
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 free production of ethylene oxide via liquid phase epoxidation of ethylene using niobium oxide incorporated mesoporous silica material as the catalyst†
Muhammad Maqboola,
Toheed Akhter *a,
Muhammad Faheema,
Sohail Nadeema,
Chan Ho Park*b and
Asif Mahmood*c
*a,
Muhammad Faheema,
Sohail Nadeema,
Chan Ho Park*b and
Asif Mahmood*c
aDepartment of Chemistry, University of Management and Technology, C-II, Johar Town, Lahore, 54770, Pakistan. E-mail: Toheed.akhter@umt.edu.pk
bDepartment of Chemical and Biological Engineering, Gachon University, Seongnam, 13120, Republic of Korea. E-mail: chhopark@gachon.ac.kr
cCollege of Engineering, Department of Chemical Engineering, King Saud University, 11421 Riyadh, Saudi Arabia. E-mail: ahayat@ksu.edu.sa
First published on 10th January 2023
Abstract
Ethylene Oxide (EO) is an essential raw material used in various consumer products like different glycol derivatives, ethoxylates, and polymers. We hydrothermally synthesize niobium oxide incorporated with mesoporous silica material (Nb/MSM), an efficient catalyst for CO2 free-ethylene oxide (EO) production via partial oxidation of ethylene. The structural properties of Nb/MSM catalysts were characterized using XRD, TEM, and N2 adsorption–desorption. The catalytic activity of synthesized materials in liquid phase epoxidation (LPE) of ethylene was evaluated in the presence of peracetic acid (PAA) as an oxidant to avoid the production of CO2 and also minimize metal leaching. GC chromatography was used to investigate the successful production of EO, and a peak with a retention time (RT) of 9.01 min served as confirmation. Various reaction parameters viz. temperature, catalyst concentration, ethylene to PAA molar ratio, and solvent effect were investigated in order to optimize the reaction conditions for enhancing the ethylene conversion and selectivity for EO production. By this approach, the challenges of greenhouse gas production and metal leaching were addressed which were associated with previously reported catalysts.
1. Introduction
In the field of epoxidation, various materials have been explored to assess their potential as efficient catalysts for polyethylene, polyvinyl chloride, and ethylene oxide (EO) production. In particular, EO is the third-largest ethylene derivative which is used as raw/starting material for the synthesis of a wide range of commodities.1 The traditional method of producing EO is to use oxygen (O2) as an oxidant at a temperature of 392–500 °F under a pressure of 10–30 bar in the presence of a silver-based catalyst.2,3 However, the epoxidation reaction's selectivity is very poor (around 85%) in this method leading to the production of a huge amount of CO2 as a by-product making it the second-highest emitter of CO2 as a by-product among all chemical processes after ammonia synthesis.2,3 Researchers have tried to change the CO2-emitting gas-phase epoxidation (GPE) to a CO2-free liquid-phase epoxidation (LPE).3 The LPE employs a homogeneous methyltrioxorhenium (MTO) catalyst and H2O2 as an oxidant, resulting in a high catalytic performance with nearly 100% EO selectivity.4 However, there are two disadvantages of MTO used in this method: (1) the catalyst is deactivated by the accumulation of water (as a by-product); (2) the cost of Re metal is high, and its supply is unstable. Thus, using less expensive catalytic materials with high H2O2 usage efficiency instead of Re could increase process economics and practical possibilities.5 As a result, further research should be carried out to develop a different heterogeneous epoxidation catalyst.6In this quest, previous studies have paid attention to the catalysts consisting of transition metals in high oxidation states supported on mesoporous silica material (MSM), specifically MCM-41 and SBA-15.7–11 It was observed that the substrate was catalyzed via the redox reaction by facilitating oxygen transfer from oxidant to alkene double bond.12–16 Among the various metals for the efficient catalyst, niobium (Nb)-based catalysts have been shown significant catalytic activity in the epoxidation of cyclic alkenes, propene, and other chemical substrates such as carvone and limonene with excellent thermal stability and resistance to metal leaching and hydrolysis suitable for LPE process.17–25 A combination of Nb/MSM has been reported by using Nb/MCM-41 and MCM-48 during the oxidation of cyclohexene. Nevertheless, only at low incorporation of Nb metal into MCM-48 support shown efficient selectivity of epoxidation.17,26–31 Also, there has been rarely reported about the Nb/MSM-based EO production: Yan et al. studied tungsten (W) and Nb based MSM as ethylene epoxidation catalysts with H2O2 as the oxidant.32 It was observed that both W-KIT-6 and Nb-KIT-6 metals catalysts are efficient for the production of EO under normal conditions. However, significant metal leaching and H2O2 decomposition was the main drawback associated with W-KIT-6, and Nb-KIT-6-based catalysts.33
Here, we have developed an EO production method with zero greenhouse gas emission. For this purpose, Nb/MSM heterocatalyst was used along with peracetic acid (PAA) as an oxidant. This catalyst was prepared via a hydrothermal procedure in which NbCl5 was used to incorporate Nb metal into ordered cubic large pore mesoporous silicate (SiO2) which was generated from TEOS. The PAA was chosen as an oxidant because it reacts with ethylene gas to produce EO along with by-products such as acetic acid (AA), ethylene glycol (EG), and its derivatives. These by-products are also quite significant and have their own applications in different fields.
The heteroatom structure of Nb/MSM was confirmed by XRD analysis while the porosity was also investigated by BET analysis. The morphology of the catalyst was evaluated by TEM. The prepared catalyst was used for EO production via liquid phase epoxidation process in which the ethylene gas was compressed (under pressure, 50 bars) and dissolved in a liquid reaction medium of PAA. This LPE process was used for the production of EO and acetic acid which was confirmed by gas chromatography (GC). The GC analysis confirmed that the conversion of ethylene to EO was 79.5% and selectivity was found to be 84.3%. Thus, in this whole process, highly desirable EO was produced along with useful by-products with zero emission of CO2 making it an environment-friendly method of EO production.
2. Experimental
2.1 Materials
Tetraethylorthosilicate (TEOS) (Sigma-Aldrich, Germany 98%), Pluronic P123 (Sigma-Aldrich, Germany), niobium(V) chloride (Sigma-Aldrich, Germany), sodium thiosulphate (Sigma-Aldrich, Germany), concentrated sulphuric acid (Merck, Germany), 1-ethanol (Sigma-Aldrich, Germany), peracetic acid 32% (Sigma-Aldrich, Germany), and distilled water. All the chemicals and reagents were used as received without any further purification.2.2 Synthesis of catalyst (Nb/MSM)
For the synthesis of the Nb/MSM, 6.0 g of Pluronic P123 was dissolved in a solvent mixture containing 218 mL of distilled water and 7.92 mL of 35% conc. HCl at 35 °C. After complete dissolution, 7.41 mL of n-butanol was added to this solution with continuous stirring at 35 °C over 1 hour. Then, 1.539 g of NbCl5 and TEOS (14.849 mL) was poured into this solution dropwise under the same conditions. The resulting mixture was stirred for 24 h at 35 °C and then heated at 100 °C for the next 24 h in a closed flask (hydrothermal process), which resulted in the formation of precipitates in the reaction mixture.39 These precipitates were filtered off and dried at 100 °C overnight. Then, the crude product was washed with a mixture of ethanol and HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The purified product was then calcined at 550 °C for 5 hours leading to the preparation of MSM which was the support of the catalyst.
:1). The purified product was then calcined at 550 °C for 5 hours leading to the preparation of MSM which was the support of the catalyst.
2.3 Synthesis of the MSM
For the evaluation of the properties, MSM (support of the catalyst) alone was prepared using the same synthesis procedure of Nb/MSM, except NbCl5 was not added to the reaction mixture with TEOS.2.4 EO production
The synthesized catalyst was used for the production of ethylene oxide via liquid-phase epoxidation of ethylene as shown in Fig. S1.† The peracetic acid, acetonitrile, and catalyst were added to the reactor. Subsequently, pure ethylene gas was introduced from a cylinder. The consumption of ethylene gas was monitored by a weighing balance. The pressure in the reactor was controlled by the pressure controller mounted on the tail of the gas line. The reactor temperature was controlled using an oil bath (temperature range = −20 to 200 °C) equipped with a digital heater, cooler, and a variable speed pump. The reaction was carried out under different sets of parameters, including temperature (40 °C, 50 °C, and 60 °C) and reaction time (1 h, 2 h, 3 h, 4 h, 5 h, and 6 h). In all these conditions, the pressure was kept constant at 50 bars.At the end of each experiment, a certain amount of the sample was taken and injected to GC. As a reference, all these experiments were also carried out using prepared MSM alone instead of the NB/MSM.
The reaction procedures are also shown in Scheme 1.
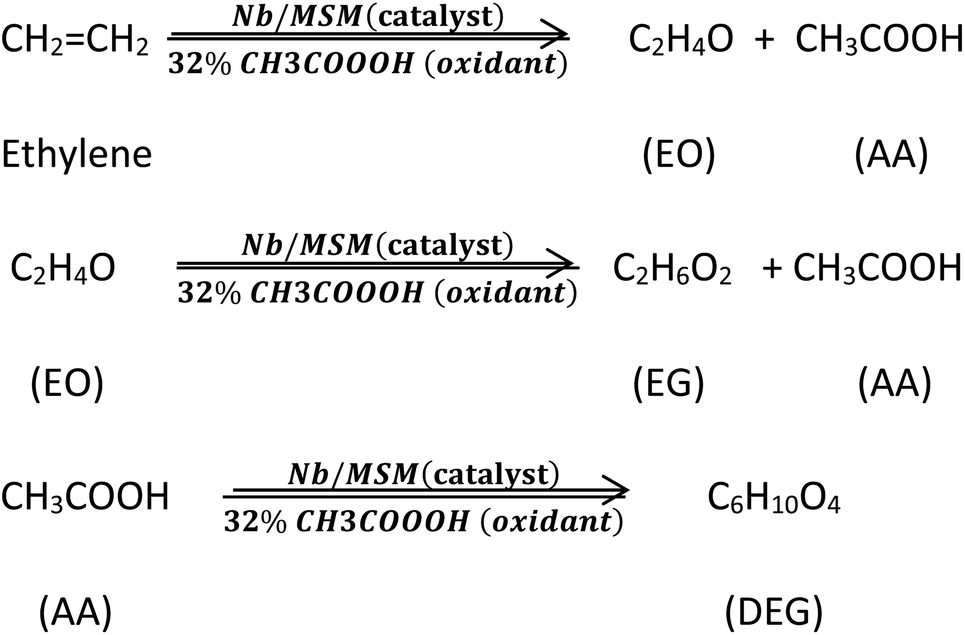 | ||
| Scheme 1 Production of EO and its derivatives from ethylene. | ||
The concentration of produced EO, and EO selectivity was calculated by given equations.
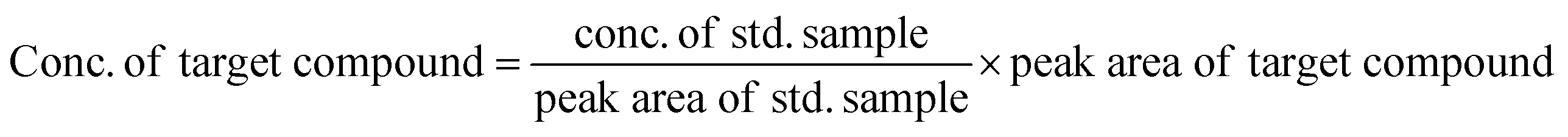 | (1) |
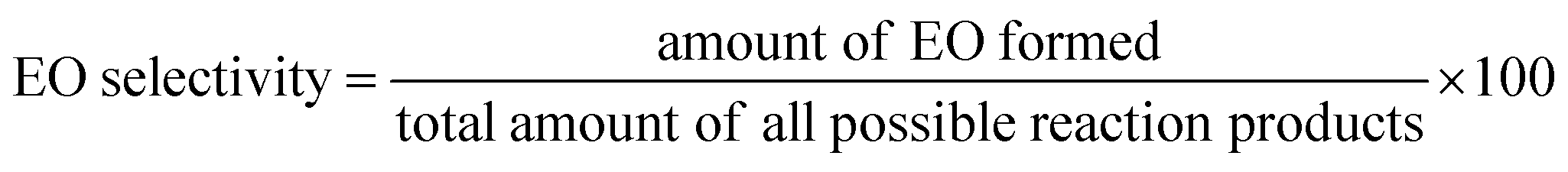 | (2) |
The utilization (UPAA) and conversion (XPAA) efficiencies of PAA were calculated by a reported method.34
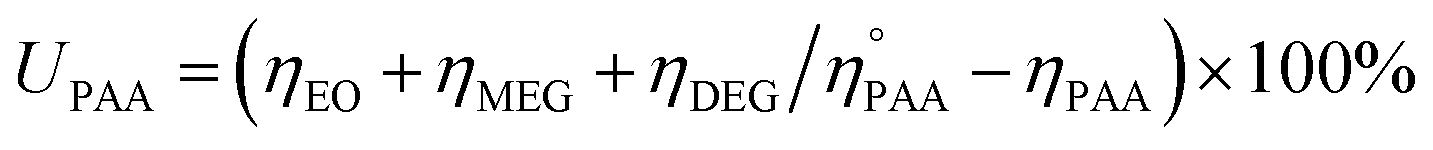
ηEO, ηMEG, ηDEG = moles of EO, monoethylene glycol (MEG), and diethylene glycol (DEG), respectively.
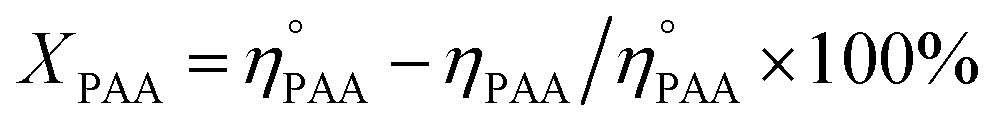
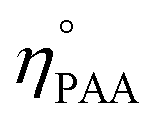 and ηPAA = initial and final moles of PAA.
and ηPAA = initial and final moles of PAA.
3. Characterization
The formation of the support (MSM) and the catalyst (Nb/MSM) was confirmed by Miniflex Benchtop powder X-ray diffractometer (Rigaku, Japan) with Cu Kα irradiation (λ = 1.5418 Å, 200 kV, 50 mA) in the range of 2θ = 2° and 8° for the support and 10°–80° for the catalyst Nb/MSM. The average size of crystallites (size error 0.5 nm) was calculated from the XRD diffractogram using Scherrer equation.35,36 The surface area of the support and catalyst was measured via N2 adsorption–desorption process using Micro-metrics Gemini VII 2390t Automated Gas Sorption Instrument at 77 K and a pressure range of 0.05–0.30 bars. The Brunauer–Emmett–Teller (BET) technique was used to determine the pore size distributions of the samples based on the desorption values. Prior to analysis, the samples were heated at 150 K under a vacuum for 4 h.37 TEM microscope (JEOL 2010F) was used for obtaining TEM images at a higher resolution, working with a high voltage of 200 kV. The samples for the TEM analysis were prepared using a suspension of samples, which was diluted with methanol and then drop-dried on the holey carbon-coated copper TEM grids. Gas chromatography was carried out using Agilent GC-7890A equipped with GC Column DB-WAX capillary column (30 μm, 320 μm, 0.25 μm).4. Results and discussion
The successful synthesis of the catalyst and its support was confirmed by XRD analysis and their diffractograms are shown in Fig. 1. In the XRD pattern of MSM (Fig. 1a), a single broad peak is present at 2θ = 1.7° which exhibits that MSM is completely amorphous after calcination at 550 °C for 5 h. In the case of Nb/MSM (Fig. 1b), the XRD diffractogram is of a typical mesoporous silica material with a broad peak at 2θ = 26.5° in XRD pattern that can be attributed to Nb2O5. This result suggests that Nb ions are well bonded and uniformly distributed on the inner and outer channels of MSM without any changes in the amorphous nature of the MSM framework. These results are in good agreement with previously published literature.38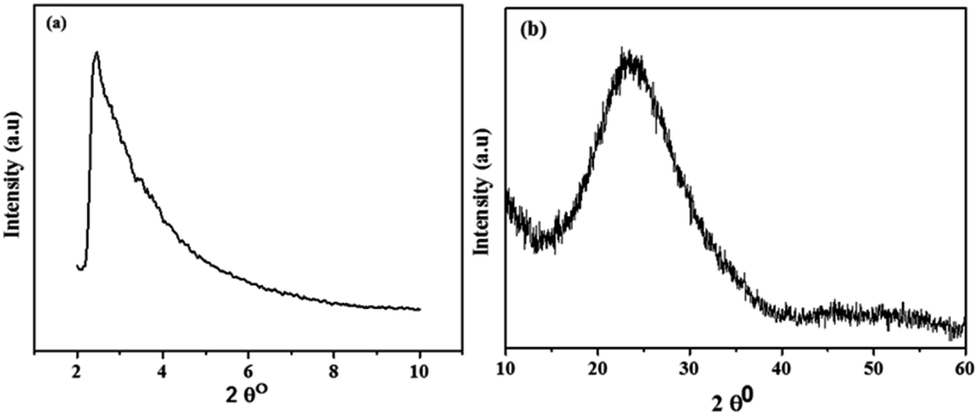 | ||
| Fig. 1 Low-angle and wide-angle X-ray diffraction (a) of MSM and (b) of Nb/MSM. | ||
The surface property of Nb/MSM and MSM was analyzed by BET analysis at 77 K. According to IUPAC Nomenclature, Nb/MSM and MSM can be classified as type IV isotherm as shown in Fig. 2. The hysteresis loops show that these isotherms are characteristic of highly organized mesoporous silica materials. The support MSM has a large surface area of 270.48 m2 g−1 and a pore volume of 0.434 cm3 g−1. After loading of Nb metal on MSM, the significant reduction in the surface area was observed from 270.475 to 129.400 m2 g−1. Also, the pore volume was decreased from 0.434 to 0.163 cm3 g−1 which indicates the partial blockage of mesoporous silica material as shown in Fig. 2b. The pore size distribution of Nb/MSM and MSM was estimated by Barrett–Joyner–Halenda (BJH) method.39,40 Furthermore, the capillary condensation of Nb/MSM and MSM was also measured from the adsorption–desorption curve (Fig. 2b) to assess the uniformity of the pore size. This capillary condensation of nitrogen within the pores is primarily responsible for detecting the sharp inflection at a relative pressure (p/p0) between 0.5 and 0.7. The degree of uniformity of pore size is determined by the height and sharpness of the inflection. It was observed that the average pore diameter has a value of 4.54 nm in the case of Nb/MSM, while MSM has a pore diameter of 4.15 nm. These results are comparable to previous literature.41,42 In BET analysis, surface area (SBET), cumulative pore volume (VBJH), and mesoporous diameter (DBJH) are among the textural parameters listed in Table 1.
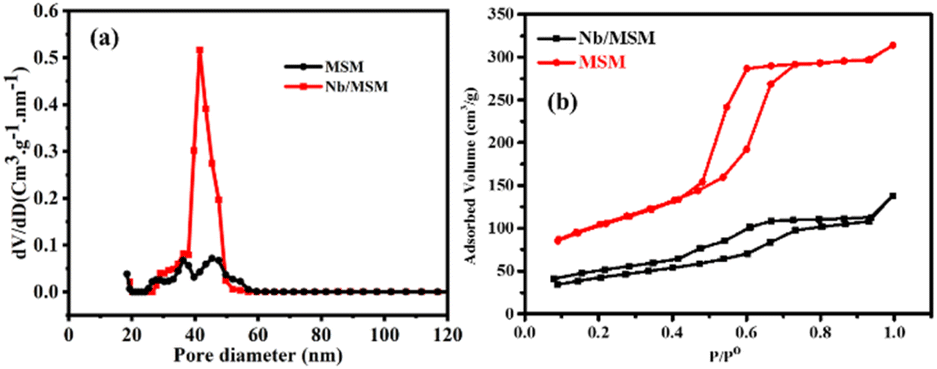 | ||
| Fig. 2 (a) Pore size distribution (b) N2 adsorption–desorption isotherm. | ||
| Entry | Material | SBET (m2 g−1) | VBJH (mL g−1) | (DBJH) (nm) |
|---|---|---|---|---|
| 1 | MSM support | 270.475 | 0.434 | 4.152 |
| 2 | Nb/MSM | 129.400 | 0.163 | 4.543 |
The porous nature and morphology of the Nb/MSM catalyst was evaluated by TEM and micrographs at a resolution of 50 and 100 nm are shown in Fig. 3. These TEM images confirm the mesoporous structure of the Nb/MSM. It can be observed from the TEM images that Nb2O5 nanoparticles, having a size of 20–30 nm, are well dispersed in the MSM support. It shows that the incorporation of Nb2O5 nanoparticles into MSM support does not damage the geometry of the silica framework.27 This fact was also exhibited by XRD and BET analysis.
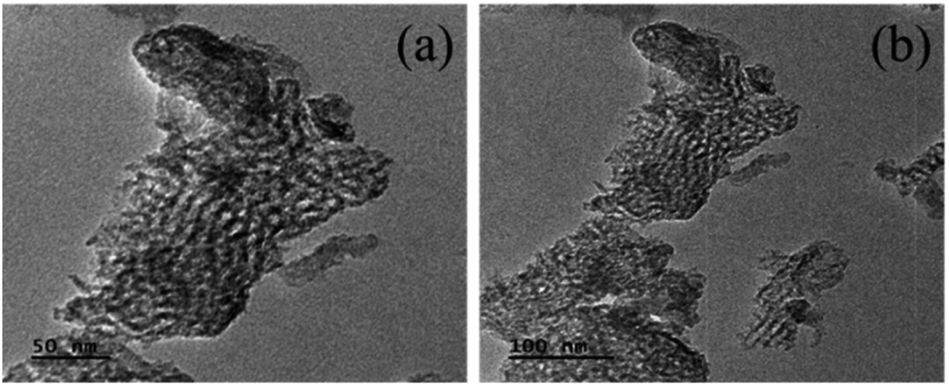 | ||
| Fig. 3 TEM images of Nb/MSM catalyst. | ||
4.1 Catalytic activity
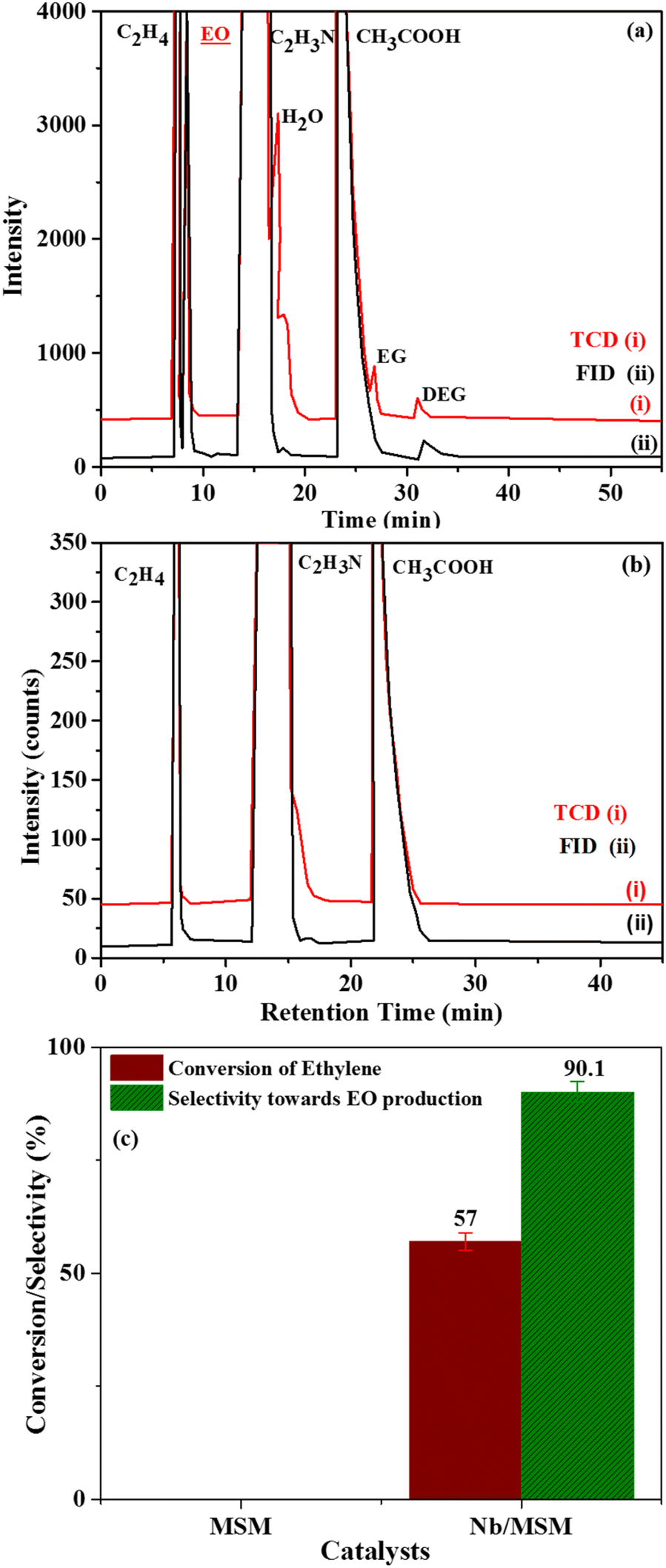 | ||
| Fig. 4 GC chromatogram of (a) Nb/MSM, (b) MSM support and (c) ethylene conversion and selectivity of Nb/MSM towards EO production [reaction condition: ethylene = 3.28 mmol, (32%) PAA = 5.5 mmol, temperature = (50 °C), amount of catalyst (10 wt%) = 0.3 g, solvent = C2H3N (20 mL), time = 6 h]. | ||
Data derived from epoxidation of ethylene using Nb/MSM, MSM and a blank is presented in Table 2. As obvious from the data, 14.3% conversion efficiency and 17.21% utilization efficiency of PAA was achieved using Nb/MSM catalyst.
When MSM was used instead of Nb/MSM, no decomposition of PAA was observed and also no EO was produced. Whereas in the blank run a conversion efficiency of PAA of 11.4% was noted. In the blank run, the decomposition of PAA can be attributed to the acidity of the catalyst caused by the incorporation of Nb into MSM which is in good agreement to previously published literature.32
Furthermore, TOF of Nb/MSM was also calculated by the reported method.44 In our case where peracetic acid was used as an oxidant and Nb/MSM as catalyst, TOF was found to be 163.
In comparison, TOF has a value of 89 in a research report by Wu et al. where H2O2 was used as an oxidant along with Nb/MSM catalyst.44 Thus, it can be inferred that peracetic acid is more potential oxidant for EO formation as compared to H2O2.
Moreover, in order to achieve optimum selectivity of catalyst towards EO production and conversion of ethylene to EO, LPE was carried out at various reaction parameters like temperature, catalyst amount, ethylene to PAA molar ratio, and in the presence of different solvents.
:2 molar ratio and catalyst amount. The reaction was carried out for 6 h using acetonitrile as a solvent and the results of this study are shown in Fig. 5. When the reaction was carried out at 50 °C, the ethylene conversion was 57%, while the selectivity of the catalyst was 90.1%. At 60 °C, ethylene conversion increased from 57 to 73.6%, while selectivity decreased from 90.1% to 83.2%. Similarly at 70 °C, the ethylene conversion showed a slight improvement with a value of 74.8%, but the selectivity of the catalyst was further declined to 79.6%. The decreased selectivity of catalyst towards EO production at elevated temperature can be attributed to the formation of the acetic acid and derivatives of glycol as by-products. It is well established that this reaction proceeds via the formation of ethylene oxide intermediate, and at higher temperature C–C bond is more stable as compared to C–O bond.45,46 Moreover, the accelerated decomposition of peracetic acid at higher temperatures lead to the enhanced ethylene conversion and decreased catalyst selectivity towards EO production.47 Thus, it can be inferred that 60 °C is the optimum temperature where highest possible ethylene conversion and catalyst selectivity towards EO production in the presence of Nb/MSM can be achieved.
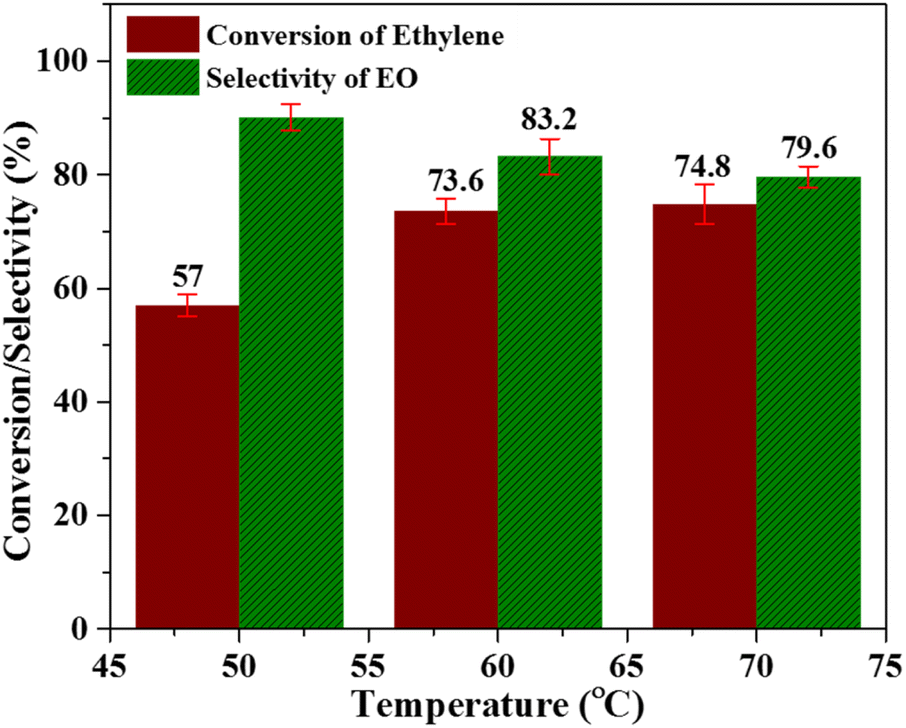 | ||
| Fig. 5 Effect of temperature [reaction conditions: ethylene = 3.28 mmol (32%) PAA = 6.5 mmol, time = 6 h, catalyst amount = 0.3 g, solvent = C2H3N (20 mL)]. | ||
:2 in acetonitrile solvent for 6 h (Fig. 6). An increase in the catalyst amount resulted in a significant improvement in the ethylene conversion and a marginal enhancement in the selectivity towards the EO production. These results can be interpreted on the basis of an increase in the active species of metal oxide (with increasing catalyst amount), which, in turn, expedited the rate of the reaction. As it is clear from Fig. 6, when the catalyst amount was increased from 0.1 to 0.3 g, the ethylene conversion was remarkably improved from 53.6% to 73.6%. Further increase in catalyst amount resulted in only a small improvement in the ethylene conversion. However, an increase in the catalyst amount from 0.1–0.5 g could only increase the value of selectivity from 82.2–83.9%. Thus, a catalyst amount of 0.3 g can be considered as an optimized catalyst amount under the presented reaction conditions.
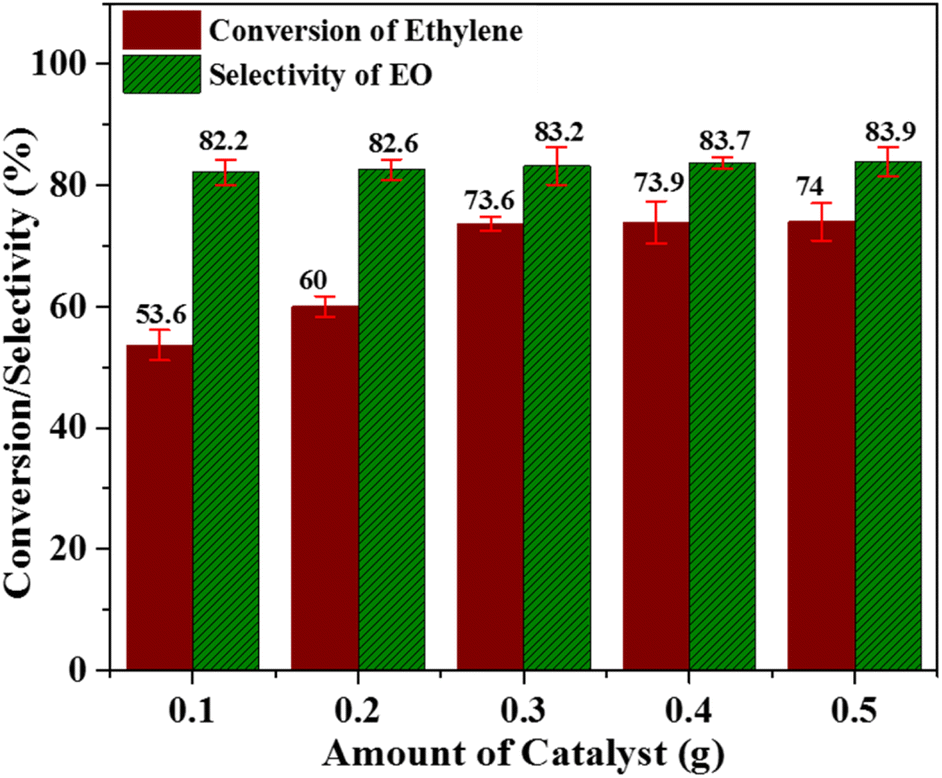 | ||
| Fig. 6 Effect of catalyst amount; [reaction conditions: ethylene = 3.28 mmol, (32%) PAA = 6.56 mmol, time = 6 h, solvent = C2H3N (20 mL), temperature = 60 °C]. | ||
:1, 1:2, 1:3, 1:6. As obvious from Fig. 7, the ethylene conversion has a value of 60.1% at the ethylene: PAA molar ratio of 1:1. This continuously increased with increasing amount of PAA and reached a maximum value of 80.6% at the molar ratio of 1:6. However, the selectivity towards EO production displayed the opposite trend i.e. it continuously decreased with increasing amount of PAA. This can be related with pronounced decomposition of PAA at higher concentration instead of its attachment with catalyst active sites. It can be inferred from these results that using excessive amount of the oxidant is not helpful in improving the catalyst activity. Therefore, ethylene to PAA molar ratio of 1:3 is the optimum molar ratio where both ethylene conversion and selectivity of EO production are having trade-off values.
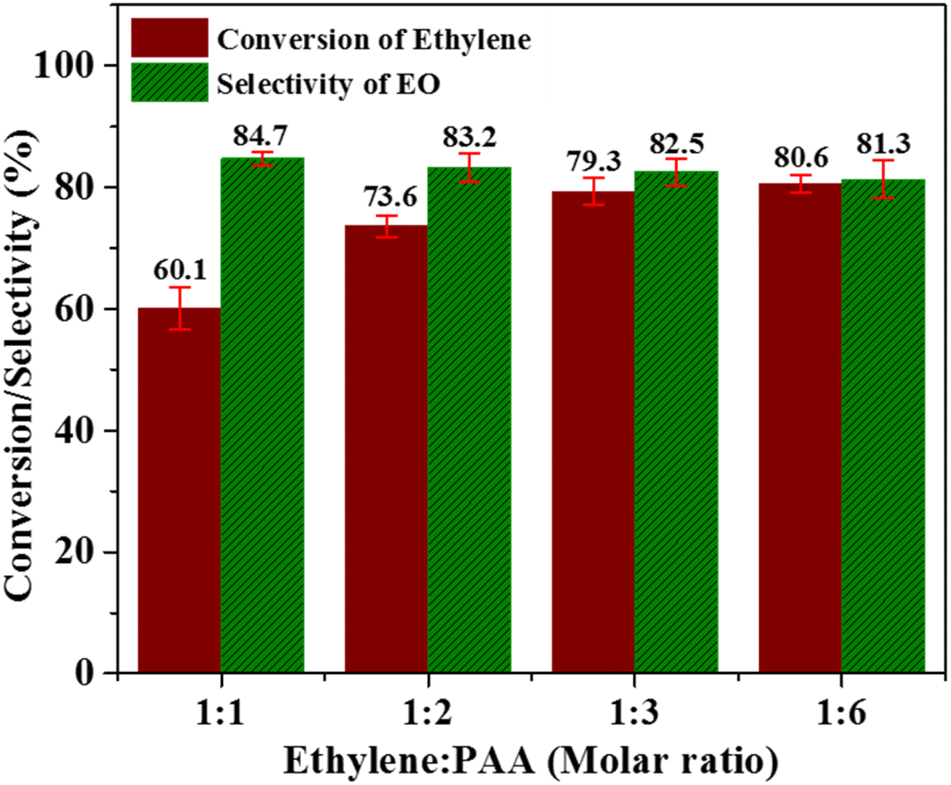 | ||
| Fig. 7 Effect of ethylene to PPA molar ratio; [reaction conditions: ethylene = 3.28 mmol, catalyst amount = 0.3 g, time = 6 h, solvent = C2H3N (20 mL), temperature = 60 °C]. | ||
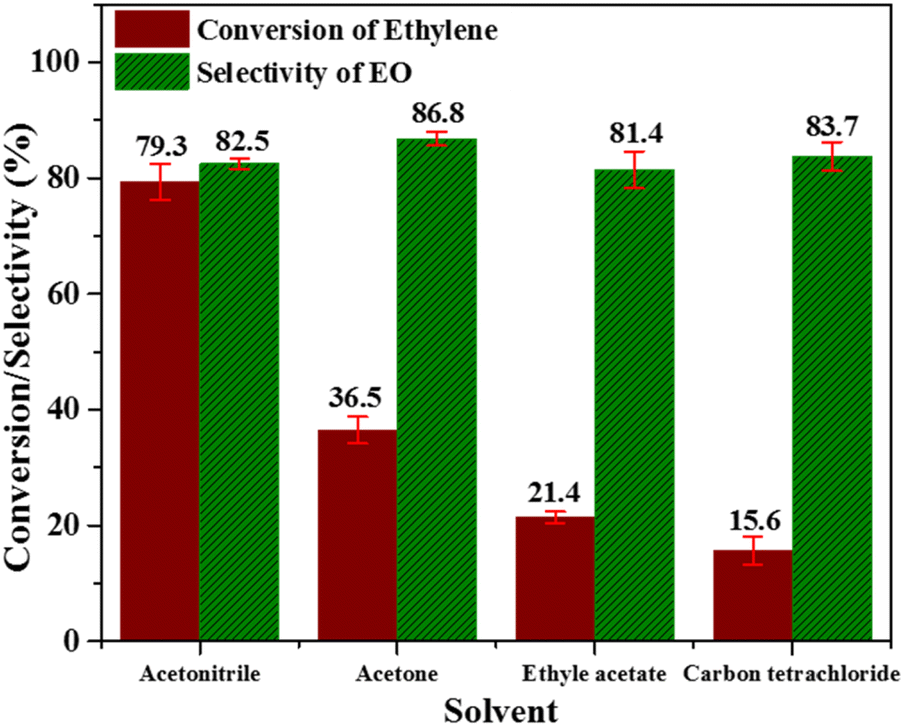 | ||
| Fig. 8 Effect of several solvents; [reaction conditions: ethylene = 3.28 mmol, (32%) PAA = 8.78 mmol, temperature = 60 °C, time = 6 h, catalyst = 0.3 g, solvent = C2H3N (20 mL)]. | ||
From all these studies, it can be concluded that the trade-off conditions for having optimum EO selectivity and ethylene conversion are; temperature (60 °C), catalyst amount (0.3 g), and ethylene:PAA molar ratio (1:3). Acetonitrile is the best solvent for EO formation in LPE using Nb/MSM catalyst along with PAA oxidant. To the best of our knowledge no other literature report is available where peracetic acid is used as an oxidant with Nb/MSM catalyst for EO formation.
We speculate that peracetic acid may contribute to hydrolysis of EO which can reduce the EO selectivity. In order to decrease opening of the epoxide ring, epoxidation should be carried out at a possible lower catalyst loading on the support because higher catalyst/acidity in the catalyst can result in increased decomposition of oxidant and more hydrolysis of EO ring.34 Also it is well reported that water that is produced in the process can increase Brønsted acid sites by blocking Lewis acid sites. These Brønsted acid sites result in increased hydrolysis of epoxide ring.32 However, it is difficult to manage the water, which is produced in the process. The investigations for reducing the hydrolysis of EO are currently under process.
Both the samples, collected at 6 hours and 12 hours, were analysed by GC technique to assess any further conversion of ethylene. The analysis revealed no further conversion of ethylene as shown in Fig. 9. This data demonstrates well that Nb/MSM heterocatalyst (when used with peracetic acid oxidant) is also very effective in alleviating the problem of leaching encountered in case of other metal based catalysts used for EO production.
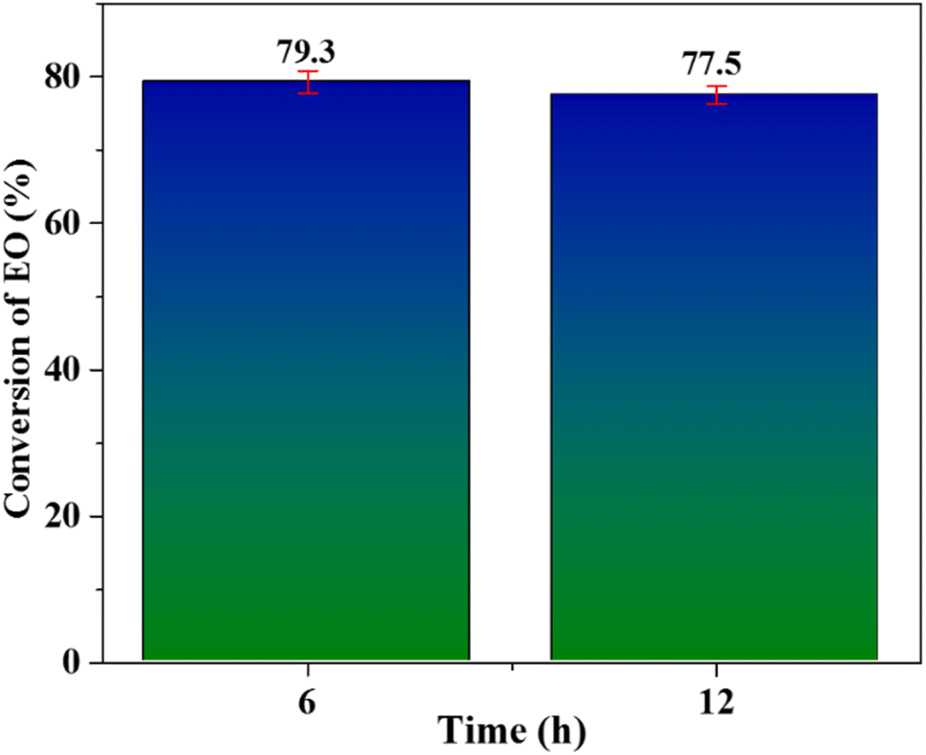 | ||
| Fig. 9 Leaching test for Nb/MSM catalyst for conversion of ethylene. | ||
This leaching resistance/no epoxidation due to some leached species (if any) can be explained according to reported literature.48 In the epoxidation process oligomerized metal oxide species are produced on the surface of the catalyst in the presence of oxidant. According to Das et al., these oligomerized metal oxide species cannot catalyze the epoxidation process.48 We assume that such oligomerized metal oxide can also be present in the supernatant solution which was collected for the leaching test. The investigation of the nature of the metal species (if present in supernatant) are outside the scope of the presented study, however, these investigations are being carried out separately.
5. Conclusions
An environmentally friendly production of EO was achieved by the preparation of Nb/MSM catalyst by liquid phase epoxidation of ethylene. To synthesize the catalyst, niobium oxide was supported on mesoporous silica. By using the XRD technique, the hetero atom structure in Nb/MSM was confirmed, and N2 adsorption isotherms were used to evaluate the mesoporous structure. Catalytic activity of Nb/MSM towards EO production was examined using liquid phase epoxidation of ethylene in the presence of peracetic acid as an oxidant. The 79.3% conversion of ethylene with 82.5% selectivity, towards ethylene oxide, was achieved by Nb/MSM metal catalyst. For this purpose, various reaction conditions were optimized including temperature (60 °C), catalyst dosage (0.3 g), and ethylene to PAA molar ratio (1:3), and reaction time (6 h) using acetonitrile as an organic solvent. These investigations and obtained results confirmed that the prepared Nb/MSM is a potential catalyst for an efficient EO production.
Abbreviations
| EO | Ethylene oxide |
| CO2 | Carbon dioxide |
| GPE | Gas phase epoxidation |
| LPE | Liquid phase epoxidation |
| MTO | Methyltrioxorhenium |
| H2O2 | Hydrogen peroxide |
| Re | Rhenium |
| MSM | Mesoporous silica material |
| MCM-41 | Mobile composition of matter no. 1 |
| SBA-15 | Santa Barbara amorphous-15 |
| Nb | Niobium |
| W | Tungsten |
| PAA | Peracetic acid |
| NbCl5 | Niobium(V) chloride |
| SiO2 | Mesoporous silicate |
| TEOS | Tetraethylorthosilicate |
| EG | Ethylene glycol |
| AA | Acetic acid |
| XRD | X-ray diffraction |
| BET | Brunauer–Emmett–Teller |
| TEM | Transmission electron microscope |
| GC | Gas chromatography |
| P123 | Pluronic |
| ACN | Acetonitrile (C2H3N) |
| AA | CH3COOH |
| EO | C2H4O |
| MEG | C2H6O2 |
| DEG | C6H10O4 |
| PAA | CH3COOOH |
| BJH | Barrett–Joyner–Halenda |
| SBET | Surface area |
| VBJH | Cumulative pore volume |
| DBJH | Mesoporous diameter |
| RT | Retention time |
Author contributions
Conceptualization, Toheed Akhter, Asif Mahmood; methodology, Muhammad Maqbool and Asif Mahmood; validation, Toheed Akhter and Asif Mahmood; formal analysis, Muhammad Maqbool and Toheed Akhter; investigation, Muhammad Maqbool; resources, Sohail Nadeem, Muhammad Faheem and Chan Ho Park; writing—original draft preparation, Toheed Akhter and Muhammad Maqbool; writing—review and editing, Toheed Akhter and Chan Ho Park; supervision, Toheed Akhter and Asif Mahmood; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- V. Dal Santo, M. Guidotti, R. Psaro, L. Marchese, F. Carniato and C. Bisio, Proc. R. Soc. A, 2012, 468, 1904–1926 CrossRef CAS.
- L. Pinaeva and A. Noskov, Pet. Chem., 2020, 60, 1191–1206 CrossRef CAS.
- V. K. Aggarwal, R. S. Grainger, G. K. Newton, P. L. Spargo, A. D. Hobson and H. Adams, Org. Biomol. Chem., 2003, 1, 1884–1893 RSC.
- H.-J. Lee, M. Ghanta, D. H. Busch and B. Subramaniam, Chem. Eng. Sci., 2010, 65, 128–134 CrossRef CAS.
- M. Ghanta, T. Ruddy, D. Fahey, D. Busch and B. Subramaniam, Ind. Eng. Chem. Res., 2013, 52, 18–29 CrossRef CAS.
- D. M. Minahan and G. B. Hoflund, J. Catal., 1996, 158, 109–115 CrossRef CAS.
- F. Carniato, C. Bisio, E. Boccaleri, M. Guidotti, E. Gavrilova and L. Marchese, Eur. J. Chem., 2008, 14, 8098–8101 CrossRef CAS PubMed.
- M. Selvaraj, D.-W. Park, I. Kim, S. Kawi and C. Ha, Dalton Trans., 2012, 41, 9633–9638 RSC.
- M. Selvaraj, M. Kandaswamy, D. Park and C. Ha, Catalysis, 2010, 158, 286–295 CAS.
- M. Selvaraj and P. Sinha, New J. Chem., 2010, 34, 1921–1929 RSC.
- M. Selvaraj, M. Kandaswamy, D. Park and C. Ha, Catalysis, 2010, 158, 377–384 CAS.
- R. A. Sheldon, M. Wallau, I. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., 2003, 42, 156–181 CrossRef PubMed.
- J. M. Thomas, Chem. Phys., 2008, 128, 182502 Search PubMed.
- D. J. Xuereb and R. Raja, Catal. Sci. Technol., 2011, 1, 517–534 RSC.
- V. Dal Santo, M. Guidotti, R. Psaro, L. Marchese, F. Carniato and C. Bisio, Proc. R. Soc. A, 2012, 468, 1904–1926 CrossRef CAS.
- Y. Liu, K. Murata and M. Inaba, Chem. Lett., 2003, 32, 992–993 CrossRef CAS.
- F. Somma, P. Canton and G. Strukul, J. Catal., 2005, 229, 490–498 CrossRef CAS.
- M. Trejda, A. Tuel, J. Kujawa, B. Kilos and M. Ziolek, Microporous Mesoporous Mater., 2008, 110, 271–278 CrossRef CAS.
- J. V. Coelho, L. C. Oliveira, F. C. Moura, P. P. de Souza, C. A. Silva, K. B. Batista and M. J. da Silva, Appl. Catal., A, 2012, 419, 215–220 CrossRef.
- A. Feliczak-Guzik, A. Wawrzyńczak and I. Nowak, Microporous Mesoporous Mater., 2015, 202, 80–89 CrossRef CAS.
- P. Chagas, H. S. Oliveira, R. Mambrini, M. Le Hyaric, M. V. de Almeida and L. C. Oliveira, Appl. Catal., A, 2013, 454, 88–92 CrossRef CAS.
- S. L. Barbosa, G. R. Hurtado, S. I. Klein, V. L. Junior, M. J. Dabdoub and C. F. Guimaraes, Appl. Catal., A, 2008, 338, 9–13 CrossRef CAS.
- V. S. Braga, I. C. Barros, F. A. Garcia, S. C. Dias and J. A. Dias, Catalysis, 2008, 133, 106–112 Search PubMed.
- A. Feliczak-Guzik and I. Nowak, Catalysis, 2009, 142, 288–292 CAS.
- I. Nowak, A. Feliczak, I. Nekoksová and J. Čejka, Appl. Catal., A, 2007, 321, 40–48 CrossRef CAS.
- F. Quignard, A. Choplin and R. Teissier, J. Mol. Catal. A: Chem., 1997, 120, L27–L31 CrossRef CAS.
- E. Gianotti, M. E. Raimondi, L. Marchese, G. Martra, T. Maschmeyer, J. M. Seddon and S. Coluccia, Catal. Lett., 2001, 76, 21–26 CrossRef CAS.
- K. Kang and W. Ahn, J. Mol. Catal. A: Chem., 2000, 159, 403–410 CrossRef CAS.
- W. Adam, W. Malisch, K. J. Roschmann, C. R. Saha-Möller and W. A. Schenk, J. Organomet. Chem., 2002, 661, 3–16 CrossRef CAS.
- A. Held and P. Florczak, Catalysis, 2009, 142, 329–334 CAS.
- W. Yan, A. Ramanathan, M. Ghanta and B. Subramaniam, Catal. Sci. Technol., 2014, 4, 4433–4439 RSC.
- J. Zhang, Z. Xin, X. Meng, Y. Lv and M. Tao, Fuel, 2014, 116, 25–33 CrossRef CAS.
- W. Yan, A. Ramanathan, P. D. Patel, S. K. Maiti, B. B. Laird, W. H. Thompson and B. Subramaniam, J. Catal., 2016, 336, 75–84 CrossRef CAS.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2003, 125, 4034–4035 CrossRef CAS PubMed.
- S. Linic and P. Christopher, ChemCatChem, 2010, 2, 1061–1063 CrossRef CAS.
- X. Zhao, S. Veintemillas-Verdaguer, O. Bomati-Miguel, M. Morales and H. Xu, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 024106 CrossRef.
- D. R. Das and A. K. Talukdar, ChemistrySelect, 2017, 2, 8983–8989 CrossRef CAS.
- a. C. Kresge, M. Leonowicz, W. J. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. Kresge, K. Schmitt, C. Chu, D. H. Olson, E. Sheppard and S. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- G. Somorjai and R. Rioux, Catal. Today, 2005, 100, 201–215 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- B. Kalita, P. Phukan and A. K. Talukdar, Catal. Sci. Technol., 2012, 2, 2341–2350 RSC.
- X. Lu, W.-J. Zhou, Y. Guan, A. Liebens and P. Wu, Catal. Sci. Technol., 2017, 7, 2624–2631 RSC.
- S. Narayanan, J. J. Vijaya, S. Sivasanker, C. Ragupathi, T. Sankaranarayanan and L. J. Kennedy, J. Porous Mater., 2016, 23, 741–752 CrossRef CAS.
- J. Liu, Z. Wang, P. Jian and R. Jian, J. Colloid Interface Sci., 2018, 517, 144–154 CrossRef CAS PubMed.
- W. Zhan, Y. Guo, Y. Wang, X. Liu, Y. Guo, Y. Wang, Z. Zhang and G. Lu, J. Phys. Chem. B, 2007, 111, 12103–12110 CrossRef CAS PubMed.
- D. R. Das, P. Kalita and A. K. Talukdar, J. Porous Mater., 2020, 27, 893–903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07240h |
| This journal is © The Royal Society of Chemistry 2023 |