | 4 mol% | 3 | 98 |
| 6 | 2 mmol K2CO3 | Toluene | 4 mol% | 3 | 30 |
| 7 | 2 mmol K2CO3 | Dichloromethane | 4 mol% | 3 | 28 |
| 8 | 2 mmol K2CO3 | Ethanol | 4 mol% | 3 | 60 |

The use of K2CO3 gave supreme results among the inorganic bases applied (Table 7, entry 5). Entries 6 and 7 reveal the effect of different solvents.
The results prove that PdIIL2NIS complex is an excellent catalyst for the Suzuki coupling of aryl halide substrates at room temperature. The coupling of a range of aryl bromides and iodides with phenylboronic acid are presented in Table 8.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yield (%) |
| 1 | 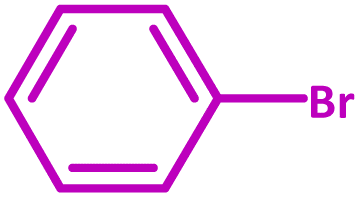 |
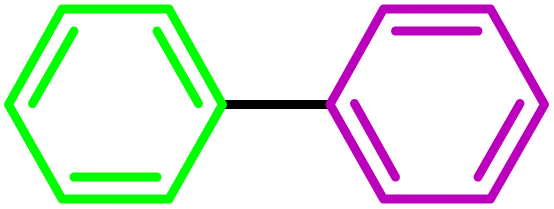 |
3 | 100 |
| 2 | 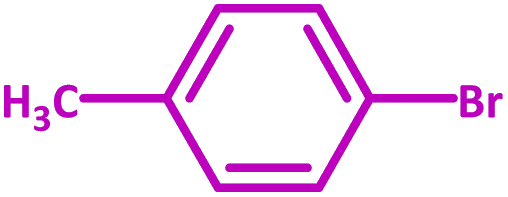 |
 |
2 | 100 |
| 3 |  |
 |
2.5 | 100 |
| 4 |  |
 |
3.5 | 100 |
| 5 |  |
 |
5 | 100 |
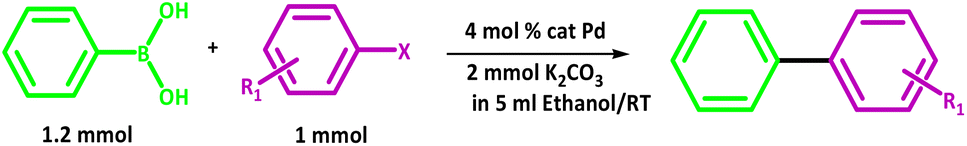
In all cases, the reactions were carried out in ethanol with potassium carbonate as a base. In general, biaryl derivatives were obtained with high efficiency for all the reactions. Different 4-substituted aryl bromides owning either electron-donating or electron-withdrawing groups, such as Me or NO2 gave the corresponding products with good efficiency. The cross-coupling reaction also launched quickly, even with the significant steric effect of aryl bromide. From both academic and industrial viewpoints, other reaction media are of significant concern for making this palladium-catalysed cross-coupling process “greener” by minimizing the use of organic solvents. In this work, ethanol has been used as the reaction solvent. Ethanol is the apparent prime option regarding cost, environmental benefits, and safety. A comparison of the PdIIL2NIS efficiency with the literature data (Table 9) shows that the complex reported here is the excellent catalyst in the Suzuki reaction, even with the mild conditions used.
| Catalyst | Solvent/T (°C) | Time (h) | Yield (%) | Additive | Ref. (year) | |
|---|---|---|---|---|---|---|
| 1 | PdNPs/TMC | Water/80 | 14 | 99 | K2CO3 | RSC Adv.,30 2015 |
| 2 | Pd-basic zeolites | Ethanol–water/50 | 24 | 85 | — | J. Catal.,31 2005 |
| 3 | Ph-SBA-15-PPh3-Pd | Supercritical carbon dioxide/90 | 24 | NR | KOH | Green Chem.,32 2010 |
| 4 | β-CD-capped Pd nanoparticles | Water/reflux | 2 | 98 | Na2CO3 | Langmuir,33 2003 |
| 5 | Pd2dba3/phosphine ligand | Dioxane/80 | 24 | 75 | Cs2CO3 | J. Org. Chem.,34 2003 |
| 6 | Pd(OAc)2 | Water/100 | 24 | 42 | — | New J. Chem.,35 2017 |
| 7 | PdIIL2NIS | Ethanol/RT | 3 | 100 | K2CO3 | This work |
The mechanism postulated for the Suzuki coupling with the use of PdIIL2NIS seems to be different. In the first step, in the presence of aryl bromide, PdIIL2NIS converts to the Pd0 concomitant with the change of the o-iminobenzosemiquinone ligands oxidation state to o-iminoquinone form of Pd0L2Q which reacts with aryl bromide. Aryl bromide is added through an oxidative addition concomitant with the change of Pd0L2Q to PdIIL2Q centre. Starting from PdIIL2NIS in this kind of Suzuki coupling lead us to this idea that possibly the regulation of the oxidation state of the Pd metal centre will be facilitated by its coordinated non-innocent redox active ligands. Therefore, contrary to other catalysts, the use of Pd(0) complex or any reducing agents is not required in this case. Then, aryl group is transferred from aryl boronic acid to the Pd(II) centre of PdIIL2Q via a transmetalation reaction.
Two aryl molecules are combined on Pd(II) centre via the formation of metal-carbon bonds. Next, the aryl groups bound to the palladium(II) are positioned next to each other during the trans/cis isomerization, and the coupling occurs, leading to the new carbon–carbon bond formation. Finally, the reductive elimination produces the final product, also changing of PdIIL2Q to Pd0L2Q. Variation of two o-iminoquinone ligands to their corresponding oxidized form of o-iminobenzosemiquinone ligands of Pd0L2Q lead to restoring the PdIIL2NIS initial catalyst. Thus, the oxidation state tuning might be focussed of on both ligands and central metal. Therefore, unlike for other catalysts, we have not used any additive in this reaction. The proposed mechanism is shown in Scheme 5.
 | ||
| Scheme 5 The suggested mechanism for Suzuki reaction with PdIIL2NIS | ||
Experimental
Materials and methods
All chemicals were purchased from commercial companies and used without further purification. Elemental analyses (CHN) were carried out on Thermo-Finnigan Flash EA1112. IR spectra were recorded on a Shimadzu FT-IR 8300 infrared spectrophotometer. Electronic spectra were recorded on a Shimadzu UV-1280. The 1H (250 MHz) spectra were recorded on a Bruker Advance DPX 250 MHz spectrometer in CDCl3 solvent, using tetramethylsilane (TMS) as an internal standard. X-ray single-crystal diffraction experiment was performed on an Oxford Sapphire CCD diffractometer. The progress of the reactions was monitored by TLC using silica gel Polygram's SIL G/UV 254 plates. All reaction yields refer to the isolated products.Synthesis
The ligand H2LNAP was synthesized according to the literature method.18 2-Aminobenzonitrile (0.118 g, 1 mmol) was added to a solution of 3,5-di-tert-butylcatechol (0.222 g, 1 mmol) and 0.05 mL of Net3 in n-hexane (4 mL), and the suspension was refluxed for two days and then stirred at room temperature under air for four days. The yellow solution converted to red after stirring. Afterward, the reaction mixture was retained for crystallization, and then the product was recrystallized from a 1:1 CH2Cl2:MeOH solvent mixture for purification. H2LNAP, a colorless crystalline solid emerged. Yield: 2.18 g (69%). The IR vmax(KBr)/cm−1: 3421 (O–H), 3355 (N–H), and 2222 (–CN) (Spectral data in Fig. S1, ESI†). Elemental analysis, found (calc.)%: C21H26N2O (MW = 322.44 g mol−1) C: 76.15 (78.22); H: 9.87 (8.13); N: 8.66 (8.69).
PdIIL2NIS. In a round-bottomed flask, H2LNAP (0.162 g, 0.5 mmol) and triethylamine (0.14 mL, 1 mmol) were dissolved in a 2:1 dichloromethane/methanol mixture (15 mL), and then palladium(II) acetate (0.112 g, 0.5 mmol) was added. Afterward, the reaction mixture was stirred at 298 K for 24 hours in the presence of air. After that, a green precipitate of PdIIL2NIS was filtered off. X-ray quality dark green single crystals were grown from a 1:1 (v/v) dichloromethane/methanol. Elemental analysis: calculated (found) for C42H48N4O2Pd (747.28 g mol−1): C, 78.22 (76.14); H, 8.13 (9.8); N, 8.69 (8.65). νmax(KBr)/cm−1: 3075 (CH2 aromatic), 2954, 2906 and 2870 (tert-butyl groups), 2229 (–CN), 1610 and 1479 (CC) (spectral data in Fig. S2, ESI†). NMR data in CDCl3; δ(1H) = 1.10–1.13 [s, 12H, tert-butyl groups]; 7.39 [t, 1H, CH trans to CN in benzonitrile ring]; 7.55 [d, 1H, CH group in benzonitrile ring]; 7.66 [t, 1H, CH]; 7.77 [d, 1H, CH group in the vicinity of the carbon atom containing the nitrile group]. δ(13C) = 29.00–35.15 [s, tert-butyl groups]; 111.57–148.90 [s, carbon atoms corresponding to two aromatic rings] (spectral data in Fig. S3–S5, ESI†).
Homo-coupling of phenylacetylene derivatives using PdIIL2NIS complex
A mixture of a typical substrate of phenylacetylene (1 mmol), KOH (0.112 g, 2 mmol) and PdIIL2NIS (0.0149 g, 2 mol%) was stirred in THF (2 mL) at room temperature. The reaction progress was monitored by TLC till the disappearance of phenylacetylene. The product was separated by silica gel plates and characterized by 1H NMR (Fig. S6–S9, ESI†). (4-Phenyl-1,3-butadiynyl)benzene: 1H NMR (250 MHz, CDCl3): δ = 7.56–7.51 (m, 2H), 7.38–7.33 (m, 3H), 4-methyl-1-[4-(4-methylphenyl)buta-1,3-diynyl]benzene: 1H NMR (250 MHz, CDCl3): δ = 7.16 (dd, J = 7.55, 1.67 Hz, 2H), 7.44 (dd, J = 7.55 Hz, 2H), 2.37 (s, 3H), 1,1′-(1,3-butadiyne-1,4-diyl)bis(2-methoxybenzene): 1H NMR (250 MHz, CDCl3): δ = 6.94–6.87 (m, 2H), 7.36–7.29 (m, 1H), 7.48–7.45 (m, 1H), 3.90 (s, 3H), 1,1′-(1,3-butadiyne-1,4-diyl)bis(4-fluorobenzene): 1H NMR (250 MHz, CDCl3): δ = 7.54–7.48 (m, 2H), 7.07–7.00 (m, 2H).Catalytic activity of the PdIIL2NIS complex in Suzuki reaction
Suzuki reactions were performed under air. A mixture of aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2.0 mmol) and 4 mol% of Pd catalyst in EtOH (5.0 mL) was stirred at room temperature. The reaction progress was monitored by TLC. After completion, the product was extracted using H2O/EtOAc (10 mL). The organic layer was separated. The pure products were isolated by the column chromatography on silica gel eluted with n hexane–EtOAc mixture. The prepared products were characterized by 1H NMR (Fig. S10–S14, ESI†). Biphenyl: 1H NMR (250 MHz, CDCl3): δ = 7.23–7.29 (t, 2H), 7.33–7.39 (t, 4H), 7.50–7.53 (d, 4H), 4-methylbiphenyl: 1H NMR (250 MHz, CDCl3): δ = 2.41 (s, 3H), 7.25–7.28 (d, 2H), 7.33–7.36 (m, 1H), 7.41–7.47 (m, 2H), 7.46–7.52 (d, 2H), 7.58–7.60 (d, 2H), 4-nitrobiphenyl: 1H NMR (250 MHz, CDCl3): δ = 7.45–7.53 (m, 3H), 7.62–7.65 (d, 2H), 7.73–7.76 (d, 2H), 8.29–8.32 (d, 2H), 2,4-dinitrobiphenyl: 1H NMR (250 MHz, CDCl3): δ = 7.25–7.29 (m, 2H), 7.40–7.43 (m, 3H), 7.59–7.62 (d, 1H), 8.37–8.42 (d, 1H), 8.63–8.64 (d, 1H), 4-biphenylcarbaldehyde: 1H NMR (250 MHz, CDCl3): δ = 7.45 (s, 2H), 7.42–7.47 (m, 5H), 7.59–7.62 (m, 6H).X-ray analysis
The X-ray diffraction data were collected on an Oxford Sapphire CCD diffractometer using MoKα radiation λ = 0.71073 Å, at 293(2) K, by ω–2θ method. The structure has been solved by direct methods and refined with the full-matrix least-squares method on F2 using SHELX2017 (ref. 36) program package. The analytical absorption correction was applied36,37 (Table 1). No extinction correction was applied. Hydrogen atoms were positioned from the electron density maps, and their positions were constrained in the refinement.Conclusion
In this study, the geometrical details of the diamagnetic, O,N-coordinated iminobenzosemiquinone [(ISQ)1−] radical form of the ligand have been investigated by X-ray crystallography. Analysis reveals that the ligand has semiquinone character with different C–C bond lengths. The obtained neutral complex has the formula [PdII(LISQ)2]. We have shown that the Pd(II) is found in a square-planar environment composed of two o-iminobenzosemiquinonato(1−) [N, O] donor radical ligands. This complex can perform the C–C coupling reactions. The experiments prove that complex [PdII(LISQ)2] catalyzes the homocoupling of phenyl acetylenes to their corresponding diynes in excellent yields (100%) with less catalyst and lower temperature compared to other catalysts reported in the literature. In addition, we have developed a simple and highly effective phosphine-free protocol for the palladium-catalyzed Suzuki coupling of aryl bromides and iodides with arylboronic acids in ethanol at room temperature and reached 100% yields.The DFT theoretical calculations confirm the role of electron reservoir properties of the ligand on the efficiency of complexes. Insights into the electronic structure of the non-innocent ligand complex, which can tune its oxidation states, helps us choose this interesting type of redox-active complex for designing the catalyst. It is worth saying that most Suzuki reactions have been reported to use Pd(0) and Pt(0) complexes. We have performed this reaction by using Pd(II) complex without any reducing agent, and we have shown that non-innocent ligands can play an important role in oxidative addition reactions. In other words, despite classic oxidative addition reactions which proceeds concomitant with the metal oxidation state variation, the oxidation state of ligand is changed in the reported case. Therefore, in reactions where the metal oxidation state variation was a limiting factor in classic complexes, this limitation is removed in complexes with non-innocent ligands.
In addition, the C–C coupling by the Ni and Pd complexes is different. In classic complexes, we can say that Pd(0) can catalyze C–C coupling more efficiently than Ni(0), since Pd(0) can achieve the Pd(II) oxidation state necessary for an oxidative addition more easily than Ni. Here, we can attribute the difference between the reactivity of our Pd(II) and Ni(II) complex to the differences in formation energy of non-innocent ligand oxidation states.
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors are grateful to Shiraz, Yasouj Universities, and Nicolaus Copernicus University for partial support under Center for Excellence in Research and the BRAIN interdisciplinary research group (AW).References
- P. Chaudhuri, K. Wieghardt, T. Weyhermüller, T. K. Paine, S. Mukherjee and C. Mukherjee, 2005.
- B. Hirani, J. Li, P. I. Djurovich, M. Yousufuddin, J. Oxgaard, P. Persson, S. R. Wilson, R. Bau, W. A. Goddard and M. E. Thompson, Inorg. Chem., 2007, 46, 3865–3875 CrossRef CAS PubMed.
- M. Nasibipour, E. Safaei, M. S. Masoumpour and A. Wojtczak, RSC Adv., 2020, 10, 24176–24189 RSC.
- S. El Ghachtouli, B. Lassalle-Kaiser, R. Guillot and A. Aukauloo, Eur. J. Inorg. Chem., 2014, 2014, 4750–4755 CrossRef CAS.
- Y. CHEN, W. LAI, L. XIE and W. HUANG, Chin. Sci. Bull., 2011, 56, 995–1006 CrossRef.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- R. B. Bedford, S. L. Hazelwood, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2003, 4164–4174 RSC.
- S. Paul and J. H. Clark, Green Chem., 2003, 5, 635–638 RSC.
- C. Liu, Q. Ni, F. Bao and J. Qiu, Green Chem., 2011, 13, 1260–1266 RSC.
- M. Pittelkow, K. Moth-Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682–7684 CrossRef CAS.
- D. Seigler, Acetylenes, Academic, New York, 1973 Search PubMed.
- S. Nakayama, Y. Uto, K. Tanimoto, Y. Okuno, Y. Sasaki, H. Nagasawa, E. Nakata, K. Arai, K. Momose and T. Fujita, Bioorg. Med. Chem., 2008, 16, 7705–7714 CrossRef CAS PubMed.
- F. Cataldo, Polyynes: synthesis, properties, and applications, CRC Press, 2005 Search PubMed.
- W.-Y. Wong, J. Inorg. Organomet. Polym. Mater., 2005, 15, 197–219 CrossRef CAS.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- S. Ghorai and C. Mukherjee, Chem. Commun., 2012, 48, 10180–10182 RSC.
- M. Nasibipour, E. Safaei, G. Wrzeszcz and A. Wojtczak, New J. Chem., 2020, 44, 4426–4439 RSC.
- C. Mukherjee, U. Pieper, E. Bothe, V. Bachler, E. Bill, T. Weyhermüller and P. Chaudhuri, Inorg. Chem., 2008, 47, 8943–8956 CrossRef CAS PubMed.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, J. L. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian09, Wallingford, CT, 2016 Search PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- K. Sindhu and G. Anilkumar, RSC Adv., 2014, 4, 27867–27887 RSC.
- R. Rossi, A. Carpita and C. Bigelli, Tetrahedron Lett., 1985, 26, 523–526 CrossRef CAS.
- Q. Liu and D. J. Burton, Tetrahedron Lett., 1997, 38, 4371–4374 CrossRef CAS.
- L. Yin and J. Liebscher, Synthesis, 2005, 2005, 131–135 Search PubMed.
- A. Affrose, P. Suresh, I. A. Azath and K. Pitchumani, RSC Adv., 2015, 5, 27533–27539 RSC.
- A. Corma, D. Das, H. García and A. Leyva, J. Catal., 2005, 229, 322–331 CrossRef CAS.
- X. Feng, M. Yan, T. Zhang, Y. Liu and M. Bao, Green Chem., 2010, 12, 1758–1766 RSC.
- L. Strimbu, J. Liu and A. E. Kaifer, Langmuir, 2003, 19, 483–485 CrossRef CAS.
- T. E. Pickett, F. X. Roca and C. J. Richards, J. Org. Chem., 2003, 68, 2592–2599 CrossRef CAS PubMed.
- Z. Li, C. Gelbaum, Z. S. Campbell, P. C. Gould, J. S. Fisk, B. Holden, A. Jaganathan, G. T. Whiteker, P. Pollet and C. L. Liotta, New J. Chem., 2017, 41, 15420–15432 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2157991. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra07252a |
| This journal is © The Royal Society of Chemistry 2023 |